[level-membership-for-pathology-category]
Pathology of Liver and Hematopoietic Stem Cell Transplantation
Anthony J. Demetris
James M. Crawford
Kumiko Isse
Marta I. Minervini
Michael A. Nalesnik
Erin Rubin
Parmjeet S. Randhawa
Eizaburo Sasatomi
Introduction
Liver transplantation is used worldwide to treat a broad spectrum of end-stage liver diseases. Hepatitis C virus (HCV) infection, alcohol-induced liver disease, and nonalcoholic fatty liver disease–induced cirrhosis are the leading indications in North America, Europe, and South America. In Asia, hepatitis B virus (HBV)–induced cirrhosis is responsible for most liver transplantation, followed by the previously listed causes. Recurrence of the original disease is common after transplantation, and liver allografts are susceptible to a variety of technical complications (e.g., blood component sludging) that cause dysfunction and affect the morphologic diagnosis.
This chapter focuses on aspects of common diseases in the transplantation setting and on conditions unique to allografts, such as infection, rejection, small-for-size syndrome, preservation/reperfusion injury, and minimization of immunosuppression. Most disorders are discussed in the order in which specimens are received, from donor and then recipient, after transplantation. The terminology and acronyms used in liver transplantation are listed in Table 52.1.
Table 52.1
Acronyms Used in Liver Transplantation
| Acronym | Definition |
| ACR | Acute cellular rejection |
| AHR | Acute humoral rejection |
| AMR | Antibody-mediated rejection |
| ASTS | American Society of Transplant Surgeons |
| CIT | Cold ischemia time |
| DCD | Donation after cardiac death |
| DSA | Donor-specific antibodies |
| ECD | Extended criteria donors |
| FCH | Fibrosing cholestatic hepatitis |
| GVHD | Graft-versus-host disease |
| HAT | Hepatic artery thrombosis |
| HSC(T) | Hematopoietic stem cell (transplantation) |
| IPTH | Idiopathic posttransplantation hepatitis |
| IS | Immunosuppression |
| NRH | Nodular regenerative hyperplasia |
| PHP | Portal hyperperfusion |
| PTLD | Posttransplantation lymphoproliferative disorder |
| SFSS | Small-for-size (graft) syndrome |
| SOS | Sinusoidal obstruction syndrome |
| VOD | Veno-occlusive disease |
Donor Evaluation
Cadavers
Gross and frozen section examination can assist in evaluation of nonideal or extended criteria donors (ECDs) as defined by various characteristics.1–3 Included are advanced donor age (>60 years), hypernatremia (>155 meq/L), macrovesicular steatosis (>40%), cold ischemia time (>12 hours), partial liver allografts, and donation after cardiac death (DCD), hemodynamic instability, use of vasopressors, hypernatremia, HBV or HCV infection or hepatitis B core antibody (anti-HBc) positivity, history of cancer, or finding of a liver mass, fibrosis, or other focal lesions.
Feng and colleagues4 introduced the concept of a donor risk score based on a study of more than 20,000 transplants; the scores inversely correlate with 1- and 3-year recipient survival times. The overall score is the sum of component scores for the following parameters: donor age greater than 60 years, anoxic and cerebrovascular causes of death, black race, short height, DCD, split or partial grafts, regional or national sharing, and cold ischemic time.
Evaluation of donor livers by a pathologist is most often requested because of the gross appearance, texture or color of the donor liver, known preexisting donor disease (e.g., HCV infection), and the clinical history or circumstances of the donor’s death or harvesting procedure. Considering the importance of gross examination, the tissue for frozen section evaluation should be fresh, preferably obtained in the presence of the pathologist, who should also inspect the organ to ensure that the tissue sample represents the liver as a whole. Three tissue samples should be collected if the gross appearance is uniform: two 2.0-cm, 16-gauge needle cores, one each from the right and left lobes, and one 2.0-cm2 subcapsular right lobe wedge biopsy. The wounds are closed by sutures. The core biopsies are also useful for staging the degree of fibrosis. The wedge biopsy can be helpful to evaluate arterial or arteriolar disease and steatosis.
A few guidelines can help the pathologist to avoid introduction of artifacts during sample preparation. Fresh liver tissue should be immediately transported to the frozen section room on a paper towel moistened with preservation solution or in a plastic specimen container. Storage in physiologic saline, air drying, and placement of the tissue sample on an absorbent substrate should be avoided. Air-drying and storage in physiologic saline can cause hepatocytes to appear shrunken or necrotic, which can lead to overestimation of ischemic injury. Absorbent substrates also blot fat out of the tissue, resulting in underestimation of the extent of fatty infiltration.
Difficulty cutting the frozen section should alert the pathologist to the possibility of a steatotic donor liver. Recognition of hepatocytes in various stages of injury or necroapoptosis because of ischemic damage can be enhanced by staining several sections with eosin for increasing lengths of time. This enhances the contrast between viable and damaged or nonviable hepatocytes; the latter are hypereosinophilic and often show early nuclear karyorrhexis.
Histopathologic findings should be correlated with the donor’s history and laboratory values. Because partial or fragmented clinical histories can be misleading, the pathologist should aggressively request additional information if the biopsy findings do not correlate with the known history or events. However, donor evaluation by biopsy is only one laboratory test. Pathologists are unable to predict the adequacy of organ function after transplantation based solely on the absence of significant histopathologic findings on frozen section evaluation of the donor organ.
Organs are usually disqualified for transplantation if the donor is positive for certain serologically confirmed infections (e.g., human immunodeficiency virus [HIV], HCV, rabies) or has had a high-risk malignancy.5,6 Biopsy findings that usually disqualify organs include diffuse necrosis involving more than 10% of hepatocytes, severe macrovesicular steatosis involving 50% or more hepatocytes, moderate or severe atherosclerosis of intrahepatic artery branches, and definite evidence of bridging fibrosis.7–10 In our experience, polarized light microscopy at the time of frozen section examination offers a rapid, easy, and accurate estimate of liver fibrosis without the need for a trichrome stain.
Some ECD factors that may have histopathologic manifestations are advanced donor age (>60 years), macrovesicular steatosis (>40%), HCV infection, cardiovascular instability or ischemic injury, and occasionally DCD. Other ECD factors are not reliably associated with specific histopathologic findings and do not justify biopsy evaluation: black race, short stature, cerebrovascular cause of death, hypernatremia (>155 mEq/L), cold ischemia time exceeding 12 hours, and partial-liver allografts.
DCDs are the only growing source of donor organs. DCDs represent approximately 4% to 5% of the total donor pool,11 but their use is fraught with potential pitfalls. The American Society of Transplant Surgeons (ASTS) best practice guidelines have been reviewed by Reich and colleagues.11 DCD livers are exposed to significant warm ischemia, which optimally should be limited to less than 20 minutes. Even under ideal circumstances, DCDs are still susceptible to ischemic cholangiopathy that can develop several weeks to months after transplantation. It is thought that suboptimal flushing of the peribiliary capillary plexus leads to microvascular thrombosis and subsequently to poor reperfusion and ischemic injury to the biliary tree.
A grossly fatty appearance of the organ is the most common reason for requesting frozen section evaluation of a cadaveric donor liver (Fig. 52.1). Experienced donor surgeons are usually able to accurately estimate the severity of steatosis before biopsy evaluation, except for donors with small vacuolar steatosis. Lighting conditions in operating rooms can significantly influence the gross appearance of donor livers and can lead to underestimation of the degree of steatosis.
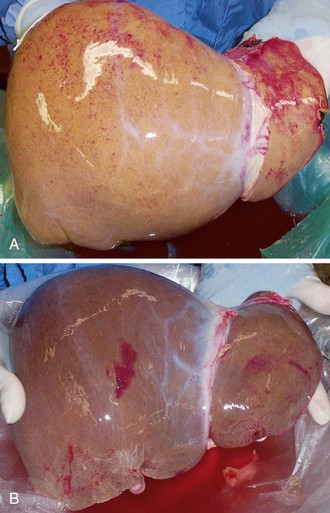
Large vacuolar (macrovesicular) steatosis is defined as fat globules greater than the nuclear diameter and associated with peripheral nuclear displacement. Small vacuolar steatosis (previously known as microvesicular steatosis) is defined as multiple, small fat globules less than the nuclear diameter and associated with a centrally placed nucleus. Moderate macrovesicular steatosis (>30%) increases susceptibility to preservation/reperfusion injury, impairs regeneration, and is associated with decreased graft survival.2,3,12 Small vacuolar steatosis is often found after a short period of warm ischemia or other insults and usually does not adversely affect outcome. One study, however, associated high-grade, small vacuolar steatosis with delayed graft function.13 In our opinion, the severity of macrovesicular steatosis can be roughly estimated on hematoxylin and eosin (H&E)–stained slides alone. Fat stains are unnecessary.
Most studies confirm the reproducibility of identifying donor macrovesicular steatosis of 50% or more of affected hepatocytes by frozen section evaluation before transplantation,14,15 but reproducibility is decreased at the lower cutoff value of 30%.14,15 One study recommended that pathologic evaluation be abandoned16 because of poor intrapathologist and interpathologist reproducibility for microvesicular and macrovesicular steatosis, inflammation, and hepatocyte ballooning and because of poor correlation between pathologists and computerized morphometric fat assessment. Other modalities used to assess steatosis include clinical and biochemical parameters17 and hepatic computed tomography (CT) in conjunction with other noninvasive clinical data,18 and magnetic resonance imaging (MRI).19
The use of steatotic donor livers is controversial and varies among transplantation centers. Most disqualify donor livers when macrovesicular steatosis exceeds 50% of hepatocyte volume (Fig. 52.2) because it has been reliably associated with an increased risk of early graft dysfunction and failure.2,3,12,20 This practice, however, has been questioned,13,20,21 particularly if other risk factors (e.g., cold ischemia time) or complications are absent or have been mitigated.21,22
Some studies have applied an evenly distributed range for scoring macrovesicular steatosis (mild < 30%, moderate = 30% to 60%, and severe > 60%),23 but we and others3,24 use a scale that more closely reflects the triage algorithm for ECD at our center. In our algorithm, mild donor macrovesicular steatosis (<10%) does not influence the decision-making process. Livers with moderate macrovesicular steatosis (10% to 30%) typically are used for transplantation, but other criteria (e.g., ECD characteristics) are also taken into consideration in the decision-making process. Livers with more than 30% (severe) steatosis are used only in special circumstances, such as when the cold ischemic time is kept to a minimum (<9 hours) and there are few or no other ECD risk factors. The outcomes in these situations are comparable to those for nonsteatotic donor livers.21,22
Necrosis in donor biopsies has negatively impacted recipient outcomes in some23,25 but not all studies.13,15 An algorithmic and reproducible method of quantifying the necrosis has not been defined, which may account for differences in observations. In our experience, the liver is usually disqualified if more than 10% of hepatocytes are necrotic and the necrosis diffusely involves the wedge and needle cores. However, assessment should not be based on necrosis limited only to subcapsular areas, because this finding is quite common, especially when the harvesting operation is associated with vigorous manipulation. Correlation with serial preharvest donor serum liver injury test profiles can be used as an additional gauge of the extent of necrosis.
Many centers use mildly diseased HCV-positive donors to prolong the life of a HCV-positive recipient with end-stage HCV-induced liver failure and in HCV-negative patients with fulminant hepatic failure.26,27 Graft and patient outcomes have been minimally impacted by donor HCV status,26,27 but more rapid progression of fibrosis can occur.28 All HCV-positive donors at our institution are subjected to frozen section biopsy analysis. Those with nonbridging fibrosis (<3 of 6 on the Ishak scale) are offered to potential recipients after informed consent is obtained. Other groups report a fibrosis cutoff value of one lower stage (<2 of 6).28
Anti-HBc–positive donors can transmit HBV to naïve and unvaccinated recipients.29 The risk is lower in vaccinated and anti-HBc–positive recipients and can be further reduced by anti-HBV medications and passive antibodies.30 Donor biopsy evaluation usually is not helpful in this circumstance because most biopsies are normal in the absence of other diseases.
Neoplastic, infectious, and metabolic diseases have been inadvertently transferred from donors to recipients.6 Examples that may be detectable by pathologists include various cancers, amyloidosis, hemochromatosis, and fungal, viral, and parasitic diseases.6 Metabolic diseases such as familial amyloid polyneuropathy,31 oxalosis,32 and possibly α1-antitrypsin deficiency33 can be transferred with the donor organ in so-called domino transplants, with the expectation that the latency period between transplantation and onset of disease in the recipient constitutes a gain in life span. Post hoc analysis of donor data for recipients with unexpected complications may provide valuable insights.
Living Donors
Living donor operations account for approximately 5% or less of all livers transplanted in North America and Europe but represent most transplantations performed in Asia.34 Because major liver resection is risky (mortality rate of approximately two deaths per 700 patients),35 most centers routinely subject potential living donors to a rigorous screening protocol. Liver biopsy evaluation is included at some centers to further minimize donor risk.36–39
A thorough stepwise medical and surgical donor evaluation screens for any major medical diseases, obesity, previous major abdominal surgery, anatomic compatibility between the donor and recipient, infectious diseases that could be transmitted to the recipient, psychosocial instability, and any liver function abnormality or disease that may put the donor at risk.35,39 Abnormalities detected during the workup can disqualify a potential donor, signal the need for a liver biopsy, or require further evaluation.
Several groups have reported histopathologic findings for likely living donors.39 Most biopsies from living donors are normal or show mild steatosis, but 20% to 50% show mild abnormalities. The most common pathology is some degree of macrovesicular steatosis. It is identified in 14% to 53% of candidates and is the most common reason for donor disqualification.36–41 Disqualification rates based on biopsy findings alone vary from 3% to 21%.39 Most programs try to limit the severity of macrovesicular steatosis in living donors to less than 30%, because this level does not adversely impact the postoperative course of the donor or recipient.38 Candidates with biopsies showing more than 30% of macrovesicular steatosis undergo diet modification and other treatments to reduce hepatic steatosis. Some programs limit macrovesicular steatosis to less than 10% or 20%.41,42
Other biopsy findings include low-grade chronic hepatitis of undetermined origin, granulomas, and a variety of unexpected findings (e.g., early-stage primary biliary cirrhosis [PBC]).39 Mild (1+ to 2+ on a scale of 0 to 4+) periportal hepatocellular iron deposits occur in approximately 17% of mostly male potential donors. Hepatocyte iron deposits probably represent a normal finding in men that does not preclude transplantation. Unexplained portal tract eosinophilia may be seen, and in two cases, it did not adversely affect the postoperative clinical course of the donor or recipient.39
Sources of Graft Dysfunction
Graft Dysfunction After Transplantation
Operative Timing and Methods
Accurate biopsy interpretation requires familiarity with the donor and the type of operation, because many causes of dysfunction are attributable to agonal events in the donor and to complications during harvesting of organs. In Western countries, orthotopic liver transplantation with a whole cadaveric donor liver is the most common procedure. The native liver is replaced in an anatomically correct fashion after resection of the donor gallbladder. End-to-end anastomoses connect the recipient and donor portal vein, hepatic artery, bile duct, and vena cava.43 Donor and recipient are usually matched for size and ABO blood group, unless the recipient is critically ill, or the donor pool is limited by blood type.
Surgical variations include the use of live donors, split livers, and alternate vena caval anastomoses.44 For example, living donor operations are common in Asia. Because the operative technique can influence graft dysfunction, it is important for the pathologist to understand the histopathologic effects of transplantation and the methods used for all vascular and biliary anastomoses.
More technically demanding operations usually deviate from reconstruction of normal anatomy and increase the risk of complications (e.g., suboptimal venous drainage). Split donor livers, reduced-size livers, and living donor transplantations increase the risk of vascular and biliary tract complications.44
Accurate biopsy interpretation requires an understanding of the characteristic time periods during which certain causes of allograft dysfunction occur (Table 52.2). Information about the original disease, time from transplantation to graft failure, and liver injury test profile often is enough to generate a reasonably accurate diagnosis, even without biopsy evaluation. Because liver injury often has more than one cause, all clinical parameters should be explored before the final histopathologic diagnosis is given.
Table 52.2
Timing of Common Allograft Syndromes
| Syndrome | Clinical Associations and Observations | Peak Period |
| Preservation/reperfusion injury | Long cold (>12 hr) or warm (>120 min) ischemic time; older donor age (>60 yr), hemodynamically unstable, DCD, repeat anastomosis; poor bile production; prolonged cholestatic phase predisposes to biliary sludge syndrome | Recognized primarily in postreperfusion biopsies and biopsies obtained during the first few weeks after OLT; changes may persist for several months, depending on severity of the initial injury |
| Antibody-mediated rejection | ABO-incompatible donor; high-titer (>1 : 32) lymphocytotoxic crossmatch DSAs; persistently low platelet counts and complement levels during first several weeks after transplantation | First several weeks to months after transplantation; later onset less common and not well defined |
| Acute rejection | Younger, healthier female and inadequately immunosuppressed recipients, long cold ischemic times, and disorders of dysregulated immunity (e.g., PSC, AIH, PBC) | Peak depends on IS regimen; usually 3-40 days; later onset usually associated with inadequate IS |
| Chronic rejection | Usually occurs in inadequately immunosuppressed patients (e.g., infections, tumors, PTLD); patients have a history of moderate or severe or persistent acute rejection episodes or are noncompliant | Bimodal distribution; early peak during first year and later increase in noncompliant and inadequately immunosuppressed patients |
| Hepatic artery thrombosis | Suboptimal anastomosis; pediatric or small-caliber vessels; donor and/or recipient atherosclerosis; suboptimal or difficult arterial anastomosis; large difference in vessel caliber across anastomosis; hypercoagulopathy; suboptimal arterial flow (vasospasm caused by small-for-size syndrome) | Bimodal distribution; early peak at 0-4 wk and later peak at 18-36 mo |
| Biliary tract obstruction or stricturing | Arterial insufficiency or thrombosis; long cold ischemia, DCD, difficult biliary anastomosis; AMR; original disease of PSC | Varies but timing can be used to determine cause: <6 mo, usually mechanical, preservation/reperfusion injury (ischemic cholangiopathy), or AMR; >6 mo, recurrent disease or mechanical |
| Venous outflow obstruction | Difficult piggyback hepatic vein reconstruction; cardiac failure | Usually during the first several months |
| Opportunistic viral (e.g., CMV, EBV, adenovirus) and fungal infections | Seropositive donors to seronegative recipients (often pediatric); excessive IS | 0-8 wk, much less common thereafter except for EBV-related PTLDs and other EBV-related tumors |
| Recurrent or new-onset viral hepatitis (e.g., HBV, HCV, HEV). | Original disease HBV, HCV, or acquired HEV-induced hepatitis in patients with contact with animals or culinary exposures | Usually first becomes apparent 4-6 wk after transplantation and persists thereafter; earlier onset (<2 wk) in aggressive cases |
| Recurrent AIH, PBC, and PSC | Original disease of AIH, PBC, or PSC | Usually > 6 mo after transplantation; incidence of recurrence increases with time after transplantation |
| Alcohol abuse | Recipient psychiatric comorbidity or social instability; noncompliance with treatment protocols; GGT/ALP ratio > 1.4 | Usually > 6 mo |
| NASH | Original disease of NASH or cryptogenic cirrhosis; persistent or worsening risk factors for NASH in the general population | Usually 3-4 wk and increases with time if risk factors persist |
ABO, Blood group system; AIH, autoimmune hepatitis; ALP, alkaline phosphatase; AMR, antibody-mediated rejection; CMV, Cytomegalovirus; DCD, donation after cardiac death; DSAs, donor-specific antibodies; EBV, Epstein-Barr virus; GGT, γ-glutamyltransferase; HBV, hepatitis B virus; HCV, hepatitis C virus; HEV, hepatitis E virus; HLA, human leukocyte antigen; HSV, herpes simplex virus; IS, immunosuppression; NASH, nonalcoholic steatohepatitis; OLT, orthotopic liver transplantation; PBC, primary biliary cirrhosis; PTLD, posttransplantation lymphoproliferative disorder; PSC, primary sclerosing cholangitis.
Adapted from Demetris AJ, Nalesnik M, Randhawa P, et al. Histologic patterns of rejection and other causes of liver dysfunction. In: Busuttil RW, Klintmalm GB, eds. Transplantation of the Lveri. Philadelphia: Saunders; 2005:1057-1128.
Posttransplantation Needle Biopsies of Allografts
Biopsy of the allograft is used to determine the cause of dysfunction, to assess the effect of therapy or progression of disease, and to document the immunologic and architectural tissue status to help guide immunosuppressive therapy. Tissue triage depends on the reason for the biopsy, the clinical differential diagnosis, and the time after transplantation. For native livers, the American Association for the Study of Liver Diseases has recommended two passes with a 16-gauge needle for assessment of fibrosis, because staging is subject to sampling error for small biopsies (<20 mm long), and those containing fewer than 11 portal tracts may not be representative.45 Similar guidelines should be followed for liver allograft biopsies, particularly those obtained late after transplantation, when assessment of fibrosis is important.
Most diagnostically important pathologic studies can be completed on routinely processed, formalin-fixed, paraffin-embedded (FFPE) sections. A clinical differential that incorporates antibody-mediated rejection (AMR) optimally includes fresh frozen tissue for immunoglobulin and complement immunofluorescence examination.46,47 FFPE samples can also be used for C4d immunofluorescence staining.47,48 For each biopsy, we routinely review two H&E-stained slides, each containing two- to four-step sections. The most frequently used special stains, which are ordered only after review of the H&E slides, include trichrome, iron, and copper to detect chronic cholestasis. Cytokeratin 7 or 19 immunohistochemistry can be used to help localize bile ducts and ductular metaplasia of periportal hepatocytes in cases with suspected ductopenia and chronic rejection.49 Sign-out stations should have access to the electronic medical records, serial laboratory results, immunosuppression drug levels, and donor-specific antibody (DSA) data.
Optimal information needed for interpretation of posttransplantation allograft biopsies includes the original disease, ABO compatibility, DSA status, time after transplantation, and transplant source (e.g., standard whole organ cadaveric, DCD livers, reduced-size cadaveric, living related). These variables influence susceptibility to specific complications and consequently affect the morphologic differential diagnosis.
Although a clinical differential diagnosis is valuable, complete clinical information can bias biopsy interpretation. The slide review should be completed before the clinician correlates the findings with the clinical history and laboratory results to generate the differential diagnosis. We routinely evaluate any previous biopsies, which greatly assists with interpretation of the current biopsy, including assessment of the effects of therapeutic intervention and disease progression or response. Post hoc multidisciplinary conference review of all liver allograft biopsies is an essential quality assessment tool to track outcomes of clinical interventions based on liver biopsy interpretations, providing feedback to the clinicians and pathologists.
Evaluation of the Failed Allograft
Gross examination of failed allografts should use an approach similar to that for native hepatectomy specimens.50 Special attention should be paid to dissection and inspection of the biliary, hepatic artery, portal vein, and hepatic vein anastomotic sites. This procedure may require assistance of the surgeon to explain the anatomy. Routine tissue sampling for microscopy should include anastomoses in the resected specimen, superficial and deep sections of the right and left lobe, at least one deep hilar section with cross sections of medium-sized bile ducts and arteries, and grossly obvious defects.
The most common causes of allograft failure vary according to the time after transplantation. Preservation/reperfusion injury or primary nonfunction, vascular thrombosis, and death of the patient are the leading causes of allograft failure within the first several weeks after transplantation.51,52 Allograft failure because of acute cellular rejection (ACR) or AMR has become rare.53,54 Recurrent disease, delayed manifestations of technical complications (e.g., vascular thrombosis, biliary sludge syndrome), and patient death are most commonly responsible for late graft failures (>1 year after transplantation).55,56 Chronic rejection has become an uncommon cause of graft failure, and its incidence continues to decrease.55,57 Recurrent HCV-induced cirrhosis is a leading cause of allograft failure,58 but newer forms of antiviral therapy may help in the future. Review of prior graft biopsies is usually of value in correlating the clinical course with the findings in the failed allograft.
Reduced-Size and Living Related Allografts
Normal liver structure and function depends on optimal portal venous and hepatic artery inflow and adequate venous outflow and biliary drainage. Transplantation of a portion of the liver (e.g., living donors, cadaveric splits) necessarily compromises at least one of these vascular or biliary conduits, especially near the cut edge of the residual liver fragment. The pathologist should be aware of the technical details of the operation and the exact origin of the posttransplantation biopsy, because sampling errors in reduced-size allografts can be quite misleading. The clinical and laboratory context is important to determine whether there is a sampling artifact.
For example, infarcted parenchyma or morphologic features of high-grade biliary or venous outflow obstruction can be seen in biopsies obtained near the cut surface in otherwise well recipients with normal or near-normal liver injury test results. The histopathologic changes are attributable to localized defects of blood or bile flow and are not representative of the entire organ. If the patient has more than one biliary anastomosis, biopsies from one lobe may show obstructive cholangiopathic changes, whereas the other lobe may be normal.
Early after transplantation, reduced-size or living donor allografts usually undergo rapid growth and may be more susceptible to damage from needle biopsies59 and AMR.60 Late after transplantation, portal venopathy, low-grade ductular reactions, and nodular regenerative hyperplasia are fairly common.61
Preservation/Reperfusion Injury
The term preservation/reperfusion injury describes donor organ damage that occurs during agonal events in the donor, cold preservation of the graft, warm sanguineous reperfusion in the recipient, and perioperative events.62,63 The term cold ischemia refers to damage that occurs when the donor organ is stored in preservation fluid and immersed in an ice bath. It preferentially damages sinusoidal endothelial cells, causing them to lift from the underlying matrix. Cold ischemic time should be less than 12 hours. The term warm ischemia refers to the time the organ is exposed to blood but is suboptimally perfused because of hypotension or death of the patient. Pathophysiologic mechanisms of warm and cold ischemia and preservation/reperfusion injury have been reviewed elsewhere.62,63 Preexisting steatosis increases susceptibility to warm and cold ischemic injury.62,63
DCD donors merit special mention because these organs suffer from a different type of warm ischemia insult. Instead of a relatively smooth transition from adequate blood perfusion to cold preservation solution, blood flow stops completely in the DCD donor. Ideally, the time from pronouncement of cardiac death to infusion with preservation solution should be less than 20 minutes. Even in the best circumstances, considerable blood component sludging and subsequent damage occurs in the peribiliary plexus, which often results in ischemic cholangiopathy,64–66 which is the bane of DCD donor livers.
Dysfunction attributable to preservation/reperfusion injury usually occurs shortly after transplantation. Technical or vascular insults, such as arterial or venous thrombosis, alloimmunologic or adverse drug reactions, toxin exposure, and infections, must be excluded. Donor and recipient hypotension, warm ischemia, metabolic abnormalities, cold ischemia during organ preservation, and reperfusion injury contribute to the syndrome of preservation/reperfusion injury.
Clinical Features
Reliable early signs of significant preservation/reperfusion injury after complete revascularization include poor bile production and persistent elevation of the serum lactate level. This condition is usually associated with marked (>2500 IU/mL) elevations of the serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels during the first few days after transplantation,9 followed by a rapid normalization of the ALT/AST rato during the first week and a prolonged cholestatic phase characterized by persistent elevation of total bilirubin and GTP values. Grafts that recover from early significant injury have gradual resolution of abnormal liver injury test results, but they are also at risk for ischemic cholangiopathy, which causes biliary sludge syndrome.67,68
Pathologic Features
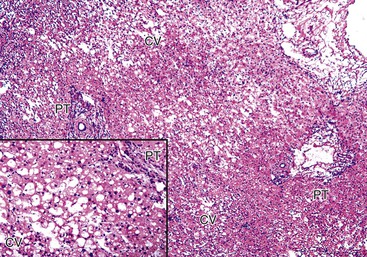
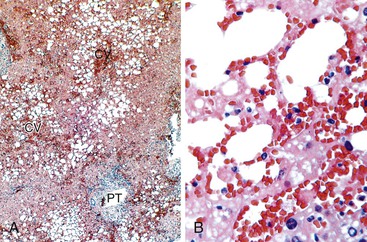
Postreperfusion needle biopsies obtained within several hours of complete revascularization can reliably gauge the extent of preservation/reperfusion injury.9,69 The mild damage that occurs in many liver allografts includes microvesicular steatosis, which is usually attributable to warm ischemia, hepatocellular cytoaggregation (i.e., detachment of individual hepatocytes from each other and accumulation of the cytoplasm so that the cell assumes a rounded appearance), and hepatocellular swelling.9,50 Severe injury is characterized by zonal or confluent coagulative necrosis, particularly if it is periportal or bridging, and severe neutrophilic inflammation.9,69 The pathologist should not overinterpret surgical hepatitis, which is characterized by perivenular sinusoidal neutrophilia without necrosis, or manipulation injury, which manifests as necrosis and neutrophilia in the immediate subcapsular parenchyma in wedge biopsies, as a preservation/reperfusion injury.
Repair responses usually begin 1 to 2 days after injury and are directly proportional to the severity of the insult. Mild injury is usually followed by hepatocellular mitosis, thickening of the plates, and nuclear enlargement. Persistent injury manifests as mild centrilobular hepatocellular swelling, and hepatocanalicular cholestasis often coexists and persists for several weeks (Fig. 52.3). Severe preservation/reperfusion injury is usually accompanied by marked centrilobular hepatocellular swelling and hepatocanalicular and cholangiolar cholestasis.9,50 These features often persist for 1 or 2 months (Fig. 52.4). Cholangiolar proliferation is usually triggered by periportal and confluent bridging necrosis with collapse of the reticulin framework.9,50 Coexistent sepsis, which is common during this period, can contribute to the pattern of injury. If the graft recovers, normal architecture can be restored, but patients are at risk for ischemic cholangiopathy.
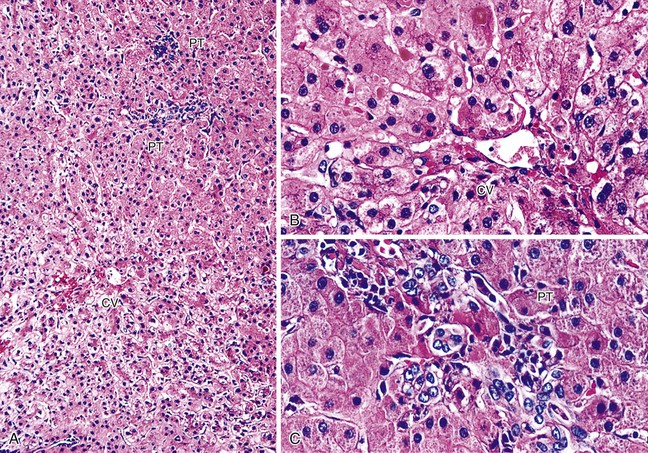
Preservation/reperfusion injury of donor livers with more than 20% macrovesicular steatosis causes death of some fat-containing hepatocytes, which then release large lipid droplets into the sinusoids. These droplets coalesce into even larger globules, which trigger local fibrin deposition, neutrophilia, red blood cell congestion, and local obstruction of sinusoidal blood flow (Fig. 52.5).7 If the liver recovers, the large fat globules become surrounded by macrophages and eventually resolve over several weeks. Because normal hepatocytes require only 4 to 6 hours to undergo the entire apoptotic cycle, recognition of apoptotic hepatocytes or coagulative necrosis in a biopsy obtained more than several days after transplantation should arouse suspicion of another, usually ischemic, insult.70
Differential Diagnosis
Biliary obstruction, pancreatitis, sepsis, AMR, and cholestatic hepatitis can produce histopathologic changes that resemble preservation/reperfusion injury. Detailed donor information, including age and organ type (e.g., ECD, DCD), cold and warm ischemic times, operative difficulties, recipient’s clinical profile, blood culture, blood crossmatch, and results for DSAs and C4d staining help to determine the likely source of injury.53 Preservation/reperfusion injury and operative technical difficulties most often are the causes of liver injury early after transplantation.
Examination of the true bile ducts (not cholangioles) contained within the original portal tract stroma and cholangioles at the interface zone provide useful clues for distinguishing preservation/reperfusion injury from biliary tract obstruction or stricturing. The latter usually cause at least some degree of periductal lamellar edema or produce stellate septal bile duct lumens with neutrophils in the lumen or infiltrating between biliary epithelial cells. Preservation/reperfusion injury does not usually show true bile duct changes unless it is associated with or leads to ischemic cholangiopathy. Instead, neutrophils surround the interface zone cholangioles. Centrilobular hepatocanalicular, cholangiolar cholestasis and intralobular neutrophil clusters are common to both disorders. The clinical context, history, and laboratory results are often needed to distinguish sepsis from preservation/reperfusion injury.
ACR superimposed on preservation injury is recognized by the typical rejection-type inflammatory infiltrate in the portal and perivenular regions. The infiltrate consists of blastic and smaller lymphocytes and especially eosinophils, which are an excellent early marker of an emerging rejection reaction, as are findings of lymphocytic cholangitis and lymphocytic central perivenulitis. Distinguishing preservation injury from AMR is discussed later (see Antibody-Mediated Rejection).
Cholestatic hepatitis can be difficult to distinguish from preservation injury and cholestatic hepatitis without knowledge of the clinical history. Cholestatic hepatitis has been reported only for patients infected with HBV or HCV, and it is distinctly unusual during the first 3 to 4 weeks after transplantation. Cholestatic hepatitis usually worsens with time, unless the patient is treated with decreased immunosuppression or antiviral therapy, whereas the trend is gradual improvement for patients with a preservation/reperfusion injury.
Small-for-Size Syndrome
Pathophysiology
Adequate liver structure, function, and growth depend on finely balanced portal and hepatic artery inflow and hepatic venous outflow and biliary tract drainage. It is difficult for surgeons to divide a reduced-size donor liver (e.g., living donor, split) with sufficient precision such that all of these requirements are satisfied in the donor and recipient. This is especially true when a reduced-size or living donor allograft (<30% of the expected recipient’s liver volume or <0.8% of the recipient’s body weight) is placed into the hyperdynamic and hypertensive portal circulation characteristic of a cirrhotic recipient. The donor liver fragment may be unable to accommodate the increased portal inflow, especially if hepatic venous drainage is compromised.71,72 Too much portal venous inflow can injure portal venous and periportal sinusoidal endothelium61,73 and contribute to allograft dysfunction. This is referred to as portal hyperperfusion or small-for-size syndrome (PHP/SFSS).
The arterial buffer response, which refers to reciprocal regulation of portal venous and hepatic artery hepatic blood flow, is an important aspect of PHP/SFSS.74,75 Increased portal venous flow, as occurs in SFSS, causes arterial constriction and diminishes hepatic arterial flow, which results in suboptimal oxygenation. This predisposes the patient to arterial thrombosis and ischemic cholangitis, particularly if the arterial anastomosis is imperfect. Conversely, decreased portal venous flow, as occurs in cases of portal venopathy, causes hepatic arterial dilation and increased arterial flow.
The arterial buffer response is thought to be mediated by portal venous flow and the relative washout rate of adenosine,74,75 an arterial vasodilator produced in the portal tracts. Other compounds, such as adenosine triphosphate (ATP) and hydrogen sulfide (H2S), and sensory innervation are likely to be involved.75 Temporarily reducing portal venous flow by transient portal-caval shunting, banding, splenic artery ligation, and pharmacologic manipulation can ameliorate some PHP/SFSS manifestations.71,75–77 Conversely, low portal inflow can impair liver regeneration and cause graft steatosis because the hepatic parenchyma depends on portal venous blood.78,79
Reduced-size or living donor allografts are usually required to grow after transplantation and are likely to experience PHP/SFSS to some extent, although it may not be clinically evident. Portal hyperperfusion is an important physiologic stimulus of liver regeneration.61 Portal pressure elevation after partial hepatectomy inversely depends on the size of the graft.80 The rate of subsequent hepatocyte regeneration is directly proportional to increased portal pressure and flow.81,82 Failure of liver regeneration is usually not the major clinical problem associated with PHP/SFSS, although regeneration can be impeded by suboptimal hepatic venous drainage in severe cases.83 Instead, PHP/SFSS becomes significant when the structural integrity of the hepatic vasculature is compromised by the arterial buffer response, in which hepatic arterial flow counteracts changes in portal venous flow. Decreased arterial flow causes parenchymal or biliary ischemia and infarction. Splanchnic venous stasis and perfusion of the liver with endotoxin-rich blood may contribute to the evolving cholestasis.61
The optimal situation is to gradually titrate venous inflow and have adequate venous drainage. This creates venous inflow high enough to trigger and sustain regeneration but not directly cause injury or indirectly compromise arterial flow because of sustained spasm as part of the arterial buffer response.
Clinical Features
Dahm and colleagues defined PHP/SFSS as at least two of the following complications occurring on 3 consecutive days in the first several weeks after transplantation: elevated bilirubin (>100 µmol/L), international normalized ratio (INR) higher than 2, and grade 3 or 4 encephalopathy after exclusion of technical, immunologic, or infectious complications.84 Surgeons are usually aware of the potential for PHP/SFSS, but the diagnosis is difficult to establish with certainty. Clinical manifestations are not specific and may be caused by other insults (e.g., portal vein thrombosis).
It is often difficult to determine whether PHP/SFSS contributes to complications that may otherwise be deemed technical, especially because the latter are encountered with increased frequency in cases of reduced-size allografts. For example, a strong arterial buffer response can lead to significant arterial vasospasm and predispose to thrombosis, especially if blood vessels are a smaller caliber than normal.61 This event may be misinterpreted as a technical complication.
Pathologic Features
Initial events in PHP/SFSS have been observed reliably in experimental animal models73,81,85,86 and occasionally in postreperfusion or early posttransplantation allograft biopsies obtained within the first several days in humans. Denudation of portal vein and periportal sinusoidal endothelium occurs as early as 5 minutes after transplantation.61,73,81,85,86 In severe cases, microvascular rupture at the portal vein–sinusoidal junction can result in hemorrhage into the portal and periportal connective tissue and dissect into hepatic parenchyma.87,88
If the allograft survives the initial crisis, reparative changes occur, including endothelial cell hypertrophy and subendothelial edema accompanied by growth of myofibroblasts and endothelial cells into the subendothelial space. Eventually, this process leads to fibrointimal hyperplasia or intimal thickening and luminal obliteration or recanalization of thrombi (Fig. 52.6). Venous findings typical of PHP/SFSS are uncommon in peripheral core-needle biopsies. The pathologist instead often observes a constellation of nonspecific findings that may include centrilobular hepatocanalicular cholestasis, centrilobular hepatocyte steatosis or hepatocyte atrophy with sinusoidal dilation, centrilobular hepatocyte necrosis, and a low-grade ductular reaction (Fig. 52.7).
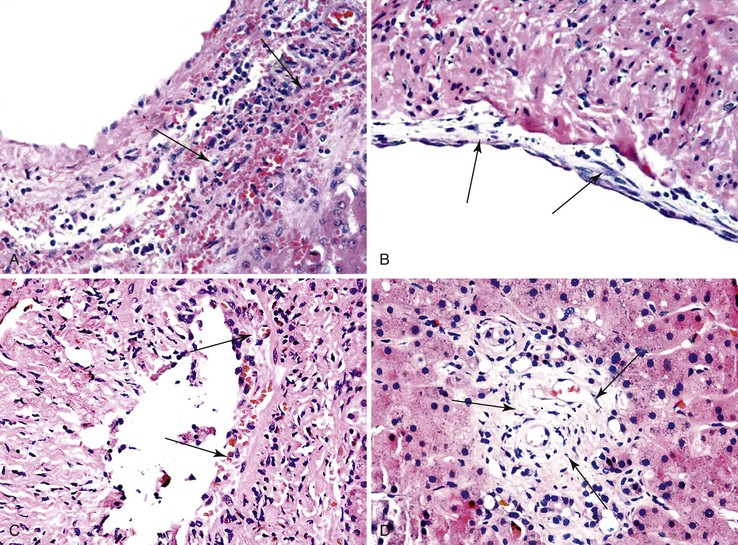
Review of the operative and radiographic reports and clinical history and a discussion with the surgeon help to determine whether pathologic changes in peripheral needle biopsy can be attributed to PHP/SFSS. Hilar sections of allografts that fail because of PHP/SFSS frequently show changes of traumatic injury to large portal vein endothelium, focal fibrointimal hyperplasia of vein branches, evidence of arterial vasospasm, and, in some cases, ischemic cholangitis, particularly if the hepatic artery has thrombosed.61
If the graft recovers, portal hypertension and ascites resolve during a period of several weeks. This is accompanied by restoration of normal architecture, except that some grafts eventually develop significant nodular regenerative hyperplasia as a result of portal venopathy from the initial injury or from suboptimal perfusion with associated collapse.61
Differential Diagnosis
Suboptimal arterial flow resulting from arterial thrombosis or stricturing, sepsis, hypotension, biliary tract obstruction or stricturing, or ischemic cholangitis can cause pathologic changes similar to those of PHP/SFSS. Although preservation injury is a differential diagnostic consideration, living donor grafts are usually not affected significantly.
Because portal hyperperfusion can lead to diminished arterial flow and thrombosis, it is not surprising that more than one complication may occur. Biliary tract obstruction or stricturing alone is usually not accompanied by portal tract connective tissue hemorrhage, centrilobular hepatocyte ischemic changes, or significant nodular regenerative hyperplasia. When these changes are found, PHP/SFSS should be considered.
Arterial vasospasm is usually detected microscopically only in failed allografts and only when severe. The vessels most commonly affected are medium-sized perihilar arteries.
Complications of Transplantation
Vascular Complications
Most vascular complications occur within the first several months after transplantation. They are related to anastomotic imperfections (e.g., narrowing, flaps, dramatic caliber reductions), preexisting atherosclerotic disease, manipulation of the vascular tree, creation of kinks or abnormal tortuosity, metabolic or physiologic abnormalities that predispose to thrombosis, or a combination of these factors. Potential problems often manifest because of factors that increase the technical difficulty of the vascular anastomoses (e.g., small-caliber vessels in pediatric recipients, reduced-size living donor grafts, abnormal anatomy such as piggyback venal caval anastomosis). Physiologic or metabolic abnormalities that decrease hepatic blood flow or promote coagulation, such as cardiac failure, clotting abnormalities, rejection, and infections, also increase the risk of complications.
Vascular interposition arterial grafts or small venous segments, which are used to link the donor and recipient arteries or veins, respectively, can be a significant source of problems. Vascular grafts increase the need for anastomoses, are often cryopreserved or stored in preservation fluid for one to several days before implantation, and may be marginally viable. These factors increase the risk of thrombosis, stimulate atherogenesis, and promote infection.
Hepatic Artery Thrombosis
Pathophysiology
Hepatic artery thrombosis is the most common major vascular complication and an important cause of allograft damage and failure. It occurs in 2% to 20% of transplants, usually within 30 days of transplantation.89,90 Allografts are more susceptible to arterial ischemia than native livers because they are devoid of a collateral arterial circulation.
The hepatic artery supplies the extrahepatic and intrahepatic bile ducts, hilar and portal tract connective tissue, and hilar lymph nodes, and these structures are preferentially damaged by inadequate arterial flow or by arterial thrombosis. This often results in ischemic cholangitis50,67 or ischemic cholangiopathy,68 terms that denote ischemic damage to the biliary tract. These conditions manifest as frank necrosis, poor anastomotic healing, biliary leaks, and cholangitic abscesses, resulting in biliary sludge syndrome.91
Most arterial thromboses occur early (within the first month after transplantation) at or near the suture line. A second, smaller wave of technically related arterial thromboses occurs 1 to 3 years after transplantation.90,92 Suboptimal arterial anastomoses can cause turbulent arterial flow downstream from the suture line, which eventually leads to arterial fibrointimal hyperplasia, luminal narrowing, and thrombosis. Fibrointimal hyperplasia often develops more quickly in arterial interposition grafts, which predisposes them to thrombosis.
Reported risk factors for early hepatic artery thrombosis include cytomegalovirus (CMV) mismatch (i.e., seropositive donor liver in a seronegative recipient), low weight of a pediatric recipient, procoagulant activity, retransplantation, use of arterial conduits, prolonged operation time, variant arterial anatomy, and low-volume transplantation centers.89 Risk factors for late hepatic artery thrombosis are difficult to define because this event is uncommon, but technical factors and procoagulant activity are suspected.
Clinical Features
Most early hepatic artery thromboses manifest clinically with fever, leukocytosis, septic shock, and severe elevations of liver injury laboratory test results.89,90 Late hepatic artery thrombosis may be asymptomatic, or it may maniest insidiously along with cholangitis, relapsing fever, sepsis (related to hepatic infarcts), abscesses, and ischemic cholangiopathy with subsequent impaired bile flow.89,90
Ultrasonography is often used as a screening tool to inspect hepatic arterial blood flow, but angiography is the most reliable method of establishing a diagnosis. Urgent revascularization is often attempted to salvage the graft, but even if successful, the patient can develop ischemic cholangiopathy.
Pathologic Features
Hepatic artery thrombosis can be diagnosed on peripheral core-needle biopsies. However, this approach is often not reliable, because needle biopsies sample subcapsular parenchyma, which is only sometimes affected.50,93 The structures more susceptible to ischemic injury are the perihilar tissue and large bile ducts, which are not routinely sampled.
Peripheral core-needle biopsies show frank centrilobular coagulative necrosis, marked centrilobular hepatocyte swelling, and biliary tract complications such as cholangiolar proliferation, acute cholangiolitis, and obstruction or stricturing. The peripheral biopsy may be unremarkable because vascular collaterals render the thrombosis inconsequential or the necrosis of bile ducts and biliary sludging have not developed in the peripheral liver parenchyma. Occasionally, hepatic artery thrombosis can lead to spotty acidophilic necrosis of hepatocytes, called ischemic hepatitis, and can mimic acute viral hepatitis. Chronic suboptimal arterial flow can cause centrilobular hepatocellular atrophy and sinusoidal widening.
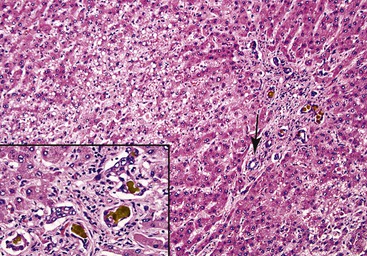
Examination of failed allografts with hepatic artery thrombosis (Fig. 52.8) often reveals necrosis of the hilar or perihilar bile ducts (Fig. 52.9) and leakage of bile into the surrounding connective tissue. Biliary sludge, biliary abscesses seeded with fungi and bacteria, infarction of hepatic hilar lymph nodes, and patchy parenchymal infarction may also be seen.
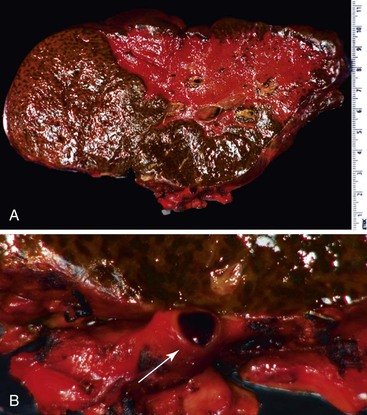
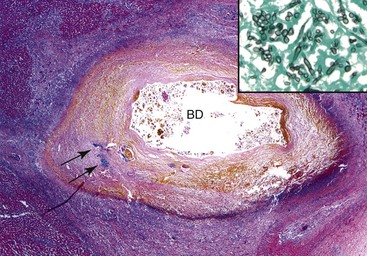
Differential Diagnosis
Because hepatic artery thrombosis or arterial narrowing can mimic almost every type of liver allograft syndrome, uncommon manifestations can easily be misdiagnosed. Ischemic necrosis, which is often centrilobular, most reliably indicates hepatic artery thrombosis, but suboptimal flow can also result in centrilobular hepatocyte swelling and biliary tract obstruction or cholangitis. Chronic suboptimal arterial flow can cause biliary epithelial cell senescence that resembles chronic rejection. Ischemic hepatitis can appear similar to the lobular phase of acute or recurrent viral hepatitis (e.g., HCV infection). However, Liu and colleagues suggest that a combination of apoptosis and mitosis (i.e., low ratio of apoptosis to mitosis) without inflammation is more common in ischemic hepatitis than in recurrent hepatitis C, in which apoptosis predominates.94 The relationship between arterial thrombosis and biliary tract complications is common, and examination of hepatic arterial patency should be performed when biliary tract complications are encountered.
Portal Vein Thrombosis
Pathophysiology
Portal vein complications, which affect 1% to 2% of transplant recipients, are less common than hepatic artery complications.95 Complications include portal vein thrombosis, strictures, and poor flow because of persistent collateral circulation or hypotension. The incidence is increased in reduced-size and living donor transplants and when cryopreserved venous interposition grafts are used.96 Risk factors include problems with the portal venous anastomosis, small portal vein diameter, pediatric recipients, previous portal vein thrombosis, surgical shunting before transplantation, and splenectomy. Long-surviving liver allografts with cirrhosis are also susceptible to portal vein thrombosis.95
Clinical Features
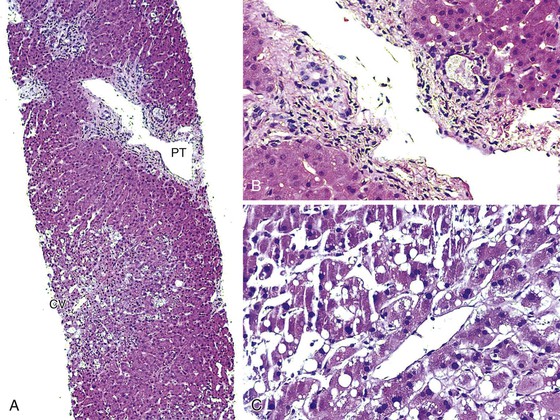
Portal vein thrombosis in a noncirrhotic allograft can cause widespread necrosis (Fig. 52.10, A). It manifests with bleeding varices, fulminant hepatic failure, and portal hypertension with massive ascites and edema.95 If the thrombus is partial, small infarcts can develop, or the liver can become seeded by intestinal bacteria, which may cause relapsing fever. Suboptimal flow can result in periportal and midzonal hepatocyte atrophy, necrosis, and diffuse macrovesicular steatosis (see Fig. 52.10, B).97 The clinical presentation of portal vein thrombosis in a cirrhotic allograft is the same as that in a native liver with cirrhosis: variceal hemorrhage, splenomegaly, and ascites.
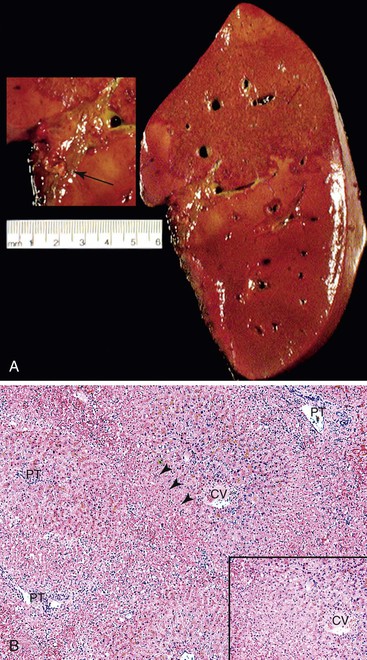
Pathologic Features
Findings depend on the severity of portal vein flow compromise, time after transplantation, and the condition or structure of the allograft. Complete portal vein obstruction early after transplantation in a noncirrhotic allograft often causes massive coagulative necrosis. Suboptimal portal vein flow because of strictures, kinks, or persistent collateral circulation can cause periportal or midzonal hepatocyte atrophy, coagulative necrosis (see Fig. 52.5), unexplained zonal or panlobular steatosis, and nodular regenerative hyperplasia. Suboptimal portal vein blood flow, which is usually associated with persistent venous collaterals that have failed to close or have reopened after transplantation, can also manifest with hepatic steatosis. Bacterial or fungal infection of a partial portal vein thrombus can result in miliary abscesses of the liver.
Differential Diagnosis
Suboptimal portal vein blood flow can be difficult to distinguish from suboptimal hepatic venous drainage. Linear zones of ischemic necrosis or hepatocellular atrophy favor the former, whereas red blood cell congestion in central veins and centrilobular sinusoids and obliterative central venopathy suggest suboptimal hepatic venous drainage. Ultrasonography and angiography often are needed to specify the cause of the vascular abnormality. Cases of suboptimal portal vein flow that manifest with intrahepatic steatosis need to be distinguished from recurrent or de novo steatosis and steatohepatitis.
Hepatic Vein and Vena Cava Complications
Pathophysiology
Complications involving hepatic venous outflow, including the hepatic veins and vena cava, are relatively uncommon. Risk factors include reduced-size or living donor allografts without inclusion of the middle hepatic vein,71 difficulties with reconstruction of the venous outflow tract, and alternative anastomoses such as the piggyback approach.44 Significant stenosis or thrombosis of the outflow tract is associated with significant clinical and pathologic manifestations.
Clinical Features
The clinical presentation depends on the severity of outflow tract compromise. Severe stenosis or thrombosis leads to Budd-Chiari syndrome, with hepatic enlargement, tenderness, ascites, and edema. Less severe stenosis may cause only pathologic manifestations or an increase in the portal vein–vena cava pressure gradient. In less severe cases, it is often difficult to determine whether surgical intervention is required.
Pathologic Features
Congestion and hemorrhage involving the hepatic venules and surrounding perivenular sinusoids are the most reliable pathologic findings in patients with suboptimal hepatic venous drainage (Fig. 52.11). Bland centrilobular hepatocyte necrosis and dropout are usually identified. Chronic changes include nodular regenerative hyperplasia, perivenular fibrosis, and central vein occlusion with venocentric cirrhosis, particularly if outflow obstruction is severe or of long duration. Chronic changes can also be accompanied by a prominent ductular reaction at the interface zone. Ductular reaction can also occur in perivenular zones, which can make it difficult to recognize architectural landmarks and to exclude biliary obstruction.
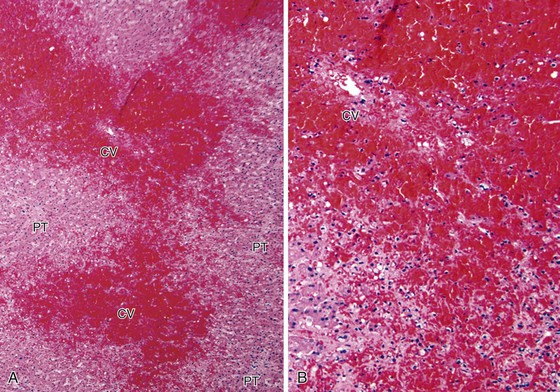
Differential Diagnosis
If any of the previously described perivenular changes are not accompanied by noticeable inflammation, a mechanical or outflow obstruction should be suspected. Studies of long-surviving allografts, especially in children, show that acute or chronic AMR may contribute to noninflamed perivenular subsinusoidal fibrosis.98–100 Recognition of an immune-mediated cause may be aided by the finding of increased perivenular and central vein endothelial C4d and CD20 cell staining and by detection of serum DSAs of relatively high titers (>10,000 median fluorescence intensity [MFI]).98–100 Many affected children had previous episodes of ACR. If acute or chronic centrilobular vascular changes are accompanied by significant lymphocytic, histiocytic, or lymphoplasmacytic inflammation, an immunologically mediated cause of injury such as acute or chronic rejection, plasma cell–rich or autoimmune hepatitis (AIH), or an adverse drug reaction should be suspected.
Perivenular inflammation can be transient in some cases of immune-mediated centrilobular injury. It can manifest at a later stage, when it is indistinguishable from mechanical venous outflow obstruction.101
Bile Duct Reconstruction
Pathophysiology
The biliary tract continues to be the Achilles heel of liver transplantation. Complications occur in approximately 20% of recipients, and an even higher incidence is observed for living and reduced-size allografts.102 Biliary tract reconstruction varies considerably. The most common anastomoses used for cadaveric whole organs are end-to-end, duct-to-duct, and donor biliary–recipient enteric anastomosis. Most biliary tract complications are attributable to ischemic or traumatic injury, immunologic injury, or surgically introduced abnormal anatomy. These factors predispose to poor biliary tract wound healing, inadequate drainage, and inordinate reflux, occurring singly or in combination.102,103
The portion of the extrahepatic donor bile duct immediately adjacent to the anastomosis is particularly vulnerable to ischemic injury. Its peribiliary arterial plexus was supplied by arterial channels originating proximal to the anastomotic margin and left behind in the donor, putting this short segment at risk for ischemia. To ascertain adequate arterial flow and duct viability after the hepatic arterial anastomosis has been made, the terminal end of the donor extrahepatic bile duct is trimmed back toward the liver until bleeding occurs from the cut surface.
The peribiliary arterial plexus is particularly vulnerable to preservation/reperfusion injury during operative manipulation.104 For example, in DCD donors, sludging of blood or use of the more viscous University of Wisconsin solution105,106 can prevent adequate reperfusion of the peribiliary plexus in the recipient.102 Perfusion can be improved by arterial and peribiliary plexus infusion with thrombolytic agents or less viscous preservation solutions (e.g., histidine-tryptophan-ketoglutarate [HTK]) before reperfusion in the recipient.91,102,103, 105,106
Other causes of biliary tract ischemia resulting from arterial or peribiliary plexus injury include small-for-size syndrome and severe arterial vasospasm,61 prolonged cold ischemia, older donors with atherosclerotic disease, and preformed or de novo donor antibodies (e.g., ABO isoagglutinin, anti–human leukocyte antigen [HLA] antibodies).102,107 Once the myriad other causes of biliary tract complications have been excluded, recurrent primary disorders such as recurrent primary sclerosing cholangitis (PSC) should be considered.
Complications include anastomotic dehiscence, transmural necrosis, bile leakage, cholangitic abscesses, ascending cholangitis, bile casts (Fig. 52.12), strictures, obstruction, ampullary dysfunction, and biliary-vascular fistulas.102,108–111 They occur in approximately 15% of whole cadaveric allografts and as many as 30% to 40% of reduced-size or living donor allografts.102,112,113
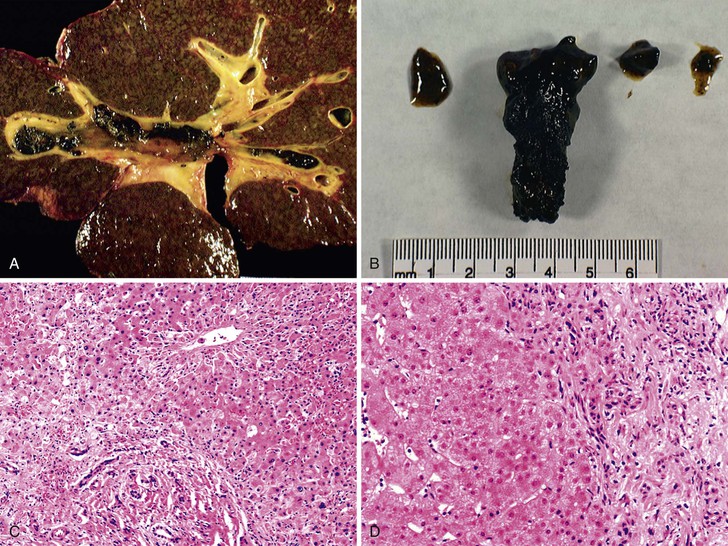
Strictures are the most common complication. They are categorized according to location and the time after transplantation as intrahepatic or extrahepatic, nonanastomotic or anastomotic, and early or late.108–112 Intrahepatic strictures are further categorized as hilar or peripheral.
Anastomotic strictures usually appear within the first several months after transplantation, but they appear at a reduced rate for many years.108,112 Risk factors for anastomotic strictures include postoperative bile leaks, female donor–male recipient combinations, and a more recent year of transplantation.102,108,112 Compared with nonanastomotic strictures, anastomotic strictures are more amenable to radiographically directed or surgical intervention, and they have less of a negative impact on long-term graft and patient survival.102,108,112
Nonanastomotic strictures usually occur later after transplantation (>1 year), are usually untreatable, are progressive, and negatively impact graft and patient survival.108–112 These strictures are usually peripheral and associated with immunologic risk factors such as PSC as the original disease. Other risk factors for nonanastomotic strictures include use of high-viscosity preservation solution, Roux-en-Y biliary anastomoses, and CMV infection.102,109–111
Nonanastomotic strictures occurring earlier (<1 year) are often associated with preservation-related injury111,114 and are usually located in perihilar bile ducts. Similar to the nonanastomotic strictures that occur later, risk factors for early stricture include long cold and warm ischemic times, high-viscosity preservation solution, older recipient age, a duct-to-duct biliary anastomosis, and bile leaks.111
Clinical Features
Liver injury tests that show selective elevation of γ-glutamyltransferase (GGT) and alkaline phosphatase levels are the usual methods of detecting early or minor biliary tract complications such as strictures and stones. Clinical signs and symptoms are uncommon unless the patient is jaundiced or develops acute cholangitis. Biliary tract problems are also often first suspected on routine biopsy evaluation, but cholangiography (e.g., magnetic resonance cholangiopancreatography [MRCP], duct-to-duct endoscopic retrograde cholangiopancreatography [ERCP], percutaneous transhepatic cholangiography [PTC]) is needed to confirm the diagnosis and localize the defects.108,112,115
More serious biliary tract complications, such as obstruction, cholangitic abscesses, and ascending cholangitis, usually manifest with fever, jaundice, right upper quadrant pain, and intermittent bacteremia. During the first several months after transplantation, T-tube stents provide access for cholangiograms, which are routinely performed before clamping at 1 week postoperatively and again at the time of T-tube removal.102
Pathologic Features
Allograft biliary tract complications are pathologically identical to those seen in native livers. Portal, periportal, periductal, and pericholangiolar edema; predominantly neutrophilic portal inflammation; intraepithelial and intraluminal neutrophils in true bile ducts; an interface ductular reaction of some severity; centrilobular hepatocanalicular cholestasis; and small clusters of neutrophils throughout the lobules are typical findings of stricturing or obstructive cholangiopathy (see Fig. 52.12). With time, chronic biliary tract strictures are often associated with mixed or predominantly chronic portal inflammation, biliary epithelial cell senescence changes, and low-grade ductopenia involving small bile ducts. More than 1 year after transplantation, in addition to the classic features described previously, biliary strictures are a relatively common cause of portal eosinophilia.
Biliary-vascular fistulas are characterized by red blood cells in the lumen of bile ducts or by bile concretions in blood vessels. Occasionally, an inordinate elevation of the serum bilirubin level is associated with a fistula. In contrast, periductal hemorrhage surrounding small interlobular bile ducts is an inconsequential finding in asymptomatic patients when a biopsy is obtained within 1 or 2 days after transhepatic cholangiography.116
Differential Diagnosis
The differential diagnosis is heavily influenced by the time after transplantation and the patient’s original disease. Within the first several weeks after transplantation, biliary obstruction or cholangitis can be difficult to distinguish from preservation/reperfusion injury and ACR, particularly if the patient was treated with increased immunosuppression before the biopsy was obtained.
Features that favor biliary tract obstruction or stricturing to ACR include neutrophilic-predominant portal inflammation, periductal edema, retention of the normal nucleus-to-cytoplasm ratio in biliary epithelial cells, and an absence of perivenular mononuclear inflammation. Acute rejection is favored when the mixed portal inflammation is composed of blastic and small lymphocytes, plasma cells, and eosinophils. Lymphocytic cholangitis, an increased nucleus-to-cytoplasm ratio in biliary epithelial cells, and perivenular inflammation also are seen. Portal eosinophilia in early acute rejection can be quite striking, especially in patients treated with steroid-sparing immunosuppressive regimens.117
Late-onset chronic biliary tract complications occasionally manifest with predominantly mononuclear portal inflammation, biliary epithelial cell senescence changes, and low-grade ductopenia.118 Late-onset chronic biliary strictures can also cause portal eosinophilia and can mimic acute and chronic rejection, viral hepatitis, and recurrent autoimmune disorders.
It can be difficult to differentiate chronic rejection caused by biliary strictures from other causes, especially recurrent PSC, in peripheral core-needle biopsies.119 The at-risk populations are similar. Both conditions can cause intrahepatic cholestasis, biliary epithelial cell senescence changes, and small bile duct loss. Careful examination of the clinical history, evaluation of serial biopsies, and analysis of the pathology are needed to distinguish between biliary strictures and chronic rejection.119
In analyzing needle biopsies, features that favor biliary strictures or recurrent primary sclerosis cholangitis instead of chronic rejection include a history of biliary tract complications, PSC as the original disease, periductal lamellar edema involving true bile ducts, stellate portal expansion, portal neutrophilia, a ductular reaction affecting at least some portal tracts, and deposition of copper or copper-associated protein in periportal hepatocytes. Features that favor acute or chronic rejection include a previous history of rejection, inadequate immunosuppression, lymphoplasmacytic portal inflammation, small portal tracts, absence of a ductular reaction, active central perivenulitis, and perivenular fibrosis.
Additional findings in failed allografts can be used to distinguish biliary strictures or recurrent PSC from chronic rejection.119 Chronically rejected livers are usually of normal or slightly increased size and weight, whereas those with obstructive cholangiopathy are usually significantly enlarged. In hilar lymph nodes, obstructive cholangiopathy usually causes bile-pigmented sinus histiocytosis, whereas chronically rejected nodes are usually atrophic and fibrotic. Perihilar arteries are normal or show mild focal eccentric fibrointimal hyperplasia. Foam cell arteriopathy and significant concentric fibrointimal hyperplasia are typical of chronic rejection. In obstructive cholangiopathy, extrahepatic or large intrahepatic bile ducts often show focal ulceration, periductal lymphoplasmacytic inflammation, and fibrosis. In chronic rejection, large duct inflammation and ulceration are unusual. When the infiltrate shows plasma cell–rich infiltrates, the possibility of recurrent or de novo immunoglobulin G4 (IgG4)–associated sclerosing disease should be considered.120
Cholangitis favors a biliary tract complication, whereas cholangiolitis and lobular disarray favor chronic hepatitis. A cholestatic liver laboratory test profile favors obstructive cholangiopathy unless the patient has the cholestatic variant of viral hepatitis. Review of HBV or HCV nucleic acid levels in blood helps to distinguish between the two, because cholestatic viral hepatitis can be associated with very high levels of viral replication (e.g., 20 to 30 million IU/mL of HCV RNA). Deposits of copper or copper-associated protein in periportal hepatocytes indicate obstructive cholangiopathy because they are not seen in chronic rejection alone or in cholestatic hepatitis.
Angiography and cholangiography (e.g., ERCP) are useful in making the distinction between obstructive cholangiopathy and chronic rejection. Pruning of peripheral arterial and biliary trees and poor peripheral filling are seen in chronic rejection. In cases of obstructive cholangiopathy, some intrahepatic duct dilation is observed, but arterial changes are not seen or are insignificant.
Clinicopathologic correlation can help to distinguish late-onset biliary tract complications from late-onset acute rejection. Adequate immunosuppressive drug levels, with preferential elevation of the GGT and alkaline phosphatase levels,121 favors obstructive cholangiopathy because late-onset acute rejection is unusual in this circumstance. It is not possible to reliably distinguish recurrent PSC from other causes of biliary tract obstruction or stricturing on the basis of needle biopsy evaluation alone.
Allograft Rejection
Liver allograft rejection is broadly categorized as AMR, ACR, and chronic rejection.122 Relatively isolated AMR in ABO-incompatible allografts usually occurs within the first several weeks after transplantation. It can also occur in recipients with positive lymphocytotoxic crossmatch123 and DSAs, but it is less common in this circumstance.124 DSAs are also associated with uncontrolled ACR and chronic rejection.125
Late-onset or chronic AMR has not been adequately defined morphologically in liver allografts. This is an area in need of further study. Effective immunosuppression has made otherwise typical acute and chronic rejection reactions uncommon as a cause of allograft failure.57,126,127
Antibody-Mediated Rejection
Crossing ABO blood group barriers causes predictable and severe liver allograft injury.46 It is usually avoided in most North American programs because it leads to an approximately 60% incidence of significant AMR and graft failure.107 However, ABO-incompatible liver allografts are still used in Asian programs, for which the donor pool is more limited. Plasmapheresis and vigorous antirejection and microcirculatory protective therapy are needed to achieve reasonable results in these circumstances,128,129 but ABO-incompatible recipients are still at risk for late biliary tract complications such as ischemic cholangitis.107,130
Liver allografts are much less susceptible than other solid organ allografts to damage from AMR because of anti-HLA class I and II antibodies.53,107 However, depending on the circumstances, the liver is not completely spared from injury by these and other antibodies.131 DSAs do not routinely influence organ triage or recipient selection at most centers,53,54,123 but they are associated with higher rejection rates and decreased survival in some centers.132 In cases of late onset, combined AMR and ACR have been reported.133,134 Several studies have associated DSAs with the development of progressive fibrosis in pediatric liver allografts.98–100
Living donor liver allografts are more susceptible to AMR than whole liver cadaveric donors,135 but ABO-incompatible allografts are still routinely used in Japan.128,129 This practice is probably related to several factors unique to reduced-sized or living donor allografts, including smaller-caliber blood vessels; enhanced microvascular injury and arterial vasospasm because of a combination of portal hyperperfusion and pathophysiologic mediators of AMR (see Reduced-Size and Living Related Allografts), and upregulation of major histocomatibility complex (MHC) antigens on the microvasculature and bile ducts.
Pathophysiology
The consequences of anti-donor antibodies depend on the subclass, titer, and timing of the antibody response and on the density and distribution of target antigens in the liver.107,136,137 Antibodies can cause various degrees of damage and can even be protective in some circumstances.138–140 Complement-fixing, high-titer, preformed antibodies directed at antigens expressed on endothelial cells, such as isoagglutinins and lymphocytotoxic (anti-HLA class I and II) antibodies, are the most dangerous.141 MHC class I antigens and ABO antigens are moderately to strongly expressed on the vasculature in normal livers. The former can be upregulated when the graft is inflamed for other reasons. MHC class II antigens are not expressed or are expressed at low levels in the hepatic microvasculature in the normal liver. However, they are upregulated significantly after transplantation.142–144
ABO incompatibility places recipients at risk for AMR, especially when isoagglutinins are present in high titers.123,129 Isoagglutinins also cause more severe damage than lymphocytotoxic antibodies; the latter do not reliably cause clinically relevant allograft dysfunction unless present at relatively high titer (>1 : 32) and do not routinely influence short-term survival.53,54,145,146
Resistance of liver allografts to AMR from anti-HLA antibodies has been attributed to (1) secretion of soluble MHC class I antigens that bind to and neutralize anti-class I HLA147; (2) Kupffer cell phagocytosis of immune complexes and activated platelet aggregates; (3) a dual afferent hepatic blood supply; (4) unique hepatic sinusoidal microvasculature (which is devoid of a conventional basement membrane)107; and (5) a homologous source of complement in xenografts.148
Positive lymphocytotoxic crossmatches (mostly anti-HLA antibodies) are encountered in 8% to 12% of recipients.53,54,145,146 Most recipients are female. Only 5% to 20% of recipients with a positive crossmatch have titers high enough to cause clinically significant liver injury.53,123 IgG usually causes more damage than immunoglobulin M (IgM) lymphocytotoxic antibodies.107,149
Because the patient population at risk for severe AMR because of anti-HLA antibodies is small, AMR is often overlooked as a cause of liver dysfunction or failure. This occurs because the incidence of AMR in ABO-compatible allografts early after transplantation is relatively low (approximately 5% among patients harboring anti-HLA antibodies124); many programs do not routinely conduct pretransplantation crossmatching or DSA determinations; and even if crossmatches are performed, the titer and specificity are not determined, and posttransplantation monitoring is not routinely carried out. The long-term consequences of intermittent or persistent DSAs require further study.53,150
Antibodies cause damage by binding to allograft endothelium and fixing complement. This triggers the complement cascade and causes endothelial damage, deposition of platelet-fibrin thrombi, and initiation of the clotting and fibrinolytic cascades. In severe, rapidly evolving cases, this leads to microvascular thrombosis, arterial vasospasm, and coagulopathy that act in concert to impair blood flow and cause hemorrhagic necrosis of the liver.138,139 Less precipitous injury can occur during the transition from acute to chronic rejection when destruction of peribiliary capillary plexuses contributes to ductopenic or chronic rejection.141,151–153
Clinical Presentation
Hyperacute AMR, the most severe form, has largely been eliminated by avoiding ABO-incompatible transplants without pretreatment.128,129 When it does occur, hyperacute rejection begins immediately after reperfusion. Even in severe cases, allograft dysfunction evolves during a period of hours to days, as with hyperacute rejection in other organs.107 Initial signs of dysfunction usually develop in the operating room after complete revascularization. It can be recognized by uneven reperfusion, swelling, a dusky appearance, and cessation of bile flow. These signs are often accompanied by coagulopathy, which manifests as difficulty in achieving hemostasis and an inordinate need for platelets and other blood components.46 Precipitous allograft failure is unusual.
A more common manifestation of AMR is a persistent elevation of serum bilirubin and ALT/AST levels (depending on whether the antibodies are isoagglutinins or anti-HLA) during the first several days to weeks after transplantation. This is often accompanied by persistence of the positive crossmatch, the presence of DSAs, unexplained and often refractory thrombocytopenia, and low serum complement activity levels.47,123,124,149 In severe cases, hepatic angiograms, obtained to exclude an arterial thrombosis, usually show segmental narrowing (indicative of immunologically mediated arterial vasospasm) and diffuse luminal narrowing with poor peripheral blood flow.107
Pathologic Features
Pathologic manifestations of AMR depend on the timing of the biopsy; the subclass, titer, and specificity of the anti-donor antibodies; and whether ACR coexists.107 Isoagglutinins usually cause more red blood cell congestion and necrosis than anti-HLA or lymphocytotoxic antibodies (Fig. 52.13).
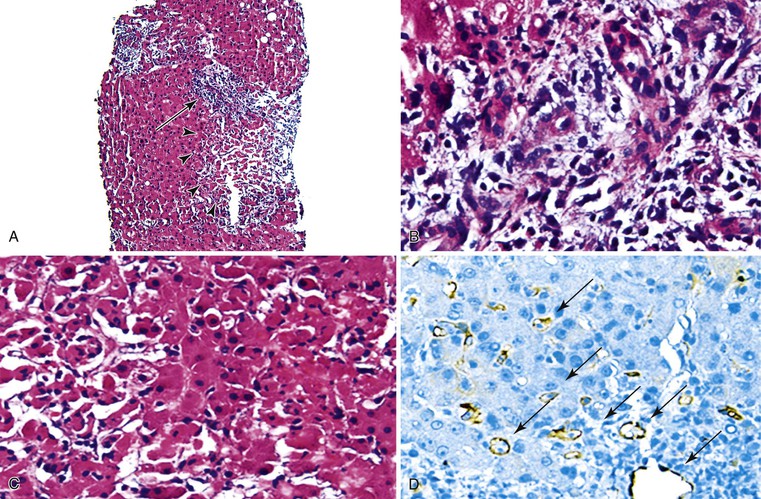
High-titer isoagglutinins (>1 : 64) often cause prominent red blood cell and focal neutrophil sludging in the sinusoids and platelet-fibrin thrombi in periportal sinusoids, portal veins, and central veins.46,154,155 Acidophilic hepatocyte necrosis develops within 2 to 6 hours after reperfusion in untreated recipients. This is often quickly followed by portal or periportal edema, necrosis, focal hemorrhage, C4d deposits in portal stroma,154 and diffuse endothelial C4d deposits in portal veins, capillaries, and sinusoids.53,154
In severe untreated cases, confluent coagulative necrosis, prominent sinusoidal and venous congestion, and edema and hemorrhage into the portal tracts appear within 1 to 5 days.46,53,154 Portal veins often show fibrin deposition. If sampled, medium-size arteries show endothelial cell hypertrophy and evidence of arterial vasospasm, such as mural myocyte vacuolization, wrinkling of the elastic lamina, and thickening of the wall with narrowing of the lumen. Neutrophilic or necrotizing arteritis is only occasionally observed. Portal neutrophilia, cholangiolar proliferation, and small areas of confluent hepatic necrosis begin to appear at 2 to 3 days after transplantation. If untreated, progressive hemorrhagic infarction occurs in ABO-incompatible organs during the next 1 to 2 weeks.
ABO-incompatible allografts that fail because of untreated AMR are often grossly enlarged, cyanotic, and mottled and show areas of geographic necrosis. Capsular ruptures, hepatic artery, or portal vein thrombosis can develop in extreme cases. Microscopically, changes in the hilum or perihilar region can be particularly helpful in recognizing that AMR caused or contributed to allograft failure. These changes include congestion and leukocyte margination in the peribiliary vascular plexus, partially organized thrombi in arterial branches, focal mural necrosis of large septal bile ducts, and inflammatory and necrotizing arteritis. Late sequelae of AMR include biliary sludge and stricturing caused by obstructive cholangiopathy, obliterative arteriopathy, loss of small bile ducts, and chronic rejection.46,128,130,156,157
Reperfusion biopsies from patients with high-titer (>1 : 32) lymphocytotoxic antibodies show platelet aggregates in the portal and terminal hepatic veins more often than crossmatch-negative controls.9,123 Spotty acidophilic necrosis of hepatocytes and centrilobular hepatocellular swelling, accompanied by cholangiolar proliferation and hepatocanalicular cholestasis, occurs during the first week after transplantation in patients with high-titer antibodies. Inflammatory and necrotizing arteritis is rare, but it is almost diagnostic when detected. Coexistent ACR with portal eosinophilia is common.124 The pathologic changes closely resemble those of preservation/reperfusion injury. Clinicopathologic correlation with exclusion of other causes of dysfunction and staining for C4d and other immune deposits are needed to confirm the diagnosis.47,48,53,153
An AMR diagnosis in ABO-compatible liver allografts should be based on a biopsy showing changes consistent with an AMR-related pattern of injury, clinicopathologic exclusion of causes of a similar pattern of injury, serologic evidence of DSAs, and evidence of strong and diffuse antibody or complement (C4d) deposition within the injured allograft (Fig. 52.14).46,48,53,123,158 The latter is defined by strong portal vein, capillary, and usually periportal sinusoidal endothelial staining involving most portal tracts.124 Compared with other allografts, a diagnosis of isolated liver allograft AMR because of anti-HLA antibodies is difficult to establish with certainty.
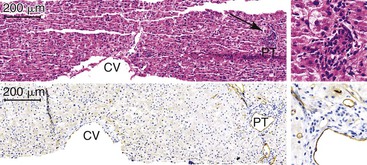
Difficulties can be encountered in establishing a correct diagnosis when liver allografts are large and able to absorb high antibody loads (especially anti–class I HLA antibodies)147 and are therefore resistant to AMR-related damage.9,107,123,136,159 In some cases, the immune deposits are ephemeral46,123 or can be associated with other insults.134 Clinicopathologic similarities exist between AMR and preservation injury, sepsis, and biliary or vascular complications.46,123
Intrahepatic immune deposits are ephemeral in AMR, even when evaluated by immunofluorescence in frozen tissue sections.46–48,123,158 In severe cases, selective deposits of IgG, IgM, C3, or C4 are usually detectable in the sinusoids and in perihilar arteries, portal veins, and the peribiliary plexus.46,47,123 Thereafter, the distribution of immune deposits becomes patchy, and they may be difficult to distinguish from background staining unless antibody titers are high. If AMR is suspected, saving frozen tissue for immunofluorescence testing is important.
Recognition that C4d persists for several days, can be detected in FFPE tissues and correlates with circulating DSAs makes an AMR diagnosis easier to establish in kidney allografts.160,161 Interpretation and utility of C4d staining in liver allografts is less clearcut.154,158,162–169
Bellamy48 and Hubscher158 reviewed the literature on C4d immunohistochemistry in liver allografts. Normal liver and allograft biopsies are usually negative for C4d staining. Portal vein, arterial, and capillary C4d and sinusoidal endothelial C4d have been detected in crossmatch-positive recipients more often than in crossmatch-negative controls167 and in patients with isolated AMR.124,134,169 Endothelial cell C4d staining is most specific for AMR, but portal vein C4d stromal staining has also been described in cases of ABO-incompatible AMR,154 ACR,167 and chronic rejection.153,170 Kozlowski and coworkers134 suggest that C4d staining of FFPE tissue sections is nonspecific and that linear sinusoidal C4d deposits in frozen tissue specimens are needed to establish AMR. We found that C4d staining in FFPE tissue is reliable and that endothelial deposits are most specific. However, clinicopathologic correlation is needed to establish a diagnosis of AMR with certainty. In contrast, portal and perivenular stromal staining is commonly observed and difficult to interpret. In nonliver allografts, capillary microvascular staining is used mainly to determine whether antibodies contribute to allograft injury.
As in other allografts, C4d deposits are often accompanied by ACR.53,153,154,162–169 In some studies, the burden of C4d deposits is directly proportional to the Banff grade.154,162–169 Necrotic and steatotic hepatocytes can show nonspecific C4d staining.
Portal vein and capillary C4d deposits can be detected in other causes of allograft dysfunction, including biliary obstruction,162 recurrent HBV infection,163 recurrent HCV infection,165 and de novo autoimmune hepatitis.171 C4d staining has also been described in portal vein and capillary, sinusoidal, central vein, and arterial endothelial cells; in lymphoid nodules; and in periductal and portal stromal cells in native pediatric livers with hepatitis B, hepatitis C, autoimmune hepatitis, and overlap syndromes that include autoimmune hepatitis and PSC.172 Endothelial C4d deposits in non–rejection-related allograft disorders are less widespread than in severe AMR or ACR. Similar to kidney and heart allografts, liver C4d deposits have been associated with macrophage and plasma cell infiltrates.164
The possibility that chronic or low-grade, persistent AMR may contribute to indolent but progressive fibrosis has received increased attention, particularly for pediatric recipients.98–100 Evidence supporting this association includes increased prevalence and titer of DSAs associated with tissue C4d deposits and suboptimal immunosuppression.98–100
Differential Diagnosis
Distinguishing AMR from preservation/reperfusion injury can be difficult. The presensitization state, persistence of circulating antibodies, diffuse C4d staining,53 and posttransplantation clinical and laboratory profile can provide discriminating information. AMR should be suspected in female liver allograft recipients with high-titer DSAs or lymphocytotoxic antibodies who have received a liver with a short cold ischemic time but who show persistent DSAs after transplantation and develop graft dysfunction, refractory and otherwise unexplained thrombocytopenia, and circulating low complement levels in the first several weeks after transplantation.47,53,123,149
Alloantibody-mediated AMR should be favored instead of preservation/reperfusion injury when any of the following histopathologic findings are detected: (1) Neutrophils, macrophages, eosinophils, and to a lesser extent, lymphocytes may be marginating on the luminal aspect and beneath the portal and central veins; this finding may be combined with diffuse endothelial cell reactivity. (2) Occasional blastic lymphocytes and eosinophils and other features of ACR may be seen in the portal or perivenular areas. (3) Diffuse C4d staining may be detected in the microvasculature.53 Diffuse staining is defined as positive staining in most portal veins and capillaries, with extension into the periportal sinusoids. Portal or periportal edema, necrosis, and hemorrhage should raise the possibility of ABO incompatibility.
Precipitous allograft failure from severe AMR is rare, except in ABO-incompatible livers. When it occurs, it is difficult to distinguish from hemorrhagic liver necrosis caused by hypotension, poor perfusion, sepsis, or vascular thrombosis. Unless unequivocal evidence of AMR is detected, such as inflammatory or necrotizing arteritis or diffuse immunoglobulin and complement (C4d) deposition combined with serologic evidence of anti-donor antibodies, the cause of allograft failure can be extremely difficult to determine with certainty. More commonly, changes diagnostic of AMR are not identified. The diagnosis of AMR requires thorough clinicopathologic correlation.
Acute Cellular Rejection
ACR is defined as inflammation of the allograft elicited by a genetic disparity between the donor and recipient that primarily affects interlobular bile ducts and vascular endothelia, including portal and hepatic veins and occasionally the hepatic artery and its branches.122 Most episodes occur within 30 days after transplantation because the reaction is precipitated by mass migration of donor cells into recipient lymphoid tissues.173–175 Early acute rejection rarely leads to allograft failure or permanent allograft damage, because it usually is easily controlled by immunosuppression.57,176 Acute rejection occurring late after transplantation has a different histopathologic appearance. It more closely resembles chronic hepatitis and is more difficult to control, Acute rejection occurring later more often produces permanent allograft damage, possibly due to a delay in treatment.175,177–179
Clinical Features
ACR usually manifests clinically between 5 and 30 days after transplantation, depending on the immunosuppressive regimen. ACR affects approximately 30% of liver transplant recipients.175 Earlier manifestations can be seen among presensitized patients or patients who receive less than optimal baseline immunosuppression. Later manifestations can occur in recipients who have been treated with lymphocyte-depleting antibodies or recipients with inadequate maintenance immunosuppression. Clinical findings are often absent early in the course of disease, when tissue damage is mild. Later in the course or in cases of severe acute rejection, fever and allograft enlargement, cyanosis, and tenderness may occur.
Early after transplantation, when biliary T-tubes are in place, decreased bile flow and thin, pale bile are commonly seen and do not necessarily denote acute rejection. Likewise, ascites may develop because of increased intrahepatic hydrostatic pressure that results in liver swelling and increased lymph production.180 These clinical findings alone are not sufficient to implicate acute rejection.
Biochemical evidence of liver dysfunction because of acute rejection usually includes nonselective elevation of some or all of the standard liver injury test values. Leukocytosis and eosinophilia are frequently identified.180 The levels of a variety of proteins (e.g., interleukins, neopterin, amyloid A protein) are increased in the peripheral blood, but none of the proteins is routinely assayed to monitor recipients. Instead, ACR is usually suspected on clinical grounds and then confirmed by a core-needle biopsy.
Risk factors for the development of early acute rejection depend on the type of immunosuppressive regimen. This condition can affect any recipient subgroup, including young recipients, those with advanced-age donors, healthy recipients (e.g., normal serum creatinine level, low Child-Pugh classification), donor-recipient HLA-DR mismatches, patients with immune dysregulated syndromes (e.g., PSC, autoimmune hepatitis, PBC), those with long cold ischemic times, and those with an HLA-C genotype.175,181 Late-onset acute rejection more than 1 year after transplantation may have a different pathologic appearance. It is more often associated with inadequate immunosuppression, is more difficult to control, and more frequently leads to allograft injury and failure.177,181,182
Pathologic Features and Grading
ACR is characterized by predominantly mononuclear but mixed portal inflammation containing blastic or activated lymphocytes, neutrophils, and eosinophils; by subendothelial inflammation of portal and terminal hepatic venules; and by bile duct inflammation and damage.180,183 Minimal diagnostic criteria needed to establish a diagnosis of acute rejection include at least two of these pathologic findings. The diagnosis is strengthened if more than 50% of the ducts or terminal hepatic veins are damaged or unequivocal endotheliitis of portal or terminal hepatic vein branches are identified. Pathologic evidence of severe injury, which is used for grading, includes perivenular inflammation, centrilobular necrosis, arteritis, and inflammatory features such as central-to-central, bridging inflammation and necrosis.176,180
Acute rejection-type infiltrates are encountered mostly in portal tracts but occasionally surround central veins. Endotheliitis (i.e., endothelialitis) refers to lymphocytes located underneath the portal and central vein endothelium. This characteristic feature of acute rejection may be seen in other types of allograft dysfunction.184 The lymphocytes are predominantly T cells, including CD8-positive cells that often surround and invade damaged bile ducts.185,186 B cells usually comprise a minor fraction of the infiltrate. Macrophages and other leukocytes are present and can predominate in severe acute rejection.185–187 Immunophenotypic analysis is not prognostically or clinically useful for establishing a diagnosis of acute rejection, except when attempting to distinguish acute rejection (T-cell predominant) from a posttransplantation lymphoproliferative disorder (B-cell predominant).
Lymphocytic cholangitis involving small bile ducts (<30 µm in diameter) is an important feature of ACR. Lymphocytes are located inside ductal basement membranes in association with biliary epithelial cell injury. The morphologic features of acute injury include paranuclear vacuolization, apoptotic bodies, increased nucleus-to-cytoplasm ratio, increased mitoses, and prominent nucleoli. Cytoplasmic eosinophilia and multinucleation usually signal senescence-related changes and chronic injury.118 Breaks in the basement membrane indicate severe bile duct damage. Portal or peribiliary granulomas are not a feature of acute or chronic rejection. When encountered, a non–rejection-related cause of duct injury, such as recurrent PBC, a coexistent mycobacterial or fungal infection, or sarcoidosis, should be suspected.
Inflammatory, necrotizing arteritis is a defining feature of severe acute rejection, but it is rarely detected in needle biopsies because the arteries that are most commonly affected are located in the hilum of the liver. Recognition of arteritis in peripheral needle biopsies is a poorly reproducible finding.188 Arteritis is not usually included in grading schemes. When present, this feature indicates a severe rejection episode. The portal tract–parenchyma interface zone is usually unremarkable in cases of early mild and moderate ACR. In severe cases, portal inflammatory cells involve the periportal parenchyma.178
Rejection-type infiltrates can be seen in the perivenular sinusoids and extracellular matrix surrounding terminal hepatic venules (i.e., central perivenulitis).178 The infiltrates are identified in as many as 30% cases of acute rejection. Perivenulitis occurs later (>100 days) after transplantation. Severe ACR is diagnosed only when typical portal changes of acute rejection are accompanied by perivenular inflammation and zonal centrilobular congestion, hemorrhage, and hepatocyte necrosis and dropout.
The Banff grading scheme for acute and chronic rejection was constructed on data generated by recognized experts in liver transplant pathology, hepatology, and surgery from many of the major hepatic transplantation centers in North America, Europe, and Asia (Tables 52.3 and 52.4).183 It is widely used189 easy to apply, and reproducible.190 It is scientifically correct and has prognostic significance.176,191
Table 52.3
Banff Working Group Criteria for Acute Liver Allograft Rejection
| Global Assessment* | Criteria |
| Indeterminate | Portal inflammatory infiltrate that fails to meet the criteria for acute rejection |
| Mild or grade I | Rejection infiltrate in a minority of portal tracts that is usually mild and confined within the portal spaces |
| Moderate or grade II | Rejection infiltrate expanding most or all portal tracts |
| Severe or grade III | Similar to moderate, with spillover into periportal areas and moderate to severe perivenular inflammation that extends into the hepatic parenchyma and is associated with perivenular hepatocyte necrosis |
* Global assessment of rejection grade is made on review of the entire biopsy specimen and only after a diagnosis of rejection has been established. It is inappropriate to provide a rejection grade when the diagnosis of rejection is uncertain.
Adapted from Demetris AJ, Nalesnik M, Randhawa P, et al. Histologic patterns of rejection and other causes of liver dysfunction. In: Busuttil RW, Klintmalm GB, eds. Transplantation of the Liver. Philadelphia: Saunders; 2005:1057-1128.
Table 52.4
Acute Rejection Activity Index
| Category | Criteria | Score* |
| Portal inflammation | Mostly lymphocytic inflammation involving but not noticeably expanding a minority of portal tracts | 1 |
| Expansion of most or all portal tracts by a mixed infiltrate containing lymphocytes with occasional blasts, neutrophils, and eosinophils | 2 | |
| Marked expansion of most or all portal tracts by a mixed infiltrate containing numerous blasts and eosinophils, with inflammatory spillover into the periportal parenchyma | 3 | |
| Bile duct inflammation damage | A minority of ducts are cuffed and infiltrated by inflammatory cells and show only mild reactive changes, such as increased nucleus-to-cytoplasm ratio of the epithelial cells | 1 |
| Most or all ducts are infiltrated by inflammatory cells. Some ducts show degenerative changes, such as nuclear pleomorphism, disordered polarity, or cytoplasmic vacuolization of the epithelium. | 2 | |
| As above, with most or all ducts showing degenerative changes or focal luminal disruption | 3 | |
| Venous endothelial inflammation | Subendothelial lymphocytic infiltration involving some portal and/or hepatic venules | 1 |
| Subendothelial infiltration involving most or all portal and/or hepatic venules | 2 | |
| As above, with moderate or severe perivenular inflammation that extends into the perivenular parenchyma and association with perivenular hepatocyte necrosis | 3 |
* Total rejection activity index (RAI) score = sum of all component scores for portal inflammation, bile duct inflammation or damage, and venous endothelial inflammation.
Adapted from Demetris AJ, Nalesnik M, Randhawa P, et al. Histologic patterns of rejection and other causes of liver dysfunction. In: Busuttil RW, Klintmalm GB, eds. Transplantation of the Liver. Philadelphia: Saunders; 2005:1057-1128.
The Banff system uses descriptive grades of indeterminate, mild (Fig. 52.15), moderate (Fig. 52.16), and severe (Fig. 52.17 and Table 52.3). It also uses a semiquantitative rejection activity index (RAI) (Table 52.4),183 which is a remnant of the European grading system192 and equivalent to the hepatitis activity index.193 RAI semiquantitatively scores the prevalence and severity of three separate pathologic features, each on a scale of 0 to 3: portal inflammation, bile duct damage, and subendothelial inflammation. The individual components are then summed to determine a total RAI score.
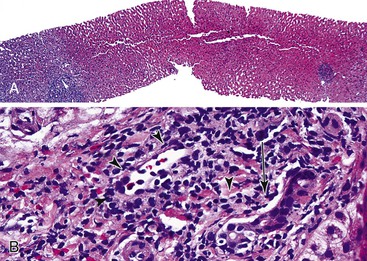
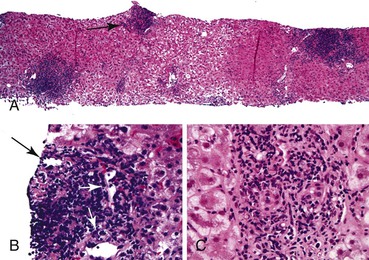
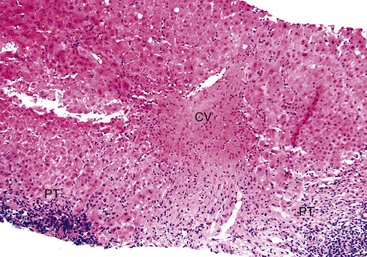
There is direct correlation between the total RAI score and rejection grade and correlation with increased risk of persistent or recurrent acute rejection, chronic rejection, and graft failure.176 The ranges of RAI scores are indeterminate (1 to 2), mild (3 to 4), moderate (5 to 6), and severe (>6). The maximum possible total RAI score is 9, but biopsies rarely achieve this score.176 Instead, most episodes are mild, have a total RAI score of less than 6, and respond to increased immunosuppression. These cases do not lead to significant fibrosis, bile duct loss, or arteriopathy as determined in subsequent or follow-up biopsies.176 Graft failure from ACR is distinctly unusual.
Additional immunosuppression causes some pathologic changes more difficult to detect due to resolution of some characteristic findings, such as subendothelial infiltration of veins, can occur within 24 hours. Immunosuppressive treatment before biopsy can lead to centrilobular hepatocyte swelling and hepatocanalicular cholestasis, causing further confusion. Typically, 7 to 10 days are required for rejection-related changes to resolve after therapy.
Late acute rejection (>100 days) is characterized by features found in early ACR. However, slightly atypical features can develop,178 such as fewer blastic lymphocytes, more necroinflammatory interface activity, less venous subendothelial inflammation, a higher incidence of perivenular inflammation, and slightly more lobular activity. The features more closely resemble those of chronic hepatitis.178
Late ACR can manifest with prominent perivenular inflammation, hepatocyte dropout, and minimal or no portal tract changes (i.e., isolated central perivenulitis) (Fig. 52.18).194–196 These cases may evolve into typical chronic rejection with ductopenia and perivenular fibrosis. Subendothelial inflammation of the portal or central veins is not a required finding. Perivenular fibrosis and a Budd-Chiari syndrome or a veno-occlusive disease–like clinical syndrome can develop as a consequence of the severe perivenular injury.101,197 Evidence suggests that chronic AMR may contribute to perivenular sinusoidal fibrosis.98–100 The Banff Working Group proposal is described in Table 52.5.
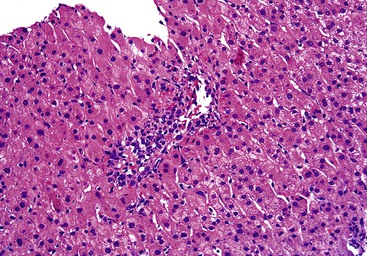
Table 52.5
Banff Working Group: Biopsy Findings for Late Allograft Dysfunction
| Description | Biopsy Findings |
| Minimal or indeterminate | Perivenular inflammation involving a minority of terminal hepatic veins, with patchy perivenular hepatocyte loss but without confluent perivenular necrosis |
| Mild | As above, but involving most terminal hepatic veins |
| Moderate | As above, with focal confluent perivenular hepatocyte dropout and mild or moderate inflammation but without bridging necrosis |
| Severe | As above, with confluent perivenular hepatocyte dropout and inflammation involving most hepatic venules and with central-to-central bridging necrosis |
Data from Banff Working Group, Demetris AJ, Adeyi O, et al. Liver biopsy interpretation for causes of late liver allograft dysfunction. Hepatology. 2006;44:489-501.
Minimal and mild cases may resolve spontaneously.196 Severe perivenular changes probably warrant more aggressive treatment, but no prospective studies have been performed.
Differential Diagnosis
The differential diagnosis of ACR depends on the time since transplantation. ACR should be distinguished from preservation injury and obstructive cholangiopathy or cholangitis during the first several months after transplantation.
Distinguishing acute rejection from recurrent hepatitis B or C and autoimmune hepatitis can be difficult. Hepatitis and rejection show predominantly mononuclear portal inflammation, bile duct damage, and acidophilic necrosis of hepatocytes. The severity and prevalence of bile duct damage, interface activity, lobular changes, and perivenular inflammation and hepatocyte dropout should be assessed.198 Features that favor acute rejection include inflammatory bile duct damage and perivenular inflammation involving most ducts and central veins, respectively, and low-grade or absent lobular and interface necroinflammatory activity. Conversely, recurrent or new-onset viral hepatitis or autoimmune hepatitis is favored when interface and lobular necroinflammatory activity predominate over bile duct and perivenular changes.
Allograft autoimmune hepatitis199 resembles viral hepatitis in most aspects, except that autoimmune hepatitis more often shows conspicuous plasma cells as a component of the infiltrate and often shows more prominent interface activity. Some allograft recipients with autoimmune hepatitis have a prominent plasma cell–rich central perivenulitis that involves most central veins (similar to autoimmune hepatitis affecting native livers). Severe interface activity, plasma cell predominance in the inflammatory infiltrate, and relatively mild bile duct damage favor allograft autoimmune hepatitis over acute rejection.
Chronic Rejection
Chronic rejection is an immunologic injury that evolves from severe or persistent acute rejection and results in potentially irreversible damage to bile ducts, arteries, and veins.200 This condition usually occurs within several months after transplantation, but allograft failure may occur in the first year after transplantation.200
Chronic rejection (e.g., ductopenia, obliterative arteriopathy, perivenular fibrosis) affects approximately 3% to 5% of liver allograft recipients 5 years after transplantation, which is a dramatic decrease in incidence since the 1980s, when the incidence was 15% to 20%.200 The rate of chronic rejection does not increase with time after transplantation, unless idiopathic posttransplantation hepatitis (IPTH) and perivenular subsinusoidal, portal, or periportal fibrosis are included in this category. Better recognition and control of acute rejection, reversibility of the early phases of chronic rejection, the unique immunologic properties of liver allografts, and the ability of the liver to regenerate without fibrosis after recovery from acute rejection have contributed to the decreased incidence of chronic rejection.
Nevertheless, chronic rejection is an important cause of late liver allograft dysfunction and failure, particularly if patients with IPTH and otherwise unexplained fibrosis are included. Chronic rejection is seen mostly in noncompliant patients, HCV-positive recipients treated with activating drugs such as interferon-α,201,202 and recipients who have immunosuppression lowered because of adverse consequences such as a lymphoproliferative disorder.203
Risk factors for chronic rejection are divided into alloantigen-dependent and nonalloantigen-dependent categories. Alloantigen-dependent, immunologic, or rejection-related factors are the most important, especially the number and severity of acute rejection episodes.181,200 In cyclosporine-treated cohorts, risk factors for late-onset acute rejection include younger recipient age; male-to-female sex mismatch; a primary diagnosis of autoimmune hepatitis or biliary disease; baseline immunosuppression; interactions between HLA-DR3, tumor necrosis factor status, and CMV infection204; nonwhite recipient200; and use of interferon-α to treat recurrent HCV infection.201,202 The effects of histocompatibility differences and CMV infection are controversial, but prospective studies suggest that persistent, strong DSAs are associated with late graft loss.125,141,151,205 In a large tacrolimus-treated cohort, most of the matching factors described for the cyclosporine-treated patients were eliminated as significant risk factors, but the influence of the number and severity of acute rejection episodes remained significant.203 Nonalloantigen-dependent or nonimmunologic risk factors include donor age older than 40 years.203
Pathophysiology
Immunologic mechanisms of injury that contribute to allograft damage during acute rejection also contribute to the development of chronic rejection,176,200,203,206–209 including an antibody component.125,141,151,205 Bile duct loss in chronic rejection has been attributed to a combination of direct immunologic and indirect ischemic damage resulting from obliterative arteriopathy, small artery or arteriolar loss, and destruction of the peribiliary capillary plexus,152,210 with a possible contribution from alloantibodies.123,125,151 Cumulative damage triggers biliary epithelial cell senescence.118 Several studies have shown that the early phase of chronic rejection is potentially reversible,118,208,211,212 provided that ductules and surrounding microvasculature are preserved.213
Clinical Features
Chronic rejection usually occurs in patients with a history of acute rejection in whom progressive cholestasis and an increase in canalicular enzymes unresponsive to antirejection treatment develops.180 Two clinical settings are typical: culmination of multiple or unresolved episodes of ACR and AMR and an indolent manifestation without preceding clinically recognizable episodes of acute rejection. The first scenario is most common, especially in the first year after transplantation. The latter presentation is relatively uncommon and likely reflects inadequate monitoring. Late-onset chronic rejection (>1 year after transplantation) typically occurs in inadequately immunosuppressed patients as a result of noncompliance or because of infectious, neoplastic, or toxic complications of chronic immunosuppression.203
Standard liver function tests usually show a cholestatic or biliary pattern of injury, with preferential elevation of GGT and alkaline phosphatase levels.121,180,214 Persistent elevation of ALT and total bilirubin levels usually marks the transition from acute to chronic rejection and can presage allograft failure.194,203,208 Clinical symptoms resemble those of acute rejection until dysfunction becomes significant enough to cause jaundice. Biliary sludging, biliary strictures, hepatic infarcts, and loss of hepatic synthetic function are other late findings that often occur immediately before allograft failure.180 Hepatic angiograms show pruning of the intrahepatic arteries with poor peripheral filling and segmental narrowing, reflecting the obliterative arteriopathy of chronic rejection.180,215
Pathologic Features and Staging
Chronic rejection primarily affects portal tracts and the perivenular parenchyma (Fig. 52.19). It is divided into early (Fig. 52.20) and late (Fig. 52.21) stages according to the Banff scheme (Table 52.6).200 Early-stage chronic rejection is characterized by mild portal inflammation, lymphocytic cholangitis, biliary epithelial cell senescence changes involving most small bile ducts, and some degree of small bile duct loss (usually involving less than 50% of portal tracts). Compared with acute rejection, chronic rejection usually shows less severe inflammation and fewer eosinophils. The inflammatory infiltrate is composed primarily of lymphocytes, plasma cells, and mast cells.216
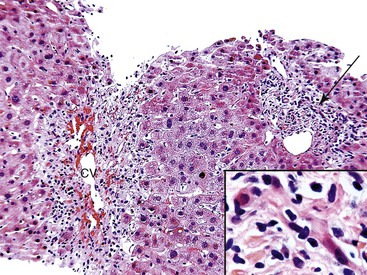
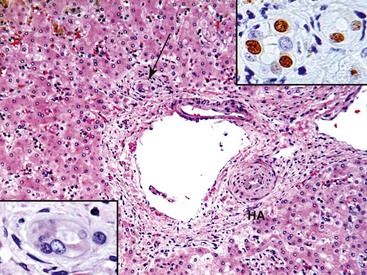
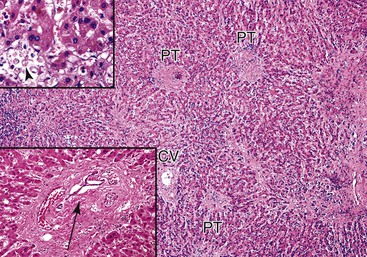
Table 52.6
Features of Early and Late Chronic Liver Allograft Rejection
| Structure | Early Chronic Rejection | Late Chronic Rejection |
| Small bile ducts (<60 µm) | Bile duct loss in <50% of portal tracts | Loss in ≥50% of portal tracts |
| Degenerative change involving most ducts: eosinophilic transformation of the cytoplasm; nuclear hyperchromasia; uneven nuclear spacing; ducts only partially lined by biliary epithelial cells | Degenerative changes in remaining bile ducts | |
| Terminal hepatic venules and zone 3 hepatocytes | Intimal or luminal inflammation | Focal obliteration |
| Lytic zone 3 necrosis and inflammation | Inflammation varies | |
| Mild perivenular fibrosis | Severe perivenular fibrosis, defined as central-to-central bridging fibrosis | |
| Portal tract hepatic arterioles | Occasional loss involving < 25% of portal tracts | Loss involving > 25% of portal tracts |
| Large perihilar hepatic artery branches | Intimal inflammation, focal foam cell deposition without luminal compromise | Luminal narrowing by subintimal foam cells; fibrointimal proliferation |
| Large perihilar bile ducts | Inflammation, damage, and focal foam cell deposition | Mural fibrosis |
| Other | Transitional hepatitis* with spotty necrosis of hepatocytes | Sinusoidal foam cell accumulation; marked cholestasis |

* Transitional hepatitis is characterized by mild lobular disarray and spotty, acidophilic necrosis of hepatocytes that can occur during evolution from early to late stages of chronic rejection.
Data from Demetris A, Adams D, Bellamy C, et al. Update of the International Banff Schema for Liver Allograft Rejection: working recommendations for the histopathologic staging and reporting of chronic rejection. An International Panel. Hepatology. 2000;31:792-799.
Recognition of biliary epithelial cell senescence is critical to the diagnosis of early chronic rejection.118 Features include nuclear enlargement, hyperchromasia, and uneven spacing resembling cytologic dysplasia; syncytia formation; eosinophilic transformation of the cytoplasm; and bile ducts only partially lined by biliary epithelial cells (Figs. 52.19 and 52.20). Immunohistochemical markers of senescence and downregulated cell cycle progression, such as CDKN2A (p16) and CDKN1A (p21), become upregulated in cells under severe stress.118,217 Downregulation of epithelial junctional proteins combined with upregulation of mesenchymal proteins may occur in response to injury.218
Late-stage chronic rejection is characterized by bile duct loss involving more than 50% of portal tracts. Arteriolar loss can also be seen.
Crawford and colleagues219 defined a portal tract as “a focus within the parenchyma containing connective tissue (by Masson trichrome stain) and at least two luminal structures, each with a continuous connective tissue circumference.” In normal liver sampled by percutaneous liver biopsy, bile ducts and hepatic artery branches are seen in 93% ± 6% and 91% ± 7% of portal tracts, respectively.219 These percentages may be lower in larger tissue samples.152 With two standard deviations from the normal as a cutoff, bile duct loss is diagnosed when less than 80% of portal tracts contain bile ducts. Arterial loss is diagnosed when less than 77% of portal tracts contain hepatic artery branches.
A similar approach uses the concept of bile duct–artery parallelism. Ductopenia is defined by at least one unpaired artery in more than 10% of all portal tracts or two unpaired arteries in different portal tracts.220 Unpaired arteries were defined as arteries without an accompanying bile duct within a distance of 10 hepatic artery diameters from the edge of the artery.
Late chronic rejection can cause bile duct and arterial loss,152,210 which can present difficulties when trying to apply these diagnostic algorithms. Portal tract recognition should be based primarily on the parenchymal location of the putative structure; cholestasis in chronic rejection is centrilobular.
A ductular reaction at the interface zone is unusual in chronic rejection, unless the liver is in a recovery phase.208,211,212 Staining with cytokeratins 19 and 7 can help to document bile duct loss in chronic rejection and detect ductular metaplasia of periportal hepatocytes during recovery.49 When a ductular reaction is easily noticeable by routine light microscopy, it signals the regrowth of bile ducts208,211,212 or a downstream biliary stricture. The latter can be documented by identifying periportal hepatocyte copper deposits, which are not seen in chronic rejection.
Early chronic rejection in the terminal hepatic venules and surrounding perivenular parenchyma is characterized by subendothelial and perivenular mononuclear inflammation (see Fig. 52.19). The perivenular infiltrate usually consists of lymphocytes, pigment-laden macrophages, and plasma cells,200,209 often accompanied by hepatocyte dropout and mild perivenular fibrosis.200 Spotty acidophilic hepatocytes elsewhere in the parenchyma can be seen during evolution from early to late chronic rejection.221
Perivenular changes in late chronic rejection are characterized by severe (bridging) perivenular fibrosis, at least focal central-to-central or central-to-portal bridging, and sometimes obliteration of terminal hepatic venules (Fig. 52.21).200 Well-developed cirrhosis arising from chronic rejection is unusual, until the very late stages, when venous obliteration leads to parenchymal extinction and venocentric cirrhosis.206 True regenerative nodules are uncommon, presumably because coexistent obliterative arteriopathy blunts any regenerative response.222 Perivenular hepatocyte ballooning and dropout, centrilobular hepatocanalicular cholestasis, nodular regenerative hyperplasia, and intrasinusoidal foam cell clusters are other common findings in late chronic rejection.
The final diagnosis of chronic rejection should be based on a combination of clinical, radiologic, laboratory, and pathologic findings. In a biopsy specimen, minimal diagnostic criteria for chronic rejection are senescent changes affecting most bile ducts with or without bile duct loss, convincing foam cell obliterative arteriopathy, or bile duct loss affecting more than 50% of portal tracts.200
A diagnosis of chronic rejection is much easier to establish in explanted failed allograft specimens because diagnostic findings can be seen in first-, second-, and third-order branches of the hepatic artery (Fig. 52.22). Obliterative arteriopathy is usually found in at least some of the perihilar arteries, except in cases characterized by bile duct loss and perivenular fibrosis alone. Accumulation of foamy macrophages usually occurs initially in the intima, which triggers proliferation of intimal and migration of medial donor-derived myofibroblasts. This causes intimal thickening, luminal narrowing, and medial thinning as arteries attempt to dilate and compensate for reduced arterial flow. Eventually, compensatory mechanisms fail, and the entire arterial wall may be replaced by foam cells. The artery may undergo thrombosis, causing necrosis of large bile ducts and ischemic cholangiopathy.
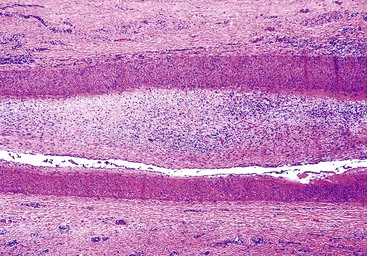
Foamy macrophages may be seen around bile ducts and veins and within the connective tissue. Large perihilar bile ducts can show sloughing of the epithelium, papillary hyperplasia, mural fibrosis, and acute and chronic inflammation.
Staging of chronic rejection assumes that the diagnosis has already been established.200 The term early chronic rejection implies that a significant potential for recovery exists if the source of immunologic injury can be controlled or reversed. The term late chronic rejection implies that the potential for recovery is limited and that retransplantation should be considered. It has not been established that all patients proceed sequentially from early to late stage chronic rejection. Some patients persist in the early stage for months or years, whereas severe fibrosis and late changes rapidly develop within the first year after transplantation or within weeks or months after the initial onset of chronic rejection. Some cases show predominantly or exclusively bile duct loss or arteriopathy alone. Both features usually occur together.200
One important practical implication of chronic rejection staging is that biopsy findings do not define the point of no return. Instead, the biopsy provides information about the likelihood of reversal, which should be correlated with other clinical and laboratory parameters, such as persistently elevated serum levels of bilirubin (>20 mg/dL), progressive decline in synthetic function, superimposed hepatic artery thrombosis, and bile duct necrosis or biliary sludging.
Differential Diagnosis
In percutaneous needle biopsy specimens, a diagnosis of chronic rejection diagnosis is primarily based on damage and loss of small bile ducts and perivenular fibrosis. Arteries with pathognomonic changes are rarely seen in biopsy specimens.200 Duct injury and ductopenia can also occur because of non–rejection-related complications, such as obstructive cholangiopathy, hepatic artery stricturing or thrombosis, cholangitic adverse drug reactions, and CMV infection. Perivenular fibrosis can also be caused by suboptimal hepatic venous drainage and other causes of perivenular injury. A diagnosis of chronic rejection based on biliary epithelial senescence or loss or on perivenular fibrosis alone should first exclude other non–rejection-related causes of ductal injury and perivenular fibrosis.
Chronic rejection is very difficult to distinguish from biliary tract obstruction or stricturing and recurrent PSC.119 Features that favor obstructive cholangiopathy are bile duct loss in some portal tracts, accompanied by a ductular reaction in others; neutrophil clusters within the lobules; bile infarcts; deposition of copper or copper-associated protein in periportal hepatocytes; and hepatocanalicular cholestasis out of proportion to the prevalence of ductopenia (<50%). Features that favor chronic rejection are bile duct changes combined with central perivenulitis or fibrosis, an absence of changes typical of recurrent PSC, and a history of persistent acute rejection or suboptimal immunosuppression. Cholangiography or angiography may be required to distinguish chronic rejection from biliary obstruction. Studies usually show pruning and poor peripheral filling in patients with chronic rejection.
Isolated ductopenia involving less than 50% of portal tracts can be seen without significant elevations of liver function tests. Whether these uncommon cases represent an early phase of chronic rejection remains uncertain. Isolated perivenular fibrosis can be caused by mechanical outflow obstruction, adverse drug reactions, chronic and persistent DSAs,98–100 and nonrejection causes of hepatic outflow obstruction.
The safest approach to diagnosing chronic rejection is to review prior biopsies and correlate the pathologic findings with the clinical course. Particular attention should be paid to a history of severe or unresolved acute rejection preceding the development of histologic findings interpreted as chronic rejection.
Posttransplantation Infections
The pathologist should always consider infection when reviewing tissue specimens from liver allograft recipients. Stress and tissue damage from the operation combined with the high levels of immunosuppression needed to prevent acute rejection in the first 2 months after transplantation places the recipient at high risk for serious opportunistic fungal and viral infections. Bacterial infections usually occur more than 6 months after transplantation. Fever, anastomotic or wound dehiscence, retransplantation, persistent abdominal pain, and vascular thrombosis are clinical signs and symptoms that should arouse suspicion of infection.
Bacterial and fungal infections often arise in nonviable tissue. Necrotic tissue should be routinely subjected to special stains for bacteria and fungi. Granulomas and coexistent acute and chronic inflammation are surrogate markers of infection, but they may not occur because of immunosuppression. Pathologic manifestations of deep fungal and bacterial infections are familiar to most pathologists and are beyond the scope of this chapter.
Infections resulting from CMV, Epstein-Barr virus (EBV), herpes simplex (HSV) or varicella-zoster virus (VZV), and adenovirus do not usually cause clinically significant acute hepatitis in immunocompetent individuals but can in immunodeficient patients.
Reactivation infections develop in patients (usually adults) carrying latent infections, whereas primary infections that are more severe than reactivation infections develop in naïve, usually pediatric recipients. An important aspect of latent infection is that it results in periodic viral shedding into the peripheral circulation, where it can be monitored by measuring viral antigens or nucleic acids. When viral markers surpass certain empirically set thresholds, preemptive lowering of immunosuppression and treatment with specific antiviral agents can prevent active disease.223 Monitoring and preemptive therapy has dramatically reduced the incidence of diseases caused by these viruses. A tissue diagnosis of CMV, EBV, HSV, or VZV hepatitis is becoming rare for routine adult cases and uncommon for pediatric cases.
Cytomegalovirus Hepatitis
Effective prophylactic and preemptive therapy have greatly decreased the incidence of CMV hepatitis, but among seronegative recipients, prophylaxis has also been associated with suboptimal long-term outcomes.224,225 Even though clinically significant CMV disease is uncommon, it has been associated with an increased risk of acute and chronic rejection, biliary strictures, vascular thrombosis, accelerated HCV disease recurrence, other opportunistic infections, and decreased patient and graft survival.224,225
Risk factors for CMV hepatitis include seronegativity before transplantation, rejection, viral replication (usually associated with overimmunosuppression), use of mycophenolate mofetil, anti-leukocyte antibody therapy, human herpesvirus 6 (HHV-6) and HHV-7 infections, gene polymorphisms for toll-like receptors, mannose-binding lectin deficiency, chemokine and cytokine defects (e.g., interleukin-10 [IL-10], CC2 [formerly MCP1], CCR5), and deficiency of viral-specific T cells.224,225 An emerging challenge is compartmentalized disease, in which CMV is detectable in tissue biopsies but not in serologic or molecular assays.224,225
Clinical Features
Depending on the extent of viral dissemination, any organ system can be involved. The most common signs and symptoms of active CMV infection are gastrointestinal. They include fever, diarrhea, ulcers, leukopenia, and low-grade hepatitis; the latter has modestly elevated liver function test results.224,225 Respiratory insufficiency and retinitis are signs of severe disseminated disease. CMV can cause a syndrome that mimics EBV-associated posttransplantation lymphoproliferative disorder, with lymphadenopathy, fever, and atypical lymphocytosis.226
Pathologic Features
CMV hepatitis is characterized by spotty lobular necrosis, Kupffer cell hypertrophy, mild lobular disarray, and patchy lobular inflammation. Under current management schemes, infected hepatocytes rarely contain diagnostic nuclear or cytoplasmic inclusions. Instead, inclusions are usually limited to patients who are overimmunosuppressed and not adequately monitored or treated. Any cell type can be infected. Diagnostic features include large eosinophilic intranuclear inclusions surrounded by a clear halo and accompanied by small basophilic or amphophilic cytoplasmic inclusions. In severe cases, which are largely of historical significance, numerous cells containing CMV inclusions can be found throughout the liver. CMV infection alone does not cause submassive or massive necrosis.226
Infected cells are often surrounded by neutrophils, microabscesses, or clusters of macrophages and lymphocytes, called microgranulomas. CMV hepatitis can be associated with mild lymphoplasmacytic portal inflammation with bile duct infiltration and damage that can resemble acute or early chronic rejection (Fig. 52.23). Bile duct loss and chronic rejection have been associated with persistent CMV infection in allografts.224,225
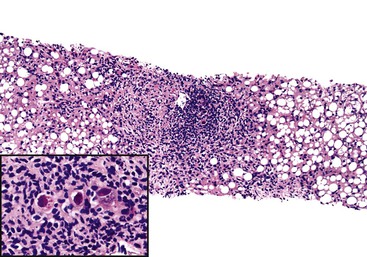
Viral inclusions are rare. CMV hepatitis often manifests with parenchymal alterations but without inclusions. Instead, fragmented nuclear CMV inclusions occur but are often difficult to recognize without immunoperoxidase staining or in situ hybridization. Rapidly dividing tissues such as young granulation tissue, proliferating cholangioles, the edges of infarcts, abscesses, or other intraparenchymal defects are fertile soil for CMV growth.50 Immunoperoxidase stain for an epitope of the CMV matrix phosphoprotein 65 (pp65) may be more sensitive, but it requires frozen tissue. It is used less frequently than the monoclonal antibody against CMV p52, an early nuclear protein that works better in FFPE specimens.224
Differential Diagnosis
CMV hepatitis can be difficult to distinguish from the early phase of HBV or HCV recurrence and EBV hepatitis. CMV can also be difficult to distinguish from HSV hepatitis because both disorders can cause multinucleation and intranuclear eosinophilic inclusions surrounded by halos. CMV inclusion–containing cells can show small basophilic or amphophilic cytoplasmic inclusions, which helps to distinguish them from HSV-infected cells. Circumscribed foci of coagulative necrosis characteristic of HSV is not a feature of CMV hepatitis.
In CMV cases without inclusions, subtle clues that enable distinction from early acute HBV or HCV hepatitis include less lobular disarray and hepatocyte swelling. Microabscesses and microgranulomas are not usually seen in the early lobular phase of HBV or HCV infection. A definitive diagnosis of CMV hepatitis requires characteristic inclusions, which are rare, or demonstration of viral antigens or nucleic acids with immunostaining or in situ hybridization.
CMV hepatitis can resemble EBV hepatitis because both may show mild lymphoplasmacytic portal and lobular inflammation and contain blastic and atypical lymphocytes. EBV hepatitis usually causes more cytologic atypia of the lymphocytes, whereas CMV hepatitis causes more intralobular foci of inflammation (e.g., microgranulomas, microabscesses). Deeper sections, staining for EBV and CMV viral antigens, and in situ hybridization for EBV nucleic acid are usually required to make the distinction.
CMV hepatitis most commonly develops in patients who have recently completed an augmented immunosuppressive regimen for rejection. In some cases, it is difficult to determine whether the liver injury is attributable to residual CMV hepatitis, relapse of acute rejection, or development of chronic rejection. O’Grady and colleagues showed an association between CMV infection and chronic rejection,227,228 further complicating the issue. Others have not found this association.229 Priority may be given to the finding of CMV inclusions, antigens, or nucleic acid, followed by anti-CMV therapy and follow-up biopsy after 1 to 2 weeks, if liver function abnormalities persist.
Herpes Simplex and Varicella-Zoster Viral Hepatitis
Type 1 and 2 HSV and VZV hepatitis may occur any time after transplantation.226 Patients often have fever, vesicular rashes, fatigue, body pain, and elevated liver function test results. Undetected HSV hepatitis can rapidly lead to submassive or massive hepatic necrosis, hypotension, disseminated intravascular coagulation, metabolic acidosis, and death.230 Fulminant cases occur more often as a result of a primary infection in patients without evidence of prior immunity. VZV-induced hepatitis is rare because of effective vaccines and anti-VZV hepatitis immunoglobulin.231
Pathologic Features
Recognition and prompt reporting of HSV and VZV hepatitis is essential because effective pharmacologic therapy is available. Without treatment, the infection can be rapidly fatal. Localized and diffuse patterns of HSV hepatitis have been described.230 Both pathologic patterns cause circumscribed areas of coagulative-type necrosis (Fig. 52.24).226,230 The center of the necrotic zones is typically occupied by ghosts of hepatocytes intermixed with neutrophils and nuclear debris. Viable hepatocytes rim the periphery and usually contain HSV or VZV hepatitis inclusions. Infected cells are usually slightly enlarged and contain ground-glass nuclei or characteristic Cowdry type A eosinophilic inclusions. Multinucleate smudgy cells are occasionally seen. However, diagnostic inclusions of HSV or VZV hepatitis may be absent on H&E-stained slides.
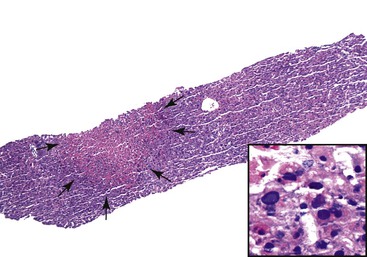
Immunoperoxidase stains for HSV antigens are confirmatory. In our experience, antibody preparations used to detect HSV 1 and 2 can show cross-reactivity with each other and with VZV hepatitis, making it difficult to distinguish these viruses. In our experience, antibodies used to detect the VZV hepatitis precursor and mature glycoprotein are more discriminating. If a histopathologic diagnosis of HSV or VZV hepatitis is considered on the basis of H&E staining, the physicians should be immediately notified.232 This will prompt effective antiviral therapy that can be discontinued if the diagnosis is not confirmed on immunostaining.
Human Herpesvirus 6
HHV-6, a member of the β-Herpesviridae subfamily of human herpesviruses, is a ubiquitous virus that usually infects humans during the first 2 years of life and then remains dormant in more than 90% of adults. Reactivation infection occurs in 15% to 80% of liver allograft recipients, usually during the first 2 to 8 weeks after transplantation. It is often precipitated by heavy immunosuppression.233
Direct HHV-6 clinical manifestations include fever with or without a rash, myelosuppression, hepatitis, pneumonitis, and neurologic dysfunction. Indirect effects attributed to HHV-6 include exacerbation of CMV disease, increased severity of HCV recurrence, increased risk of other opportunistic infections, allograft dysfunction, and ACR.233
Liver allograft biopsy findings include moderate lymphocytic portal inflammation and patchy lobular inflammation, including microabscesses234,235 and syncytial giant cell hepatitis.236 ACR can also occur at the time of disease.234,235 Immunoperoxidase stains for HHV-6 localize viral antigens primarily to mononuclear inflammatory cells234 and hepatocyte syncytial giant cells.236
Human Herpesvirus 8
HHV-8 is a lymphotropic γ-herpesvirus family member that exerts a pathogenic role in development of Kaposi sarcoma, multicentric Castleman disease, and primary effusion lymphoma in the general population and among liver allograft recipients.237–239 This infection is prevalent in sub-Saharan Africa and in southern Italy and is less prevalent elsewhere. Primary and reactivation infection occurs after liver transplantation. Primary infection is often more severe and can result in liver dysfunction, multiorgan failure, multicentric Castleman disease, and Kaposi sarcoma.237–239 Liver biopsy findings resemble those of acute viral hepatitis (Fig. 52.25), showing lobular disarray, ballooning, hepatocyte swelling and apoptosis, and a ductular reaction. When multicentric Castleman disease, Kaposi sarcoma, or an unexplained acute hepatitis is recognized, the pathologist should include HHV-6 and HHV-8 in the differential diagnosis.
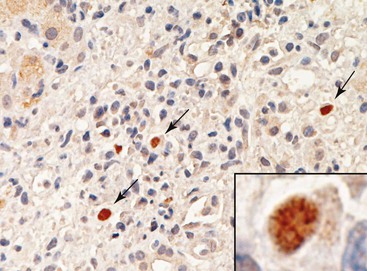
Epstein-Barr Virus
EBV infection immortalizes B lymphocytes in vitro but lies dormant in B lymphocytes and some epithelial cells in vivo because the immune system effectively controls viral replication.240–242 Potent immunosuppression depresses the T-cell immune surveillance that normally keeps EBV replication and B-cell proliferation in check. This enhances EBV replication and increases the risk of clinically evident EBV infection, ranging from a self-limited EBV-like syndrome to aggressive posttransplantation lymphoproliferative disease.240–242 Disease incidence is higher among seronegative, usually pediatric recipients, but the incidence varies from 1% to 5% among liver allograft recipients. The likelihood of infection increases with time after transplantation and is strongly associated with high-dose immunosuppression.240–242
Clinical Features
Clinical manifestations include hepatitis; gastroenteritis; B, T, and natural killer (NK) cell posttransplantation lymphoproliferative diseases; Hodgkin disease; smooth muscle stromal tumors; and a variety of cancers (e.g., nasopharyngeal, gastric).240–244 EBV-related syndromes often resemble infectious mononucleosis, manifesting with fever, lymphadenitis, pharyngitis, and jaundice. Atypical signs and symptoms include jaw pain, arthralgia, joint space effusions, diarrhea, encephalitis, pneumonitis, mediastinal lymphadenopathy, and ascites. Liver allograft hepatitis or posttransplantation lymphoproliferative disease (PTLD) involvement usually produces mildly elevated ALT and AST values, and atypical lymphocytes are see in the circulation. Pancytopenia is occasionally found.245–247
Risk factors for the development of serious EBV-associated disease include primary infection, high-dose immunosuppression, underlying Langerhans cell histiocytosis, and coexistent CMV disease. The risk of developing PTLD late after transplantation is not influenced by the type of immunosuppressive agents but is increased by the duration and intensity of immunosuppression.240,241,248
Peripheral blood monitoring for EBV nucleic acid is used to monitor viral replication.240 When levels are elevated, the patient is examined for lymphadenopathy and other signs of EBV-related disease, and a preemptive reduction in immunosuppression is usually made before more serious EBV-related disease develops.249 The treatment for significant EBV-related disease depends on the clinicopathologic presentation and clonality of the proliferating lymphocytes. Polyclonal infectious mononucleosis-like syndromes usually respond to a reduction of immunosuppression, whereas monomorphic PTLD resembling clonal lymphomas often requires more aggressive immunotherapy or chemotherapy, or both.240–242,244
Pathologic Features
EBV causes morphologic changes ranging from acute hepatitis characterized by a mild nonspecific portal and sinusoidal lymphocytosis to PTLDs (Fig. 52.26) that resemble diffuse large B-cell lymphomas.245–247,250 Even patients with enhanced EBV replication and viremia have increased intragraft T and B cells,251 with the latter occasionally containing EBV-encoded small RNA (EBER) sequences. Even in patients without overt EBV disease, EBER-positive cells may be admixed with other inflammatory cells associated with other causes of allograft dysfunction.250 The contention that EBV causes chronic hepatitis or recurrent acute relapses is controversial.252
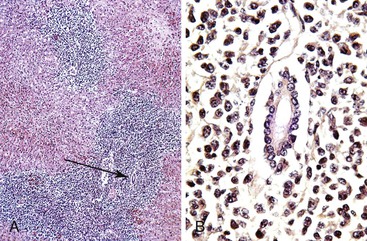
EBV hepatitis manifests as mild portal lymphoplasmacytic inflammation combined with sinusoidal lymphocytosis composed of small or mildly atypical lymphocytes. Lymphocytes lining up in the sinusoids should suggest an EBV-related disorder, as in native livers. Other lobular changes include focal hepatocellular swelling, mild acidophilic necrosis of hepatocytes, mild lobular disarray, and regenerative activity.
In the liver, PTLDs show cytologically atypical lymphoplasmacytic portal infiltrates. In early or polymorphic lesions, atypical cells are usually mixed with small and blastic lymphocytes, plasmacytoid lymphocytes, and plasma cells. In monomorphic lesions, atypical cells predominate and manifest as maplike enlargement of portal tracts due to sheets of atypical immunoblastic cells that obscure the normal portal anatomic landmarks. Atypical cells can be seen in the sinusoids. Some cases have significant hepatic necrosis. Hodgkin lymphoma–like PTLDs with classic Reed-Sternberg cells can occur, and they may be associated with bile duct loss. Subendothelial localization of lymphocytes in portal and central veins can mimic acute rejection.
Outside of the liver, PTLDs usually occur in the lymph nodes and intestines and are categorized according to the World Health Organization (WHO) classification scheme253 as early lesions (i.e., reactive plasmacytic hyperplasia and infectious mononucleosis-like), polymorphic (i.e., hyperplasia and lymphoma), monomorphic (i.e., T- and B-cell subtypes), and Hodgkin and Hodgkin-like lesions.
Diagnosis of any EBV-related disorder is confirmed by in situ hybridization for EBV RNA (i.e., EBER sequence). Routine workup should include immunohistochemical stains or in situ hybridization for κ and λ light chains and CD20 to determine possible responsiveness to anti-CD20 antibodies.242 If fresh tissue is available, a portion may be submitted for flow cytometry and molecular analyses, which enable a more detailed phenotypic characterization and study of immunoglobulin gene rearrangement, respectively.
Differential Diagnosis
EBV hepatitis and PTLD are difficult to distinguish from severe ACR. Features that favor acute rejection include pleomorphic, rejection-type portal or perivenular inflammatory infiltrates, conspicuous eosinophils, and severe bile duct damage that is proportional to the severity of inflammation. Features favoring EBV include a relatively monomorphic portal infiltrate, consisting primarily of activated and immunoblastic mononuclear cells. Many of the cells show plasmacytic differentiation, and some show atypical cytologic features. Patchy and mild inflammatory bile duct damage is less severe than would be expected based on the severity of the portal inflammatory infiltrate. Eosinophils are usually less conspicuous.
EBV hepatitis can be difficult to distinguish from nonspecific reactive hepatitis and acute HBV, HCV, or CMV hepatitis. HCV and EBV hepatitis can show sinusoidal lymphocytosis, but EBV-related disorders usually contain atypical cells. In contrast, lymphocytes associated with HCV hepatitis are usually small, round, and inactive appearing, and they form nodular aggregates in the portal tracts.245–247,250 Clinical suspicion and increased peripheral blood EBV levels also point to the correct diagnosis.
In most cases, the final diagnosis of any type of EBV-related disorder depends on in situ hybridization for EBV RNA (i.e., EBER probe). However, results should be interpreted with caution,254 because rare EBER-positive cells are found in lymphoid tissues from the general population. These cells are found with slightly increased frequency in allograft recipients, the significance of which is controversial.254 However, clustering of EBER-positive cells or the presence of EBER-positive cells in tissues that show other pathologic features of EBV-associated disease indicates enhanced EBV replication. These patients are at increased risk for EBV-related disease.
Adenovirus Hepatitis
Pathophysiology
Adenovirus is a double-stranded DNA virus with 51 serotypes. It causes a substantial number of respiratory infections in children younger than 5 years of age, including conjunctivitis, pharyngitis, croup, bronchiolitis, bronchitis, pneumonia, and diarrhea. Adenovirus-related disease after liver transplantation is largely restricted to primary infections in pediatric recipients.255,256 Some adult cases have been reported,257,258 and they mimic those seen in the general population.259
Adenovirus hepatitis usually manifests 50 to 100 days after transplantation.259 Fever, respiratory distress, elevated liver function test values, diarrhea, and leukocytosis develop in patients. The diagnosis is confirmed by liver biopsy examination.255–258 Adenovirus subtypes 1, 2, and 5 have been isolated from the lung and gastrointestinal tract.255,256,260 Allograft hepatitis is most often caused by viral subtype 5, but in the general population, hepatitis has also been caused by subtypes 2, 11, and 16.255,256
Pathologic Features
Adenovirus hepatitis is uncommon, and nuclear inclusions are difficult to recognize with certainty. Typical cases show poxlike granulomas, consisting of macrophages with or without neutrophils that are spread randomly throughout the liver parenchyma (Fig. 52.27). In other cases, the granulomas surround small, maplike areas of necrosis.255–257 Viral inclusions are usually found in nuclei of viable hepatocytes near the edge of necrotic zones or granulomas. Infected cells have smudgy appearance with crowding of chromatin near the nuclear membrane, which makes the nucleus appear like a baked muffin. Immunohistochemical staining is usually required to confirm the diagnosis. Given that there are 51 serotypes, it is prudent to use antibodies reactive with the hexon protein common to all serotypes (e.g., MAB805, blend of clones 20/11 and 2/6, Chemicon International, Temecula, Calif.).258
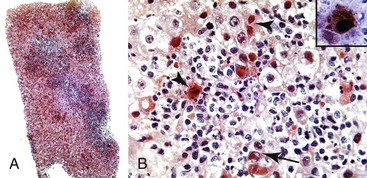
[/level-membership-for-pathology-category][not-level-membership-for-pathology-category]<
[/not-level-membership-for-pathology-category]
