[level-membership-for-basic-science-category]
33 Pain
Acute pain may be thought of as a physiological process having a biological function, allowing the patient to avoid or minimise injury. Persistent pain, on the other hand, may be described more as a disease than a symptom (Woolf, 2004).
Aetiology and neurophysiology
Neuroanatomy of pain transmission
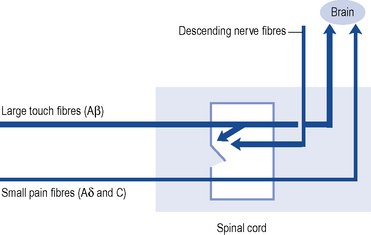
Pain transmission further within the Central Nervous System (CNS) is far more complex and understood less well. The most important parts of this process are the wide dynamic range cells in the spinothalamic tract that project to the thalamus and to the somatosensory cortex beyond. Modulation or inhibition of these neurones within the spinal cord result in less activity in the pain pathway. This modulatory action can be activated by stress or certain analgesic drugs such as morphine and is an important component of the gate theory of pain (Fig. 33.1). The gate control theory recognises the pivotal role the spinal cord plays in the continual modulation of neuronal activity by the relative activity of large (Aβ) and small (Aδ and C) fibres and by descending messages from the brain. Conversely, other influences can lead to an increased sensitivity to noxious stimuli. The most important of these is pain itself and further painful stimuli can lead to increased pain from relatively trivial insults. This occurs through neurochemical and anatomical changes within the CNS that have been termed central sensitisation.
Assessment of pain
Pain is a subjective phenomenon and quantitative assessment is difficult (Breivik et al., 2008). The most commonly used instruments are visual analogue and verbal rating scales. Visual analogue scales are 10 cm long lines labelled with an extreme at each end; usually ‘no pain at all’ and ‘worst pain imaginable’. The patient is required to mark the severity of the pain between the two extremes of the scale. Verbal rating scales use descriptors such as ‘none’, ‘mild’, ‘moderate’ and ‘excruciating’. More elaborate questionnaires such as the Brief Pain Inventory and the McGill Pain Questionnaire help to describe other aspects of the pain, and pain diaries record the influence of activity and medication on pain.
Management
Analgesic ladder
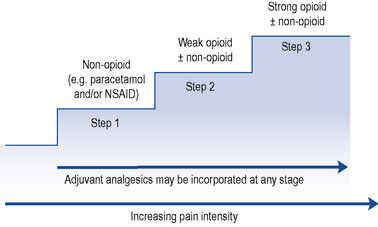
The World Health Organization (WHO) analgesic ladder (Fig. 33.2) forms the basis of many approaches to the use of analgesic drugs. There are essentially three steps: non-opioid analgesics, weak opioids and strong opioids. The analgesic efficacy of non-opioids, such as paracetamol and non-steroidal anti-inflammatory drugs (NSAIDs) (e.g. aspirin, ibuprofen and diclofenac), is limited by side effects and ceiling effects, that is, beyond a certain dose, no further pharmacological effect is seen. If pain remains uncontrolled, then a weak opioid, such as codeine or dihydrocodeine, may be helpful. There may be additional benefit in combining a weak opioid with a non-opioid drug, although many commercial preparations contain inadequate quantities of both components and are no more effective than a non-opioid alone. Strong opioids, of which morphine is considered the gold standard, have no ceiling effect and therefore increased dosage continues to give increased analgesia but side effects often limit effectiveness. Adjuvant drugs, such as corticosteroids, antidepressants or anti-epileptics, may be considered at any step of the ladder.
Analgesic drugs
Paracetamol
Despite being used in clinical practice for over 50 years and much investigation, the mechanism by which paracetamol exerts its analgesic effect remains uncertain. Inhibition of prostaglandin synthesis within the CNS has been proposed, although this is probably not the only mechanism. Interaction with the serotonin (Tjolsen et al., 1991) and endocannabinoid (Högestätt et al., 2005) neurotransmitter systems have been demonstrated in animal models.
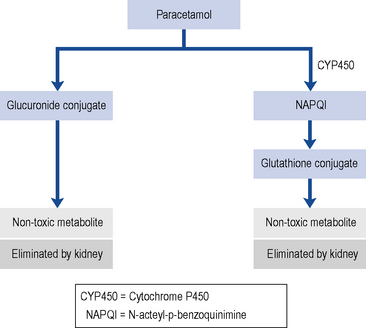
With normal doses, the majority of paracetamols are metabolised and inactivated in the liver, undergoing a phase II conjugation reaction with glucuronic acid (Fig. 33.3). A small P450 mediated reaction that forms a reactive intermediate, N-acteyl-p-benzoquinimine (NAPQI). Usually, NAPQI can be deactivated by conjugation with glutathione in the liver. However, following ingestion of a large amount of paracetamol the hepatic stores of both glucuronic acid and glutathione become depleted leaving free NAPQI, which causes liver damage.
The usual therapeutic dose for adults is paracetamol 1 g taken four times daily. It is very important that this dose is not exceeded, otherwise hepatotoxicity is more common. This may be particularly problematic for malnourished adults with low body weight (Claridge et al., 2010). A reduced maximum daily infusion dose (3 g/24 hours) is recommended for patients with hepatocellular insufficiency, chronic alcoholism or dehydration. Paracetamol is also available as an over-the-counter (OTC) medicine and is a component of many cold and influenza remedies. Compared with other analgesics, paracetamol is not as potent; however, when taken in combination with a NSAID or opioid, there is an additive analgesic effect.
Non-steroidal anti-inflammatory drugs
Mode of action
NSAIDs exert their analgesic and anti-inflammatory effects through inhibition of the enzyme cyclo-oxygenase. NSAIDs are used widely to relieve pain, with or without inflammation, in people with acute and persistent musculoskeletal disorders. In single doses, NSAIDs have superior analgesic activity compared to paracetamol (Hyllested et al., 2002). In regular higher dosages, they have both analgesic and anti-inflammatory effects, which makes them particularly useful for the treatment of continuous or regular pain associated with inflammation. NSAIDs have been shown to be suitable for the relief of pain in dysmenorrhoea, toothache and some headaches and to treat pain caused by secondary bone tumours, which result from lysis of bone and release of prostaglandins.
Guidance on NSAID use
The lowest effective dose of NSAID or COX-2 selective inhibitor should be prescribed for the shortest time necessary. The need for long-term treatment should be reviewed periodically. Prescribing should be based on the safety profiles of individual NSAIDs or COX-2 selective inhibitors, on individual patient risk profiles, for example, gastro-intestinal and cardiovascular. Prescribers should not switch between NSAIDs without careful consideration of the overall safety profile of the products and the patient’s individual risk factors, as well as the patient’s preference (Medicines and Healthcare Regulatory Agency, 2006).
Weak opioids
Dextropropoxyphene
Historically, dextropropoxyphene was prescribed in combination with other analgesics such as paracetamol (co-proxamol). There are few data on its therapeutic value, and at least one review concluded that analgesic efficacy is less than aspirin and barely more than placebo. At best, dextropropoxyphene failed to show any superiority over paracetamol (Li Wan Po and Zhang, 1997). At worst, it is a dangerous drug which has the potential for steadily developing toxicity. Patients with hepatic dysfunction and poor renal function are particularly at risk. It is associated with problems in overdose, notably a non-naloxone reversible depression of the cardiac conducting system. Dextropropoxyphene interacts unpredictably with a number of drugs, including carbamazepine and warfarin. In 2005, the Medicines and Healthcare products Regulatory Agency (MHRA) announced concerns about the safety and effectiveness of co-proxamol and directed that it should be withdrawn from clinical use in the UK; however, it still remains available as an unlicensed medicine for the small number of patients who do not obtain analgesia with other analgesic medicines.
Strong opioids
Other strong opioids
The relative potencies of the commonly used opioids are summarised in Table 33.1.
| Drug | Potency (morphine = 1) |
|---|---|
| Codeine | 0.1 |
| Dihydrocodeine | 0.1 |
| Tramadol | 0.2 |
| Pethidine | 0.1 |
| Morphine | 1 |
| Diamorphine | 2.5 |
| Hydromorphone | 7 |
| Methadone | 2–10 (with repeat dosing) |
| Fentanyl (transdermal) | 150 |
Clinical considerations
Use of opioids is almost universally accepted in cancer pain but many patients with persistent non-cancer pain can find considerable relief with strong opioids; however, barriers to their use in this context appear to be based more on ignorance and political fashions than clinical evidence (Ballantyne and Mao, 2003). As a general rule, strong opioids effective in the management of neuropathic and musculoskeletal pain, including osteoarthritis, are less effective for sympathetically maintained pain.
Adverse effects of opioids
Tolerance, dependence and addition
Persistent treatment with opioids often causes tolerance to the analgesic effect, although the mechanism remains unclear (Holden et al., 2005). When this occurs the dosage should be increased or, alternatively, another opioid can be substituted, since cross-tolerance is not usually complete. Addiction is very rare when opioids are prescribed for pain relief.
Smooth muscle spasm
Opioids cause spasm of the sphincter of Oddi in the biliary tract and may cause biliary colic, as well as urinary sphincter spasm and urinary retention. Thus, in biliary or renal colic, it may be preferable to use another drug without these effects. Pethidine was believed to be the most effective in these circumstances but the evidence for this has been questioned (Thompson, 2001).
Adjuvant medication
To be an analgesic, a drug must relieve pain in animal models and give demonstrable and reliable pain relief in patients. Drugs such as the opioids and the NSAIDs clearly are analgesics. In some types of pain, such as cancer pain or neuropathic pain, the addition of non-analgesic drugs to analgesic therapy can enhance pain relief. A list of some adjuvant drugs is given in Table 33.2. It should be remembered that some drugs such as tricyclic antidepressants (TCAs) have intrinsic analgesic activity, perhaps related to their ability to affect 5-HT and noradrenergic neurotransmission.
| Drug class | Type of pain | Example |
|---|---|---|
| Anti-epileptics | Neuropathic pain | Carbamazepine |
| Migraine | Sodium valproate | |
| Cluster headache | Gabapentin | |
| Pregabalin | ||
| Lamotrigine | ||
| Antidepressants | Neuropathic pain | Amitriptyline |
| Musculoskeletal pain | Imipramine | |
| Venlafaxine | ||
| Duloxetine | ||
| Intravenous anaesthetic agents | Neuropathic pain | Ketamine |
| Burn pain | ||
| Cancer pain | ||
| Skeletal muscle relaxants | Muscle spasm | Baclofen |
| Spasticity | Dantrolene | |
| Botulinum toxin (type A) | ||
| Steroids | Raised intracranial pressure | Dexamethasone |
| Prednisolone | ||
| Nerve compression | ||
| Antibiotics | Infection | As indicated by culture and sensitivity |
| Antispasmodics | Colic | Hyoscine butylbromide |
| Smooth muscle spasm | Loperamide | |
| Hormones/hormonal analogues | Malignant bone pain | Calcitonin |
| Spinal stenosis | Octreotide | |
| Intestinal obstruction | ||
| Bisphosphonates | Bone pain (caused by cancer or osteoporosis) | Pamidronate (for cancer pain) |
| Alendronate |
Antidepressants
The TCAs are frequently used for the treatment of persistent pain conditions with and without the anti-epileptics, and there is a substantial body of literature about their analgesic action (McQuay et al., 1996).
Clinical use of antidepressants in persistent pain
Tricyclic antidepressants have a wide range of adverse effects due to interaction with histamine and muscarinic acetylcholine receptors and these may cause a marked reduction in patient adherence. Newer antidepressant drugs have generally been disappointing from the analgesic perspective. However, much of the research has looked at the selective serotonin reuptake inhibitors (SSRIs). Scientific (Sindrup and Jensen, 1999) and clinical evidence (Sindrup et al., 2005) suggest that a combination of noradrenergic and serotonergic transmission both need to be enhanced for an analgesic effect to be seen. The serotonin/noradrenaline reuptake inhibitors (SNRIs) venlafaxine and duloxetine have effects on both monoamines and appear to possess analgesic activity in neuropathic pain models. A number of antidepressant compounds, including trazodone and mirtazepine, do not act via monoamine reuptake inhibition and do not appear to possess intrinsic analgesic activity. They are effective antidepressants and may have a place in the treatment of co-existing depression but analgesia should be treated separately.
Anti-epileptics
The usefulness of this group of drugs is well established for the treatment of neuropathic pain (McQuay et al., 1995). Conditions which may respond to anti-epileptics include trigeminal neuralgia, glossopharyngeal neuralgia, various neuropathies, lancinating pain arising from conditions such as postherpetic neuralgia and multiple sclerosis and similar pains that may follow amputation or surgery. Several classes of drugs show anti-epileptic activity and these can be broadly classed as sodium channel blockers (carbamazepine, phenytoin), glutamate inhibitors (lamotrigine), voltage gated calcium channel ligands (gabapentin, pregabalin), GABA potentiators (sodium valproate, tiagabine) or drugs showing a mixture of these effects (topiramate). Failure to respond to one particular drug does not indicate that anti-epileptics as a broad class will be ineffective. A drug with a different mechanism of action or combination therapy could be considered.
Ketamine
Ketamine is an intravenous anaesthetic agent with a variety of actions within the CNS. Many of its effects are related to its activity at central glutamate receptors, although it also has actions at certain voltage-gated ion channels and opioid receptors. Low doses of ketamine (0.1–0.3 mg/kg/h via the intravenous route) can produce profound analgesia, even in situations where opioids have been ineffective, such as neuropathic pain. Despite its variable oral availability, oral administration of ketamine can be surprisingly effective (Annetta et al., 2005; Mercadante, 1996). Its usefulness is limited by troublesome psychotropic side effects, although the simultaneous administration of benzodiazepines or antipsychotics can reduce these problems.
Cannabinoids
Cannabis has been used as an analgesic for hundreds of years. Problems concerning the legal status of cannabis in most countries have hindered scientific investigation of its analgesic properties. The active ingredient in preparations made from the hemp plant, Cannabis sativa, is δ-9 tetrahydrocannabinol. This compound has analgesic activity in animal models of experimental pain as well as in the clinical situation (Burns and Ineck, 2006). Overall, analgesic activity appears relatively weak and it has not been possible to separate the analgesic activity from the potent psychotropic effects characteristic of these drugs but a commercial preparation is now licensed for the management of spasticity in multiple sclerosis. There may be a clearer analgesic effect in neuropathic pain but the evidence for this remains anecdotal.
Stimulation-produced analgesia
Treatment of selected pain syndromes
Postoperative pain
Apart from the obvious humanitarian benefit of relieving suffering, pain relief is desirable for a number of physiological reasons after surgery or any form of major tissue injury. For example, poor-quality analgesia reduces respiratory function, increases heart rate and blood pressure, and amplifies the stress response to surgery. The use of intermittent and patient-controlled intermittent intravenous opioids injections has been described earlier in this chapter. However, opioids themselves may delay recovery and are associated with adverse events in the postoperative period (Kehlet et al., 1996). It is now common to treat postoperative pain using a ‘multimodal approach’, consisting of paracetamol, NSAIDs, opioids and local anaesthetic blocks or wound infiltration. NSAIDs such as diclofenac and ketorolac are used frequently, but must be used with caution in the postoperative period where there is a possibility of renal stress, such as blood loss, and the normal protective effect of prostaglandins on the kidney will be lost, culminating in acute renal failure. There is no evidence to support the pre-emptive use of either NSAIDs or local anaesthetic techniques, although there is some theoretical and clinical evidence suggesting that opioids given prior to surgery may be more effective than when given postoperatively.
Cancer pain
Cancer and pain are not synonymous. One-third of patients with cancer do not experience severe pain. Of the remaining two-thirds that do, about 88% can be controlled using basic principles of pain management (Scottish Intercollegiate Guidelines Network, 2008). Pain associated with cancer may arise from many different sources, and may exhibit the characteristics of both acute and persistent pain. The mechanisms and sources of cancer pain may change with time and regular assessment is required. Cancer occurs more frequently in the elderly, who may have a larger proportion of painful conditions than the general population. Pain may be arising from these sources too, and these require treatment at the same time.
Although this chapter is concerned only with the management of pain, care of the patient with a terminal illness requires management of all aspects of the patient. The Liverpool Care Pathway (LCP) is a resource recommended to promote high-quality care in the last days of life (Ellershaw and Wilkinson, 2003). At a basic level, the LCP is a way of acknowledging that death is imminent and ensuring the patient’s comfort by omitting long-term non-essential medication and anticipatory prescribing in case the patient experiences pain, delirium, vomiting or breathlessness.
Neuropathic pain
Neuropathic pain may be defined as ‘pain arising as a direct consequence of a disease or lesion affecting the somatosensory system’ and may occur as a result of pathological damage to nerve fibres in a peripheral nerve or in the CNS (see Table 33.3). Neuropathic pain may be spontaneous in nature (continuous or paroxysmal) or evoked by sensory stimuli. As the underlying aetiology is different to inflammatory types of pain, patients typically present with disturbances in sensory function often describing their pain as tingling, shooting or electric shocks. It is possible for patients to present with pain in the context of sensory loss. Unlike inflammatory pain, neuropathic pain serves no biological advantage and can be described as an illness in its own right.
| Cause of neuropathy | Examples |
|---|---|
| Trauma | Phantom limb |
| Peripheral nerve injury | |
| Spinal cord injury | |
| Surgical | |
| Infection/inflammation | Post-herpetic neuralgia |
| HIV | |
| Compression | Trigeminal neuralgia |
| Sciatica | |
| Cancer | Invasion/compression of nerve tissue by tumour |
| Ischaemia | Post-stroke pain |
| Metabolic neuropathies (e.g. diabetic peripheral neuropathy) | |
| Demyelination | Multiple sclerosis |
| Drugs | Vinca alkaloids |
| Ethanol | |
| Taxols | |
| Anti-bacterials for TB and HIV |
Typically, neuropathic pain does not respond as well to conventional analgesics, such as paracetamol and NSAIDs. Guidelines for the pharmacological management of neuropathic pain in the non-specialist setting have been published (National Institute for Health and Clinical Excellence, 2010).
Specific neuropathic pain syndromes
Postherpetic neuralgia
The pain associated with herpes zoster infection is severe, continuous and often described as burning and lancinating. Antiviral therapy, such as aciclovir, initiated at the first sign of the rash can reduce the duration of the pain, particularly postherpetic pain, which follows the disappearance of the rash. Tricyclic antidepressants such as amitriptyline are the mainstay of treatment, commencing with low doses (e.g. amitriptyline 10–25 mg at night) and gradually increased according to pain relief (usual dose amitriptyline 50–75 mg at night). This may be combined with anti-epileptic drugs if the response is poor or incomplete. Carbamazepine is historically important but newer anti-epileptic drugs, such as gabapentin and pregabalin, are considered first-line therapy and may be better tolerated. One study has demonstrated a significant difference in the incidence, and to a lesser extent the intensity, of pain in patients who received a single epidural methylprednisolone and bupivacaine injection, compared with those who received antiviral therapy and analgesia as ‘standard care’ (van Wijck et al., 2006). However, given the modest clinical effects on acute pain and no effect on the incidence of postherpetic neuralgia, the routine use of epidural local anaesthetic and steroid injection is not widely supported.
Diabetic peripheral polyneuropathy
Nerve damage and neuropathy is one of the long-term complications of diabetes mellitus (see Chapter 44) and is most prevalent in elderly patients with type II diabetes. Often patients describe numbness but also experience a burning sensation on their feet. The sensory loss can result in painless foot ulcers. Tricyclic antidepressants or serotonin noradrenaline reuptake inhibitors (duloxetine or venlafaxine), and anti-epileptics, such as gabapentin and pregabalin, have been demonstrated to be beneficial.
Peripheral nerve injury and neuropathy
Damage to, or entrapment of, nerves can cause pain, unpleasant sensations and paraesthesiae. Tricyclic antidepressants and anti-epileptic drugs, such as gabapentin, have been used with some success to treat neuropathic pain (Moore et al., 2011). A neuroma occurs when damaged or severed nerve fibres sprout new small fibres in an attempt to regenerate. Pain develops several weeks after the nerve injury, and is often due to the neuroma growing into scar tissue, causing pain as it is stretched or mobilised. Treatment of neuroma is very difficult and few treatments are successful. Options include surgery and injections of steroid and local anaesthetic agents.
Back and neck pain
Back pain is one of the commonest reasons for presentation to a medical practitioner. Despite this, the problem is poorly understood. The most practical classification is based on the duration of symptoms (BenDebba et al., 1997). Acute low pain is generally defined as pain that lasts for a few days or weeks. The majority of these problems tend to be self-limiting and resolve spontaneously. Typical treatments include rest, adequate analgesia with paracetamol, combined with a NSAID and/or a weak opioid, and physiotherapy.
Persistent back pain lasts for much longer and progressively leads to a chronic state associated with pain, depression, anxiety and disability. Early intervention is necessary to ensure good functional and vocational outcomes. If a patient is off work for as much as 6 months, then there is a less than 50% chance of them ever returning to work. The likelihood of returning to work falls to less than 25% after 1 year and is almost zero after 2 years. Although pharmacological therapies may aid rehabilitation, other treatment strategies have a greater role to play in the management of persistent back pain. Guidelines for the management of non-specific persistent low back pain have been developed (National Institute for Health and Clinical Excellence, 2009). It is essential for the patient to develop self-management skills, and current recommendations emphasise the importance of using a biopsychosocial approach to manage this problem. Other treatment options include exercise programmes, manual therapy and acupuncture.
Osteoarthritis and rheumatoid arthritis
Pain often is a presenting symptom in osteoarthritis or the inflammatory arthritidies, which include rheumatoid arthritis. The pathophysiology and management of osteoarthritis and rheumatoid arthritis is covered in Chapter 53.
Headache
Migraine
Most migraine attacks respond to simple analgesics such as aspirin or paracetamol. Soluble preparations are best, as gut motility is reduced during a migraine attack and absorption of oral medication may be delayed. Migraine treatment has improved markedly with the development of the triptan drugs such as almotriptan, eletriptan, rizatriptan, sumatriptan, naratriptan and zolmitriptan (Goadsby, 2005). These are 5HT1B/1D-agonists that will often abort an attack, especially when given by the subcutaneous route. Their vasoconstrictor activity precludes their use in patients with angina or cerebrovascular disease but side effects are less serious than with the ergot derivatives they have replaced.
Burn pain
A summary of medicine indications and common therapeutic problems associated with analgesic use are presented in Table 33.4.
| Problem | Solution | Example |
|---|---|---|
| Neuropathic pain | Anti-epileptics | Carbamazepine |
| Sodium valproate | ||
| Gabapentin | ||
| Lamotrigine | ||
| Antidepressants | Amitriptyline | |
| Imipramine | ||
| Intravenous anaesthetic agents | Ketamine | |
| Malignant bone pain | Bisphosphonates | Pamidronate |
| Calcitonin | ||
| Muscle spasm/spasticity | Skeletal muscle relaxants | Baclofen |
| Dantrolene | ||
| Botulinum toxin (type A) | ||
| Raised intracranial pressure | Corticosteroids | Dexamethasone |
| Prednisolone | ||
| Nausea with morphine | Antiemetic | Cyclizne |
| Droperidol | ||
| Ondansetron | ||
| Use an alternative route of administration | Topical or subcutaneous | |
| Constipation | Determine if drug induced, for example, opioids or tricyclic antidepressant | Co-prescribe laxatives (e.g. docusate sodium and senna) |
| Antidepressants in patients with ischaemic heart disease | Use a less cardiotoxic antidepressant | Duloxetine, venlafaxine |
| Drug interactions with carbamazepine | Use an anti-epileptic which does not affect hepatic enzymes | Gabapentin |
| Renal failure | Morphine accumulates; use lower dose | Fentanyl |
| Buprenorphine | ||
| Use a drug which is not eliminated by kidney | ||
| Sedation/ impaired cognition | Identify any drug-related causes and adjust dose or stop drug |
Answer
Mrs NP has developed diabetic peripheral neuropathy, a type of neuropathic pain. She is experiencing intolerable side effects from the increased dose of amitriptyline and therefore does not take it regularly. There are several options to improve tolerability. Firstly, Mrs NP should consider increasing the dose of amitriptyline more slowly. She may also benefit from taking her amitriptyline dose earlier in the evening, approximately 60–90 min before retiring to bed, so that it results in less hangover effect the following day. If neither of these strategies is beneficial, she should consult her primary care doctor about switching to an alternative drug to manage her neuropathic symptoms. Recent guidance (National Institute for Health and Clinical Excellence, 2010) recommends either pregabalin or duloxetine as alternatives to a tricyclic anti-depressant as first-line therapy for neuropathic pain in the non-specialist setting.
Annetta M.G., Iemma D., Garisto C., et al. Ketamine: new indications for an old drug. Curr. Drug Targets. 2005;6:789-794.
Ballantyne J.C., Mao J. Opioid therapy for chronic pain. N. Engl. J. Med.. 2003;349:1943-1953.
BenDebba M., Torgerson W.S., Long D.M. Personality traits, pain duration, and severity, functional impairment, and psychological distress in patients with persistent low back pain. Pain. 1997;72:115-125.
Breivik H., Borchgrevink, P.C., et al. Assessment of pain. British Journal of Anaesthesia. 2008;101:17-24.
Burns T.L., Ineck J.R. Cannabinoid analgesia as a potential new therapeutic option in the treatment of persistent pain. Ann. Pharmacother.. 2006;40:251-260.
Claridge L.C., Eksteen B., Smith A., et al. Acute liver failure after administration of paracetamol at the maximum recommended daily dose in adults. Br. Med. J. 341, c6764,. doi: 10.1136/bmj.c6764, 2010.
Ellershaw J., Wilkinson S. Care of the Dying: A Pathway to Excellence. Oxford: Oxford University Press; 2003.
Goadsby P.J. Advances in the understanding of headache. Br. Med. Bull.. 2005;73:83-92.
Högestätt E.D., Jonsson B.A., Ermund A., et al. Conversion on acetaminophen to the bioactive N-acylphenolamine AM404 via fatty acid hydrolase-dependent arachadonic acid conjugation in the nervous system. J. Biol. Chem.. 2005;280:405-412.
Holden J.E., Jeong Y., Forrest J.M. The endogenous opioid system and clinical pain management. AACN Clin. Issues. 2005;16:291-301.
Hyllested M., Jones S., Pedersen J.L., et al. Comparative effect of paracetamol, NSAIDs or their combination in postoperative pain management: a qualitative review. Br. J. Anaesth.. 2002;88:199-214.
Kehlet H., Rung G.W., Callesen T. Postoperative opioid analgesia: time for a reconsideration? J. Clin. Anesth.. 1996;8:441-445.
Li Wan Po A., Zhang W.Y. Systematic overview of co-proxamol to assess analgesic effects of addition of dextropropoxyphene to paracetamol. Br. Med. J.. 1997;315:1565-1571.
McQuay H., Carroll D., Jadad A.R., et al. Anticonvulsant drugs for management of pain: a systematic review. Br. Med. J.. 1995;311:1047-1052.
McQuay H.J., Tramer M., Nye B., et al. A systematic review of antidepressants in neuropathic pain. Pain. 1996;68:217-227.
Medicines and Healthcare Regulatory Agency. Updated safety information for non-steroidal anti-inflammatory drugs (NSAIDs). Available at http://www.mhra.gov.uk/NewsCentre/Pressreleases/CON2025039, 2006.
Mercadante S. Ketamine in cancer pain: an update. Palliat. Med.. 1996;10:225-230.
Moore, R.A., Wiffen, P.J., Derry, S., McQuay, H.J. Gabapentin for chronic neuropathic pain and fibromyalgia in adults. Cochrane Database of Systematic Reviews 2011, Issue 3. Art. No.: CD007938. DOI: 10.1002/14651858.CD007938.pub2.
National Institute for Health and Clinical Excellence. Low back pain. Early management of persistent non-specific low back pain, Clinical Guideline 88. London: NICE. 2009. Available at http://www.nice.org.uk/nicemedia/pdf/CG88NICEGuideline.pdf
National Institute for Health and Clinical Excellence. Neuropathic pain: the pharmacological management of neuropathic pain in adults in non-specialist setting, Clinical Guideline 96. London: NICE. 2010. Available at http://www.nice.org.uk/nicemedia/live/12948/47949/47949.pdf
Scottish Intercollegiate Guidelines Network. Control of pain in adults with cancer. Edinburgh: SIGN; 2008. Available at http://www.sign.ac.uk/pdf/SIGN106.pdf
Sindrup S.H., Jensen T.S. Efficacy of pharmacological treatments of neuropathic pain: an update and effect related to mechanism of drug action. Pain. 1999;83:389-400.
Sindrup S.H., Otto M., Finnerup N.B., et al. Antidepressants in the treatment of neuropathic pain. Basic Clin. Pharmacol. Toxicol.. 2005;96:399-409.
Tjolsen A., Lund A., Hole K. Antinociceptive effect of paracetamol in rats is partly dependent on spinal serotonergic systems. Eur. J. Pharmacol.. 1991;193:193-201.
Thompson D.R. Narcotic analgesic effects on the sphincter of Oddi: a review of the data and therapeutic implications in treating pancreatitis. Am. J. Gastroenterol.. 2001;96:1266-1272.
van Wijck A.J., Opstelten W., Moons K.G., et al. The PINE study of epidural steroids and local anaesthetics to prevent postherpetic neuralgia: a randomised controlled trial. Lancet. 2006;367:219-224.
Woolf C.J. Pain: moving from symptom control toward mechanism-specific pharmacologic management. Ann. Intern. Med.. 2004;140:441-451.
British Association for the Study of Headache. Guidelines for all healthcare professionals in the diagnosis and management of migraine, tension-type, cluster and medication-overuse headache. Available at http://216.25.88.43/upload/NS_BASH/BASH_guidelines_2007.pdf, 2007.
MacIntyre P., Schug S.A. Acute Pain Management: A Practical Guide, third ed. Philadelphia: Elsevier; 2007.
Macintyre P.E., Scott D.A., Schug S.A., et al, editors. Acute Pain Management: Scientific Evidence, third ed., Melbourne: Australia and New Zealand College of Anaeshetists and Faculty of Pain Medicine, 2010. Available at http://www.nhmrc.gov.au/_files_nhmrc/file/publications/synopses/cp104_3.pdf
Marcus D.A., editor. Chronic Pain: A Primary Care Guide to Practical Management. Totowa: Humana Press, 2005.
McMahon S., Koltzenburg M., editors. Melzack and Wall’s Textbook of Pain, fifth ed., Edinburgh: Churchill Livingstone, 2005.
Melzack R., Wall P.D. Handbook of Pain Management: A Clinical Companion to Melzack and Wall’s Textbook of Pain. Edinburgh: Churchill Livingstone; 2003.
Moore R.A., McQuay H.J. Prevalence of opioid adverse events in persistent non-malignant pain: systematic review of randomized trials of oral opioids. Arthritis Res. Therapy. 2005;7:R1046-R1051.
National Institute for Health and Clinical Excellence. Improving supportive and palliative care for adults with cancer. London: NICE; 2004. Available at http://www.nice.org.uk/nicemedia/live/10893/28816/28816.pdf
http://www.painradar.co.uk/acute-pain-management-consensus.aspx The Radar Approach to Pain. http://www.painradar.co.uk/acute-pain-management-consensus.aspx.
http://www.medicine.ox.ac.uk/bandolier/booth/painpag/index.html Oxford Pain Internet site. http://www.medicine.ox.ac.uk/bandolier/booth/painpag/index.html.
http://www.change-pain.co.uk/ Change Pain. http://www.change-pain.co.uk/.
http://www.palliativedrugs.com/ PalliativeDrugs.com. http://www.palliativedrugs.com/.
http://www.britishpainsociety.org The British Pain Society. http://www.britishpainsociety.org.
[/level-membership-for-basic-science-category][not-level-membership-for-basic-science-category]
33 Pain
Acute pain may be thought of as a physiological process having a biological function, allowing the patient to avoid or minimise injury. Persistent pain, on the other hand, may be described more as a disease than a symptom (Woolf, 2004).
Aetiology and neurophysiology
Neuroanatomy of pain transmission
Pain transmission further within the Central Nervous System (CNS) is far more complex and understood less well. The most important parts of this process are the wide dynamic range cells in the spinothalamic tract that project to the thalamus and to the somatosensory cortex beyond. Modulation or inhibition of these neurones within the spinal cord result in less activity in the pain pathway. This modulatory action can be activated by stress or certain analgesic drugs such as morphine and is an important component of the gate theory of pain (Fig. 33.1). The gate control theory recognises the pivotal role the spinal cord plays in the continual modulation of neuronal activity by the relative activity of large (Aβ) and small (Aδ and C) fibres and by descending messages from the brain. Conversely, other influences can lead to an increased sensitivity to noxious stimuli. The most important of these is pain itself and further painful stimuli can lead to increased pain from relatively trivial insults. This occurs through neurochemical and anatomical changes within the CNS that have been termed central sensitisation.
Assessment of pain
Pain is a subjective phenomenon and quantitative assessment is difficult (Breivik et al., 2008). The most commonly used instruments are visual analogue and verbal rating scales. Visual analogue scales are 10 cm long lines labelled with an extreme at each end; usually ‘no pain at all’ and ‘worst pain imaginable’. The patient is required to mark the severity of the pain between the two extremes of the scale. Verbal rating scales use descriptors such as ‘none’, ‘mild’, ‘moderate’ and ‘excruciating’. More elaborate questionnaires such as the Brief Pain Inventory and the McGill Pain Questionnaire help to describe other aspects of the pain, and pain diaries record the influence of activity and medication on pain.
Management
Analgesic ladder
The World Health Organization (WHO) analgesic ladder (Fig. 33.2) forms the basis of many approaches to the use of analgesic drugs. There are essentially three steps: non-opioid analgesics, weak opioids and strong opioids. The analgesic efficacy of non-opioids, such as paracetamol and non-steroidal anti-inflammatory drugs (NSAIDs) (e.g. aspirin, ibuprofen and diclofenac), is limited by side effects and ceiling effects, that is, beyond a certain dose, no further pharmacological effect is seen. If pain remains uncontrolled, then a weak opioid, such as codeine or dihydrocodeine, may be helpful. There may be additional benefit in combining a weak opioid with a non-opioid drug, although many commercial preparations contain inadequate quantities of both components and are no more effective than a non-opioid alone. Strong opioids, of which morphine is considered the gold standard, have no ceiling effect and therefore increased dosage continues to give increased analgesia but side effects often limit effectiveness. Adjuvant drugs, such as corticosteroids, antidepressants or anti-epileptics, may be considered at any step of the ladder.
Analgesic drugs
Paracetamol
Despite being used in clinical practice for over 50 years and much investigation, the mechanism by which paracetamol exerts its analgesic effect remains uncertain. Inhibition of prostaglandin synthesis within the CNS has been proposed, although this is probably not the only mechanism. Interaction with the serotonin (Tjolsen et al., 1991) and endocannabinoid (Högestätt et al., 2005) neurotransmitter systems have been demonstrated in animal models.
With normal doses, the majority of paracetamols are metabolised and inactivated in the liver, undergoing a phase II conjugation reaction with glucuronic acid (Fig. 33.3). A small P450 mediated reaction that forms a reactive intermediate, N-acteyl-p-benzoquinimine (NAPQI). Usually, NAPQI can be deactivated by conjugation with glutathione in the liver. However, following ingestion of a large amount of paracetamol the hepatic stores of both glucuronic acid and glutathione become depleted leaving free NAPQI, which causes liver damage.
The usual therapeutic dose for adults is paracetamol 1 g taken four times daily. It is very important that this dose is not exceeded, otherwise hepatotoxicity is more common. This may be particularly problematic for malnourished adults with low body weight (Claridge et al., 2010). A reduced maximum daily infusion dose (3 g/24 hours) is recommended for patients with hepatocellular insufficiency, chronic alcoholism or dehydration. Paracetamol is also available as an over-the-counter (OTC) medicine and is a component of many cold and influenza remedies. Compared with other analgesics, paracetamol is not as potent; however, when taken in combination with a NSAID or opioid, there is an additive analgesic effect.
Non-steroidal anti-inflammatory drugs
Mode of action
NSAIDs exert their analgesic and anti-inflammatory effects through inhibition of the enzyme cyclo-oxygenase. NSAIDs are used widely to relieve pain, with or without inflammation, in people with acute and persistent musculoskeletal disorders. In single doses, NSAIDs have superior analgesic activity compared to paracetamol (Hyllested et al., 2002). In regular higher dosages, they have both analgesic and anti-inflammatory effects, which makes them particularly useful for the treatment of continuous or regular pain associated with inflammation. NSAIDs have been shown to be suitable for the relief of pain in dysmenorrhoea, toothache and some headaches and to treat pain caused by secondary bone tumours, which result from lysis of bone and release of prostaglandins.
Guidance on NSAID use
The lowest effective dose of NSAID or COX-2 selective inhibitor should be prescribed for the shortest time necessary. The need for long-term treatment should be reviewed periodically. Prescribing should be based on the safety profiles of individual NSAIDs or COX-2 selective inhibitors, on individual patient risk profiles, for example, gastro-intestinal and cardiovascular. Prescribers should not switch between NSAIDs without careful consideration of the overall safety profile of the products and the patient’s individual risk factors, as well as the patient’s preference (Medicines and Healthcare Regulatory Agency, 2006).
Weak opioids
Dextropropoxyphene
Historically, dextropropoxyphene was prescribed in combination with other analgesics such as paracetamol (co-proxamol). There are few data on its therapeutic value, and at least one review concluded that analgesic efficacy is less than aspirin and barely more than placebo. At best, dextropropoxyphene failed to show any superiority over paracetamol (Li Wan Po and Zhang, 1997). At worst, it is a dangerous drug which has the potential for steadily developing toxicity. Patients with hepatic dysfunction and poor renal function are particularly at risk. It is associated with problems in overdose, notably a non-naloxone reversible depression of the cardiac conducting system. Dextropropoxyphene interacts unpredictably with a number of drugs, including carbamazepine and warfarin. In 2005, the Medicines and Healthcare products Regulatory Agency (MHRA) announced concerns about the safety and effectiveness of co-proxamol and directed that it should be withdrawn from clinical use in the UK; however, it still remains available as an unlicensed medicine for the small number of patients who do not obtain analgesia with other analgesic medicines.
Strong opioids
Other strong opioids
The relative potencies of the commonly used opioids are summarised in Table 33.1.
| Drug | Potency (morphine = 1) |
|---|---|
| Codeine | 0.1 |
| Dihydrocodeine | 0.1 |
| Tramadol | 0.2 |
| Pethidine | 0.1 |
| Morphine | 1 |
| Diamorphine | 2.5 |
| Hydromorphone | 7 |
| Methadone | 2–10 (with repeat dosing) |
| Fentanyl (transdermal) | 150 |
Clinical considerations
Use of opioids is almost universally accepted in cancer pain but many patients with persistent non-cancer pain can find considerable relief with strong opioids; however, barriers to their use in this context appear to be based more on ignorance and political fashions than clinical evidence (Ballantyne and Mao, 2003). As a general rule, strong opioids effective in the management of neuropathic and musculoskeletal pain, including osteoarthritis, are less effective for sympathetically maintained pain.
Adverse effects of opioids
Tolerance, dependence and addition
Persistent treatment with opioids often causes tolerance to the analgesic effect, although the mechanism remains unclear (Holden et al., 2005). When this occurs the dosage should be increased or, alternatively, another opioid can be substituted, since cross-tolerance is not usually complete. Addiction is very rare when opioids are prescribed for pain relief.
Smooth muscle spasm
Opioids cause spasm of the sphincter of Oddi in the biliary tract and may cause biliary colic, as well as urinary sphincter spasm and urinary retention. Thus, in biliary or renal colic, it may be preferable to use another drug without these effects. Pethidine was believed to be the most effective in these circumstances but the evidence for this has been questioned (Thompson, 2001).
