Chapter 10 Pathophysiology of Cervical Myelopathy
Biomechanics and Deformative Stress
Clinical Patterns of Cervical Spondylotic Myelopathy
Clinical myelopathy typically appears in late adulthood in the setting of progressive degenerative changes, including cervical disc degeneration, osteophytic spur and transverse bar formation, posterior longitudinal ligament calcification, ligamentum flavum thickening, and osteoarthritic facet hypertrophy.1–3 Progressive encroachment on the spinal canal by ventral and dorsal anatomic structures may first lead to spinal cord compression—compression that occurs only transiently during physiologic cervical range of motion. The appearance of clinical signs and symptoms arising from this condition has been described as “dynamic stenosis.” With progressive narrowing of the spinal canal, dynamic compression may eventually evolve into static compression of the enclosed spinal cord and the appearance of classic CSM.
Retrospective observational studies indicate that development of CSM is more common in patients with underlying congenital stenosis of the spinal canal. A sagittal spinal canal diameter of less than 12 mm is strongly associated with signs and symptoms of myelopathy, whereas a diameter greater than 16 mm confers a low risk.4–8
Histopathology of Cervical Spondylotic Myelopathy
The theory that ischemic injury is the pathophysiologic basis of CSM originates in early histologic studies of cervical myelopathy, which revealed several changes consistent with ischemic tissue damage. These include cystic cavitation, gliosis, anterior horn cell dropout, and prominent involvement of the central gray matter, as well as wallerian degeneration of the posterior columns and corticospinal tracts.2,9–11 In these studies, the most severe histologic changes were observed at the level of ventral spondylotic bars, with the most visible histologic changes occurring in the lateral funiculi of the spinal cord, particularly the corticospinal tracts. The anterior columns and dorsal region of the dorsal columns appeared to demonstrate the least extent of injury-related change.
Spinal Cord Ischemia and Cervical Spondylotic Myelopathy
The anatomic basis for the ischemic insult proposed in CSM has been attributed to various mechanisms, including compression of radicular feeders in the neuroforamina, compromise of venous drainage by ventral spondylotic bars, and compression of the anterior spinal artery, as well as its ventral branches.12,13 Several animal studies support the concept of a potential role for compressive ischemia in the pathogenesis of CSM.14–16
Cadaver studies have demonstrated that flattening of the cervical spinal cord is associated with elongation of the laterally directed terminal branches of the central arteries arising from the anterior spinal artery, as well as elongation of the penetrating branches of the lateral pial plexus (corona radiata). It is hypothesized that attenuation of these transversely directed arteries results in decreased arterial blood flow to the corticospinal tracts. Shortening of the ventral-dorsal dimension of the spinal cord, however, results in widening of the arteries directed in the ventral-dorsal direction and relative preservation of blood flow to the anterior columns. These findings might explain the relative vulnerability to injury of the laterally positioned corticospinal tracts, compared with the anterior columns.17
Recent clinical studies strongly suggest that compression and ischemia alone do not fully explain the pathogenesis of CSM. Despite observational studies associating CSM with various anatomic factors, such as the presence of decreased ventral-dorsal spinal canal diameter, subluxation, and dorsal osteophytes, at least one study has demonstrated that these factors hold no significant predictive value in terms of identifying which patients are at risk for clinical progression of their myelopathy.18 Several other studies have also failed to identify an association between the degree of spinal stenosis and spinal cord compression and clinical prognosis.7,12,19
Moreover, surgical decompression that results in expansion of the spinal canal and relief of compressive pressures does not consistently alter the natural history of CSM.20 Ebersold et al.21 performed a retrospective review of 100 patients with CSM undergoing surgical decompression, with an average 7-year follow-up, and concluded that decompression alone resulted in no clear, long-term improvement. Two thirds of patients experienced initial clinical improvement, but half of these demonstrated subsequent clinical deterioration. At final follow-up, only a third of the original group were improved, leading the authors to conclude that long-term outcome was not predicated on the presence or severity of spinal cord compression and ischemia, but on other, “nonvascular” factors.
Biomechanical Factors and Cervical Spondylotic Myelopathy
There is a growing body of evidence indicating that abnormal or excessive motion of the cervical spine is strongly associated with clinical progression of CSM. In a retrospective clinical review, Adams and Logue12 demonstrated a cervical flexion-extension arc in excess of 40 degrees was the most significant variable in predicting poor clinical outcome in patients with CSM. Similar retrospective studies have been performed by Barnes and Saunders,18 as well as by Yonenobu et al.,19 in which patients with a flexion-extension arc of greater than 60 degrees after laminectomy were at increased risk for development of progressive myelopathy.
In contrast to the relatively poor results after simple decompression for CSM, several studies demonstrate excellent clinical results associated with the elimination of abnormal cervical motion. Using a simple neck brace to restrict cervical motion often leads to improvement in patients with cervical myelopathy from disc protrusions.22 The largest series of patients undergoing ventral decompression and fusion for CSM demonstrated an 86% improvement rate, with no significant deterioration.23 Most recently, Uchida et al.24 discovered that among patients with CSM who had kyphotic deformity in excess of 10 degrees, correction of sagittal alignment of the vertebrae significantly improved neurologic outcomes. Uchida et al. state that “ kyphotic alignment may contribute to cervical myelopathy,” that longitudinal distraction is a factor in progressive spinal cord dysfunction, and that the pathophysiologic mechanism is similar to that of tethered cord syndrome.24 Overall, surgical fusion through a variety of approaches has been associated with favorable clinical results, including ventral decompression and fusion without instrumentation21 or with ventral plating, 25–29 and dorsal decompression with instrumented fusion.30–33
The significant clinical recovery experienced by most myelopathic patients after decompression and fusion indicates that neurologic deficits resulting from cervical myelopathy are recoverable.23,25,26,29–31 Moreover, the rapid improvement experienced by many patients after surgery suggests that these patients do not have irreversible, ischemic histologic changes demonstrated in many early pathologic studies. In contrast, failure of some patients to improve clinically after decompression and fusion may be a result of irreversible spinal cord injury. Histologic examination of spinal cord tissue from these patients may reveal severe ischemic injury.2
Pathophysiology of Deformative Stress Injury of the Cervical Spinal Cord
The significance of spinal stenosis and spinal cord compression in early CSM may not be the generation of local ischemia, but rather the creation of a tethering effect, which results in production of local, potentially injurious, tissue strain and shear forces. The concept that increased cervical mobility, coupled with kyphotic deformity, results in spinal cord elongation and increased axial strain forces is well documented.12,13,17,18,24,34–41 Several studies have demonstrated the adverse effects of even low-grade mechanical stretching on neural tissues. During normal motion, large axial strains occur in the cervical spinal cord.42 The white matter of the spinal cord can be viewed as an axial array of parallel fibers, with individual fibers demonstrating variable levels of crimping. As a whole the cord is initially compliant to stretch, but it becomes progressively stiffer as the fibers straighten and begin to bear tensile load.35 Rapid occurrence of these strains can exceed the material properties of the tissue, leading to tissue disruption and transient or permanent neurologic injury. The degree of injury appears to be related to the peak strain of the tissue and the loading rate.43
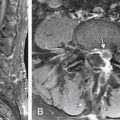
Cadaver studies suggest that even physiologic flexion of the cervical spine leads to stretching and the production of strain forces in the neuraxis.17 Flexion of the spinal column has been found to result in significant elongation of the spinal canal, with concomitant stretching of the spinal cord. During physiologic flexion of the head and trunk in rhesus monkeys, net movement of the spinal cord occurs from the upper spine downward to the level of C4-5, whereas net movement of the spinal cord occurs upward below this level.34 Net movement occurs to a greater extent below C4-5, with 1.6 mm of movement at C1 and 6 mm of movement at T3. The amount of spinal cord stretch occurring at each level is proportional to the degree of flexion at the adjacent intervertebral disc space. Thus, forces that are generated in the spinal cord upon flexion can be visualized with neutral and flexion MRIs of the cervical spine. Flexion of the neck results in significant elongation of the enclosed spinal cord (Fig. 10-1). The increase in length (l) over the original length of the same section of the spinal cord (lo) provides the strain (ε), thus:
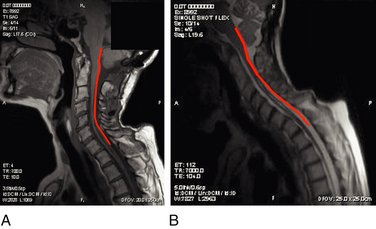
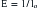
At the lower cervical and upper thoracic spine, where the amount of flexion tends to be greatest, local spinal cord strain can reach 24%. Thus, the strain produced at the cervicothoracic junction can exceed 0.2, the strain level at which the giant squid axon ceases to function.43 This phenomenon might explain the clinical observation that signs are often localized to levels apparently remote from the level of stenosis (e.g., hand intrinsic muscle wasting with high cervical stenosis).
In the absence of a compressive pathologic process, the natural elongation of the spinal cord that occurs with neck flexion and hyperextension is distributed over the entire length of the spinal cord. However, with tethering of the spinal cord, as a result of local compression, the axial strain cannot be distributed throughout the cord and is instead limited to the segment of cord between the distracting force and the tethering point. Local spinal cord degenerative changes are frequently identified adjacent to thickened dentate ligaments, which suggests that localization of injurious mechanical forces at these levels may be associated with the tethering effect of the ligaments.36,44 A biomechanical study of the material properties of the dura mater indicates that elastic behavior is uniform throughout the length of the spinal canal; however, strain forces are significantly greater in the cervical region than in either the thoracic or lumbar region.45
The tethering action of the dentate ligaments may be responsible for accentuating the effect of tensile spinal cord stress and exacerbating local tissue injury. Moreover, it has been suggested that dorsal displacement of the spinal cord, as a result of the presence of ventral spondylotic bars, may lead to stretching of the dentate ligaments and tethering of the cervical cord through the ventrolaterally positioned nerve root sleeves. Repetitive and persistent microtrauma to these nerve root sleeves may lead to the progressive thickening that has been observed with age.44 Therefore, axial tension generated in the spinal cord during physiologic motion may be amplified at certain levels, as a result of two separate factors—overall spinal canal lengthening and the local tethering effects of the dentate ligaments.
Several investigators have attributed delayed, progressive cervical myelopathy to a combination of underlying structural kyphosis and abnormal or excessive cervical motion.12,13,24,38 Dynamic lengthening of the cervical spinal cord that occurs during neck flexion is magnified in patients with cervical kyphosis. Conversely, kinematic MRI studies have demonstrated that lengthening of the spinal cord also occurs during neck extension in some patients with fixed kyphotic deformity of the cervical spine. In the setting of static spinal cord compression and superimposed instability, cervical extension can also lead to aggravation of the cord impingement and significant upper cervical spinal cord elongation.46
Mathematical Models of Spinal Cord Stretch Injury
Numerous mathematical models for spinal cord stretch injury have been developed. Levine36 represented the spinal cord as a simplified solid material with uniform elastic properties to predict the three-dimensional stresses experienced during physiologic motion and in spondylosis. According to this model, flattening of the cord is not a result of ventral-dorsal compression, but rather the consequence of laterally directed tension arising from the dentate ligaments, which tighten in flexion. This model, with a ventral spondylotic bar and tethering dentate ligaments, predicts maximal stresses in the lateral funiculi. The model provides a possible explanation for the characteristic histologic findings in CSM, in which there is relative sparing of the anterior and posterior funiculi. It also explains why histopathologic changes are found over a relatively extended segment of spinal cord tissue, as opposed to being limited to the point of compression. However, the importance of the dentate ligaments in the etiology of CSM is brought into question by the inconsistent results of sectioning these ligaments at the time of surgery.47
Breig38 also developed a mechanical model to explain some of the apparent inconsistencies found in histologic studies of CSM. For instance, in addressing the question of why some chronic, ventral compression injuries result in predominantly dorsal cord injury, cadaver models demonstrated that a compression force applied ventrally to the spinal cord in the presence of stenosis creates a pincer mechanism, resulting in increased axial tension in the cord and fissuring opposite the side of compression. In this model the spinal cord is represented as a viscoelastic cylinder that, when compressed from the sides, exhibits net tissue creep to the free ends of the cylinder. As a result, tension forces are created perpendicular to the plane of compression. With mild compressive deformation of the spinal cord, elastic stretch of the axis cylinders occurs. However, when the ventral-dorsal diameter of the spinal cord is reduced by 20% to 30%, axial tension forces exceed the material properties of the tissue and result in tearing of axial fibers. The stress field produced by this pincer mechanism is multidirectional, and secondary shearing forces are also created. This model explains how ventral compression of the spinal cord in the presence of stenosis might result in stretch and shear injury to myelin and neural elements.
Finite Element Models of Spinal Cord Stretch Injury
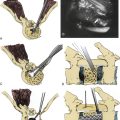
More recently, researchers have produced mathematical models of the cord using finite element analysis, a method adapted from materials science and fluid mechanics. Finite element analysis reduces a continuous structure into discrete, finite “brick” elements. This allows the approximation of partial differential equations by a linear system of ordinary differential equations, which can then be solved by numerical methods with the appropriate boundary conditions.48 In this particular case, the equations concern mechanical strain (stretch), “out of plane” loading (shear due to transverse compression, such as from a retroflexed odontoid process), and material properties such as Young’s modulus of elasticity or Poisson’s ratio. Ichihara et al.40 used finite element analysis to simulate the cervical spinal cord under compression and showed different amounts of stress at a given strain rate were to be expected owing to the differing material properties of gray and white matter. Kato et al.41 showed that the addition of a small amount of flexion to a model with static compression significantly increased predicted stresses, with the majority of stresses in the anterior and posterior horns. Henderson et al.49 demonstrated that increased deformative stresses in the corticospinal tracts, as predicted by the finite element analysis, were strongly correlated with neurologic deficits in a cohort of children with cervical and medullary symptoms. Elevated stress levels due to strain occurred during normal neck flexion in the spinal cord at the C1 level of one patient (MRIs from this patient are shown in Fig. 10-1); the addition of compression (shear) from a retroflexed odontoid process generates much higher stress levels with the same degree of flexion (Fig. 10-2).
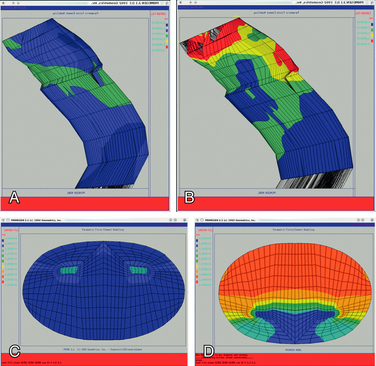
FIGURE 10-2 Finite element analysis of a portion of the cervical spine of the patient whose MRIs are shown in Figure 10-1. A, Sagittal view demonstrating the stresses on flexion. B, Sagittal view demonstrating more severe stresses on addition of local compression due to retroflexed odontoid with same degree of flexion as in A. C, Axial view at C1 of A. D, Axial view at C1 of B.
Spinal Cord Tethering and Shear Injury
Studies involving the tethered spinal cord syndrome may also contribute to a better understanding of the pathogenesis of CSM. Stretch injury is now widely accepted as the principal cause of myelopathy in tethered cord syndrome. The symptoms and clinical findings of pain, numbness, weakness, pes cavus, scoliosis, and bowel and bladder dysfunction have all been attributed to stretching injury of the spinal cord.50–56 The degree or amount of traction on the conus medullaris determines the age of onset of symptoms. Extensive tethering and severe stretching of the conus medullaris results in neurologic disturbances in infancy, whereas a lesser degree of tethering often remains subclinical until adulthood, when symptoms may become manifest in the setting of an acute event (i.e., hyperflexion injury) or chronic process (e.g., development of ventral disc or bone protrusions).57 Although clinical manifestations of tethered cord syndrome are more commonly referable to the lumbosacral spinal cord, many neurologic findings are referable to the cervical cord. For example, long tract involvement in tethered cord syndrome may lead to hand numbness and poor coordination, as well as upper extremity hyper-reflexia and even speech difficulties. Quadriparesis has also been reported.58 The phenomenon of increased strain supports the hypothesis that tension in the spinal cord might be transmitted to the brainstem and remote segments of the cord. Injury to the large-diameter fibers of the corticospinal tracts may occur some distance from actual tethering, and result in mixed upper and lower motor neuron deficits.57
Experimental studies involving the lumbar and sacral spinal cord of cats have demonstrated that acute tethering is very traumatic to spinal cord tissue, particularly when stretching occurs repeatedly.59 Spinal cord elongation is most pronounced immediately adjacent to the point of application of the tethering force. Under low levels of tension the spinal cord demonstrates purely elastic behavior and returns to normal resting length. At greater tension, plastic deformation occurs. Portions of the spinal cord near the point of application of stretch remain elongated by 7% over the original length, even after release of tension.
Tissue dysfunction in tethered cord syndrome has been associated with impairment of oxidative metabolism. The relationship of tissue ischemia to spinal cord stretching in this syndrome is unclear. Although a tethered cord may result in permanent neurologic deficit, the fact that surgical untethering usually results in significant improvement of sensorimotor and bladder function indicates a degree of reversibility that militates against a purely ischemic etiology.
A guinea pig model of spinal cord stretch injury has been developed in which the filum terminale was tethered and attached to a 5-g weight. Tethering resulted in significant delay and decreased amplitude of somatosensory evoked potentials. Lipid peroxidation and hypoxanthine levels were significantly increased. Electron microscopic examination of tissue revealed potentially reversible histologic changes, such as edema, destruction of the gray-white junction, axonal injury with loss of neurofilaments, and evidence of myelin sheath damage.60
Demyelinization of corticospinal tracts in trauma is similar to the demyelinization and edema seen in the posterolateral funiculi of patients with CSM.11,13,61–64 Autopsy studies of patients with rheumatoid arthritis and myelopathy have revealed edema localized to the posterolateral funiculi, as well as axonal retraction balls, suggestive of stretch-related injury without evidence of significant ischemia.37
The finding that tethering of the spinal cord in one region leads to generation of stretch and shear forces remote from the site of tethering or compression is directly applicable to numerous pathologic processes throughout the spine. The spinal cord can be tethered at any level by scarring, external compression, or spinal deformity. Spinal cord deformation over a large disc herniation at the apex of a thoracic kyphosis can contribute to stretch and shear injury remote from the locus of deformation.38 Similarly, deformation of the medullospinal junction over the odontoid process in basilar invagination results in both local and remote neurologic dysfunction (e.g., diplopia, dysphagia, dysarthria, vertigo), as well as sensorimotor deficits.65 Although these effects may also be explained by ischemic injury, local ischemia has not been found.37 Again, correction of medullospinal deformity through surgical removal of the odontoid process or traction/reduction and occipitocervical stabilization typically results in significant clinical recovery.66–68 Disturbances of sleep and alterations in central respiratory function have been attributed to ventral deformity of the upper spinal cord and lower brainstem in basilar invagination, and these disturbances have been reversed by correction of the ventral cervicomedullary deformity.39
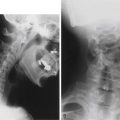
The neurologic dysfunction observed in association with an abnormally acute clivo-axial angle (CAA) is the result of deformation and deformative stress injury of the neuraxis. Kim et al.69 determined that an abnormal CAA caused subtle deformity of the upper spinal cord and medulla, resulting in headache, weakness, and sensory changes, as well as brainstem-related symptoms. Kubota et al.70 found that syringomyelia was more likely to resolve after treatment of a Chiari malformation if the CAA was more obtuse and the brainstem therefore straighter. Henderson et al.49 found that normalizing the CAA (increasing the angle to the normal 160 degrees) significantly improved neurologic function in a cohort of children with cervicomedullary syndrome due to an abnormal CAA. There is significant evidence that an abnormally acute CAA is indicative of a specialized form of brainstem tethering, which may produce the pattern of elevated stresses observed throughout the cord in cervical flexion myelopathy and CSM (Fig. 10-3).
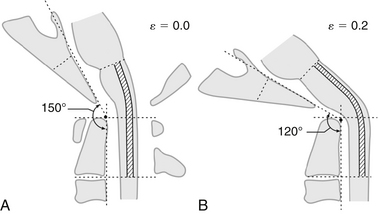
Histopathology of Spinal Cord Shear Injury
DAI is the most common brain injury resulting from blunt head trauma, and patient morbidity has been directly associated with the extent of DAI.71,72 Experimental primate models have demonstrated that the location and quantity of axonal changes directly correlate with observed morbidity.73,74 Clinical and pathologic studies have revealed that axonal injury is a component of traumatic brain injury throughout the spectrum of severity, from concussion to severe forms of prolonged coma.75 Despite these histopathologic observations, the pathogenesis of DAI remains unclear. An early hypothesis speculated that tearing of the axon at the time of injury resulted in expulsion of a ball of axoplasm into the brain parenchyma.76,77 However, recent DAI studies have demonstrated that axons undergoing shear strain do not undergo immediate disruption, but rather a nondestructive injury manifests as axonal swelling in internodal regions.78 Axonal stretch at the time of injury results in axolemmal damage, disruption of axon transport and metabolism, and the delayed formation of a retraction ball or reactive axonal swelling.78,79 This focal swelling is thought to be a prestage secondary axotomy.80–83
Studies have demonstrated that traumatic axonal injury results in impairment of anterograde axonal transport. In a guinea pig optic nerve model, 17% of axons demonstrated injury within 15 minutes of an applied stretch injury. The cell body of injured axons retained the ability to incorporate and transport horseradish peroxidase, but local interruption of axonal transport was demonstrated.84 In a separate study, axonal injury was localized to the nodes of Ranvier and manifested as axolemmal blebs, loss of subaxolemmal density, loss of nodal gap substance, and neurofilament disarray.82 Although distended, the axolemma remained intact. These findings suggest the possibility that stretch injury disrupts unidentified structural elements located at the node (i.e., membrane-associated proteins) that associate with the cytoskeleton and maintain nodal architecture. Furthermore, the study investigators speculated that nodal disruption leads to local cytoskeletal collapse and impairment of anterograde transport in a grossly intact axon.82
The cell ultrastructural events proceeding from axonal injury have been well characterized.80 Continued anterograde transport to the site of focal impairment appears to result in localized ballooning of the axon and formation of a reactive axon bulb, or “Strich ball.” Over the ensuing 1 to 3 days, the proximal axon segment containing the axon bulb continues to expand because of persistent anterograde transport and deposition of smooth endoplasmic reticulum and other intracellular organelles. These deposits become dispersed peripherally around an enlarged neurofilamentous core within the axon bulb. With further enlargement of the bulb, thinning of the overlying axolemma and myelin sheath occurs. Eventually, anatomic disconnection occurs between axonal segments proximal and distal to the original site of injury. The overlying myelin sheath is disrupted and then reforms to enclose the axon bulb, while the distal axonal segment undergoes wallerian degeneration. Meanwhile, the proximal axon bulb continues to expand as a result of continued anterograde transport of intracellular contents from the neuronal soma. In rodent studies, by 14 days, most reactive axons degenerate, become electron dense, and are eventually phagocytized by microglia. By contrast, in studies of mild to moderate head trauma in cats, some reactive axons have been observed to undergo a regenerative process, with outgrowth of regenerative sprouts and growth cones.85,86
Axon cytoskeletal collapse and rapid loss of the microtubular network appear to underlie the observed impairment of axoplasmic transport after injury.87 A quantitative analysis of injury-associated changes in the axoskeleton identified evidence of injury throughout the length of the axon: small axons demonstrated compaction of neurofilaments, larger axons demonstrated enlargement of the para-axonal space, compaction of neurofilaments, loss of microtubules, and reduction in axonal caliber. Neurofilaments have been implicated in maintenance of axon caliber, whereas microtubules are thought to provide the mechanism for fast axonal transport. Neurofilament compaction is thought to precede the cytoskeletal disappearance accompanying wallerian degeneration. Collapse of neurofilaments into tightly packed bundles in the center of the axon may precede secondary axotomy in nondisruptive stretch injury of central nerves.88
Injury-associated changes in the axonal cytoskeleton are preceded by alterations in axolemmal permeability. Intra-axonal accumulation of calcium has been demonstrated in focal spinal cord injury.89–92 Recently, increased calcium influx has been demonstrated in axons suffering stretch injury.93 Using a guinea pig optic nerve model, a characteristic sequence of cellular events has been observed to occur over 24 hours. Initially, tensile strain leads to mechanical disruption of the myelin lamellae surrounding the nerve. Presumed loss of activity of the ecto-Ca-ATPase pump at sites of myelin disruption is then thought to allow increased calcium influx into the myelin, possibly mediating myelin dissociation, and increased periaxonal space over several hours. Increased calcium influx into the injured axon results in proteolysis of neurofilaments and dephosphorylation of neurofilament side arms.94 In severe spinal cord injury, calcium-induced neurofilamentous degradation can be detected within 30 minutes.95
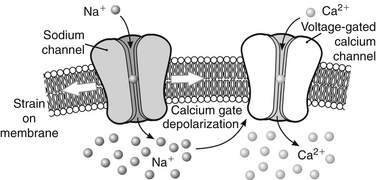
Abnormal strains in the spinal cord and brainstem from medullary kinking and basilar invagination result in predictable biomolecular changes: altered conformation of the Na+ mechanoreceptors causes increased intra-axonal Na+, which can be blocked with tetrodotoxin. The increased Na+ results in depolarization of the voltage-gated Ca2+ channels and reversal of the Na+/Ca2+ exchange pumps, with the consequence of abnormal influx of Ca2+ and activation of a deleterious cascade of reactions93,96–98 (Fig. 10-4).
Although increased calcium influx has been strongly implicated in neurofilamentous degradation by calcium-activated neutral proteases, some investigators question the relationship between calcium influx and the reactive axonal changes seen in stretch-related injury.80 Povlishock80 contends that neurofilamentous disarray is either a direct mechanical effect of trauma on the cytoskeleton or the result of increased neurofilament subunit exchange between stable neurofilaments and a pool of soluble kinetically active subunits.
Although changes in axolemmal permeability and cytoskeletal disruption appear to trigger a cascade of intra-axonal changes in moderate to severe injury, in mild injury reactive axonal changes and retraction balls have been demonstrated in the absence of any change in axolemmal permeability and without evidence of neurofilament or microtubule loss. In these instances it has been speculated that a “focal misalignment” occurs at the time of injury, resulting in impaired axoplasmic flow and delayed axotomy.99 It is conceivable that two different injury patterns exist and that the specific mechanism depends on the severity of tensile strain. In vitro studies have shown that axons under low tensile load undergo disruption of axoplasmic flow without evidence of axolemmal permeability change. High-tensile loading leads to immediate changes in axolemmal permeability and rapid disruption of axoplasmic flow.100
Anatomically, axons appear to be disrupted at sites of maximal tension. Large-caliber axons with a long intra-axial course appear to be more susceptible to tensile injury.57 Reactive changes have been observed in which axons change course, cross blood vessels, and decussate.101
Relationship between Ischemia and Shear Injury
Stretch injury renders axons more susceptible to secondary injury from other processes, including ischemia.101 However, the role of ischemia in stretch-related injury is unclear. Reactive axonal swelling occurs against a histologic background that lacks strong evidence of ischemic change. Microscopic studies have failed to identify correlative changes in microvasculature or ischemia-related changes in the neuronal soma, axonal processes, or dendritic processes.78 Iodoantipyrine studies have revealed no significant changes in regional blood flow.102,103 Moreover, axons undergoing reactive change are frequently found surrounded by intact neurons, without any evidence of ischemia or injury. When axonal injury is observed near the soma, central chromatolysis has been observed but may be secondary to pathologic processes within the axon. The rapid onset of axonal changes weighs heavily against a process originating in the neuronal soma.
The fact that some axons undergo reactive change while immediately adjacent axons appear uninjured is difficult to explain. It is conceivable, as Povlishock80 speculates, that specific differences in axonal anatomy, such as location of intra-axial turns, crossing points, and decussations, may make certain axons uniquely susceptible to injury at specific levels.
In the peripheral nervous system, axonal swelling can be seen in response to various insults (e.g., ischemia, severance, and crushing).80 Caution must be used before assigning a given etiology to the morphologic finding of axonal swelling.
Apoptosis
The pathogenesis of myelopathy is beginning to be investigated on a molecular level. Recent studies suggest that a significant portion of cell loss in chronic compression-related myelopathy is caused by apoptosis.104 The process of apoptosis is biologically distinct from necrosis and refers to a well-defined sequence of intracellular events that are characterized by internucleosomal chromosome fragmentation, membrane blebbing, and phagocytosis, without generation of an inflammatory response.105 In contrast, cell necrosis typically involves random DNA cleavage, membrane disruption, mitochondrial swelling, and local inflammation.106
Although necrosis and apoptosis often occur concurrently, identifying the dominant biologic process can provide important insight into the causes of specific disorders. In the case of CSM, the identification of primarily apoptotic cell death is significant. Although ischemia is one of numerous triggers associated with apoptotic cell death, severe ischemia such as that implied in the pathogenesis of CSM is more characteristically thought to cause cell death through necrosis. Therefore, the fact that oligodendrocyte disappearance in CSM appears to be apoptotic in nature suggests that a mechanism other than pure ischemia is involved.107 A prominent role for apoptosis has already been implicated in the secondary cell loss that occurs after traumatic spinal cord injury.107–111
Cell loss occurs in spinal cord injury, both at the time of injury and secondarily over a period of days to weeks. At the injury epicenter, most cell death occurs through necrosis and leads to active clearance of necrotic cell debris through macrophage and microglial phagocytosis.112 However, white matter cell loss continues through a longer segment of the spinal cord for several weeks in a process called secondary injury. Animal studies have demonstrated that compressive cord injury leads to apoptosis of oligodendrocytes along degenerating white matter tracts.107,109 These studies indicate that apoptosis begins within 24 hours of injury and continues for at least 3 weeks.
Strong evidence for the occurrence of apoptotic cell death in chronic compression-related cervical myelopathy comes from studies of an animal model of chronic compression-related cervical myelopathy, the tiptoe-walking Yoshimura mouse.113 The Yoshimura mouse is an inbred strain that characteristically develops quadriparesis 4 to 8 months after birth because of severe spinal cord compression, a result of hyperostosis along the posterolateral margins of the C1 and C2 vertebrae.114 Histopathologic examination of cord tissue from Yoshimura mice has demonstrated descending degeneration affecting the anterior and lateral columns, ascending degeneration along the posterior columns, as well as severe injury at the level of compression.104 Glial cell apoptosis mirrored the pattern of white column degeneration. Histologic staining using cell type-specific markers confirmed that the apoptotic cells were oligodendrocytes. The study investigators also performed an autopsy of a patient with cervical myelopathy from ossification of the posterior longitudinal ligament, and reported discovering a similar pattern of neuronal loss, demyelination, and apoptosis.
Stretch and strain are major epigenetic factors in trauma. For example, stretch results in the up-regulation of N-methyl-d-aspartate receptors. This renders the neuron more susceptible to ischemic insults and the effects of nitrous oxide and free radical species.115
Summary
Strong support for the shear and strain injury theory of CSM pathogenesis comes from several recent developments, including the clinical concept of “dynamic stenosis,” an increased neurobiologic understanding of the pathophysiology of stretch-related myelin and axonal injury, insight into the pathogenesis of spinal cord tethering, histologic studies revealing reactive axonal injury in the spinal cord of patients with CSM, and mathematical and finite element analysis modeling of the neuraxis under conditions of deformative stress.
Agrawal S., Fehlings M. Mechanisms of secondary injury to spinal cord axons in vitro: role of Na+, Na(+)-K(+)-ATPase, the Na(+)-H+ exchanger, and the Na(+)-Ca2+ exchanger. J Neurosci. 1996;16(2):545-552.
Arundine M., Aarts M., Lau A., et al. Vulnerability of central neurons to secondary insults after in vitro mechanical stretch. J Neurosci. 2004;24(37):8106-8123.
Breig A. Overstretching of the spinal cord: a basic cause of symptoms in cord disorders. J Biomech. 1970;3:7-9.
Chung R.S., Staal J.A., McCormack G.H., et al. Mild axonal stretch injury in vitro induces a progressive series of neurofilament alterations ultimately leading to delayed axotomy. J Neurotrauma. 2005;22(10):1081-1091.
Henderson F.C., Wilson W.A., Mott S., et al. Deformative stress associated with an abnormal clivo-axial angle: a finite element analysis. Surg Neurol Int. 2010;1:30.
Ichihara K., Taguchi T., Sakuramoto I., et al. Mechanism of the spinal cord injury and the cervical spondylotic myelopathy: new approach based on the mechanical features of the spinal cord white and gray matter. J Neurosurg. 2003;99(Suppl 3):278-285.
Jafari S., Maxwell W.L., Neilson M., et al. Axonal cytoskeletal changes after non-disruptive axonal injury. J Neurocytol. 1997;26:207-221.
Wolf J.A., Stys P.K., Lusardi T., et al. Traumatic axonal injury induces calcium influx modulated by tetrodotoxin-sensitive sodium channels. J Neurosci. 2001;21(6):1923-1930.
1. Taylor A.R. The mechanism of injury to the spinal cord in the neck without injury to the vertebral column. J Bone Joint Surg [Br]. 1951;33:543-547.
2. Payne E.E., Spillane J.D. The cervical spine: an anatomico-pathological study of 70 specimens (using a special technique) with a particular reference to the problem of cervical spondylosis. Brain. 1957;80:571-617.
3. Stoltman H.F., Blackwood W. The role of the ligamenta flava in the pathogenesis of myelopathy in cervical spondylosis. Brain. 1964;87:47-50.
4. Arnold J.G. The clinical manifestations of spondylochondrosis (spondylosis) of the cervical spine. Ann Surg. 1955;141:872-889.
5. Crandall P.H., Batzdorf U. Cervical spondylotic myelopathy. J Neurosurg. 1966;25:57-66.
6. Dunsker S.B. Cervical spondylotic myelopathy: pathogenesis and pathophysiology. In: Dunsker S.B., editor. Cervical spondylosis. New York: Raven Press; 1981:119-134.
7. Hayashi H., Okada K., Hashimoto J., et al. A radiographic evaluation of the aging changes in the cervical spine and etiologic factors of myelopathy. Spine (Phila Pa 1976). 1988;13:618-625.
8. Wolf B.S., Khilnani M., Malis L. The sagittal diameter of the bony cervical canal and its significance in cervical spondylosis. J Mt Sinai Hosp. 1956;23:283-292.
9. Hoff J., Nishimura M., Pitts L., et al. The role of ischemia in the pathogenesis of cervical spondylotic myelopathy: a review and new microangiopathic evidence. Spine (Phila Pa 1976). 1972;2:100-108.
10. Hughes J.T. Pathology of the spinal cord. Philadelphia: WB Saunders; 1978. pp 176–179
11. Ogino H., Tada K., Okada K., et al. Canal diameter, antero-posterior compression ratio, and spondylitic myelopathy of the cervical spine. Spine (Phila Pa 1976). 1983;8:1-15.
12. Adams C.B.T., Logue V. The movement and contour of the spine in relation to the neural complications of cervical spondylosis. Brain. 1971;94:569-586.
13. Mair W.G.P., Druckman R. The pathology of spinal cord lesions and their relation to the clinical features in protrusion of cervical intervertebral discs. Brain. 1953;76:70-91.
14. Al-Mefty O., Harkey H.L., Marawi I., et al. Experimental chronic compressive cervical myelopathy. J Neurosurg. 1993;79:550-561.
15. Doppman J.L. The mechanism of ischemia in anteroposterior compression of the spinal cord. Invest Radiol. 1975;10:543-551.
16. Gooding M.R., Wilson C.B., Hoff J.T. Experimental cervical myelopathy: effects of ischemia and compression on the canine cervical spinal cord. J Neurosurg. 1975;43:9-17.
17. Breig A., Turnbull I., Hassler O. Effects of mechanical stresses upon the cervical cord in cervical spondylosis: a study on fresh cadaver material. J Neurosurg. 1966;25:45-56.
18. Barnes M.P., Saunders M. The effect of cervical mobility on the natural history of cervical spondylotic myelopathy. J Neurol Neurosurg Psychiatry. 1985;47:17-20.
19. Yonenobu K., Okada K., Fuji T., et al. Causes of neurological deterioration following surgical treatment of cervical myelopathy. Spine (Phila Pa 1976). 1986;11:818-823.
20. Nurick S. The pathogenesis of the spinal cord disorder associated with cervical spondylosis. Brain. 1972;95:87-100.
21. Ebersold M.J., Pare M.C., Quast L.M. Surgical treatment for cervical spondylitic myelopathy. J Neurosurg. 1995;82:745-751.
22. Campbell A.M.G., Phillips D.G. Cervical disc lesions with neurologic disorder. Br Med J. 1960;2:481-485.
23. Eleraky M.A., Llanos C., Sonntag V.K.H. Cervical corpectomy: report of 185 cases and review of the literature. J Neurosurg. 1999;90:35-41.
24. Uchida K., Nakajima H., Sato R., et al. Cervical spondylotic myelopathy associated with kyphosis or sagittal sigmoid alignment: clinical article. J Neurosurg Spine. 2009;11(5):521-528.
25. Saunders R.L., Bernini P.M., Shireffs T.G.J., et al. Central corpectomy for cervical spondylotic myelopathy: a consecutive series with long-term follow-up evaluation. J Neurosurg. 1991;74:163-170.
26. Zdeblick T.A., Bohlmann H.H. Cervical kyphosis and myelopathy: treatment by anterior corpectomy and strut grafting. J Bone Joint Surg [Am]. 1989;71:170-182.
27. Bernard T.N.J., Whitecloud T.S.I. Cervical spondylotic myelopathy and myeloradiculopathy: anterior decompression and stabilization with autogenous fibular strut graft. Clin Orthop Relat Res. 1987;128:149-160.
28. Seifert V., Stolke D. Multisegmental cervical spondylosis: treatment by spondylectomy, microsurgical decompression and osteosynthesis. Neurosurgery. 1991;29:498-503.
29. Kojima T., Waga S., Kubo Y., et al. Anterior cervical vertebrectomy and interbody fusion for multilevel spondylosis and ossification of the posterior longitudinal ligament. Neurosurgery. 1989;24:864-872.
30. Maurer P.K., Ellenbogen R.G., Ecklund J., et al. Cervical spondylotic myelopathy: treatment with posterior decompression and Luck rectangle bone fusion. Neurosurgery. 1991;29:680-684.
31. Kumar V.G.R., Rea G.L., Mervis L.J., et al. Cervical spondylotic myelopathy: functional and radiographic long-term outcome after laminectomy and posterior fusion. Neurosurgery. 1999;44:771-777.
32. Cusik J.F., Steiner R.E., Berns T. Total stabilization of the cervical spine in patients with cervical spondylitic myelopathy. Neurosurgery. 1986;18:491-495.
33. Gonzalez-Feria L. The effect of surgical immobilization after laminectomy in the treatment of advanced cases of cervical myelopathy. Acta Neurochir (Wien). 1975;31:185-193.
34. Smith C.G. Changes in the length and position of the spinal cord with changes in posture in the monkey. Radiology. 1956;66:259-266.
35. Bilston L.E., Thibault L.E. The mechanical properties of the human cervical spinal cord in vitro. Ann Biomed Eng. 1996;24:67-74.
36. Levine D.N. The pathogenesis of cervical spondylotic myelopathy. J Neurol Neurosurg Psychiatry. 1997;62:334-340.
37. Henderson F.C., Geddes J.F., Crockard H.A. Neuropathology of the brainstem and spinal cord in end stage rheumatoid arthritis: implications for treatment. Ann Rheum Dis. 1993;52:629-637.
38. Breig A. Adverse mechanical tension in the central nervous system. New York: Wiley; 1978.
39. Howard R.S., Henderson F.C., Hirsch N.P., et al. Respiratory abnormalities due to craniovertebral junction compression in rheumatoid disease. Ann Rheum Dis. 1994;53:134-136.
40. Ichihara K., Taguchi T., Sakuramoto I., et al. Mechanism of the spinal cord injury and the cervical spondylotic myelopathy: new approach based on the mechanical features of the spinal cord white and gray matter. J Neurosurg. 2003;99(Suppl 3):278-285.
41. Kato Y., Kataoka H., Ichihara K., et al. Biomechanical study of cervical flexion myelopathy using a three-dimensional finite element method. J Neurosurg Spine. 2008;8:436-441.
42. Margulies S., Meaney D.F., Bilston L.B., et al. In vivo motion of the human cervical spinal cord during extension and flexion. In Proceedings of the IRCOBI Conference. Verona, Italy. 1992:213-224.
43. Galbraith J.A., Thibault L.E., Matteson D.R. Mechanical and electrical responses in the giant squid axon to simple elongation. J Biomech Eng. 1993;115:13-22.
44. Bedford P.D., Bosanquet F.D., Russell W.R. Degeneration of the spinal cord associated with cervical spondylosis. Lancet. 1952;2(6724):55-59.
45. Tencer A.F., Allen B.L., Ferguson R.L. A biomechanical study of thoracolumbar spine fractures with bone in the canal: III. Mechanical properties of the dura and its tethering ligaments. Spine (Phila Pa 1976). 1985;10:741-747.
46. Muhle C., Metzner J., Weinert D., et al. Kinematic imaging in surgical management of cervical disc disease, spondylosis and spondylotic myelopathy. Acta Radiol. 1999;40:146-153.
47. Bishara S.N. The posterior operation in treatment of cervical spondylosis with myelopathy: a long-term follow-up study. J Neurol Neurosurg Psychiatry. 1971;34:393-398.
48. Huebner K.H., Dewhirst D.L., Smith D.E., et al. The finite element method for engineers, ed 4. New York: Wiley-Interscience; 2001.
49. Henderson F.C., Wilson W.A., Mott S., et al. Deformative stress associated with an abnormal clivo-axial angle: a finite element analysis. Surg Neurol Int. 2010;1:30.
50. Sostrin R.D., Thompson J.R., Roue S.A., et al. Occult spinal dysraphism in the geriatric patient. Radiology. 1977;125:165-169.
51. Kaplan J.O., Quencer R.M. The occult tethered cord syndrome in the adult. Radiology. 1980;137:387-391.
52. Hoffman H.J., Hendrick E.B., Humphreys R.P. The tethered spinal cord: its protean manifestations, diagnosis and surgical correction. Childs Brain. 1976;2:145-155.
53. Garceau J.G. The filum terminale syndrome. J Bone Joint Surg [Am]. 1953;35:711-716.
54. Fitz C.R., Nash D.C. The tethered conus. Am J Neuroradiol. 1975;125:515-523.
55. Breig A. Overstretching of the spinal cord: a basic cause of symptoms in cord disorders. J Biomech. 1970;3:7-9.
56. Pierre-Kahn E., Lacombe J., Pichon J., et al. Intraspinal lipomas with spina bifida. J Neurosurg. 1986;65:756-761.
57. Pang D., Wilberger J.E. Tethered cord syndrome in adults. J Neurosurg. 1982;57:32-47.
58. Rogers H.R., Long D.M., Chou S.N., et al. Lipomas of the spinal cord and the cauda equina. J Neurosurg. 1971;34:349-354.
59. Tani S., Yamada S., Knighton R.S. Extensibility of the lumbar and sacral cord: pathophysiology of the tethered spinal cord in cats. J Neurosurg. 1987;66:116-123.
60. Kocak A., Kilik A., Nurlu G., et al. A new model for tethered cord syndrome: a biochemical, electrophysiological and electron microscopic study. Pediatr Neurosurg. 1997;34:120-126.
61. Wilkinson M. The morbid anatomy of cervical spondylosis and myelopathy. Brain. 1960;83:589-617.
62. Dastur D.K., Wadia N.H., Desai A.D., et al. Medullospinal compression due to atlantoaxial dislocation and sudden hematomyelia during decompression. Brain. 1965;88:897-927.
63. Wadia N.H. Myelopathy complicating congenital atlantoaxial dislocation. Brain. 1967;90:449-470.
64. Nakano K.K., Schoene C.W., Baker R.A., et al. The cervical myelopathy associated with rheumatoid arthritis: analysis of 32 patients with two postmortem cases. Ann Neurol. 1978;3:144-151.
65. Menezes A.H., van Gilder J.C., Clark C.R., et al. Odontoid upward migration in rheumatoid arthritis. J Neurosurg. 1985;63:500-509.
66. Kohno K., Sakaki S., Shiraishi T., et al. Successful treatment of Arnold-Chiari malformation associated with basilar impression and syringomyelia by the transoral approach. Surg Neurol. 1990;33:284-287.
67. Levy W.J., Mason L., Hahn J.F. Chiari malformation presenting in adults: a surgical experience in 127 cases. Neurosurgery. 1983;12:377-390.
68. Paul K.S., Lye R.H., Strange F.A., et al. Arnold-Chiari malformation: review of 71 cases. J Neurosurg. 1983;58:183-187.
69. Kim L.J., Rekate H.L., Klopfenstein J.D., Sonntag K.V.H. Treatment of basilar invagination associated with Chiari I malformations in the pediatric population: cervical reduction and posterior occipitocervical fusion. J Neurosurg. 2004;101(Suppl 2):189-195.
70. Kubota M., Yamauchi T., Saeki N., et al. Surgical results of foramen magnum decompression for Chiari type 1 malformation associated with syringomyelia: a retrospective study on neuroradiological characters influencing shrinkage of syringes. Spinal Surg. 2004;18(2):81-86.
71. Adams J.H., Mitchell D.E., Graham D.I., et al. Diffuse brain damage of intermediate impact type: its relationship to “primary brainstem damage” in head injury. Brain. 1977;100:487-502.
72. Adams J.H., Graham D.I., Murray L.S., et al. Diffuse axonal injury due to non-missile injury in humans: an analysis of 45 cases. Ann Neurol. 1982;12:557-563.
73. Gennarelli T.A., Thibault L.E., Adams J.H., et al. Diffuse axonal injury and traumatic coma in the primate. Ann Neurol. 1982;12:564-574.
74. Adams J.H., Doyle D., Ford I., et al. Diffuse axonal injury in head injury: definition, diagnosis, and grading. Histopathology. 1989;15:49-59.
75. Gennarelli T.A. Cerebral concussion and diffuse brain injuries. In: Cooper P.R., editor. Head injury. Baltimore: Williams & Wilkins; 1993:137-158.
76. Cajal S.R.Y. Degeneration and regeneration of the central nervous system. Oxford: Oxford University Press; 1928.
77. Strich S.J. Shearing of nerve fibers as a cause of brain damage due to head injury. Lancet. 1961;2:443-448.
78. Povlishock J.T., Becker D.P., Cheng C.L.Y., et al. Axonal change in minor head injury. J Neuropathol Exp Neurol. 1983;42:225-242.
79. Erb D.E., Povlishock J.T. Axonal damage in severe traumatic brain injury: an experimental study in cat. Acta Neuropathol. 1988;76:347-358.
80. Povlishock J.T. Traumatically induced axonal injury: pathogenesis and pathobiological implications. Brain Pathol. 1992;2:1-12.
81. Christman C.W., Grady M.S., Walker S.A., et al. Ultrastructural studies of diffuse axonal injury in humans. J Neurotrauma. 1994;11:173-186.
82. Maxwell W.L., Irvine A., Graham D.I., et al. Focal axonal injury: the early response to stretch. J Neurocytol. 1991;20:157-164.
83. Pettus E.H., Povlishock J.T. Characterization of a distinct set of intraaxonal ultrastructural changes associated with traumatically induced alteration in axolemmal permeability. Brain Res. 1996;722:1-11.
84. Gennarelli T.A., Thibault L.E., Tipperman R., et al. Axonal injury in the optic nerve: a model of diffuse axonal injury in the brain. J Neurosurg. 1989;17:244-253.
85. Lampert P.W. A comparative electron microscopic study of reactive, degenerating, and dystrophic axons. J Neuropathol Exp Neurol. 1967;26:345-368.
86. Friede R.L., Bischausen R. The fine structure of stumps of transected nerve fibers in subserial sections. J Neurol Sci. 1980;44:181-187.
87. Maxwell W.L. Microtubular changes in axons after stretch injuries. J Neurotrauma. 1995;12:363.
88. Jafari S., Maxwell W.L., Neilson M., et al. Axonal cytoskeletal changes after non-disruptive axonal injury. J Neurocytol. 1997;26:207-221.
89. Schlaepfer W.W. Calcium induced degeneration of axoplasm in isolated segments of rat peripheral nerve. Brain Res. 1974;69:203-215.
90. Ballentine J.D., Spector M.S. Calcification of axons in experimental spinal cord trauma. Ann Neurol. 1977;2:520-523.
91. Ballentine J.D. Pathology of experimental spinal cord trauma: II. Ultrastructure of axons and myelin. Lab Invest. 1978;39:254-266.
92. Ballentine J.D. Spinal cord trauma in search of the meaning of granular axoplasm and vesicular myelin. J Neuropathol Exp Neurol. 1988;47:77-92.
93. Maxwell W.L., McGreath B.J., Graham D.I., et al. Cytochemical evidence for redistribution of membrane pump calcium-ATPase and ecto-Ca-ATPase activity, and calcium influx into myelinated nerve fibers of the optic nerve after stretch injury. J Neurocytol. 1995;24:925-942.
94. Hall G.F., Lee V.M. Neurofilament sidearm proteolysis is a prominent effect of axotomy in lamprey giant central neurons. J Comp Neurol. 1995;353:38-49.
95. Chung R.S., Staal J.A., McCormack G.H., et al. Mild axonal stretch injury in vitro induces a progressive series of neurofilament alterations ultimately leading to delayed axotomy. J Neurotrauma. 2005;22(10):1081-1091.
96. Agrawal S., Fehlings M. Mechanisms of secondary injury to spinal cord axons in vitro: role of Na+, Na(+)-K(+)-ATPase, the Na(+)-H+ exchanger, and the Na(+)-Ca2+ exchanger. J Neurosci. 1996;16(2):545-552.
97. Wolf J.A., Stys P.K., Lusardi T., et al. Traumatic axonal injury induces calcium influx modulated by tetrodotoxin-sensitive sodium channels. J Neurosci. 2001;21(6):1923-1930.
98. Lusardi T.A., Smith D.H., Wolf J.A., et al. The separate roles of calcium and mechanical forces in mediating cell death in mechanically injured neurons. Biorheology. 2003;40:401-409.
99. Pettus E.H., Christman C.W., Giebel M.L., et al. Traumatically induced altered membrane permeability: its relationship to traumatically induced reactive axonal change. J Neurotrauma. 1994;11:507-522.
100. Galant P.E. The direct effects of graded axonal compression on axoplasm and fast axoplasmic transport. J Neuropathol Exp Neurol. 1992;51:220-230.
101. Povlishock J.T., Jenkins L.W. Are the pathobiological changes evoked by traumatic brain injury immediate and reversible? Brain Pathol. 1995;5:415-426.
102. Povlishock J.T. Diffuse deafferentation as the major determinant of morbidity and recovery following traumatic brain injury. Adv Neurotrauma Res. 1990;2:1-11.
103. Povlishock J.T. Current concepts on axonal damage due to head injury. In: Proceedings of the 11th International Congress of Neuropathology. Japan: Kyoto; 1991:749-753.
104. Yamaura I., Yone K., Nakahara S., et al. Mechanism of destructive pathologic changes in the spinal cord under chronic mechanical compression. Spine (Phila Pa 1976). 2002;27:21-26.
105. Cohen J.J. Apoptosis. Immunol Today. 1993;14:126-130.
106. Majno G., Joris I. Apoptosis, oncosis, and necrosis: an overview of cell death. Am J Pathol. 1995;146:3-15.
107. Li G.L., Brodin G., Farooque M., et al. Apoptosis and expression of Bcl-2 after compression trauma to rat spinal cord. J Neuropathol Exp Neurol. 1996;55:280-289.
108. Katoh K., Ikata T., Katoh S., et al. Induction and its spread of apoptosis in rat spinal cord after mechanical trauma. Neurosci Lett. 1996;216:9-12.
109. Crowe M.J., Bresnahan J.C., Shuman S.L., et al. Apoptosis and delayed degeneration after spinal cord injury in rats and monkeys. Nat Med. 1997;3:73-76.
110. Liu X.Z., Xu X.M., Hu R., et al. Neuronal and glial apoptosis after traumatic spinal cord injury. J Neurosci. 1997;17:5395-5406.
111. Springer J.E., Azbill R.D., Knapp P.E. Activation of the caspase-3 apoptotic cascade in traumatic spinal cord injury. Nat Med. 1999;5:943-946.
112. Grossman S.D., Rosenberg L.J., Wrathall J.R. Temporal-spatial pattern of acute neuronal and glial loss after spinal cord contusion. Exp Neurol. 2001;168:273-282.
113. Baba H., Maezawa Y., Imura S., et al. Quantitative analysis of the spinal cord motoneuron under chronic compression: an experimental observation. J Neurol. 1996;243:109-116.
114. Yamazaki M., Moriya H., Goto S., et al. Increased type XI collagen expression in the spinal hyperostotic mouse (twy/twy). Calcif Tissue Int. 1991;48:182-189.
115. Arundine M., Aarts M., Lau A., et al. Vulnerability of central neurons to secondary insults after in vitro mechanical stretch. J Neurosci. 2004;24(37):8106-8123.