[level-membership-for-cardiovascular-category]
Chapter 22 Pathophysiology of Renal Artery Disease
Vascular disease affecting the renal arteries presents complex challenges to clinicians. Thanks to recent advances in vascular imaging, more patients than ever before are being identified with some degree of atherosclerotic or fibromuscular renovascular disease. Many of these lesions are of minor hemodynamic importance at the time of detection. Some reach a degree at which perfusion pressures and intrarenal hemodynamics are altered, leading to changes in blood pressure regulation and renal function. These can produce a variety of recognizable clinical syndromes illustrated in Figure 22-1. These range from modest changes in systemic arterial pressure to impaired volume control associated with congestive heart failure (CHF) to threatened viability of the kidney, sometimes designated ischemic nephropathy. Understanding the pathways by which renovascular disease affects cardiovascular and renal disease is important for both diagnosis and for defining optimal management using tools both to block the renin-angiotensin system and to restore the circulation.
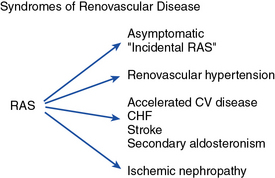
CHF, congestive heart failure; CV, cardiovascular.
(Modified with permission from Garovic V, Textor SC: Renovascular hypertension and ischemic nephropathy. Circulation 112:1362–1374, 2005.)89
Most renovascular lesions are the result of atherosclerosis. With the aging of the U.S. and other Western populations and reduced mortality from stroke and coronary disease, the prevalence of vascular disease in other vascular beds reaching clinically critical levels appears to be increasing.1 Understanding the variety of clinical manifestations of these lesions, the potential for disease progression, and the benefits and limitations of vascular repair are essential for vascular medicine specialists. This chapter will examine the pathophysiology of renovascular lesions regarding blood pressure control, ischemic nephropathy, and clinical syndromes such as flash pulmonary edema. Specific issues regarding diagnostic evaluation and management are addressed elsewhere (see Chapter 23).
A wide range of lesions can affect the renal blood supply, some of which are summarized in Box 22-1. Historically, recognition of renovascular disease resulted from searching for underlying causes of hypertension. This followed the seminal observations of Goldblatt more than 70 years ago2 that renal artery constriction produced a rise in arterial pressure in the dog. These studies were among the first to establish a primary role of the kidney in overall blood pressure regulation. Renovascular hypertension produced by a “clipped” renal artery remains among the most widely studied experimental forms of angiotensin-dependent hypertension.3,4
 Box 22-1 Vascular Lesions That Produce Renal Hypoperfusion and Renovascular Hypertension Syndrome
Box 22-1 Vascular Lesions That Produce Renal Hypoperfusion and Renovascular Hypertension Syndrome
Unilateral Disease (Analogous to Two-Kidney, One-Clip [2K1C] Hypertension)
Epidemiology of Renal Artery Disease
Fibromuscular disease may be identified in 1% to 3% of normal kidney donors subjected to angiography before donor nephrectomy.5 Of those developing clinical hypertension and referred for revascularization, more than 85% are females with a predilection for disease in the right renal artery.6 The location of these lesions is most commonly in the midportion and distal segments of the renal artery. A variety of fibromuscular lesions have been described, but the most common is medial fibroplasia. Occasionally, such lesions may be found in the carotid and other vascular beds, but most commonly they are limited to the renal arteries. Most do not progress to impair renal function, although some lead to arterial dissection and/or thrombosis with loss of the kidney.
Atherosclerosis is the most common cause of renal artery disease. Its presence and severity are related to age and the presence of other atherosclerotic disease of the descending aorta and lower extremities. Population-based series, such as one from North Carolina, indicate that among 834 subjects older than 65 years, significant renal artery stenosis (RAS; defined as Doppler peak systolic velocity (PSV) above 1.8 m/s) can be identified in 6.8% of the general population, regardless of race.7 Recent series of carotid, coronary, and peripheral angiography indicate that the prevalence of renovascular disease corresponds to overall atherosclerotic burden. Incidental renal artery occlusive disease (> 50% stenosis) has been reported in 11% to 18% of patients with coronary artery disease (CAD), particularly when significant hypertension is present.8 Peripheral vascular and aortic disease is associated with higher prevalence (25%-33%). As expected, risk factors predicting the presence of RAS include smoking, hyperlipidemia, hypertension, and diabetes. A corollary observation is that renovascular hypertension resulting from these lesions is now most commonly superimposed gradually upon preexisting essential hypertension. Hence, the blood pressure response and “cure” rates after successful restoration of blood flows to the kidney are limited by preexisting conditions.
Pathophysiological Consequences of Renovascular Disease
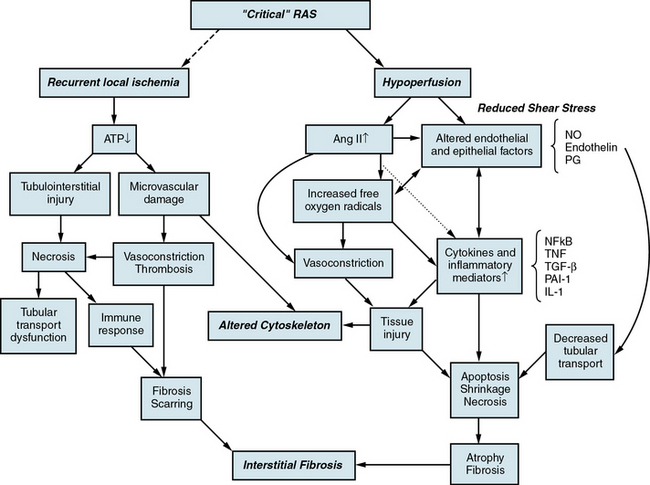
Under basal conditions, renal blood flow is among the highest of all organs. This feature reflects the kidney’s filtration function, and less than 10% of delivered oxygen is sufficient to maintain renal metabolic needs. Importantly, a fall in renal blood flow is accompanied by decreased oxygen consumption, partly due to reduced metabolic demands of filtration and tubular solute reabsorption. Reduced renal blood flow can be sustained without measurable change in total kidney oxygen levels (as assessed by renal vein oxygen tension),9 stimulation of erythropoietin release,10 or reduced medullary and cortical tissue oxygenation as measured in human subjects using blood oxygen level–dependent (BOLD) magnetic resonance (MR).11 These observations argue against an overall lack of oxygen as a primary stimulus for either hypertension or renal tissue injury and cast some doubt on the term ischemic nephropathy. Alternative terms proposed included azotemic renovascular disease and hypoperfusion injury.12 Nonetheless, severe vascular stenosis leading to diminished renal perfusion eventually does lead to renal tissue injury and interstitial fibrosis.
Subcritical Levels of Stenosis
The majority of renal artery lesions that compromise renal function are caused by gradually developing atherosclerosis of the renal vascular bed. As noted earlier, some patients undergoing cardiac catheterization have “incidental” renal lesions producing more than 50% cross-sectional stenosis,13 for whom the presence of RAS is a strong independent predictor of mortality. Moreover, nonobstructive RAS (20%-50% decrease in renal arterial luminal diameter) can be found in an additional 28% of patients undergoing cardiac catheterization13 and 48% of patients undergoing aortography for peripheral vascular disease.14 A recent systematic analysis of these reports including more than 15,000 subjects confirmed this overall range of disease prevalence and the rise with increasing atherosclerotic burden.8 Although lesions producing less than 50% in arterial luminal diameter are not considered hemodynamically significant, the relationships between resting pressure gradients and angiographic degree of stenosis are curvilinear and only approximate at best. As a predictor of mortality, even low-grade atherosclerotic lesions denote a hazard nearly equal to more advanced disease.15 Estimating severity of vascular occlusion from angiographic images is notoriously unreliable. It should be emphasized that activation of pressor mechanisms depends upon the presence of a pressure gradient between the aorta and distal renal vasculature16 (Fig. 22-2). There is a general relationship between estimated diameter stenosis and peak translesional pressure gradients, but the relationship is not linear. In some instances, an abrupt fall in poststenotic pressure develops beyond a subcritical range of stenosis.17 Even moderate stenosis, especially when superimposed on intrarenal microvascular disease, may contribute to adverse renal outcomes. Kidneys with a baseline renal artery disease classification of less than 60% stenosis have 11.7% 2-year cumulative incidence of renal atrophy (defined radiologically as a loss of kidney size)18 and 28% cumulative incidence of renal artery disease progression, although progression to total renal artery occlusion is uncommon.19 Increased severity of RAS in patients undergoing cardiac catheterization has an adverse effect on survival, with the 4-year adjusted survival for patients with a 50% stenosis decreasing to 70%, compared to 89% in patients without RAS.20 Therefore, even relatively minor stenosis in the renal artery might have long-term functional implications, especially in the presence of additional risk factors or coexisting renal disease.
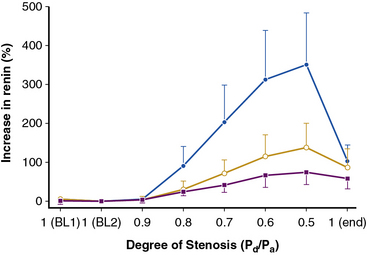
(Reproduced with permission from De Bruyne B, Manoharan G, Pijls NH, et al: Assessment of renal artery stenosis severity by pressure gradient measurements. J Am Coll Cardiol 48:1851–1855, 2006.)16
Renal Microvascular Disease
Lesions in the main renal artery may be superimposed upon or confused with other causes for ischemic renal injury. Intrarenal vascular lesions are commonly observed in the course of various nephropathies, many of which have an ischemic component.21 Risk factors including diabetes, hypertension, atherosclerosis, and aging elicit vasoconstriction or structural changes leading to intrarenal small-vessel disease and ischemic injury similar to that observed in large-vessel disease. Loss of microvessels and impaired capillary repair correlate with development of glomerular and tubulointerstitial scarring,22,23 and may lead to end-stage renal failure. Renal microvascular disease distal to a stenosis in the renal artery may perpetuate and exacerbate renal parenchymal injury and may blunt renal recovery. The presence of small microvessel injury is difficult to verify but may account for changes in diastolic blood flow such as that producing changes in renal resistance index. Elevations of renal resistance index have been proposed to predict poor outcomes in many renal diseases, including renovascular disease.24
Critical Renal Artery Stenosis
High-grade vascular stenosis eventually leads to a decrease in renal perfusion pressure. Critical stenosis is identified when it produces a fall in renal blood flow and glomerular filtration rate (GFR). During experimental renal artery occlusion, the kidney sustains autoregulation of blood flow through a range of perfusion pressures from 200 mmHg to approximately 80 mmHg. Mechanisms underlying autoregulation include myogenic responses to changes in wall tension, release of vasoactive substances, and the tubuloglomerular feedback. The latter responds to decreased renal perfusion pressure and salt delivery by decreasing vascular resistance distal to the obstruction. In addition, during a fall in renal perfusion pressure, the kidney activates multiple pathways that elevate systemic blood pressure, an effect that tends to restore renal perfusion pressure and sustain renal blood flow at the expense of arterial hypertension (Fig. 22-3). Consequently, as long as systemic arterial pressure is allowed to rise, a fall in renal blood flow does not occur until renal arterial diameter is reduced by 65% to 75%. Recent clinical studies suggest that noninvasive radiological imaging commonly overstates the degree of stenosis. Measurement of physiological stimuli, such as the release of renin, indicate that a translesion gradient of at least 10% to 20% reduction is necessary for biological responses to occur in humans.16 To achieve such a gradient, luminal occlusion may need to exceed 80% stenosis. Under some conditions, gradients above 20 mmHg that develop during intrarenal hyperemic challenge with dopamine may disclose hemodynamic significance of lesions under 60% in severity.25
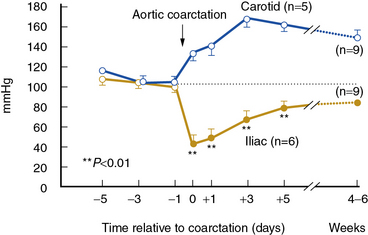
(Reproduced with permission from Textor SC, Smith-Powell L: Post-stenotic arterial pressure, renal haemodynamics and sodium excretion during graded pressure reduction in conscious rats with one- and two-kidney coarctation hypertension. J Hypertens 6:311–319, 1988.)90
When renal perfusion pressure falls gradually, additional mechanisms are recruited that protect the kidney from the functional and morphological consequences observed after acute ischemic injury. These include development of collateral vessels and redistribution of intrarenal blood flow from the cortex to the medulla. Renal cortical blood flow autoregulates more efficiently than the outer medulla, which is continuously on the verge of anoxia. During chronic reduction of renal blood flow, medullary perfusion and oxygenation are relatively maintained by adaptive mechanisms at the expense of cortical blood flow.26 When poststenotic renal artery pressures eventually fall further, either due to progressive vascular occlusion or reduction of systemic blood pressures by drug therapy, renal volume decreases.
In clinical terms, renal atrophy can be defined as a loss of renal length by at least 1 centimeter, and a difference in size between the two kidneys is suggestive of unilateral RAS (or a higher grade of stenosis in one of the kidneys). A decrease in renal volume results from a decrease in filling pressure, filtrate, and blood content of the kidney, as well as structural atrophy of the renal tubules due to apoptosis and necrosis. Apoptosis is an active, pre-programmed form of cell death that is intricately regulated and distinct from cellular necrosis and likely serves as a protective mechanism to allow renal “hibernation.” These changes may be reversible, since tubular cells show vigorous potential for regeneration. Loss of intrarenal microvessels that accompanies the ongoing scarring process may also contribute to renal shrinkage27 (Fig. 22-4), but might be partly reversible upon enhancement of angiogenic signaling.28 However, if a blood flow deficit persists, permanent damage to the kidney may occur. As mentioned, decreased renal blood flow is often accompanied by a decline in GFR and inhibition of tubular epithelial transport that limit renal oxygen consumption and maintain oxygen saturation. Hence, the kidney does not actually develop “ischemia” until an extreme decrease in renal blood flow develops.
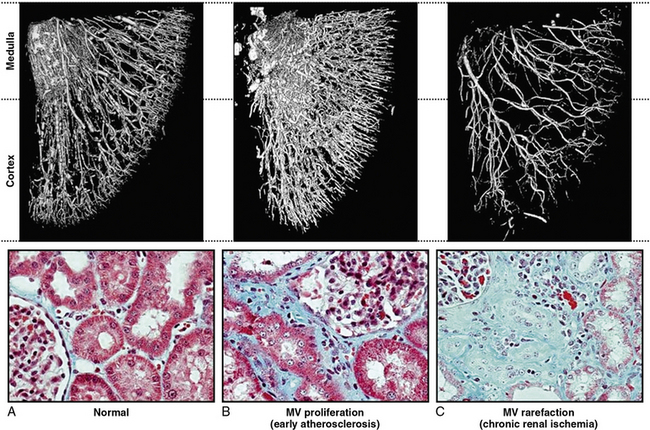
Figure 22-4 Microcomputed tomography imaging of vascular structures in kidney cortex and medulla in a swine model.
(Reproduced with permission from Lerman LO, Chade AR: Angiogenesis in the kidney: a new therapeutic target? Curr Opin Nephrol Hypertens 18:160–165, 2009.)91
Renovascular Hypertension
Goldblatt et al and Loesch were the first to show in the 1930s that obstruction of the renal artery is followed by an increase in systemic blood pressure.2,29 The characteristics of renovascular hypertension depend to a large extent on the status of the kidneys. Unilateral RAS may be present with an intact contralateral renal artery (the experimental form is termed two-kidney, one-clip, or 2K1C). This model is characterized by counterregulatory processes in the contralateral kidney leading to sodium excretion in response to elevated arterial pressure (pressure natriuresis; Fig. 22-5A-B). Alternatively, RAS may affect a solitary kidney (one-kidney, one-clip, or 1K1C; Fig. 22-6A-B). Bilateral RAS and 1K1C lead to more severe renovascular hypertension, although bilateral RAS may behave similarly to 2K1C if one kidney is significantly less ischemic than the other. Patients with this constellation of findings have higher mortality, are more prone to circulatory congestion, and are more likely to experience deterioration of kidney function during administration of antihypertensive agents, including angiotensin-converting enzyme (ACE) inhibitors or angiotensin II (AngII) receptor blockers (ARBs).
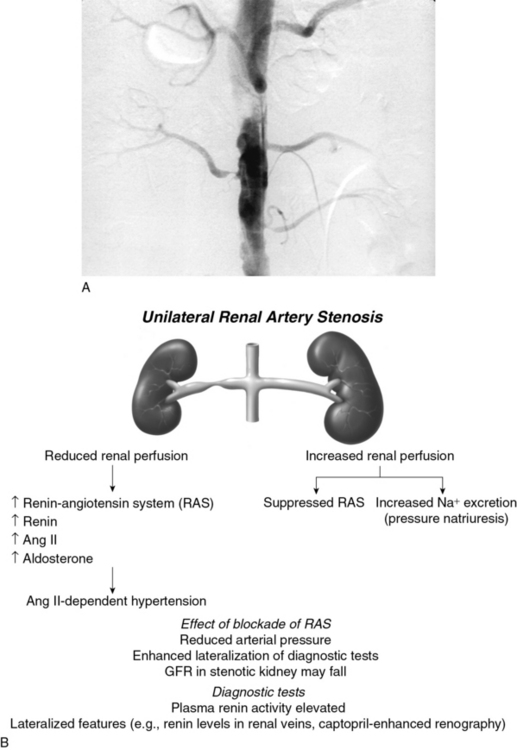
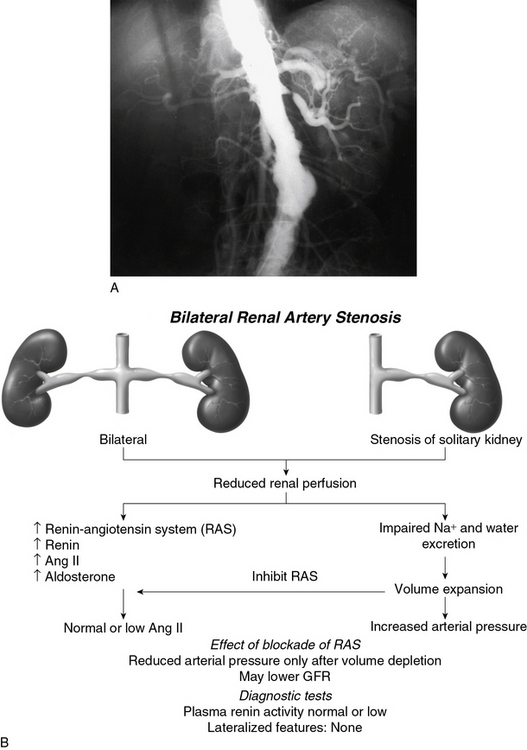
The exact mechanisms responsible for renovascular hypertension have long been debated. The immediate increase in blood pressure in RAS results from release of renin from the stenotic kidney. This leads to increased formation of Ang II, which increases peripheral vascular resistance, plasma aldosterone, sodium retention, extracellular volume, and cardiac output (Fig. 22-7). Early studies using ACE inhibitors30 and more recent studies in an AT1A receptor knockout mouse model of 2K1C confirm the essential role of Ang II in mediating Goldblatt hypertension during its initial phase.31 Experiments with kidney transplantation in these knockout strains indicate that both renal and extrarenal angiotensin receptors participate in regulation of blood pressure.4 Blockade of angiotensin action in experimental models prevents the initial series of events and delays the development of renovascular hypertension indefinitely. Activation of the sympathetic nervous system also plays an important role in the pathogenesis of renovascular hypertension32 primarily via the renal afferent nerves. Both the peripheral and central aspects of the autonomic system are also under the influence of Ang II. If the increase in pressure restores renal perfusion pressure distal to the stenosis, most of these alterations return to baseline levels, with the exception of peripheral vascular resistance.
It is important to recognize that activation of the systemic renin-angiotensin system is temporary in renovascular hypertension. After a period of time, circulating levels of plasma renin activity and angiotensin fall despite sustained elevation of peripheral vascular resistance. This may be the result of both (1) a slow response to Ang II, through which low levels of angiotensin have pressor actions, and (2) recruitment of additional mechanisms of vasoconstriction. The latter include activation of vasoconstrictor lipoxygenase products, oxidative stress, and endothelin. Additional rise in pressure results from an imbalance between vasoconstrictors and vasodilators, such as that derived from decreased bioavailability of nitric oxide (NO). An important role is ascribed to dissociation between systemic blood pressure, extracellular volume, and inappropriate levels of Ang II.33 The complexity of these relationships partly explains the failure of measuring any single pathway to predict blood pressure responses to renal revascularization.34
Accelerated Hypertension and Pulmonary Edema
Series of patients referred for renal revascularization in the last decade have included older patients with more widespread atherosclerotic disease than ever before.35,36 This reflects both improved medical care leading to better blood pressure control and reduced mortality from coronary and cerebrovascular disease. Patient demographics commonly include more women than men and a high prevalence of coronary disease, CHF, and known cerebrovascular disease. In some cases, suspicion arises regarding RAS because of rapid acceleration of these processes, particularly the rapid rise in arterial pressure in a previously stable patient. When untreated, a cycle of malignant-phase hypertension and hyponatremia (attributed to the dipsogenic action of Ang II) may ensue. In other cases, presenting symptoms include recent progression of hypertension followed by neurological symptoms of an acute stroke.
Some patients develop cycles of worsening CHF out of proportion to left ventricular (LV) dysfunction. This sometimes has been designated flash pulmonary edema.37 Many of these patients have bilateral disease or stenosis to a solitary functioning kidney. When volume expanded, renal function may improve slightly at the price of hypertension and circulatory congestion. Sudden pulmonary edema partly reflects diastolic dysfunction precipitated by a rapid rise in afterload38 in addition to impaired sodium excretion as a result of renal hypoperfusion. During volume depletion, serum creatinine commonly rises with evidence of prerenal azotemia. This condition warrants recognition because several series indicate that cycles of symptomatic exacerbation and hospitalization can be improved with successful renal revascularization.39,40
Renal Vasoactive Hormonal Systems
Angiotensin II
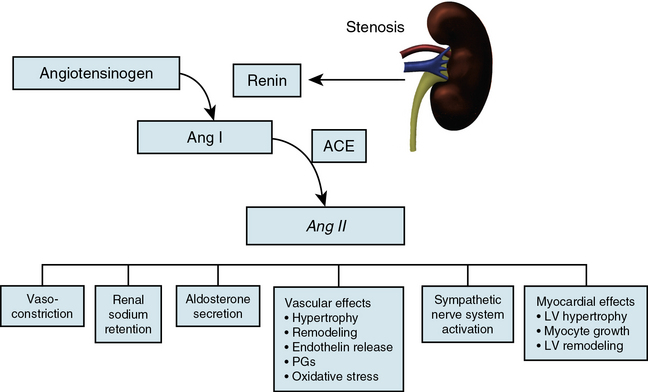
Renal hypoperfusion is accompanied by activation of the renin-angiotensin system, a mechanism normally designed to regulate volume homeostasis and maintain GFR during a transient decrease in renal perfusion pressure. Angiotensin II maintains glomerular capillary pressure and GFR by way of its predominant vasoconstrictor effect on the efferent arteriole. The importance of Ang II for preserving GFR is most evident under conditions of reduced preglomerular arterial pressures, particularly under conditions of volume depletion.41 This feature underlies the fall in GFR sometimes observed following administration of ACE inhibitors to patients with RAS, particularly when the entire renal mass is affected.
Angiotensin II effects in the kidney include induction of cell hypertrophy and hyperplasia, and stimulation of hormone synthesis and ion transport. It also seems to contribute to the pathogenesis of renal fibrosis by recruiting bone marrow–derived fibrocytes, circulating cells that contribute to the pathogenesis of fibrotic diseases.42 Its renal actions are mediated primarily through AT1 receptors expressed on endothelial, epithelial, and vascular smooth muscle cells (VSMCs). Chronic activation of AT1 receptors in renal ischemic injury may elicit local inflammatory and fibrogenic responses. Angiotensin II has been implicated in stimulation of vascular smooth muscle and mesangial cell growth, platelet aggregation, generation of superoxide, activation of adhesion molecules and macrophages, infiltration of inflammatory cells, increased expression of extracellular matrix (ECM) proteins, and induction of proto-oncogenes.
The intrarenal effects of Ang II during renal ischemia are modulated by interactions with other humoral systems (Fig. 22-8). Vasodilator prostaglandins (PGs) attenuate vasoconstriction caused by Ang II and may limit ischemia due to elevated levels of this hormone.43 Nitric oxide negates many actions of Ang II, modulates the effects of Ang II on the afferent arteriole and the proximal tubule, and down-regulates ACE and AT1 gene expression.44 On the other hand, endothelin-1 (ET-1) regulates renal ACE expression, mediates some of the vascular effects of Ang II and amplifies its pressor effects, and activates a similar signal transduction pathway for growth- and differentiation-related genes. Thromboxane A2, TxA2, a vasoconstrictor metabolite of arachidonic acid, is also released within the kidney by Ang II and mediates much of the pressor and renal hemodynamic responses to Ang II.45
Nitric Oxide
Nitric oxide is synthesized from L-arginine within the kidney by a family of nitric oxide synthases (NOS) and plays a crucial role in the regulation of renal hemodynamics and excretory function. Differential expression, localization, and regulation of three isoforms of NOS expressed in the kidney—neuronal (nNOS), inducible (iNOS), and endothelial (eNOS)—contribute to diverse intrarenal actions.46 Consequently, NO reduces vascular tone, increases sodium excretion, modulates tubuloglomerular feedback, has antithrombotic protection, inhibits growth-related responses to injury, and modulates the aforementioned renal actions of Ang II. Nitric oxide further buffers many processes implicated in the pathogenesis of tissue injury in renovascular disease, including growth of (VSMCs), mesangial cell hypertrophy and hyperplasia, and synthesis of ECM. However, regulation of renal blood flow becomes less dependent on eNOS-derived NO and more dependent on PGs as RAS progresses47 because of a decrease in renal perfusion pressure and vascular shear stress distal to the stenosis, which are primary stimuli to eNOS. On the other hand, the contralateral kidney continues to rely on NO to negate the actions of Ang II.48
The role of NO in renal tissue ischemia is complex, however. The iNOS isoform is up-regulated during renal ischemia49 and generates NO that can be cytotoxic to renal epithelial cells and contributes to tubular injury, both by decreasing activity of the eNOS isoform and by formation of the oxidant peroxynitrite.
Endothelins
The endothelin peptides comprise a family of peptides produced and released from endothelial cells (ECs), which have potent and long-lasting vasoconstrictor effects on the renal microcirculation and modify tubular function. Endothelin release can be stimulated by Ang II, thrombin, transforming growth factor (TGF)-β, and other cytokines (e.g., tumor necrosis factor [TNF]-α, interleukin [IL]-1β). Tissue levels of ET-1 are increased in the stenotic kidney,50 and in fact in most forms of renal failure, and may persist for days after resolution of the initial injury. Its involvement in renal ischemic injury is underscored by the observation that ET-1 blockade is more efficient in improving the early course of postischemic renal injury than inhibition of Ang II.51 Chronic blockade of the endothelin-A receptor directly inhibits cellular growth and gene expression, and in ischemic acute renal failure provides long-term functional and morphological benefits. These effects are greater than those observed during simultaneous blockade of both the A and B receptor,52 likely because of the role of the latter in eliminating salt, although the endothelin-B receptor has also been implicated in inflammation and fibrosis in progressive renal injury.53
Prostaglandins
Prostaglandins are cyclooxygenase (COX) derivatives of arachidonic acid that have important roles in maintaining renal blood flow and glomerular filtration. Biosynthesis of vasodilator PGs like prostacyclin (PGI2) and prostaglandin E2 protects the kidney against the effects of prolonged ischemia and Ang II43 and prevents hypoxic tissue injury. In RAS, they selectively prevent preglomerular constriction and thus limit a fall in GFR in the stenotic kidney,54 potentially through regulatory interactions with NO. Conversely, TxA2 is an endothelium-derived vasoconstrictor PG that is up-regulated in kidneys with renovascular disease.55 It is released within the kidney by reactive oxygen species (ROS) or Ang II, modulates some of the deleterious effects of Ang II and ET-1, and contributes to kidney disease. Blockade of TxA2 receptors thus improves urine volume, GFR, and renal plasma flow in ischemic kidneys and exerts a variety of beneficial effects that reduce the severity of ischemic damage. Furthermore, vascular expression of COX-2 is up-regulated in kidneys with arterial stenosis,56 and COX-2-derived prostaglandin I2 regulates renin release and renovascular hypertension in severe and moderate RAS.
Oxidative Stress
A growing body of evidence implicates increased generation of reactive radical species in the mechanisms of renal injury in renovascular disease.3,57 Angiotensin II is a potent stimulus for superoxide production via the membrane NADH/NADPH oxidase system, and xanthine oxidase is an important source of oxygen free radicals during renal ischemia. Increased oxidative stress can promote formation of a variety of vasoactive mediators including ET-1, leukotrienes, and prostaglandin F2α isoprostanes, endogenous products of lipid peroxidation. In addition, a chemical reaction between superoxide anion and NO not only decreases bioavailability of NO but also leads to production of toxic species (e.g., peroxynitrite [ONOO−]). Functionally, ROS have been implicated in decreasing stenotic kidney blood flow and sustaining renovascular hypertension.58,59
Furthermore, oxidative stress contributes to progressive tissue damage in the stenotic kidney. In mesangial cells, superoxide promotes hypertrophy and ECM production by both interaction with NO and by acting as an intracellular signal for growth-related responses, which may lead to microvascular and tissue remodeling.60 In addition, ROS are implicated in the pathogenesis of ischemic renal injury by causing lipid peroxidation of cell and organelle membranes and disrupting the structural integrity and capacity for cell transport and energy production, especially in the proximal tubule. Activation of growth factors and cytokines like nuclear factor (NF)-κB61 may also play an important role in the mechanism of action of Ang II and ROS. Studies in humans confirm that oxidative stress contributes to the impairment in endothelium-dependent vasodilation observed in patients with renovascular hypertension, which can be reversed with successful renal revascularization.62
The fibrogenic factors TGF-β, tissue inhibitor of matrix metalloproteinases (TIMP)-1, and plasminogen activator inhibitor (PAI)-1, which are up-regulated in stenotic kidneys,61 are important mediators of ECM synthesis that characterizes progression of renal tissue injury. Early induction of TGF-β via the AT1 receptor plays a major role in tissue fibrosis63 by increasing type IV collagen deposition and may play a role in interstitial scarring observed in chronic renal injury characterized by increased activity of intrarenal Ang II.64 It interacts with endothelin and several growth factors and cytokines in promoting progressive interstitial fibrosis, primarily via its downstream effectors from the Smad family, but also participates in postischemic renal healing.
Tubular Cells
Acute ischemic renal failure is characterized by a rapid decline in adenosine triphosphate (ATP) that leads to secondary cascades of cellular injury, including increases in intracellular calcium, activation of phospholipases, and generation of oxygen radicals, which cause significant surface membrane damage.65 The susceptible proximal tubule cells are primarily responsible for the pathophysiological and clinical aspects of ischemic acute renal failure. Of central importance is disruption and dissociation of the actin cytoskeleton and associated surface membrane structures that occur rapidly and are dependent on the severity and duration of ischemic injury.66 These alterations may be secondary to activation and relocation of the actin-associated protein actin depolymerizing factor/cofilin and β1-integrin to the apical membrane. Adenosine triphosphate depletion also induces necrotic cell death67 by opening a plasma membrane “death channel” normally kept closed in ischemic tissue by tissue glycine and decreased pH. The epithelial brush border may disappear in association with apical membrane blebbing, interruption of cell-to-cell junctions, and subsequently epithelial desquamation. Detachment of tubule cells and microvilli contributes to backleak of glomerular filtrate and formation of intraluminal aggregations of exfoliated cells, proteins, and glycoproteins (GPs) such as fibronectin (FN), resulting in tubular obstruction. The functional ramifications of these changes are substantial in terms of tubular reabsorption, function of the intercellular tight junction, impaired cell substrate adhesion, and integral membrane protein function.
Tubulointerstitial Injury
In patients with atherosclerotic RAS, the severity of pathological tissue damage is an important determinant and predictor of renal functional outcome.68 The earliest and most prominent pathological feature in renal ischemia is tubulointerstitial injury, which is considered to be the best prognostic factor in all nephropathies and may subsequently contribute to development of hypertension. The early phase of tubulointerstitial injury involves cellular activation, migration of mononuclear cells into the interstitium, leukocyte-endothelial interactions, and release of inflammatory products by myofibroblasts/activated fibroblasts. Altered antigenic profile of the tubular epithelium may initiate a cell-mediated immune response and be accompanied by interstitial inflammatory infiltrates composed of B lymphocytes, T-helper lymphocytes, and macrophages.69 Subsequently, immunosuppressive regulatory T cells (Tregs) promote repair during the healing process, possibly by regulating proinflammatory cytokine production of other T-cell subsets.70 Although the tubular lesions are initially reversible, tubulointerstitial injury may lead to irreversible fibrosis. A plethora of fibrogenic factors have been implicated in development of renal fibrosis following ischemic injury, such as TGF-β1, PAI-1, TIMP-1, α-1(IV) collagen, fibronectin-EIIIA (FN-EIIIA), tissue transglutaminase,71 and others, which may increase synthesis of ECM. Recent evidence suggests that in the context of atherosclerosis, matrix degradation is also impaired so that the overall matrix turnover balance favors fibrosis.
Glomerulosclerosis
In human atherosclerotic RAS, glomerulosclerosis is a relatively late sequela and is exacerbated by long duration, preexisting renal injury, and comorbid clinical conditions. In experimental models of chronic moderate RAS, glomerular lesions are initially minimal. Ischemia may elicit global or focal segmental glomerulosclerosis, manifested as segmental collapse or sclerosis, with or without reactive podocyte hypertrophy and proliferation. Initiation of glomerular cell apoptosis, thickening of the basement membrane, and expansion of the mesangial ECM involve progression of glomerulosclerosis.72 Presence of glomeruli that are not connected to normal tubule segments correlates with the concomitant decrease in GFR.
Cellular Death and Repair
Reactive oxygen species produced in renal proximal tubule epithelium under conditions of ischemia/reperfusion or hypoxia/reoxygenation are partly responsible for the apoptotic death of these cells.73 This process is partly mitigated by autophagy, a process of degradation and recycling of cytoplasmic constituents that may either contribute to cell death or ameliorate further cellular damage. During hypoxic and ischemic renal injury, autophagy seems to provide a protective mechanism and enhance cell survival.74
Despite various defense mechanisms that are activated during an ischemic insult, cell loss by apoptosis or necrosis does occur when renal hypoxia is severe. For reconstitution of its function, successful repair of the kidney requires rapid replacement of injured cells.75 Cell loss during kidney injury is followed by dedifferentiation and proliferation of adjacent surviving tubular cells, which are the chief contributors to tubular repair, and to a lesser degree interstitial kidney stem cells are stimulated to divide, migrate, and undergo phenotypical changes that allow them to replace lost cells. Bone marrow–derived cells likely contribute relatively little to this process.76 Nevertheless, exogenous administration of progenitor cells has been shown to improve renal function and attenuate renal damage in the chronic RAS in swine28,77 and ischemia/reperfusion injury in mice,78 suggesting a potential therapeutic utility for this experimental approach to preserve the ischemic kidney.
Renal Artery Disease and Mortality
Role of Disease Progression
Follow-up studies of patients with incidentally identified renal artery disease20 indicate that RAS independently predicts subsequent mortality. Rarely is this risk due to progressive renal disease alone, but more commonly to associated cardiovascular events. Mortality is remarkably similar in those treated with either medical management or renal revascularization.79–81 Data from recent prospective randomized trials with atherosclerotic RAS indicate that progression of renal dysfunction is relatively uncommon.82 Death is most commonly related to cardiovascular events, and only infrequently is progressive renal failure the primary cause of death.79,81 Some authors suggest that atherosclerotic disease affecting the kidney is a general marker of the degree of atherosclerotic burden. Others argue that renovascular disease augments these conditions and directly accelerates cardiovascular mortality.83 Available data do not support a direct role for renal revascularization to improve overall survival,84 although observational series indicate that patients experiencing an improvement in GFR after successful revascularization do in fact have reduced cardiovascular mortality over several years of follow-up.85,86
Survival is reduced in patients with bilateral renal artery disease or stenosis to a solitary functioning kidney. Prospective studies using Doppler ultrasound indicate that atherosclerotic lesions can progress in severity over periods of 3 to 5 years.87 Risks of progression are related to initial severity of the stenotic lesion and systolic blood pressure levels. It must be emphasized that clinical manifestations of renal artery disease within an individual patient may change over time. It is important that clinicians identify these transitions to consider interventions timed to when they are most likely to be effective.88
1 Fatica RA, Port FK, Young EW. Incidence trends and mortality in end-stage renal disease attributed to renovascular disease in the United States. Am J Kidney Dis. 2001;37:1184–1190.
2 Goldblatt H, Lynch J, Hanzal RE, et al. Studies on experimental hypertension I: the production of persistent elevation of systolic blood pressure by means of renal ischemia. J Exp Med. 1934;59:347–379.
3 Welch WJ, Mendonca M, Aslam S, et al. Roles of oxidative stress and AT1 receptors in renal hemodynamics and oxygenation in the postclipped 2K,1C kidney. Hypertension. 2003;41:692–696.
4 Crowley SD, Gurley SB, Oliverio MI, et al. Distinct roles for the kidney and systemic tissues in blood pressure regulation by the renin-angiotensin system. J Clin Invest. 2005;115(4):1092–1099.
5 Lorenz EC, Vrtiska TJ, Lieske JC, et al. Prevalence of renal artery and kidney abnormalities by computed tomography among healthy adults. Clin J Am Soc Nephrol. 2010;5:431–438.
6 Slovut DP, Olin JW. Current concepts: fibromuscular dysplasia. N Engl J Med. 2004;350:1862–1871.
7 Hansen KJ, Edwards MS, Craven TE, et al. Prevalence of renovascular disease in the elderly: a population based study. J Vasc Surg. 2002;36:443–451.
8 de Mast Q, Beutler JJ. The prevalence of atherosclerotic renal artery stenosis in risk groups: a systematic literature review. J Hypertens. 2009;27:1333–1340.
9 Gloviczki ML, Glockner JF, Lerman LO, et al. Preserved oxygenation despite reduced blood flow in poststenotic kidneys in human atherosclerotic renal artery stenosis. Hypertension. 2010;55:961–966.
10 Wiecek A, Kokot F, Kuczera M, et al. Plasma erythropoietin concentration in renal venous blood of patients with unilateral renovascular hypertension. Nephrol Dial Transplant. 1992;7:221–224.
11 Textor SC, Glockner JF, Lerman LO, et al. The use of magnetic resonance to evaluate tissue oxygenation in renal artery stenosis. J Am Soc Nephrol. 2008;19:780–788.
12 Textor SC, Wilcox CS. Renal artery stenosis: a common, treatable cause of renal failure? Annu Rev Med. 2001;52:421–442.
13 Rihal CS, Textor SC, Breen JF, et al. Incidental renal artery stenosis among a prospective cohort of hypertensive patients undergoing coronary angiography. Mayo Clin Proc. 2002;77:309–316.
14 Iglesias JI, Hamburger RJ, Feldman L, et al. The natural history of incidental renal artery stenosis in patients with aortoiliac vascular disease. Am J Med. 2000;109:642–647.
15 Dechering DG, Kruis HM, Adiyaman A, et al. Clinical significance of low-grade renal artery stenosis. J Intern Med. 2010;267:305–315.
16 De Bruyne B, Manoharan G, Pijls NHJ, et al. Assessment of renal artery stenosis severity by pressure gradient measurements. J Am Coll Cardiol. 2006;48:1851–1855.
17 Gross CM, Kramer J, Weingartner O, et al. Determination of renal arterial stenosis severity: comparison of pressure gradient and vessel diameter. Radiology. 2001;220:751–756.
18 Caps MT, Zierler RE, Polissar NL, et al. Risk of atrophy in kidneys with atherosclerotic renal artery stenosis. Kidney Int. 1998;53:735–742.
19 Pearce JD, Craven BL, Craven TE, et al. Progression of atherosclerotic renovascular disease: a prospective, population-based study. J Vasc Surg. 2006;44:955–963.
20 Conlon PJ, Little MA, Pieper K, et al. Severity of renal vascular disease predicts mortality in patients undergoing coronary angiography. Kidney Int. 2001;60:1490–1497.
21 Meyrier A, Hill GW, Simon P. Ischemic renal diseases: new insights into old entities. Kidney Int. 1998;54:2–13.
22 Kang DH, Kanellis J, Hugo C, et al. Role of the microvascular endothelium in progressive renal disease. J Am Soc Nephrol. 2002;13:806–816.
23 Urbieta-Caceres VH, Lavi R, Zhu XY, et al. Early atherosclerosis aggravates the effect of renal artery stenosis on the swine kidney. Am J Physiol Renal Physiol. 2010;299:F135–F140.
24 Radermacher J, Chavan A, Bleck J, et al. Use of Doppler ultrasonography to predict the outcome of therapy for renal-artery stenosis. N Engl J Med. 2001;344:410–417.
25 Mangiacapra F, Trana C, Sarno G, et al. Translesional pressure gradients to predict blood pressure response after renal artery stenting in patients with renovascular hypertension. Circ Cardiovasc Interven. 2010;99:999.
26 Evans RG, Gardiner BS, Smith DW, et al. Intrarenal oxygenation: unique challenges and the biophysical basis of homeostasis. Am J Physiol Renal Physiol. 2008;295:F1259–F1270.
27 Zhu XY, Chade AR, Rodriquez-Porcel M, et al. Cortical microvascular remodeling in the stenotic kidney. Role of increased oxidative stress. Arterioscler Thromb Vasc Biol. 2004;24:1854–1859.
28 Chade AR, Zhu X, Lavi R, et al. Endothelial progenitor cells restore renal function in chronic experimental renovascular disease. Circulation. 2009;119:547–557.
29 Loesch J. Ein Beitrag zur experimentellen Nephritis und zum arteriellen Hochdruck I. Die Veranderungen im Blutdruck II. Die Veranderungen in der Blutchemie. Zentralblatt fur Innere Medizin. 1933;7:144–169.
30 DeForrest JM, Knappenberger RC, Antonaccio MJ, et al. Angiotensin II is a necessary component for the development of hypertension in the two-kidney, one clip rat. Am J Cardiol. 1982;49:1515–1517.
31 Cervenka L, Horacek V, Vaneckova I, et al. Essential role of AT1-A receptor in the development of 2K1C hypertension. Hypertension. 2002;40:735–741.
32 Grisk O, Rettig R. Interactions between the sympathetic nervous system and the kidneys in arterial hypertension. Cardiovasc Res. 2004;61:238–246.
33 Reckelhoff JF, Romero JC. Role of oxidative stress in angiotensin-induced hypertension. Am J Physiol. 2003;284:R893–R912.
34 Safian RD, Textor SC. Medical progress: renal artery stenosis. N Engl J Med. 2001;344:431–442.
35 Conlon PJ, O’Riordan E, Kalra PA. Epidemiology and clinical manifestations of atherosclerotic renal artery stenosis. Am J Kidney Dis. 2000;35:573–587.
36 Textor SC, McKusick M. Renovascular hypertension and ischemic nephropathy: angioplasty and stenting. In: Brady HR, Wilcox CS. Therapy in nephrology and hypertension. London: WB Saunders; 2003:599–609.
37 Messina LM, Zelenock GB, Yao KA, et al. Renal revascularization for recurrent pulmonary edema in patients with poorly controlled hypertension and renal insufficiency: a distinct subgroup of patients with arteriosclerotic renal artery occlusive disease. J Vasc Surg. 1992;15:73–82.
38 Gandhi SK, Powers JC, Nomeir AM, et al. The pathogenesis of acute pulmonary edema associated with hypertension. N Engl J Med. 2001;344:17–22.
39 Missouris CG, Belli AM, MacGregor Ga. “Apparent” heart failure: a syndrome caused by renal artery stenoses. Heart. 2000;83:152–155.
40 Pickering TG, Herman L, Devereux RB, et al. Recurrent pulmonary oedema in hypertension due to bilateral renal artery stenosis: treatment by angioplasty or surgical revascularisation. Lancet. 1988;2:551–552.
41 Hall JE. Renal function in one-kidney, one-clip hypertension and low renin essential hypertension. Am J Hypertens. 1991;4:523s–533s.
42 Sakai N, Wada T, Matsushima K, et al. The renin-angiotensin system contributes to renal fibrosis through regulation of fibrocytes. J Hypertens. 2008;26:780–790.
43 Stebbins CL, Symons JD, Hageman KS, et al. Endogenous prostaglandins limit angiotensin-II induced regional vasoconstriction in conscious rats. J Cardiovasc Pharmacol. 2003;42:10–16.
44 Singh P, Deng A, Weir MR, et al. The balance of angiotensin II and nitric oxide in kidney diseases. Curr Opin Nephrol Hypertens. 2010;17:51–56.
45 Wilcox CS, Welch WJ. Angiotensin II and thromboxane in the regulation of blood pressure and renal function. Kidney Int. 1990;38:S81–S83.
46 Kone BC, Baylis C. Biosynthesis and homeostatic roles of nitric oxide in the normal kidney. Am J Physiol. 1997;272:F561–F578.
47 Tokuyama H, Hayashi K, Matsuda H, et al. Stenosis-dependent role of nitric oxide and prostaglandins in chronic renal ischemia. Am J Physiol. 2002;282:F859–F865.
48 Helle F, Hultstrom M, Skogstrand T, et al. Angiotensin II induced contraction is attenuated by nitric oxide in afferent arterioles from the non-clipped kidney in 2K1C. Am J Physiol Renal Physiol. 2010;296:F78–F86.
49 Chade AR, Rodriguez-Porcel M, Grande JP, et al. Distinct renal injury in early atherosclerosis and renovascular disease. Circulation. 2002;106:1165–1171.
50 Firth JD, Ratcliffe PJ. Organ distribution of the three rat endothelin messenger RNAs and the effects of ischemia on renal gene expression. J Clin Invest. 1992;90:1023–1031.
51 Jerkic M, Miloradovic Z, Jovovic D, et al. Relative roles of endothelin-1 and angiotensin II in experimental post-ischaemic acute renal failure. Nephrol Dial Transplant. 2004;19:83–94.
52 Forbes JM, Hewitson TD, Becker GJ, et al. Simultaneous blockade of endothelin A and B receptors in ischemic acute renal failure is detrimental to long-term kidney function. Kidney Int. 2001;59:1333–1341.
53 Neuhofer W, Pittrow D. Role of endothelin and endothelin receptor antagonists in renal disease. Eur J Clin Invest. 2006;36(Suppl 3):78–88.
54 Milot A, Lambert R, Lebel M, et al. Prostaglandins and renal function in hypertensive patients with unilateral renal artery stenosis and patients with essential hypertension. J Hypertens. 1996;14:765–771.
55 Welch WJ, Patel K, Modlinger P, et al. Roles of vasoconstrictor prostaglandins, COX-1 and -2, and AT1, AT2, and TP receptors in a rat model of early 2K,1C hypertension. Am J Physiol Heart Circ Physiol. 2007;293:H2633–H2699.
56 Therland KL, Stubbe J, Thiesson HC, et al. Cycloxygenase-2 is expressed in vasculature of normal and ischemic adult human kidney and is co-localized with vascular prostaglandin E2 EP4 receptors. J Am Soc Nephrol. 2004;15:1189–1198.
57 Lerman LO, Nath KA, Rodriguez-Porcel M, et al. Increased oxidative stress in experimental renovascular hypertension. Hypertension. 2001;37:541–546.
58 Chade AR, Krier JD, Rodgriguez-Porcel M, et al. Comparison of acute and chronic antioxidant interventions in experimental renovascular disease. Am J Physiol. 2004;286:F1079–F1086.
59 Palm F, Onozato M, Welch WJ, et al. Blood pressure, blood flow, and oxygenation in the clipped kidney of chronic 2-kidney, 1-Clip rats: effects of tempol and angiotensin blockade. Hypertension. 2010;55:298–304.
60 Carlstrom M, Lai EY, Ma Z, et al. Superoxide dismutase 1 limits renal microvascular remodeling and attenuates arteriole and blood pressure responses to angiotensin II via modulation of nitric oxide bioavailability. Hypertension. 2010;56:907–913.
61 Chade AR, Rodriguez-Porcel M, Grande JP, et al. Mechanisms of renal structural alterations in combined hypercholesterolemia and renal artery stenosis. Arterioscler Thromb Vasc Biol. 2003;23:1295–1301.
62 Higashi Y, Sasaki S, Nakagawa K, et al. Endothelial function and oxidative stress in renovascular hypertension. N Engl J Med. 2002;346:1954–1962.
63 Leask A. Potential therapeutic targets for cardiac fibrosis: TGF beta, angiotensin, endothelin, CCN2, and PDGF: partners in fibroblast activation. Circ Res. 2010;106:1675–1680.
64 Yang F, Chung AC, Huang XR, et al. Angiotensin II induces connective tissue growth factor and collagen I expression via transforming growth factor beta-dependent and -independent Smad pathways: the role of Smad3. Hypertension. 2009;54:877–884.
65 Kellerman PS. Cellular and metabolic consequences of chronic ischemia on kidney function. Semin Nephrol. 1996;16:33–42.
66 Sutton TA, Molitoris BA. Mechanisms of cellular injury in ischemic acute renal failure. Semin Nephrol. 1998;18:490–497.
67 Bonventre JV, Weinberg JM. Recent advances in the pathophysiology of ischemic acute renal failure. J Am Soc Nephrol. 2003;14:199–2210.
68 Wright JR, Duggal A, Thomas R, et al. Clinicopathological correlation in biopsy-proven atherosclerotic nephropathy: implications for renal functional outcome in atherosclerotic renovascular disease. Nephrol Dial Transplant. 2001;16:765–770.
69 Truong LD, Farhood A, Tasby J, et al. Experimental chronic renal ischemia: morphologic and immunologic studies. Kidney Int. 1992;41:1676–1689.
70 Gandolfo MT, Jang HR, Bagnasco SM, et al. Foxp3 + regulatory T-cells participate in repair of ischemic acute kidney injury. Kidney Int. 2009;76:717–729.
71 Johnson TS, El-Koriaie AF, Skill NJ, et al. Tissue transglutaminase and the progression of human renal scarring. J Am Soc Nephrol. 2003;14:2052–2062.
72 Makino H, Sugiyama H, Kashihara N. Apoptosis and extracellular matrix-cell interactions in kidney disease. Kidney Int. 2000;77(Supplement):S67–S75.
73 Chien CT, Lee PH, Chen CF, et al. De novo demonstration and co-localization of free radical production and apoptosis formation in rat kidney subjected to ischemia/reperfusion. J Am Soc Nephrol. 2001;12:973–982.
74 Jiang M, Liu K, Luo J, et al. Autophagy is a renoprotective mechanism during in-vitro hypoxia and in-vivo ischemia-reperfusion injury. Am J Pathol. 2010;176:1181–1190.
75 Guo JK, Cantley LG. Cellular maintenance and repair of the kidney. Annu Rev Physiol. 2010;72:357–376.
76 Lin F, Moran A, Igarashi P. Intrarenal cells, not bone marrow-derived cells, are the major source for regeneration in postischemic kidney. J Clin Invest. 2005;115:1756–1764.
77 Chade AR, Zhu XY, Krier JD, et al. Endothelial progenitor cells homing and renal repair in experimental renovascular disease. Stem Cells. 2010;28:1039–1047.
78 Li B, Cohen A, Hudson TE, et al. Mobilized human hematopoietic stem/progenitor cells promote kidney repair after ischemia/reperfusion injury. Circulation. 2010;121:2211–2220.
79 Chabova V, Schirger A, Stanson AW, et al. Outcomes of atherosclerotic renal artery stenosis managed without revascularization. Mayo Clin Proc. 2000;75:437–444.
80 Dorros G, Jaff M, Mathiak L, et al. Multicenter Palmaz stent renal artery stenosis revascularization registry report: four-year follow-up of 1,058 successful patients. Catheter Cardiovasc Interv. 2002;55:182–188.
81 Uzzo RG, Novick AC, Goormastic M, et al. Medical versus surgical management of atherosclerotic renal artery stenosis. Transplant Proc. 2002;34:723–725.
82 The ASTRAL Investigators. Revascularization versus medical therapy for renal-artery stenosis. N Engl J Med. 2009;361:1953–1962.
83 Rimmer JM, Plante DA, Madias NE. Therapeutic decision making in renal vascular hypertension. In: Novick AC, Scoble J, Hamilton G. Renal vascular disease. London: W.B. Saunders; 1996:245–266.
84 Textor SC, Lerman L, McKusick M. The uncertain value of renal artery interventions: where are we now? J Am Coll Cardiol Cardiovasc Interv. 2009;2:175–182.
85 Kennedy DJ, Colyer WR, Brewster PS, et al. Renal insufficiency as a predictor of adverse events and mortality after renal artery stent placement. Am J Kidney Dis. 2003;14:926–935.
86 Kalra PA, Chrysochou C, Green D, et al. The benefit of renal artery stenting in patients with atheromatous renovascular disease and advanced chronic kidney disease. Catheter Cardiovasc Interv. 2010;75:1–10.
87 Caps MT, Perissinotto C, Zierler RE, et al. Prospective study of atherosclerotic disease progression in the renal artery. Circulation. 1998;98:2866–2872.
88 Textor SC. Progressive hypertension in a patient with “incidental” renal artery stenosis. Hypertension. 2002;40:595–600.
89 Garovic V, Textor SC. Renovascular hypertension and ischemic nephropathy. Circulation. 2005;112:1362–1374.
90 Textor SC, Smith-Powell L. Post-stenotic arterial pressure, renal haemodynamics and sodium excretion during graded pressure reduction in conscious rats with one- and two-kidney coarctation hypertension. J Hypertens. 1988;6:311–319.
91 Lerman LO, Chade AR. Angiogenesis in the kidney: a new therapeutic target? Curr Opin Nephrol Hypertens. 2009;18:160–165.
92 Lerman L, Textor SC. Pathophysiology of ischemic nephropathy. Urol Clin North Am. 2001;28:793–803.
[/level-membership-for-cardiovascular-category][not-level-membership-for-cardiovascular-category]
Chapter 22 Pathophysiology of Renal Artery Disease
Vascular disease affecting the renal arteries presents complex challenges to clinicians. Thanks to recent advances in vascular imaging, more patients than ever before are being identified with some degree of atherosclerotic or fibromuscular renovascular disease. Many of these lesions are of minor hemodynamic importance at the time of detection. Some reach a degree at which perfusion pressures and intrarenal hemodynamics are altered, leading to changes in blood pressure regulation and renal function. These can produce a variety of recognizable clinical syndromes illustrated in Figure 22-1. These range from modest changes in systemic arterial pressure to impaired volume control associated with congestive heart failure (CHF) to threatened viability of the kidney, sometimes designated ischemic nephropathy. Understanding the pathways by which renovascular disease affects cardiovascular and renal disease is important for both diagnosis and for defining optimal management using tools both to block the renin-angiotensin system and to restore the circulation.
CHF, congestive heart failure; CV, cardiovascular.
(Modified with permission from Garovic V, Textor SC: Renovascular hypertension and ischemic nephropathy. Circulation 112:1362–1374, 2005.)89
Most renovascular lesions are the result of atherosclerosis. With the aging of the U.S. and other Western populations and reduced mortality from stroke and coronary disease, the prevalence of vascular disease in other vascular beds reaching clinically critical levels appears to be increasing.1 Understanding the variety of clinical manifestations of these lesions, the potential for disease progression, and the benefits and limitations of vascular repair are essential for vascular medicine specialists. This chapter will examine the pathophysiology of renovascular lesions regarding blood pressure control, ischemic nephropathy, and clinical syndromes such as flash pulmonary edema. Specific issues regarding diagnostic evaluation and management are addressed elsewhere (see Chapter 23).
A wide range of lesions can affect the renal blood supply, some of which are summarized in Box 22-1. Historically, recognition of renovascular disease resulted from searching for underlying causes of hypertension. This followed the seminal observations of Goldblatt more than 70 years ago2 that renal artery constriction produced a rise in arterial pressure in the dog. These studies were among the first to establish a primary role of the kidney in overall blood pressure regulation. Renovascular hypertension produced by a “clipped” renal artery remains among the most widely studied experimental forms of angiotensin-dependent hypertension.3,4
Box 22-1 Vascular Lesions That Produce Renal Hypoperfusion and Renovascular Hypertension Syndrome
Unilateral Disease (Analogous to Two-Kidney, One-Clip [2K1C] Hypertension)
Epidemiology of Renal Artery Disease
Fibromuscular disease may be identified in 1% to 3% of normal kidney donors subjected to angiography before donor nephrectomy.5 Of those developing clinical hypertension and referred for revascularization, more than 85% are females with a predilection for disease in the right renal artery.6 The location of these lesions is most commonly in the midportion and distal segments of the renal artery. A variety of fibromuscular lesions have been described, but the most common is medial fibroplasia. Occasionally, such lesions may be found in the carotid and other vascular beds, but most commonly they are limited to the renal arteries. Most do not progress to impair renal function, although some lead to arterial dissection and/or thrombosis with loss of the kidney.
Atherosclerosis is the most common cause of renal artery disease. Its presence and severity are related to age and the presence of other atherosclerotic disease of the descending aorta and lower extremities. Population-based series, such as one from North Carolina, indicate that among 834 subjects older than 65 years, significant renal artery stenosis (RAS; defined as Doppler peak systolic velocity (PSV) above 1.8 m/s) can be identified in 6.8% of the general population, regardless of race.7 Recent series of carotid, coronary, and peripheral angiography indicate that the prevalence of renovascular disease corresponds to overall atherosclerotic burden. Incidental renal artery occlusive disease (> 50% stenosis) has been reported in 11% to 18% of patients with coronary artery disease (CAD), particularly when significant hypertension is present.8 Peripheral vascular and aortic disease is associated with higher prevalence (25%-33%). As expected, risk factors predicting the presence of RAS include smoking, hyperlipidemia, hypertension, and diabetes. A corollary observation is that renovascular hypertension resulting from these lesions is now most commonly superimposed gradually upon preexisting essential hypertension. Hence, the blood pressure response and “cure” rates after successful restoration of blood flows to the kidney are limited by preexisting conditions.
Pathophysiological Consequences of Renovascular Disease
Under basal conditions, renal blood flow is among the highest of all organs. This feature reflects the kidney’s filtration function, and less than 10% of delivered oxygen is sufficient to maintain renal metabolic needs. Importantly, a fall in renal blood flow is accompanied by decreased oxygen consumption, partly due to reduced metabolic demands of filtration and tubular solute reabsorption. Reduced renal blood flow can be sustained without measurable change in total kidney oxygen levels (as assessed by renal vein oxygen tension),9 stimulation of erythropoietin release,10 or reduced medullary and cortical tissue oxygenation as measured in human subjects using blood oxygen level–dependent (BOLD) magnetic resonance (MR).11 These observations argue against an overall lack of oxygen as a primary stimulus for either hypertension or renal tissue injury and cast some doubt on the term ischemic nephropathy. Alternative terms proposed included azotemic renovascular disease and hypoperfusion injury.12 Nonetheless, severe vascular stenosis leading to diminished renal perfusion eventually does lead to renal tissue injury and interstitial fibrosis.
Subcritical Levels of Stenosis
The majority of renal artery lesions that compromise renal function are caused by gradually developing atherosclerosis of the renal vascular bed. As noted earlier, some patients undergoing cardiac catheterization have “incidental” renal lesions producing more than 50% cross-sectional stenosis,13 for whom the presence of RAS is a strong independent predictor of mortality. Moreover, nonobstructive RAS (20%-50% decrease in renal arterial luminal diameter) can be found in an additional 28% of patients undergoing cardiac catheterization13 and 48% of patients undergoing aortography for peripheral vascular disease.14 A recent systematic analysis of these reports including more than 15,000 subjects confirmed this overall range of disease prevalence and the rise with increasing atherosclerotic burden.8 Although lesions producing less than 50% in arterial luminal diameter are not considered hemodynamically significant, the relationships between resting pressure gradients and angiographic degree of stenosis are curvilinear and only approximate at best. As a predictor of mortality, even low-grade atherosclerotic lesions denote a hazard nearly equal to more advanced disease.15 Estimating severity of vascular occlusion from angiographic images is notoriously unreliable. It should be emphasized that activation of pressor mechanisms depends upon the presence of a pressure gradient between the aorta and distal renal vasculature16 (Fig. 22-2). There is a general relationship between estimated diameter stenosis and peak translesional pressure gradients, but the relationship is not linear. In some instances, an abrupt fall in poststenotic pressure develops beyond a subcritical range of stenosis.17 Even moderate stenosis, especially when superimposed on intrarenal microvascular disease, may contribute to adverse renal outcomes. Kidneys with a baseline renal artery disease classification of less than 60% stenosis have 11.7% 2-year cumulative incidence of renal atrophy (defined radiologically as a loss of kidney size)18 and 28% cumulative incidence of renal artery disease progression, although progression to total renal artery occlusion is uncommon.19 Increased severity of RAS in patients undergoing cardiac catheterization has an adverse effect on survival, with the 4-year adjusted survival for patients with a 50% stenosis decreasing to 70%, compared to 89% in patients without RAS.20 Therefore, even relatively minor stenosis in the renal artery might have long-term functional implications, especially in the presence of additional risk factors or coexisting renal disease.
(Reproduced with permission from De Bruyne B, Manoharan G, Pijls NH, et al: Assessment of renal artery stenosis severity by pressure gradient measurements. J Am Coll Cardiol 48:1851–1855, 2006.)16
Renal Microvascular Disease
Lesions in the main renal artery may be superimposed upon or confused with other causes for ischemic renal injury. Intrarenal vascular lesions are commonly observed in the course of various nephropathies, many of which have an ischemic component.21 Risk factors including diabetes, hypertension, atherosclerosis, and aging elicit vasoconstriction or structural changes leading to intrarenal small-vessel disease and ischemic injury similar to that observed in large-vessel disease. Loss of microvessels and impaired capillary repair correlate with development of glomerular and tubulointerstitial scarring,22,23 and may lead to end-stage renal failure. Renal microvascular disease distal to a stenosis in the renal artery may perpetuate and exacerbate renal parenchymal injury and may blunt renal recovery. The presence of small microvessel injury is difficult to verify but may account for changes in diastolic blood flow such as that producing changes in renal resistance index. Elevations of renal resistance index have been proposed to predict poor outcomes in many renal diseases, including renovascular disease.24
Critical Renal Artery Stenosis
High-grade vascular stenosis eventually leads to a decrease in renal perfusion pressure. Critical stenosis is identified when it produces a fall in renal blood flow and glomerular filtration rate (GFR). During experimental renal artery occlusion, the kidney sustains autoregulation of blood flow through a range of perfusion pressures from 200 mmHg to approximately 80 mmHg. Mechanisms underlying autoregulation include myogenic responses to changes in wall tension, release of vasoactive substances, and the tubuloglomerular feedback. The latter responds to decreased renal perfusion pressure and salt delivery by decreasing vascular resistance distal to the obstruction. In addition, during a fall in renal perfusion pressure, the kidney activates multiple pathways that elevate systemic blood pressure, an effect that tends to restore renal perfusion pressure and sustain renal blood flow at the expense of arterial hypertension (Fig. 22-3). Consequently, as long as systemic arterial pressure is allowed to rise, a fall in renal blood flow does not occur until renal arterial diameter is reduced by 65% to 75%. Recent clinical studies suggest that noninvasive radiological imaging commonly overstates the degree of stenosis. Measurement of physiological stimuli, such as the release of renin, indicate that a translesion gradient of at least 10% to 20% reduction is necessary for biological responses to occur in humans.16 To achieve such a gradient, luminal occlusion may need to exceed 80% stenosis. Under some conditions, gradients above 20 mmHg that develop during intrarenal hyperemic challenge with dopamine may disclose hemodynamic significance of lesions under 60% in severity.25
(Reproduced with permission from Textor SC, Smith-Powell L: Post-stenotic arterial pressure, renal haemodynamics and sodium excretion during graded pressure reduction in conscious rats with one- and two-kidney coarctation hypertension. J Hypertens 6:311–319, 1988.)90
When renal perfusion pressure falls gradually, additional mechanisms are recruited that protect the kidney from the functional and morphological consequences observed after acute ischemic injury. These include development of collateral vessels and redistribution of intrarenal blood flow from the cortex to the medulla. Renal cortical blood flow autoregulates more efficiently than the outer medulla, which is continuously on the verge of anoxia. During chronic reduction of renal blood flow, medullary perfusion and oxygenation are relatively maintained by adaptive mechanisms at the expense of cortical blood flow.26 When poststenotic renal artery pressures eventually fall further, either due to progressive vascular occlusion or reduction of systemic blood pressures by drug therapy, renal volume decreases.
In clinical terms, renal atrophy can be defined as a loss of renal length by at least 1 centimeter, and a difference in size between the two kidneys is suggestive of unilateral RAS (or a higher grade of stenosis in one of the kidneys). A decrease in renal volume results from a decrease in filling pressure, filtrate, and blood content of the kidney, as well as structural atrophy of the renal tubules due to apoptosis and necrosis. Apoptosis is an active, pre-programmed form of cell death that is intricately regulated and distinct from cellular necrosis and likely serves as a protective mechanism to allow renal “hibernation.” These changes may be reversible, since tubular cells show vigorous potential for regeneration. Loss of intrarenal microvessels that accompanies the ongoing scarring process may also contribute to renal shrinkage27 (Fig. 22-4), but might be partly reversible upon enhancement of angiogenic signaling.28 However, if a blood flow deficit persists, permanent damage to the kidney may occur. As mentioned, decreased renal blood flow is often accompanied by a decline in GFR and inhibition of tubular epithelial transport that limit renal oxygen consumption and maintain oxygen saturation. Hence, the kidney does not actually develop “ischemia” until an extreme decrease in renal blood flow develops.
Figure 22-4 Microcomputed tomography imaging of vascular structures in kidney cortex and medulla in a swine model.
(Reproduced with permission from Lerman LO, Chade AR: Angiogenesis in the kidney: a new therapeutic target? Curr Opin Nephrol Hypertens 18:160–165, 2009.)91
Renovascular Hypertension
Goldblatt et al and Loesch were the first to show in the 1930s that obstruction of the renal artery is followed by an increase in systemic blood pressure.2,29 The characteristics of renovascular hypertension depend to a large extent on the status of the kidneys. Unilateral RAS may be present with an intact contralateral renal artery (the experimental form is termed two-kidney, one-clip, or 2K1C). This model is characterized by counterregulatory processes in the contralateral kidney leading to sodium excretion in response to elevated arterial pressure (pressure natriuresis; Fig. 22-5A-B). Alternatively, RAS may affect a solitary kidney (one-kidney, one-clip, or 1K1C; Fig. 22-6A-B). Bilateral RAS and 1K1C lead to more severe renovascular hypertension, although bilateral RAS may behave similarly to 2K1C if one kidney is significantly less ischemic than the other. Patients with this constellation of findings have higher mortality, are more prone to circulatory congestion, and are more likely to experience deterioration of kidney function during administration of antihypertensive agents, including angiotensin-converting enzyme (ACE) inhibitors or angiotensin II (AngII) receptor blockers (ARBs).
[/not-level-membership-for-cardiovascular-category]
