[level-membership-for-critical-care-medicine-category]
129 Pathophysiology of Sepsis and Multiple Organ Dysfunction
 Pathophysiology of Sepsis
Pathophysiology of Sepsis
Sepsis has been defined as an invasion of microorganisms or their toxins into the bloodstream, together with the host response to this invasion.1 Thus, the pathophysiology of sepsis combines the impact of infection with the host response of generalized inflammation, which finally leads to multiorgan dysfunction and death. This definition has been extended by the addition of several terms to more carefully describe the disease and its pathophysiology (Table 129-1). The American College of Chest Physicians/Society of Critical Care Medicine (ACCP/SCCM) Consensus Conference defined sepsis as a systemic inflammatory response syndrome (SIRS) caused by infection.2 More recently it has been recognized that SIRS is counteracted by a hypoinflammatory state that also plays an important role in the further development of organ dysfunction.3
| Term | Definition |
|---|---|
| Bacteremia | Presence of viable bacteria in the blood |
| Systemic inflammatory response syndrome (SIRS) | Generalized hyperinflammatory response to several impacts |
| Sepsis | SIRS caused by infection |
| Severe sepsis | Sepsis associated with organ dysfunction |
| Septic shock | Sepsis associated with arterial hypotension |
Data from ACCP/SCCM Consensus Conference Committee. Definition for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. Crit Care Med 1992;20:864–74.
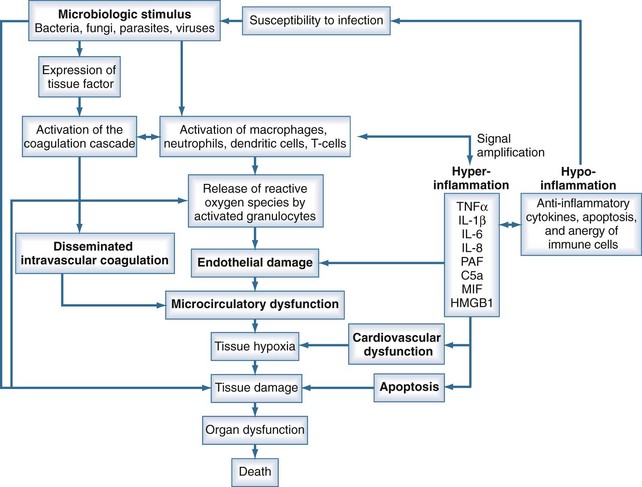
Sepsis is characterized by loss of hemostatic balance and endothelial dysfunction, which in turn severely compromise the cardiocirculatory system as well as intracellular homeostasis. Cellular hypoxia and apoptosis (programmed cell death) then contribute to organ dysfunction and death. The network of organ systems affected by sepsis is depicted in Figure 129-1.
Microbiological Stimulus
By definition, infection is a fundamental part of the pathophysiology of sepsis. Any microorganism able to induce infection in humans may be complicated by sepsis. Bacteria as well as fungi, parasites, and to a lesser degree viruses can trigger the mechanisms that lead to sepsis. Although SIRS is the final common pathway of this process, the signal transduction pathway from infection to complex host response differs with the microbiological stimuli. Induction of an innate immune response is triggered by specific microbial molecules (e.g., bacterial wall components, exotoxins, bacterial DNA, viral RNA) called pathogen-associated molecular patterns (PAMPs). Damage-associated molecular patterns (DAMPs) are the noninfectious equivalents to PAMPs. DAMPs are released after cellular injury of the host (i.e., trauma) and can also induce the innate immune response.4
The presence of PAMPs is sensed by recognition molecules called pattern-recognition proteins (PRR), which are able to initiate a host response. These proteins may be categorized into secreted, transmembrane, and cytosolic PRRs. The Toll-like receptors (TLRs) represent the membrane PRRs. Eleven different TLRs have been discovered in mammals, whereas TLR-11 is not expressed in humans (Table 129-2). Retinoic acid-inducible gene I (RIG-I)–like receptors (RLRs) and the nucleotide-binding domain and leucine-rich repeat-containing receptors (NLRs) are cytosolic PRRs. RLRs recognize viral RNA and some double-stranded DNA. NLRs represent a large family of intracellular sensors that can detect pathogens and stress signals. NLRs detect microbiological products such as peptidoglycans and other degradation products of microorganisms as well as stress-related substances.5,6
TABLE 129-2 Human Toll-Like Receptors and Their Natural Ligands
| TLR Type | Related Pathogen-Associated Molecular Pattern | Location |
|---|---|---|
| TLR1 (via TLR2) | Bacterial products such as tri-acyl lipopeptides | Cell surface |
| TLR2 | Gram-positive bacterial products, including peptidoglycans; some virus-related proteins | Cell surface |
| TLR3 | Viral double-stranded RNA | Endosomal |
| TLR4 | Endotoxin, other bacterial products, some fungal products | Cell surface |
| TLR5 | Flagellin | Cell surface |
| TLR6 (via TLR2) | Some bacterial products | Cell surface |
| TLR7 | Single-stranded RNA | Endosomal |
| TLR8 | Viral single-stranded RNA | Endosomal |
| TLR9 | Viral and bacterial DNA | Endosomal |
| TLR10 | Unknown |
TLR, Toll-like receptor.
Gram-Negative Sepsis
In gram-negative bacteremia, initiation of the immune response is mediated primarily by lipopolysaccharide (LPS), a bacterial cell wall product. In plasma, LPS is bound to the LPS binding protein (LBP). Bound LPS is transported to the opsonic receptor, CD14, which is located on several cell membranes including on monocytes.7 A soluble form of CD14 interacts with CD14-negative cells (e.g., dendritic cells). However, CD14 alone cannot explain the actions of LPS, because CD14 does not have an intracellular tail.
Another binding site of LPS is the transmembranous receptor, TLR4, which exists in combination with the accessory protein, MD2.8 The binding of LPS to CD14 and TLR4 induces, via other molecules, activation of the transcription factor, nuclear factor kappa-B (NF-κB). Activated NF-κB migrates into the nucleus where it binds to and activates gene promoters, resulting in the transcription and expression of genes for cytokines and other proinflammatory mediators.9 In monocytes, LPS also induces cytokine transcription via the triggering receptor expressed on myeloid cells-1 and the myeloid DAP12-associated lectin.10 Intracellular pattern-recognition proteins in monocytes for LPS have recently been identified as another pathway of cytokine expression and include nucleotide-binding oligomerization domain 1 and 2 as LPS binding sites.11
Gram-Positive Sepsis
During the last decade, gram-positive bacteria have gained greater importance as causative organisms for sepsis.12 Gram-positive bacteria lack endotoxin and are recognized by cell wall components such as peptidoglycans and released bacterial toxins (exotoxins). Recently, lipoteichoic acid (LTA), a component of the cell wall in all gram-positive bacteria, has been recognized as the main pattern for recognition of gram-positive bacteria.13 TLR2 has been identified as the only pattern-recognition protein for gram-positive bacteria.14 The relationship between LTA and TLR2 is not completely clarified. Although LTA clearly interacts with TLR2, TLR2 is not a specific receptor for LTA, because TLR2 can recognize several other components of gram-positive bacteria.15 Gram-positive and gram-negative sepsis are indistinguishable clinically, suggesting a similar pathway of signal transduction. Indeed, peptidoglycans and LTA stimulate the release of tumor necrosis factor alpha (TNF-α), interleukin (IL)-6, and IL-10. It has been speculated that CD14 is also involved in the signaling of gram-positive infections.
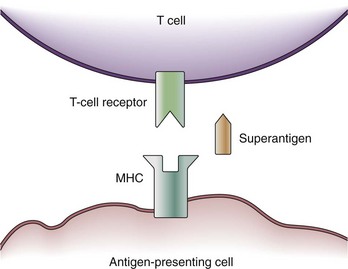
Some exotoxins cause a special type of septic shock called the toxic shock syndrome (TSS). TSS may be caused by the exotoxin, TSS toxin-1, staphylococcal enterotoxins from Staphylococcus aureus, or streptococcal pyogenic exotoxins.16 These toxins are capable of acting as so-called superantigens which deploy their effects via the T-cell antigen receptor (TCR). The TCR consists of five variable elements: Vβ, Dβ, Jβ, Vα, and Jα. Conventionally, the T cell is activated if the major histocompatibility complex (MHC) of an antigen-presenting cell matches all five elements. Thus, T cells are activated by proper antigen contact only. This results in the stimulation of about 1 in 10,000 T cells. However, a superantigen such as TSS toxin-1 works as a bridge between the MHC and the Vβ chain of the TCR only (Figure 129-2).17 Because T-cell activation now occurs independently of a match between the MHC and TCR, about 20% of the entire T-cell pool may be activated at once. Besides further T-cell proliferation, T-cell activation causes the release of several cytokines (i.e., interferon gamma [IFN-γ], IL-2, TNF-α) from T cells, as well as IL-1β and TNF-α from macrophages. Thus, the presence of superantigens results in a release of cytokines similar to gram-negative sepsis. It is assumed that actions other than cytokine production may be responsible for actions of superantigens in TSS; for example, superantigens may amplify the effects of LPS.
Other Microbiological Stimuli of Sepsis
Sepsis can also be induced by fungi, viruses, and parasites. Signal transduction by nonbacterial products, however, is not as well characterized as bacterial sepsis. In part, this may be due to the fact that induction of cytokine release differs markedly not only among different microorganisms but also among species. Nevertheless, the release of proinflammatory mediators has been demonstrated during infections with nonbacterial infections such as Candida albicans18 and Plasmodium falciparum.19 The signal transduction in viral infections is complicated by the fact that viruses can interfere with TNF-related cytokine release to avoid the host’s antiviral activities.20 Pattern recognition of viruses occurs mainly via endosomal TLR receptors which detect single- and double-stranded RNA or DNA (see Table 129-2).
The Immune Response in Sepsis
The cytokines TNF-α and IL-1β are released by activated macrophages and CD4 T cells within the first hour after infection. These primary mediators induce the release of several secondary mediators that amplify inflammation (Table 129-3). An important step in signal amplification is activation of the complement system. Besides being activated by antigen-antibody complexes, the complement system may be stimulated by bacterial surface sugars and endotoxin. The complement fragment C5a, a cleavage product of the complement cascade, is a strong chemoattractant. C5a appears about 2 hours after the initiation of sepsis and stimulates macrophages to further produce proinflammatory mediators. Another mediator that amplifies the immune response is macrophage migration inhibitory factor (MIF), which is produced by T cells, macrophages, monocytes, and pituitary cells in response to an infectious stimulus. MIF appears about 8 hours after the onset of sepsis and activates T cells and macrophages to produce proinflammatory mediators. About 24 hours after the initiation of sepsis, levels of high-mobility group box 1 (HMGB1) protein increase and appear to play a role in endotoxin-related sepsis. HMGB1 is a nuclear binding protein that, among other things, is capable of activating NF-κB. As a rather late mediator in sepsis, it is produced by macrophages and neutrophils and stimulates phagocytic cells.21
TABLE 129-3 Macrophage Mediators Involved in the Pathogenesis of Sepsis
| Mediator | Typical Effects |
|---|---|
| Cytokines: IL-1, IL-6, IL-12, IL-15, IL-18, TNF, MIF, HMGB1, IL-10 | Activate neutrophils, lymphocytes, and vascular endothelium; up-regulate cellular adhesion molecules; induce prostaglandins, nitric oxide synthase, and acute-phase proteins; induce fever |
| IL-10 is predominantly a negative regulator of these effects. | |
| Chemokines: IL-8, MIP-1α, MIP-1β, MCP-1, MCP-3 | Mobilize and activate inflammatory cells, especially neutrophils; activate macrophages |
| Lipid mediators: platelet-activating factor, prostaglandins, leukotrienes, thromboxane, tissue factor | Activate vascular endothelium; regulate vascular tone; activate extrinsic coagulation cascade |
| Oxygen radicals: superoxide and hydroxyl radicals, nitric oxide | Antimicrobial properties; regulation of vascular tone |
HMGB, high-mobility group B protein; IL, interleukin; MCP, monocyte chemoattractant protein; MIF, migration inhibitory factor; MIP, macrophage inflammatory protein; TNF, tumor necrosis factor.
Data from Cohen J. The immunopathogenesis of sepsis. Nature 2002;420:885–91.
It is not completely understood why inflammation becomes generalized in some patients but stays localized in others. Genetic variants of cytokines may play a role in this issue. Single nucleotide polymorphisms (SNPs) are single-base changes in the DNA which do not cause obvious changes in the function of the respective cytokine. However, SNPs in some cytokines are associated with a worse outcome from septic shock or an increased risk for developing sepsis.22–24 Among several others, such variants have been described in TNF-α, IL-6, and CD14.25 However, results from these studies are difficult to interpret because of contradictory results and differences in populations of different ethnicities.
The immune response in sepsis does not involve only proinflammatory mediators. As in many other physiologic processes, the organism produces inhibitors to control certain reactions. Proinflammatory mediators are counteracted by antiinflammatory molecules such as IL-4 and IL-10 because CD4 T cells can switch from the production of inflammatory cytokines (type 1 helper T cells [TH1]) to the production of antiinflammatory cytokines (type 2 helper T cells [TH2]). Soluble TNF receptors and IL-1 receptor antagonists (IL-1Ra) are released to inhibit the actions of TNF and IL-1 in their roles as primary mediators of sepsis. T cells, neutrophils, and macrophages also may become unresponsive to infectious stimuli (anergy).26 Another mechanism of the antiinflammatory response is the onset of apoptosis, a genetically programmed autodestructive release of proteases that induces cell death. In sepsis, enhanced apoptosis causes loss of immune effector cells, including CD4 and CD8 T cells, B cells, and dendritic cells.27 Absolute lymphocyte counts are significantly decreased in patients with sepsis.28 Further, apoptotic cells impair the function of surviving immune cells.29
Results from animal studies suggest that the autonomic nervous system is also involved in suppression of cytokine release during sepsis. In the experimental setting, vagal stimulation can inhibit TNF expression. It is hypothesized that an inflammatory reflex, with the afferent vagal nerve sensing cytokine release and an efferent immunosuppressing cholinergic arm, exists.30 The importance of such a reflex in humans merits further investigation.
The antiinflammatory response in sepsis has been termed the compensatory antiinflammatory response syndrome (CARS).3 It has been suggested that the first response to infection is hyperinflammation, which is followed by a hypoimmune state. From there, recovery would be possible, but the prolonged inability to eradicate microorganisms might result in the death of the patient.31 However, serum levels of antiinflammatory cytokines are increased in parallel with the increase of proinflammatory mediators.32,33 Thus, diminished inflammation develops at the same time as the process of hyperinflammation. Although the persistence of high levels of antiinflammatory mediators may contribute to mortality in septic patients, the clinical role of change between hyperinflammatory and hypoinflammatory states remains unclear.
Loss of Hemostatic Balance
Under normal conditions, the vascular luminal surface has anticoagulant properties. Tissue factor is a 4.5-kD protein that is bound to cell membranes which are normally not in contact with blood. Expression of tissue factor mainly depends on release of IL-6.34,35 Tissue factor expression occurs on mononuclear cells, but endothelial cells, polymorphonuclear cells, and other cell types may be additional sources. The expression of tissue factor induces intravascular thrombin formation initiated by the extrinsic coagulation pathway. Because this process is not restricted to a local area, it is called disseminated intravascular coagulation (DIC); DIC causes a consumption of coagulation factors.
Physiologically, excessive coagulation is counteracted by several natural anticoagulants including antithrombin, the thrombomodulin/protein C/protein S system, and tissue factor pathway inhibitor. In addition to the activation of tissue factor-dependent thrombin generation, anticoagulant function is attenuated in sepsis. Patients with sepsis demonstrate reduced levels of protein C and antithrombin due to consumption and reduced synthesis.36 Thus, the physiologic balance between procoagulant and anticoagulant substances is altered in sepsis as there is a shift of the hemostatic balance toward a procoagulant state (Figure 129-3).
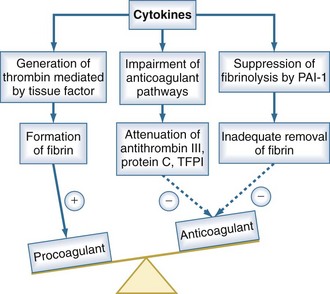
Besides its anticoagulant actions, the protein C pathway is an important link between coagulation and inflammation, because activated protein C has antiinflammatory properties. Protein S can bind to receptors that mediate an antiinflammatory regulatory loop of dendritic cell and monocyte inflammatory function. Thrombomodulin has been described to prevent excessive complement activation.37 Thus, there is considerable cross-talk between inflammation and coagulation which is impaired in sepsis as a result of depletion of the thrombomodulin/protein C/protein S system. There is also evidence that antithrombin blunts activation of several cytokines, suggesting that low antithrombin levels also affect the relationship between coagulation and inflammation.38
Endothelial Dysfunction
The endothelium produces several vasoactive mediators, including nitric oxide (NO), prostacyclins, and endothelin. NO is a potent vasodilator produced by NO synthase (NOS) from the amino acid, L-arginine. NO directly relaxes the vessel’s smooth muscle. There are two different forms of endothelial NOS: the constitutional form (cNOS) and the inducible form (iNOS). Physiologically, cNOS—also referred to as endothelial NOS (eNOS)—produces only small amounts of NO, and iNOS is expressed at low levels.39 In sepsis, iNOS expression is stimulated by cytokines such as IL-1β and TNF-α.40 This is followed by massive NO production and profound vasodilatation. Whether increased activity of cNOS also plays a role in sepsis is currently a matter of debate.
During inflammation, endothelial cells express adhesion molecules on their surface, which causes the adherence of leukocytes. These adhesion molecules include endothelial leukocyte adhesion molecule-1, intracellular adhesion molecule-1, and vascular cell adhesion molecule-1. Endothelial leukocyte adhesion molecule-1 is a selectin that mediates the initial step of leukocyte adhesion, followed by leukocyte rolling along the endothelial surface. The leukocyte finally migrates through the endothelial layer into the tissue, mediated by intracellular adhesion molecule-1 and vascular cell adhesion molecule-1 expression on both endothelial cells and leukocytes.41
Migration of leukocytes into the tissue is a physiologic mechanism to move immune cells to the site of infection. However, in generalized inflammation such as in sepsis, endothelial cells in several organs remote from the site of infection express adhesion molecules, inducing a generalized rolling and sticking of circulating leukocytes to the vascular surface. Adherence to endothelial cells activates leukocytes and induces a respiratory burst.42 The respiratory burst involves the release of cytotoxic substances such as elastase, myeloperoxidase, and reactive oxygen species. These products are capable of damaging endothelial cells and the surrounding tissue. Endothelial cell damage causes capillary leakage whereby intravascular fluid penetrates the extracellular space, leading to tissue edema.
Cardiac and Circulatory Dysfunction
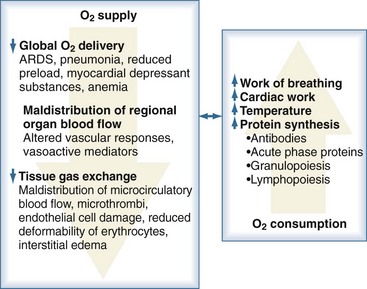
Sepsis is frequently complicated by organ dysfunction and shock. Shock occurs when the cardiovascular system is unable to transport sufficient amounts of oxygen to the tissues. In fact, sepsis compromises all levels of the cardiovascular system, resulting in cardiac dysfunction, vascular dysregulation, and microcirculatory damage. Impairment of the cardiovascular system causes a characteristic hemodynamic pattern that in cases of adequate fluid loading and the absence of severe preexisting cardiac dysfunction, consists of a high cardiac output, arterial hypotension, and low systemic oxygen (O2) extraction. In early sepsis, O2 consumption is increased owing to higher metabolic needs (i.e., tachypnea, fever, increased cardiac work, increased rate of protein synthesis), further compromising the relationship between O2 supply and demand (Figure 129-4). The hepatic and splanchnic region is markedly affected by these changes associated with sepsis. Hepatosplanchnic O2 uptake increases markedly during fever and bacteremia.43
Cardiac Dysfunction
In experimental septic shock, myocardial contractility is compromised shortly after the induction of sepsis.44 This finding is confirmed in septic patients when a reduced ejection fraction is observed by echocardiography, especially in patients with elevated troponin levels.45 The drop in myocardial contractility is accompanied by diastolic dilatation of the left ventricle, which causes the left ventricular end-diastolic volume to rise. This mechanism allows the heart to maintain a sufficient stroke volume despite impaired contractility. Clinically, a rightward shift of the Frank-Starling curve occurs. Thus, compared with healthy humans, patients with sepsis require greater cardiac filling pressures to maintain a similar stroke volume.46 Septic patients without compensatory left ventricular dilatation have a significantly greater risk of death.47 Cardiac dysfunction is reversible if the patient recovers from sepsis.
The presence of myocardial depressant substances was initially proposed in the 1980s because the serum of septic patients was able to suppress the contractility of rat myocytes in vitro.48,49 Cytokines induce increased activation of iNOS, with subsequent enhanced NO production. NO affects myocytes in several ways: NO stimulates guanylate cyclase, and its product, 3′,5′-cyclic guanosine monophosphate, interferes with intracellular myocardial calcium metabolism. This includes a reduction in calcium’s affinity to the contractile apparatus and inhibition of the α-adrenergic-mediated increase in the slow inward calcium current. NO may directly damage myocardial cells by the formation of peroxynitrite via combination with superoxide ions. Peroxynitrite deploys toxic effects on many intracellular molecules by means of oxidation.50
Sepsis is associated with alterations of regional and microregional blood flow, which results in a mismatch between regional O2 supply and demand and, subsequently, multiple organ dysfunction. It was therefore hypothesized that the heart shares in this type of injury. Although there were hints from experimental work that the coronary circulatory reserve is altered in sepsis,51,52 clinical studies did not show a compromised coronary blood flow.53,54 However, more recently it was demonstrated that patients with sepsis show elevated serum levels of troponin.55 Elevated troponin values are associated with a higher incidence of regional wall motion abnormalities and death.
Vascular Dysfunction and Hypovolemia
In cardiogenic or hypovolemic shock, vasoconstriction is a common mechanism to avoid arterial hypotension. In sepsis, however, profound arterial vasodilatation occurs. Endothelial cells play an important role in the regulation of vascular tone because they release several vasoactive substances such as NO and endothelin. Sepsis shifts the balance of these substances toward a vasodilatory state by uncontrolled NO production, as discussed earlier. Severe arterial hypotension due to profound systemic vasodilatation is one of the characteristic hemodynamic features of sepsis. The mechanism by which NO induces vasodilatation is complex. Important pathways in which NO participates include the activation of potassium channels and hyperpolarization of the plasma membrane of smooth muscle cells. These mechanisms in turn inhibit the actions of vasopressors such as norepinephrine and angiotensin II, so that vasoconstriction does not occur despite high serum concentrations of these substances.56
Endothelial cells regulate vascular tone not only to maintain systemic blood pressure but also to control blood flow to single organs. Several mechanisms to preserve organ blood flow are impaired in sepsis. For example, there is a loss of coupling between the hepatic artery flow and the portal blood flow in endotoxic shock.50 Similarly, the coronary circulatory reserve necessary to quickly adjust myocardial O2 supply based on changes in myocardial O2 requirements is reduced in sepsis.57 The autoregulation of perfusion of the intestinal mucosa is also depressed in experimental models of sepsis.58
Hypovolemia is another characteristic of sepsis. Sepsis is accompanied by the development of significant tissue edema. The underlying mechanism is capillary leakage, which is another effect of endothelial damage.59 This leakage also allows for the extravasation of albumin,60 which reduces the intravascular oncotic pressure. Under conditions of capillary leakage, the Starling forces cannot counteract the development of tissue edema or reduce existing edema. Because endothelial damage affects all parts of the capillary network throughout the body, large amounts of intravascular fluid are shifted into the extravascular space.
Microcirculatory Dysfunction
Severe sepsis and septic shock may be associated with high lactate levels and metabolic acidosis despite a low systemic O2 extraction. These signs of tissue hypoxia, which may be observed despite adequate fluid resuscitation, are interpreted as microcirculatory failure. Some parts of the microcirculation are extremely sensitive to physiologic stress, including hypoxia or ischemia. These areas are referred to as weak microcirculatory units.61,62
An increase in the number of weak microcirculatory units—and therefore increased microcirculatory shunting—is thought to play a major role in the O2 extraction deficit in sepsis. This hypothesis has been confirmed in experimental sepsis. By using intravital microscopy, an increased number of capillaries with lack of flow was observed in septic animals.63 More recently, capillary red blood cell oxygenation was measured in vivo by a spectrophotometric functional imaging system.64 In the presence of sepsis, an increased proportion of perfused capillaries showed very high red blood cell velocities. These high-flow capillaries were interpreted as microcirculatory shunts. The remaining capillaries with normal flow had a fivefold increase in O2 extraction. However, this increase in O2 extraction was insufficient to maintain O2 supply to all regions, given the number of capillaries without flow.
Several factors may be responsible for microcirculatory shunting in sepsis. The underlying mechanism is the hindrance of blood flow by microvascular obstruction, which has several causes: (1) the onset of intravascular coagulation due to a shift to a procoagulant state causes the development of microthrombi; (2) activation and damage of endothelial cells lead to endothelial cell swelling, narrowing the capillary lumen; (3) activated leukocytes hinder red blood cell flow by rolling and sticking to endothelial cells; and (4) red blood cells have reduced deformability in sepsis, which causes them to be captured in capillaries.65
Although there is good evidence from experimental work supporting the hypothesis of microcirculatory dysfunction in sepsis, there is considerable debate whether the O2 extraction deficit is due to a derangement of intracellular metabolic pathways rather than to microcirculatory dysfunction.63 Some capillaries are able to increase their O2 extraction,66 which argues against this hypothesis. However, the assessment of tissue oxygenation on the cellular level is problematic, even in the experimental setting. Currently it is thought that microcirculatory dysfunction shares in the development of tissue hypoxia in sepsis. Clinically this hypothesis is supported by the finding that early resuscitation guided by central venous O2 saturation improves survival in these patients.67
Endocrine Dysfunction
As depicted in Table 129-4, critical illness is associated with alterations in several endocrine functions. It is not clear whether these changes represent a physiologic response to critical illness or reflect a complex picture of endocrine dysfunction that needs diagnostic and treatment strategies. In sepsis, adrenal insufficiency and vasopressin deficiency might contribute to the loss of vasomotor control. However, current studies do not support treatment of endocrine dysfunction in severe sepsis or septic shock.
TABLE 129-4 Changes in Hormone Concentrations in Critically Ill Patients
| Hormone | Acute Critical Illness | Prolonged Critical Illness |
|---|---|---|
| Catecholamines | ++ | + |
| Cortisol | ++ | + |
| Adrenocorticotropic hormone | Ø + | Ø − |
| Growth hormone | Ø − | − |
| Thyroid hormones | Ø − | − |
| Thyroid-stimulating hormone | Ø − | − |
| Androgen hormones | − | − |
| Prolactin | – | Unknown |
Data from Ligtenberg JJ, Girbes AR, Beentjes JA et al. Hormones in the critically ill patient: to intervene or not to intervene? Intensive Care Med 2001;27:1567–77.
Adrenal Insufficiency
Adrenal corticosteroids are involved in several physiologic pathways in the human body, including maintenance of vascular tone, vascular permeability, and distribution of total body water. In the clinical setting, corticosteroids also augment the effects of vasopressors.68 Under normal conditions, corticosteroids are secreted by the adrenal cortex in a diurnal pattern. Corticosteroid secretion is tightly controlled by a feedback mechanism involving the hypothalamic-pituitary-adrenal axis. However, several mechanisms may impair the physiologic stress response of the hypothalamic-pituitary-adrenal axis in critically ill patients (Figure 129-5), resulting in an inadequate increase of serum cortisol levels. This condition is referred to as relative adrenal insufficiency.69
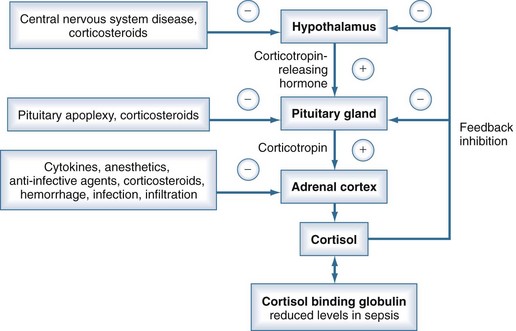
The concept of relative adrenal insufficiency as a clinically relevant condition is a matter of debate. It has been demonstrated that an inadequate rise of cortisol after the corticotropin stimulation test is associated with increased mortality in patients with septic shock.70 However, most studies measured total cortisol levels, which are reduced due to hypoalbuminemia, while the concentrations of free cortisol may be adequate.71 Administration of low-dose corticosteroids does not affect survival from septic shock.72
Vasopressin Deficiency
Vasopressin is excreted from the neurohypophysis in response to arterial hypotension or hypovolemia. Because septic shock is characterized by both arterial hypotension and hypovolemia, one would expect plasma vasopressin levels to be high; however, they are low in patients with septic shock73 and do not adequately respond when arterial hypotension occurs.74 Indeed, the administration of vasopressin or analogs can quickly restore blood pressure in these patients.75 Inadequate vasopressin levels may be caused by depression of the baroreflex, increased metabolism of vasopressin, and depletion of vasopressin stores in the pituitary gland.
Vasopressin is metabolized by plasma vasopressinase and by renal and hepatic clearance. Increased vasopressin metabolism seems unlikely in sepsis, because renal and hepatic functions are often compromised in this setting, and there is no evidence of increased vasopressinase activity in this disease. Depression of the baroreflex may play a role in vasopressin depletion. The baroreflex is mediated by sympathetic stimulation, and there is some evidence that sympathetic function might be impaired in sepsis.76 Depletion of pituitary vasopressin stores was found by magnetic resonance imaging in a case series of three septic shock patients with low plasma vasopressin levels.77 The clinical relevance of the low vasopressin response to septic shock remains unclear, except in a subgroup with less severe septic shock, because administration of low-dose vasopressin was not associated with better survival.78
Insulin Deficiency
Hyperglycemia is a common feature in critically ill patients, such as those with severe sepsis or septic shock, and is caused by the endocrine response to stress. This includes activation of the hypothalamic-pituitary-adrenal axis, with the release of cortisol. Additionally, secretion of epinephrine, glucagon, and growth hormone is increased. All these hormones counteract effects of insulin. Besides the endocrine stress response, several cytokines additionally increase insulin resistance by inhibiting intracellular pathways normally activated by the insulin receptor.79 There is also evidence that pancreatic insulin secretion is impaired in sepsis.80,81
Endocrine stress response, insulin resistance, and impaired insulin secretion are responsible for the hyperglycemic state of patients with sepsis. Besides hyperglycemia, insulin deficiency may also be unfavorable in sepsis, since insulin has several beneficial effects such as potent antiinflammatory and anabolic actions. However, insulin administration to achieve normal glucose levels is not supported by clinical trials; the risk of hypoglycemia seems to outweigh any possible beneficial effects of insulin.82,83
 Pathophysiology of Multiorgan Dysfunction
Pathophysiology of Multiorgan Dysfunction
Multiorgan dysfunction is the parallel or sequential failure of at least two organs. It is a frequent complication of sepsis. Clinically, multiorgan dysfunction is termed the multiple organ dysfunction syndrome (MODS). MODS can involve any organ of a critically ill patient, even a remote organ that was not originally affected by the underlying disease. The development of MODS significantly contributes to ICU mortality. Scoring systems such as the Sequential Organ Failure Assessment (SOFA) or the Multiorgan Dysfunction Score, which assess the severity of MODS, correlate well with mortality.4
Some major components of the pathophysiology of MODS are depicted in Figure 129-6. The development of MODS includes a complicated network of inter- and intracellular actions. Inflammation seems to be a major trigger for the induction of the processes that lead to MODS. Both infectious and noninfectious stimuli may be responsible for activation of the innate immune response. As previously discussed, PAMPs activate the immune response and induce complex metabolic and circulatory changes in the host. Correspondingly, cellular trauma causes the release of DAMPs such as mitochondrial peptides and mitochondrial DNA into the circulation. DAMPs may induce systemic inflammation similar to sepsis.84
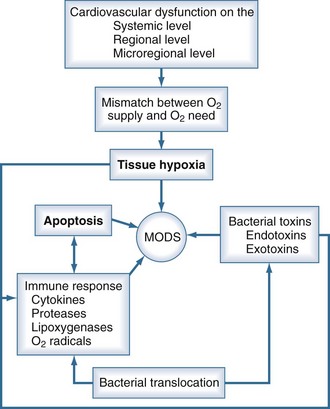
Because MODS can involve a variety of pathologic changes, different concepts of the pathophysiology of MODS have been generated (Table 129-5).2,85 Cellular dysfunction due to tissue hypoxia is likely an important factor in the onset of MODS. However, other factors also play a role, including the onset of programmed cell death (apoptosis) and the direct toxic effects of substances such as endotoxin and reactive oxygen species. The development of SIRS does not require the presence of infection; severe trauma, burns, pancreatitis, and cardiac surgery with cardiopulmonary bypass are also associated with SIRS and increase the risk for MODS.86
| Pathologic Process | Manifestation |
|---|---|
| Uncontrolled infection | Persistent infection, nosocomial acquired infection, endotoxemia |
| Systemic inflammation | Cytokinemia (particularly IL-6, IL-8, TNF), leukocytosis, increased capillary permeability |
| Immune paralysis | Nosocomial infection, increased antiinflammatory cytokine levels (IL-10), decreased HLA-DR expression; shift from type 1 to type 2 helper T-cells |
| Tissue hypoxia | Increased lactate, low central venous O2 saturation |
| Microvascular coagulopathy and endothelial dysfunction | Increased procoagulant activity, decreased anticoagulant activity (antithrombin III ↓, protein C ↓), high levels of fibrin derivatives, increased von Willebrand factor, soluble thrombomodulin, increased capillary permeability |
| Dysregulated apoptosis | Increased epithelial and lymphoid apoptosis, decreased neutrophil apoptosis |
| Gut-liver axis | Increased infection with gut organisms, endotoxemia, Kupffer cell activation |
HLA, human leukocyte antigen; IL, interleukin; TNF, tumor necrosis factor.
Modified from Marshall JC. Inflammation, coagulopathy, and the pathogenesis of multiple organ dysfunction syndrome. Crit Care Med 2001;29:S99–106.
Tissue Hypoxia
Tissue hypoxia is difficult to assess in a critically ill patient. It is therefore uncertain whether tissue hypoxia plays a leading role in organ dysfunction, because other mechanisms such as apoptosis have been identified as well.87 In addition, it has been suggested that disturbance of mitochondrial O2 utilization rather than tissue hypoxia is the motor of organ dysfunction. This hypothesis is supported by the observation that depletion of enzymes of the respiratory chain is associated with the development of MODS in septic patients.66 Nevertheless, data are available that demonstrate the importance of tissue oxygenation in the development of MODS. For example, when central venous O2 saturation was used to guide aggressive treatment to maintain O2 delivery, there was a significant reduction in mortality,88 supporting the concept of tissue hypoxia as an important mechanism in MODS. Likewise, patients with early lactate clearance have less severe organ dysfunction and improved outcome.89
Apoptosis
Apoptosis is a physiologic mechanism whereby activation of a specific intracellular program induces cell death. Apoptosis is therefore a regulatory process for the proliferation and differentiation of cells. However, pathologic activation of apoptosis seems to be involved in the pathogenesis of MODS. Apoptosis is induced by a cascade system through either an extrinsic (receptor-dependent) or an intrinsic (receptor-independent) pathway. The extrinsic pathway is activated by the so-called death receptor superfamily, consisting of receptors such as the Fas receptor (CD95) or the TNF receptor. Thus, cytokines are able to induce apoptosis. The intrinsic pathway may be induced by DNA damage. The process of apoptosis is mediated by an enzymatic cascade system in which active caspase-3 is the executioner protein that finally starts apoptosis. The receptors of the extrinsic pathway mediate the activation of procaspase-8 to active caspase-8 via several signaling proteins (Figure 129-7). The intrinsic pathway works by altering the mitochondrial membrane potential through the signal protein, p53, which mediates the activation of caspase-9. Both active caspase-8 and active caspase-9 activate the final common step in the apoptosis pathway (caspase-3).90
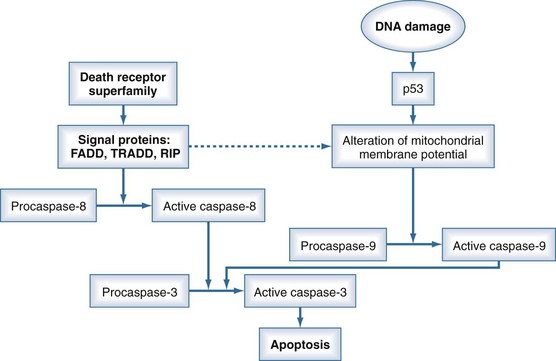
The extent to which apoptosis-related cell death contributes to the development of MODS is difficult to assess because this process has been investigated primarily in the experimental setting. However, apoptosis of intestinal epithelial cells has been shown in trauma patients shortly after injury.91 The importance of increased apoptosis in the clinical setting warrants further elucidation.
The “Two-Hit” Theory
The two-hit theory has been criticized for being somewhat arbitrary, because the differentiation between primary and secondary MODS is not always possible in the clinical setting.92 However, our current understanding of the inflammatory response to injury supports the concept of priming. Neutrophils are known to produce greater amounts of reactive oxygen species and have increased adhesion properties after they have been exposed to proinflammatory mediators. It has been demonstrated, at least in vitro, that priming followed by activation of neutrophils increases the extent of endothelial damage.
Key Points
Cohen J. The immunopathogenesis of sepsis. Nature. 2002;420:885-891.
van der Poll T, Opal SM. Host pathogen interaction in sepsis. Lancet Infect Dis. 2008;8:32-43.
Hotchkiss RS, Nicholson DW. Apoptosis and caspases regulate death and inflammation in sepsis. Nat Rev Immunol. 2006;6:813-822.
Levi M, van der Poll T. Inflammation and coagulation. Crit Care Med. 2010;38:S26-S34.
Cunnion RE, Parrillo JE. Myocardial dysfunction in sepsis. Crit Care Clin. 1989;5:99-117.
Landry DW, Oliver JA. The pathogenesis of vasodilatory shock. N Engl J Med. 2001;345:588-595.
Trzeciak S, Rivers EP. Clinical manifestations of disordered microcirculatory perfusion in severe sepsis. Crit Care. 2005;9:S20-S26.
Marshall JC. Inflammation, coagulopathy, and the pathogenesis of multiple organ dysfunction syndrome. Crit Care Med. 2001;29:S99-106.
1 Bone RC, Fisher CJJr, Clemmer TP, et al. Sepsis syndrome: A valid clinical entity. Crit Care Med. 1989;17(5):389-393.
2 ACCP/SCCM Consensus Conference Committee. Definition for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. Crit Care Med. 1992;20(6):864-874.
3 Hotchkiss RS, Karl IE. The pathophysiology and treatment of sepsis. N Engl J Med. 2003;348(2):138-150.
4 Zhang Q, Raoof M, Chen Y, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464(7285):104-107.
5 Iwasaki A, Medzhitov R. Regulation of adaptive immunity by the innate immune system. Science. 2010;327(5963):291-295.
6 van der Poll T, Opal SM. Host-pathogen interactions in sepsis. Lancet Infect Dis. 2008;8(1):32-43.
7 Medzhitov R, Preston-Hurlburt P, Janeway CAJr. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature. 1997;388(6640):394-397.
8 Macdonald J, Galley HF, Webster NR. Oxidative stress and gene expression in sepsis. Br J Anaesth. 2003;90(2):221-232.
9 Cohen J. The immunopathogenesis of sepsis. Nature. 2002;420(6917):885-891.
10 Inohara N, Ogura Y, Nunez G. Nods: a family of cytosolic proteins that regulate the host response to pathogens. Curr Opin Microbiol. 2002;5(1):76-80.
11 Martin GS, Mannino DM, Eaton S, Moss M. The epidemiology of sepsis in the United States from 1979 through 2000. N Engl J Med. 2003;348(16):1546-1554.
12 Draing C, Sigel S, Deininger S, et al. Cytokine induction by gram-positive bacteria. Immunobiology. 2008;213(3-4):285-296.
13 Yoshimura A, Lien E, Ingalls RR, Tuomanen E, Dziarski R, Golenbock D. Cutting edge: recognition of gram-positive bacterial cell wall components by the innate immune system occurs via Toll-like receptor 2. J Immunol. 1999;163(1):1-5.
14 Wang JE, Dahle MK, McDonald M, Foster SJ, Aasen AO, Thiemermann C. Peptidoglycan and lipoteichoic acid in gram-positive bacterial sepsis: receptors, signal transduction, biological effects, and synergism. Shock. 2003;20(5):402-414.
15 McCormick JK, Yarwood JM, Schlievert PM. Toxic shock syndrome and bacterial superantigens: an update. Annu Rev Microbiol. 2001;55:77-104.
16 Fraser JD, Proft T. The bacterial superantigen and superantigen-like proteins. Immunol Rev. 2008;225:226-243.
17 Presterl E, Lassnigg A, Mueller-Uri P, El-Menyawi I, Graninger W. Cytokines in sepsis due to Candida albicans and in bacterial sepsis. Eur Cytokine Netw. 1999;10(3):423-430.
18 Abraham E, Glauser MP, Butler T, et al. p55 Tumor necrosis factor receptor fusion protein in the treatment of patients with severe sepsis and septic shock. a randomized controlled multicenter trial. Ro 45-2081 Study Group. JAMA. 1997;277(19):1531-1538.
19 Benedict CA. Viruses and the TNF-related cytokines, an evolving battle. Cytokine Growth Factor Rev. 2003;14(3-4):349-357.
20 Riedemann NC, Guo RF, Ward PA. Novel strategies for the treatment of sepsis. Nat Med. 2003;9(5):517-524.
21 Sutherland AM, Walley KR. Bench-to-bedside review: Association of genetic variation with sepsis. Crit Care. 2009;13(2):210.
22 Gordon AC, Lagan AL, Aganna E, et al. TNF and TNFR polymorphisms in severe sepsis and septic shock: a prospective multicentre study. Genes Immun. 2004;5(8):631-640.
23 Sutherland AM, Walley KR, Manocha S, Russell JA. The association of interleukin 6 haplotype clades with mortality in critically ill adults. Arch Intern Med. 2005;165(1):75-82.
24 D’Avila LC, Albarus MH, Franco CR, et al. Effect of CD14 -260C>T polymorphism on the mortality of critically ill patients. Immunol Cell Biol. 2006;84(4):342-348.
25 Heidecke CD, Hensler T, Weighardt H, et al. Selective defects of T-lymphocyte function in patients with lethal intraabdominal infection. Am J Surg. 1999;178(4):288-292.
26 Hotchkiss RS, Nicholson DW. Apoptosis and caspases regulate death and inflammation in sepsis. Nat Rev Immunol. 2006;6(11):813-822.
27 Hotchkiss RS, Swanson PE, Freeman BD, et al. Apoptotic cell death in patients with sepsis, shock, and multiple organ dysfunction. Crit Care Med. 1999;27(7):1230-1251.
28 Voll RE, Herrmann M, Roth EA, Stach C, Kalden JR, Girkontaite I. Immunosuppressive effects of apoptotic cells. Nature. 1997;390(6658):350-351.
29 Bone RC. Sir Isaac Newton, sepsis, SIRS, and CARS. Crit Care Med. 1996;24(7):1125-1128.
30 Tracey KJ. The inflammatory reflex. Nature. 2002;420(6917):853-859.
31 Loisa P, Rinne T, Laine S, Hurme M, Kaukinen S. Anti-inflammatory cytokine response and the development of multiple organ failure in severe sepsis. Acta Anaesthesiol Scand. 2003;47(3):319-325.
32 van der Poll T, Levi M, Hack CE, et al. Elimination of interleukin 6 attenuates coagulation activation in experimental endotoxemia in chimpanzees. J Exp Med. 1994;179(4):1253-1259.
33 Levi M, van der Poll T, ten Cate H, van Deventer SJ. The cytokine-mediated imbalance between coagulant and anticoagulant mechanisms in sepsis and endotoxaemia. Eur J Clin Invest. 1997;27(1):3-9.
34 Massignon D, Lepape A, Bienvenu J, Barbier Y, Boileau C, Coeur P. Coagulation/fibrinolysis balance in septic shock related to cytokines and clinical state. Haemostasis. 1994;24(1):36-48.
35 Fourrier F, Chopin C, Goudemand J, et al. Septic shock, multiple organ failure, and disseminated intravascular coagulation. Compared patterns of antithrombin III, protein C, and protein S deficiencies. Chest. 1992;101(3):816-823.
36 Weiler H. Regulation of inflammation by the protein C system. Crit Care Med. 2010;38(2 Suppl):S18-S25.
37 Roemisch J, Gray E, Hoffmann JN, Wiedermann CJ. Antithrombin: a new look at the actions of a serine protease inhibitor. Blood Coagul Fibrinolysis. 2002;13(8):657-670.
38 Titheradge MA. Nitric oxide in septic shock. Biochem Biophys Acta. 1999;1411(2-3):437-455.
39 Taylor BS, Geller DA. Molecular regulation of the human inducible nitric oxide synthase (iNOS) gene. Shock. 2000;13(6):413-424.
40 Reinhart K, Bayer O, Brunkhorst F, Meisner M. Markers of endothelial damage in organ dysfunction and sepsis. Crit Care Med. 2002;30(5 Suppl):S302-S312.
41 Elliott MJ, Finn AH. Interaction between neutrophils and endothelium. Ann Thorac Surg. 1993;56(6):1503-1508.
42 Wilmore DW, Goodwin CW, Aulick LH, Powanda MC, Mason ADJr, Pruitt BAJr. Effect of injury and infection on visceral metabolism and circulation. Ann Surg. 1980;192(4):491-504.
43 Natanson C, Fink MP, Ballantyne HK, MacVittie TJ, Conklin JJ, Parrillo JE. Gram-negative bacteremia produces both severe systolic and diastolic dysfunction in a canine model that simulates human septic shock. J Clin Invest. 1986;78:259-270.
44 Bouhemad B, Nicolas-Robin A, Arbelot C, Arthaud M, Feger F, Rouby JJ. Acute left ventricular dilatation and shock-induced myocardial dysfunction. Crit Care Med. 2009;37(2):441-447.
45 Ognibene FP, Parker MM, Natanson CN, Shelhamer JH, Parrillo JE. Depressed left ventricular performance. Response to volume infusion in patients with sepsis and septic shock. Chest. 1988;93(5):903-910.
46 Cunnion RE, Parrillo JE. Myocardial dysfunction in sepsis. Critical Care Clinics. 1989;5(1):99-117.
47 Parrillo JE, Burch C, Shelhamer JH, Parker MM, Natanson C, Schuette W. A circulating myocardial depressant substance in humans with septic shock. J Clin Invest. 1985;76:1539-1553.
48 Ungureanu-Longrois D, Balligand JL, Kelly RA, Smith TW. Myocardial contractile dysfunction in the systemic inflammatory response syndrome: role of a cytokine-inducible nitric oxide synthase in cardiac myocytes. J Mol Cell Cardiol. 1995;27(1):155-167.
49 Szabo C, Cuzzocrea S, Zingarelli B, O’Connor M, Salzman AL. Endothelial dysfunction in a rat model of endotoxic shock. Importance of the activation of poly (ADP-ribose) synthetase by peroxynitrite. J Clin Invest. 1997;100(3):723-735.
50 Bloos FM, Morisaki HM, Neal AM, et al. Sepsis depresses the metabolic oxygen reserve of the coronary circulation in mature sheep. Am J Respir Crit Care Med. 1996;153(5):1577-1584.
51 Cunnion RE, Schaer GL, Parker MM, Natanson C, Parrillo JE. The coronary circulation in human septic shock. Circulation. 1986;73(4):637-644.
52 Dhainaut JF, Huyghebaert M-F, Monsallier JF, et al. Coronary hemodynamics and myocardial metabolism of lactate, free fatty acids, glucose, and ketones in patients with septic shock. Circulation. 1987;75(3):533-541.
53 Ammann P, Fehr T, Minder EI, Gunter C, Bertel O. Elevation of troponin I in sepsis and septic shock. Intensive Care Med. 2001;27(6):965-969.
54 Mehta NJ, Khan IA, Gupta V, Jani K, Gowda RM, Smith PR. Cardiac troponin I predicts myocardial dysfunction and adverse outcome in septic shock. Int J Cardiol. 2004;95(1):13-17.
55 Landry DW, Oliver JA. The pathogenesis of vasodilatory shock. N Engl J Med. 2001;345(8):588-595.
56 Ayuse T, Brienza N, Revelly J, O’Donnel CP, Robotham JL. Alterations in liver hemodynamics in an intact porcine model of endotoxin shock. Am J Physiol. 1995;268:H1106-H1114.
57 Withworth PW, Cryer HM, Garrison RN, Baumgarten TE, Harris PD. Hypoperfusion of the intestinal microcirculation without decreased cardiac output during life Escherichia coli sepsis in rats. Circ Shock. 1989;27:111-122.
58 Yi ES, Ulich TR. Endotoxin, interleukin-1, and tumor necrosis factor cause neutrophil-dependent microvascular leakage in postcapillary venules. Am J Pathol. 1992;140:659-663.
59 Dormehl IC, Hugo N, Pretorius JP, Redelinghuys IF. In vivo assessment of regional microvascular albumin leakage during E. coli septic shock in the baboon model. Circulatory Shock. 1992;38:9-13.
60 Ince C, Sinaasappel M. Microcirculatory oxygenation and shunting in sepsis and shock. Crit Care Med. 1999;27(7):1369-1377.
61 Farquhar I, Martin CM, Lam C, Potter R, Ellis CG, Sibbald WJ. Decreased capillary density in vivo in bowel mucosa of rats with normotensive sepsis. J Surg Res. 1996;61(1):190-196.
62 Lam C, Tyml K, Martin C, Sibbald W. Microvascular perfusion is impaired in a rat model of normotensive sepsis. J Clin Invest. 1994;94(5):2077-2083.
63 Ellis CG, Bateman RM, Sharpe MD, Sibbald WJ, Gill R. Effect of a maldistribution of microvascular blood flow on capillary O2 extraction in sepsis. Am J Physiol Heart Circ Physiol. 2002;282(1):H156-H164.
64 Trzeciak S, Rivers EP. Clinical manifestations of disordered microcirculatory perfusion in severe sepsis. Crit Care. 2005;9(Suppl 4):S20-S26.
65 Boulos M, Astiz ME, Barua RS, Osman M. Impaired mitochondrial function induced by serum from septic shock patients is attenuated by inhibition of nitric oxide synthase and poly(ADP-ribose) synthase. Crit Care Med. 2003;31(2):353-358.
66 Rivers E, Nguyen B, Havstad S, et al. Early goal-directed therapy in the treatment of severe sepsis and septic shock. N Engl J Med. 2001;345(19):1368-1377.
67 Ligtenberg JJ, Girbes AR, Beentjes JA, Tulleken JE, van der Werf TS, Zijlstra JG. Hormones in the critically ill patient: to intervene or not to intervene? Intensive Care Med. 2001;27(10):1567-1577.
68 Cooper MS, Stewart PM. Corticosteroid insufficiency in acutely ill patients. N Engl J Med. 2003;348(8):727-734.
69 Annane D, Sebille V, Troche G, Raphael JC, Gajdos P, Bellissant E. A 3-level prognostic classification in septic shock based on cortisol levels and cortisol response to corticotropin. JAMA. 2000;283(8):1038-1045.
70 Beishuizen A, Thijs LG, Vermes I. Patterns of corticosteroid-binding globulin and the free cortisol index during septic shock and multitrauma. Intensive Care Med. 2001;27(10):1584-1591.
71 Sprung CL, Annane D, Keh D, et al. Hydrocortisone therapy for patients with septic shock. N Engl J Med. 2008;358(2):111-124.
72 Landry DW, Levin HR, Gallant EM, et al. Vasopressin deficiency contributes to the vasodilation of septic shock. Circulation. 1997;95(5):1122-1125.
73 Jochberger S, Dorler J, Luckner G, et al. The vasopressin and copeptin response to infection, severe sepsis, and septic shock. Crit Care Med. 2009;37(2):476-482.
74 O’Brien A, Clapp L, Singer M. Terlipressin for norepinephrine-resistant septic shock. Lancet. 2002;359(9313):1209-1210.
75 Garrard CS, Kontoyannis DA, Piepoli M. Spectral analysis of heart rate variability in the sepsis syndrome. Clin Auton Res. 1993;3(1):5-13.
76 Sharshar T, Carlier R, Blanchard A, et al. Depletion of neurohypophyseal content of vasopressin in septic shock. Crit Care Med. 2002;30(3):497-500.
77 Russell JA, Walley KR, Singer J, et al. Vasopressin versus norepinephrine infusion in patients with septic shock. N Engl J Med. 2008;358(9):877-887.
78 Marik PE, Raghavan M. Stress-hyperglycemia, insulin and immunomodulation in sepsis. Intensive Care Med. 2004;30(5):748-756.
79 Mizock BA. Alterations in fuel metabolism in critical illness: hyperglycaemia. Best Pract Res Clin Endocrinol Metab. 2001;15(4):533-551.
80 Brunkhorst FM, Engel C, Bloos F, et al. Intensive insulin therapy and pentastarch resuscitation in severe sepsis. N Engl J Med. 2008;358(2):125-139.
81 Finfer S, Chittock DR, Su SY, et al. Intensive versus conventional glucose control in critically ill patients. N Engl J Med. 2009;360(13):1283-1297.
82 Vincent JL, Moreno R, Takala J, et al. The SOFA (Sepsis-related Organ Failure Assessment) score to describe organ dysfunction/failure. On behalf of the Working Group on Sepsis-Related Problems of the European Society of Intensive Care Medicine. Intensive Care Med. 1996;22(7):707-710.
83 Marshall JC, Cook DJ, Christou NV, Bernard GR, Sprung CL, Sibbald WJ. Multiple Organ Dysfunction Score: A reliable descriptor of a complex clinical outcome. Crit Care Med. 1995;23:1638-1652.
84 Marshall JC. Inflammation, coagulopathy, and the pathogenesis of multiple organ dysfunction syndrome. Crit Care Med. 2001;29(7 Suppl):S99-106.
85 Laffey JG, Boylan JF, Cheng DC. The systemic inflammatory response to cardiac surgery: implications for the anesthesiologist. Anesthesiology. 2002;97(1):215-252.
86 Suter PM, Romand J-A. Multiple organ failure due to tissue hypoxia: Myth or reality. Réan Urg. 1996;5(2):243.
87 Brealey D, Brand M, Hargreaves I, et al. Association between mitochondrial dysfunction and severity and outcome of septic shock. Lancet. 2002;360(9328):219-223.
88 Nguyen HB, Loomba M, Yang JJ, et al. Early lactate clearance is associated with biomarkers of inflammation, coagulation, apoptosis, organ dysfunction and mortality in severe sepsis and septic shock. J Inflamm (Lond). 2010;7:6.
89 Duprez L, Wirawan E, Vanden Berghe T, Vandenabeele P. Major cell death pathways at a glance. Microbes Infect. 2009;11(13):1050-1062.
90 Hotchkiss RS, Schmieg REJr, Swanson PE, et al. Rapid onset of intestinal epithelial and lymphocyte apoptotic cell death in patients with trauma and shock. Crit Care Med. 2000;28(9):3207-3217.
91 Saadia R, Schein M. Multiple organ failure. How valid is the “two hit” model? J Accid Emerg Med. 1999;16(3):163-166. discussion 166-167
92 Partrick DA, Moore FA, Moore EE, Barnett CCJr, Silliman CC. Neutrophil priming and activation in the pathogenesis of postinjury multiple organ failure. New Horiz. 1996;4(2):194-210.
[/level-membership-for-critical-care-medicine-category][not-level-membership-for-critical-care-medicine-category]
129 Pathophysiology of Sepsis and Multiple Organ Dysfunction
Pathophysiology of Sepsis
Sepsis has been defined as an invasion of microorganisms or their toxins into the bloodstream, together with the host response to this invasion.1 Thus, the pathophysiology of sepsis combines the impact of infection with the host response of generalized inflammation, which finally leads to multiorgan dysfunction and death. This definition has been extended by the addition of several terms to more carefully describe the disease and its pathophysiology (Table 129-1). The American College of Chest Physicians/Society of Critical Care Medicine (ACCP/SCCM) Consensus Conference defined sepsis as a systemic inflammatory response syndrome (SIRS) caused by infection.2 More recently it has been recognized that SIRS is counteracted by a hypoinflammatory state that also plays an important role in the further development of organ dysfunction.3
| Term | Definition |
|---|---|
| Bacteremia | Presence of viable bacteria in the blood |
| Systemic inflammatory response syndrome (SIRS) | Generalized hyperinflammatory response to several impacts |
| Sepsis | SIRS caused by infection |
| Severe sepsis | Sepsis associated with organ dysfunction |
| Septic shock | Sepsis associated with arterial hypotension |
Data from ACCP/SCCM Consensus Conference Committee. Definition for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. Crit Care Med 1992;20:864–74.
Sepsis is characterized by loss of hemostatic balance and endothelial dysfunction, which in turn severely compromise the cardiocirculatory system as well as intracellular homeostasis. Cellular hypoxia and apoptosis (programmed cell death) then contribute to organ dysfunction and death. The network of organ systems affected by sepsis is depicted in Figure 129-1.
Microbiological Stimulus
By definition, infection is a fundamental part of the pathophysiology of sepsis. Any microorganism able to induce infection in humans may be complicated by sepsis. Bacteria as well as fungi, parasites, and to a lesser degree viruses can trigger the mechanisms that lead to sepsis. Although SIRS is the final common pathway of this process, the signal transduction pathway from infection to complex host response differs with the microbiological stimuli. Induction of an innate immune response is triggered by specific microbial molecules (e.g., bacterial wall components, exotoxins, bacterial DNA, viral RNA) called pathogen-associated molecular patterns (PAMPs). Damage-associated molecular patterns (DAMPs) are the noninfectious equivalents to PAMPs. DAMPs are released after cellular injury of the host (i.e., trauma) and can also induce the innate immune response.4
The presence of PAMPs is sensed by recognition molecules called pattern-recognition proteins (PRR), which are able to initiate a host response. These proteins may be categorized into secreted, transmembrane, and cytosolic PRRs. The Toll-like receptors (TLRs) represent the membrane PRRs. Eleven different TLRs have been discovered in mammals, whereas TLR-11 is not expressed in humans (Table 129-2). Retinoic acid-inducible gene I (RIG-I)–like receptors (RLRs) and the nucleotide-binding domain and leucine-rich repeat-containing receptors (NLRs) are cytosolic PRRs. RLRs recognize viral RNA and some double-stranded DNA. NLRs represent a large family of intracellular sensors that can detect pathogens and stress signals. NLRs detect microbiological products such as peptidoglycans and other degradation products of microorganisms as well as stress-related substances.5,6
TABLE 129-2 Human Toll-Like Receptors and Their Natural Ligands
| TLR Type | Related Pathogen-Associated Molecular Pattern | Location |
|---|---|---|
| TLR1 (via TLR2) | Bacterial products such as tri-acyl lipopeptides | Cell surface |
| TLR2 | Gram-positive bacterial products, including peptidoglycans; some virus-related proteins | Cell surface |
| TLR3 | Viral double-stranded RNA | Endosomal |
| TLR4 | Endotoxin, other bacterial products, some fungal products | Cell surface |
| TLR5 | Flagellin | Cell surface |
| TLR6 (via TLR2) | Some bacterial products | Cell surface |
| TLR7 | Single-stranded RNA | Endosomal |
| TLR8 | Viral single-stranded RNA | Endosomal |
| TLR9 | Viral and bacterial DNA | Endosomal |
| TLR10 | Unknown |
TLR, Toll-like receptor.
Gram-Negative Sepsis
In gram-negative bacteremia, initiation of the immune response is mediated primarily by lipopolysaccharide (LPS), a bacterial cell wall product. In plasma, LPS is bound to the LPS binding protein (LBP). Bound LPS is transported to the opsonic receptor, CD14, which is located on several cell membranes including on monocytes.7 A soluble form of CD14 interacts with CD14-negative cells (e.g., dendritic cells). However, CD14 alone cannot explain the actions of LPS, because CD14 does not have an intracellular tail.
Another binding site of LPS is the transmembranous receptor, TLR4, which exists in combination with the accessory protein, MD2.8 The binding of LPS to CD14 and TLR4 induces, via other molecules, activation of the transcription factor, nuclear factor kappa-B (NF-κB). Activated NF-κB migrates into the nucleus where it binds to and activates gene promoters, resulting in the transcription and expression of genes for cytokines and other proinflammatory mediators.9 In monocytes, LPS also induces cytokine transcription via the triggering receptor expressed on myeloid cells-1 and the myeloid DAP12-associated lectin.10 Intracellular pattern-recognition proteins in monocytes for LPS have recently been identified as another pathway of cytokine expression and include nucleotide-binding oligomerization domain 1 and 2 as LPS binding sites.11
Gram-Positive Sepsis
During the last decade, gram-positive bacteria have gained greater importance as causative organisms for sepsis.12 Gram-positive bacteria lack endotoxin and are recognized by cell wall components such as peptidoglycans and released bacterial toxins (exotoxins). Recently, lipoteichoic acid (LTA), a component of the cell wall in all gram-positive bacteria, has been recognized as the main pattern for recognition of gram-positive bacteria.13 TLR2 has been identified as the only pattern-recognition protein for gram-positive bacteria.14 The relationship between LTA and TLR2 is not completely clarified. Although LTA clearly interacts with TLR2, TLR2 is not a specific receptor for LTA, because TLR2 can recognize several other components of gram-positive bacteria.15 Gram-positive and gram-negative sepsis are indistinguishable clinically, suggesting a similar pathway of signal transduction. Indeed, peptidoglycans and LTA stimulate the release of tumor necrosis factor alpha (TNF-α), interleukin (IL)-6, and IL-10. It has been speculated that CD14 is also involved in the signaling of gram-positive infections.
Some exotoxins cause a special type of septic shock called the toxic shock syndrome (TSS). TSS may be caused by the exotoxin, TSS toxin-1, staphylococcal enterotoxins from Staphylococcus aureus, or streptococcal pyogenic exotoxins.16 These toxins are capable of acting as so-called superantigens which deploy their effects via the T-cell antigen receptor (TCR). The TCR consists of five variable elements: Vβ, Dβ, Jβ, Vα, and Jα. Conventionally, the T cell is activated if the major histocompatibility complex (MHC) of an antigen-presenting cell matches all five elements. Thus, T cells are activated by proper antigen contact only. This results in the stimulation of about 1 in 10,000 T cells. However, a superantigen such as TSS toxin-1 works as a bridge between the MHC and the Vβ chain of the TCR only (Figure 129-2).17 Because T-cell activation now occurs independently of a match between the MHC and TCR, about 20% of the entire T-cell pool may be activated at once. Besides further T-cell proliferation, T-cell activation causes the release of several cytokines (i.e., interferon gamma [IFN-γ], IL-2, TNF-α) from T cells, as well as IL-1β and TNF-α from macrophages. Thus, the presence of superantigens results in a release of cytokines similar to gram-negative sepsis. It is assumed that actions other than cytokine production may be responsible for actions of superantigens in TSS; for example, superantigens may amplify the effects of LPS.
Other Microbiological Stimuli of Sepsis
Sepsis can also be induced by fungi, viruses, and parasites. Signal transduction by nonbacterial products, however, is not as well characterized as bacterial sepsis. In part, this may be due to the fact that induction of cytokine release differs markedly not only among different microorganisms but also among species. Nevertheless, the release of proinflammatory mediators has been demonstrated during infections with nonbacterial infections such as Candida albicans18 and Plasmodium falciparum.19 The signal transduction in viral infections is complicated by the fact that viruses can interfere with TNF-related cytokine release to avoid the host’s antiviral activities.20 Pattern recognition of viruses occurs mainly via endosomal TLR receptors which detect single- and double-stranded RNA or DNA (see Table 129-2).
The Immune Response in Sepsis
The cytokines TNF-α and IL-1β are released by activated macrophages and CD4 T cells within the first hour after infection. These primary mediators induce the release of several secondary mediators that amplify inflammation (Table 129-3). An important step in signal amplification is activation of the complement system. Besides being activated by antigen-antibody complexes, the complement system may be stimulated by bacterial surface sugars and endotoxin. The complement fragment C5a, a cleavage product of the complement cascade, is a strong chemoattractant. C5a appears about 2 hours after the initiation of sepsis and stimulates macrophages to further produce proinflammatory mediators. Another mediator that amplifies the immune response is macrophage migration inhibitory factor (MIF), which is produced by T cells, macrophages, monocytes, and pituitary cells in response to an infectious stimulus. MIF appears about 8 hours after the onset of sepsis and activates T cells and macrophages to produce proinflammatory mediators. About 24 hours after the initiation of sepsis, levels of high-mobility group box 1 (HMGB1) protein increase and appear to play a role in endotoxin-related sepsis. HMGB1 is a nuclear binding protein that, among other things, is capable of activating NF-κB. As a rather late mediator in sepsis, it is produced by macrophages and neutrophils and stimulates phagocytic cells.21
TABLE 129-3 Macrophage Mediators Involved in the Pathogenesis of Sepsis
| Mediator | Typical Effects |
|---|---|
| Cytokines: IL-1, IL-6, IL-12, IL-15, IL-18, TNF, MIF, HMGB1, IL-10 | Activate neutrophils, lymphocytes, and vascular endothelium; up-regulate cellular adhesion molecules; induce prostaglandins, nitric oxide synthase, and acute-phase proteins; induce fever |
| IL-10 is predominantly a negative regulator of these effects. | |
| Chemokines: IL-8, MIP-1α, MIP-1β, MCP-1, MCP-3 | Mobilize and activate inflammatory cells, especially neutrophils; activate macrophages |
| Lipid mediators: platelet-activating factor, prostaglandins, leukotrienes, thromboxane, tissue factor | Activate vascular endothelium; regulate vascular tone; activate extrinsic coagulation cascade |
| Oxygen radicals: superoxide and hydroxyl radicals, nitric oxide | Antimicrobial properties; regulation of vascular tone |
HMGB, high-mobility group B protein; IL, interleukin; MCP, monocyte chemoattractant protein; MIF, migration inhibitory factor; MIP, macrophage inflammatory protein; TNF, tumor necrosis factor.
Data from Cohen J. The immunopathogenesis of sepsis. Nature 2002;420:885–91.
It is not completely understood why inflammation becomes generalized in some patients but stays localized in others. Genetic variants of cytokines may play a role in this issue. Single nucleotide polymorphisms (SNPs) are single-base changes in the DNA which do not cause obvious changes in the function of the respective cytokine. However, SNPs in some cytokines are associated with a worse outcome from septic shock or an increased risk for developing sepsis.22–24 Among several others, such variants have been described in TNF-α, IL-6, and CD14.25 However, results from these studies are difficult to interpret because of contradictory results and differences in populations of different ethnicities.
The immune response in sepsis does not involve only proinflammatory mediators. As in many other physiologic processes, the organism produces inhibitors to control certain reactions. Proinflammatory mediators are counteracted by antiinflammatory molecules such as IL-4 and IL-10 because CD4 T cells can switch from the production of inflammatory cytokines (type 1 helper T cells [TH1]) to the production of antiinflammatory cytokines (type 2 helper T cells [TH2]). Soluble TNF receptors and IL-1 receptor antagonists (IL-1Ra) are released to inhibit the actions of TNF and IL-1 in their roles as primary mediators of sepsis. T cells, neutrophils, and macrophages also may become unresponsive to infectious stimuli (anergy).26 Another mechanism of the antiinflammatory response is the onset of apoptosis, a genetically programmed autodestructive release of proteases that induces cell death. In sepsis, enhanced apoptosis causes loss of immune effector cells, including CD4 and CD8 T cells, B cells, and dendritic cells.27 Absolute lymphocyte counts are significantly decreased in patients with sepsis.28 Further, apoptotic cells impair the function of surviving immune cells.29
Results from animal studies suggest that the autonomic nervous system is also involved in suppression of cytokine release during sepsis. In the experimental setting, vagal stimulation can inhibit TNF expression. It is hypothesized that an inflammatory reflex, with the afferent vagal nerve sensing cytokine release and an efferent immunosuppressing cholinergic arm, exists.30 The importance of such a reflex in humans merits further investigation.
The antiinflammatory response in sepsis has been termed the compensatory antiinflammatory response syndrome (CARS).3 It has been suggested that the first response to infection is hyperinflammation, which is followed by a hypoimmune state. From there, recovery would be possible, but the prolonged inability to eradicate microorganisms might result in the death of the patient.31 However, serum levels of antiinflammatory cytokines are increased in parallel with the increase of proinflammatory mediators.32,33 Thus, diminished inflammation develops at the same time as the process of hyperinflammation. Although the persistence of high levels of antiinflammatory mediators may contribute to mortality in septic patients, the clinical role of change between hyperinflammatory and hypoinflammatory states remains unclear.
Loss of Hemostatic Balance
Under normal conditions, the vascular luminal surface has anticoagulant properties. Tissue factor is a 4.5-kD protein that is bound to cell membranes which are normally not in contact with blood. Expression of tissue factor mainly depends on release of IL-6.34,35 Tissue factor expression occurs on mononuclear cells, but endothelial cells, polymorphonuclear cells, and other cell types may be additional sources. The expression of tissue factor induces intravascular thrombin formation initiated by the extrinsic coagulation pathway. Because this process is not restricted to a local area, it is called disseminated intravascular coagulation (DIC); DIC causes a consumption of coagulation factors.
Physiologically, excessive coagulation is counteracted by several natural anticoagulants including antithrombin, the thrombomodulin/protein C/protein S system, and tissue factor pathway inhibitor. In addition to the activation of tissue factor-dependent thrombin generation, anticoagulant function is attenuated in sepsis. Patients with sepsis demonstrate reduced levels of protein C and antithrombin due to consumption and reduced synthesis.36 Thus, the physiologic balance between procoagulant and anticoagulant substances is altered in sepsis as there is a shift of the hemostatic balance toward a procoagulant state (Figure 129-3).
Besides its anticoagulant actions, the protein C pathway is an important link between coagulation and inflammation, because activated protein C has antiinflammatory properties. Protein S can bind to receptors that mediate an antiinflammatory regulatory loop of dendritic cell and monocyte inflammatory function. Thrombomodulin has been described to prevent excessive complement activation.37 Thus, there is considerable cross-talk between inflammation and coagulation which is impaired in sepsis as a result of depletion of the thrombomodulin/protein C/protein S system. There is also evidence that antithrombin blunts activation of several cytokines, suggesting that low antithrombin levels also affect the relationship between coagulation and inflammation.38
Endothelial Dysfunction
The endothelium produces several vasoactive mediators, including nitric oxide (NO), prostacyclins, and endothelin. NO is a potent vasodilator produced by NO synthase (NOS) from the amino acid, L-arginine. NO directly relaxes the vessel’s smooth muscle. There are two different forms of endothelial NOS: the constitutional form (cNOS) and the inducible form (iNOS). Physiologically, cNOS—also referred to as endothelial NOS (eNOS)—produces only small amounts of NO, and iNOS is expressed at low levels.39 In sepsis, iNOS expression is stimulated by cytokines such as IL-1β and TNF-α.40 This is followed by massive NO production and profound vasodilatation. Whether increased activity of cNOS also plays a role in sepsis is currently a matter of debate.
During inflammation, endothelial cells express adhesion molecules on their surface, which causes the adherence of leukocytes. These adhesion molecules include endothelial leukocyte adhesion molecule-1, intracellular adhesion molecule-1, and vascular cell adhesion molecule-1. Endothelial leukocyte adhesion molecule-1 is a selectin that mediates the initial step of leukocyte adhesion, followed by leukocyte rolling along the endothelial surface. The leukocyte finally migrates through the endothelial layer into the tissue, mediated by intracellular adhesion molecule-1 and vascular cell adhesion molecule-1 expression on both endothelial cells and leukocytes.41
Migration of leukocytes into the tissue is a physiologic mechanism to move immune cells to the site of infection. However, in generalized inflammation such as in sepsis, endothelial cells in several organs remote from the site of infection express adhesion molecules, inducing a generalized rolling and sticking of circulating leukocytes to the vascular surface. Adherence to endothelial cells activates leukocytes and induces a respiratory burst.42 The respiratory burst involves the release of cytotoxic substances such as elastase, myeloperoxidase, and reactive oxygen species. These products are capable of damaging endothelial cells and the surrounding tissue. Endothelial cell damage causes capillary leakage whereby intravascular fluid penetrates the extracellular space, leading to tissue edema.
