[level-membership-for-cardiovascular-category]
Chapter 37 Pathophysiology, Epidemiology, and Prognosis of Aortic Aneurysms
Aortic aneurysms result in significant morbidity and mortality, accounting for nearly 13,000 deaths and 55,000 hospital discharges per year in the United States.1 Although aneurysms may affect any part of the aorta from the aortic root down to the abdominal aorta, the prognosis and outcome in patients with aortic aneurysms vary based on location and underlying etiology. Timely and appropriate intervention may improve the natural history of the disease process. This chapter reviews the pathophysiology, epidemiology, and prognosis of aortic aneurysms.
The Normal Aorta
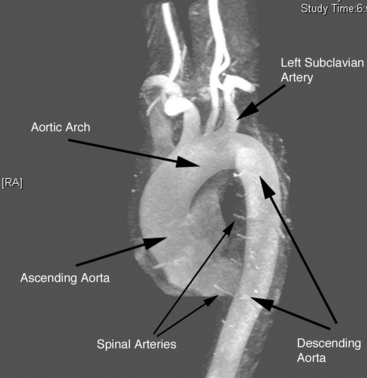
The aorta is the large conduit vessel through which the heart delivers blood to the entire body. It courses from the heart through the thorax and abdomen, and ultimately bifurcates into the common iliac arteries (CIAs) in the abdomen. In the thorax, the aorta can be subdivided into three segments: ascending aorta (from the base of the heart to the innominate artery), transverse aorta or aortic arch (including the great vessels and extending to the left subclavian artery), and descending aorta (from the distal edge of the subclavian artery to the level of the diaphragm) (Fig. 37-1).
Like other arterial structures, the aorta is composed of three layers: tunica intima, tunica media, and adventitia. The innermost surface of the tunica intima is lined by a single-cell-thick layer of endothelial cells (ECs). The intima is bound by the internal elastic lamina. The tunica media is composed of smooth muscle cells (SMCs), collagen, fibroblasts, elastin fibers, and ground substance, which together control the degree of vessel constriction and vasodilation. The presence of elastin fibers in the media defines the aorta as an elastic artery and provides the tensile strength that permits the aorta to withstand pulsatile delivery of blood from the heart. Elastin content gradually decreases with distance from the heart.2 The outermost layer, the adventitia, is a thin layer that contains connective tissue, fibroblasts, and the nutritive vasa vasorum.
Definition of Aortic Aneurysm
In adults, the normal diameter of the aorta is approximately 3 cm at the origin, 2.5 cm in the descending thoracic aorta, and 1.8 to 2 cm in the abdominal aorta. Aortic aneurysm is defined as a maximal aortic dimension greater than 3.0 cm, or a 50% increase in size compared with the normal segment proximal to the aneurysm. Mild expansion that does not meet these criteria may be referred to as aortic ectasia. True aneurysms are classified into two major groups on the basis of morphology: (1) fusiform (Figs. 37-2 and 37-3), defined as a circumferential expansion of the aorta, and (2) saccular, representing a focal outpouching of a segment of the aorta (Fig. 37-4). Fusiform aneurysms are the most common manifestation. In contrast to true aneurysms, which involve expansion of all three layers of the aortic wall, a pseudoaneurysm, also known as a false aneurysm, results from a disruption of the aortic wall and essentially represents a contained rupture of the aorta.
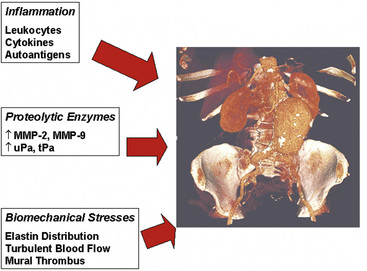
Figure 37-2 Three pathophysiological mechanisms that best characterize the process of aneurysm formation.
(Volume-rendered computed tomography image of abdominal aortic aneurysm used with permission of Joseph Schoepf, MD.)
Pathophysiology of Aortic Aneurysms
A wide variety of pathological states are associated with aortic aneurysms (Box 37-1). These include degenerative diseases, inherited disorders, infections, inflammatory conditions (i.e., vasculitis), and trauma. Specific disorders associated with aortic aneurysms are discussed later in this chapter. Important determinants of aortic aneurysm formation include inflammation, proteolysis of the structural components of the aortic wall, and abnormal biomechanical forces3 (see Fig. 37-2). Understanding the underlying pathophysiology of aneurysm formation is critical not only for prevention of initial aneurysm formation but also for limiting aneurysm growth and expansion.
 Box 37-1
Box 37-1Traditionally, pathological aortic aneurysm formation was ascribed to a process akin to atherogenesis. Although advances in basic and clinical investigation in both lesion types have revealed some common themes, newer studies suggest that aneurysm formation is fundamentally different from atherosclerosis. Preferential weakening of the adventitia and media—rather than an intimal proliferative process, as in atherosclerosis—results in diminished aortic resilience and tensile strength, culminating in aortic wall thinning, dilation, and increased wall stress, all of which may result in rupture. Although atherosclerotic changes may be seen in the wall of aneurysms, these changes may be a consequence of local turbulent flow as opposed to a cause of aneurysm formation.4,5 Moreover, the degree of systemic atherosclerosis does not correlate well with the degree of aneurysm formation.6
Development of aneurysms is associated with loss of two critical structural elements in the aortic wall: elastin and collagen. Elastin provides radial and longitudinal support, enabling the aorta to respond to pulsatile flow while maintaining normal arterial dimensions. The importance of elastin in maintaining aortic structure is highlighted by animal models where elastase infusion results in elastin breakdown and experimental aortic aneurysm formation.7 However, breakdown of elastin alone appears insufficient to cause aneurysmal expansion and rupture. Loss of collagen, another important structural element, is an additional contributor, and the relative balance of elastin and collagen deposition, among other factors, may be critical for determining aneurysm formation.8 Early in aneurysm formation, the aorta compensates for loss of elastin by increasing production of collagen,8 but as elastin content decreases, collagen (as the major source of tensile strength) is overwhelmed, and aortic expansion occurs. This is exacerbated by up-regulation of collagenases, resulting in further collagen degradation as described later.9 Structural changes in each layer of the aortic wall develop that together promote aortic stiffness. As a consequence, decrease in the vessel’s ability to distend normally with left ventricular (LV) contraction, weakening of the vessel wall, and increase in the tendency for dilation and ectasia follow.10 Some of the changes in aortic structure that promote aneurysm formation may arise as a result of the normal aging process. With normal aging, aortic stiffness due to fragmentation of elastin fibers, deposition of glycosaminoglycans, fibronectin (FN), and collagen, and reduced bioavailability of endothelium-derived nitric oxide (NO) occurs.11–14
Pathophysiologically, the major determinants of aortic aneurysm formation include proteolysis of the structural components of the aortic wall, inflammation, and abnormal biomechanical forces3 (see Fig. 37-2). Pathology of aortic aneurysms varies in different segments of the aorta and in different predisposing diseases. Frequently observed histological features include cystic medial necrosis, mucoid infiltration, and cyst formation in the setting of elastin necrosis and vascular smooth muscle apoptosis. In patients with Marfan’s syndrome (MFS), bicuspid aortic valve (BAV), or Turner’s syndrome, cystic medial necrosis is a common feature, but in contrast, inflammation is less prominent than in abdominal aortic aneurysms (AAAs).15,16 On the other hand, cystic medial necrosis is less likely to be observed in AAAs. Instead, AAAs typically have disrupted elastin fibers, inflammation, greater vascular smooth muscle cell (VSMC) apoptosis, and deficient glycosaminoglycan production.17 Elastin fragmentation occurs adjacent to the inflammatory cells. Despite differences in pathophysiology due to location and underlying etiology, formation of all aortic aneurysms involves to some degree the processes described in the following discussions (i.e., proteolytic degradation, inflammation, changes in biomechanical forces) that together facilitate aneurysm formation.
Proteolytic Degradation
Several proteolytic enzymes contribute to degradation of structural components of the arterial wall, ultimately increasing risk of aneurysm formation. Matrix metalloproteinases (MMPs) are endopeptidases that degrade one or more components of the extracellular matrix (ECM). Thus far, several classes of MMPs, comprising nearly 30 individual proteinases, have been characterized. They include collagenases, gelatinases, stromelysins, matrilysins, membrane-type (MT)-MMPs, and other MMPs.18 Typically produced as proenzymes, MMPs may be activated both intracellularly and extracellularly, and may be secreted by endothelial cells, vascular smooth muscle cells, or adventitial fibroblasts. Extracellular regulation of activation may occur as a result of an MMP–MT-MMP interaction or via interaction with plasmin or reactive oxygen species (ROS).19–21 The inhibitors of MMPs are the tissue inhibitors of matrix metalloproteinases (TIMPs) and plasminogen activator inhibitors (PAI) 1 and 2.18
Increased local production of MMPs in aortic aneurysms was first reported nearly 20 years ago.22–24 Although elevations of several MMPs have been noted, MMP-1 (collagenase) and MMP-3 (stromelysin), MMP-2 (gelatinase A), and MMP-9 (gelatinase B) represent the principal proteinases in aortic aneurysms that result in elastin and collagen degradation.25,26 MMP-2 and MMP-9 gelatinases specifically break down collagen. They are synthesized by local cells in the aortic wall, including infiltrating macrophages and resident aortic VSMCs.27,28 MMP-9 is typically found in the adventitia near the vasa vasorum, localizing to infiltrating macrophages.29 MMP-2 is synthesized constitutively by VSMCs30 but can also be synthesized by infiltrating leukocytes.31 Interestingly, MMP-2 production is increased in the vasculature remote from the aorta, suggesting a systemic underlying disease process that manifests with aortic aneurysmal disease.32
The centrality of these enzymes in aneurysm formation is supported by several lines of evidence. Studies have demonstrated that MMP-2 and MMP-9 levels are higher in tissue obtained from aortic aneurysms than in atherosclerotic plaque or normal arterial tissue.30,31 Furthermore, MMP-9 and MMP-2 knockout mice do not form aortic aneurysms in experimental models.33 Reinfusion of competent macrophages from wild-type mice into MMP-9 knockout mice enables aneurysm formation.33 In addition, elevated levels of MMP-1, MMP-8 (a neutrophil collagenase), and MMP-9 have been associated with aneurysm rupture, and levels of these MMPs may vary with aneurysm size.34–36 Relationship to aneurysm size is less clear for MMP-2.37–40
Elevated levels of other proteolytic enzymes such as human macrophage metalloelastase (MMP-12) and membrane type-1 metalloproteinase (MT1-MMP) have also been demonstrated in aortic aneurysms.41 Expression of MMP-12 is increased in aortic aneurysms as a result of macrophage infiltration. However, the relevance of MMP-12 to aneurysm formation is less clear, based on the finding that MMP-12 knockout mice are not completely protected from aneurysm formation.42 MT1-MMP, a collagenase produced by macrophages and increased in aortic aneurysms, likely has its greatest effect as an activator of the proenzyme form of MMP-2.43,44
In addition to increased expression of MMPs, aneurysm formation is associated with abnormal regulation of MMP levels in tissues. Matrix metalloproteinase levels are increased in states of inflammation and oxidant stress, both known to play a role in aneurysm formation. Compared with nonaneurysmal sections of aorta, superoxide anion and markers of oxidative stress are increased in aneurysmal segments, and ROS can convert proenzymes of MMPs to their active form.45–47 In addition, MMP levels can be augmented by plasminogen activators, such as urokinase-type plasminogen activator (uPA) and tissue-type plasminogen activator (tPA), which are specific physiological regulators of MMP-2 and MMP-9 activation and overexpressed in aortic aneurysms but not in healthy aortic specimens.48
Matrix metalloproteinase levels are also normally regulated by TIMPs. Although early work reported decreased concentrations of TIMPs in aneurysmal tissue,49 more recent data have been less clear, showing no difference in certain TIMP levels between aneurysmal and healthy aortic tissue.36,43,50,51 However, the relative imbalance in MMPs to TIMPs may be the more relevant factor.41 Nonetheless, experimental data have clearly demonstrated the significance of TIMPs to aneurysm formation. Local overexpression of TIMP-1 prevented aneurysm formation in a rat model of aortic aneurysm,52 but TIMP knockout mice have increased MMP activity and greater aneurysm formation.53 Similarly, lower expression of PAI-1, a regulator of plasminogen activator activity, has been reported in aneurysmal disease.49,54 Increased expression of plasminogen activators without reciprocal changes in their inhibitors alters the balance toward fibrinolysis, MMP activation, and tissue degradation. Experimental overexpression of PAI-1 prevents aneurysm formation in a rat model of aortic aneurysm,55 confirming the importance of relative rather than absolute concentrations.
The contribution of MMPs to the pathophysiology of aortic aneurysms is highlighted by the beneficial effect of medications known to reduce MMP levels on aneurysm expansion. For example, tetracyclines (e.g., doxycycline) have long been recognized as generalized inhibitors of metalloproteinases.56 Their potential value in aortic aneurysmal disease has been demonstrated in both organ culture and rodent models of aneurysm formation.36,57–59 In humans, doxycycline limits the activity of both MMP-2 and MMP-9 in aortic wall specimens, both by reducing macrophage MMP-9 messenger ribonucleic acid (mRNA) expression and diminishing activation of the proenzyme form of MMP-2.60–62 In a few small randomized phase 2 trials, doxycycline has been shown to decrease aneurysm expansion rate.63,64 Although these data await confirmation in larger trials, they do support the conceptual framework of the importance of proteolytic enzymes in aortic aneurysm formation and expansion.
By reducing levels of proteolytic enzymes, several other therapies may potentially be of therapeutic benefit in aneurysmal disease. HMG-CoA reductase inhibitors, or statins, have antianeurysm properties in experimental models by virtue of reducing oxidant stress and macrophage production of MMPs.65–67 However, data in humans have been inconsistent. A meta-analysis of five studies including 697 patients with small aortic aneurysms (<55 mm) treated with or without statins suggested that statin therapy was associated with lower rates of expansion.68 On the other hand, other human studies have not shown a benefit of preoperative statin treatment on MMP or TIMP levels in aneurysm specimens, and no difference in aneurysm expansion rate.69–71 Indomethacin also appears to prevent aortic aneurysm formation in a rat model,72 and the mechanism may be related to inhibition of cyclooxygenase (COX) 2, prostaglandin E2 (PGE2), and reductions in MMP-9.73–75 Indeed, PGE2 expression is up-regulated more than 30-fold in aortic aneurysms.76 Prostaglandin E2 localizes to infiltrating macrophages, and its expression is dependent on COX activity.77 Prostaglandin E2, through activation of interleukin (IL)-6, may increase VSMC apoptosis, further weakening the structural elements of the aorta.75,76,78
Inflammation
The contribution of inflammation to many pathological arterial processes has been well established.79 In 1981, Rose and Dent80 reported mild chronic inflammation in 72.5% and moderate inflammation in 15.7% in 51 consecutively resected AAAs. Subsequently it was discovered that lymphocytes and macrophages are found in greater quantity in the adventitia and media of AAAs than of atherosclerotic or normal aortas.81 In addition, surgical explant specimens from patients with aortic aneurysms demonstrated higher levels of adhesion molecules, including intracellular adhesion molecule (ICAM)-1 and vascular cell adhesion molecule (VCAM)-1, than seen in atherosclerotic and normal aortas. Similarly, tissue levels of proinflammatory cytokines, such as tumor necrosis factor (TNF)-α, interleukin (IL)-1β, IL-8, monocyte chemoattractant protein (MCP)-1, interferon (IFN)-γ, and IL-6, have all been noted to be elevated in patients with aneurysms compared with control subjects.82 Some studies have even suggested cytokine levels are higher in ruptured aortic aneurysms than in asymptomatic aortic aneurysms,83 although the data have been inconsistent.84 Finally, other acute-phase proteins, including C-reactive protein (CRP), D-dimer, and ceruloplasmin, are also present at increased levels in plasma and in the vessel wall.82,85–87
These inflammatory mediators largely derive from infiltrating macrophages, but lymphocytes and aortic ECs and SMCs also contribute to the inflammatory milieu. The presence of inflammation in aortic aneurysms is supported by positron emission tomographic imaging with18 fluorodeoxyglucose (FDG-PET) showing greater FDG uptake in aneurysmal compared to nonaneurysmal aortic segments from matched control subjects, and by advanced magnetic resonance imaging (MRI) techniques.88,89 Thus, immune cells invade the aortic wall, become activated, and create an inflammatory environment that engages the activity of local stabilizing cells, initiating the process of elastin and collagen breakdown and aneurysm formation. However, the initial signals that drive inflammatory cell recruitment remain unclear. Animal studies with experimental aneurysm models have confirmed the human studies and demonstrated that increased inflammation promotes aneurysm formation.90
Given the contribution of inflammation to aortic aneurysms, it follows that strategies to reduce inflammation might reduce aneurysm formation or limit aneurysm growth. Indeed, as mentioned previously, some data suggest that statins, known to have beneficial antiinflammatory properties beyond their effect on cholesterol lowering, may limit aneurysm growth and expansion.67,91,92 Recent studies have also shown that limiting inflammation can reduce aneurysm formation in animal models.93–97 Future studies will be required to clarify whether strategies to target inflammation can prevent formation of aortic aneurysms or limit expansion in humans.
Increases in Biomechanical Wall Stress
Several specific structural changes may predispose the abdominal aorta to aneurysm formation. For example, elastin within the aortic wall is organized into circumferential plates, or lamellae, that respond to the pulsatile load created by the heart. Each lamellar unit consists predominantly of two elastin bundles and vascular smooth muscle. However, deposition of elastin is not uniform along the aorta, with the thoracic aorta incorporating 35 to 56 lamellar units compared to only 28 in the abdominal aorta.98 The abdominal aorta may therefore be more susceptible to elastin breakdown due to a relative increase in pressure withstood per lamellar unit, compared with the rest of the vessel. In addition, the abdominal aorta has a decreased concentration of nutritive vasa vasorum compared to more proximal aortic segments.99 Reductions in aortic tissue perfusion stiffen the vessel, reducing compliance and ability to withstand pulsatile stress.100
Vascular cells in the aorta attempt to restore elastin content in the setting of elastin degradation to compensate for reductions in tensile and radial strength. Human AAA samples show a four- to sixfold increase in tropoelastin protein compared with control arteries.101,126 Elastin is produced by SMCs and macrophages. In areas of macrophage infiltration, elastin deposition is not organized into mature effective bundles. Indeed, compared with normal specimens, aneurysm specimens have a ninefold reduction in desmosine, a marker for mature elastin cross-linking.101 Thus, compensatory elastin replacement is disordered and does not improve aortic compliance.
Another factor that may make the abdominal segment of the aorta more prone to aneurysm formation is blood flow patterns specific to that segment. In experimental models, the infrarenal segment of the aorta is subject to much higher levels of oscillating flow and reflected pressure waves compared with the suprarenal segment,102 resulting in higher levels of aortic wall tension. Turbulence and pressure are exacerbated by the aneurysm’s morphology, which promotes development of local vortices and turbulent flow patterns.103 Excluding these flow patterns from the aneurysm by placing an aortic endograft rapidly reduces plasma MMP-9 levels in patients.104,105 In a rodent elastase infusion model of aortic aneurysm, flow conditions were examined by creating a left femoral arteriovenous fistula (AVF) or left iliac artery ligation. Increases in shear stress due to fistula formation resulted in a more stable aortic phenotype with decreased oxidative stress, decreased macrophage density in the media, increased aortic ECs and vascular smooth muscle cells, and reduced apoptosis compared to the lower shear stress introduced by femoral artery ligation.4,106,107 Improved flow decreased aortic expansion by 26%.106
In addition to adverse biomechanical forces creating an environment permissive for aneurysm formation, the role of intraluminal thrombus on wall stress and aneurysm expansion has recently been a focus of much study. Using finite element analysis in a three-dimensional (3D) model of the aorta derived from computed tomography (CT) scans, intraluminal thrombus was found to lower peak wall stress by up to 38%108 relatively independent of thrombus constituents.109 Thrombus decreases transmission of luminal pressure to the aneurysm wall and may prevent aneurysm rupture by reducing wall strain.110–115 On the other hand, intraluminal thrombus may also contribute to further aneurysm formation,111 given that thrombus has been demonstrated to have higher levels of proteolytic enzymes and enzyme activators than the aneurysm itself.116 Intraluminal thrombus may act as a proteolytic enzyme reservoir through polymorphonuclear leukocytes, and enzyme accumulation provides a ready source of destructive elements for the adjacent aneurysm.116
Understanding biomechanical factors may help improve assessment of risk of aneurysm growth and rupture above and beyond the predictive value of lumen diameter alone.117 Several studies have used novel computational models to incorporate biomechanical factors such as wall stress, wall strength, and extent of intraluminal thrombus for assessment of abdominal aneurysms.112,118–123 Some studies have suggested that assessment of wall stress, wall strength, and the ratio of wall stress to wall strength may be better predictors of rupture risk than diameter.124,125
Epidemiology and Prognosis of Aortic Aneurysms
Abdominal Aortic Aneurysms
Prevalence
The prevalence of aneurysms of the abdominal aorta has been determined on the basis of several large screening studies and autopsy series (Table 37-1). In an early series of 24,000 consecutive autopsies performed over 23 years, 1.97% of the subjects were found to have an AAA.126 Of the 473 aneurysms found, 58% were larger than 4 cm in diameter, nearly three quarters of the patients were men, and one fourth of the aneurysms had ruptured. More recent large screening programs in targeted populations have further evaluated the prevalence of AAA. The largest screening program performed was the Aneurysm Detection and Management (ADAM) Study Screening Program, which studied 126,196 veterans 50 to 79 years of age.127 In this cohort of predominantly male American veterans, 3.6% of subjects had an infrarenal aortic diameter greater than 3 cm, and an AAA 4 cm or larger was found in 1.2%. The Multicentre Aneurysm Screening Study (MASS) also screened 27,147 of 33,830 invited men aged 65 to 74 and reported a 4.9% prevalence of AAA 3 cm or larger.128
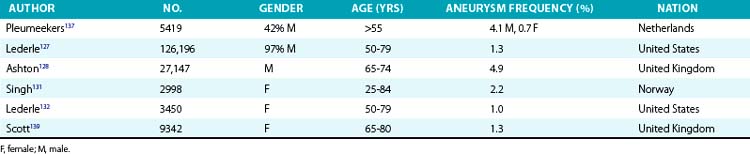
Several studies have demonstrated lower prevalence of AAA in women. The largest and most recent of these studies screened nearly 10,012 women (mean age, 69.6 years) and found an AAA prevalence rate of 0.7%, with only 4 of 74 detected aneurysms measuring larger than 5 cm.129 These low prevalence rates were consistent with findings from earlier studies. Among 4237 subjects aged 65 to 80 who participated in a screening study among general practitioners in West Sussex, United Kingdom, 2290 women agreed to undergo abdominal ultrasonography.130 Only 1.4% of the women had an AAA 3 cm or larger, and only 0.3% had an AAA 4 cm or larger. This dramatically lower prevalence in women has been confirmed in subsequent studies. In the Norwegian Tromso study, 2% of 2943 women aged 55 to 84 had an AAA 3 cm or larger, and 0.5% had an abdominal aneurysm 4 cm or larger.131 Finally, among female American veterans, the prevalence of an AAA 3 cm or larger was just 1%.132 However, an increasing number of cardiovascular risk factors does increase the risk of AAA in women, with a prevalence rate as high as 6.4% in this higher-risk group.129
Risk factors
Three risk factors predict the vast majority of AAAs: age, gender, and cigarette smoking. Aneurysms usually affect the elderly, seldom occurring in those younger than 60 years of age, and there is a clear increase in incidence with increasing age, even when limiting the studies to older individuals.133–135 In a Norwegian population-based study of 6386 men and women aged 25 to 84, the incidence of AAA in men increased from 0% in those aged 25 to 44, to 6% in those aged 55 to 64, and to 18.5% in those aged 75 to 84.131 Large North American epidemiological studies have demonstrated an increase in AAA risk ranging from 58% to 300% with each additional decade of life.136 Gender is also an important predictor of AAA formation; in all age groups, risk of AAA is two- to sixfold higher in men than women.131,132,137–140 Cigarette smoking is the most potent modifiable risk factor and increases the risk of AAA by 60% to 850%.141–147 In the ADAM study and the Edinburgh Artery Study, risk of an aneurysm increased threefold with any smoking history.127,148 Risk of AAA development further increases with number of cigarettes smoked, duration of smoking, and lack of filtration, indicating a dose-response relationship.149 In the Whitehall study of 18,403 male civil servants examined at age 40 to 64 years, aneurysm frequency increased from sixfold with manufactured cigarettes with filters to 25-fold with hand-rolled cigarettes.150 Smoking cessation can reduce the risk of aneurysm formation, with former smokers having a lower AAA risk than current smokers.136,149,151
Risk factors for cardiovascular disease in general (e.g., hypertension, hyperlipidemia) also increase the risk of AAA formation but are less potent risk factors than age, gender, and smoking.127,136,152–154 In the REACH registry, there was a clear modest association of both hypertension and hyperlipidemia with AAA.155 Earlier studies had suggested a less consistent relationship with hypertension,138,156 but aneurysm formation seems to correlate best with diastolic blood pressure157 or use of an antihypertensive medication.140 Some data suggest that hypertension may be a greater risk factor for aneurysm rupture than for initial aneurysm formation.158 The association between cholesterol and AAA is clear, although hyperlipidemia is a less potent risk factor than those mentioned previously.135,137,159 Risk of AAA formation increased 30% per 40 mg/dL total cholesterol in the Chicago Heart Association Detection Project in Industry cohort.138 For specific components of the lipid profile, higher levels of low-density lipoprotein (LDL) and lower levels of high-density lipoprotein (HDL) cholesterol are both associated with aneurysm formation.133,159 Similarly, the presence of atherosclerosis increases risk of aneurysm formation,133,134,137 although as mentioned previously, aneurysm formation is likely a process distinct from atherosclerosis. In the ADAM study of more than 100,000 subjects, hypertension, elevated cholesterol, and presence of other vascular disease increased risk of aneurysm formation by 15%, 44%, and 66%, respectively.127 In contrast, diabetes and black race appear protective against formation of an aneurysm.127,132,136,160 Diabetes decreases risk of aneurysm formation by 30% to 50%.136,157
A dramatic increase in frequency of AAA formation in relatives of patients with aortic aneurysm suggests a genetic component to the disease. Although cigarette smoking numerically accounts for the vast majority of AAAs in the population,136 the most potent risk factor for aneurysm formation is a history of aneurysm in a first-degree relative. Norgaard et al.161 identified an 18% incidence of aneurysms in first-degree relatives of patients with AAA. In the ADAM study, a family history doubled the risk of AAA, but was reported in only 5.1% of more than 100,000 participants.162 Investigations specific to the impact of family history demonstrate a larger risk. Several studies have demonstrated that a family history of AAA increases the risk of AAA four- to fivefold.157,163 Family history of AAA was also related to earlier AAA formation and rupture by nearly a decade.164 Rate of rupture was nearly fourfold higher in patients with a family history than in sporadic AAA patients. Frydman et al.165 screened the siblings of 400 AAA patients and found an AAA in 43% of male siblings and 16% of female siblings. More specifically, the risk of AAA formation consistently rises above 20% for men older than 50 who have a first-degree relative with AAA.161,166–169 Using segregation analysis, Majumder et al.170 reported that the relative risk of developing an AAA is 3.97 and 4.03 with paternal and maternal history, respectively. Risk increases to nearly 10-fold with an affected male sibling and 23-fold when a female sibling is affected.170 Twin studies further support a genetic component to AAA formation, with one study reporting an odds ratio (OR) of 71 (95% confidence interval [CI], 27-183) for monozygotic twins and 7.6 (95% CI, 3.0-19) for dizygotic twins.171
Despite the wealth of data supporting a genetic component to AAA formation, no clear mode of inheritance and no single candidate gene has been identified. Early studies suggested evidence of both sex-linked and autosomal dominant patterns of inheritance. Associations have been made with blood types, haptoglobin variations, α1-antitrypsin, and human leukocyte antigen class II (HLA-II) immune response genes.172–174 More than 100 reports on genetic associations with AAA have appeared in the literature,175 including genes related to cardiovascular disease, inflammation, and related signaling pathways. One study suggested an association between reduced AAA growth and five single-nucleotide polymorphisms (SNPs) in latent TGF-β binding protein (LTBP4), as well as an allelic variant of TGFB3.176 However, another study showed no association between genetic polymorphisms in the main receptors for TGF-β and AAA formation.177 Two genome-wide association studies (GWAS) have suggested an association between AAA and a SNP located on chromosome 3p12.3 in a region near the gene CNTN3 for contactin-3, a lipid-anchored cell adhesion molecule.178 Another GWAS in a population from Iceland and the Netherlands found a SNP on 9q33 associated with AAA with an OR of 1.21. The same SNP has been associated with coronary artery disease (CAD), peripheral artery disease (PAD), and pulmonary embolism (PE).179 The SNP resides in the gene coding for DAB2IP, a gene encoding a cell growth and survival inhibitor. In addition, the same gene variant associated with myocardial infarction (MI) at locus 9p21 has been associated with AAA.180,181 Finally, of interest but of unclear significance is the association between telomere length and AAA in a small cohort in the United Kingdom.182 This may represent the association of aging, telomere length, and aneurysm formation, but the direct pathophysiological link remains unclear.
Prognosis
The natural history of AAA is one of silent coexistence and sudden lethal rupture. In a large autopsy study performed over a quarter of a century, one fourth of abdominal aneurysms were ruptured on postmortem examination.126 The frequency of rupture was dependent largely on size in this study population, ranging from 9.5% in aneurysms smaller than 4 cm to 45.6% in aneurysms 7.1 to 10 cm in diameter. In a comparison of patients with aortic aneurysms divided into two groups at a cutoff point of 6 cm, survival was markedly decreased in the patients with larger aneurysms.183 In a single-center study that included 60 AAA ruptures over a 30-year period, only 2 occurred in patients with an aneurysm diameter smaller than 5 cm.184 Similarly, in a study in Rochester, Minnesota, no ruptures occurred in aneurysms smaller than 5 cm, whereas rupture occurred in 25% of AAAs larger than 5 cm.185 In 198 patients with aneurysms 5.5 cm in diameter or larger, but who were deemed too risky for surgery, 23% had presumed rupture over a mean follow-up of 1.6 years.186 Mortality rates as high as 64% are observed in patients who present with a ruptured aortic aneurysm.187
On the basis of these data, two large trials have been conducted to determine whether early recognition and treatment can alter the natural history of AAA. The U.K. Small Aneurysm Trial randomized 1090 patients aged 60 to 76 years with asymptomatic AAAs 4 to 5.5 cm in diameter to undergo early elective open surgery or ultrasonographic surveillance.188 In the surveillance group, surgery was performed when the aneurysm reached 5.5 cm. Early surgery did not affect overall mortality. At 3 years, nearly 20% of both groups had died, although abdominal aneurysms accounted for only a quarter of the deaths in both groups. Cardiovascular mortality unrelated to the aneurysm accounted for 40% of total mortality, and cancer caused slightly more than 20% of the deaths. In a similar study performed by the U.S. Veterans Administration, 1136 subjects aged 50 to 79 years with asymptomatic AAA 4 to 5.5 cm in diameter were randomized to undergo either early elective open surgery or ultrasonographic surveillance153 (Fig. 37-5). After 5 years, there was no significant difference in survival between the groups, each with a near 25% mortality rate. In this study, aneurysm-related deaths accounted for only 3% of total mortality. More recently, the PIVOTAL study demonstrated no difference between surveillance and early endovascular repair in patients with small aneurysms (measuring 4-5 cm), and no difference in aneurysm-related death in the two groups after 3 years of follow-up.189 Together, these data suggest that early repair of small aneurysms does not alter outcomes.
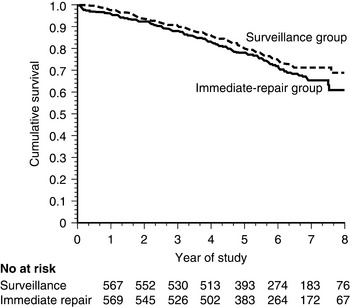
No significant difference in mortality was found between the two groups.
(From Lederle FA, Wilson SE, Johnson GR, et al: Immediate repair compared with surveillance of small abdominal aortic aneurysms. N Engl J Med 346:1437, 2002.)153
Several factors can predict the likelihood of expansion and rupture of AAAs and can help identify which patients require intervention. The factor most predictive of rupture is initial size of the aneurysm. In one study of patients too ill for surgery, aneurysm rupture rates ranged from 9.4% for AAA of 5.5 to 5.9 cm to 32.5% for AAA of 7 cm or more.186 In the U. K. Small Aneurysm Study, rate of rupture was 0.9% in aneurysms 3 to 3.9 cm, 2.7% in aneurysms 4 to 5.5 cm, and 27.8% in aneurysms 5.6 cm and larger.158 Larger aneurysm size also predicted a more rapid increase in diameter.190 Similar to factors that predispose to initial development of AAA, cigarette smoking and higher blood pressure increase the risks of rapid expansion and rupture. In contrast to the decreased risk of AAA development in women, being female actually increases risk of rupture and death after rupture in those with established AAA.158,191,192 In the U.K. Small Aneurysm Study, women had a threefold higher risk of AAA rupture than men.158 As mentioned previously, some have advocated use of biomechanical factors (wall stress, wall strength) to help with risk prediction. Prospective studies will be required to determine whether these factors can improve assessment of patients at high risk for aneurysm rupture and can improve selection of patients for aneurysm repair.
Thoracic and Thoracoabdominal Aortic Aneurysms
Prevalence
The most common location of involvement of thoracic aortic aneurysms (TAAs) is the ascending aorta and/or aortic root (noted in 60%), with the descending aorta affected in approximately 40% of cases and the aortic arch in 10%.193 Thoracoabdominal aortic aneurysms are defined by contiguous involvement of the descending thoracic aorta and abdominal aorta and account for 5% to 10% of all aortic aneurysms.193,194 The prevalence of isolated TAA is poorly defined but estimated at 6 persons per 100,000 per year.195 In autopsy records reported from 63% of 70,368 deaths between 1958 and 1985 in the city of Malmo, Sweden, TAAs were diagnosed in 205 of the deceased, 53% of whom were men.196 Of the 44,332 autopsies performed, 63 individuals (0.14%) had died of a ruptured TAA. The relative infrequency of TAA is confirmed by a retrospective analysis in Rochester, Minnesota.197 Over 30 years, of approximately 45,000 residents, 72 (0.16%) were diagnosed with a TAA; 61% were women, and 67 patients had thoracic aortic involvement only. Most studies, however, suggest that men are twice as likely to develop a TAA as women.197,198 Thoracoabdominal aortic aneurysms occur even more infrequently. Of the 44,332 people to undergo necropsy in Malmo, Sweden, a mere 10 had TAAA.196 In the Rochester, Minnesota, experience the incidence was 0.37 per 100,000 person-years of follow-up.197 Because of the relatively small sample sizes reported in the literature, risk factors for TAAA development are less well defined. Risk factors currently associated with development of TAAA include smoking, hypertension, and atherosclerotic vascular disease.193
Etiology and pathophysiology
As with AAA, age is a significant risk factor for development of TAA; median age for presentation varies from 64 to 69 years.193 Cystic medial necrosis develops to some degree as a natural consequence of aging and may be accelerated by comorbid conditions such as hypertension.199 Although a degenerative aneurysmal process is found in the majority of patients with TAAA, several other etiologies should be considered, including inherited collagen vascular disorders (e.g., MFS), BAV disease, aortic dissection, infection, and vasculitis (e.g., giant cell or Takayasu’s arteritis). Rarer causes of TAA include coarctation of the aorta and trauma. Finally, some aneurysms develop because of a familial predisposition termed familial thoracic aortic syndrome.
Natural history and prognosis
The natural history and prognosis for TAAs and TAAAs remain poor but not completely defined, in part because modern imaging techniques now allow for early detection and intervention of aneurysm before rupture, and because the natural history is dependent in large part on underlying etiology. However, in a study accumulated over 25 years that followed 94 patients with TAAAs who did not undergo operative repair, 76% died within 2 years of follow-up, with half of the deaths resulting from aneurysm rupture.200 A more recent experience with 57 TAAA patients managed without operation revealed a 69% 2-year survival, with aneurysm rupture as the cause of death in 19%.201 Features associated with aneurysm expansion and rupture include smoking, chronic obstructive pulmonary disease (COPD), and renal insufficiency.202 Dissection as an underlying cause of aneurysm formation is also associated with a greater risk of rupture compared with degenerative causes.203 Extension of a TAA into the abdomen is associated with a 50% relative increase in risk of rupture, compared with those limited to the thoracic aorta alone.204
The natural history of TAA depends in large part on rate of expansion. Initial aneurysm size at the time of diagnosis is the most important predictor of thoracic aneurysm growth.193 Longitudinal data have suggested that the mean rate of growth of thoracic aneurysms averages 0.1 cm/yr.205 Several factors also contribute to the rate of growth. Growth rate is increased in the setting of an aortic dissection. In one study, presence of a chronic aortic dissection was associated with a 0.37 cm/yr rate of growth.205 Descending TAAs tend to increase in size more rapidly than ascending TAAs.205,206 Those who smoke have a twofold increased rate of growth over nonsmokers.207
Survival rates for TAA range from 39% to 87% at 1 year and from 13% to 46% at 5 years.201,207,208 In a study of 67 patients with TAA (mean age, 65 years), those with an aortic diameter less than 5 cm had a 90% 3-year survival rate, compared to 60% for patients with a TAA larger than 5 cm.207 Similarly, in two other studies, patients with TAAs 6 cm or larger had a higher mortality rate than those with smaller aneurysms.206 Based on these data, referral for surgery is recommended when the aneurysm reaches 5.5 cm or greater in the ascending aorta.209
However, several factors increase risk of aneurysm rupture and may prompt earlier referral for intervention. Guidelines recommend that patients with MFS or other genetic disorders (e.g., vascular Ehlers-Danlos’, Loeys-Dietz’s, and Turner’s syndromes, BAV disease, or familial aortic aneurysm) may require intervention at smaller diameters, such as 4.0 cm to 5.0 cm, depending on clinical circumstances.206,209–214 In addition, those with a more rapid rate of growth than expected (i.e., ≥0.5 cm/yr) may be at increased risk of rupture and require repair at diameters less than 5.5 cm.209 Finally, repair should be considered in patients with ascending aortic diameter greater than 4.5 cm if undergoing aortic valve surgery.209
For aneurysms involving the aortic arch, surgery should be considered when the diameter reaches 5.5 cm or greater.209 For degenerative descending TAAs, repair is recommended when the diameter exceeds 5.5 cm, although in individuals with TAAAs or those with high surgical risk, elective surgery is recommended when the diameter exceeds 6.0 cm.209 Other factors reported to significantly increase rate of rupture or need for surgery include older age, history of COPD, pain possibly related to the aneurysm, higher blood pressure, and extension of the aneurysm into the abdomen.202,204
Inherited and Developmental Disorders
Marfan’s Syndrome
Marfan’s syndrome is an autosomal dominant inherited disorder of connective tissue arising from mutations in FBN1, a gene on chromosome 15 encoding the ECM protein fibrillin-1 (FBN1). Abnormalities in fibrillin synthesis may affect multiple tissues in patients with Marfan syndrome, including the cardiovascular, skeletal, and ocular systems. Excessive signaling through the TGF-β cascade has been suggested as a contributing factor. This is supported by experiments showing that TGF-β-neutralizing antibodies reverse aortic disease in a mouse model of Marfan syndrome, and by the demonstration that losartan, an angiotensin receptor blocker (ARB) with anti-TGF-β properties, can partially reverse aortic wall defects in these mice.215 Clinical manifestations of MFS include ectopia lentis, hyperelasticity and ligamentous redundancy, valvular heart disease, and abnormalities in skin, fascia, skeletal muscle, and adipose tissue.
However, potentially the most lethal complication in MFS is disease of the ascending aorta resulting in aneurysm, dissection, and rupture. Dilation of the aortic root has been demonstrated early in childhood in patients with Marfan syndrome. Histologically, changes in the media seen in patients with MFS include cystic medial necrosis with fragmentation and disarray of elastic fibers, a paucity of smooth muscle cells, and separation of muscle fibers by collagen and glycosaminoglycans.216
Ehlers-Danlos’ Syndrome
Ehlers-Danlos’ syndrome type 4 (vascular type) is a rare congenital defect in the synthesis of type 3 collagen resulting from a mutation in the COL3A1 gene.217 Patients with EDS typically present with acrogeria (distinctive facial appearance), bruising, thin skin, and vascular or visceral rupture. Histological examination reveals a thinned, fragmented internal elastic lamina.218 Moreover, deposition of glycosaminoglycans in the media of major arteries and intima of smaller arteries, with intimal thickening, has been noted.219 Abnormalities in type 3 collagen fiber formation reduce stability or prevent formation of collagen, decreasing vascular wall stability.218 In a study of 199 patients with confirmed Ehlers-Danlos syndrome, 25% of patients suffered a ruptured vessel or viscus by age 20 and 80% by age 40.220 Mean survival was 48 years. There were 131 deaths, 103 of which were due to vascular rupture. Complications of pregnancy caused the death of 15% of the women who became pregnant.
Loeys-Dietz’s Syndrome
Loeys-Dietz’s syndrome is an autosomal dominant condition arising from mutations in either the type I or type II receptor for TGF-β (TGFBR1 or TGFBR2). Individuals with Loeys-Dietz’s syndrome have several characteristic features, including abnormal uvula, hypertelorism (increased space between the eyes), and arterial aneurysms, among other abnormalities, some of which are similar to Marfan syndrome.214 The syndrome is characterized by particularly aggressive arterial disease manifested as aortic aneurysm with high risk of aortic dissection and rupture. As such, the average age of death of 26 years. Because of the high morbidity and mortality and the high rate of aortic dissection, even with aneurysms of less than 5.0 cm in size, early repair at smaller diameters is recommended.209,214
Bicuspid Aortic Valve
Presence of a BAV increases the risk of ascending aortic aneurysm formation.193 Although it was once thought that the aneurysmal dilation was a “post-stenotic” phenomenon arising secondary to abnormal flow through a diseased aortic valve, more recent data support the notion that aortic expansion occurs independently of valvular dysfunction, severity, age, and body size.221 Further evidence that aortic dilation is not dependent on valve dysfunction is found in a study of 118 consecutive patients with BAV in whom the diameter of the ascending aorta was not correlated with severity of aortic stenosis.222 Moreover, abnormalities in the aortic wall can arise even when there is no hemodynamically significant aortic valve disease, and replacement of a diseased valve does not change the rate of aortic expansion.223
In one study comparing aorta and pulmonary artery specimens in patients with BAV and tricuspid aortic valve disease, those with BAV had decreased FBN1 concentrations in both the aortic and pulmonary specimens, suggesting a systemic disorder.224 Vascular smooth muscle cells from patients with BAV show intracellular accumulation and reduction of extracellular distribution of several structural elements including fibrillin, fibronectin, and tenascin.16 Surgical specimens demonstrate greater amounts of inflammation and increased expression of MMP-2 and MMP-9 in patients with BAV compared to those with tricuspid aortic valve disease.225,226 Patients with Turner’s syndrome, who are at increased risk of developing BAV, also exhibit an increased rate of aneurysm formation.227
Aortic Coarctation
Coarctation of the aorta represents 5% of congenital heart disease. The clinical consequences of aortic coarctation are varied, ranging from being life-threatening in infancy to remaining unappreciated until adulthood.228 Coarctation has long been recognized as associated with de novo aortic aneurysm development, and aneurysms can also develop at the site of coarctation repair—specifically patch angioplasty repair—in up to 20% of patients.229,230 Some reports indicate aneurysm formation can even occur several decades after initial repair. Intermediate follow-up studies suggest that percutaneous balloon angioplasty repair results in a 2% to 5% rate of repair-site aortic aneurysm formation.231,232 A potential explanation for the relationship between coarctation and aortic aneurysm is the common link with the presence of BAV in approximately 15% of patients with aortic coarctation.212
Other Conditions Associated with Aortic Aneurysm
Although most aortic aneurysms occur as a result of degenerative processes in the aortic wall as described earlier, certain disease states including vasculitis, infection, and inherited abnormalities of structural proteins predispose patients to aortic aneurysm formation (see Box 37-1).
Vasculitides
Giant cell arteritis
Giant cell (temporal) arteritis (GCA; see Chapter 43) is a medium-vessel chronic inflammatory vasculitis that affects the aorta and its branch vessels. It most commonly occurs in patients older than 55 years of age and is twice as common in women as men.233 Between 1% and 20% of GCA patients develop aneurysms, most commonly in the thoracic aorta.234 In a series of 41 patients with GCA-related TAAs, 16 developed aortic dissection, 15 had valvular annular expansion causing symptomatic aortic valve insufficiency, and 18 required surgery.235 In a series of 168 patients with GCA, 18% developed aortic aneurysm or dissection, and these occurrences were inversely associated with development of intracranial disease manifestations.234 Presence of an aortic aneurysm itself was not associated with increased mortality; however, the nine individuals who developed aortic dissection had significantly increased mortality.236 The mechanism of aneurysm formation seems to be similar to patients without GCA, with increases in MMP-2- and MMP-9-associated destruction of the vessel wall.237,238
Takayasu’s arteritis
Named for a Japanese professor of ophthalmology, Takayasu’s arteritis (TA; see Chapter 42) is a large-vessel vasculitis that typically has its onset between the age of 10 and 30 years. The most common vascular presentation is occlusive disease, found in 80% to 94% of patients; however, aortic aneurysms may be found in up to one fourth of patients with TA.239,240 Development of aneurysmal disease has been associated with worse outcome in a series of 120 Japanese patients followed for 13 years.241 Blood levels of MMP-2 and MMP-9 are elevated in TA, but the mechanism of aneurysm formation remains unknown.242
Behçet’s disease
Behçet’s disease (see Chapter 41) is a small-vessel vasculitis originally characterized by a set of three symptoms: aphthous stomatitis, genital ulcers, and uveitis.243 Involvement of medium-sized and large arteries, as well as veins, arises not from direct vascular inflammation but rather due to vasculitis of the small arteries of the vasa vasorum that supply the vessel wall.244 Vascular involvement, including aneurysms, can be found in 7% to 38% of patients.243,245 Management of aneurysmal disease in Behçet’s disease depends in large part on the location of the abnormality and clinical circumstances. First-line therapy includes an antiinflammatory regimen with corticosteroids. As with other vasculitides, risk of intervention is greatest during the state of active inflammation, and there is an increased risk of rupture, dissection, and/or future aneurysmal dilation at the site of revascularization.
Seronegative spondyloarthropathies
The spondyloarthropathies (see Chapter 41) are characterized by inflammation of the spine and sacroiliac joints, association with HLA-B27, and absence of rheumatoid factor (RF). These disorders are known to be associated with an increased risk of aortic aneurysm formation. Specific spondyloarthropathies include ankylosing spondylitis, Reiter’s syndrome, and relapsing polychondritis.
Ankylosing spondylitis is an HLA-B27 disease that requires the presence of four of the five following features: onset younger than 40 years of age, back pain for more than 3 months, insidious onset of symptoms, morning stiffness, and improvement with exercise. Aortic root and valve disease is present in up to 80% of patients.242 In a series of 44 outpatients, aortic root disease and valve disease were found in 82%; thickening of the aortic valve was noted in 41% of patients, and aortic dilation in 25%.246 Aortic valve thickening manifested as nodularities of the aortic cusps, forming a characteristic subaortic bump.242 Valve regurgitation was seen in almost half of patients, and 40% had moderate lesions.
Reiter’s syndrome is a reactive arthritis that affects the lower limbs, causing an asymmetrical oligoarthritis. To make the diagnosis, patients must have evidence of a preceding infection, diarrhea, or urethritis 4 weeks preceding the syndrome.247 Less than 1% of Reiter’s syndrome patients develop cardiovascular complications. Among this group, aortic insufficiency is a late finding.
Relapsing polychondritis is a paroxysmal and progressive inflammatory disease of the cartilaginous structures, affecting the ear, nose, and hyaline cartilage of the tracheobronchial tree. Cardiovascular disease, including aortic aneurysms, is found in 25% to 50% of patients.248
Infectious Aortic Aneurysms
Mycotic aneurysms
Also known as infective endarteritis (see Chapter 59), mycotic aneurysms are rare phenomena. Two large necropsy studies including 22,000 and 20,000 patients, respectively, revealed a combined incidence of 0.03% in the United States.249,250 The average age of patients with mycotic aneurysms is 65, and men are threefold more likely to develop mycotic aneurysms than women.251,252 Hematogenous seeding, such as occurs in patients with endocarditis, affects a vessel that may be “at risk” because of preexisting atherosclerosis or previous damage and represents the most common cause of mycotic aneurysms.253 Indeed, as many as 15% of patients with endocarditis developed mycotic aneurysm before the antibiotic era.254 Other etiologies include septic microemboli, contiguous extension, and trauma with direct contamination. In contrast to the typical degenerative or vasculitic fusiform expansion, mycotic aneurysms are more likely to be saccular (see Fig. 37-4). The outpouching may range in size from 1 mm to 10 cm and include components of acute and chronic inflammation, hemorrhage, abscess formation, and necrosis.
Clinical manifestations of mycotic aneurysm most commonly include pain and fever and, if related to a new aneurysm, should prompt directed investigation. The organisms that most commonly cause mycotic aneurysms include Staphylococcus and Salmonella species, which cause 40% and 20% of mycotic aneurysms, respectively.255,256 Surgery should be prompt, since rupture occurs in up to 80%.257,258 Prognosis for cerebral vascular infection is dire, with 1-year mortality for patients who have cerebrovascular mycotic aneurysms reaching as high as 90%.258
Tuberculous aneurysms
Aortic aneurysm due to tuberculosis is quite rare. In a series of more than 22,000 autopsies performed at one urban medical center in the first half of the 20th century, only 1 of 308 aortic aneurysms had tuberculous aneurysms,249 whereas there were no tuberculous aneurysms among 20,000 autopsies performed in a rural setting.250 Three mechanisms have been postulated to facilitate tuberculous adhesion and endarteritis. It is thought that direct extension from a contiguous source, such as the spine or lung, may cause 75% of tuberculous aneurysms.259 Other possibilities include adhesion to a vessel damaged by atherosclerosis or infiltration of the inner layers of the aorta via the vasa vasorum.259 The abdominal and thoracic portions of the aorta are affected similarly. The presentation of the patient with a tuberculous aneurysm varies significantly. The patient may be asymptomatic, have a palpable or radiologically visible paraaortic mass, complain of chest or abdominal pain, or present with aortic rupture and hypovolemic shock. Tuberculous aneurysms that are symptomatic or rapidly expanding and pseudoaneurysms typically require surgical repair.
Syphilitic aneurysms
Although syphilis may once have been a common cause of aortic disease, antibiotics have greatly diminished the incidence of syphilitic aortic aneurysm (luetic aneurysm), such that fewer than 50 cases have been reported in the antibiotic era.260 Central nervous system (CNS) and cardiovascular complications denote the tertiary stage of syphilis. Classically, this arises after a latent phase of roughly 10 to 30 years from initial spirochete infection. Syphilitic aortitis may occur in up to 10% of patients with tertiary syphilis (Fig. 37-6). Destruction of the elastic lamina occurs as a consequence of lymphoplasmacytic infiltrate around the vasa vasorum, owing to direct spirochete infection of the aortic media. This ultimately leads to expansion but also fibrosis and calcification, producing the classic “tree bark” radiographic pattern. Luetic aortic aneurysms commonly involve the ascending aorta and are saccular. Involvement of the coronary ostia may result in coronary stenosis and resultant anginal symptoms. Survival with syphilitic aortic aneurysm is worse than the general population.
1 Roger V.L., Go A.S., Lloyd-Jones D.M., et al. Heart disease and stroke statistics–2011 update: a report from the American Heart Association. Circulation. 2011;123(4):e18–e209.
2 Wolinsky H., Glagov S. A lamellar unit of aortic medial structure and function in mammals. Circ Res. 1967;20(1):99–111.
3 Wassef M., Baxter B.T., Chisholm R.L., et al. Pathogenesis of abdominal aortic aneurysms: a multidisciplinary research program supported by the National Heart, Lung, and Blood Institute. J Vasc Surg. 2001;34(4):730–738.
4 Hoshina K., Sho E., Sho M., et al. Wall shear stress and strain modulate experimental aneurysm cellularity. J Vasc Surg. 2003;37(5):1067–1074.
5 Golledge J., Norman P.E. Atherosclerosis and abdominal aortic aneurysm: cause, response, or common risk factors? Arterioscler Thromb Vasc Biol. 2010;30(6):1075–1077.
6 Johnsen S.H., Forsdahl S.H., Singh K., et al. Atherosclerosis in abdominal aortic aneurysms: a causal event or a process running in parallel? The Tromso study. Arterioscler Thromb Vasc Biol. 2010;30(6):1263–1268.
7 Halpern V.J., Nackman G.B., Gandhi R.H., et al. The elastase infusion model of experimental aortic aneurysms: synchrony of induction of endogenous proteinases with matrix destruction and inflammatory cell response. J Vasc Surg. 1994;20(1):51–60.
8 Baxter B.T., Davis V.A., Minion D.J., et al. Abdominal aortic aneurysms are associated with altered matrix proteins of the nonaneurysmal aortic segments. J Vasc Surg. 1994;19(5):797–802. discussion 803
9 Dobrin P.B., Mrkvicka R. Failure of elastin or collagen as possible critical connective tissue alterations underlying aneurysmal dilatation. Cardiovasc Surg. 1994;2(4):484–488.
10 Groenink M., Langerak S.E., Vanbavel E., et al. The influence of aging and aortic stiffness on permanent dilation and breaking stress of the thoracic descending aorta. Cardiovasc Res. 1999;43(2):471–480.
11 Movat H.Z., More R.H., Haust M.D. The diffuse intimal thickening of the human aorta with aging. Am J Pathol. 1958;34(6):1023–1031.
12 Kawasaki T., Sasayama S., Yagi S., et al. Non-invasive assessment of the age related changes in stiffness of major branches of the human arteries. Cardiovasc Res. 1987;21(9):678–687.
13 Breithaupt-Grogler K., Belz G.G. Epidemiology of the arterial stiffness. Pathol Biol (Paris). 1999;47(6):604–613.
14 Lakatta E.G. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: part III: cellular and molecular clues to heart and arterial aging. Circulation. 2003;107(3):490–497.
15 Lin A.E., Lippe B.M., Geffner M.E., et al. Aortic dilation, dissection, and rupture in patients with Turner syndrome. J Pediatr. 1986;109(5):820–826.
16 Nataatmadja M., West M., West J., et al. Abnormal extracellular matrix protein transport associated with increased apoptosis of vascular smooth muscle cells in Marfan syndrome and bicuspid aortic valve thoracic aortic aneurysm. Circulation. 2003;108(Suppl 1):II329–II334.
17 Zhang J., Schmidt J., Ryschich E., et al. Increased apoptosis and decreased density of medial smooth muscle cells in human abdominal aortic aneurysms. Chin Med J (Engl). 2003;116(10):1549–1552.
18 Visse R., Nagase H. Matrix metalloproteinases and tissue inhibitors of metalloproteinases: structure, function, and biochemistry. Circ Res. 2003;92(8):827–839.
19 Louwrens H.D., Kwaan H.C., Pearce W.H., et al. Plasminogen activator and plasminogen activator inhibitor expression by normal and aneurysmal human aortic smooth muscle cells in culture. Eur J Vasc Endovasc Surg. 1995;10(3):289–293.
20 Allaire E., Hasenstab D., Kenagy R.D., et al. Prevention of aneurysm development and rupture by local overexpression of plasminogen activator inhibitor-1. Circulation. 1998;98(3):249–255.
21 Reilly J.M., Sicard G.A., Lucore C.L. Abnormal expression of plasminogen activators in aortic aneurysmal and occlusive disease. J Vasc Surg. 1994;19(5):865–872.
22 Vine N., Powell J.T. Metalloproteinases in degenerative aortic disease. Clin Sci (Lond). 1991;81(2):233–239.
23 Brophy C.M., Marks W.H., Reilly J.M., et al. Decreased tissue inhibitor of metalloproteinases (TIMP) in abdominal aortic aneurysm tissue: a preliminary report. J Surg Res. 1991;50(6):653–657.
24 Herron G.S., Unemori E., Wong M., et al. Connective tissue proteinases and inhibitors in abdominal aortic aneurysms. Involvement of the vasa vasorum in the pathogenesis of aortic aneurysms. Arterioscler Thromb. 1991;11(6):1667–1677.
25 Newman K.M., Malon A.M., Shin R.D., et al. Matrix metalloproteinases in abdominal aortic aneurysm: characterization, purification, and their possible sources. Connect Tissue Res. 1994;30(4):265–276.
26 Newman K.M., Ogata Y., Malon A.M., et al. Identification of matrix metalloproteinases 3 (stromelysin-1) and 9 (gelatinase B) in abdominal aortic aneurysm. Arterioscler Thromb. 1994;14(8):1315–1320.
27 Newman K.M., Jean-Claude J., Li H., et al. Cellular localization of matrix metalloproteinases in the abdominal aortic aneurysm wall. J Vasc Surg. 1994;20(5):814–820.
28 Patel M.I., Melrose J., Ghosh P., et al. Increased synthesis of matrix metalloproteinases by aortic smooth muscle cells is implicated in the etiopathogenesis of abdominal aortic aneurysms. J Vasc Surg. 1996;24(1):82–92.
29 Thompson R.W., Holmes D.R., Mertens R.A., et al. Production and localization of 92-kilodalton gelatinase in abdominal aortic aneurysms. An elastolytic metalloproteinase expressed by aneurysm-infiltrating macrophages. J Clin Invest. 1995;96(1):318–326.
30 Davis V., Persidskaia R., Baca-Regen L., et al. Matrix metalloproteinase-2 production and its binding to the matrix are increased in abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol. 1998;18(10):1625–1633.
31 McMillan W.D., Patterson B.K., Keen R.R., et al. In situ localization and quantification of seventy-two-kilodalton type IV collagenase in aneurysmal, occlusive, and normal aorta. J Vasc Surg. 1995;22(3):295–305.
32 Goodall S., Crowther M., Hemingway D.M., et al. Ubiquitous elevation of matrix metalloproteinase-2 expression in the vasculature of patients with abdominal aneurysms. Circulation. 2001;104(3):304–309.
33 Longo G.M., Xiong W., Greiner T.C., et al. Matrix metalloproteinases 2 and 9 work in concert to produce aortic aneurysms. J Clin Invest. 2002;110(5):625–632.
34 Wilson W.R., Anderton M., Choke E.C., et al. Elevated plasma MMP1 and MMP9 are associated with abdominal aortic aneurysm rupture. Eur J Vasc Endovasc Surg. 2008;35(5):580–584.
35 Wilson W.R., Anderton M., Schwalbe E.C., et al. Matrix metalloproteinase-8 and -9 are increased at the site of abdominal aortic aneurysm rupture. Circulation. 2006;113(3):438–445.
36 Petersen E., Wagberg F., Angquist K.A. Proteolysis of the abdominal aortic aneurysm wall and the association with rupture. Eur J Vasc Endovasc Surg. 2002;23(2):153–157.
37 Nishimura K., Ikebuchi M., Kanaoka Y., et al. Relationships between matrix metalloproteinases and tissue inhibitor of metalloproteinases in the wall of abdominal aortic aneurysms. Int Angiol. 2003;22(3):229–238.
38 Papalambros E., Sigala F., Georgopoulos S., et al. Immunohistochemical expression of metalloproteinases MMP-2 and MMP-9 in abdominal aortic aneurysms: correlation with symptoms and aortic diameter. Int J Mol Med. 2003;12(6):965–968.
39 Petersen E., Gineitis A., Wagberg F., et al. Activity of matrix metalloproteinase-2 and -9 in abdominal aortic aneurysms. Relation to size and rupture. Eur J Vasc Endovasc Surg. 2000;20(5):457–461.
40 Petersen E., Wagberg F., Angquist K.A. Serum concentrations of elastin-derived peptides in patients with specific manifestations of atherosclerotic disease. Eur J Vasc Endovasc Surg. 2002;24(5):440–444.
41 Tamarina N.A., McMillan W.D., Shively V.P., et al. Expression of matrix metalloproteinases and their inhibitors in aneurysms and normal aorta. Surgery. 1997;122(2):264–271. discussion 271–262
42 Pyo R., Lee J.K., Shipley J.M., et al. Targeted gene disruption of matrix metalloproteinase-9 (gelatinase B) suppresses development of experimental abdominal aortic aneurysms. J Clin Invest. 2000;105(11):1641–1649.
43 Annabi B., Shedid D., Ghosn P., et al. Differential regulation of matrix metalloproteinase activities in abdominal aortic aneurysms. J Vasc Surg. 2002;35(3):539–546.
44 Crowther M., Goodall S., Jones J.L., et al. Localization of matrix metalloproteinase 2 within the aneurysmal and normal aortic wall. Br J Surg. 2000;87(10):1391–1400.
45 Henrotin Y.E., Bruckner P., Pujol J.P. The role of reactive oxygen species in homeostasis and degradation of cartilage. Osteoarthritis Cartilage. 2003;11(10):747–755.
46 Miller F.J.Jr, Sharp W.J., Fang X., et al. Oxidative stress in human abdominal aortic aneurysms: a potential mediator of aneurysmal remodeling. Arterioscler Thromb Vasc Biol. 2002;22(4):560–565.
47 Siwik D.A., Colucci W.S. Regulation of matrix metalloproteinases by cytokines and reactive oxygen/nitrogen species in the myocardium. Heart Fail Rev. 2004;9(1):43–51.
48 Carmeliet P., Moons L., Lijnen R., et al. Urokinase-generated plasmin activates matrix metalloproteinases during aneurysm formation. Nat Genet. 1997;17(4):439–444.
49 Defawe O.D., Colige A., Lambert C.A., et al. TIMP-2 and PAI-1 mRNA levels are lower in aneurysmal as compared to athero-occlusive abdominal aortas. Cardiovasc Res. 2003;60(1):205–213.
50 Saito S., Zempo N., Yamashita A., et al. Matrix metalloproteinase expressions in arteriosclerotic aneurysmal disease. Vasc Endovasc Surg. 2002;36(1):1–7.
51 Ailawadi G., Knipp B.S., Lu G., et al. A nonintrinsic regional basis for increased infrarenal aortic MMP-9 expression and activity. J Vasc Surg. 2003;37(5):1059–1066.
52 Allaire E., Forough R., Clowes M., et al. Local overexpression of TIMP-1 prevents aortic aneurysm degeneration and rupture in a rat model. J Clin Invest. 1998;102(7):1413–1420.
53 Silence J., Collen D., Lijnen H.R. Reduced atherosclerotic plaque but enhanced aneurysm formation in mice with inactivation of the tissue inhibitor of metalloproteinase-1 (TIMP-1) gene. Circ Res. 2002;90(8):897–903.
54 Falkenberg M., Holmdahl L., Tjarnstrom J., et al. Abnormal levels of urokinase plasminogen activator protein and tissue plasminogen activator activity in human aortic aneurysms. Eur J Surg. 2001;167(1):10–14.
55 Qian H.S., Gu J.M., Liu P., et al. Overexpression of PAI-1 prevents the development of abdominal aortic aneurysm in mice. Gene Ther. 2008;15(3):224–232.
56 Thompson R.W., Liao S., Curci J.A. Therapeutic potential of tetracycline derivatives to suppress the growth of abdominal aortic aneurysms. Adv Dent Res. 1998;12(2):159–165.
57 Manning M.W., Cassis L.A., Daugherty A. Differential effects of doxycycline, a broad-spectrum matrix metalloproteinase inhibitor, on angiotensin II-induced atherosclerosis and abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol. 2003;23(3):483–488.
58 Bartoli M.A., Parodi F.E., Chu J., et al. Localized administration of doxycycline suppresses aortic dilatation in an experimental mouse model of abdominal aortic aneurysm. Ann Vasc Surg. 2006;20(2):228–236.
59 Chung A.W., Yang H.H., Radomski M.W., et al. Long-term doxycycline is more effective than atenolol to prevent thoracic aortic aneurysm in Marfan syndrome through the inhibition of matrix metalloproteinase-2 and -9. Circ Res. 2008;102(8):e73–e85.
60 Curci J.A., Mao D., Bohner D.G., et al. Preoperative treatment with doxycycline reduces aortic wall expression and activation of matrix metalloproteinases in patients with abdominal aortic aneurysms. J Vasc Surg. 2000;31(2):325–342.
61 Baxter B.T., Pearce W.H., Waltke E.A., et al. Prolonged administration of doxycycline in patients with small asymptomatic abdominal aortic aneurysms: report of a prospective (phase II) multicenter study. J Vasc Surg. 2002;36(1):1–12.
62 Lindeman J.H., Abdul-Hussien H., van Bockel J.H., et al. Clinical trial of doxycycline for matrix metalloproteinase-9 inhibition in patients with an abdominal aneurysm: doxycycline selectively depletes aortic wall neutrophils and cytotoxic T cells. Circulation. 2009;119(16):2209–2216.
63 Mosorin M., Juvonen J., Biancari F., et al. Use of doxycycline to decrease the growth rate of abdominal aortic aneurysms: a randomized, double-blind, placebo-controlled pilot study. J Vasc Surg. 2001;34(4):606–610.
64 Dodd B.R., Spence R.A. Doxycycline inhibition of abdominal aortic aneurysm growth–a systematic review of the literature. Curr Vasc Pharmacol. 2011;9:471–478.
65 Nagashima H., Aoka Y., Sakomura Y., et al. A 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitor, cerivastatin, suppresses production of matrix metalloproteinase-9 in human abdominal aortic aneurysm wall. J Vasc Surg. 2002;36(1):158–163.
66 Ejiri J., Inoue N., Tsukube T., et al. Oxidative stress in the pathogenesis of thoracic aortic aneurysm: protective role of statin and angiotensin II type 1 receptor blocker. Cardiovasc Res. 2003;59(4):988–996.
67 Schouten O., van Laanen J.H., Boersma E., et al. Statins are associated with a reduced infrarenal abdominal aortic aneurysm growth. Eur J Vasc Endovasc Surg. 2006;32(1):21–26.
68 Takagi H., Matsui M., Umemoto T. A meta-analysis of clinical studies of statins for prevention of abdominal aortic aneurysm expansion. J Vasc Surg. 2010;52(6):1675–1681.
69 Rahman M.N., Khan J.A., Mazari F.A., et al. A randomized placebo controlled trial of the effect of preoperative statin use on matrix metalloproteinases and tissue inhibitors of matrix metalloproteinases in areas of low and peak wall stress in patients undergoing elective open repair of abdominal aortic aneurysm. Ann Vasc Surg. 2011;25(1):32–38.
70 Hurks R., Hoefer I.E., Vink A., et al. Different effects of commonly prescribed statins on abdominal aortic aneurysm wall biology. Eur J Vasc Endovasc Surg. 2010;39(5):569–576.
71 Ferguson C.D., Clancy P., Bourke B., et al. Association of statin prescription with small abdominal aortic aneurysm progression. Am Heart J. 2010;159(2):307–313.
72 Holmes D.R., Petrinec D., Wester W., et al. Indomethacin prevents elastase-induced abdominal aortic aneurysms in the rat. J Surg Res. 1996;63(1):305–309.
73 Franklin I.J., Walton L.J., Greenhalgh R.M., et al. The influence of indomethacin on the metabolism and cytokine secretion of human aneurysmal aorta. Eur J Vasc Endovasc Surg. 1999;18(1):35–42.
74 Miralles M., Wester W., Sicard G.A., et al. Indomethacin inhibits expansion of experimental aortic aneurysms via inhibition of the cox2 isoform of cyclooxygenase. J Vasc Surg. 1999;29(5):884–892. discussion 892–883
75 Walton L.J., Franklin I.J., Bayston T., et al. Inhibition of prostaglandin E2 synthesis in abdominal aortic aneurysms: implications for smooth muscle cell viability, inflammatory processes, and the expansion of abdominal aortic aneurysms. Circulation. 1999;100(1):48–54.
76 Reilly J.M., Miralles M., Wester W.N., et al. Differential expression of prostaglandin E2 and interleukin-6 in occlusive and aneurysmal aortic disease. Surgery. 1999;126(4):624–627. discussion 627–628
77 Holmes D.R., Wester W., Thompson R.W., et al. Prostaglandin E2 synthesis and cyclooxygenase expression in abdominal aortic aneurysms. J Vasc Surg. 1997;25(5):810–815.
78 Bayston T., Ramessur S., Reise J., et al. Prostaglandin E2 receptors in abdominal aortic aneurysm and human aortic smooth muscle cells. J Vasc Surg. 2003;38(2):354–359.
79 Libby P. Inflammation in atherosclerosis. Nature. 2002;420(6917):868–874.
80 Rose A.G., Dent D.M. Inflammatory variant of abdominal atherosclerotic aneurysm. Arch Pathol Lab Med. 1981;105(8):409–413.
81 Satta J., Laurila A., Paakko P., et al. Chronic inflammation and elastin degradation in abdominal aortic aneurysm disease: an immunohistochemical and electron microscopic study. Eur J Vasc Endovasc Surg. 1998;15(4):313–319.
82 Golledge A.L., Walker P., Norman P.E., et al. A systematic review of studies examining inflammation associated cytokines in human abdominal aortic aneurysm samples. Dis Markers. 2009;26(4):181–188.
83 Treska V., Kocova J., Boudova L., et al. Inflammation in the wall of abdominal aortic aneurysm and its role in the symptomatology of aneurysm. Cytokines Cell Mol Ther. 2002;7(3):91–97.
84 Wilson W.R., Wills J., Furness P.N., et al. Abdominal aortic aneurysm rupture is not associated with an up-regulation of inflammation within the aneurysm wall. Eur J Vasc Endovasc Surg. 2010;40(2):191–195.
85 Parry D.J., Al-Barjas H.S., Chappell L., et al. Markers of inflammation in men with small abdominal aortic aneurysm. J Vasc Surg. 2010;52(1):145–151.
86 Hellenthal F.A., Buurman W.A., Wodzig W.K., et al. Biomarkers of abdominal aortic aneurysm progression. Part 2: inflammation. Nat Rev Cardiol. 2009;6(8):543–552.
87 Vainas T., Lubbers T., Stassen F.R., et al. Serum C-reactive protein level is associated with abdominal aortic aneurysm size and may be produced by aneurysmal tissue. Circulation. 2003;107(8):1103–1105.
88 Truijers M., Kurvers H.A., Bredie S.J., et al. In vivo imaging of abdominal aortic aneurysms: increased FDG uptake suggests inflammation in the aneurysm wall. J Endovasc Ther. 2008;15(4):462–467.
89 Howarth S.P., Tang T.Y., Graves M.J., et al. Non-invasive MR imaging of inflammation in a patient with both asymptomatic carotid atheroma and an abdominal aortic aneurysm: a case report. Ann Surg Innov Res. 1:4, 2007.
90 Tang E.H., Shvartz E., Shimizu K., et al. Deletion of EP4 on bone marrow-derived cells enhances inflammation and angiotensin II-induced abdominal aortic aneurysm formation. Arterioscler Thromb Vasc Biol. 2011;31(2):261–269.
91 Shiraya S., Miyake T., Aoki M., et al. Inhibition of development of experimental aortic abdominal aneurysm in rat model by atorvastatin through inhibition of macrophage migration. Atherosclerosis. 2009;202(1):34–40.
92 Kalyanasundaram A., Elmore J.R., Manazer J.R., et al. Simvastatin suppresses experimental aortic aneurysm expansion. J Vasc Surg. 2006;43(1):117–124.
93 Schultz G., Tedesco M.M., Sho E., et al. Enhanced abdominal aortic aneurysm formation in thrombin-activatable procarboxypeptidase B-deficient mice. Arterioscler Thromb Vasc Biol. 2010;30(7):1363–1370.
94 Leeper N.J., Tedesco M.M., Kojima Y., et al. Apelin prevents aortic aneurysm formation by inhibiting macrophage inflammation. Am J Physiol Heart Circ Physiol. 2009;296(5):H1329–H1335.
95 Onoda M., Yoshimura K., Aoki H., et al. Lysyl oxidase resolves inflammation by reducing monocyte chemoattractant protein-1 in abdominal aortic aneurysm. Atherosclerosis. 2010;208(2):366–369.
96 Kaneko H., Anzai T., Morisawa M., et al. Resveratrol prevents the development of abdominal aortic aneurysm through attenuation of inflammation, oxidative stress, and neovascularization. Atherosclerosis. 2011;217:350–357.
97 Hannawa K.K., Cho B.S., Sinha I., et al. Attenuation of experimental aortic aneurysm formation in P-selectin knockout mice. Ann N Y Acad Sci. 2006;108:353–359.
98 Wolinsky H., Glagov S. Comparison of abdominal and thoracic aortic medial structure in mammals. Deviation of man from the usual pattern. Circ Res. 1969;25(6):677–686.
99 Benjamin H.B., Becker A.B. Etiologic incidence of thoracic and abdominal aneurysms. Surg Gynecol Obstet. 1967;125(6):1307–1310.
100 Stefanadis C., Vlachopoulos C., Karayannacos P., et al. Effect of vasa vasorum flow on structure and function of the aorta in experimental animals. Circulation. 1995;91(10):2669–2678.
101 Krettek A., Sukhova G.K., Libby P. Elastogenesis in human arterial disease: a role for macrophages in disordered elastin synthesis. Arterioscler Thromb Vasc Biol. 2003;23(4):582–587.
102 Moore J.E.Jr, Ku D.N., Zarins C.K., et al. Pulsatile flow visualization in the abdominal aorta under differing physiologic conditions: implications for increased susceptibility to atherosclerosis. J Biomech Eng. 1992;114(3):391–397.
103 Egelhoff C.J., Budwig R.S., Elger D.F., et al. Model studies of the flow in abdominal aortic aneurysms during resting and exercise conditions. J Biomech. 1999;32(12):1319–1329.
104 Sangiorgi G., D’Averio R., Mauriello A., et al. Plasma levels of metalloproteinases-3 and -9 as markers of successful abdominal aortic aneurysm exclusion after endovascular graft treatment. Circulation. 2001;104(12 Suppl 1):I288–I295.
105 Lorelli D.R., Jean-Claude J.M., Fox C.J., et al. Response of plasma matrix metalloproteinase-9 to conventional abdominal aortic aneurysm repair or endovascular exclusion: implications for endoleak. J Vasc Surg. 2002;35(5):916–922.
106 Nakahashi T.K., Hoshina K., Tsao P.S., et al. Flow loading induces macrophage antioxidative gene expression in experimental aneurysms. Arterioscler Thromb Vasc Biol. 2002;22(12):2017–2022.
107 Sho E., Sho M., Hoshina K., et al. Hemodynamic forces regulate mural macrophage infiltration in experimental aortic aneurysms. Exp Mol Pathol. 2004;76(2):108–116.
108 Wang D.H., Makaroun M.S., Webster M.W., et al. Effect of intraluminal thrombus on wall stress in patient-specific models of abdominal aortic aneurysm. J Vasc Surg. 2002;36(3):598–604.
109 Di Martino E.S., Vorp D.A. Effect of variation in intraluminal thrombus constitutive properties on abdominal aortic aneurysm wall stress. Ann Biomed Eng. 2003;31(7):804–809.
110 Thubrikar M.J., Robicsek F., Labrosse M., et al. Effect of thrombus on abdominal aortic aneurysm wall dilation and stress. J Cardiovasc Surg (Torino). 2003;44(1):67–77.
111 Speelman L., Schurink G.W., Bosboom E.M., et al. The mechanical role of thrombus on the growth rate of an abdominal aortic aneurysm. J Vasc Surg. 2010;51(1):19–26.
112 Bluestein D., Dumont K., De Beule M., et al. Intraluminal thrombus and risk of rupture in patient specific abdominal aortic aneurysm–FSI modelling. Comput Methods Biomech Biomed Engin. 2009;12(1):73–81.
113 Georgakarakos E., Ioannou C., Kostas T., et al. Inflammatory response to aortic aneurysm intraluminal thrombus may cause increased 18F-FDG uptake at sites not associated with high wall stress: comment on “high levels of 18F-FDG uptake in aortic aneurysm wall are associated with high wall stress”. Eur J Vasc Endovasc Surg. 2010;39(6):795. author reply 795–796
114 Li Z.Y., U-King-Im J., Tang T.Y., et al. Impact of calcification and intraluminal thrombus on the computed wall stresses of abdominal aortic aneurysm. J Vasc Surg. 2008;47(5):928–935.
115 Wiernicki I., Cnotliwy M., Baranowska-Bosiacka I., et al. Elastin degradation within the abdominal aortic aneurysm wall–relationship between intramural pH and adjacent thrombus formation. Eur J Clin Invest. 2008;38(12):883–887.
116 Fontaine V., Jacob M.P., Houard X., et al. Involvement of the mural thrombus as a site of protease release and activation in human aortic aneurysms. Am J Pathol. 2002;161(5):1701–1710.
117 Breeuwer M., de Putter S., Kose U., et al. Towards patient-specific risk assessment of abdominal aortic aneurysm. Med Biol Eng Comput. 2008;46(11):1085–1095.
118 Sheidaei A., Hunley S.C., Zeinali-Davarani S., et al. Simulation of abdominal aortic aneurysm growth with updating hemodynamic loads using a realistic geometry. Med Eng Phys. 2011;33(1):80–88.
119 Georgakarakos E., Ioannou C.V., Papaharilaou Y., et al. Computational evaluation of aortic aneurysm rupture risk: what have we learned so far? J Endovasc Ther. 2011;18(2):214–225.
120 Reeps C., Gee M., Maier A., et al. The impact of model assumptions on results of computational mechanics in abdominal aortic aneurysm. J Vasc Surg. 2010;51(3):679–688.
121 Malkawi A.H., Hinchliffe R.J., Xu Y., et al. Patient-specific biomechanical profiling in abdominal aortic aneurysm development and rupture. J Vasc Surg. 2010;52(2):480–488.
122 Maier A., Gee M.W., Reeps C., et al. A comparison of diameter, wall stress, and rupture potential index for abdominal aortic aneurysm rupture risk prediction. Ann Biomed Eng. 2010;38(10):3124–3134.
123 Georgakarakos E., Ioannou C.V., Kamarianakis Y., et al. The role of geometric parameters in the prediction of abdominal aortic aneurysm wall stress. Eur J Vasc Endovasc Surg. 2010;39(1):42–48.
124 Vande Geest J.P., Di Martino E.S., Bohra A., et al. A biomechanics-based rupture potential index for abdominal aortic aneurysm risk assessment: demonstrative application. Ann N Y Acad Sci. 2006;1085:1–21.
125 Vande Geest J.P., Dillavou E.D., Di Martino E.S., et al. Gender-related differences in the tensile strength of abdominal aortic aneurysm. Ann N Y Acad Sci. 2006;1085:400–402.
126 Darling R.C., Messina C.R., Brewster D.C., et al. Autopsy study of unoperated abdominal aortic aneurysms. The case for early resection, Circulation. 1977;56(3 Suppl):II161–II164.
127 Lederle F.A., Johnson G.R., Wilson S.E., et al. The aneurysm detection and management study screening program: validation cohort and final results. Aneurysm Detection and Management Veterans Affairs Cooperative Study Investigators. Arch Intern Med. 2000;160(10):1425–1430.
128 Ashton H.A., Buxton M.J., Day N.E., et al. The Multicentre Aneurysm Screening Study (MASS) into the effect of abdominal aortic aneurysm screening on mortality in men: a randomised controlled trial. Lancet. 2002;360(9345):1531–1539.
129 Derubertis B.G., Trocciola S.M., Ryer E.J., et al. Abdominal aortic aneurysm in women: prevalence, risk factors, and implications for screening. J Vasc Surg. 2007;46(4):630–635.
130 Scott R.A., Ashton H.A., Kay D.N. Abdominal aortic aneurysm in 4237 screened patients: prevalence, development and management over 6 years [see comments]. Br J Surg. 1991;78(9):1122–1125.
131 Singh K., Bonaa K.H., Jacobsen B.K., et al. Prevalence of and risk factors for abdominal aortic aneurysms in a population-based study: the Tromso Study. Am J Epidemiol. 2001;154(3):236–244.
132 Lederle F.A., Johnson G.R., Wilson S.E. Abdominal aortic aneurysm in women. J Vasc Surg. 2001;34(1):122–126.
133 Alcorn H.G., Wolfson S.K.Jr, Sutton-Tyrrell K., et al. Risk factors for abdominal aortic aneurysms in older adults enrolled in the Cardiovascular Health Study. Arterioscler Thromb Vasc Biol. 1996;16(8):963–970.
134 Kurvers H.A., van der Graaf Y., Blankensteijn J.D., et al. Screening for asymptomatic internal carotid artery stenosis and aneurysm of the abdominal aorta: comparing the yield between patients with manifest atherosclerosis and patients with risk factors for atherosclerosis only. J Vasc Surg. 2003;37(6):1226–1233.
135 Tornwall M.E., Virtamo J., Haukka J.K., et al. Life-style factors and risk for abdominal aortic aneurysm in a cohort of Finnish male smokers. Epidemiology. 2001;12(1):94–100.
136 Lederle F.A., Johnson G.R., Wilson S.E., et al. Prevalence and associations of abdominal aortic aneurysm detected through screening. Aneurysm Detection and Management (ADAM) Veterans Affairs Cooperative Study Group. Ann Intern Med. 1997;126(6):441–449.
137 Pleumeekers H.J., Hoes A.W., van der Does E., et al. Aneurysms of the abdominal aorta in older adults. The Rotterdam Study. Am J Epidemiol. 1995;142(12):1291–1299.
138 Rodin M.B., Daviglus M.L., Wong G.C., et al. Middle age cardiovascular risk factors and abdominal aortic aneurysm in older age. Hypertension. 2003;42(1):61–68.
139 Scott R.A., Bridgewater S.G., Ashton H.A. Randomized clinical trial of screening for abdominal aortic aneurysm in women. Br J Surg. 2002;89(3):283–285.
140 Vardulaki K.A., Walker N.M., Day N.E., et al. Quantifying the risks of hypertension, age, sex and smoking in patients with abdominal aortic aneurysm. Br J Surg. 2000;87(2):195–200.
141 Doll R., Peto R., Wheatley K., et al. Mortality in relation to smoking: 40 years’ observations on male British doctors. BMJ. 1994;309(6959):901–911.
142 Goldberg R.J., Burchfiel C.M., Benfante R., et al. Lifestyle and biologic factors associated with atherosclerotic disease in middle-aged men. 20-year findings from the Honolulu Heart Program. Arch Intern Med. 1995;155(7):686–694.
143 Hammond E.C. Smoking in relation to the death rates of one million men and women. Natl Cancer Inst Monogr. 1966;19:127–204.
144 Nilsson S., Carstensen J.M., Pershagen G. Mortality among male and female smokers in Sweden: a 33-year follow-up. J Epidemiol Community Health. 2001;55(11):825–830.
145 Rogot E., Murray J.L. Smoking and causes of death among U.S. veterans: 16 years of observation. Public Health Rep. 1980;95(3):213–222.
146 Tang J.L., Morris J.K., Wald N.J., et al. Mortality in relation to tar yield of cigarettes: a prospective study of four cohorts. BMJ. 1995;311(7019):1530–1533.
147 Weir J.M., Dunn J.E.Jr. Smoking and mortality: a prospective study. Cancer. 1970;25(1):105–112.
148 Lee A.J., Fowkes F.G., Carson M.N., et al. Smoking, atherosclerosis and risk of abdominal aortic aneurysm. Eur Heart J. 1997;18(4):671–676.
149 Wilmink T.B., Quick C.R., Day N.E. The association between cigarette smoking and abdominal aortic aneurysms. J Vasc Surg. 1999;30(6):1099–1105.
150 Strachan D.P. Predictors of death from aortic aneurysm among middle-aged men: the Whitehall study. Br J Surg. 1991;78(4):401–404.
151 Lederle F.A., Nelson D.B., Joseph A.M. Smokers’ relative risk for aortic aneurysm compared with other smoking-related diseases: a systematic review. J Vasc Surg. 2003;38(2):329–334.
152 Powell J.T., Greenhalgh R.M. Clinical practice. Small abdominal aortic aneurysms. N Engl J Med. 2003;348(19):1895–1901.
153 Lederle F.A., Wilson S.E., Johnson G.R., et al. Immediate repair compared with surveillance of small abdominal aortic aneurysms. N Engl J Med. 2002;346(19):1437–1444.
154 Brady A.R., Thompson S.G., Fowkes F.G., et al. Abdominal aortic aneurysm expansion: risk factors and time intervals for surveillance. Circulation. 2004;110(1):16–21.
155 Baumgartner I., Hirsch A.T., Abola M.T., et al. Cardiovascular risk profile and outcome of patients with abdominal aortic aneurysm in out-patients with atherothrombosis: data from the Reduction of Atherothrombosis for Continued Health (REACH) Registry. J Vasc Surg. 2008;48(4):808–814.
156 Franks P.J., Edwards R.J., Greenhalgh R.M., et al. Risk factors for abdominal aortic aneurysms in smokers. Eur J Vasc Endovasc Surg. 1996;11(4):487–492.
157 Blanchard J.F., Armenian H.K., Friesen P.P. Risk factors for abdominal aortic aneurysm: results of a case-control study. Am J Epidemiol. 2000;151(6):575–583.
158 Brown L.C., Powell J.T. Risk factors for aneurysm rupture in patients kept under ultrasound surveillance. UK Small Aneurysm Trial Participants. Ann Surg. 1999;230(3):289–296. discussion 296–287
159 Hobbs S.D., Claridge M.W., Quick C.R., et al. LDL cholesterol is associated with small abdominal aortic aneurysms. Eur J Vasc Endovasc Surg. 2003;26(6):618–622.
160 Gillum R.F. Epidemiology of aortic aneurysm in the United States. J Clin Epidemiol. 1995;48(11):1289–1298.
161 Norrgard O., Rais O., Angquist K.A. Familial occurrence of abdominal aortic aneurysms. Surgery. 1984;95(6):650–656.
162 Lederle F.A. Ultrasonographic screening for abdominal aortic aneurysms. Ann Intern Med. 2003;139(6):516–522.
163 Baird P.A., Sadovnick A.D., Yee I.M., et al. Sibling risks of abdominal aortic aneurysm. Lancet. 1995;346(8975):601–604.
164 Verloes A., Sakalihasan N., Koulischer L., et al. Aneurysms of the abdominal aorta: familial and genetic aspects in three hundred thirteen pedigrees. J Vasc Surg. 1995;21(4):646–655.
165 Frydman G., Walker P.J., Summers K., et al. The value of screening in siblings of patients with abdominal aortic aneurysm. Eur J Vasc Endovasc Surg. 2003;26(4):396–400.
166 Adams D.C., Tulloh B.R., Galloway S.W., et al. Familial abdominal aortic aneurysm: prevalence and implications for screening. Eur J Vasc Surg. 1993;7(6):709–712.
167 Cole C.W., Barber G.G., Bouchard A.G., et al. Abdominal aortic aneurysm: consequences of a positive family history. Can J Surg. 1989;32(2):117–120.
168 Webster M.W., Ferrell R.E., St. Jean P.L., et al. Ultrasound screening of first-degree relatives of patients with an abdominal aortic aneurysm. J Vasc Surg. 1991;13(1):9–13. discussion 13–14
169 Webster M.W., St. Jean P.L., Steed D.L., et al. Abdominal aortic aneurysm: results of a family study. J Vasc Surg. 1991;13(3):366–372.
170 Majumder P.P., St Jean P.L., Ferrell R.E., et al. On the inheritance of abdominal aortic aneurysm. Am J Hum Genet. 1991;48(1):164–170.
171 Wahlgren C.M., Larsson E., Magnusson P.K., et al. Genetic and environmental contributions to abdominal aortic aneurysm development in a twin population. J Vasc Surg. 2010;51(1):3–7. discussion 7
172 Wiernicki I., Gutowski P., Ciechanowski K., et al. Abdominal aortic aneurysm: association between haptoglobin phenotypes, elastase activity, and neutrophil count in the peripheral blood. Vasc Surg. 2001;35(5):345–350. discussion 351
173 St Jean P., Hart B., Webster M., et al. Alpha-1-antitrypsin deficiency in aneurysmal disease. Hum Hered. 1996;46(2):92–97.
174 Schardey H.M., Hernandez-Richter T., Klueppelberg U., et al. Alleles of the alpha-1-antitrypsin phenotype in patients with aortic aneurysms. J Cardiovasc Surg (Torino). 1998;39(5):535–539.
175 Hinterseher I., Tromp G., Kuivaniemi H. Genes and abdominal aortic aneurysm. Ann Vasc Surg. 2011;25(3):388–412.
176 Thompson A.R., Cooper J.A., Jones G.T., et al. Assessment of the association between genetic polymorphisms in transforming growth factor beta, and its binding protein (LTBP), and the presence, and expansion, of abdominal aortic aneurysm. Atherosclerosis. 2010;209(2):367–373.
177 Golledge J., Clancy P., Jones G.T., et al. Possible association between genetic polymorphisms in transforming growth factor beta receptors, serum transforming growth factor beta1 concentration and abdominal aortic aneurysm. Br J Surg. 2009;96(6):628–632.
178 Elmore J.R., Obmann M.A., Kuivaniemi H., et al. Identification of a genetic variant associated with abdominal aortic aneurysms on chromosome 3p12.3 by genome wide association. J Vasc Surg. 2009;49(6):1525–1531.
179 Gretarsdottir S., Baas A.F., Thorleifsson G., et al. Genome-wide association study identifies a sequence variant within the DAB2IP gene conferring susceptibility to abdominal aortic aneurysm. Nat Genet. 2010;42(8):692–697.
180 Helgadottir A., Thorleifsson G., Magnusson K.P., et al. The same sequence variant on 9p21 associates with myocardial infarction, abdominal aortic aneurysm and intracranial aneurysm. Nat Genet. 2008;40(2):217–224.
181 Thompson A.R., Golledge J., Cooper J.A., et al. Sequence variant on 9p21 is associated with the presence of abdominal aortic aneurysm disease but does not have an impact on aneurysmal expansion. Eur J Hum Genet. 2009;17(3):391–394.
182 Atturu G., Brouilette S., Samani N.J., et al. Short leukocyte telomere length is associated with abdominal aortic aneurysm (AAA). Eur J Vasc Endovasc Surg. 2010;39(5):559–564.
183 Szilagyi D.E., Elliott J.P., Smith R.F. Clinical fate of the patient with asymptomatic abdominal aortic aneurysm and unfit for surgical treatment. Arch Surg. 1972;104(4):600–606.
184 Bickerstaff L.K., Hollier L.H., Van Peenen H.J., et al. Abdominal aortic aneurysms: the changing natural history. J Vasc Surg. 1984;1(1):6–12.
185 Nevitt M.P., Ballard D.J., Hallett J.W.Jr. Prognosis of abdominal aortic aneurysms. A population-based study. N Engl J Med. 1989;321(15):1009–1014.
186 Lederle F.A., Johnson G.R., Wilson S.E., et al. Rupture rate of large abdominal aortic aneurysms in patients refusing or unfit for elective repair. JAMA. 2002;287(22):2968–2972.
187 Harris L.M., Faggioli G.L., Fiedler R., et al. Ruptured abdominal aortic aneurysms: factors affecting mortality rates. J Vasc Surg. 1991;14(6):812–818. discussion 819–820
188 Mortality results for randomised controlled trial of early elective surgery or ultrasonographic surveillance for small abdominal aortic aneurysms. The UK Small Aneurysm Trial Participants. Lancet. 1998;352(9141):1649–1655.
189 Ouriel K., Clair D.G., Kent K.C., et al. Endovascular repair compared with surveillance for patients with small abdominal aortic aneurysms. J Vasc Surg. 2010;51(5):1081–1087.
190 Brown P.M., Sobolev B., Zelt D.T. Selective management of abdominal aortic aneurysms smaller than 5.0 cm in a prospective sizing program with gender-specific analysis. J Vasc Surg. 2003;38(4):762–765.
191 Powell J.T., Brown L.C. The natural history of abdominal aortic aneurysms and their risk of rupture. Acta Chir Belg. 2001;101(1):11–16.
192 Evans S.M., Adam D.J., Bradbury A.W. The influence of gender on outcome after ruptured abdominal aortic aneurysm. J Vasc Surg. 2000;32(2):258–262.
193 Isselbacher E.M. Thoracic and abdominal aortic aneurysms. Circulation. 2005;111(6):816–828.
194 Svensson L.G. Natural history of aneurysms of the descending and thoracoabdominal aorta. J Card Surg. 1997;12(2 Suppl):279–284.
195 Ince H., Nienaber C.A. Etiology, pathogenesis and management of thoracic aortic aneurysm. Nat Clin Pract Cardiovasc Med. 2007;4(8):418–427.
196 Svensjo S., Bengtsson H., Bergqvist D. Thoracic and thoracoabdominal aortic aneurysm and dissection: an investigation based on autopsy. Br J Surg. 1996;83(1):68–71.
197 Bickerstaff L., Pairolero P., Hollier L., et al. Thoracic aortic aneurysms: a population-based study. Surgery. 1982;92(6):1103–1108.
198 Svensson L.G., Crawford E.S., Hess K.R., et al. Experience with 1509 patients undergoing thoracoabdominal aortic operations. J Vasc Surg. 1993;17(2):357–368. discussion 368–370
199 Guo D., Hasham S., Kuang S.Q., et al. Familial thoracic aortic aneurysms and dissections: genetic heterogeneity with a major locus mapping to 5q13–14. Circulation. 2001;103(20):2461–2468.
200 Crawford E.S., DeNatale R.W. Thoracoabdominal aortic aneurysm: observations regarding the natural course of the disease. J Vasc Surg. 1986;3(4):578–582.
201 Cambria R.A., Gloviczki P., Stanson A.W., et al. Outcome and expansion rate of 57 thoracoabdominal aortic aneurysms managed nonoperatively. Am J Surg. 1995;170(2):213–217.
202 Griepp R.B., Ergin M.A., Galla J.D., et al. Natural history of descending thoracic and thoracoabdominal aneurysms. Ann Thorac Surg. 1999;67(6):1927–1930. discussion 1953–1928
203 Pitt M.P., Bonser R.S. The natural history of thoracic aortic aneurysm disease: an overview. J Card Surg. 1997;12(2 Suppl):270–278.
204 Juvonen T., Ergin M.A., Galla J.D., et al. Prospective study of the natural history of thoracic aortic aneurysms. Ann Thorac Surg. 1997;63(6):1533–1545.
205 Davies R.R., Goldstein L.J., Coady M.A., et al. Yearly rupture or dissection rates for thoracic aortic aneurysms: simple prediction based on size. Ann Thorac Surg. 2002;73(1):17–27. discussion 27–18
206 Elefteriades J.A. Natural history of thoracic aortic aneurysms: indications for surgery, and surgical versus nonsurgical risks. Ann Thorac Surg. 2002;74(5):S1877–S1880. discussion S1892–1878
207 Dapunt O.E., Galla J.D., Sadeghi A.M., et al. The natural history of thoracic aortic aneurysms. J Thorac Cardiovasc Surg. 1994;107(5):1323–1332. discussion 1332–1323
208 Pressler V., McNamara J.J. Aneurysm of the thoracic aorta. Review of 260 cases. J Thorac Cardiovasc Surg. 1985;89(1):50–54.
209 Hiratzka L.F., Bakris G.L., Beckman J.A., et al. 2010 ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM guidelines for the diagnosis and management of patients with thoracic aortic disease: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines, American Association for Thoracic Surgery, American College of Radiology, American Stroke Association, Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, Society of Interventional Radiology, Society of Thoracic Surgeons, and Society for Vascular Medicine. Circulation. 2010;121(13):e266–e369.
210 Gott V.L., Greene P.S., Alejo D.E., et al. Replacement of the aortic root in patients with Marfan’s syndrome. N Engl J Med. 1999;340(17):1307–1313.
211 Svensson L.G., Kouchoukos N.T., Miller D.C., et al. Expert consensus document on the treatment of descending thoracic aortic disease using endovascular stent-grafts. Ann Thorac Surg. 2008;85(1 Suppl):S1–S41.
212 Tzemos N., Therrien J., Yip J., et al. Outcomes in adults with bicuspid aortic valves. JAMA. 2008;300(11):1317–1325.
213 Vallely M.P., Semsarian C., Bannon P.G. Management of the ascending aorta in patients with bicuspid aortic valve disease. Heart Lung Circ. 2008;17(5):357–363.
214 Loeys B.L., Schwarze U., Holm T., et al. Aneurysm syndromes caused by mutations in the TGF-beta receptor. N Engl J Med. 2006;355(8):788–798.
215 Habashi J.P., Judge D.P., Holm T.M., et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science. 2006;312(5770):117–121.
216 El-Hamamsy I., Yacoub M.H. Cellular and molecular mechanisms of thoracic aortic aneurysms. Nature Reviews Cardiology. 2009;6(12):771–786.
217 Germain D.P. Clinical and genetic features of vascular Ehlers-Danlos syndrome. Ann Vasc Surg. 2002;16(3):391–397.
218 Arteaga-Solis E., Gayraud B., Ramirez F. Elastic and collagenous networks in vascular diseases. Cell Struct Funct. 2000;25(2):69–72.
219 Nishiyama Y., Manabe N., Ooshima A., et al. A sporadic case of Ehlers-Danlos syndrome type IV: diagnosed by a morphometric study of collagen content. Pathol Int. 1995;45(7):524–529.
220 Pepin M., Schwarze U., Superti-Furga A., et al. Clinical and genetic features of Ehlers-Danlos syndrome type IV, the vascular type. N Engl J Med. 2000;342(10):673–680.
221 Nistri S., Sorbo M.D., Marin M., et al. Aortic root dilatation in young men with normally functioning bicuspid aortic valves. Heart. 1999;82(1):19–22.
222 Keane M.G., Wiegers S.E., Plappert T., et al. Bicuspid aortic valves are associated with aortic dilatation out of proportion to coexistent valvular lesions. Circulation. 2000;102(19 Suppl 3):III35–III39.
223 Yasuda H., Nakatani S., Stugaard M., et al. Failure to prevent progressive dilation of ascending aorta by aortic valve replacement in patients with bicuspid aortic valve: comparison with tricuspid aortic valve. Circulation. 2003;108(Suppl 1):II291–II294.
224 Fedak P.W., de Sa M.P., Verma S., et al. Vascular matrix remodeling in patients with bicuspid aortic valve malformations: implications for aortic dilatation. J Thorac Cardiovasc Surg. 2003;126(3):797–806.
225 Schmid F.X., Bielenberg K., Holmer S., et al. Structural and biomolecular changes in aorta and pulmonary trunk of patients with aortic aneurysm and valve disease: implications for the Ross procedure. Eur J Cardiothorac Surg. 2004;25(5):748–753.
226 Boyum J., Fellinger E.K., Schmoker J.D., et al. Matrix metalloproteinase activity in thoracic aortic aneurysms associated with bicuspid and tricuspid aortic valves. J Thorac Cardiovasc Surg. 2004;127(3):686–691.
227 Sybert V.P. Cardiovascular malformations and complications in Turner syndrome. Pediatrics. 1998;101(1):E11.
228 Jenkins N.P., Ward C. Coarctation of the aorta: natural history and outcome after surgical treatment. QJM. 1999;92(7):365–371.
229 Knyshov G.V., Sitar L.L., Glagola M.D., et al. Aortic aneurysms at the site of the repair of coarctation of the aorta: a review of 48 patients. Ann Thorac Surg. 1996;61(3):935–939.
230 Benzaquen B.S., Therrien J. Thoracic aortic aneurysm occurring at a coarctation repair site. Can J Cardiol. 2003;19(5):561–562.
231 Rao P.S., Galal O., Smith P.A., et al. Five- to nine-year follow-up results of balloon angioplasty of native aortic coarctation in infants and children. J Am Coll Cardiol. 1996;27(2):462–470.
232 Fletcher S.E., Nihill M.R., Grifka R.G., et al. Balloon angioplasty of native coarctation of the aorta: midterm follow-up and prognostic factors. J Am Coll Cardiol. 1995;25(3):730–734.
233 Beckman J.A. Giant cell arteritis. Curr Treat Options Cardiovasc Med. 2000;2(3):213–218.
234 Nuenninghoff D.M., Hunder G.G., Christianson T.J., et al. Incidence and predictors of large-artery complication (aortic aneurysm, aortic dissection, and/or large-artery stenosis) in patients with giant cell arteritis: a population-based study over 50 years. Arthritis Rheum. 2003;48(12):3522–3531.
235 Evans J.M., Bowles C.A., Bjornsson J., et al. Thoracic aortic aneurysm and rupture in giant cell arteritis. A descriptive study of 41 cases, [published erratum appears in Arthritis Rheum 38(2):290, 1995]. Arthritis Rheum, 37;10. 1994:1539–1547.
236 Nuenninghoff D.M., Hunder G.G., Christianson T.J., et al. Mortality of large-artery complication (aortic aneurysm, aortic dissection, and/or large-artery stenosis) in patients with giant cell arteritis: a population-based study over 50 years. Arthritis Rheum. 2003;48(12):3532–3537.
237 Nikkari S.T., Hoyhtya M., Isola J., et al. Macrophages contain 92-kd gelatinase (MMP-9) at the site of degenerated internal elastic lamina in temporal arteritis. Am J Pathol. 1996;149(5):1427–1433.
238 Tomita T., Imakawa K. Matrix metalloproteinases and tissue inhibitors of metalloproteinases in giant cell arteritis: an immunocytochemical study. Pathology. 1998;30(1):40–50.
239 Subramanyan R., Joy J., Balakrishnan K.G. Natural history of aortoarteritis (Takayasu’s disease). Circulation. 1989;80(3):429–437.
240 Kerr G.S., Hallahan C.W., Giordano J., et al. Takayasu arteritis. Ann Intern Med. 1994;120(11):919–929.
241 Ishikawa K., Maetani S. Long-term outcome for 120 Japanese patients with Takayasu’s disease. Clinical and statistical analyses of related prognostic factors. Circulation. 1994;90(4):1855–1860.
242 Roldan C.A., Chavez J., Wiest P.W., et al. Aortic root disease and valve disease associated with ankylosing spondylitis. J Am Coll Cardiol. 1998;32(5):1397–1404.
243 Sakane T., Takeno M., Suzuki N., et al. Behcet’s disease. N Engl J Med. 1999;341(17):1284–1291.
244 Yazici H., Yurdakul S., Hamuryudan V. Behcet disease. Curr Opin Rheumatol. 2001;13(1):18–22.
245 Ehrlich G.E. Vasculitis in Behcet’s disease. Int Rev Immunol. 1997;14(1):81–88.
246 Roldan C.A., Chavez J., Wiest P.W., et al. Aortic root disease and valve disease associated with ankylosing spondylitis. J Am Coll Cardiol. 1998;32(5):1397–1404.
247 Amor B. Reiter’s syndrome. Diagnosis and clinical features. Rheum Dis Clin North Am. 1998;24(4):677–695. vii
248 Letko E., Zafirakis P., Baltatzis S., et al. Relapsing polychondritis: a clinical review. Semin Arthritis Rheum. 2002;31(6):384–395.
249 Parkhurst G.F., Dekcer J.P. Bacterial aortitis and mycotic aneurysm of the aorta; a report of twelve cases. Am J Pathol. 1955;31(5):821–835.
250 Sommerville R.L., Allen E.V., Edwards J.E. Bland and infected arteriosclerotic abdominal aortic aneurysms: a clinicopathologic study. Medicine (Baltimore). 1959;38:207–221.
251 Bennett D.E., Cherry J.K. Bacterial infection of aortic aneurysms. A clinicopathologic study. Am J Surg. 1967;113(3):321–326.
252 Sedwitz M.M., Hye R.J., Stabile B.E. The changing epidemiology of pseudoaneurysm. Therapeutic implications. Arch Surg. 1988;123(4):473–476.
253 Mansur A.J., Grinberg M., Leao P.P., et al. Extracranial mycotic aneurysms in infective endocarditis. Clin Cardiol. 1986;9(2):65–72.
254 Anderson C.B., Butcher H.R.Jr, Ballinger W.F. Mycotic aneurysms. Arch Surg. 1974;109(5):712–717.
255 Jarrett F., Darling R.C., Mundth E.D., et al. The management of infected arterial aneurysms. J Cardiovasc Surg (Torino). 1977;18(4):361–366.
256 Vogelzang R.L., Sohaey R. Infected aortic aneurysms: CT appearance. J Comput Assist Tomogr. 1988;12(1):109–112.
257 Taylor L.M.Jr, Deitz D.M., McConnell D.B., et al. Treatment of infected abdominal aneurysms by extraanatomic bypass, aneurysm excision, and drainage. Am J Surg. 1988;155(5):655–658.
258 Johansen K., Devin J. Mycotic aortic aneurysms. A reappraisal. Arch Surg. 1983;118(5):583–588.
259 Long R., Guzman R., Greenberg H., et al. Tuberculous mycotic aneurysm of the aorta: review of published medical and surgical experience. Chest. 1999;115(2):522–531.
260 Pugh P.J., Grech E.D. Syphilitic aortitis. N Engl J Med. 2002;346(9):676.
[/level-membership-for-cardiovascular-category][not-level-membership-for-cardiovascular-category]
Chapter 37 Pathophysiology, Epidemiology, and Prognosis of Aortic Aneurysms
Aortic aneurysms result in significant morbidity and mortality, accounting for nearly 13,000 deaths and 55,000 hospital discharges per year in the United States.1 Although aneurysms may affect any part of the aorta from the aortic root down to the abdominal aorta, the prognosis and outcome in patients with aortic aneurysms vary based on location and underlying etiology. Timely and appropriate intervention may improve the natural history of the disease process. This chapter reviews the pathophysiology, epidemiology, and prognosis of aortic aneurysms.
The Normal Aorta
The aorta is the large conduit vessel through which the heart delivers blood to the entire body. It courses from the heart through the thorax and abdomen, and ultimately bifurcates into the common iliac arteries (CIAs) in the abdomen. In the thorax, the aorta can be subdivided into three segments: ascending aorta (from the base of the heart to the innominate artery), transverse aorta or aortic arch (including the great vessels and extending to the left subclavian artery), and descending aorta (from the distal edge of the subclavian artery to the level of the diaphragm) (Fig. 37-1).
Like other arterial structures, the aorta is composed of three layers: tunica intima, tunica media, and adventitia. The innermost surface of the tunica intima is lined by a single-cell-thick layer of endothelial cells (ECs). The intima is bound by the internal elastic lamina. The tunica media is composed of smooth muscle cells (SMCs), collagen, fibroblasts, elastin fibers, and ground substance, which together control the degree of vessel constriction and vasodilation. The presence of elastin fibers in the media defines the aorta as an elastic artery and provides the tensile strength that permits the aorta to withstand pulsatile delivery of blood from the heart. Elastin content gradually decreases with distance from the heart.2 The outermost layer, the adventitia, is a thin layer that contains connective tissue, fibroblasts, and the nutritive vasa vasorum.
Definition of Aortic Aneurysm
In adults, the normal diameter of the aorta is approximately 3 cm at the origin, 2.5 cm in the descending thoracic aorta, and 1.8 to 2 cm in the abdominal aorta. Aortic aneurysm is defined as a maximal aortic dimension greater than 3.0 cm, or a 50% increase in size compared with the normal segment proximal to the aneurysm. Mild expansion that does not meet these criteria may be referred to as aortic ectasia. True aneurysms are classified into two major groups on the basis of morphology: (1) fusiform (Figs. 37-2 and 37-3), defined as a circumferential expansion of the aorta, and (2) saccular, representing a focal outpouching of a segment of the aorta (Fig. 37-4). Fusiform aneurysms are the most common manifestation. In contrast to true aneurysms, which involve expansion of all three layers of the aortic wall, a pseudoaneurysm, also known as a false aneurysm, results from a disruption of the aortic wall and essentially represents a contained rupture of the aorta.
Figure 37-2 Three pathophysiological mechanisms that best characterize the process of aneurysm formation.
(Volume-rendered computed tomography image of abdominal aortic aneurysm used with permission of Joseph Schoepf, MD.)
Pathophysiology of Aortic Aneurysms
A wide variety of pathological states are associated with aortic aneurysms (Box 37-1). These include degenerative diseases, inherited disorders, infections, inflammatory conditions (i.e., vasculitis), and trauma. Specific disorders associated with aortic aneurysms are discussed later in this chapter. Important determinants of aortic aneurysm formation include inflammation, proteolysis of the structural components of the aortic wall, and abnormal biomechanical forces3 (see Fig. 37-2). Understanding the underlying pathophysiology of aneurysm formation is critical not only for prevention of initial aneurysm formation but also for limiting aneurysm growth and expansion.
Traditionally, pathological aortic aneurysm formation was ascribed to a process akin to atherogenesis. Although advances in basic and clinical investigation in both lesion types have revealed some common themes, newer studies suggest that aneurysm formation is fundamentally different from atherosclerosis. Preferential weakening of the adventitia and media—rather than an intimal proliferative process, as in atherosclerosis—results in diminished aortic resilience and tensile strength, culminating in aortic wall thinning, dilation, and increased wall stress, all of which may result in rupture. Although atherosclerotic changes may be seen in the wall of aneurysms, these changes may be a consequence of local turbulent flow as opposed to a cause of aneurysm formation.4,5 Moreover, the degree of systemic atherosclerosis does not correlate well with the degree of aneurysm formation.6
Development of aneurysms is associated with loss of two critical structural elements in the aortic wall: elastin and collagen. Elastin provides radial and longitudinal support, enabling the aorta to respond to pulsatile flow while maintaining normal arterial dimensions. The importance of elastin in maintaining aortic structure is highlighted by animal models where elastase infusion results in elastin breakdown and experimental aortic aneurysm formation.7 However, breakdown of elastin alone appears insufficient to cause aneurysmal expansion and rupture. Loss of collagen, another important structural element, is an additional contributor, and the relative balance of elastin and collagen deposition, among other factors, may be critical for determining aneurysm formation.8 Early in aneurysm formation, the aorta compensates for loss of elastin by increasing production of collagen,8 but as elastin content decreases, collagen (as the major source of tensile strength) is overwhelmed, and aortic expansion occurs. This is exacerbated by up-regulation of collagenases, resulting in further collagen degradation as described later.9 Structural changes in each layer of the aortic wall develop that together promote aortic stiffness. As a consequence, decrease in the vessel’s ability to distend normally with left ventricular (LV) contraction, weakening of the vessel wall, and increase in the tendency for dilation and ectasia follow.10 Some of the changes in aortic structure that promote aneurysm formation may arise as a result of the normal aging process. With normal aging, aortic stiffness due to fragmentation of elastin fibers, deposition of glycosaminoglycans, fibronectin (FN), and collagen, and reduced bioavailability of endothelium-derived nitric oxide (NO) occurs.11–14
Pathophysiologically, the major determinants of aortic aneurysm formation include proteolysis of the structural components of the aortic wall, inflammation, and abnormal biomechanical forces3 (see Fig. 37-2). Pathology of aortic aneurysms varies in different segments of the aorta and in different predisposing diseases. Frequently observed histological features include cystic medial necrosis, mucoid infiltration, and cyst formation in the setting of elastin necrosis and vascular smooth muscle apoptosis. In patients with Marfan’s syndrome (MFS), bicuspid aortic valve (BAV), or Turner’s syndrome, cystic medial necrosis is a common feature, but in contrast, inflammation is less prominent than in abdominal aortic aneurysms (AAAs).15,16 On the other hand, cystic medial necrosis is less likely to be observed in AAAs. Instead, AAAs typically have disrupted elastin fibers, inflammation, greater vascular smooth muscle cell (VSMC) apoptosis, and deficient glycosaminoglycan production.17 Elastin fragmentation occurs adjacent to the inflammatory cells. Despite differences in pathophysiology due to location and underlying etiology, formation of all aortic aneurysms involves to some degree the processes described in the following discussions (i.e., proteolytic degradation, inflammation, changes in biomechanical forces) that together facilitate aneurysm formation.
Proteolytic Degradation
Several proteolytic enzymes contribute to degradation of structural components of the arterial wall, ultimately increasing risk of aneurysm formation. Matrix metalloproteinases (MMPs) are endopeptidases that degrade one or more components of the extracellular matrix (ECM). Thus far, several classes of MMPs, comprising nearly 30 individual proteinases, have been characterized. They include collagenases, gelatinases, stromelysins, matrilysins, membrane-type (MT)-MMPs, and other MMPs.18 Typically produced as proenzymes, MMPs may be activated both intracellularly and extracellularly, and may be secreted by endothelial cells, vascular smooth muscle cells, or adventitial fibroblasts. Extracellular regulation of activation may occur as a result of an MMP–MT-MMP interaction or via interaction with plasmin or reactive oxygen species (ROS).19–21 The inhibitors of MMPs are the tissue inhibitors of matrix metalloproteinases (TIMPs) and plasminogen activator inhibitors (PAI) 1 and 2.18
Increased local production of MMPs in aortic aneurysms was first reported nearly 20 years ago.22–24 Although elevations of several MMPs have been noted, MMP-1 (collagenase) and MMP-3 (stromelysin), MMP-2 (gelatinase A), and MMP-9 (gelatinase B) represent the principal proteinases in aortic aneurysms that result in elastin and collagen degradation.25,26 MMP-2 and MMP-9 gelatinases specifically break down collagen. They are synthesized by local cells in the aortic wall, including infiltrating macrophages and resident aortic VSMCs.27,28 MMP-9 is typically found in the adventitia near the vasa vasorum, localizing to infiltrating macrophages.29 MMP-2 is synthesized constitutively by VSMCs30 but can also be synthesized by infiltrating leukocytes.31 Interestingly, MMP-2 production is increased in the vasculature remote from the aorta, suggesting a systemic underlying disease process that manifests with aortic aneurysmal disease.32
The centrality of these enzymes in aneurysm formation is supported by several lines of evidence. Studies have demonstrated that MMP-2 and MMP-9 levels are higher in tissue obtained from aortic aneurysms than in atherosclerotic plaque or normal arterial tissue.30,31 Furthermore, MMP-9 and MMP-2 knockout mice do not form aortic aneurysms in experimental models.33 Reinfusion of competent macrophages from wild-type mice into MMP-9 knockout mice enables aneurysm formation.33 In addition, elevated levels of MMP-1, MMP-8 (a neutrophil collagenase), and MMP-9 have been associated with aneurysm rupture, and levels of these MMPs may vary with aneurysm size.34–36 Relationship to aneurysm size is less clear for MMP-2.37–40
Elevated levels of other proteolytic enzymes such as human macrophage metalloelastase (MMP-12) and membrane type-1 metalloproteinase (MT1-MMP) have also been demonstrated in aortic aneurysms.41 Expression of MMP-12 is increased in aortic aneurysms as a result of macrophage infiltration. However, the relevance of MMP-12 to aneurysm formation is less clear, based on the finding that MMP-12 knockout mice are not completely protected from aneurysm formation.42 MT1-MMP, a collagenase produced by macrophages and increased in aortic aneurysms, likely has its greatest effect as an activator of the proenzyme form of MMP-2.43,44
In addition to increased expression of MMPs, aneurysm formation is associated with abnormal regulation of MMP levels in tissues. Matrix metalloproteinase levels are increased in states of inflammation and oxidant stress, both known to play a role in aneurysm formation. Compared with nonaneurysmal sections of aorta, superoxide anion and markers of oxidative stress are increased in aneurysmal segments, and ROS can convert proenzymes of MMPs to their active form.45–47 In addition, MMP levels can be augmented by plasminogen activators, such as urokinase-type plasminogen activator (uPA) and tissue-type plasminogen activator (tPA), which are specific physiological regulators of MMP-2 and MMP-9 activation and overexpressed in aortic aneurysms but not in healthy aortic specimens.48
Matrix metalloproteinase levels are also normally regulated by TIMPs. Although early work reported decreased concentrations of TIMPs in aneurysmal tissue,49 more recent data have been less clear, showing no difference in certain TIMP levels between aneurysmal and healthy aortic tissue.36,43,50,51 However, the relative imbalance in MMPs to TIMPs may be the more relevant factor.41 Nonetheless, experimental data have clearly demonstrated the significance of TIMPs to aneurysm formation. Local overexpression of TIMP-1 prevented aneurysm formation in a rat model of aortic aneurysm,52 but TIMP knockout mice have increased MMP activity and greater aneurysm formation.53 Similarly, lower expression of PAI-1, a regulator of plasminogen activator activity, has been reported in aneurysmal disease.49,54 Increased expression of plasminogen activators without reciprocal changes in their inhibitors alters the balance toward fibrinolysis, MMP activation, and tissue degradation. Experimental overexpression of PAI-1 prevents aneurysm formation in a rat model of aortic aneurysm,55 confirming the importance of relative rather than absolute concentrations.
The contribution of MMPs to the pathophysiology of aortic aneurysms is highlighted by the beneficial effect of medications known to reduce MMP levels on aneurysm expansion. For example, tetracyclines (e.g., doxycycline) have long been recognized as generalized inhibitors of metalloproteinases.56 Their potential value in aortic aneurysmal disease has been demonstrated in both organ culture and rodent models of aneurysm formation.36,57–59 In humans, doxycycline limits the activity of both MMP-2 and MMP-9 in aortic wall specimens, both by reducing macrophage MMP-9 messenger ribonucleic acid (mRNA) expression and diminishing activation of the proenzyme form of MMP-2.60–62 In a few small randomized phase 2 trials, doxycycline has been shown to decrease aneurysm expansion rate.63,64 Although these data await confirmation in larger trials, they do support the conceptual framework of the importance of proteolytic enzymes in aortic aneurysm formation and expansion.
By reducing levels of proteolytic enzymes, several other therapies may potentially be of therapeutic benefit in aneurysmal disease. HMG-CoA reductase inhibitors, or statins, have antianeurysm properties in experimental models by virtue of reducing oxidant stress and macrophage production of MMPs.65–67 However, data in humans have been inconsistent. A meta-analysis of five studies including 697 patients with small aortic aneurysms (<55 mm) treated with or without statins suggested that statin therapy was associated with lower rates of expansion.68 On the other hand, other human studies have not shown a benefit of preoperative statin treatment on MMP or TIMP levels in aneurysm specimens, and no difference in aneurysm expansion rate.69–71 Indomethacin also appears to prevent aortic aneurysm formation in a rat model,72 and the mechanism may be related to inhibition of cyclooxygenase (COX) 2, prostaglandin E2 (PGE2), and reductions in MMP-9.73–75 Indeed, PGE2 expression is up-regulated more than 30-fold in aortic aneurysms.76 Prostaglandin E2 localizes to infiltrating macrophages, and its expression is dependent on COX activity.77 Prostaglandin E2, through activation of interleukin (IL)-6, may increase VSMC apoptosis, further weakening the structural elements of the aorta.75,76,78
Inflammation
The contribution of inflammation to many pathological arterial processes has been well established.79 In 1981, Rose and Dent80 reported mild chronic inflammation in 72.5% and moderate inflammation in 15.7% in 51 consecutively resected AAAs. Subsequently it was discovered that lymphocytes and macrophages are found in greater quantity in the adventitia and media of AAAs than of atherosclerotic or normal aortas.81 In addition, surgical explant specimens from patients with aortic aneurysms demonstrated higher levels of adhesion molecules, including intracellular adhesion molecule (ICAM)-1 and vascular cell adhesion molecule (VCAM)-1, than seen in atherosclerotic and normal aortas. Similarly, tissue levels of proinflammatory cytokines, such as tumor necrosis factor (TNF)-α, interleukin (IL)-1β, IL-8, monocyte chemoattractant protein (MCP)-1, interferon (IFN)-γ, and IL-6, have all been noted to be elevated in patients with aneurysms compared with control subjects.82 Some studies have even suggested cytokine levels are higher in ruptured aortic aneurysms than in asymptomatic aortic aneurysms,83 although the data have been inconsistent.84 Finally, other acute-phase proteins, including C-reactive protein (CRP), D-dimer, and ceruloplasmin, are also present at increased levels in plasma and in the vessel wall.82,85–87
These inflammatory mediators largely derive from infiltrating macrophages, but lymphocytes and aortic ECs and SMCs also contribute to the inflammatory milieu. The presence of inflammation in aortic aneurysms is supported by positron emission tomographic imaging with18 fluorodeoxyglucose (FDG-PET) showing greater FDG uptake in aneurysmal compared to nonaneurysmal aortic segments from matched control subjects, and by advanced magnetic resonance imaging (MRI) techniques.88,89 Thus, immune cells invade the aortic wall, become activated, and create an inflammatory environment that engages the activity of local stabilizing cells, initiating the process of elastin and collagen breakdown and aneurysm formation. However, the initial signals that drive inflammatory cell recruitment remain unclear. Animal studies with experimental aneurysm models have confirmed the human studies and demonstrated that increased inflammation promotes aneurysm formation.90
Given the contribution of inflammation to aortic aneurysms, it follows that strategies to reduce inflammation might reduce aneurysm formation or limit aneurysm growth. Indeed, as mentioned previously, some data suggest that statins, known to have beneficial antiinflammatory properties beyond their effect on cholesterol lowering, may limit aneurysm growth and expansion.67,91,92 Recent studies have also shown that limiting inflammation can reduce aneurysm formation in animal models.93–97 Future studies will be required to clarify whether strategies to target inflammation can prevent formation of aortic aneurysms or limit expansion in humans.
Increases in Biomechanical Wall Stress
Several specific structural changes may predispose the abdominal aorta to aneurysm formation. For example, elastin within the aortic wall is organized into circumferential plates, or lamellae, that respond to the pulsatile load created by the heart. Each lamellar unit consists predominantly of two elastin bundles and vascular smooth muscle. However, deposition of elastin is not uniform along the aorta, with the thoracic aorta incorporating 35 to 56 lamellar units compared to only 28 in the abdominal aorta.98 The abdominal aorta may therefore be more susceptible to elastin breakdown due to a relative increase in pressure withstood per lamellar unit, compared with the rest of the vessel. In addition, the abdominal aorta has a decreased concentration of nutritive vasa vasorum compared to more proximal aortic segments.99 Reductions in aortic tissue perfusion stiffen the vessel, reducing compliance and ability to withstand pulsatile stress.100
Vascular cells in the aorta attempt to restore elastin content in the setting of elastin degradation to compensate for reductions in tensile and radial strength. Human AAA samples show a four- to sixfold increase in tropoelastin protein compared with control arteries.101,126 Elastin is produced by SMCs and macrophages. In areas of macrophage infiltration, elastin deposition is not organized into mature effective bundles. Indeed, compared with normal specimens, aneurysm specimens have a ninefold reduction in desmosine, a marker for mature elastin cross-linking.101 Thus, compensatory elastin replacement is disordered and does not improve aortic compliance.
Another factor that may make the abdominal segment of the aorta more prone to aneurysm formation is blood flow patterns specific to that segment. In experimental models, the infrarenal segment of the aorta is subject to much higher levels of oscillating flow and reflected pressure waves compared with the suprarenal segment,102 resulting in higher levels of aortic wall tension. Turbulence and pressure are exacerbated by the aneurysm’s morphology, which promotes development of local vortices and turbulent flow patterns.103 Excluding these flow patterns from the aneurysm by placing an aortic endograft rapidly reduces plasma MMP-9 levels in patients.104,105 In a rodent elastase infusion model of aortic aneurysm, flow conditions were examined by creating a left femoral arteriovenous fistula (AVF) or left iliac artery ligation. Increases in shear stress due to fistula formation resulted in a more stable aortic phenotype with decreased oxidative stress, decreased macrophage density in the media, increased aortic ECs and vascular smooth muscle cells, and reduced apoptosis compared to the lower shear stress introduced by femoral artery ligation.4,106,107 Improved flow decreased aortic expansion by 26%.106
In addition to adverse biomechanical forces creating an environment permissive for aneurysm formation, the role of intraluminal thrombus on wall stress and aneurysm expansion has recently been a focus of much study. Using finite element analysis in a three-dimensional (3D) model of the aorta derived from computed tomography (CT) scans, intraluminal thrombus was found to lower peak wall stress by up to 38%108 relatively independent of thrombus constituents.109 Thrombus decreases transmission of luminal pressure to the aneurysm wall and may prevent aneurysm rupture by reducing wall strain.110–115 On the other hand, intraluminal thrombus may also contribute to further aneurysm formation,111 given that thrombus has been demonstrated to have higher levels of proteolytic enzymes and enzyme activators than the aneurysm itself.116 Intraluminal thrombus may act as a proteolytic enzyme reservoir through polymorphonuclear leukocytes, and enzyme accumulation provides a ready source of destructive elements for the adjacent aneurysm.116
Understanding biomechanical factors may help improve assessment of risk of aneurysm growth and rupture above and beyond the predictive value of lumen diameter alone.117 Several studies have used novel computational models to incorporate biomechanical factors such as wall stress, wall strength, and extent of intraluminal thrombus for assessment of abdominal aneurysms.112,118–123 Some studies have suggested that assessment of wall stress, wall strength, and the ratio of wall stress to wall strength may be better predictors of rupture risk than diameter.124,125
Epidemiology and Prognosis of Aortic Aneurysms
Abdominal Aortic Aneurysms
Prevalence
The prevalence of aneurysms of the abdominal aorta has been determined on the basis of several large screening studies and autopsy series (Table 37-1). In an early series of 24,000 consecutive autopsies performed over 23 years, 1.97% of the subjects were found to have an AAA.126 Of the 473 aneurysms found, 58% were larger than 4 cm in diameter, nearly three quarters of the patients were men, and one fourth of the aneurysms had ruptured. More recent large screening programs in targeted populations have further evaluated the prevalence of AAA. The largest screening program performed was the Aneurysm Detection and Management (ADAM) Study Screening Program, which studied 126,196 veterans 50 to 79 years of age.127 In this cohort of predominantly male American veterans, 3.6% of subjects had an infrarenal aortic diameter greater than 3 cm, and an AAA 4 cm or larger was found in 1.2%. The Multicentre Aneurysm Screening Study (MASS) also screened 27,147 of 33,830 invited men aged 65 to 74 and reported a 4.9% prevalence of AAA 3 cm or larger.128
Several studies have demonstrated lower prevalence of AAA in women. The largest and most recent of these studies screened nearly 10,012 women (mean age, 69.6 years) and found an AAA prevalence rate of 0.7%, with only 4 of 74 detected aneurysms measuring larger than 5 cm.129 These low prevalence rates were consistent with findings from earlier studies. Among 4237 subjects aged 65 to 80 who participated in a screening study among general practitioners in West Sussex, United Kingdom, 2290 women agreed to undergo abdominal ultrasonography.130 Only 1.4% of the women had an AAA 3 cm or larger, and only 0.3% had an AAA 4 cm or larger. This dramatically lower prevalence in women has been confirmed in subsequent studies. In the Norwegian Tromso study, 2% of 2943 women aged 55 to 84 had an AAA 3 cm or larger, and 0.5% had an abdominal aneurysm 4 cm or larger.131 Finally, among female American veterans, the prevalence of an AAA 3 cm or larger was just 1%.132 However, an increasing number of cardiovascular risk factors does increase the risk of AAA in women, with a prevalence rate as high as 6.4% in this higher-risk group.129
Risk factors
Three risk factors predict the vast majority of AAAs: age, gender, and cigarette smoking. Aneurysms usually affect the elderly, seldom occurring in those younger than 60 years of age, and there is a clear increase in incidence with increasing age, even when limiting the studies to older individuals.133–135 In a Norwegian population-based study of 6386 men and women aged 25 to 84, the incidence of AAA in men increased from 0% in those aged 25 to 44, to 6% in those aged 55 to 64, and to 18.5% in those aged 75 to 84.131 Large North American epidemiological studies have demonstrated an increase in AAA risk ranging from 58% to 300% with each additional decade of life.136 Gender is also an important predictor of AAA formation; in all age groups, risk of AAA is two- to sixfold higher in men than women.131,132,137–140 Cigarette smoking is the most potent modifiable risk factor and increases the risk of AAA by 60% to 850%.141–147 In the ADAM study and the Edinburgh Artery Study, risk of an aneurysm increased threefold with any smoking history.127,148 Risk of AAA development further increases with number of cigarettes smoked, duration of smoking, and lack of filtration, indicating a dose-response relationship.149 In the Whitehall study of 18,403 male civil servants examined at age 40 to 64 years, aneurysm frequency increased from sixfold with manufactured cigarettes with filters to 25-fold with hand-rolled cigarettes.150 Smoking cessation can reduce the risk of aneurysm formation, with former smokers having a lower AAA risk than current smokers.136,149,151
Risk factors for cardiovascular disease in general (e.g., hypertension, hyperlipidemia) also increase the risk of AAA formation but are less potent risk factors than age, gender, and smoking.127,136,152–154 In the REACH registry, there was a clear modest association of both hypertension and hyperlipidemia with AAA.155 Earlier studies had suggested a less consistent relationship with hypertension,138,156 but aneurysm formation seems to correlate best with diastolic blood pressure157 or use of an antihypertensive medication.140 Some data suggest that hypertension may be a greater risk factor for aneurysm rupture than for initial aneurysm formation.158 The association between cholesterol and AAA is clear, although hyperlipidemia is a less potent risk factor than those mentioned previously.135,137,159 Risk of AAA formation increased 30% per 40 mg/dL total cholesterol in the Chicago Heart Association Detection Project in Industry cohort.138 For specific components of the lipid profile, higher levels of low-density lipoprotein (LDL) and lower levels of high-density lipoprotein (HDL) cholesterol are both associated with aneurysm formation.133,159 Similarly, the presence of atherosclerosis increases risk of aneurysm formation,133,134,137 although as mentioned previously, aneurysm formation is likely a process distinct from atherosclerosis. In the ADAM study of more than 100,000 subjects, hypertension, elevated cholesterol, and presence of other vascular disease increased risk of aneurysm formation by 15%, 44%, and 66%, respectively.127 In contrast, diabetes and black race appear protective against formation of an aneurysm.127,132,136,160 Diabetes decreases risk of aneurysm formation by 30% to 50%.136,157
A dramatic increase in frequency of AAA formation in relatives of patients with aortic aneurysm suggests a genetic component to the disease. Although cigarette smoking numerically accounts for the vast majority of AAAs in the population,136 the most potent risk factor for aneurysm formation is a history of aneurysm in a first-degree relative. Norgaard et al.161 identified an 18% incidence of aneurysms in first-degree relatives of patients with AAA. In the ADAM study, a family history doubled the risk of AAA, but was reported in only 5.1% of more than 100,000 participants.162 Investigations specific to the impact of family history demonstrate a larger risk. Several studies have demonstrated that a family history of AAA increases the risk of AAA four- to fivefold.157,163 Family history of AAA was also related to earlier AAA formation and rupture by nearly a decade.164 Rate of rupture was nearly fourfold higher in patients with a family history than in sporadic AAA patients. Frydman et al.165 screened the siblings of 400 AAA patients and found an AAA in 43% of male siblings and 16% of female siblings. More specifically, the risk of AAA formation consistently rises above 20% for men older than 50 who have a first-degree relative with AAA.161,166–169 Using segregation analysis, Majumder et al.170 reported that the relative risk of developing an AAA is 3.97 and 4.03 with paternal and maternal history, respectively. Risk increases to nearly 10-fold with an affected male sibling and 23-fold when a female sibling is affected.170 Twin studies further support a genetic component to AAA formation, with one study reporting an odds ratio (OR) of 71 (95% confidence interval [CI], 27-183) for monozygotic twins and 7.6 (95% CI, 3.0-19) for dizygotic twins.171
Despite the wealth of data supporting a genetic component to AAA formation, no clear mode of inheritance and no single candidate gene has been identified. Early studies suggested evidence of both sex-linked and autosomal dominant patterns of inheritance. Associations have been made with blood types, haptoglobin variations, α1-antitrypsin, and human leukocyte antigen class II (HLA-II) immune response genes.172–174 More than 100 reports on genetic associations with AAA have appeared in the literature,175 including genes related to cardiovascular disease, inflammation, and related signaling pathways. One study suggested an association between reduced AAA growth and five single-nucleotide polymorphisms (SNPs) in latent TGF-β binding protein (LTBP4), as well as an allelic variant of TGFB3.176 However, another study showed no association between genetic polymorphisms in the main receptors for TGF-β and AAA formation.177 Two genome-wide association studies (GWAS) have suggested an association between AAA and a SNP located on chromosome 3p12.3 in a region near the gene CNTN3 for contactin-3, a lipid-anchored cell adhesion molecule.178 Another GWAS in a population from Iceland and the Netherlands found a SNP on 9q33 associated with AAA with an OR of 1.21. The same SNP has been associated with coronary artery disease (CAD), peripheral artery disease (PAD), and pulmonary embolism (PE).179 The SNP resides in the gene coding for DAB2IP, a gene encoding a cell growth and survival inhibitor. In addition, the same gene variant associated with myocardial infarction (MI) at locus 9p21 has been associated with AAA.180,181 Finally, of interest but of unclear significance is the association between telomere length and AAA in a small cohort in the United Kingdom.182 This may represent the association of aging, telomere length, and aneurysm formation, but the direct pathophysiological link remains unclear.
Prognosis
The natural history of AAA is one of silent coexistence and sudden lethal rupture. In a large autopsy study performed over a quarter of a century, one fourth of abdominal aneurysms were ruptured on postmortem examination.126
[/not-level-membership-for-cardiovascular-category]
