[level-membership-for-basic-science-category]Michael C. Lee, Mark Abrahams






















Pain and analgesics
Definition of pain
The International Association for the Study of Pain defines pain as ‘an unpleasant sensory and emotional experience associated with actual or potential tissue damage, or described in terms of such damage’. This implies that the degree of pain experienced by the patient may be unrelated to the extent of underlying tissue damage, and that emotional or spiritual distress can add to the patient’s experience of pain (Fig. 18.1).
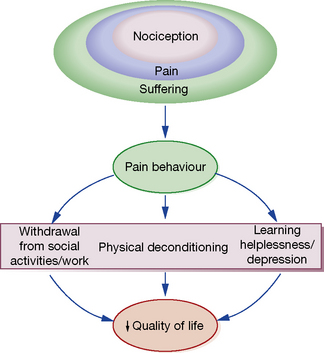
This chapter focuses on the use of drugs for pain relief and illustrates the use of many analgesics that may be encountered in clinical practice. However, clinicians should recognise that the experience of pain is influenced by physical, emotional and psychological factors. While drug therapy is an expedient (and familiar) form of treatment, successful management of pain requires a more holistic approach that addresses all the components of pain.2
Nociception
Pain alerts us to ongoing or potential tissue damage and the ability to sense pain is vital to our survival. The physiological process by which pain is perceived is known as nociception. While the neurobiology of nociception is complex, its appreciation provides a useful framework for understanding the way analgesics work (Fig. 18.2).
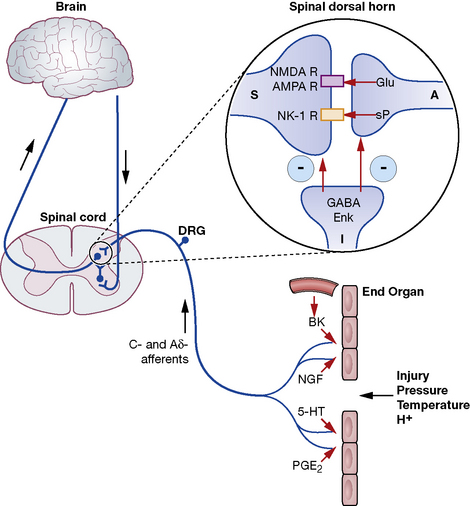
Fig. 18.2 Schematic representation of nociceptive pathways. Noxious stimuli such as protons (H +), temperature (temp) etc. applied to end-organs activate nociceptors. Injury leads to the release of prostaglandins such as prostaglandin E2 (PGE2), serotonin (5-HT), nerve growth factor (NGF) etc. from damaged cells, bradykinin (BK) from blood vessels and substance P (sP) from nociceptors. These agents either activate nociceptors directly or sensitise them to subsequent stimuli by parallel activation of intracellular kinases by G-protein-coupled receptors and tyrosine kinase receptors. Primary nociceptive afferents (C-fibres, Ad-fibres) of dorsal root ganglion (DRG) neurones synapse on second order neurones (S) in the spinal dorsal horn (magnified in inset). Here, glutamate (Glu) and sP released from primary afferent terminals (A) activate glutamate receptors (NMDA R, AMPA R, mGluRs) and neurokinin-1 (NK-1) receptors, respectively, located postsynaptically on spinal neurones. These synapses are negatively modulated by spinal inhibitory interneurones (I), which employ enkephalins (Enk) or γ-aminobutyric acid (GABA) as neurotransmitters. Spinal neurones convey nociceptive information to the brain and brainstem. Activation of descending noradrenergic/norepinergic and/or serotonergic systems, which originate in the brain and brainstem, leads to the activation of spinal inhibitory interneurones (I) thereby resulting in antinociception (http://encref.springer.de/mp/0002.htm).
Evaluation of pain
• Allodynia – pain due to a stimulus which does not normally provoke pain.
• Hyperalgesia – an increased response to a stimulus which is normally painful.
• Paraesthesia – abnormal sensation, e.g. ‘pins and needles’.
• Dyaesthesia – a painful paraesthesia, e.g. burning foot pain in diabetic neuropathy.
Non-opioid analgesics
NSAIDs (non-steroidal anti-inflammatory drugs)
Mechanism of analgesia
Prostanoid is a major sensitiser that is produced at the site of tissue injury. NSAIDs act by inhibiting cyclo-oxygenase, an enzyme involved in the production of prostanoid, as well as other prostaglandins. This enzyme has a number of isoforms, the most studied being cyclo-oxgenase-1 (COX-1) and cyclo-oxygenase-2 (COX-2). Their actions are inhibited by NSAIDs (see Ch. 16). Increased COX-2 production is induced by tissue injury and accounts for the efficacy of COX-2-specific inhibitor drugs (COXIBs). The inhibitory effect of NSAIDs on the production of other prostaglandins is responsible for the common side-effects of these drugs. Among other functions, the prostaglandins produced by cyclo-oxygenase act to protect the gastric mucosa, maintain normal blood flow in the kidney and preserve normal platelet function. Inhibition of prostaglandin production, therefore, can cause gastric irritation, damage to the kidney and an increased risk of bleeding.
Side-effects
All NSAIDs are associated with dose-dependent side-effects. In particular, there is a risk of gastrointestinal bleed, renal toxicity and a possibility of cardiac-related complications. The morbidity related to gastrointestinal adverse effects is considerable (~ 5 per 1000 patients per year of treatment) and catastrophic bleeding can occur without any preceding warning symptoms. For this reason, it is recommended that, when used in the longer term, NSAIDs should be prescribed along with an appropriate gastro-protective agent (see p. 531).
Opioid analgesics
Classification of opioid drugs
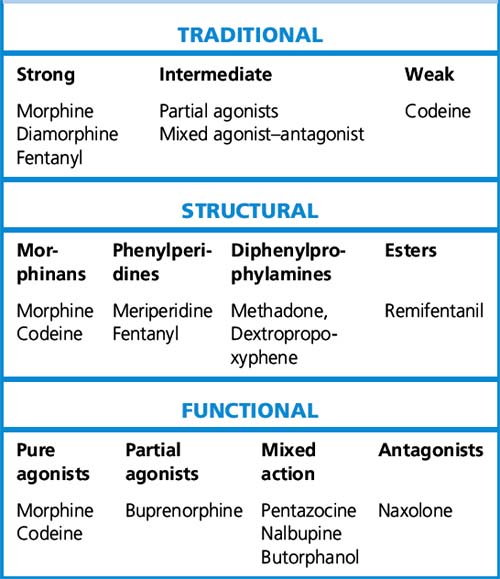
Opioids have been traditionally classified as strong, intermediate and weak, according to their perceived analgesic properties and propensity for addiction. This approach can be misleading, as it implies that weak opioids such as codeine are less effective but safe. Codeine may be less potent than morphine but can cause respiratory depression if given in sufficient quantities. Codeine-like drugs are also frequently abused. Opioids may also be classified according to their structure. As described later, the properties of opioids may be predicted on the basis of activity on opioid and other receptor systems. The functional classification is probably of most clinical use (Table 18.1).
Table 18.1 Classification of opioids
Systemic effects of opioid analgesics
Pharmacokinetics
Bio-availability varies between opioids after oral administration but is generally poor. Methadone is an exception (80%) (Table 18.2).
| Opioid | Approximate oral bio-availability % |
|---|---|
| Hydromorphone | 20 |
| Morphine | 30 |
| Diamorphine | 30 |
| Meperidine | 30 |
| Codeine | 60 |
| Oxycodone | 60 |
| Levorphanol | 70 |
| Tramadol | 80 |
| Methadone | 80 |
Pharmacology of individual opioids
Opioid agonist drugs
Opioids with action on other systems
Pethidine (meperidine)
Pethidine is eight to ten times less potent than morphine, has poor and variable oral absorption, with a short duration of action in the range of 2–3 h. For all these reasons, it is recommended that pethidine should be avoided if alternatives are available.3
Choice of opioid analgesic
Knowledge of equi-analgesic doses of opioids is essential when changing drugs or routes of administration (Tables 18.3 and 18.4). Cross-tolerance between drugs is incomplete, so when one drug is substituted for another, the equi-analgesic dose should be reduced by 50%. The only exception is methadone, which should be reduced by 75–90%. Opioid rotation is commonly used in cancer-related and chronic non-malignant pain as a means of reducing side-effects and limiting the development of tolerance.
| Drug | Oral:parenteral potency ratio* | Parenteral potency relative to morphine** |
|---|---|---|
| Morphine | 1:6 | 1.0 |
| Codeine | 2:3 | 0.1 |
| Hydromorphone | 1:5 | 6.0 |
| Meperidine | 1:4 | 0.15 |
| Oxycodone | 1:2 | 1.0 |
| Methadone | 1:2 | 1.0 |
* Oral–parenteral ratio: for example, morphine is six times more potent parenterally than orally.
** Potency relative to morphine: for example, hydromorphone is six times more potent than an equal dose of morphine when given parenterally.
(Mitchell J P 1989 General care of the patient. In: Claiborne D, Ridner M (eds) Manual of Medical Therapeutics, 26th edn. Little, Brown, p. 5.)
Table 18.4 Opioid oral analgesic equivalents
| Analgesic | Single dose | Equi-analgesic dose Oral morphine |
|---|---|---|
| Codeine | 60 mg | 5 mg |
| Dihydrocodeine | 60 mg | 8 mg |
| Tramadol | 50 mg | 10 mg |
| Meptazinol | 200 mg | 8 mg |
| Buprenorphine sublingual |
200 micrograms | 10 mg |
| Hydromorphone | 1.3 mg | 10 mg |
| Methadone | 1 mg | 10 mg |
| Oxycodone | 5 mg | 10 mg |
Tolerance, dependence and addiction
Addiction
is a behavioural problem characterised by drug-seeking activity in the individual in order to experience its psychotropic effects. This drug-seeking behaviour may persist despite the knowledge that continued use of the drug will result in considerable physical, emotional, social or economic harm. The incidence of addiction in patients taking opioid medications for acute pain is negligible, and is low even in patients on long-term opioids for chronic non-malignant pain (< 10%), provided that the patients are carefully screened for drug misuse potential and adequately monitored.4 The risk of iatrogenic addiction in patients prescribed opioids for cancer-related pain is believed to be extremely low.
Co-analgesics
Adjuvant analgesics used in neuropathic pain
Anticonvulsants
Gabapentin and pregabalin are newer anticonvulsant agents that show good efficacy in clinical trials of neuropathic pain. These drugs bind to the α2δ-1 subunit of voltage-dependent calcium channels and may work by preventing the formation of excitatory synapses within the central nervous system.5 Gabapentin is generally better tolerated than the older anticonvulsants and has a licence in the UK for the treatment of neuropathic pain. It is not metabolised by the liver and has few clinically significant drug interactions. It should be started at a dose of 300 mg at night (100 mg in the elderly) and titrated upwards as tolerated or to a dose of 600–1200 mg three times daily. A saturable gut transport mechanism limits bio-availability at high oral doses (but also protects against overdosage). Pregabalin shares a similar mode of action to gabapentin, but has the advantage of more linear pharmacokinetics and can be given as a twice daily preparation (normal maintenance dose up to 300 mg twice daily).
Pharmacotherapy of acute migraine headaches
The pathophysiology of migraine is complex but a likely causative factor is the release of vasoactive peptides from the sensory nerve terminals that innervate meningeal blood vessels, causing dilatation of the arteries in the meninges, perivascular inflammation and amplification of the nociceptive afferent nerve supply. Sensory input from dural and cerebrovascular sensory fibres is amplified and perceived as pain (allodynia). Activation of the sympathetic nervous system is the likely cause of autonomic symptoms such as nausea and vomiting. Sensory symptoms (aura) are produced by a transient, spreading disturbance in cortical function. Migraine possesses features of inflammatory and functional pain, as well as objective neurologic dysfunction. Diagnosis is based on the headache’s characteristics and associated symptoms.6
Selective 5-HT1 agonists (triptans)
The onset of action of most triptans is 20–60 min (10 min for sumatriptan). If necessary, patients can take another dose after 2 or 4 h. If the appropriate dose of triptan is ineffective or has unacceptable side-effects, consider a switch to an alternative triptan formulation. The drugs can be used in combination with other simple analgesics, NSAIDs and antiemetics. There is a risk of developing serotonin syndrome (see p. 318) if given in combination with other serotonin-reuptake inhibitor drugs (e.g. SSRI or MAOI antidepressants). While the risk of causing birth defects is probably low, triptans should not be used routinely during pregnancy.
Goadsby P.J., Sprenger T. Current practice and future directions in the prevention and acute management of migraine. Lancet Neurol.. 2010;9(3):285–298.
Portenoy R.K. Treatment of cancer pain. Lancet. 2011;377:2236–2247.
Tracey I., Mantyh P.W. The cerebral signature for pain perception and its modulation. Neuron. 2007;55(3):377–391.
Turk D.C., Wilson H.D., Cahana A. Treatment of chronic non-cancer pain. Lancet. 2011;377:2226–2235.
Woolf C.J. Nociceptors – noxious stimulus detectors. Neuron. 2007;55(3):353–364.
Wu C.L., Raja S.N. Treatment of acute post-operative pain. Lancet. 2011;377:2226–2235.
1 Pope John Paul II. Letter handed to John Bonica on the occasion of the Fifth World Congress on Pain. In: Benedetti C, Chapman C R, Giron G (eds) 1990 Opioid Analgesia: Recent Advances in Systemic Administration. Advances in Pain Research and Therapy, vol. 14. Raven Press, New York.
2 ‘Another event at Elsterhorst had a marked effect on me. The Germans dumped a young Soviet prisoner in my ward late one night. The ward was full, so I put him in my room as he was moribund and screaming and I did not want to wake the ward. I examined him. He had obvious gross bilateral cavitation and a severe pleural rub. I thought the latter was the cause of the pain and the screaming. I had no morphia, just aspirin, which had no effect. I felt desperate. I knew very little Russian then and there was no one in the ward who did. I finally instinctively sat down on the bed and took him in my arms, and the screaming stopped almost at once. He died peacefully in my arms a few hours later. It was not the pleurisy that caused the screaming but loneliness. It was a wonderful education about the care of the dying. I was ashamed of my misdiagnosis and kept the story secret.’ Cochrane A L (with M Blythe). London: BMJ (Memoir Club), 1989, p. 82.
3 World Health Organization 1996.
4 Opioids for persistent pain. 2010 Guidelines British Pain Society.
6 Headache Classification Subcommittee of the International Headache Society 2004 International classification of headache disorders. Cephalalgia 24(Suppl.1):9–160.
[/level-membership-for-basic-science-category][not-level-membership-for-basic-science-category]Michael C. Lee, Mark Abrahams
Pain and analgesics
Definition of pain
The International Association for the Study of Pain defines pain as ‘an unpleasant sensory and emotional experience associated with actual or potential tissue damage, or described in terms of such damage’. This implies that the degree of pain experienced by the patient may be unrelated to the extent of underlying tissue damage, and that emotional or spiritual distress can add to the patient’s experience of pain (Fig. 18.1).
This chapter focuses on the use of drugs for pain relief and illustrates the use of many analgesics that may be encountered in clinical practice. However, clinicians should recognise that the experience of pain is influenced by physical, emotional and psychological factors. While drug therapy is an expedient (and familiar) form of treatment, successful management of pain requires a more holistic approach that addresses all the components of pain.2
Nociception
Pain alerts us to ongoing or potential tissue damage and the ability to sense pain is vital to our survival. The physiological process by which pain is perceived is known as nociception. While the neurobiology of nociception is complex, its appreciation provides a useful framework for understanding the way analgesics work (Fig. 18.2).
Fig. 18.2 Schematic representation of nociceptive pathways. Noxious stimuli such as protons (H +), temperature (temp) etc. applied to end-organs activate nociceptors. Injury leads to the release of prostaglandins such as prostaglandin E2 (PGE2), serotonin (5-HT), nerve growth factor (NGF) etc. from damaged cells, bradykinin (BK) from blood vessels and substance P (sP) from nociceptors. These agents either activate nociceptors directly or sensitise them to subsequent stimuli by parallel activation of intracellular kinases by G-protein-coupled receptors and tyrosine kinase receptors. Primary nociceptive afferents (C-fibres, Ad-fibres) of dorsal root ganglion (DRG) neurones synapse on second order neurones (S) in the spinal dorsal horn (magnified in inset). Here, glutamate (Glu) and sP released from primary afferent terminals (A) activate glutamate receptors (NMDA R, AMPA R, mGluRs) and neurokinin-1 (NK-1) receptors, respectively, located postsynaptically on spinal neurones. These synapses are negatively modulated by spinal inhibitory interneurones (I), which employ enkephalins (Enk) or γ-aminobutyric acid (GABA) as neurotransmitters. Spinal neurones convey nociceptive information to the brain and brainstem. Activation of descending noradrenergic/norepinergic and/or serotonergic systems, which originate in the brain and brainstem, leads to the activation of spinal inhibitory interneurones (I) thereby resulting in antinociception (http://encref.springer.de/mp/0002.htm).
Evaluation of pain
• Allodynia – pain due to a stimulus which does not normally provoke pain.
• Hyperalgesia – an increased response to a stimulus which is normally painful.
• Paraesthesia – abnormal sensation, e.g. ‘pins and needles’.
• Dyaesthesia – a painful paraesthesia, e.g. burning foot pain in diabetic neuropathy.
Non-opioid analgesics
NSAIDs (non-steroidal anti-inflammatory drugs)
Mechanism of analgesia
Prostanoid is a major sensitiser that is produced at the site of tissue injury. NSAIDs act by inhibiting cyclo-oxygenase, an enzyme involved in the production of prostanoid, as well as other prostaglandins. This enzyme has a number of isoforms, the most studied being cyclo-oxgenase-1 (COX-1) and cyclo-oxygenase-2 (COX-2). Their actions are inhibited by NSAIDs (see Ch. 16). Increased COX-2 production is induced by tissue injury and accounts for the efficacy of COX-2-specific inhibitor drugs (COXIBs). The inhibitory effect of NSAIDs on the production of other prostaglandins is responsible for the common side-effects of these drugs. Among other functions, the prostaglandins produced by cyclo-oxygenase act to protect the gastric mucosa, maintain normal blood flow in the kidney and preserve normal platelet function. Inhibition of prostaglandin production, therefore, can cause gastric irritation, damage to the kidney and an increased risk of bleeding.
Side-effects
All NSAIDs are associated with dose-dependent side-effects. In particular, there is a risk of gastrointestinal bleed, renal toxicity and a possibility of cardiac-related complications. The morbidity related to gastrointestinal adverse effects is considerable (~ 5 per 1000 patients per year of treatment) and catastrophic bleeding can occur without any preceding warning symptoms. For this reason, it is recommended that, when used in the longer term, NSAIDs should be prescribed along with an appropriate gastro-protective agent (see p. 531).
Opioid analgesics
Classification of opioid drugs
Opioids have been traditionally classified as strong, intermediate and weak, according to their perceived analgesic properties and propensity for addiction. This approach can be misleading, as it implies that weak opioids such as codeine are less effective but safe. Codeine may be less potent than morphine but can cause respiratory depression if given in sufficient quantities. Codeine-like drugs are also frequently abused. Opioids may also be classified according to their structure. As described later, the properties of opioids may be predicted on the basis of activity on opioid and other receptor systems. The functional classification is probably of most clinical use (Table 18.1).
Table 18.1 Classification of opioids
