[level-membership-for-endocrinology-diabetes-and-metabolism-category]Chapter 1
Parathyroid Hormone and Parathyroid Hormone–Related Peptide In The Regulation of Calcium Homeostasis and Bone Development
Regulation of Mineral Ion Homeostasis—General Considerations
PTH Biosynthesis and Intraglandular Processing
Regulation of PTH Gene Expression
PTH-Dependent Regulation of Mineral Ion Homeostasis Mediated Through the PTH/PTHrP Receptor (Type 1 Receptor)
Parathyroid hormone (PTH) and PTH-related peptide (PTHrP), along with other calciotropic hormones, play critical roles in calcium homeostasis and bone biology. First discovered as a calcium-regulating hormone in the 1920s,1–3 PTH is secreted by the parathyroid glands and is one of the most important regulators of blood calcium concentration in all terrestrial vertebrate species, from amphibians to mammals. PTHrP, a slightly larger molecule than PTH, was discovered more recently through efforts to identify the factor that causes, when produced in excess by certain tumors, the humoral hypercalcemia of malignancy syndrome. In contrast to PTH, which is produced by discrete endocrine glands, PTHrP is produced as a paracrine/autocrine factor in many different adult and fetal tissues and has, unlike PTH, multiple functions (Fig. 1-1).4–6
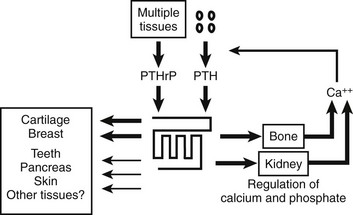
FIGURE 1-1 The parathyroid hormone (PTH)/PTH-related peptide (PTHrP) receptor interacts with indistinguishable efficiency and efficacy with PTH and PTHrP, and it activates at least two distinct second messenger systems, cyclic adenosine monophosphate and Ca2+/inositol 1,4,5-triphosphate. The receptor is abundantly expressed in bone and kidney, where it mediates the endocrine actions of PTH, and in the metaphyseal growth plate and numerous other tissues, where it mediates the autocrine/paracrine actions of PTHrP.
PTH and PTHrP most likely evolved from a common ancestral precursor. Despite this common evolutionary origin, both peptides share only limited overall amino acid sequence identity, yet at least their N-terminal regions are sufficiently homologous to enable them to bind to and activate a common G protein–coupled receptor, the PTH/PTHrP receptor (also referred to as PTH1R).7–9 This receptor mediates the most important biological actions of both peptides: PTH-dependent regulation of calcium and phosphorous homeostasis and PTHrP-dependent regulation of endochondral bone formation.10–14
This chapter reviews (1) the comparative chemistry of PTH and PTHrP, their genes, and their interactions with the PTH1R; (2) the current molecular models of productive interactions of the two ligands with their common receptor; and (3) the different biological roles of both peptides on target tissues, such as the role of PTH in calcium homeostasis and bone turnover, the role of PTHrP in bone and cartilage development, and the role of PTH in regulating renal phosphate excretion (see Fig. 1-1). However, the chapter does not review the potentially numerous and still incompletely characterized biological roles of PTHrP outside the field of mineral ion homeostasis and bone biology. The evolutionary history of the principal PTH/PTHrP receptor is reviewed, as well as the functional characteristics of two novel, closely related receptors and the pharmacologic and physicochemical evidence for several additional, still incompletely characterized, receptors for PTH and PTHrP.
Regulation of Mineral Ion Homeostasis—General Considerations
To ensure a multitude of essential cellular functions, the extracellular concentration of calcium (Ca2+o) is maintained within narrow limits.15,16 In terrestrial vertebrates, calcium is necessary for adequate mineralization of the skeleton, which provides mechanical support and protection for internal organs and acts as levers for the various muscle groups involved in locomotion. Because of its high calcium content, 99% of the body’s supply, the skeleton also serves as the most important reservoir from which calcium can be rapidly mobilized. Because food intake and thus the nutritional supply of calcium are usually discontinuous, intestinal calcium absorption occurs only intermittently. Maintenance of a constant blood calcium concentration thus constitutes a major homeostatic challenge, which during evolution led to the development of highly efficient mechanisms to increase intestinal calcium absorption, reduce urinary calcium losses, and facilitate, if necessary, rapid mobilization of calcium from the skeletal reservoir (see Chapter 5).16
In contrast to these environmental challenges of most terrestrial vertebrates, marine animals, which are usually exposed to the high environmental calcium concentration of seawater (10 mM) had to adopt mechanisms by which extracellular calcium could be reduced.17,18 Unlike the diet of terrestrial animals, seawater provides only a very limited supply of phosphate, and this environmental deficiency resulted in the development of mechanisms to conserve phosphate. It thus appears plausible that the efficient intestinal absorption of phosphate and the impressive capacity of the mammalian kidney to retain phosphate15,19 are remnants of earlier evolutionary adaptations to life in the low-phosphate environment of the oceans. To reduce blood calcium concentrations, fish use stanniocalcin, which is produced by the corpuscles of Stannius, as well as several other hormonal factors.17,18,20 Some data indicate that the mammalian homologue of stanniocalcin has similar properties when tested in rodents, but it remains uncertain whether this peptide hormone has a significant physiologic role in mammalian mineral ion homeostasis.21 A widely expressed mammalian peptide, stanniocalcin 2, that was discovered because of its structural homology with stanniocalcin, appears to inhibit phosphate uptake in renal epithelial cells.22 However, newer data (see later) indicate that fibroblast growth factor-23 (FGF-23) and perhaps soluble frizzled-related protein 4 (sFRP4), dentin matrix protein 1 (DMP-1), and matrix extracellular phosphoglycoprotein (MEPE)23 are more likely to be involved physiologically in the regulation of mammalian phosphate homeostasis (see Chapter 6). Calcitonin, made by the ultimobranchial bodies in fish, has a calcium-lowering function in these vertebrate species, but its biological role in mammals remains uncertain (for review, see Chapter 2).24
Parathyroid Hormone
PTH and the active form of vitamin D, 1,25-dihydroxyvitamin D3 [1,25(OH)2D3], are the principal physiologic regulators of calcium homeostasis in humans and all terrestrial vertebrates.11,25,26 Synthesis and secretion of PTH are stimulated by any decrease in blood calcium, and conversely, secretion of the hormone is inhibited by an increase in blood calcium.27–29 This rapid negative feedback regulation of PTH production, along with the biological actions of the hormone on different target tissues, represents the most important homeostatic mechanism for minute-to-minute control of calcium concentration in the extracellular fluid (ECF) (Fig. 1-2).30–32 In contrast to the rapid actions of PTH, 1,25(OH)2D3 is of critical importance for long-term, day-to-day, and week-to-week calcium balance (see Chapter 3). The actions of both hormones are coordinated, and each influences the synthesis and secretion of the other. Calcitonin, the third of the calciotropic hormones known to be important in the regulation of vertebrate mineral ion homeostasis (see Chapter 2), may be vestigial in humans with respect to calcium homeostasis and will not be discussed in this brief review of physiology.
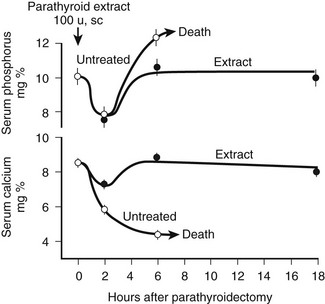
FIGURE 1-2 Rate of change in blood phosphorus and calcium levels in rats with stressed calcium homeostasis (low-calcium diet) after parathyroidectomy without treatment or with 100 U of parathyroid hormone extract given in addition at the time of parathyroidectomy. Rapid and usually fatal hypocalcemia and hyperphosphatemia result within hours unless hormone is given. (Data from Munson PL: Studies on the role of the parathyroids in calcium and phosphorus metabolism, Ann N Y Acad Sci 60:776–796, 1955.)
At least three distinct but coordinated actions of PTH increase the flow of calcium into the ECF and thus increase the concentration of blood calcium (see Fig. 1-2).27–29 Through its rapid actions on the kidney and bone, which are all mediated through the PTH/PTHrP receptor and subsequent secondary messages in specific and highly specialized cells, PTH increases the release of calcium from bone, reduces the renal clearance of calcium, and stimulates the production of 1,25(OH)2D3 by activating the gene encoding 25-hydroxyvitamin D-1α-hydroxylase (1α-hydroxylase) in the kidney. The relative importance of the first two actions of PTH on the rapid, minute-to-minute regulation of calcium is not definitively resolved, but most physiologists have stressed the importance of the effects of PTH on bone in maintaining hour-to-hour calcium homeostasis in the ECF. Several lines of evidence, such as that provided by calcium kinetic analysis, indicate a transfer between ECF and bone of as much as 500 mg of calcium daily, which is equivalent to one-fourth to one-half the total ECF calcium content.15 Besides regulating this transfer of calcium from bone through direct breakdown of bone tissue (mineral and matrix), PTH influences the rates of exchange of calcium adsorbed to the surface of bone; this exchangeable calcium pool can be stimulated to provide a rapid and substantial rate of entry of calcium into blood. In addition to these PTH-dependent actions on bone, actions of PTH on the kidney may also be extremely important in the precise hourly regulation of ECF calcium. The third action of PTH on calcium homeostasis—namely, enhancement of intestinal calcium absorption—is indirect and involves the synthesis of 1,25(OH)2D3 from the biologically inactive precursor 25(OH)D3. However, it is difficult to quantitatively analyze or to proportionately contrast the relative physiologic importance of the direct and indirect actions of PTH on the three principal target tissues: kidney, bone, and intestine.
The complexity of bone as a tissue and the many detectable rates of exchange of calcium between the skeleton and the ECF have made the action of PTH on the skeleton difficult to analyze. The state of calcium in blood is complex; much of the calcium is present as chelates or is bound to plasma proteins (for detailed review, see Chapter 5). Because actual filtered loads depend on the ratio of free and bound forms of calcium, it is difficult to calculate renal calcium clearance accurately. The different PTH-dependent actions to promote calcium entry into the ECF are most clearly defined in conditions of deficiency or excess of PTH, such as during experiments in animals or during controlled observations in patients with disorders of parathyroid gland function. The experimental data in these extremes abundantly affirm the crucial calcium homeostatic role of PTH. However, because of continuous and rapid adjustments in mineral ion concentration, it can be difficult to observe the consequences of hormone action under normal physiologic conditions. For example, the rate of PTH secretion changes continually and rapidly so that the controlled variable, calcium, remains constant, and it may therefore be difficult to experimentally detect small corrective changes.
Teleologically, the action of PTH on the regulation of blood phosphate concentration in terrestrial species is best understood as a secondary, rather than a homeostatic, action. Phosphate is abundant in the food chain in terrestrial existence. Phosphate deficiency, unlike calcium deficiency, in the absence of specific organ dysfunction is, therefore, an unlikely environmental challenge (see Chapter 6 for detailed review of the regulation of phosphate homeostasis). To correct a deficiency in calcium, mineral stores in bone can be rapidly dissolved; such activity results, however, in the simultaneous liberation of ionic calcium and phosphate. Because a high blood phosphate level tends to lower the calcium concentration through multiple mechanisms, the rise in blood calcium that occurs after bone dissolution (desirable homeostatically) is beneficial only if the concomitant increase in blood phosphate concentration (undesirable) can be rapidly corrected. To maximize the control of calcium homeostasis, PTH thus has divergent actions on renal tubular handling of the two mineral ions: It increases the retention of calcium and at the same time diminishes reabsorption of phosphate. Through these mechanisms—namely, increased renal phosphate clearance to prevent hyperphosphatemia and increased tubular calcium reabsorption—PTH guarantees that an elevation in blood calcium results from the increased release of calcium from bone. The renal action of PTH on phosphate homeostasis is biologically predominant over the increased phosphate flux from bone. Consequently, parathyroidectomy (experimentally, in animals) or renal resistance to PTH, as in patients with pseudohypoparathyroidism or renal failure, leads not only to hypocalcemia but also to an increase in blood phosphate and a marked reduction in urinary phosphate excretion (see Fig. 1-2). This finding demonstrates the importance of the PTH-dependent action on phosphate homeostasis in the kidney, which becomes particularly important in disease states when high bone turnover is the result of dietary calcium deficiency or lack of biologically active vitamin D.
Chemistry
The first extracts from bovine parathyroid glands were described in 1925, and the content of biologically active PTH was assessed by their hypercalcemic and phosphaturic properties.2,3 However, it was not until 1959, when Aurbach33 and Rasmussen and Craig34 developed improved extraction procedures, that it became possible to isolate and purify sufficient quantities to determine the primary structure of bovine, porcine, and human PTH through the protein sequence determination methods.35–40 Two groups independently determined the sequences of human and bovine hormones.35–40 Shown in Fig. 1-3A are the sequences of the bovine, porcine, and human hormones determined by one group.36,37,39,40 Discordant sequences for the human PTH polypeptide, and in one position for the bovine hormone, published by the other group,35,38 are not shown in Fig. 1-3A, since nucleotide sequence analysis of genomic and complementary DNA confirmed the amino acid sequences of the first group (the only exception was residue 76 in human PTH, which was determined to be glutamine instead of glutamic acid).36,37,40 Based on these amino acid sequences, the PTH(1–34) fragments of the different species were synthesized, and their biological activities were compared in vitro and in vivo with those of highly purified intact PTH from the same species (Table 1-1). Molecular cloning techniques then led to the deduction of the amino acid sequences of rat, chicken, and dog PTHs,41–44 followed more recently by the identification of PTH molecules in other mammals and fish, as shown in Fig. 1-3A and discussed later. The synthetic peptides used in parathyroid hormone research today are based largely on the (1-34) regions of the mammalian hormone sequences shown in Fig. 1-3A.45,46
Table 1-1
Comparison of the Biological Activity of Parathyroid Peptides from Different Species
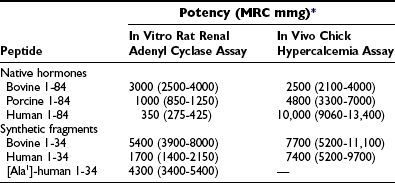
*Values are expressed as mean potency with 95% confidence intervals and are based on Medical Research Council research standard A for parathyroid hormone.
From Rosenblatt M, Kronenberg HM, Potts JT Jr: Parathyroid hormone. In DeGroot L (ed): Endocrinology, ed 2, Philadelphia, 1989, WB Saunders, p 853.

FIGURE 1-3 Sequence Relationships in PTH and PTHrP peptides. Comparisons of amino Acid sequences of PTH and PTHrP peptides from different species are shown in A and B, respectively. Comparison of human TIP39 and a PTH-Like peptide from the puffer fish (Takifugu rubripes) to human PTH and PTHrP (the (1–84) region only) is shown in C. In A-C, amino acid identities are shown on black field and similarities on gray field. A phylogenetic tree of the ligands is shown in D, with other family B receptor ligands, secretin, calcitonin, and cotricotropin-releasing factor (CRF) included as marginally homologous ligands. Fugu PTHL is speculated to be a precurssor to PTH and PTHrP.
Extensive sequence homology is present in the mammalian PTH species (see Fig. 1-3A and D); these molecules consist of a single-chain polypeptide with 84 amino acids and a molecular weight of approximately 9400 daltons (that of human PTH[1–84] is 9425 D). The N-terminal region of PTH, which is necessary and sufficient for the regulation of mineral ion homeostasis, shows high sequence conservation among all the vertebrate species (see Fig. 1-3A). The middle portions of the different molecules exhibit the most structural variation, which could suggest that this region of PTH is only of limited functional importance. The nonmammalian PTH homologues of chicken42,43 and fish species Danio rerio (zebrafish)47 and Takifugu ruberipes (puffer fish)48,49 diverge considerably from the mammalian hormones C-terminal of amino acid residue His32. Interestingly, both fish species have two distinct genes encoding two separate PTH molecules, called PTH1 and PTH2.47,48 The zebrafish peptides are considerably shorter than mammalian PTH (67 and 68 residues), while fugu PTH1 is predicted to be 81 residues in length and fugu PTH2 is predicted to be 63 residues.48
After the original work establishing that the first 34 amino acids of mammalian PTH were sufficient to produce a fully active synthetic peptide,45,46 much work has centered on defining the minimum pharmacophore essential for biological activity. In a following section, we describe how sites of ligand interaction with the PTH/PTHrP receptor were defined by performing assays with products of various combinations of shortened and modified PTH ligands and mutagenized receptors. Furthermore, a fusion protein consisting of ligand substituted for most of the receptor amino-terminal extracellular domain was generated, and this helped define a much smaller minimum chain length of PTH peptide needed for biological activity.50 As also discussed later in the section on hormone/receptor interactions, it has been determined that substitutions of non-naturally occurring amino acids (e.g., α-aminoisobutyric acid at positions 1 and 3 in the primary ligand structure) favoring formation of an α-helix, even in short peptides, such as PTH(1–14), produce peptides that are highly potent when tested in vitro using cell-based assays and have highly stabilized helical structure in solution51–55(Fig. 1-4).
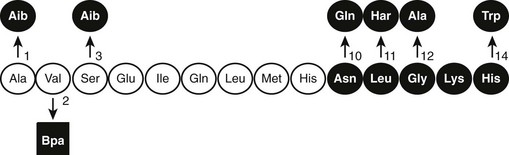
FIGURE 1-4 The native (rat) PTH(1-14) sequence and activity-modifying substitutions. The six substitutions shown above the sequence, when combined, enhance activity by as much as 100,000-fold; the Bpa2 substitution shown below confers antagonist properties to the peptide. The (1-9) region (nonshaded circles) is thought to be the minimum-length agonist pharmacophore. Nonencoded amino acids include: α-amino-isobutyric acid (Aib), homoarginine (Har), and parabenzoyl-l-phenylalanine (Bpa).
The in vitro activity of the native PTH(1-14), which is quite weak, is improved about 100,000-fold by the modifications indicated in Fig. 1-4. Shorter PTH peptides have also been shown to be active in vivo, since some cause hypercalcemia and are anabolic on bone, although their potency is much less than that of PTH(1-34) due to a more rapid clearance.56 Replacement of valine-2 in these peptides with bulky amino acids, such as tryptophan or parabenzoyl-l-phenylalanine (Bpa), results in competitive antagonist peptides defective for AC/cAMP and PLC/IP3/Ca2+ signaling, thus confirming the critical role that this conserved valine plays in receptor activation.57 Other longer-length PTH or PTHrP analogs having residue-1 (serine or alanine) replaced by glycine,58 Bpa,59 or tryptophan60 exhibit signal-selective properties in that they efficiently stimulate the cAMP cellular pathway but not the inositol tri-phosphate/intracellular Ca2+ pathway.
The three-dimensional structures of intact PTH(1-84) and the N-terminal biologically active fragments of both PTH and PTHrP have been analyzed by various solution-based methods, including nuclear magnetic resonance (NMR) spectroscopy. Interpretation of these results in terms of biological mode of action is not straightforward, because the ligand interacts with a membrane-embedded receptor, and the biophysical properties of this environment, as well as the receptor-induced conformational changes that occur, are mostly unknown. However, the bioactive (1-34) portions of the ligands are generally found to contain helical structure within their N-terminal and especially C-terminal portions, with some flexibility in the midregion.61–64 An x-ray crystallographic study of PTH(1-34) revealed α-helical structure extending nearly the full length of the peptide.65 A persisting question is thus whether the ligand bound to the receptor adopts a linear and extended62 or “U-shaped” folded structure, the latter suggested by tertiary interactions seen in some solution-phase biophysical studies.61,63,64 Of particular interest has been the question of whether common structural features would be discerned for PTH and PTHrP that could explain their use of apparently overlapping binding sites on the common PTH/PTHrP receptor; to some extent, the helical propensities of at least the C-terminal domains of the peptides are consistent with this possibility.62,66,67
Therefore, consistent with their rather limited homology in primary structure (see Fig. 1-3), convincing evidence has not yet been provided for the conclusion that the N-terminal fragments of PTH and PTHrP display a similar secondary structure in solution. Because of their generally demonstrated similar potencies at the PTH/PTHrP receptor, it seemed likely that both ligands would adopt very similar conformations when part of the active hormone-receptor complex. However, recent data (discussed later) suggest that each ligand selectively binds to or induces a distinct receptor confirmation. Ideally, each hormone should be co-crystallized with the PTH/PTHrP receptor to permit analysis by x-ray diffraction of those intermolecular interactions that are characteristic of the biologically active hormone-receptor complexes. G protein–coupled receptors such as the PTH/PTHrP receptor have multiple membrane-embedded domains and are likely to have complex three-dimensional structures. Interaction with either PTH or PTHrP appears to involve several distinct receptor domains (see later discussion) that may undergo significant conformational changes after ligand binding has occurred, which makes it even more challenging to conduct x-ray or multidimensional NMR analyses. Recent advances, however, have been made it possible to co-crystallize the extracellular portion of the PTH/PTHrP receptor with the carboxyl end of the PTH (1-34) peptide68 and to crystallize the membrane-spanning portion of a distantly related GPCR, the β2-adrenergic receptor.69
Evolution
To maintain extracellular calcium and phosphate concentrations within narrow limits, the intricate regulatory system outlined, in which PTH plays the most important role, developed in the terrestrial animals. In mammals, PTH is produced almost exclusively by the parathyroid glands (only small amounts of its messenger RNA [mRNA] have been detected elsewhere70,71). During evolution, these glands first appear as discrete organs in amphibians, that is, with the migration of vertebrates from an aquatic to a terrestrial existence, and their appearance most likely represents an evolutionary adaptation to an environment that is, by comparison to seawater, low in calcium.17,18,72 Parathyroid glands have not been identified in fish or invertebrate species, but earlier immunologic and RNA hybridization data from fish provided evidence for expression of PTH proteins in several tissues, including pituitary,17,18,73 plasma, brain, kidney, spinal cord, ultimobranchial gland, as well as in the ventral neural tube and mineralizing jaw during development.48,74 Now, with the rapid advances in characterization of complete genomes of multiple species, we have definitive proof of the earlier evolutionary origin of both PTH and PTHrP (see Fig. 1-3). Gene analyses of several teleost fish species, including the zebrafish, Danio rerio, and the puffer fishes Takifugu rubripes and Tetraodon fluviatilis, reveal the duplication of the PTH gene in each case.48,75 Both the PTH1 and PTH2 peptides derived from the zebrafish activate the PTH/PTHrP receptors from different species,49,75 and indeed, a fugu PTH(1-34) peptide has been shown to induce bone anabolic effects in osteopenic ovariectomized rats.76
In addition to PTH, the teleost fish also express PTHrP, again encoded by duplicate genes.75,77–80 Furthermore, PTHrP immunoreactivity has been detected in the cartilaginous sharks and rays81 and in a more primitive agnathan, the lamprey.82 The teleost PTHrPs contain some amino acid residues characteristic of mammalian PTH; for example, fish PTHrP contains Met at position 8, Trp at position 23, and Leu at position 28, which are amino acid residues found in mammalian PTH. However, there is only one amino acid residue, Gln(Q)25, in fish PTH that is found in some mammalian PTHrP species but not in mammalian PTH (see Fig. 1-3). This pattern suggests that the fish proteins may be phylogenetically closer to a common PTH/PTHrP precursor than are the mammalian proteins (see later). Indeed, in addition to duplicate copies of PTH and PTHrP genes, the puffer fish genome contains a fifth gene that encodes a protein containing amino acid residues characteristic of both PTH and PTHrP. This gene, called PTH-L, is phylogenetically an intermediary to PTH and PTHrP and may thus represent first definitive evidence for an ancestral gene from which the two divergent ligand forms evolved (see Fig. 1-3C and D).48,75
The PTH Gene and Its mRNA
The human PTH gene consists of three exons located on chromosome 11p15.83–86 The first exon is 85 nucleotides in length and is noncoding (Fig. 1-5). Exon 2 (90 bp) encodes most amino acids of the prepropeptide sequence, whereas the third exon (612 bp) encodes the remainder of the propeptide sequence and all amino acids of the mature peptide, and it constitutes the 3′ noncoding region.87 Several frequent intragenic polymorphisms (TaqI and PstI88; BstBI89; DraIII90; XmnI91), and a tetranucleotide repeat ([AAAT]n;92) have been identified in the human PTH gene, and some were shown to be informative in genetic linkage studies.93–95 Two mRNAs that are 822 and 793 bp in length are derived in the human gene from the two transcriptional start sites, which follow two different functional TATA boxes that are separated by 29 bp.87 Two closely spaced TATA boxes and two distinct transcripts are also derived from the bovine PTH gene, while rat and chicken PTH genes give rise to only one transcript; as a consequence of a long 3′ noncoding region, the transcript from the chicken PTH gene is unusually long and comprises 2.3 kb.25,96 The genes encoding zebrafish PTH1 and PTH2 have a similar overall organization as the mammalian PTH genes.47
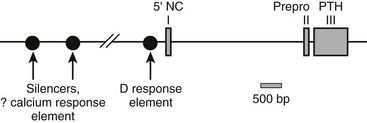
FIGURE 1-5 Schematic of the parathyroid hormone (PTH) gene along several thousand base pairs (approximate length shown by the scale marker for 500 bp). The three exons in the mRNA are represented as numbered rectangles. Control elements are identified in the 5′ noncoding region (5′ NC). A region responsive to vitamin D is within a few hundred base pairs of exon 1. Far upstream are silencers involved in calcium regulation.
PTH Biosynthesis and Intraglandular Processing
During the synthesis of the preproPTH molecule, the signal sequence, which comprises the 25-amino-acid-containing “pre” sequence, is cleaved off after entry of the nascent peptide chain into the intracisternal space bounded by the endoplasmic reticulum. A heterozygous mutation in this leader sequence, which changes a cysteine to an arginine at position −8 and thus impairs processing of preproPTH to proPTH, has been identified as the most plausible molecular cause of an autosomal dominant familial form of hypoparathyroidism.97,98 The mutant hormone was found to be trapped intracellularly, predominantly in the endoplasmic reticulum (ER), leading to a marked up-regulation of ER stress-responsive proteins (BiP and PERK) and the proapoptotic transcription factor, CHOP, indicating that apoptosis-mediated parathyroid cell death is the likely cause of the observed hypoparathyroidism.99
Subsequent to the removal of the pre-sequence, the pro-peptide is transported to the trans-Golgi network, where the pro-sequence (amino acid residues −6 through −1) is removed.100 This latter process may involve furin (paired basic amino acid cleaving enzyme) and/or proprotein convertase-7 (PC-7), which are both expressed in parathyroid tissue; their expression levels do not appear to be regulated by either calcium or 1,25(OH)2D3.101,102 After removal of the basic pro-sequence, the mature polypeptide, PTH(1-84), is packaged into secretory granules. Two proteases, cathepsins B and H, are subsequently involved in the intraglandular generation of carboxyl-terminal PTH fragments from the intact hormone; no amino-terminal PTH fragments appear to be released from the gland.103–105 Since small or intermediate-size carboxyl-terminal fragments of PTH are unlikely to be involved in the regulation of calcium homeostasis, the intraglandular degradation of intact PTH is thought to represent an inactivating pathway, at least with regard to the regulation of mineral ion homeostasis. Consistent with this conclusion, hypercalcemia results in a substantial decrease in PTH secretion and, furthermore, favors the secretion of carboxyl-terminal PTH fragments, including a previously undetected large molecular species that are truncated at the amino-terminus (see following section).105–108 However, recent studies have shown that some amino-terminally truncated PTH fragments, such as PTH(7-84), have hypocalcemic properties in vivo and can furthermore reduce the formation of osteoclasts in vitro.109
The pool of stored, intracellular PTH is small, and the parathyroid cell must therefore have mechanisms to increase hormone synthesis and release in response to sustained hypocalcemia. One such adaptive mechanism is to reduce the intracellular degradation of the hormone, thereby increasing the net amount of intact, biologically active PTH that is available for secretion. During hypocalcemia, the bulk of the hormone that is released from the parathyroid cell is intact PTH(1-84).103–105,107,108 As the level of Ca2+o increases, a greater fraction of intracellular PTH is degraded, and with overt hypercalcemia, most of the secreted immunoreactive PTH consists of biologically inactive C-terminal fragments.10,25,26
Regulation of PTH Gene Expression
Another adaptive mechanism of the parathyroid cell to sustained reductions in Ca2+o is to increase cellular levels of PTH mRNA, a response that takes several hours. A reduction in Ca2+o increases, whereas an elevation in Ca2+o reduces the cellular levels of PTH mRNA by affecting both its stability and the transcriptional rate of its gene.11,26,110,111 Available data suggest that phosphate ions also regulate, directly or indirectly, PTH gene expression. In the rat, hypophosphatemia and hyperphosphatemia, respectively, lower and raise the levels of mRNA for PTH through a mechanism that is independent of changes in Ca2+o or 1,25(OH)2D3. An elevated extracellular phosphate concentration could thus contribute importantly to the secondary hyperparathyroidism frequently encountered in patients with end-stage renal failure, who often have chronically elevated serum phosphate concentrations.
Metabolites of vitamin D, principally 1,25(OH)2D3, also play an important role in the long-term regulation of parathyroid function and may act at several levels: by affecting the secretion of PTH and regulation of its gene, by regulating transcriptional activity of the genes encoding the calcium-sensing receptor (CaSR; see later) and the vitamin D receptor (VDR), as well as by regulating parathyroid cellular proliferation.11,26,110,112 1,25(OH)2D3 is by far the most important vitamin D metabolite that modulates parathyroid function. It acts through a nuclear receptor, the VDR, often in concert with other such receptors (i.e., those for retinoic acid or glucocorticoids), on DNA sequences upstream from the PTH gene (see Chapter 3).113,114 1,25(OH)2D3-induced upregulation of VDR and CaSR expression in the parathyroid could potentiate its inhibitory action(s) on PTH synthesis and secretion.11,26,110 Noncalcemic or less calcemic analogs of 1,25(OH)2D3 inhibit PTH secretion while producing relatively little stimulation of intestinal calcium absorption and bone resorption115–117 and may thus be attractive candidates for treating the hyperparathyroidism of chronic renal insufficiency.
Regulation of Parathyroid Hormone Secretion
A large number of factors modulate PTH secretion in vitro,11,26,118 but most of these factors are not thought to control hormonal secretion in vivo in a biologically relevant manner. Therefore, we focus in this section principally on factors that are the most physiologically meaningful regulators of PTH secretion—that is, the extracellular ionized calcium concentration itself (Ca2+o), 1,25(OH)2D3, and the level of extracellular phosphate ions. Of these three, Ca2+o is most important in the minute-to-minute control of PTH secretion. Indeed, the actions of 1,25(OH)2D3 and phosphate ions on the secretion of PTH probably result at least in part from their effects on hormonal biosynthesis rather than secretion per se.11,26,118 Ca2+o also modulates several other aspects of parathyroid function that indirectly affect PTH secretion, including PTH gene expression, the hormone’s intracellular degradation, and parathyroid cellular proliferation, as described previously. Recent data have shown that novel factors playing key roles in phosphate homeostasis, especially FGF-23 and α-klotho (a coreceptor for FGF receptors), also modulate parathyroid function, inhibiting119,120 and enhancing121 parathyroid function, respectively. Our rapidly improving understanding of how these factors participate in phosphate homeostasis is described in detail in Chapter 6.
Physiologic Control of PTH Secretion by Ca2+o
As illustrated in Fig. 1-6A, the relationship between PTH and Ca2+o is represented by a steep inverse sigmoidal curve that can be quantitatively described by four parameters.122–124 These are the maximal rate of PTH secretion at low Ca2+o (parameter A); the slope of the curve at its midpoint (parameter B); the value of Ca2+o at the midpoint (e.g., the “set point” or the level of Ca2+o half-maximally suppressing PTH release; parameter C); and the minimal secretory rate at high Ca2+o (parameter D) (see Fig. 1-6B). Parameter A in vivo is the sum of the maximal rates of PTH release from all individual parathyroid chief cells, as reflected by the resultant, maximally stimulated level of circulating PTH. Because of the steepness of the Ca2+o-PTH relationship, small alterations in Ca2+o evoke large changes in PTH release, thereby contributing importantly to the near constancy of Ca2+o in vivo. Indeed, parathyroid cells can readily detect reductions in Ca2+o of a few percentage points,123 and the percent coefficient of variation in Ca2+o in humans is less than 2%.125 The set point of the parathyroid gland is the key determinant of the level at which Ca2+o is “set” in vivo, although the parathyroid set point is usually slightly lower than the ambient blood Ca2+.126 Thus, the parathyroid cell is normally more than half-maximally suppressed at normal levels of Ca2+o and has a large secretory reserve for responding to hypocalcemic stress. Nevertheless, PTH levels in vivo also fall dramatically (e.g., by 80%) when Ca2+o rises to frankly hypercalcemic levels,122,123 which is thought to contribute importantly to the mineral ion homeostatic system’s defense against hypercalcemia.126 Furthermore, elevating Ca2+o also decreases the proportion of secreted intact PTH because of increased intraglandular degradation to inactive fragments (see the earlier section, PTH Biosynthesis and Intraglandular Processing, and the later section, Metabolism of PTH).106,127 Even with severe hypercalcemia, however, some residual release of intact PTH(1-84) still occurs in vivo and persists at a level approximately 5% of that observed with a maximal hypocalcemic stimulus29,108,128 (see Fig. 1-6A). This nonsuppressible basal component of PTH release may contribute to the hypercalcemia caused by hyperparathyroidism when the mass of abnormal parathyroid tissue is very great (e.g., in patients with renal failure).124,129–131
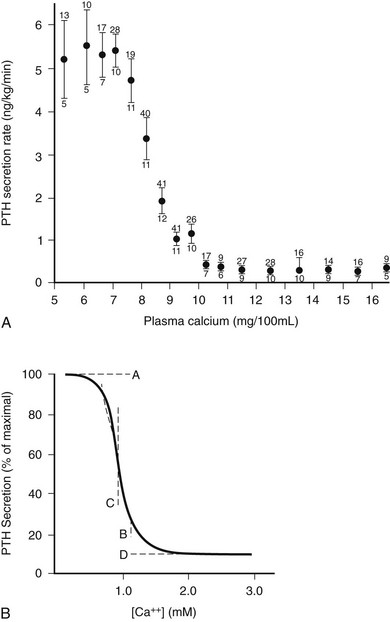
FIGURE 1-6 Inverse sigmoidal relationship between Ca2+ and parathyroid hormone (PTH) release and the four-parameter model describing these curves. A, Secretory response of bovine parathyroid glands to induced alterations in plasma calcium concentration. Calves were infused with calcium or ethylenediaminetetraacetic acid, and PTH secretion was assessed by measuring PTH levels in the parathyroid venous effluent. The symbols and vertical bars indicate the secretory rate (mean ± SE) in calcium concentration ranges of 1 or 0.5 mg/100 mL. The number of calves and samples are indicated, respectively, by numbers below and above. B, Sigmoidal curve generated by the equation Y = [(A − D)/(1 + (X/CB)] + D; the significance of A, B, C, and D are described in the text. (A, Data from Hurst JG: Sigmoidal relationship between parathyroid hormone secretion rate and plasma calcium concentration in calves. Endocrinology 10:10, 1978; B, Data from Brown EM: PTH secretion in vivo and in vitro, Miner Electrolyte Metab 8:130–150, 1982.)
The parathyroid cell has a temporal hierarchy of responses to low Ca2+o that permits it to secrete progressively larger amounts of hormone during prolonged hypocalcemia.11,26,118 To meet acute hypocalcemic challenges, PTH is released within seconds from preformed secretory vesicles by exocytosis as dictated by the sigmoidal curve (see Fig. 1-6). Sufficient PTH is stored in the parathyroid chief cell to sustain maximal, low Ca2+o-stimulated PTH release for about 60 to 90 minutes.126 Another rapid response of the parathyroid cell to hypocalcemia that enhances its net synthetic rate of PTH is reduced intracellular hormonal degradation—the opposite of what occurs at high levels of Ca2+o—which occurs within minutes to an hour.106,127 Hypocalcemia persisting for hours to days elicits increased PTH gene expression, whereas that lasting for days to weeks or longer stimulates parathyroid cellular proliferation.11,26,118,132 A greater secretory capacity for PTH on a per-cell basis (e.g., as a result of enhanced PTH gene expression) increases maximal hormonal secretion in vivo, as does an increase in cell number as a result of parathyroid cellular proliferation (see Fig. 1-6). In severe secondary hyperparathyroidism, very large increases in parathyroid cellular mass can elevate circulating PTH levels by 100-fold or more.
In addition to responding to changes in Ca2+o per se, the parathyroid cell also appears to sense the rate of change in Ca2+o such that rapid decrements in calcium promote more vigorous secretory responses than do changes of a similar magnitude occurring more slowly.133 Furthermore, during dynamic testing of parathyroid function in vivo by induced increases or decreases in Ca2+o, PTH in blood is higher at a given serum calcium concentration when Ca2+o is falling than when it is rising (e.g., hysteresis is occurring in this relationship).134,135 The latter results in an apparent direction dependence of the secretory response, which when combined with the rate dependence just described, may allow for a physiologically appropriate, more vigorous secretory response to large rapid decrements in Ca2+o. Also present are circadian136 (for review, see Diaz et al.26) and more rapid (i.e., occurring at rates of one to six pulses per hour) phasic changes in circulating PTH levels,26,137 but the physiologic significance of these changes is not known.
Mechanism of Ca2+o Sensing by Parathyroid Cells and Other Cells Involved in Mineral Ion Homeostasis
The molecular mechanism underlying Ca2+o-regulated PTH secretion involves a G protein–coupled, cell surface Ca2+o-sensing receptor (CaSR).138 The CaSR was first isolated from bovine parathyroid glands139 and subsequently from human parathyroid and several other tissues and species.140 The receptor exhibits the characteristic “serpentine” motif (seven membrane spanning domains) of the superfamily of G protein–coupled receptors (Fig. 1-7). Its long, N-terminal extracellular domain contains the major, but not all, determinants of Ca2+o binding.141–143 Changes in Ca2+o modulate a number of second messenger systems via coupling of CaSR through its intracellular domains to the relevant G proteins regulating these signaling pathways.138 These functions include activation of phospholipases C, A2, and D,144 stimulation of several mitogen-activated protein kinases,145 and inhibition of adenylyl cyclase.146 Despite numerous studies conducted over the past 25 years, a full understanding of the major second messenger pathways through which changes in Ca2+o, acting via the CaSR, regulate various aspects of the function of parathyroid and other CaSR-expressing cells remains elusive (discussed later). Recent evidence, however, indicates key roles for the G proteins, Gq and G11, but their downstream transduction pathways participating in the control of parathyroid function are uncertain.147 In the parathyroid, the CaSR mediates the inhibitory actions of Ca2+o on PTH secretion and gene expression as well as parathyroid cellular proliferation.138,148
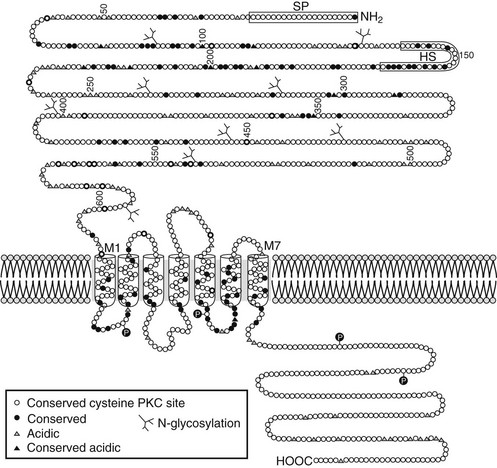
FIGURE 1-7 Schematic representation of the predicted topology of the calcium-sensing receptor cloned from human parathyroid gland. HS, Hydrophobic segment; SP, signal peptide;  , acidic residues (
, acidic residues (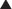 , conserved among species); branched markers, sites of glycosylation; P, sites of phosphorylation. (From Brown EM, Gamba G, Riccardi D, et al: Cloning and characterization of an extracellular Ca2+-sensing receptor from bovine parathyroid, Nature 366:575–580, 1993.)
, conserved among species); branched markers, sites of glycosylation; P, sites of phosphorylation. (From Brown EM, Gamba G, Riccardi D, et al: Cloning and characterization of an extracellular Ca2+-sensing receptor from bovine parathyroid, Nature 366:575–580, 1993.)
The CaSR is also expressed in several additional tissues involved in systemic mineral ion homeostasis, including the calcitonin-secreting C-cells of the thyroid,149 diverse cells within the kidney,150 bone cells and/or their precursors,110 and intestinal epithelial cells. In the C cell, the CaSR mediates the stimulatory action of high Ca2+o on calcitonin secretion, a Ca2+o-lowering hormone. In the kidney, the CaSR in the cortical thick ascending limb of the nephron mediates direct high-Ca2+o-induced inhibition of the tubular reabsorption of Ca2+ and Mg2+.150,151 Therefore, raising Ca2+o both directly inhibits renal tubular reabsorption of Ca2+ via actions on the CaSR expressed in nephron segments involved in hormonal regulation of Ca2+ reabsorption (e.g., by PTH) and indirectly inhibits it by reducing PTH secretion (see the later section, Renal Calcium Reabsorption). The CaSR probably also mediates the long-recognized but poorly understood inhibitory effect of hypercalcemia on renal water conservation, probably exerting this action by inhibiting vasopressin-stimulated water flow in the distal collecting duct.138 A possible physiologic relevance of this action is to prevent the development of excessively high concentrations of calcium in the distal collecting system, thereby perhaps mitigating the risk of renal stone formation.151 Elevating Ca2+o stimulates osteoblastic bone formation and inhibits osteoclastic bone resorption.110,152 The CaSR is expressed by chondrocytes as well as by osteoblasts and osteoclasts and/or their precursors,110 and recent data suggest that it plays key roles in skeletal development.153 It is not currently known whether the CaSR expressed in intestinal epithelial cells plays any role in regulating 1,25(OH)2D3-mediated absorption of calcium.
The identification of hypercalcemic (e.g., familial hypocalciuric hypercalcemia and neonatal severe hyperparathyroidism)154 and hypocalcemic (i.e., autosomal-dominant hypocalcemia)155 disorders caused by inactivating and activating mutations of the CaSR, respectively, has provided incontrovertible proof of the receptor’s central, nonredundant role in setting the serum calcium concentration.156,157 Patients with these disorders have characteristic abnormalities in parathyroid and renal Ca2+ sensing/handling that have clarified the receptor’s normal role in these tissues, outlined previously. Targeted disruption of the CASR gene has also enabled the generation of mouse models of familial hypocalciuric hypercalcemia and neonatal severe hyperparathyroidism via inactivation of one or both alleles of the CaSR158, further supporting its importance in Ca2+o homeostasis.
1,25(OH)2D3, Phosphate, and Other Factors Regulating PTH Secretion
In addition to directly inhibiting PTH gene expression, 1,25(OH)2D3 also reduces PTH secretion159,160 (for review, see11,26,118). It is not known whether this latter action is solely secondary to the effect of 1,25(OH)2D3 on biosynthesis of the hormone and/or represents a direct action on the secretory process per se. Increasing the ambient level of phosphate in vitro, independent of concomitant changes in Ca2+o, enhances parathyroid cellular proliferation, PTH gene expression, and hormonal secretion.161–163 Phosphate-induced changes in PTH secretion, however, take several hours and may result secondarily from changes in hormonal biosynthesis rather than secretion per se.163 Finally, Mg2+o clearly functions as a CaSR agonist in vitro when tested in cells containing an endogenous CaSR164 (e.g., parathyroid cells) or expressing the cloned CaSR,139 although it is twofold to threefold less potent than Ca2+o on a molar basis. Because levels of serum ionized Mg2+o are lower than those of Ca2+o, it is presently unclear whether Mg2+o acts as a physiologically relevant CaSR agonist at the parathyroid gland in vivo under normal circumstances. Patients with inactivating or activating CaSR mutations, however, can exhibit mild hypermagnesemia or hypomagnesemia,156 respectively, thus suggesting that the CaSR does contribute to setting Mg2+o in vivo, as previously suggested.165 It may do so, at least in part, in the kidney, where Mg2+o in the tubular fluid of the thick ascending limb exceeds that in blood and may be sufficient to activate the CaSR that regulates tubular reabsorption of Ca2+o and Mg2+o in this nephron segment.138,151,166,167 In addition to the inhibitory effect of elevated Mg2+o on PTH secretion, low concentrations of Mg2+o—as in patients with overt magnesium deficiency—also reduce PTH secretion.168 The mechanism(s) underlying this effect of hypomagnesemia has recently been suggested to involve increased activity of G proteins to which the CaSR normally couples, probably Gi and Gq/11, thereby leading to increased intracellular signaling and inhibition of PTH secretion.169
Metabolism of Parathyroid Hormone
Studies performed over more than 3 decades by several laboratories have focused on the heterogeneity of circulating forms of PTH, which was first identified by Berson and Yalow in 1968 (Fig. 1-8).170 From these investigations, it is now apparent that in addition to the full-length polypeptide PTH(1-84), which is the biologically active hormone, much of the circulating hormone lacks an intact N-terminus, and most of these fragments are thus devoid of biological activity, at least with regard to the PTH/PTHrP receptor-mediated regulation of mineral ion homeostasis.25 C-terminal PTH fragments are produced in and released from the parathyroid gland, but they are also derived from circulating intact hormone by efficient, high-capacity degradative systems in the liver and kidney and most likely at other peripheral sites (Fig. 1-9). However, some PTH fragments, such as PTH(7-84), appear to be generated within the gland and to have some biological activity.171–173
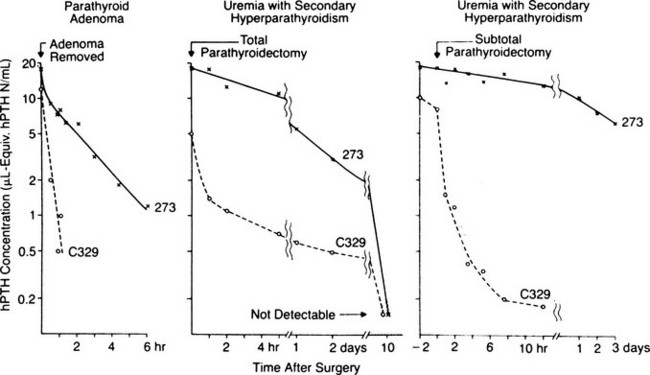
FIGURE 1-8 Disappearance of immunoreactive parathyroid hormone (PTH) from plasma after parathyroidectomy in patients with primary or secondary hyperparathyroidism. Plasma samples were assayed with antiserum C329 and antiserum 273, with an extract of a normal human parathyroid gland used as a standard (hPTH N) and 125I-bPTH used as a tracer. Plasma concentrations of hormone are given as microliter equivalents of the plasma standard of hPTH (see the text). (Data from Berson SA, Yalow RS: Immunochemical heterogenicity of parathyroid hormone in plasma, J Clin Endocrinol Metab 28:1037–1047, 1968.)
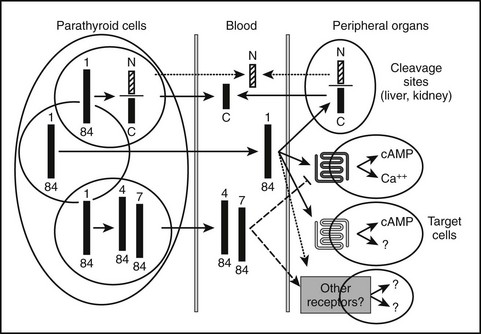
FIGURE 1-9 Scheme of parathyroid hormone (PTH) cleavage and the interaction of PTH with the PTH/PTH-related peptide receptor (PTH1R) and with other putative receptors on target cells. Within parathyroid cells, secretory vesicles (circles) and in peripheral organs various patterns of cleavages of PTH (1–84) occur, resulting in multiple circulating fragments including N-terminal (N) (diagonal stripes), suggesting rapid degradation, and C-terminal fragments (C) (solid bars). The carboxyl end of intact PTH and possibly some C fragments interact with a putative C-terminal receptor and, as well, inhibit actions of PTH on the PTH1R. cAMP, Cyclic adenosine monophosphate.
Direct measurement of arterial and venous differences in parathyroid effluent blood (with vigorous conditions to prevent any ex vivo cleavage of hormone after sample collection) were performed in cattle and confirmed that smaller C-terminal fragments and intact hormone, but not N-terminal fragments, are secreted into the circulation. The relative concentration of these C-terminal fragments released from the gland increases under conditions of systemic hypercalcemia, when overall secretion rates of intact hormone are lower.105,174 The C-terminal PTH fragments are similar to those generated by peripheral metabolism (see later) but were not chemically characterized.
The peripheral metabolism of PTH has been analyzed by injecting intact hormone into the circulation of test animals. Such experiments have not been performed in human subjects, but it is assumed that the similar metabolism of PTH in rats, dogs, and cows is reflective of its metabolism in humans.175 Clearance of intact PTH from plasma was found to be very rapid (half-life, 2 to 4 minutes),176,177 the major sites of clearance being the liver and kidney. Clearance by the liver predominates over clearance by the kidney; the two organs together account for virtually all clearance of intact hormone. Hepatic clearance of intact hormone has been estimated to be 40% to 75% and renal clearance 20% to 30%.25
In summary, current evidence indicates that intact PTH and multiple C-terminal fragments (which are derived from glandular and peripheral cleavage and may not be identical) are the principal circulating hormonal forms (however, recent data suggest the presence of a previously unrecognized large N-terminally truncated form of the hormone [discussed later]). Biologically active N-terminal fragments of PTH, if found in the circulation at all, are likely to circulate only at extremely low concentrations (<10-13 to 10-14 mol/L). More recent in vivo evidence regarding the renal clearance and metabolism of intact PTH (as distinct from C-terminal fragments) indicates a peritubular uptake process, rather than glomerular filtration followed by uptake from the tubular lumen and subsequent cleavage.178 Furthermore, other studies indicate that megalin, a multifunctional endocytic receptor expressed in the proximal renal tubules, can mediate the reuptake and subsequent degradation of the fraction of PTH that is subject to glomerular filtration.179 Megalin-mediated uptake depends on an intact N-terminus of PTH; C-terminal fragments that are eliminated by glomerular filtration are not recognized by megalin. The potential significance, quantitatively and biologically, of glomerular filtration and megalin-mediated uptake of intact PTH therefore remains uncertain. However, megalin-ablated mice excrete fourfold more N-terminal PTH in the urine than do wild-type animals.179
Because of the lack of evidence of circulating forms of biologically active, N-terminal fragments, it was feasible to introduce immunometric assays that use two different antibodies: an immobilized capturing antibody directed against the C-terminal portion of PTH(1-84) and a radiolabeled or enzyme-labeled detection antibody directed against an epitope within the N-terminal portion of the intact molecule180,181 (see Fig. 1-9).
Although these two site assays have been clinically useful and provided a great improvement over earlier assays,180 recent studies have demonstrated the presence of circulating PTH fragments that are different in character and composition from any of the hormone fragments discussed previously.108,128,182 The studies that led to the detection of these PTH species were at least partly stimulated by the clinical observations that two-site immunometric assays to measure intact PTH frequently gave high levels of nonsuppressible PTH in patients with end-stage renal disease, yet the patients had clinical and/or histologic evidence of a dynamic bone disease108,183–186 (see Chapter 14).
High-performance liquid chromatography analysis of blood samples from such uremic patients and from patients with other forms of hyperparathyroidism revealed two distinct immunoreactive peaks in column effluent that were detectable (although with differing sensitivity) by different commercial assays. One peak corresponded to intact PTH(1-84), whereas the other peak migrated close to the position occupied chromatographically by synthetic PTH(7-84).108,128 This finding suggested that the epitope of the detection antibody in these assays did not require the presence of the first six or more amino acids of PTH(1-84).128,129 The results were interpreted as being consistent with the view that the molecular entity or entities detected besides PTH(1-84) are N-terminally truncated forms of the intact molecule that are similar, but not necessarily identical, to synthetic PTH(7-84).128 In fact, more detailed chromatographic studies performed with one of these “intact” PTH assays showed significant variation in the ratio of the N-terminally truncated PTH to PTH(1-84). Individuals with normal renal function showed a lower ratio than did patients with uremia; furthermore, the percentage of immunoreactivity representing N-terminally truncated forms of PTH (PTH[7-84]) rose in both groups when hypercalcemia was present.108
The newer immunoradiometric assays that have N-terminal epitopes at the extreme amino-terminus of PTH(1-84) show significantly lower PTH concentrations in patients with chronic renal failure during stimulation and suppression of glandular activity with alterations in calcium, a result consistent with the conclusion that only full-length and therefore biologically active PTH(1-84) is detected by these assay systems.173,182,187
The findings outlined previously were expected to have considerable significance for the management of patients with parathyroid dysfunction, especially in the presence of renal failure. Treatment of patients with end-stage renal disease with large amounts of vitamin D and/or calcium (or calcimimetics) has been associated with adynamic bone disease, which can be deleterious, particularly in growing children.188–190 Particularly during hypercalcemia, N-terminally truncated forms of PTH become a significant, if not the dominant, PTH species.191,192 Measurement of “intact” PTH by earlier assays180 therefore overestimated the concentrations of biologically active PTH and could result in the overtreatment of uremic patients with vitamin D-analogs and/or calcium. The resulting suppression of the parathyroid gland, even without evoking an inhibitory effect of a fragment similar to PTH(7-84), could be acting together to excessively reduce PTH-dependent bone turnover. When secretion of PTH is suppressed, as in hypercalcemia, and the fractional concentration of the large N-truncated PTH increases, however, inhibition of the actions of native PTH on calcium or bone could occur. Recent experiments with synthetic PTH(7–84) support the conclusion that such actions occur in vivo and in vitro. For example, PTH(7-84) was shown to have hypocalcemic properties in vivo,171,173 and it reduces, presumably via receptors specific for C-terminal PTH fragments, bone resorption that may be partly due to impaired osteoclast differentiation.109,171 It is not yet established clinically whether the newer radioimmunoassays that do not detect large but amino-terminally truncated PTH molecules will have a decisive advantage in diagnosis and management of primary and secondary hyperparathyroidism187 (see Chapters 7 and 14).
By contrast to the extensive metabolism of PTH, recent evidence suggests that intact FGF23, which is most likely the most important phosphate-regulating hormone,23 does not appear to undergo significant metabolism.193 In fact, immunoreactive FGF23, which can be extraordinarily elevated in patients with end-stage renal disease, appears to be largely intact and biologically active. This makes it unlikely that FGF23, unlike PTH, is metabolized in peripheral organs.
PTH-Dependent Regulation of Mineral Ion Homeostasis Mediated Through the PTH/PTHrP Receptor (Type 1 Receptor)
Renal Calcium Reabsorption
Most of the calcium in the glomerular filtrate is reabsorbed. The bulk of this reabsorption (65%) occurs via passive, paracellular mechanisms, both in the proximal tubules and, to a lesser extent, in the thick ascending limb of Henle’s loop and the distal convoluted tubule.194–198 As noted by Diaz and colleagues,26 the calcium-sensing receptor plays an important PTH-independent role in the adjustment of renal calcium reabsorption in the cortical thick ascending limb. The physiologically important stimulation of renal calcium reabsorption by PTH occurs almost entirely in the distal nephron. In the cortical thick ascending limb, PTH increases the magnitude of the lumen positive potential that drives the passive paracellular reabsorption of calcium and magnesium. In the distal convoluted tubule, in contrast, PTH promotes increased transcellular Ca2+ reabsorption by coordinate up-regulation of molecular components of this transcellular pathway.199 PTH enhances uptake of Ca2+ into the tubular cells via the (luminal) plasma membrane channel, TRPV5,200,201 as well as its active extrusion against a steep electrochemical gradient at the basolateral membrane. This latter process of extrusion involves two types of transporters. One is the plasma membrane calcium pump (Ca2+, Mg2+-ATPase, PMCA) and the second is a Na+/Ca2+ exchanger (NCX1), which in turn is indirectly regulated by NA+/K−-ATPase(s), which maintains the transcellular Na+ gradient. Studies performed with membrane vesicles from the distal region of the kidney show increased activity of the Na+/Ca2+ exchanger in response to PTH.202 PTH also stimulates the translocation of preformed Ca2+ channels sequestered within the interior of certain distal tubular cells, presumably TRPV5, apical surface (Fig. 1-10),203 which translocate to the apical (i.e., luminal) surface of the renal tubular epithelial cell and mediate increased cellular Ca2+ uptake. Because PTH simultaneously enhances the activity of Na+/Ca2+ exchangers in the basolateral (antiluminal) membrane, the overall process promotes an increase in transcellular Ca2+ uptake from the lumen to blood, that is, from the apical to the basolateral membrane.
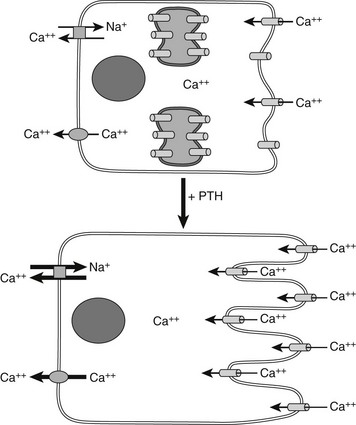
FIGURE 1-10 PTH regulation of calcium transport in renal distal tubule cells. Parathyroid hormone (PTH) triggers the translocation of preformed voltage-dependent calcium channels from sites of intracellular sequestration to the apical membrane, which also undergoes rapid morphologic changes that greatly increase its surface area. Intracellular free calcium levels rise significantly, and increased net transepithelial calcium transport occurs, mainly via enhanced Na+/Ca2+ exchange at the basolateral membrane, supported in turn by Na+/K+-ATPase.
Regulation Of 1α- and 24-Hydroxylase Activity
PTH is a major inducer of the activity of proximal tubular 1α-hydroxylase, a microsomal cytochrome P-450 enzyme that synthesizes biologically active 1,25(OH)2D3 from the substrate 25(OH)D3.204–206 This effect of PTH on synthesis of the renal enzyme shows longer lag times than its effect on renal Ca2+ transport and is mediated, at least in part, by the protein kinase A (PKA) signaling pathway of the PTH/PTHrP receptor.207,208 Although 1α-hydroxylase activity has been detected in several nonrenal tissues, its mRNA was found in abundant concentrations only in the kidney, thus confirming that this organ is the most important site for the generation of 1,25(OH)2D3. PTH-dependent synthesis of new 1α-hydroxylase protein requires several hours and is blocked by 1,25(OH)2D3 and actinomycin D, a blocker of protein synthesis.208–210 PTH administration increases the mRNA encoding the 1α-hydroxylase.208 Hypophosphatemia is, similar to PTH, a major inducer of 1α-hydroxylase, whereas hypercalcemia, as would be generated by sustained increases in circulating levels of PTH or PTHrP, suppresses synthesis of the enzyme, thus limiting overall 1,25(OH)2D3 synthesis in a homeostatic manner. The molecules 25(OH)D3 and 1,25(OH)2D3 can also be hydroxylated by the 24-hydroxylase, but the resulting metabolites, 24,25(OH)2D3 and 1,24,25(OH)3D3, appear to have no major role in the regulation of mineral ion homeostasis (see Chapter 3). However, PTH has an inhibitory effect on the 24-hydroxylase, thus reducing the inactivation of 1,25(OH)2D3; in contrast, 1,25(OH)2D3 stimulates the synthesis of 24-hydroxylase, thereby inducing its own metabolism.211,212
Renal Phosphate Transport
PTH is of major importance for maintaining normal blood calcium levels, but it is not the principal regulator of the serum phosphate concentration, which appears to depend on specific phosphaturic factors, particularly FGF-23 (see Chapter 6).19,23,213 However, when PTH causes an increase in bone resorption (as might occur with prolonged dietary calcium deprivation), calcium and phosphate increase simultaneously in the blood. Although calcium is needed, phosphate is best excreted, which is mainly accomplished by a PTH-stimulated increase in renal phosphate clearance.
PTH acts directly on proximal tubular cells, where it regulates expression of NPT2a and NPT2c in the brush border membrane. Whereas NPT2a is expressed in segments S1 through S3 of the proximal tubule, NPT2c is expressed only in the S1 segment. NPT2a protein undergoes internalization in response to PTH,214 followed by lysosomal degradation215; in contrast, recent evidence suggests that NPT2c can be recycled and reinserted into the brush border membrane.216 In the proximal renal tubules, PTH furthermore enhances the production of biologically active 1,25(OH)2 vitamin D.217–220 All these actions of PTH are mediated through the PTH/PTHrP receptor (PTH1R), which is expressed at the basolateral membrane (BLM) and at much higher levels at the apical brush border membrane (BBM) of the proximal renal tubules.217,221–223
The PTH-dependent renal actions probably involve cAMP/PKA-dependent and Ca2+/IP3/PKC-dependnent signaling events at the BLM, and both pathways appear to contribute to the reduction in proximal tubular phosphate reabsorption.217,218,220 In contrast to these dual signaling properties of the PTH1R at the BLM, there is considerable evidence to suggest that the inhibition of phosphate uptake mediated through the PTH1R at the BBM involves predominantly, if not exclusively, a pertussis-toxin-sensitive, PKC-dependent pathway.217,224–227 PTH thus activates both major signaling pathways via PTH/PTHrP receptors located at either the BLM or the BBM, and it induces phosphaturia by reducing expression of the type II sodium-phosphate cotransporters, NPT2a and NPT2c.
Activation of the cAMP/PKA pathway down-stream of the PTH1R is undoubtedly involved in the PTH-dependent regulation of NPT2a expression.226–228 In fact, patients affected by pseudohypoparathyroidism type Ia (PHP-Ia), a disease caused by inactivating mutations in GNAS, the gene encoding the alpha subunit of the stimulatory G protein (Gsα), develop hyperphosphatemia due to a lack of functional Gsα in the proximal tubules and consequently show impaired urinary cAMP and phosphate excretion in response to PTH (see Chapter 6). However, the phosphaturic response stimulated by PTH is not totally absent, since PHP-Ia patients have a small but delayed increase in urinary phosphate excretion after challenge with PTH, which suggests that signaling molecules other than cAMP could also be involved in promoting phosphate excretion.229–231
Besides regulating NPT2a expression, PTH also affects expression of the second kidney-specific sodium-dependent phosphate cotransporter, namely NPT2c.227 The mRNA encoding NTP2c is about 10-fold less abundant than that encoding NPT2a, which initially suggested that NTP2c plays only a minor biological role limited to some period during postnatal development.232 It was then found, however, that homozygous and compound heterozygous mutations in NPT2c cause hereditary hypophosphatemic rickets with hypercalciuria (HHRH), an autosomal-recessive disorder.233–236 This disease linkage makes it certain that NPT2c serves important functions in biology and is not redundant with NPT2a.218–220 Nevertheless, it is so far unknown how the two transporters differ in their functional roles. Both proteins are internalized at the BBM in response to treatment with PTH, but the time courses of these agonist-dependent processes differ, raising the possibility that different scaffolding proteins regulated by PTH underlie the differential regulation of NPT2a and NPT2c. Furthermore, activation of different signaling pathways downstream of the PTH1R—namely, the cAMP-dependent activation of PKA or the Ca2+/IP3-dependent or Ca2+/IP3-independent activation of PKC—may have different roles in the regulation of NPT2a and NPT2c expression (Fig. 1-11).
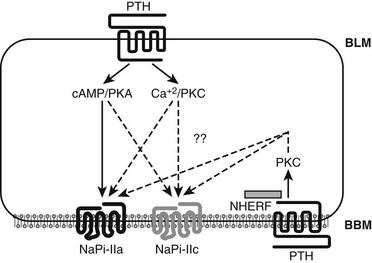
FIGURE 1-11 PTH regulation of renal phosphate transport. A proximal convoluted tubule cell is depicted. The type II sodium-dependent phosphate cotransporters NaPi-IIa and NaPi-IIc (also referred to as NPT2a and NPT2c) reabsorb phosphate from the tubule lumen. NHERF-1 promotes apical localization of NaPi-IIa (but not NaPi-IIc) and establishes a regulatory complex for PTH-mediated inhibition of phosphate reabsorption. PTH inhibits phosphate reabsorption by reducing expression of both transportes. PTH1 receptors (PTH1R) located on both the basolateral and luminal surfaces reduce NaPi-IIa and NaPi-IIc expression via signaling pathways involving protein kinase A (cAMP/PKA) (solid arrow) and other pathways (dotted arrows), still less characterized, including PLC/PKC. (Courtesy Dr. Matthew Mahon.)
These differences may involve “signalsomes,” that is, specialized intracellular domains in which proteins involved in a specific biochemical process or pathway are brought into close proximity by binding to “scaffold” proteins, thereby limiting their diffusion and altering receptor-mediated downstream signaling pathways.237 In renal proximal tubules, the sodium/hydrogen exchanger regulatory factors 1 and 2 (NHERF1/2) are prominent scaffold proteins that contain two PDZ (psd-95, discs large, ZO-1)-binding domains and a C-terminal ERM (ezrin, radixin, moesin)-binding motif (see review238). The PTH1R interacts with NHERF proteins through a C-terminal PDZ-binding motif 239,240 (see Fig. 1-11), which leads in some cell lines to NHERF-dependent suppression of PTH-stimulated cAMP accumulation.239 In opossum kidney cells (OK), NHERF1 enhances PTH1R signaling through intracellular calcium and PLC.240 In fact, anchoring the receptor to the BBM of proximal tubules appears to promote signaling through the IP3/PKC pathway, illustrating how domain-specific factors can influence receptor-dependent signaling.
Similar to the PTH1R, NPT2a also contains a C-terminal motif that binds to PDZ domains of NHERF1,241 and NHERF1-null mice display phosphate wasting due to a reduction in NPT2a expression at the BBM of proximal tubules. Thus, this transporter requires a NHERF1-assembled scaffold for proper membrane expression.242 Furthermore, NHERF1 has been shown to be an essential factor for PTH-mediated regulation of NPT2a expression in OK cells and in primary proximal tubular cell cultures isolated from NHERF1-null mice. Combined, these findings demonstrate that NHERF1 not only establishes NPT2a surface expression but also aids in the formation of a regulatory complex necessary for PTH-elicited phosphaturia. In vitro, NHERF1-assembled complexes consisting of PTH1R and NPT2a are present in apical domains of OK cells and in genetically modified LLC-PK1 cells developed for studying phosphate transport.243
The PTH fragment peptide, PTH(3-34), has been frequently used in efforts to discern the signaling events at the BBM from those at the BLM which regulate NPT2a and NPT2c; some data suggest that this analog may selectively activate the PKC signaling pathway244–246 and thus should act only at the BBM. There are, however, no data demonstrating that PTH(3-34) can directly activate the PLC-dependent formation of IP3, and in fact substitutions or deletions at positions 1 and 2 of PTH result in markedly and/or selectively impaired activation of the PLC/IP3/Ca2+ signaling response.58–60,247 PTH(3-34) has been shown to activate PKC via a non-PLC/IP3-dependent mechanism, perhaps involving another phospholipase such as PLA2.246 In any case, several investigators have shown that PTH(3-34), as well as other PTH and PTHrP fragments truncated at the amino-terminus, can inhibit the uptake of phosphate by opossum kidney cells (which do not express significant amounts of NPT2c) in the apparent absence of significant cAMP accumulation.248–252 Furthermore, PTH(3-34) was shown to promote, at least partially, urinary phosphate excretion in animals.253 This raises the possibility that PTH fragments such as PTH(7-84) and PTH(4-84), which are found in the circulation of patients with primary or secondary hyperparathyroidism, could induce phosphaturic effects.172,191,192,254,255 PTH(7-84), however, does not appear to have phosphaturic activity when tested in vivo, or in some in vitro systems. In addition, PTH(3-34) can activate at least partially the cAMP-dependent PKA in cells.256,257 This makes it plausible that the capacities of amino-terminally truncated PTH or PTHrP analogs to promote the urinary excretion of phosphate arise at least to some extent from partial activation of the cAMP/PKA pathway at the BLM.
As mentioned, PTH treatment leads to the reduction of NPT2a and NPT2c in the kidney, but effects of the hormone on the two cotransporters appear to differ, as shown in thyro-parathyroidectomized rats.218–220,227 NPT2a is expressed in the segments S1-S3 of the proximal renal tubules, whereas NPT2c is present only in S1.218–220,227 NPT2a is the more abundantly expressed cotransporter, and it handles (in the mouse) 70% to 80% of renal phosphate reabsorption, with NPT2c accounting for the remaining 20% to 30%. In response to PTH, NPT2a disappears rapidly from the BBM and translocates to lysosomes where it undergoes degradation, whereas NPT2c disappears from the BBM surface at a much slower rate and does not seem to undergo lysosomal degradation but rather may recycle back to the BBM surface.216 The mechanisms underlying these diverse response profiles are uncertain at present, but the generation of engineered LLC-PK1 cells that show PTH-dependent inhibition of phosphate transport via reductions in either NPT2a or NPT2c expression243 will likely lead to important new insights into the molecular and cellular processes involved.
Actions of Parathyroid Hormone On Bone
PTH affects a wide variety of the highly specialized bone cells, including osteoblasts, osteoclast/stromal cells, and osteocytes, the latter being the most numerous cell type in bone. Some of these effects reflect direct actions of PTH; others are indirect and mediated in an autocrine/paracrine manner through factors released by cells (osteoblasts) expressing PTH/PTHrP receptors that regulate the activity of yet other cells (osteoclasts) that lack these receptors.258–260
PTH action on bone cells can be considered from at least five distinct perspectives. First is its major physiologic role—the maintenance of calcium homeostasis. As noted in Fig. 1-2, this action can be analyzed most clearly in experimental animals through parathyroidectomy, with resultant hypocalcemia. Renal losses of calcium are significant contributors to the severe, sometimes fatal hypocalcemia, but the loss of PTH action on bone cells is the predominant cause. The traditional explanation is that the hypocalcemia is due principally to the loss of PTH stimulation of osteoclastic bone resorption, but other mechanisms may be involved (see later). A second perspective is the action of PTH when administered pharmacologically, resulting in elevation of serum calcium, immediately preceded by a short-lived fall in calcium. It is still unclear which specific cellular actions explain the physiologic actions known to follow PTH administration to animals in vivo. Earlier studies had shown that the administration of PTH leads within minutes to a transient lowering of blood calcium caused by uptake of the mineral by bone cells,261 which is rapidly followed by increased mobilization of calcium from the mineral phase into the bloodstream.261,262 Although there continues to be uncertainties regarding the cellular/anatomic basis of these rapid responses to PTH, considerable progress has been made in elucidating the mechanisms through which bone cells respond to PTH and to other autocrine/paracrine factors such as cytokines (see Chapters 4 and 12). A major pathway for calcium release from bone involves osteoclasts; these cells undergo multiple cellular changes involving the activation of cellular transporters and pumps, as well as the secretion of enzymes such as cathepsin K and collagenases, but other mechanisms of calcium release are proposed.261,262
A third perspective regarding PTH action on bone cells can be appreciated through results seen when PTH is used as a therapy for osteoporosis. Chronic administration of PTH, if given intermittently, stimulates bone formation, an action that involves a complex set of cellular responses in bone, affecting principally stromal cell/osteoblast proliferation, differentiation, and cellular actions.263–267 It is also clear that actions on osteocytes are involved in PTH actions on bone267,268; together, PTH effects on the two cell types, osteoblasts and osteocytes, stimulate bone matrix formation and bone mineral deposition, as well as bone mineral mobilization (see Chapters 4 and 5).
Fourthly, pathophysiologic actions of PTH are seen in hyperparathyroidism with chronic excess levels of PTH. In this situation, both bone formation and bone resorption are stimulated, the latter effect being predominant (see Chapters 4 and 7).
Effects on Osteoblasts
Osteoblasts express the PTH/PTHrP receptor abundantly and show vigorous responses to the hormone. PTH stimulates the generation of cAMP, inositol triphosphate, and diacylglycerol in cells expressing the cloned PTH/PTHrP receptor.8,9,269 A remarkable number of cellular activities of osteoblasts are influenced by PTH, including cellular metabolic activity, ion transport, cell shape, gene transcriptional activity, and secretion of multiple proteases (see Chapters 4 and 12). Continuous PTH administration in vivo results in decreased bone mass, whereas intermittent administration of PTH leads to an increase.25,270,271 Osteoblast synthetic activity is strongly stimulated in both situations, but osteoblast/stromal cell increased production of RANK ligand and suppression of osteoprotegerin secretion272,273 dominate through stimulation of osteoclastic activity in chronic PTH administration.
PTH administration leads to multiple osteoblastic responses, including reduced rates of cell apoptosis,263 increased expression and activity of Runx 2,274,275 and increased rates of osteoblastic cell differentiation.264,276
The exact cellular mechanisms whereby intermittent PTH administration selectively enhances bone formation remain unclear at present, as are correlations between in vivo and in vitro responses. Further exploration of these mechanisms is clearly of great interest for understanding the hormone’s therapeutic potential as a bone anabolic agent.277
Effects on Osteoclasts
It is generally agreed that osteoclasts lack PTH receptors, and hence actions of PTH are indirect, largely through stimulation of cytokines elaborated from osteoblasts/stromal cells. These molecules include osteoclast-differentiating factor, now usually called RANK-L,278,279 a membrane-associated protein with homology to the family of tumor necrosis factors (TNFs) that induces—upon cell-to-cell contact and in the presence of macrophage colony-stimulating factor—the differentiation of osteoclast precursors into mature bone-resorbing osteoclasts.280–282 These effects of RANK-L are likely to be mediated through RANK, a member of the TNF receptor family that is expressed on osteoclast precursors.279,280 However, RANK-L also interacts with osteoprotegerin, a soluble decoy receptor with homology to the TNF receptor family.283 Transgenic expression of osteoprotegerin in mice leads to impaired osteoclastogenesis and thus to osteopetrosis,284,285 whereas ablation of the osteoprotegerin gene through homologous recombination in mice results in osteoporosis associated with arterial calcifications.286 Ablation of the gene for RANK-L results in severe osteopetrosis due to lack of osteoclasts, a defect in tooth eruption, and a complete lack of lymph nodes, but without obvious abnormalities in mineral ion homeostasis.274 Similarly, ablation of the RANK-L receptor (RANK), leads to osteopetrotic changes similar to those observed in RANK-L-ablated mice, but also to hypocalcemia and secondary hyperparathyroidism and to renal phosphate wasting.275 It appears plausible that challenging the homeostatic control mechanisms of either of these knockout mice, in particular, through dietary calcium and vitamin D deficiency will further aggravate the degree of hypocalcemia and urinary phosphate excretion. However, despite the absence of such experimental data, the outlined findings further illustrate the importance of osteoclastic bone resorption for maintaining blood calcium concentrations.
Some of the factors previously noted to have an important role in the paracrine stimulation of osteoclast formation, such as interleukin 6, interleukin 11, prostaglandin E2, 1,25(OH)2D3, and other peptides, including PTH,287–289 were shown to directly stimulate the production of RANK-L by osteoblasts.290 Other cellular responses involved in bone resorption include the development of vitronectin-mediated anchorage of osteoclasts to the bone surface, acidification of the circumscribed and sealed-off extracellular environment that is created between the osteoclast and bone, and in addition, the secretion of a variety of proteases and other enzymes (see Chapter 4).
Effects on Osteocytes
Osteocytes, long neglected in bone cell biology, are the most numerous cell type in bone, and there is a growing appreciation that they play a central biological role with a myriad of functions ranging from transducing mechanical signals into bone growth and increased bone mineral density to hitherto completely unappreciated roles in phosphate homeostasis.272,273,277
Osteocytes contain abundant parathyroid hormone receptors, and there is now a clearly defined, biologically significant series of responses to parathyroid hormone action that have been described.266–268,291 Chapters 4, 5, and 6 outline the roles of the osteocyte in bone turnover and the complex, still incompletely defined role of multiple interacting factors within osteocytes or secreted by them, including matrix extracellular phosphoglycoprotein (MEPE), dentin matrix protein 1 (DMP-1), and fibroblast growth factor 23 (FGF-23), in the regulation of renal phosphate clearance.23,292
The clearly defined effect of PTH to reduce sclerostin (SOST) production by osteocytes in turn enhances osteoblastic activity by blocking the suppressive effects of SOST on osteoblast activity and the Wnt signaling pathway.266–268 Chapter 4 provides a more detailed review of the role of osteocytes as mechanosensors and sources of humoral factors in bone biology (see also Chapter 6).
Parathyroid Hormone–Related Peptide
Analysis of the physiologic actions of PTH and the molecular basis of its biological activity requires consideration of the functions of PTHrP. This peptide, discovered and characterized several decades after discovery of PTH, undoubtedly shares an evolutionary origin with PTH. PTHrP has (see Fig. 1-3), at least in mammals, both chemical and functional overlaps with PTH, but many of its biological roles are quite different. When secreted in large concentrations (e.g., by certain tumors), PTHrP has PTH-like properties. Typically, however, it functions as an autocrine/paracrine rather than an endocrine factor. PTHrP is a larger, more complex protein than PTH and is synthesized at multiple sites in different organs and tissues. The still-evolving story of this protein and its proteolytic fragments is beyond the scope of this chapter (for comprehensive review, see4,6,293). However, selective functions in the regulation of mineral ion homeostasis and bone development are reviewed here.
A substance with biological properties similar to those of PTH was first proposed in the early 1940s, when Albright discussed a patient with malignancy-associated humoral hypercalcemia.294 The clinical and biochemical characterization of similarly affected patients subsequently established the syndrome of humoral hypercalcemia of malignancy295 and eventually led to the amino acid sequence analysis and molecular cloning of PTHrP from several different tumors.296–298 It is now generally accepted that PTHrP is the most frequent humoral cause of hypercalcemia in malignancies.4,293,299 PTHrP interacts with the same receptor used by PTH, and when large amounts of the peptide are released from certain tumors, it mimics some or all of the effects of excess PTH. However, PTHrP is also expressed in a remarkable variety of normal fetal and adult tissues, 4,293,298,300–304 which suggested soon after its discovery that it has additional biological role(s) that are unrelated to calcium and phosphorus homeostasis and that these role(s) are distinct from those mediated by PTH. One of its most prominent functions is the regulation of chondrocyte proliferation and differentiation and consequently bone elongation and growth.12,13,305,306
The PTHrP Gene In Comparison To The PTH Gene
The human PTHrP gene is located on chromosome 12p12.1-11.2,298 which has a region analogous to that containing the human PTH gene on chromosome 11p15.83–85 Both the PTH and the PTHrP genes have a similar organization, including equivalent positions of the boundaries between some of the coding exons and the adjacent introns11,87,298,307 (Fig. 1-12).
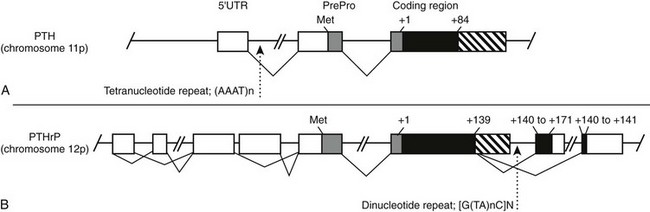
FIGURE 1-12 Schematic representation of the intron/exon organization of the genes encoding parathyroid hormone (PTH) (A) and PTH-related peptide (PTHrP) (B). The introns are represented by a solid line, and the coding and noncoding exons are shown (boxes); Met indicates the initiator methionine of the prepropeptide sequences (shaded boxes), and numbers indicate the first and the last amino acid residues of the secreted peptides that are encoded by the different exons (filled boxes). The approximate locations of frequent microsatellite polymorphisms in either the PTH 92 or the PTHrP308 gene are indicated.
Like the PTH gene, the PTHrP gene contains a single exon that encodes most amino acid residues of the prepropeptide sequence, and both genes have an exon that encodes the remainder of the propeptide sequence, that is, two basic residues (Lys and Arg) that are required for endoproteolytic cleavage of the mature peptide, and either all or most of the amino acids of the secreted peptides. The similarities in their protein sequence and in the structure of their genes, as well as the overlap in some of their functional properties, confirmed that both peptides are derived from a common ancestor.4,6,293 By now, both mammalian genes have diverged considerably; for example, in contrast to the less complex PTH gene, which gives rise to a single gene product, the PTHrP gene uses at least three different promoters and alternative splice patterns that lead to the synthesis of several different mRNA species encoding peptides with different C-terminal ends.6,293,298,307 A polymorphic dinucleotide repeat sequence that has been used for genetic linkage studies is located downstream of exon 4 (which is exon 6 according to a different nomenclature) of the human PTHrP gene308 (see Fig. 1-12); thus far, no human disorder has been discovered that is caused by mutations in the PTHrP gene.
When compared with each other, chicken PTHrP and the known mammalian species of PTHrP show strong amino acid sequence homology within the first 111 residues; the degree of amino acid sequence conservation of PTHrP is considerably higher than that of the known PTH species (see Fig. 1-3). The amino acid sequence homology between both peptide families is restricted to the N-terminal portion, where 6 of the first 12 amino acid residues are conserved in all PTH and PTHrP species (see Fig. 1-3); the middle and C-terminal regions of both peptides share no recognizable similarity.
PTHrP is likely to undergo extensive posttranslational processing, resulting in peptide fragments, some of which may have biological properties distinct from those of PTHrP(1-34) on calcium and bone metabolism. The N-terminal PTHrP fragment functionally homologous to PTH is derived through cleavage at amino acid residue Arg37.309,310 PTHrP(1-36) interacts efficiently with the PTH1R,7–9,311 and it has an in vivo efficacy similar to that of PTH(1-34).312–314 Longer, glycosylated fragments of N-terminal PTHrP were also described; however, these forms appear to be generated predominantly by skin-derived cells, and their biological role, if different from that of PTHrP(1-34) or PTHrP(1-36), remains to be established.315
In addition to the biologically active N-terminal PTHrP fragment, different C-terminal fragments are generated and accumulate in patients with end-stage renal disease.316 A PTHrP fragment consisting of amino acids 107 to 139 and a shorter peptide, PTHrP(107-111), may be relevant to the control of bone metabolism because both fragments were shown to inhibit osteoclastic bone resorption317,318 and to stimulate osteoblast activity and proliferation.319 Although some investigators, using modified experimental conditions, were unable to confirm the in vitro findings with osteoclasts,320 more recent data indicate that PTHrP(107-139) reduces the number of osteoclasts and inhibits bone resorption in vivo.321 Taken together, these findings suggest that C-terminal PTHrP fragments may have a role in regulating bone resorption and/or formation (Table 1-2).
Table 1-2
Selective Characteristics of Other Potential Receptors for PTH and/or PTHrP Distinct from the PTH1R and PTH2R
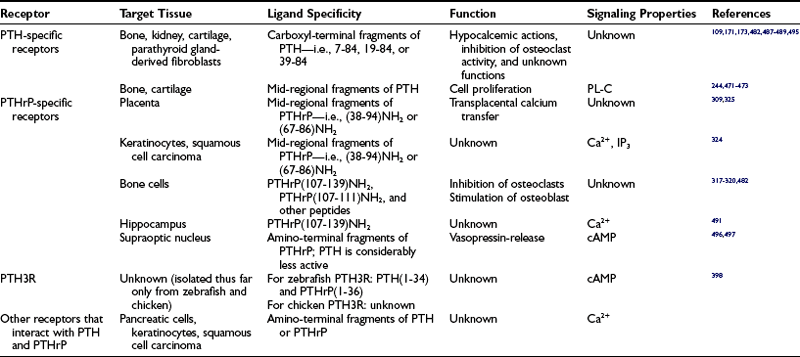
Cleavage at amino acid residue Arg37 also generates PTHrP(38-94) amide, a PTHrP fragment that could be of considerable importance in maintaining fetal calcium homeostasis.309,322 This peptide is found in the circulation323 and appears to interact with a distinct receptor that signals through changes in intracellular free calcium and is likely to be an important regulator of transplacental calcium transfer.309,322,324–326 Studies with PTHrP(38-94) amide and PTHrP(67-86) amide have shown that these fragments increase the blood calcium concentrations of parathyroidectomized fetal lambs and PTHrP-ablated murine fetuses, respectively309,325,326 (see later). These results confirmed earlier data that had provided the first evidence for an important role of PTHrP in the regulation of fetal calcium homeostasis.327
Functions of PTHrP
The first actions of PTHrP to be defined were the PTH-like actions associated with the humoral hypercalcemia of malignancy.4,6,293 In this pathologic circumstance, PTHrP acts like a hormone, that is, it is secreted from the tumor into the bloodstream and then acts on bone and kidney to raise calcium levels (see Chapter 7). Whether PTHrP circulates at high enough levels in normal adults to contribute at all as a hormone to normal calcium homeostasis is an unanswered question; the levels are certainly low, and patients with congenital or acquired hypoparathyroidism are hypocalcemic despite the presence of PTHrP. Although incomplete and somewhat conflicting, growing evidence suggests, however, that PTHrP may act as a hormone in two special circumstances: during fetal life and during lactation as outlined in the next sections.
Role of PTHrP in Placental Calcium Transport
During intrauterine life, fetal blood calcium is higher than maternal blood calcium, at least partly because of active transport of calcium across the placenta. In fetal life, in contrast to adulthood, PTHrP is made in easily detectable amounts in the parathyroid gland. Parathyroidectomy lowers the blood level of calcium in fetal sheep and abolishes active calcium transport across the experimentally perfused placenta. PTHrP from human tumors and synthetic PTHrP(1-84), PTHrP(1-108), PTHrP(1-141), and PTHrP(67-86) amide acutely restored placental transport of calcium in a perfused placenta preparation.328,329 PTHrP with an intact N-terminus, PTHrP extracts, or synthetic peptides such as PTHrP(1-34) or PTHrP(1-36), as well as intact PTH and PTH(1-34), had no effect on placental calcium transport. These results suggest that PTHrP secreted from the fetal parathyroids acts on the placenta to induce calcium transfer from the mother to the fetus.
The role of PTHrP in placental calcium transport is also supported by studies in mice missing both alleles of the PTHrP gene. The blood calcium of fetal PTHrP-ablated mice is identical to maternal blood calcium, that is, the transport of calcium from the mother into the fetus is diminished.325 The defect in placental calcium transport can be corrected acutely by injecting PTHrP(1-86) or PTHrP(67-86) into the fetal blood circulation but not by injecting PTHrP(1-34) or PTH(1-84). These studies in mice and sheep suggest that a receptor distinct from the cloned PTH/PTHrP receptor mediates the action of PTHrP on placental calcium transport. Consistent with this hypothesis, placental calcium transport is actually increased in mice missing the PTH/PTHrP receptor.325 The possible role of the fetal parathyroid gland as the crucial source of the PTHrP is suggested by the mentioned experiments in sheep,328,329 but no measurements of PTHrP in fetal sheep blood have been made.
Role of PTHrP in Lactation
The second possible setting for humoral actions of PTHrP is during pregnancy and lactation. During lactation, transfer of calcium from bone into milk results in a measurable decline in bone mineral content.326 In experimental animals, PTH and 1,25(OH)2D3 have been eliminated as possible agents responsible for directing this transfer.330,331 Furthermore, the lactating breast secretes PTHrP into the circulation, 332 and urinary cAMP rises in response to suckling.333 Postpartum lactating women have elevated levels of PTHrP in the bloodstream,334–337 and hypoparathyroid patients who are maintained normocalcemic by treatment with 1,25(OH)2D3 can become hypercalcemic during lactation. Thus, PTHrP in the bloodstream may act on bone to release calcium and on the kidney to increase reabsorption of calcium from the urine, thus retaining the calcium for transport into milk. Consistent with this hypothesis, mice missing PTHrP production specifically in breast tissue have decreased bone turnover and preservation of bone mass.338 PTH alone cannot effectively serve these roles, because the slight elevation in ionized calcium during lactation suppresses PTH levels.334–337,339 An exaggeration of this lactational elevation of PTHrP may explain the rare occurrence of hypercalcemia and high PTHrP levels in pregnant and lactating women.340,341
However, PTHrP was shown to have additional roles in the breast, and it has a major role in breast development, acting through the PTH1R.342,343 PTHrP is synthesized by breast tissue and is excreted in enormous amounts into breast milk. Thus, PTHrP in the breast appears to have both paracrine and endocrine roles. These roles may be subverted in breast cancer, a setting in which PTHrP may facilitate the growth of metastases in bone344 and also cause humoral hypercalcemia. The lactating mammary gland can sense extracellular calcium through the calcium-sensing receptor, thereby adjusting calcium and PTHrP secretion into milk. Further, in fetal life, PTHrP, acting through the PTH/PTHrP receptor, is required for the normal development of breast tissue.338,343
PTHrP in Bone and Tooth Development
PTHrP has an essential role in bone development. This role is best illustrated by the phenotype of mice missing both copies of the PTHrP gene.305,345,346 These mice die at birth and have diffuse abnormalities in all bones that form by the replacement of a cartilage mold with true bone (endochondral bone formation). The original cartilage molds form normally, but the chondrocytes within the molds stop dividing prematurely and differentiate into hypertrophic chondrocytes at an accelerated pace. Consequently, the growth plates of these bones show dramatically truncated columns of proliferating chondrocytes. The resultant bones are short and mineralize sooner than normal. Growth plates of mice missing the PTH/PTHrP receptor look similar to those of the PTHrP knockout mouse,12 which suggests that the PTH/PTHrP receptor mediates the actions of PTHrP on chondrocytes. PTHrP is made by perichondrial cells, particularly those at the ends of bones near the joint surfaces and to a lesser extent by chondrocytes, again particularly those at the ends of bones. The PTH/PTHrP receptor is expressed at low levels in adjacent chondrocytes in the proliferating columns and is dramatically expressed in chondrocytes just leaving the proliferative pool and becoming hypertrophic. The PTHrP made by the perichondrial cells and some chondrocytes thus acts on proliferating chondrocytes to keep cells in the proliferative pool to slow differentiation into hypertrophic chondrocytes, and to also slow the subsequent death of hypertrophic chondrocytes by apoptosis. This hypothesis is further supported by the phenotypes of transgenic mice that express the PTHrP gene at high levels in chondrocytes.306 In these mice, chondrocyte differentiation is dramatically slowed. An analogous phenotype is demonstrated by transgenic mice in which chondrocytes express a constitutively active PTH/PTHrP receptor.347 Both these transgenes can at least temporarily reverse the growth plate abnormalities in PTHrP knockout mice and allow the mice to live for several months postnatally.347,348 In the developing tooth bud, PTHrP via the PTH1R appears to mediate transmission of important cell-differentiation signals at the mesenchymal/epithelial interface.349
Mice with abnormal PTHrP genes or abnormal PTH/ PTHrP receptors have helped clarify the pathogenesis of two human diseases: Blomstrand’s lethal chondrodystrophy, in which homozygous or compound heterozygous inactivating PTH/PTHrP receptor mutations are found,350–353 and Jansen’s osteochondrodystrophy, in which heterozygous constitutively active PTH/PTHrP receptors lead to hypercalcemia and short stature.354–357 Both these diseases are discussed in detail in Chapter 5.
The potent effects of locally produced PTHrP on bone development and the abnormalities associated with too little or too much PTHrP action suggest a need for careful local regulation of PTHrP production (Fig. 1-13). One major determinant of PTHrP production in the growth plate is Indian hedgehog (Ihh) (see Chapter 4 for details). Ihh belongs to the hedgehog family of secreted proteins that are important for embryonic patterning358 and acts through Patched, a receptor with 12 membrane spanning helixes that associates physically with Smoothened and thereby suppresses its constitutive activity.359,360 In growth plates, expression of Ihh is restricted to the transition zone within the growth plate, where proliferating chondrocytes differentiate into hypertrophic cells.13 Ihh, directly or indirectly, stimulates the production of PTHrP by perichondrial cells and chondrocytes near the ends of long bones. This PTHrP then slows the differentiation of chondrocytes and slows the differentiation of cells capable of synthesizing Ihh. Thus, a negative feedback loop controlled by Ihh and PTHrP ensures proper pacing of the proliferation and differentiation of chondrocytes. Ihh also has actions on chondrocytes independent of the effects of PTHrP; Ihh is a powerful stimulator of chondrocyte proliferation. For a more comprehensive review of the control of the growth plate, the role of Ihh, and PTHrP, as well as their interacting cytokines, see Chapter 4.
FIGURE 1-13 Schematic regulation of chondrocyte differentiation within the metaphyseal growth plate by a paracrine feedback loop involving parathyroid hormone-related peptide (PTHrP) and Indian Hedgehog (Ihh). Ptc, Patched; Smo, smoothened.
PTHrP is made by normal cells of the osteoblast lineage361 and is likely to have a number of functions in normal osteoblast development and function. Mice missing one copy of the PTHrP gene are osteopenic. Thus, PTHrP appears to increase osteoblastic bone formation.362 One function of PTHrP in the bones adjacent to developing teeth has been clarified. The mice described earlier, in which transgenic production of PTHrP or a constitutively activated PTH/PTHrP receptor by chondrocytes reverses the growth plate abnormalities of PTHrP-ablated mice and allows postnatal survival, have failure of normal tooth eruption.347,348 Normally, the developing tooth elicits PTHrP production in the bone in which the tooth is embedded. This PTHrP then stimulates cells of the osteoblast lineage, which in turn stimulate the development and activity of osteoclasts, the bone-resorbing cells. This stimulation of bone resorption allows the tooth to erupt normally. PTH, a circulating endocrine factor, is unable to compensate for the lack of high local concentrations of PTHrP and thus cannot adequately stimulate the local bone resorption needed for tooth eruption.
It is likely that the local actions of PTHrP in the growth plate and bone are representative of the local actions of PTHrP to control cellular differentiation and proliferation in a number of organs. Still not fully characterized is the amount of cross-talk between the distinct ligands PTH and PTHrP, which can both activate the PTH/PTHrP receptor. Just as such cross-talk certainly explains PTHrP-mediated hypercalcemia of malignancy, it is likely that normally PTH and PTHrP both activate the PTH/PTHrP receptor in a number of tissues in a way that allows integration of the signals from locally produced PTHrP and systemically provided PTH (see Ligand-Induced Conformational Changes later).
Other Paracrine Actions of PTHrP
Most of the actions of PTHrP are not thought to be hormonal, but rather to be paracrine or autocrine.363 PTHrP is synthesized at one time or another during fetal life in virtually every tissue. This widespread expression of PTHrP in fetal life probably explains the extensive expression of PTHrP in a great variety of malignancies. Malignant tissues often revert to a fetal pattern of gene expression; synthesis of PTHrP may be part of this pattern. PTHrP is also synthesized by many adult tissues.4,293,363 In tissues such as skin and hair, it is likely that PTHrP regulates cell proliferation and differentiation.364 PTHrP is also synthesized widely in the smooth muscle of blood vessels and in the gastrointestinal tract, uterus, and bladder, and transgenic expression of PTHrP in smooth muscle cells leads to severe defects in cardiac development.365,366 In these tissues, PTHrP is synthesized in response to stretch and acts on smooth muscle in an autocrine fashion to relax the muscle.367 PTHrP is also widely expressed in neurons of the central nervous system; its function in the brain is unknown. In mice missing the PTHrP gene, widespread degeneration of neurons occurs postnatally; as explained earlier, these mice survive postnatally through expression of PTHrP only in cartilage cells.348
Receptors for PTH and PTHrP
Because of the diverse actions of PTH, which were shown to be either direct or indirect and to involve multiple signal transduction mechanisms, it was initially thought that several different receptors would mediate the pleiotropic actions of this peptide hormone. Although some of these actions were subsequently shown to be PTHrP-dependent rather than PTH-dependent, it was somewhat surprising that the initial cloning approaches led to the isolation of complementary DNAs (cDNAs) encoding only a single G protein–coupled receptor, the common PTH/PTHrP receptor, also called the PTH1R (Fig. 1-14). The recombinant PTH/PTHrP receptor was shown to interact about equivalently with PTH and PTHrP ligands and to activate at least two distinct second messenger pathways, the adenylyl cyclase/cAMP/PKA and phospholipase C (PLC)/IP3, Ca2+, DAG/PKC pathways, mediated by Gαs and Gαq/11, respectively.7–9,368,369 It is now clear that depending on cell type, the PTH/PTHrP receptor can activate various other signaling cascades, including the extracellular signal-regulated kinase (ERK) cascade60,370–372 and the phospholipase D/rho A cascade, 373 the former potentially involving in addition to Gαs, β-arrestins60 or transactivation of ERK via extracellularly released epidermal growth factor,371 the latter through activation of the Gα12/13 subtype of heterotrimeric G proteins.373
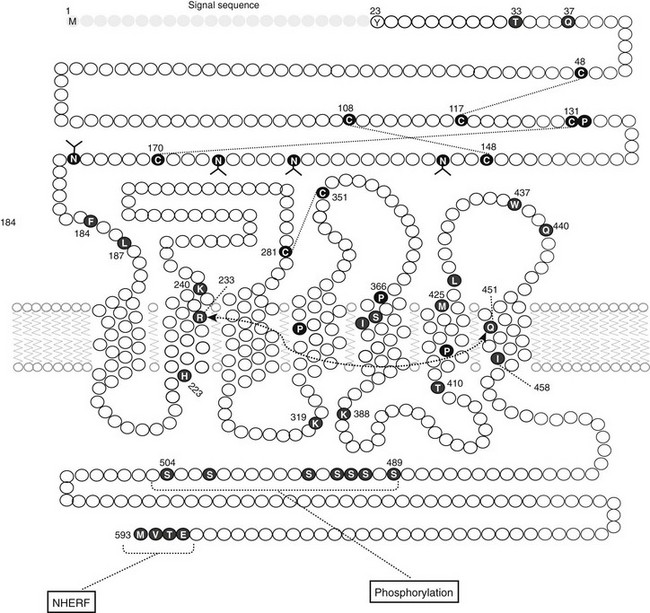
FIGURE 1-14 The PTH/PTHrP receptor: location of some key residues and crosslinking analysis. The schematic of the PTH/PTHrP receptor shows some of the residues believed to play key roles in the structure and/or function of the receptor. These include the conserved extracellular cysteines (C), which form a disulfide linkage network (dashed lines)68; the asparagines (N), modified with glycosyl groups (forks)498; Thr33 and Gln37, which contribute to PTH(15-34) binding affinity; Phe184 and Leu187, which contribute to PTH(1-14) agonist activity429; Arg233, Ser370, Ile371, Met425, Leu427, Trp437, and Gln440, which contribute to the agonist/antagonist responses mediated by residues 1 and/or 2 of the ligand430,450; a putative interhelical interaction between Arg233 and Gln451 (dashed connector)430; conserved prolines (P), including Pro132, the site of an inactivating mutation (Leu) in Blomstrand’s chondrodysplasia351; His223, Thr410, and Ile458, the sites of activating mutations in Jansen’s chondrodysplasia357; Lys319 and Lys 388, at which mutations impair Gq and Gs/Gq coupling, respectively 499,500; serine (S) residues in the carboxy-terminal tail that are phosphorylated upon agonist activation501,502; and four C-terminal residues (Glu590–Met593) that mediate binding to members of the sodium-hydrogen exchanger regulating factor (NHERF) family of proteins.239,503 The first 23 residues of the receptor (shaded) represent the signal sequence and are not present in the mature membrane-embedded receptor.
The initial finding that both PTH and PTHrP could activate the recombinant PTH/PTHrP receptor confirmed earlier studies using different clonal cell lines or renal membrane preparations that had shown that PTH and PTHrP bind to and activate a common G protein–coupled receptor with similar efficiencies and efficacies.311,374–376 Based on these and subsequent findings, such as the similar phenotypes observed in mice that are null for either PTHrP or the PTH/PTHrP receptor,12,13 it is very likely that most of the endocrine actions of PTH and most of the paracrine/autocrine actions of PTHrP are mediated through the PTH/PTHrP receptor.
Three Parathyroid Hormone Receptor Subtypes
Subsequent to cloning of the PTH/PTHrP receptor, or the PTH1R, different efforts led to the identification of two novel receptors closely related to the initially isolated receptor. One of these receptors, called the PTH2 receptor (PTH2R), was obtained from a human brain cDNA library377 and found to have reactivity towards PTH but not towards PTHrP.377–380 The PTH2R from rat, however, responded poorly to both PTH and PTHrP.381,382 A search for the true ligand for this PTH2 receptor led to the isolation of a hypothalamic peptide of 39 amino acids (TIP39) called tubular infundibular peptide which efficiently binds to and activates both rat and human PTH2Rs.383 TIP39 shows weak amino acid sequence homology to the N-terminal 34 amino acids of PTH and PTHrP, as well as some overlap in secondary structure (see Fig. 1-3).384 Intact TIP39 binds relatively weakly to the PTH1R, but deletion of the first seven residues results in a high-affinity PTH1R antagonist.385–387 The peptide TIP39 is produced at the hypothalamus and within the testes,383,388 and together with the PTH2R appears to play an autocrine/paracrine role in germ cell development, inasmuch as mice lacking the gene for TIP39 are sterile due to a failed formation of spermatatids.389
The second novel PTH receptor variant isolated, the PTH3 receptor (PTH3R), has so far been found in fish390 and birds (see NCBI chicken genome website) but not in mammals (Fig. 1-15). Functional and phylogenetic analyses indicate that the PTH3R is more closely related to the PTH1R than to the PTH2R.390 In addition to the three identified PTH receptor subtypes, there are functional and physicochemical data suggesting that there may be additional receptors that interact with portions of either PTH or PTHrP which are C-terminal of the principal bioactive region defined by the (1–34) segment (see following and Table 1-2). As of yet, however, no gene or cDNA encoding such a novel receptor has been identified.
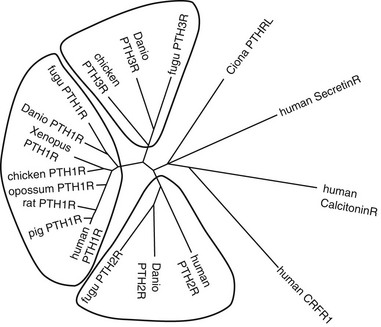
FIGURE 1-15 Phylogenetic relationships between PTH receptors from different species and other secretin receptor family members. Receptor amino acid sequences, with predicted signal peptides and exon E2-encoded segments of mammalian PTH1Rs removed, were aligned using the ClustalW(1.83) program, and an unrooted tree in which branch distances indicate amino acid sequence divergence was generated using the Phylip(3.67) DrawTree program.
The PTH/PTHrP Receptor Type-1—Evolution, Gene Structure, and Protein Topology
The PTH1R belongs to a distinct subgroup of G protein–coupled receptors called the class B, or secretin family GPCRs. The first cDNAs encoding the PTH/PTHrP receptor were isolated through expression cloning techniques from two different model cell lines: OK cells, an opossum kidney–derived proximal tubule-like cell, and ROS 17/2.8 cells, a rat osteosarcoma-derived osteoblast-like cell.7,8 Subsequently, cDNAs encoding the human,9,391,392 mouse,393 rat,394 chicken,13 porcine,395 dog,396 frog,397 and fish PTH/PTHrP receptors398 were isolated through hybridization techniques from different tissue sources, i.e., kidney, osteoblast-like cells, brain, embryonic stem cells, and/or whole embryos. Northern blot and in situ studies304,399,400 and data provided through available public EST (expressed sequence tag) databases confirmed that the PTH/PTHrP receptor is expressed in a wide variety of fetal and adult tissues. The tetraploid African clawed frog, Xenopus laevis, expresses two nonallelic isoforms of the PTH/PTHrP receptor,397 whereas all other investigated species have only one copy of the receptor gene per haploid genome.
The possible existence of PTH/PTHrP receptor subtypes with distinct, organ-specific characteristics had been suggested by distinct ligand binding properties401–403 or by the activation of distinct second messenger pathways in different clonal cell lines.251,404,405 However, the cloning of identical PTH/PTHrP receptors from human kidney, brain, and bone-derived cells9,391,392 indicated that the previously observed pharmacologic differences could likely be explained by species-based variations in receptor primary sequence, rather than to tissue-based differences in expression of various receptor subtypes.
The gene encoding the human PTH/PTHrP receptor is located on chromosome 3p (within the region 3p21.1-3p24.2). Its intron/exon structure406–408 is largely preserved in the genes encoding the rat and mouse receptor homologs.409,410 The gene spans at least 20 kb of genomic DNA and consists of 14 coding exons and at least three noncoding exons. The size of the coding exons in the human gene ranges from 42 bp (exon M7) to more than 400 bp (exon T), and the introns vary in length from 81 bp (intron between exons M6/7 and M7) to more than 10,000 bp (intron between exons S and E1). Two promoters for the PTH/PTHrP receptor, P1 and P2, have been described in rodents.409–412 The activity of P1 (also referred to as U3) is mainly restricted to the adult kidney, while that of P2 (also referred to as U1) is detected in several fetal and adult tissues, including cartilage and bone. In humans, a third promoter (P3, also referred to as S) appears to control PTH/PTHrP receptor expression in some tissues, including kidney and bone.408,413,414
The expressed human PTH/PTHrP receptor protein is 593 amino acids in length, including the 22-amino-acid N-terminal signal peptide sequence that is removed in the mature receptor. The predicted topology is defined by a relatively large amino-terminal extracellular domain (∼160 amino acids in the human PTH1R), a juxtamembrane, or J, region containing seven membrane spanning helices and interconnecting loops, and a carboxy-terminal tail of about 110 amino acids (see Fig. 1-14). In mammals, the PTH1R N-domain is encoded by five exons: S, E1, E2, E3, and G. Exon S and G encode the signal peptide and a glycosylated segment,415 respectively. Exons E1, E3, and G encode segments that include a number of conserved residues that are likely required for proper folding of the protein, surface expression, and/or ligand binding. These residues include six cysteine residues that form a disulfide bond network that stabilizes the N-domain structure.68,416,417 Exon E2 encodes a non-conserved 41-amino-acid segment that is not essential for receptor function, since it can be targeted for deletion or large insertions, such as with green fluorescent protein (GFP), without a major loss in ligand binding affinity or surface expression.416–418 Indeed, an equivalent of exon E2 is absent in the PTH1Rs from all nonmammalian species, as well as in other members of this GPCR subfamily (mammalian and nonmammalian), including the PTH2R.390,397,419 The E2 segment of the mammalian PTH1R gene might thus reflect an evolutionarily recent genetic insertion event390 or perhaps an atavistic genomic remnant. In any event, the E2 segment appears to form a nonfunctional loop that is conformationally flexible; a recent x-ray crystallographic analysis of the PTH1R N-terminal domain structure found this segment to be disordered in the crystal and hence unresolved.68
The initial cloning of the PTH1 receptor, cDN,7,8 along with the cDNAs for the receptors for secretin420 and calcitonin,421 led to the realization that these receptors form a distinct GPCR subfamily, now called the class B or secretin family receptors.419,422,423 These family B receptors share a similar overall structural topology, which is defined by the large N-terminal extracellular domain containing the conserved disulfide bond network, as well as a number of other conserved amino acids dispersed throughout the receptor protein.419 Protein alignment analyses reveal virtually no amino acid sequence homology between the family B GPCRs and the receptors comprising the four other major GPCR subgroups, including the well studied β2-adrenergic receptor and rhodopsin, representing the family A GPCRs.422 The structural similarity between these groups of receptors is therefore limited to the common use of a heptahelical domain organization for the membrane-spanning J region.422,424 As an evolutionary protein class, the secretin receptor family appears to have arisen during the divergence of the metazoans. Representatives of the receptor family are found in the genomes of a variety of invertebrate species, including the insect Drosophila melanogaster, and the nematode Caenorhabditis elegans, but not in yeast or bacteria.419 A plausible PTH/PTHrP receptor homolog is present in the genome of the tunicate sea squirt Ciona intestinalis (see Fig. 1-15) pointing to a much earlier evolutionary origin of the PTH receptor than previously known.419,425
The three-dimensional molecular structures of the receptors in the secretin family are still only poorly defined because none has been adequately isolated or crystallized in an intact form. For several members of these receptors, however, including the PTH1R,68 the CRFR1,426 the CRFR2,427 and the PACR1,428 high-resolution three-dimensional structures of the isolated N-terminal domain in complex with a cognate peptide ligand have recently been obtained, either by solution-phase NMR427,428 or x-ray crystallography.68,426 These studies reveal a similar overall fold composed of three layers of secondary structure: an upper layer formed by an N-terminal α-helix, a middle layer of β sheet, and a lower layer formed by a β strand and short C-terminal α-helix (Fig 1-16). The overall fold is stabilized by the three conserved disulfide bonds and an extensive array of hydrophobic and electrostatic packing interactions.68 The ligand, represented by the PTH(15-34) domain, binds as an amphipathic α-helix within a central groove in the structure, as described further later. The membrane-spanning helical regions of the class B receptors may be predicted to at least generally follow the heptahelical domain topology used by the class A receptors, which is now revealed by three-dimensional crystal structure of the β2-adrenergic receptor.69
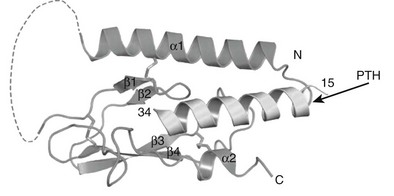
FIGURE 1-16 Crystal structure of the PTH1R amino-terminal domain in complex with PTH(15-34). The tertiary fold of the receptor N domain is defined by an upper layer of N-terminal α-helix (α1), a middle layer of beta sheet (β1, β2), and a lower layer of beta sheet (β3, β4) and short C-terminal α-helix (α2). The ligand lies as an amphipathic α-helix in a groove formed along the center of the structure. The 41-amino-acid exon E2-encoded segment is not resolved in the crystal structure and indicated here as a dashed line.68
Mechanisms of Ligand Binding and Activation at The PTH1R
The ligand-binding mechanisms used by the PTH receptor have been extensively analyzed by a combination of approaches, including functional methods that are based on receptor site–directed mutagenesis and ligand analog design strategies50,51,429–434 and biophysical methods that are based on the use of photoreactive cross-linking analogs of the ligand that permit direct identification of sites of intermolecular proximity by protein fragmentation and mapping analysis. The cross-linking analogs used in these studies have been labeled at various positions with a photoreactive benzophenone group, incorporated either directly into the peptide chain, as parabenzoyl-l-phenylalanine (BPA),434–437 or attached to the epsilon amino function of lysine-13 (BpLys).438 The combined functional and cross-linking data suggest that the ligand, as represented by the bioactive PTH(1-34) peptide, binds to the receptor via a two-step mechanism that involves two principal and somewhat autonomous components of the overall interaction.439–442 The C-terminal portion of the ligand representing the principal binding domain443,444 first contacts the N-terminal domain of the receptor to establish initial docking interactions,417 and subsequently the N-terminal portion of the ligand interacts with the J domain portion of the receptor to produce the holo-enzyme bimolecular complex that can couple to and activate G proteins (Fig. 1-17). The overall interaction is certain to be more complex than indicated here, but this general scheme of interaction is now believed to extend to most of the class B family of peptide hormone–binding G protein–coupled receptors.440,441,445,446
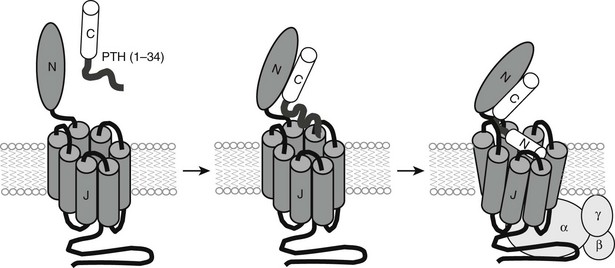
FIGURE 1-17 Two-site ligand-binding mechanisms for the PTH/PTHrP receptor. The ligand, PTH(1-34), first interacts with the PTH1R N-domain via its C-terminal helical portion and then engages the juxtamembrane (J) region via its N-terminal segment to induce the conformation changes involved in receptor activation and G-protein coupling.
Specific contacts to the receptor’s N-domain have now been elucidated by the recent crystal structure of the PTH1R N-domain in complex with PTH(15-34) (see Fig. 1-16).68 The PTH(15-34) domain binds as an amphiphilic α-helix, with the hydrophobic face of the helix formed by Trp23, Leu24, and Leu28, making extensive contacts with a hydrophobic surface formed on the floor of the central groove that runs through the N-domain structure. The guanidinium side chain of highly conserved Arg20 of the ligand makes additional contacts with at least five receptor residues that form an intricate pocket at the end of the groove. Binding studies performed using both the intact receptor and the isolated N-domain confirm the importance of the identified contacts to the development of ligand-binding affinity.68
The ligand interactions that occur with the receptor’s J domain are known with far less certainty, but these appear to principally involve ligand residues extending from position 1 to about position 19.436,441,442 This portion of the ligand contains the activation pharmacophore, which has minimally been defined by peptide functional studies as the PTH(1-9) segment.50 A contact site for conserved valine-2 in the ligand, which plays a critical role in receptor activation,57,447 has been mapped by cross-linking methods to Met425 at the extracellular end of the transmembrane domain (TM) 6.448,449 This interaction site is supported by functional data,448,449 however, other sites in the receptor (e.g., Ser370 and Ile371) have also been implicated by mutagenesis approaches to be likely contact sites for Val2.450 Other cross-linking studies identified proximities between residue 19 in the ligand and Lys240 at the extracellular end of TM2,451 and between residue 13 in the ligand and Arg186 at the boundary of the N-domain and TM1.438 These and other data combined suggest that the ligand’s N-terminal domain binds as an α-helix to lie in a groove formed at the extracellular surface of the receptor’s juxtamembrane region and in the same plane as the outer layer of the cell membrane.441,451 Such a binding mode could allow Val2, as well as other key ligand residues that define the agonist pharmacophore, such as Ile5 and Met8,50,452 to make intimate contacts with the membrane embedded core region of the receptor and thereby induce the conformational changes involved in receptor activation.59,453
The cognate interaction pocket in the receptor’s juxtamembrane region that accommodates and responds to the key pharmacophoric elements in the ligand have only been approximately defined. The development of highly potent N-terminal PTH fragment analogs, based on the weakly active native PTH(1-14) peptide scaffold, provides a class of minimized ligand structures that can used to specifically probe this component of the interaction process more effectively than is possible with PTH(1-34) (see Fig. 1-4). These minimized ligands, referred to generally as the M class of PTH ligands, maintain affinity and efficacy on the PTHR, as well as on a PTHR construct that lacks the N-terminal domain.50–53,57,429 Furthermore, the M-PTH analogs have highly stabilized N-terminal helical backbone structures, in part due to the incorporation of conformationally constrained amino acid analogs, such as α-amino-isobutyric (Aib), at positions 1 and 3.51,54,55,454 A recent use of such modified M-PTH(1-14) analogs for compound screening resulted in the identification of a new class of PTH nonpeptide mimetic ligands that bind specifically to the PTH1R J domain.455,456 Although these compounds bind with micromolar affinities and function as competitive antagonists, they represent potential leads towards new mimetic compounds that function as PTH1R agonists and are orally active.
Ligand-Induced Conformational Changes
Given the absence of a high-resolution, three-dimensional molecular structure for the intact PTH/PTHR bimolecular complex, it is difficult to describe the conformational changes that occur in the complex upon ligand binding, receptor activation, and G-protein coupling. Problems that need to be solved include determining how the N and J domains of the receptor are oriented relative to each other, whether PTH(1-34) adopts a linear or U-shaped structure as it is bound to the receptor,62–65 and the molecular movements that occur in the complex during the ligand binding, activation, and G-protein coupling cycle. Several new approaches, however, are beginning to help illuminate these aspects of PTH receptor function.
Kinetic binding assays performed in membranes with various PTH and PTHrP radioligand analogs has revealed that the PTH1R can adopt different conformations that display differential affinities for structurally diverse ligands and are differentially coupled to heterotrimeric G proteins.439,457 In particular, the studies show that PTH(1-34) can form a complex with the receptor that remains stable upon the addition of GTPγS, a reagent that at the high concentrations used is expected to fully activate the G protein and hence dissociate the receptor–G protein heterotrimer complex and thus shift the receptor into a low-affinity state.457 Moreover, the ligand can bind efficiently to the receptor in membranes prepared from cells lacking Gαs. In contrast, PTH(1-14) forms a complex that rapidly dissociates upon GTPγS addition and binds only poorly in the absence of Gαs.457
The data thus suggested that certain ligands (e.g., PTH[1-34]) preferentially bind to or induce a novel high-affinity PTH1R conformation, called R0,458 that can stably hold the ligand in place even upon G-protein activation/dissociation, whereas other ligands (e.g., M-PTH[1-14]) preferentially bind to or induce a different high-affinity conformation, called RG, that is dependent on G-protein coupling and thus reverts to a low-affinity form upon G-protein activation/dissociation. These findings predicted that ligands that bind to the R0 conformation would produce protracted signaling responses, owing to multiple or prolonged rounds of G-protein coupling. This prediction was indeed supported by data showing that PTH(1-34) produced extended cAMP responses after ligand washout, as compared to M-PTH(1-14).457 It was also found that PTHrP(1-36), in contrast to PTH(1-34), binds with relative selectivity to the RG versus R0 conformation.459 These observations suggested the hypothesis that altered modes of receptor conformational selectivity at the PTH1R, as exhibited by different ligands, may translate into altered biological actions for those ligands in vivo and may even explain the relatively deficient capacity of PTHrP(1-36), versus PTH(1-34), to induce vitamin D synthesis and increase blood calcium levels when injected into human subjects.460 This general hypothesis was tested further using a new analog, M-PTH(1-34), that has even stronger selectivity for the R0 conformation than does PTH(1-34). The analog indeed produced markedly prolonged cAMP responses in cells and markedly prolonged, even as compared to PTH(1-34), hypercalcemic (Fig. 1-18) and hypophosphatemic responses when injected into mice.461
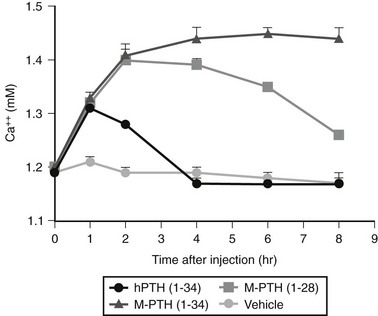
FIGURE 1-18 Actions of long-acting PTH analogs in vivo. PTH analogs were injected into mice to assess acute effects on blood ionized calcium levels. The modified ligand, M-PTH(1-34), which binds to the R0 PTHR conformation with higher affinity than does unmodified PTH(1-34), induces a markedly prolonged increase in blood ionized Ca2+. These prolonged effects were not explainable by a prolonged half-life of the modified peptide in the circulation, and they were paralleled by prolonged cAMP-stimulating activity in MC3T3 osteoblastic cells in culture. Thus, high-affinity binding to the R0 PTHR conformation can lead to prolonged PTHR signaling actions in vitro and in vivo. (Data from Okazaki M, Ferrandon S, Vilardaga JP, et al: Prolonged signaling at the parathyroid hormone receptor by peptide ligands targeted to a specific receptor conformation, Proc Natl Acad Sci U S A 105:16525–16530, 2008.)
That the PTH1R can adopt multiple conformational states and thereby display altered modes of biological action is consistent with the finding of certain PTH ligand analogs that have capacities to selectively activate one signal pathway over another58–60,247 or, when altered by certain point mutations, to mediate ligand-independent signaling activity.462 The notion of multiple PTH1R conformations and altered signaling directionality is also in line with the generally accepted views of how most if not all G protein–coupled receptors operate in their respective biological milieus.463 The recent use of fluorescent (Forster) resonance energy transfer (FRET) methodologies, utilizing PTH receptors tagged with spectral variants of green fluorescent protein together with fluorescently labeled PTH ligands, has opened new routes for defining with high temporal resolution and precision the kinetics of receptor conformational change and ligand-receptor interaction processes. Such studies show that indeed the structurally different ligands, such as PTH(1-34), PTHrP(1-36), and M-PTH(1-14), form kinetically distinguishable complexes with the PTH1R and confirm the differences in dissociation rates seen in the radioligand-based membrane binding studies (Fig. 1-19A).418,453,459
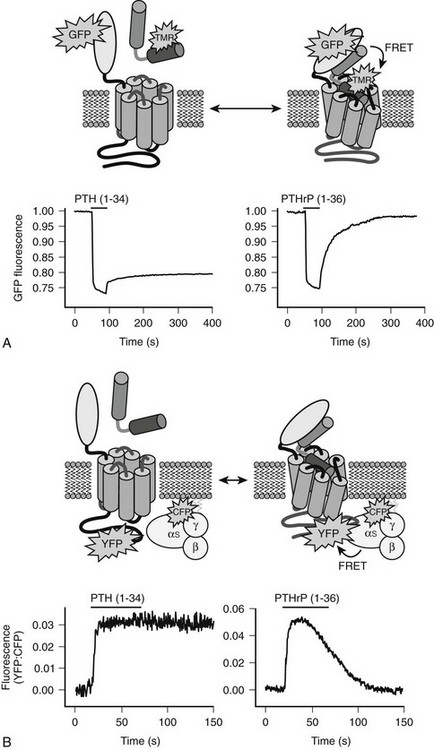
FIGURE 1-19 Differential receptor binding and G protein coupling kinetics induced by PTH and PTHrP. A, The binding of PTH and PTHrP analogs labeled on lysine13 with tetramethylrhodamine (TMR) to GFP-PTH1R was monitored using single-cell FRET recording. Upon binding, GFP fluorescence emission is reduced due to FRET-based quenching by the TMR group. The ligand was applied for 20 seconds (black bar), and then washed out. Over the time course studied, PTH remains bound to the receptor whereas PTHrP dissociates. B, Coupling to G proteins was monitored using a PTHR tagged in the cytoplasmic tail with yellow-fluorescent protein (YFP) and a Gγ subunit tagged with cyan fluorescent protein (CFP). The ratiometric FRET response (YFP emission:CFP emission upon CFP excitation) increases upon R-G coupling. The recordings show that R-G coupling following ligand wash-out is prolonged for PTH but not for PTHrP.459,464
Mechanisms of Prolonged Signaling at The PTH1R
The subcellular mechanisms and pathways that underlie the differences in the duration of the signaling responses seen for the different analogs remains to be established, but it has been found, again using the FRET approach as well as fluorescence confocal microscopy methods, that these long-acting ligands induce long-lived complexes of ligand, receptor, and G protein (see Fig. 1-19B) that remain intact and associated with adenylyl cyclase, even as the proteins move to the intracellular endosomal domain.464 This raises the intriguing possibility that in addition to, or as an extension of, the receptor-phosphorylation and arrestin-dependent internalization, desensitization and recycling processes that are known to regulate PTH1R actions,59,465–468 there may be a component of the mechanism by which the ligand•receptor•G-protein complex formed by PTH but not PTHrP remains assembled within the endosomes and catalytically active for longer time intervals than previously appreciated.464 Further studies are needed to test this hypothesis. The results could provide additional clues for designing new PTH analogs that have time-limited or prolonged actions on the PTH1R; such ligands could have improved efficacy, compared to PTH(1-34), as therapies for the diseases of osteoporosis277,469 and hypoparathyroidism,470 respectively.
Other Receptors for C-Terminal PTH or PTHrP
There is evidence for the presence of additional receptors and/or binding proteins that interact with portions of PTH or PTHrP C-terminal of the PTH(1-34), but thus far no cDNA encoding such a receptor has been identified. Considerable information has accumulated that indicates that regions of PTH(1-84) other than the N-terminal residues may be responsible for novel biological actions (see Table 1-2), although many of these effects are still largely limited to in vitro studies. A series of synthetic peptides forming the central region of PTH were reported to delineate a core domain, PTH(30–34), that stimulates the proliferation of chondrocytes but does not appear to involve the cAMP/PKA pathway.244,471–474 Early competition binding assays with renal plasma membranes and rat osteosarcoma cells demonstrated binding sites for C-terminal PTH(53-84) that are discrete from those that bind PTH(1-34).475–478 The observation that N-terminal and C-terminal fragments of PTH elicited contrasting effects on alkaline phosphatase activity479–481 supports the existence of a receptor with specificity for the C-terminal portion of PTH. Considerable evidence suggests that the C-terminal portion of PTH(1-84) has biological activity. For example, C-terminal PTH fragments such as PTH(39-84) and PTH(53-84) stimulate the formation of bone-resorbing osteoclasts from precursor cells,482 and the same PTH fragments were shown to enhance the influx of 45Ca into SaOS-2 cells.483 Several different clonal cell lines, including bone- and cartilage-derived cells in which the common PTH/PTHrP receptor had been deleted through homologous recombination, showed increased proliferative activity in response to intact PTH and C-terminal fragments.484,485 Furthermore, PTH(1-84) was more potent than PTH(1-34) in increasing the concentration of fibronectin in vivo, 486 and C-terminal PTH fragments increased type I procollagen expression.487
In direct support of a biological role for C-terminal PTH fragments, ROS 17/2.8 cells, rat PT-r3 cells, osteolytic cell lines lacking the PTH1R were shown to specifically interact with the C-terminal portion of PTH.488,489 With the use of radiolabeled [Tyr34]hPTH(19-84), which does not bind to cells that express high concentrations of the PTH/PTHrP receptor, these cells were shown to bind PTH(1-84) and PTH(19-84) with equivalent high affinity, whereas fragments of PTH(19-84) that were truncated at the N-terminal end showed a progressive loss of binding affinity.488 However, even PTH(53-84) showed some displacement of the radiolabeled [Tyr34]hPTH(19-84), whereas the midregional fragment PTH(44-68) failed to do so. Other studies using ROS 17/2.8 cells, but with [35S-Met]hPTH(1-84) instead of 125I-labeled [Tyr34]hPTH(19-84), showed similar binding characteristics of C-terminal PTH fragments, and these studies indicated that residue 84 contributes to the overall ligand-binding affinity.489 Photoaffinity cross-linking experiments led to the identification of approximately 80-kD and 30-kD proteins that show high-affinity interaction with the C-terminal portion of PTH but not with the N-terminal PTH(1-34).488 These findings suggest that the C-terminal portion of PTH interacts specifically with a novel receptor/binding protein that is distinct from the PTH1R. Complementary DNA clones encoding this putative receptor/binding protein have not yet been isolated, and little or nothing is known about the signal transduction systems that might mediate the actions of these midregional/C-terminal PTH fragments.
The development of antibodies that can distinguish truly intact PTH(1-84) from variants that are N-terminally truncated or otherwise posttranslationally modified has led to the realization that such PTH variants can be present in the circulation, and, in cases of primary or secondary hyperparathyroidism, potentially at levels exceeding those of PTH(1-84).172,191,192,255 How such PTH variants might affect calcium homeostasis and other physiologic processes remains to be determined, but in a rat model study, N-terminally truncated PTH was shown to effectively inhibit the capacity of PTH(1-34) to stimulate synthesis of 1,25(OH)2-vitamin-D3.173,490
There is also evidence to suggest that non-PTH1R receptors can mediate actions induced by the midregion of PTHrP and may thus play an important role in placental calcium transport309,325 (see earlier discussion), as well as modulate biological functions in skin.324 Likewise, actions of the C-terminal PTHrP portion on osteoclasts and osteoblasts317–319,321 and on the central nervous system491 appear to be mediated through a specific receptor, and other receptors have been characterized that interact equivalently with the N-terminal portions of PTH and PTHrP but signal only through changes in intracellular free calcium.492–494
[/level-membership-for-endocrinology-diabetes-and-metabolism-category][not-level-membership-for-endocrinology-diabetes-and-metabolism-category]Chapter 1
Parathyroid Hormone and Parathyroid Hormone–Related Peptide In The Regulation of Calcium Homeostasis and Bone Development
Regulation of Mineral Ion Homeostasis—General Considerations
PTH Biosynthesis and Intraglandular Processing
Regulation of PTH Gene Expression
PTH-Dependent Regulation of Mineral Ion Homeostasis Mediated Through the PTH/PTHrP Receptor (Type 1 Receptor)
Parathyroid hormone (PTH) and PTH-related peptide (PTHrP), along with other calciotropic hormones, play critical roles in calcium homeostasis and bone biology. First discovered as a calcium-regulating hormone in the 1920s,1–3 PTH is secreted by the parathyroid glands and is one of the most important regulators of blood calcium concentration in all terrestrial vertebrate species, from amphibians to mammals. PTHrP, a slightly larger molecule than PTH, was discovered more recently through efforts to identify the factor that causes, when produced in excess by certain tumors, the humoral hypercalcemia of malignancy syndrome. In contrast to PTH, which is produced by discrete endocrine glands, PTHrP is produced as a paracrine/autocrine factor in many different adult and fetal tissues and has, unlike PTH, multiple functions (Fig. 1-1).4–6
FIGURE 1-1 The parathyroid hormone (PTH)/PTH-related peptide (PTHrP) receptor interacts with indistinguishable efficiency and efficacy with PTH and PTHrP, and it activates at least two distinct second messenger systems, cyclic adenosine monophosphate and Ca2+/inositol 1,4,5-triphosphate. The receptor is abundantly expressed in bone and kidney, where it mediates the endocrine actions of PTH, and in the metaphyseal growth plate and numerous other tissues, where it mediates the autocrine/paracrine actions of PTHrP.
PTH and PTHrP most likely evolved from a common ancestral precursor. Despite this common evolutionary origin, both peptides share only limited overall amino acid sequence identity, yet at least their N-terminal regions are sufficiently homologous to enable them to bind to and activate a common G protein–coupled receptor, the PTH/PTHrP receptor (also referred to as PTH1R).7–9 This receptor mediates the most important biological actions of both peptides: PTH-dependent regulation of calcium and phosphorous homeostasis and PTHrP-dependent regulation of endochondral bone formation.10–14
This chapter reviews (1) the comparative chemistry of PTH and PTHrP, their genes, and their interactions with the PTH1R; (2) the current molecular models of productive interactions of the two ligands with their common receptor; and (3) the different biological roles of both peptides on target tissues, such as the role of PTH in calcium homeostasis and bone turnover, the role of PTHrP in bone and cartilage development, and the role of PTH in regulating renal phosphate excretion (see Fig. 1-1). However, the chapter does not review the potentially numerous and still incompletely characterized biological roles of PTHrP outside the field of mineral ion homeostasis and bone biology. The evolutionary history of the principal PTH/PTHrP receptor is reviewed, as well as the functional characteristics of two novel, closely related receptors and the pharmacologic and physicochemical evidence for several additional, still incompletely characterized, receptors for PTH and PTHrP.
Regulation of Mineral Ion Homeostasis—General Considerations
To ensure a multitude of essential cellular functions, the extracellular concentration of calcium (Ca2+o) is maintained within narrow limits.15,16 In terrestrial vertebrates, calcium is necessary for adequate mineralization of the skeleton, which provides mechanical support and protection for internal organs and acts as levers for the various muscle groups involved in locomotion. Because of its high calcium content, 99% of the body’s supply, the skeleton also serves as the most important reservoir from which calcium can be rapidly mobilized. Because food intake and thus the nutritional supply of calcium are usually discontinuous, intestinal calcium absorption occurs only intermittently. Maintenance of a constant blood calcium concentration thus constitutes a major homeostatic challenge, which during evolution led to the development of highly efficient mechanisms to increase intestinal calcium absorption, reduce urinary calcium losses, and facilitate, if necessary, rapid mobilization of calcium from the skeletal reservoir (see Chapter 5).16
In contrast to these environmental challenges of most terrestrial vertebrates, marine animals, which are usually exposed to the high environmental calcium concentration of seawater (10 mM) had to adopt mechanisms by which extracellular calcium could be reduced.17,18 Unlike the diet of terrestrial animals, seawater provides only a very limited supply of phosphate, and this environmental deficiency resulted in the development of mechanisms to conserve phosphate. It thus appears plausible that the efficient intestinal absorption of phosphate and the impressive capacity of the mammalian kidney to retain phosphate15,19 are remnants of earlier evolutionary adaptations to life in the low-phosphate environment of the oceans. To reduce blood calcium concentrations, fish use stanniocalcin, which is produced by the corpuscles of Stannius, as well as several other hormonal factors.17,18,20 Some data indicate that the mammalian homologue of stanniocalcin has similar properties when tested in rodents, but it remains uncertain whether this peptide hormone has a significant physiologic role in mammalian mineral ion homeostasis.21 A widely expressed mammalian peptide, stanniocalcin 2, that was discovered because of its structural homology with stanniocalcin, appears to inhibit phosphate uptake in renal epithelial cells.22 However, newer data (see later) indicate that fibroblast growth factor-23 (FGF-23) and perhaps soluble frizzled-related protein 4 (sFRP4), dentin matrix protein 1 (DMP-1), and matrix extracellular phosphoglycoprotein (MEPE)23 are more likely to be involved physiologically in the regulation of mammalian phosphate homeostasis (see Chapter 6). Calcitonin, made by the ultimobranchial bodies in fish, has a calcium-lowering function in these vertebrate species, but its biological role in mammals remains uncertain (for review, see Chapter 2).24
Parathyroid Hormone
PTH and the active form of vitamin D, 1,25-dihydroxyvitamin D3 [1,25(OH)2D3], are the principal physiologic regulators of calcium homeostasis in humans and all terrestrial vertebrates.11,25,26 Synthesis and secretion of PTH are stimulated by any decrease in blood calcium, and conversely, secretion of the hormone is inhibited by an increase in blood calcium.27–29 This rapid negative feedback regulation of PTH production, along with the biological actions of the hormone on different target tissues, represents the most important homeostatic mechanism for minute-to-minute control of calcium concentration in the extracellular fluid (ECF) (Fig. 1-2).30–32 In contrast to the rapid actions of PTH, 1,25(OH)2D3 is of critical importance for long-term, day-to-day, and week-to-week calcium balance (see Chapter 3). The actions of both hormones are coordinated, and each influences the synthesis and secretion of the other. Calcitonin, the third of the calciotropic hormones known to be important in the regulation of vertebrate mineral ion homeostasis (see Chapter 2), may be vestigial in humans with respect to calcium homeostasis and will not be discussed in this brief review of physiology.
FIGURE 1-2 Rate of change in blood phosphorus and calcium levels in rats with stressed calcium homeostasis (low-calcium diet) after parathyroidectomy without treatment or with 100 U of parathyroid hormone extract given in addition at the time of parathyroidectomy. Rapid and usually fatal hypocalcemia and hyperphosphatemia result within hours unless hormone is given. (Data from Munson PL: Studies on the role of the parathyroids in calcium and phosphorus metabolism, Ann N Y Acad Sci 60:776–796, 1955.)
At least three distinct but coordinated actions of PTH increase the flow of calcium into the ECF and thus increase the concentration of blood calcium (see Fig. 1-2).27–29 Through its rapid actions on the kidney and bone, which are all mediated through the PTH/PTHrP receptor and subsequent secondary messages in specific and highly specialized cells, PTH increases the release of calcium from bone, reduces the renal clearance of calcium, and stimulates the production of 1,25(OH)2D3 by activating the gene encoding 25-hydroxyvitamin D-1α-hydroxylase (1α-hydroxylase) in the kidney. The relative importance of the first two actions of PTH on the rapid, minute-to-minute regulation of calcium is not definitively resolved, but most physiologists have stressed the importance of the effects of PTH on bone in maintaining hour-to-hour calcium homeostasis in the ECF. Several lines of evidence, such as that provided by calcium kinetic analysis, indicate a transfer between ECF and bone of as much as 500 mg of calcium daily, which is equivalent to one-fourth to one-half the total ECF calcium content.15 Besides regulating this transfer of calcium from bone through direct breakdown of bone tissue (mineral and matrix), PTH influences the rates of exchange of calcium adsorbed to the surface of bone; this exchangeable calcium pool can be stimulated to provide a rapid and substantial rate of entry of calcium into blood. In addition to these PTH-dependent actions on bone, actions of PTH on the kidney may also be extremely important in the precise hourly regulation of ECF calcium. The third action of PTH on calcium homeostasis—namely, enhancement of intestinal calcium absorption—is indirect and involves the synthesis of 1,25(OH)2D3 from the biologically inactive precursor 25(OH)D3. However, it is difficult to quantitatively analyze or to proportionately contrast the relative physiologic importance of the direct and indirect actions of PTH on the three principal target tissues: kidney, bone, and intestine.
The complexity of bone as a tissue and the many detectable rates of exchange of calcium between the skeleton and the ECF have made the action of PTH on the skeleton difficult to analyze. The state of calcium in blood is complex; much of the calcium is present as chelates or is bound to plasma proteins (for detailed review, see Chapter 5). Because actual filtered loads depend on the ratio of free and bound forms of calcium, it is difficult to calculate renal calcium clearance accurately. The different PTH-dependent actions to promote calcium entry into the ECF are most clearly defined in conditions of deficiency or excess of PTH, such as during experiments in animals or during controlled observations in patients with disorders of parathyroid gland function. The experimental data in these extremes abundantly affirm the crucial calcium homeostatic role of PTH. However, because of continuous and rapid adjustments in mineral ion concentration, it can be difficult to observe the consequences of hormone action under normal physiologic conditions. For example, the rate of PTH secretion changes continually and rapidly so that the controlled variable, calcium, remains constant, and it may therefore be difficult to experimentally detect small corrective changes.
Teleologically, the action of PTH on the regulation of blood phosphate concentration in terrestrial species is best understood as a secondary, rather than a homeostatic, action. Phosphate is abundant in the food chain in terrestrial existence. Phosphate deficiency, unlike calcium deficiency, in the absence of specific organ dysfunction is, therefore, an unlikely environmental challenge (see Chapter 6 for detailed review of the regulation of phosphate homeostasis). To correct a deficiency in calcium, mineral stores in bone can be rapidly dissolved; such activity results, however, in the simultaneous liberation of ionic calcium and phosphate. Because a high blood phosphate level tends to lower the calcium concentration through multiple mechanisms, the rise in blood calcium that occurs after bone dissolution (desirable homeostatically) is beneficial only if the concomitant increase in blood phosphate concentration (undesirable) can be rapidly corrected. To maximize the control of calcium homeostasis, PTH thus has divergent actions on renal tubular handling of the two mineral ions: It increases the retention of calcium and at the same time diminishes reabsorption of phosphate. Through these mechanisms—namely, increased renal phosphate clearance to prevent hyperphosphatemia and increased tubular calcium reabsorption—PTH guarantees that an elevation in blood calcium results from the increased release of calcium from bone. The renal action of PTH on phosphate homeostasis is biologically predominant over the increased phosphate flux from bone. Consequently, parathyroidectomy (experimentally, in animals) or renal resistance to PTH, as in patients with pseudohypoparathyroidism or renal failure, leads not only to hypocalcemia but also to an increase in blood phosphate and a marked reduction in urinary phosphate excretion (see Fig. 1-2). This finding demonstrates the importance of the PTH-dependent action on phosphate homeostasis in the kidney, which becomes particularly important in disease states when high bone turnover is the result of dietary calcium deficiency or lack of biologically active vitamin D.
Chemistry
The first extracts from bovine parathyroid glands were described in 1925, and the content of biologically active PTH was assessed by their hypercalcemic and phosphaturic properties.2,3 However, it was not until 1959, when Aurbach33 and Rasmussen and Craig34 developed improved extraction procedures, that it became possible to isolate and purify sufficient quantities to determine the primary structure of bovine, porcine, and human PTH through the protein sequence determination methods.35–40 Two groups independently determined the sequences of human and bovine hormones.35–40 Shown in Fig. 1-3A are the sequences of the bovine, porcine, and human hormones determined by one group.36,37,39,40 Discordant sequences for the human PTH polypeptide, and in one position for the bovine hormone, published by the other group,35,38 are not shown in Fig. 1-3A, since nucleotide sequence analysis of genomic and complementary DNA confirmed the amino acid sequences of the first group (the only exception was residue 76 in human PTH, which was determined to be glutamine instead of glutamic acid).36,37,40 Based on these amino acid sequences, the PTH(1–34) fragments of the different species were synthesized, and their biological activities were compared in vitro and in vivo with those of highly purified intact PTH from the same species (Table 1-1). Molecular cloning techniques then led to the deduction of the amino acid sequences of rat, chicken, and dog PTHs,41–44 followed more recently by the identification of PTH molecules in other mammals and fish, as shown in Fig. 1-3A and discussed later. The synthetic peptides used in parathyroid hormone research today are based largely on the (1-34) regions of the mammalian hormone sequences shown in Fig. 1-3A.45,46
Table 1-1
Comparison of the Biological Activity of Parathyroid Peptides from Different Species
*Values are expressed as mean potency with 95% confidence intervals and are based on Medical Research Council research standard A for parathyroid hormone.
From Rosenblatt M, Kronenberg HM, Potts JT Jr: Parathyroid hormone. In DeGroot L (ed): Endocrinology, ed 2, Philadelphia, 1989, WB Saunders, p 853.
FIGURE 1-3 Sequence Relationships in PTH and PTHrP peptides. Comparisons of amino Acid sequences of PTH and PTHrP peptides from different species are shown in A and B, respectively. Comparison of human TIP39 and a PTH-Like peptide from the puffer fish (Takifugu rubripes) to human PTH and PTHrP (the (1–84) region only) is shown in C. In A-C, amino acid identities are shown on black field and similarities on gray field. A phylogenetic tree of the ligands is shown in D, with other family B receptor ligands, secretin, calcitonin, and cotricotropin-releasing factor (CRF) included as marginally homologous ligands. Fugu PTHL is speculated to be a precurssor to PTH and PTHrP.
Extensive sequence homology is present in the mammalian PTH species (see Fig. 1-3A and D); these molecules consist of a single-chain polypeptide with 84 amino acids and a molecular weight of approximately 9400 daltons (that of human PTH[1–84] is 9425 D). The N-terminal region of PTH, which is necessary and sufficient for the regulation of mineral ion homeostasis, shows high sequence conservation among all the vertebrate species (see Fig. 1-3A). The middle portions of the different molecules exhibit the most structural variation, which could suggest that this region of PTH is only of limited functional importance. The nonmammalian PTH homologues of chicken42,43 and fish species Danio rerio (zebrafish)47 and Takifugu ruberipes (puffer fish)48,49 diverge considerably from the mammalian hormones C-terminal of amino acid residue His32. Interestingly, both fish species have two distinct genes encoding two separate PTH molecules, called PTH1 and PTH2.47,48 The zebrafish peptides are considerably shorter than mammalian PTH (67 and 68 residues), while fugu PTH1 is predicted to be 81 residues in length and fugu PTH2 is predicted to be 63 residues.48
After the original work establishing that the first 34 amino acids of mammalian PTH were sufficient to produce a fully active synthetic peptide,45,46 much work has centered on defining the minimum pharmacophore essential for biological activity. In a following section, we describe how sites of ligand interaction with the PTH/PTHrP receptor were defined by performing assays with products of various combinations of shortened and modified PTH ligands and mutagenized receptors. Furthermore, a fusion protein consisting of ligand substituted for most of the receptor amino-terminal extracellular domain was generated, and this helped define a much smaller minimum chain length of PTH peptide needed for biological activity.50 As also discussed later in the section on hormone/receptor interactions, it has been determined that substitutions of non-naturally occurring amino acids (e.g., α-aminoisobutyric acid at positions 1 and 3 in the primary ligand structure) favoring formation of an α-helix, even in short peptides, such as PTH(1–14), produce peptides that are highly potent when tested in vitro using cell-based assays and have highly stabilized helical structure in solution51–55(Fig. 1-4).
FIGURE 1-4 The native (rat) PTH(1-14) sequence and activity-modifying substitutions. The six substitutions shown above the sequence, when combined, enhance activity by as much as 100,000-fold; the Bpa2 substitution shown below confers antagonist properties to the peptide. The (1-9) region (nonshaded circles) is thought to be the minimum-length agonist pharmacophore. Nonencoded amino acids include: α-amino-isobutyric acid (Aib), homoarginine (Har), and parabenzoyl-l-phenylalanine (Bpa).
The in vitro activity of the native PTH(1-14), which is quite weak, is improved about 100,000-fold by the modifications indicated in Fig. 1-4. Shorter PTH peptides have also been shown to be active in vivo, since some cause hypercalcemia and are anabolic on bone, although their potency is much less than that of PTH(1-34) due to a more rapid clearance.56 Replacement of valine-2 in these peptides with bulky amino acids, such as tryptophan or parabenzoyl-l-phenylalanine (Bpa), results in competitive antagonist peptides defective for AC/cAMP and PLC/IP3/Ca2+ signaling, thus confirming the critical role that this conserved valine plays in receptor activation.57 Other longer-length PTH or PTHrP analogs having residue-1 (serine or alanine) replaced by glycine,58 Bpa,59 or tryptophan60 exhibit signal-selective properties in that they efficiently stimulate the cAMP cellular pathway but not the inositol tri-phosphate/intracellular Ca2+ pathway.
The three-dimensional structures of intact PTH(1-84) and the N-terminal biologically active fragments of both PTH and PTHrP have been analyzed by various solution-based methods, including nuclear magnetic resonance (NMR) spectroscopy. Interpretation of these results in terms of biological mode of action is not straightforward, because the ligand interacts with a membrane-embedded receptor, and the biophysical properties of this environment, as well as the receptor-induced conformational changes that occur, are mostly unknown. However, the bioactive (1-34) portions of the ligands are generally found to contain helical structure within their N-terminal and especially C-terminal portions, with some flexibility in the midregion.61–64 An x-ray crystallographic study of PTH(1-34) revealed α-helical structure extending nearly the full length of the peptide.65 A persisting question is thus whether the ligand bound to the receptor adopts a linear and extended62 or “U-shaped” folded structure, the latter suggested by tertiary interactions seen in some solution-phase biophysical studies.61,63,64 Of particular interest has been the question of whether common structural features would be discerned for PTH and PTHrP that could explain their use of apparently overlapping binding sites on the common PTH/PTHrP receptor; to some extent, the helical propensities of at least the C-terminal domains of the peptides are consistent with this possibility.62,66,67
Therefore, consistent with their rather limited homology in primary structure (see Fig. 1-3), convincing evidence has not yet been provided for the conclusion that the N-terminal fragments of PTH and PTHrP display a similar secondary structure in solution. Because of their generally demonstrated similar potencies at the PTH/PTHrP receptor, it seemed likely that both ligands would adopt very similar conformations when part of the active hormone-receptor complex. However, recent data (discussed later) suggest that each ligand selectively binds to or induces a distinct receptor confirmation. Ideally, each hormone should be co-crystallized with the PTH/PTHrP receptor to permit analysis by x-ray diffraction of those intermolecular interactions that are characteristic of the biologically active hormone-receptor complexes. G protein–coupled receptors such as the PTH/PTHrP receptor have multiple membrane-embedded domains and are likely to have complex three-dimensional structures. Interaction with either PTH or PTHrP appears to involve several distinct receptor domains (see later discussion) that may undergo significant conformational changes after ligand binding has occurred, which makes it even more challenging to conduct x-ray or multidimensional NMR analyses. Recent advances, however, have been made it possible to co-crystallize the extracellular portion of the PTH/PTHrP receptor with the carboxyl end of the PTH (1-34) peptide68 and to crystallize the membrane-spanning portion of a distantly related GPCR, the β2-adrenergic receptor.69
Evolution
To maintain extracellular calcium and phosphate concentrations within narrow limits, the intricate regulatory system outlined, in which PTH plays the most important role, developed in the terrestrial animals. In mammals, PTH is produced almost exclusively by the parathyroid glands (only small amounts of its messenger RNA [mRNA] have been detected elsewhere70,71). During evolution, these glands first appear as discrete organs in amphibians, that is, with the migration of vertebrates from an aquatic to a terrestrial existence, and their appearance most likely represents an evolutionary adaptation to an environment that is, by comparison to seawater, low in calcium.17,18,72 Parathyroid glands have not been identified in fish or invertebrate species, but earlier immunologic and RNA hybridization data from fish provided evidence for expression of PTH proteins in several tissues, including pituitary,17,18,73 plasma, brain, kidney, spinal cord, ultimobranchial gland, as well as in the ventral neural tube and mineralizing jaw during development.48,74 Now, with the rapid advances in characterization of complete genomes of multiple species, we have definitive proof of the earlier evolutionary origin of both PTH and PTHrP (see Fig. 1-3). Gene analyses of several teleost fish species, including the zebrafish, Danio rerio, and the puffer fishes Takifugu rubripes and Tetraodon fluviatilis, reveal the duplication of the PTH gene in each case.48,75 Both the PTH1 and PTH2 peptides derived from the zebrafish activate the PTH/PTHrP receptors from different species,49,75 and indeed, a fugu PTH(1-34) peptide has been shown to induce bone anabolic effects in osteopenic ovariectomized rats.76
In addition to PTH, the teleost fish also express PTHrP, again encoded by duplicate genes.75,77–80 Furthermore, PTHrP immunoreactivity has been detected in the cartilaginous sharks and rays81 and in a more primitive agnathan, the lamprey.82 The teleost PTHrPs contain some amino acid residues characteristic of mammalian PTH; for example, fish PTHrP contains Met at position 8, Trp at position 23, and Leu at position 28, which are amino acid residues found in mammalian PTH. However, there is only one amino acid residue, Gln(Q)25, in fish PTH that is found in some mammalian PTHrP species but not in mammalian PTH (see Fig. 1-3). This pattern suggests that the fish proteins may be phylogenetically closer to a common PTH/PTHrP precursor than are the mammalian proteins (see later). Indeed, in addition to duplicate copies of PTH and PTHrP genes, the puffer fish genome contains a fifth gene that encodes a protein containing amino acid residues characteristic of both PTH and PTHrP. This gene, called PTH-L, is phylogenetically an intermediary to PTH and PTHrP and may thus represent first definitive evidence for an ancestral gene from which the two divergent ligand forms evolved (see Fig. 1-3C and D).48,75
The PTH Gene and Its mRNA
The human PTH gene consists of three exons located on chromosome 11p15.83–86 The first exon is 85 nucleotides in length and is noncoding (Fig. 1-5). Exon 2 (90 bp) encodes most amino acids of the prepropeptide sequence, whereas the third exon (612 bp) encodes the remainder of the propeptide sequence and all amino acids of the mature peptide, and it constitutes the 3′ noncoding region.87 Several frequent intragenic polymorphisms (TaqI and PstI88; BstBI89; DraIII90; XmnI91), and a tetranucleotide repeat ([AAAT]n;92) have been identified in the human PTH gene, and some were shown to be informative in genetic linkage studies.93–95 Two mRNAs that are 822 and 793 bp in length are derived in the human gene from the two transcriptional start sites, which follow two different functional TATA boxes that are separated by 29 bp.87 Two closely spaced TATA boxes and two distinct transcripts are also derived from the bovine PTH gene, while rat and chicken PTH genes give rise to only one transcript; as a consequence of a long 3′ noncoding region, the transcript from the chicken PTH gene is unusually long and comprises 2.3 kb.25,96 The genes encoding zebrafish PTH1 and PTH2 have a similar overall organization as the mammalian PTH genes.47
FIGURE 1-5 Schematic of the parathyroid hormone (PTH) gene along several thousand base pairs (approximate length shown by the scale marker for 500 bp). The three exons in the mRNA are represented as numbered rectangles. Control elements are identified in the 5′ noncoding region (5′ NC). A region responsive to vitamin D is within a few hundred base pairs of exon 1. Far upstream are silencers involved in calcium regulation.
PTH Biosynthesis and Intraglandular Processing
During the synthesis of the preproPTH molecule, the signal sequence, which comprises the 25-amino-acid-containing “pre” sequence, is cleaved off after entry of the nascent peptide chain into the intracisternal space bounded by the endoplasmic reticulum. A heterozygous mutation in this leader sequence, which changes a cysteine to an arginine at position −8 and thus impairs processing of preproPTH to proPTH, has been identified as the most plausible molecular cause of an autosomal dominant familial form of hypoparathyroidism.97,98 The mutant hormone was found to be trapped intracellularly, predominantly in the endoplasmic reticulum (ER), leading to a marked up-regulation of ER stress-responsive proteins (BiP and PERK) and the proapoptotic transcription factor, CHOP, indicating that apoptosis-mediated parathyroid cell death is the likely cause of the observed hypoparathyroidism.99
Subsequent to the removal of the pre-sequence, the pro-peptide is transported to the trans-Golgi network, where the pro-sequence (amino acid residues −6 through −1) is removed.100 This latter process may involve furin (paired basic amino acid cleaving enzyme) and/or proprotein convertase-7 (PC-7), which are both expressed in parathyroid tissue; their expression levels do not appear to be regulated by either calcium or 1,25(OH)2D3.101,102 After removal of the basic pro-sequence, the mature polypeptide, PTH(1-84), is packaged into secretory granules. Two proteases, cathepsins B and H, are subsequently involved in the intraglandular generation of carboxyl-terminal PTH fragments from the intact hormone; no amino-terminal PTH fragments appear to be released from the gland.103–105 Since small or intermediate-size carboxyl-terminal fragments of PTH are unlikely to be involved in the regulation of calcium homeostasis, the intraglandular degradation of intact PTH is thought to represent an inactivating pathway, at least with regard to the regulation of mineral ion homeostasis. Consistent with this conclusion, hypercalcemia results in a substantial decrease in PTH secretion and, furthermore, favors the secretion of carboxyl-terminal PTH fragments, including a previously undetected large molecular species that are truncated at the amino-terminus (see following section).105–108 However, recent studies have shown that some amino-terminally truncated PTH fragments, such as PTH(7-84), have hypocalcemic properties in vivo and can furthermore reduce the formation of osteoclasts in vitro.109
The pool of stored, intracellular PTH is small, and the parathyroid cell must therefore have mechanisms to increase hormone synthesis and release in response to sustained hypocalcemia. One such adaptive mechanism is to reduce the intracellular degradation of the hormone, thereby increasing the net amount of intact, biologically active PTH that is available for secretion. During hypocalcemia, the bulk of the hormone that is released from the parathyroid cell is intact PTH(1-84).103–105,107,108 As the level of Ca2+o increases, a greater fraction of intracellular PTH is degraded, and with overt hypercalcemia, most of the secreted immunoreactive PTH consists of biologically inactive C-terminal fragments.10,25,26
Regulation of PTH Gene Expression
Another adaptive mechanism of the parathyroid cell to sustained reductions in Ca2+o is to increase cellular levels of PTH mRNA, a response that takes several hours. A reduction in Ca2+o increases, whereas an elevation in Ca2+o reduces the cellular levels of PTH mRNA by affecting both its stability and the transcriptional rate of its gene.11,26,110,111 Available data suggest that phosphate ions also regulate, directly or indirectly, PTH gene expression. In the rat, hypophosphatemia and hyperphosphatemia, respectively, lower and raise the levels of mRNA for PTH through a mechanism that is independent of changes in Ca2+o or 1,25(OH)2D3. An elevated extracellular phosphate concentration could thus contribute importantly to the secondary hyperparathyroidism frequently encountered in patients with end-stage renal failure, who often have chronically elevated serum phosphate concentrations.
Metabolites of vitamin D, principally 1,25(OH)2D3, also play an important role in the long-term regulation of parathyroid function and may act at several levels: by affecting the secretion of PTH and regulation of its gene, by regulating transcriptional activity of the genes encoding the calcium-sensing receptor (CaSR; see later) and the vitamin D receptor (VDR), as well as by regulating parathyroid cellular proliferation.11,26,110,112 1,25(OH)2D3 is by far the most important vitamin D metabolite that modulates parathyroid function. It acts through a nuclear receptor, the VDR, often in concert with other such receptors (i.e., those for retinoic acid or glucocorticoids), on DNA sequences upstream from the PTH gene (see Chapter 3).113,114 1,25(OH)2D3-induced upregulation of VDR and CaSR expression in the parathyroid could potentiate its inhibitory action(s) on PTH synthesis and secretion.11,26,110 Noncalcemic or less calcemic analogs of 1,25(OH)2D3 inhibit PTH secretion while producing relatively little stimulation of intestinal calcium absorption and bone resorption115–117 and may thus be attractive candidates for treating the hyperparathyroidism of chronic renal insufficiency.
Regulation of Parathyroid Hormone Secretion
A large number of factors modulate PTH secretion in vitro,11,26,118 but most of these factors are not thought to control hormonal secretion in vivo in a biologically relevant manner. Therefore, we focus in this section principally on factors that are the most physiologically meaningful regulators of PTH secretion—that is, the extracellular ionized calcium concentration itself (Ca2+o), 1,25(OH)2D3, and the level of extracellular phosphate ions. Of these three, Ca2+o is most important in the minute-to-minute control of PTH secretion. Indeed, the actions of 1,25(OH)2D3 and phosphate ions on the secretion of PTH probably result at least in part from their effects on hormonal biosynthesis rather than secretion per se.11,26,118 Ca2+o also modulates several other aspects of parathyroid function that indirectly affect PTH secretion, including PTH gene expression, the hormone’s intracellular degradation, and parathyroid cellular proliferation, as described previously. Recent data have shown that novel factors playing key roles in phosphate homeostasis, especially FGF-23 and α-klotho (a coreceptor for FGF receptors), also modulate parathyroid function, inhibiting119,120 and enhancing121 parathyroid function, respectively. Our rapidly improving understanding of how these factors participate in phosphate homeostasis is described in detail in Chapter 6.
Physiologic Control of PTH Secretion by Ca2+o
As illustrated in Fig. 1-6A, the relationship between PTH and Ca2+o is represented by a steep inverse sigmoidal curve that can be quantitatively described by four parameters.122–124 These are the maximal rate of PTH secretion at low Ca2+o (parameter A); the slope of the curve at its midpoint (parameter B); the value of Ca2+o at the midpoint (e.g., the “set point” or the level of Ca2+o half-maximally suppressing PTH release; parameter C); and the minimal secretory rate at high Ca2+o (parameter D) (see Fig. 1-6B). Parameter A in vivo is the sum of the maximal rates of PTH release from all individual parathyroid chief cells, as reflected by the resultant, maximally stimulated level of circulating PTH. Because of the steepness of the Ca2+o-PTH relationship, small alterations in Ca2+o evoke large changes in PTH release, thereby contributing importantly to the near constancy of Ca2+o in vivo. Indeed, parathyroid cells can readily detect reductions in Ca2+o of a few percentage points,123 and the percent coefficient of variation in Ca2+o in humans is less than 2%.125 The set point of the parathyroid gland is the key determinant of the level at which Ca2+o is “set” in vivo, although the parathyroid set point is usually slightly lower than the ambient blood Ca2+.126 Thus, the parathyroid cell is normally more than half-maximally suppressed at normal levels of Ca2+o and has a large secretory reserve for responding to hypocalcemic stress. Nevertheless, PTH levels in vivo also fall dramatically (e.g., by 80%) when Ca2+o rises to frankly hypercalcemic levels,122,123 which is thought to contribute importantly to the mineral ion homeostatic system’s defense against hypercalcemia.126 Furthermore, elevating Ca2+o also decreases the proportion of secreted intact PTH because of increased intraglandular degradation to inactive fragments (see the earlier section, PTH Biosynthesis and Intraglandular Processing, and the later section, Metabolism of PTH).106,127 Even with severe hypercalcemia, however, some residual release of intact PTH(1-84) still occurs in vivo and persists at a level approximately 5% of that observed with a maximal hypocalcemic stimulus29,108,128 (see Fig. 1-6A). This nonsuppressible basal component of PTH release may contribute to the hypercalcemia caused by hyperparathyroidism when the mass of abnormal parathyroid tissue is very great (e.g., in patients with renal failure).124,129–131
FIGURE 1-6 Inverse sigmoidal relationship between Ca2+ and parathyroid hormone (PTH) release and the four-parameter model describing these curves. A, Secretory response of bovine parathyroid glands to induced alterations in plasma calcium concentration. Calves were infused with calcium or ethylenediaminetetraacetic acid, and PTH secretion was assessed by measuring PTH levels in the parathyroid venous effluent. The symbols and vertical bars indicate the secretory rate (mean ± SE) in calcium concentration ranges of 1 or 0.5 mg/100 mL. The number of calves and samples are indicated, respectively, by numbers below and above. B, Sigmoidal curve generated by the equation Y = [(A − D)/(1 + (X/CB)] + D; the significance of A, B, C, and D are described in the text. (A, Data from Hurst JG: Sigmoidal relationship between parathyroid hormone secretion rate and plasma calcium concentration in calves. Endocrinology 10:10, 1978; B, Data from Brown EM: PTH secretion in vivo and in vitro, Miner Electrolyte Metab 8:130–150, 1982.)
The parathyroid cell has a temporal hierarchy of responses to low Ca2+o that permits it to secrete progressively larger amounts of hormone during prolonged hypocalcemia.11,26,118 To meet acute hypocalcemic challenges, PTH is released within seconds from preformed secretory vesicles by exocytosis as dictated by the sigmoidal curve (see Fig. 1-6). Sufficient PTH is stored in the parathyroid chief cell to sustain maximal, low Ca2+o-stimulated PTH release for about 60 to 90 minutes.126 Another rapid response of the parathyroid cell to hypocalcemia that enhances its net synthetic rate of PTH is reduced intracellular hormonal degradation—the opposite of what occurs at high levels of Ca2+o—which occurs within minutes to an hour.106,127 Hypocalcemia persisting for hours to days elicits increased PTH gene expression, whereas that lasting for days to weeks or longer stimulates parathyroid cellular proliferation.11,26,118,132 A greater secretory capacity for PTH on a per-cell basis (e.g., as a result of enhanced PTH gene expression) increases maximal hormonal secretion in vivo, as does an increase in cell number as a result of parathyroid cellular proliferation (see Fig. 1-6). In severe secondary hyperparathyroidism, very large increases in parathyroid cellular mass can elevate circulating PTH levels by 100-fold or more.
In addition to responding to changes in Ca2+o per se, the parathyroid cell also appears to sense the rate of change in Ca2+o such that rapid decrements in calcium promote more vigorous secretory responses than do changes of a similar magnitude occurring more slowly.133 Furthermore, during dynamic testing of parathyroid function in vivo by induced increases or decreases in Ca2+o, PTH in blood is higher at a given serum calcium concentration when Ca2+o is falling than when it is rising (e.g., hysteresis is occurring in this relationship).134,135 The latter results in an apparent direction dependence of the secretory response, which when combined with the rate dependence just described, may allow for a physiologically appropriate, more vigorous secretory response to large rapid decrements in Ca2+o. Also present are circadian136 (for review, see Diaz et al.26) and more rapid (i.e., occurring at rates of one to six pulses per hour) phasic changes in circulating PTH levels,26,137 but the physiologic significance of these changes is not known.
Mechanism of Ca2+o Sensing by Parathyroid Cells and Other Cells Involved in Mineral Ion Homeostasis
The molecular mechanism underlying Ca2+o-regulated PTH secretion involves a G protein–coupled, cell surface Ca2+o-sensing receptor (CaSR).138 The CaSR was first isolated from bovine parathyroid glands139 and subsequently from human parathyroid and several other tissues and species.140 The receptor exhibits the characteristic “serpentine” motif (seven membrane spanning domains) of the superfamily of G protein–coupled receptors (Fig. 1-7). Its long, N-terminal extracellular domain contains the major, but not all, determinants of Ca2+o binding.141–143 Changes in Ca2+o modulate a number of second messenger systems via coupling of CaSR through its intracellular domains to the relevant G proteins regulating these signaling pathways.138 These functions include activation of phospholipases C, A2, and D,144 stimulation of several mitogen-activated protein kinases,145 and inhibition of adenylyl cyclase.146 Despite numerous studies conducted over the past 25 years, a full understanding of the major second messenger pathways through which changes in Ca2+o, acting via the CaSR, regulate various aspects of the function of parathyroid and other CaSR-expressing cells remains elusive (discussed later). Recent evidence, however, indicates key roles for the G proteins, Gq and G11, but their downstream transduction pathways participating in the control of parathyroid function are uncertain.147 In the parathyroid, the CaSR mediates the inhibitory actions of Ca2+o on PTH secretion and gene expression as well as parathyroid cellular proliferation.138,148
FIGURE 1-7 Schematic representation of the predicted topology of the calcium-sensing receptor cloned from human parathyroid gland. HS, Hydrophobic segment; SP, signal peptide; , acidic residues (, conserved among species); branched markers, sites of glycosylation; P, sites of phosphorylation. (From Brown EM, Gamba G, Riccardi D, et al: Cloning and characterization of an extracellular Ca2+-sensing receptor from bovine parathyroid, Nature 366:575–580, 1993.)
The CaSR is also expressed in several additional tissues involved in systemic mineral ion homeostasis, including the calcitonin-secreting C-cells of the thyroid,149 diverse cells within the kidney,150 bone cells and/or their precursors,110 and intestinal epithelial cells. In the C cell, the CaSR mediates the stimulatory action of high Ca2+o on calcitonin secretion, a Ca2+o-lowering hormone. In the kidney, the CaSR in the cortical thick ascending limb of the nephron mediates direct high-Ca2+o-induced inhibition of the tubular reabsorption of Ca2+ and Mg2+.150,151 Therefore, raising Ca2+o both directly inhibits renal tubular reabsorption of Ca2+ via actions on the CaSR expressed in nephron segments involved in hormonal regulation of Ca2+ reabsorption (e.g., by PTH) and indirectly inhibits it by reducing PTH secretion (see the later section, Renal Calcium Reabsorption). The CaSR probably also mediates the long-recognized but poorly understood inhibitory effect of hypercalcemia on renal water conservation, probably exerting this action by inhibiting vasopressin-stimulated water flow in the distal collecting duct.138 A possible physiologic relevance of this action is to prevent the development of excessively high concentrations of calcium in the distal collecting system, thereby perhaps mitigating the risk of renal stone formation.151 Elevating Ca2+o stimulates osteoblastic bone formation and inhibits osteoclastic bone resorption.110,152 The CaSR is expressed by chondrocytes as well as by osteoblasts and osteoclasts and/or their precursors,110 and recent data suggest that it plays key roles in skeletal development.153 It is not currently known whether the CaSR expressed in intestinal epithelial cells plays any role in regulating 1,25(OH)2D3-mediated absorption of calcium.
The identification of hypercalcemic (e.g., familial hypocalciuric hypercalcemia and neonatal severe hyperparathyroidism)154
[/not-level-membership-for-endocrinology-diabetes-and-metabolism-category]
