[level-membership-for-radiology-category]14
Paediatrics
14.1
Retarded skeletal maturation
Endocrine disorders
1. Hypothyroidism* – with granular, fragmented epiphyses. This causes severe retardation (five or more standard deviations below the mean).
2. Steroid therapy and Cushing’s disease – see Cushing’s syndrome*.
3. Hypogonadism – including older patients with Turner’s syndrome.
4. Hypopituitarism – panhypopituitarism, growth hormone deficiency and Laron dwarfism.
14.2
Generalized accelerated skeletal maturation
Congenital disorders
1. McCune–Albright syndrome – polyostotic fibrous dysplasia with precocious puberty.
2. Cerebral gigantism (Soto’s syndrome).
4. Pseudohypoparathyroidism – premature fusion of cone-shaped epiphyses.
5. Acrodysostosis – premature fusion of cone-shaped epiphyses. Acromesomelic dysplasia type Maroteaux has similar hand appearances.
Fahmy, J. L., Kaminsky, C. K., Kaufman, F., et al. The radiological approach to precocious puberty. Br J Radiol. 2000; 73(869):560–567.
Martin, D. D., Wit, J. M., Hochberg, Z., et al. The use of bone age in clinical practice – Part 1. Horm Res Paediatr. 2011; 76(1):1–9.
Martin, D. D., Wit, J. M., Hochberg, Z., et al. The use of bone age in clinical practice – Part 2. Horm Res Paediatr. 2011; 76(1):10–16.
Poznanski, A. K. The hand in radiologic diagnosis. Philadelphia, PA: WB Saunders; 1984.
Rieth, K. G., Comite, F., Dwyer, A. J., et al. CT of cerebral abnormalities in precocious puberty. AJR Am J Roentgenol. 1987; 148(6):1231–1238.
14.3
Premature closure of a growth plate
1. Local hyperaemia – juvenile idiopathic arthritides, infection, haemophilia or arteriovenous malformation.
3. Vascular occlusion – postmeningococcal septicaemia, infarcts, sickle-cell anaemia.
5. Thermal injury – burns, frostbite.
6. Multiple exostoses and enchondromatosis (Ollier’s disease).
7. Hypervitaminosis A – now more usually via vitamin A analogue treatment for dermatological conditions rather than dietary overdosage.
8. Skeletal dysplasias – including Albright’s hereditary osteodystrophy, acrodysostosis, acromesomelic dysplasia type Maroteaux, trichorhinophalangeal syndrome, all with premature fusion of cone-shaped epiphyses in the hand.
14.4
Asymmetrical maturation
Hemihypertrophy or localized gigantism
(a) Parkes–Weber syndrome – fast-flow vascular malformations and arteriovenous fistulae; red cutaneous staining and limb hypertrophy.
(b) Congenital hypertrophy associated with capillary malformation (port-wine stain).
(c) Klippel–Trénaunay–Weber syndrome – a triad of port-wine stain, anomalous veins and limb overgrowth.
2. Chronic hyperaemia – e.g. chronic arthritides (juvenile chronic arthritis or haemophilia).
3. Hemihypertrophy – M > F; R > L. May be a presenting feature of Beckwith–Wiedemann syndrome (hemihypertrophy, macroglossia, hypoglycaemia and umbilical hernia). Increased incidence of Wilms’ tumour.
5. Macrodystrophia lipomatosa.
6. Russell–Silver dwarfism – evident from birth. Triangular face with down-turned corners of the mouth, frontal bossing, asymmetrical growth and skeletal maturation.
7. Proteus syndrome – hamartomatous disorder with multiple and varied manifestations including vascular and lymphatic malformations, macrocephaly and cranial hyperostoses.
8. WAGR syndrome – Wilms’ tumour, aniridia, genitourinary abnormalities and mental retardation.
14.5
Skeletal dysplasias
Dysplasias with predominant metaphyseal involvement
Achondroplasia* – hypochondroplasia is due to mutations in the same gene, FGFR3, with milder features.
Metaphyseal chondrodysplasias
1. Jansen – severe rickets-like changes with short stature.
2. Schmid – milder than Jansen. Bowed legs.
3. McKusick (cartilage-hair hypoplasia) – associated immune deficiency and haematological problems.
4. Schwachmann – associated with pancreatic insufficiency and cyclical neutropenia.
5. Hypophosphatasia – severe forms are lethal. V-shaped metaphyseal defects. Diaphyseal spurs.
6. Jeune’s asphyxiating thoracic dystrophy – a ‘ciliopathy’: short ribs with irregular costochondral junctions, associated with renal cysts and short hands.
7. Ellis–van Creveld syndrome – another ciliopathy, short ribs, associated with congenital heart disease and polydactyly.
Dysplasias with predominant epiphyseal involvement
1. Multiple epiphyseal dysplasia – at least five different genes. Irregular epiphyseal ossification, presenting with painful joints and gait abnormalities. Epiphyses may be small and round or flat, depending on type. Normal metaphyses, mild spine changes, mild short stature.
2. Pseudoachondroplasia – a more severe epiphyseal dysplasia with short stature, proportions resemble achondroplasia but pseudoachondroplasia has a normal face. Milder mutations of same gene cause a common type of multiple epiphyseal dysplasia. Spinal radiographic changes, but usually preserved spinal height.
3. Diastrophic dysplasia – flattened epiphyses associated with joint contractures (including club feet) and kyphoscoliosis. Cauliflower ear in infancy. Hypoplastic proximally placed ‘hitch-hiker’s’ thumb. Milder mutations in same gene cause recessive form of multiple epiphyseal dysplasia.
Mesomelic dysplasias (short forearms ± shanks)
1. Dyschondrosteosis (Leri−Weill) – Short radius with Madelung deformity and dorsal subluxation of distal ulna.
2. Mesomelic dysplasia type Langer – homozygous for mutations in dyschondrosteosis gene. More severe mesomelic shortening.
3. Acromesomelic dysplasia type Maroteaux – short upper limbs with shortening more severe from distal to proximal. Associated spinal abnormalities.
Acromelic dysplasias (short hands and feet)
1. Brachydactylies A to E – abnormalities isolated to hands and feet.
2. Albright’s hereditary osteodystrophy – encompasses pseudohypoparathyroidism and pseudopseudohypoparathyroidism. Metacarpal ± phalangeal shortening identical with brachydactyly type E. Soft-tissue/basal ganglia calcifications and exostoses in some.
3. Acrodysostosis – very short metacarpals and phalanges with cone epiphyses. Radiology very similar to acromesomelic dysplasia.
4. Trichorhinophalangeal syndrome – multiple short phalanges with cone epiphyses. Sparse hair and typical facial appearances. Type 2 associated with exostoses.
Dysplasias with major involvement of the spine
1. Type 2 collagen disorders – includes spondyloepiphyseal dysplasia congenital, Kniest and Stickler type 1. Delayed appearance of epiphyseal ossification centres with progressive platyspondyly and spinal deformity. Associated ear and eye problems and micrognathia in many. Hands and feet near normal.
2. Metatropic – ‘changing form’. In infancy manifests as short-limbed dysplasia, evolving into short spine dysplasia over childhood. Epiphyseal ossification delay with marked metaphyseal flare. Characteristic pattern of platyspondyly with wide flat vertebral bodies. Some individuals have a tail. Spondylometaphyseal dysplasia type Kozlowski is a milder form due to mutations in the same gene.
14.6
Lethal neonatal dysplasia
1. Thanatophoric dysplasia – severe mutations in same gene which causes achondroplasia (homozygous achondroplasia looks similar). Short ribs, severe platyspondyly with wafer-thin vertebral bodies. Severe limb shortening. Femora and humeri may be curved. Pelvis similar to achondroplasia. Type 2 associated with craniosynsostosis.
2. Osteogenesis imperfecta – lethal forms designated type 2
(a) Type 2a – deficient skull ossification; crumpled lung bones; thick, continuously beaded ribs.
(b) Type 2b – bowed, thickened but not crumpled long bones; thin ribs with multiple fractures. Overlaps with type 3 (see Osteogenesis imperfecta in Part 2).
(c) Type 2c – very rare. Thin, twisted, paradoxically sclerotic long bones.
3. Achondrogenesis – three types
(a) Type 1a – diminished or absent vertebral body ossification; fractured ribs; extremely short long bones.
(b) Type 1b – similar to 1a, but ribs not fractured. Widening of interpedicular distances in lumbar spine. Same gene as diastrophic dysplasia.
(c) Type 2 – long bone shortening less severe than in 1a and 1b. Hands and feet look almost normal. The most severe disorder of type 2 collagen.
4. Hypochondrogenesis – radiologically milder form of achondrogenesis 2, but still lethal.
5. Short rib polydactyly syndromes – ‘ciliopathies’. Four types. Extremely severe rib shortening. Polydactyly in most with acromesomelic shortening of varying pattern.
6. Fibrochondrogenesis – short long bones with metaphyseal flaring and diamond-shaped vertebrae.
7. Campomelic dysplasia – bowed femora and tibia. Deficient ossification of thoracic pedicles and severe hypoplasia of scapular blades are most characteristic features. 11 rib pairs.
8. Chondrodysplasia punctata – see 14.16.
9. Lethal hypophosphatasia – severely deficient skull ossification. Absent pedicles in spine. Missing bones. Variable metaphyseal defects. Some bones look normal.
Hall, C. M., Offiah, A. C., Forzano, F., et al. Fetal and perinatal skeletal dysplasias, an atlas of multimodality imaging. Milton Keynes: Radcliffe; 2012.
Spranger, J. W., Brill, P. W., Poznanski, A. Bone dysplasias, an atlas of genetic disorders of skeletal development, third ed. New York: OUP; 2012.
14.7
Dumbbell-shaped long bones
Short narrow diaphyses with marked metaphyseal widening.
1. Kniest syndrome – see 14.5.
2. Metatropic dysplasia – see 14.5.
3. Diastrophic dysplasia – clubbing less severe than above, epiphyseal dysplasia, club feet. See 14.5.
4. Type 11 collagen disorders – fibrochondrogenesis (lethal), Stickler type 2, otospondylomegaepiphyseal dysplasia (OSMED).
5. Severe pseudoachondroplasia – see 14.5.
6. Dyssegmental dysplasia – mutations in perlecan gene: severe form (Silverman–Handmaker, lethal) and less severe form (Rolland–Desbuquois). Dumbbell bones and vertebral anomalies (clefts, variable vertebral body size) may be similar to Kniest.
14.9
Generalized increased bone density
Dysplasias
2. Pyknodysostosis – short stature, hypoplastic lateral ends of clavicles, hypoplastic terminal phalanges, bulging cranium and delayed closure of the anterior fontanelle. AR.
3. Dysosteosclerosis – similar to classical osteopetrosis in infancy. Progressive spinal involvement with end-plate irregularity, and marked undertubulation of long bones with submetaphyseal lucencies. Thought to be an ‘osteoclast-poor’ form of osteopetrosis.
4. Progressive diaphyseal dysplasia (Camurati–Engelmann syndrome) – presents with bone pain and weakness, with thick sclerotic long bone diaphyses.
5. Wnt-pathway disorders – including endosteal hyperostosis, sclerosteosis, osteopathia striata and Van Buchem disease.
Metabolic
Renal osteodystrophy* – rarely renal osteodystrophy causes bone sclerosis, typically seen as a ‘rugger-jersey’ spine. Oxalosis may also cause renal failure and bone sclerosis.
Poisoning
1. Lead – dense metaphyseal bands. Cortex and flat bones may also be slightly dense. Modelling deformities later, e.g. flask-shaped femora.
2. Fluorosis – more common in adults. Usually asymptomatic but may present in children with crippling stiffness and pain. Thickened cortex at the expense of the medulla. Periosteal reaction. Ossification of ligaments, tendons and interosseous membranes.
3. Hypervitaminosis D – slightly increased density of skull and vertebrae early, followed later by osteoporosis. Soft-tissue calcification. Dense metaphyseal bands and widened zone of provisional calcification.
4. Chronic hypervitaminosis A – not before 1 year of age. Failure to thrive, hepatosplenomegaly, jaundice, alopecia and haemoptysis. Cortical thickening of long and tubular bones, especially in the feet. Subperiosteal new bone. Normal epiphyses and reduced metaphyseal density. The mandible is not affected (cf. Caffey’s disease).
Idiopathic
1. Caffey’s disease (infantile cortical hyperostosis) – see 14.12.
2. Idiopathic hypercalcaemia of infancy – probably a manifestation of hypervitaminosis D. Elfin facies, failure to thrive and mental retardation. Generalized increased density or transverse dense metaphyseal bands. Increased density of the skull base.
Beighton, P., Cremin, B. J. Sclerosing bone dysplasias. Berlin: Springer; 1980.
Herman, T. E., McAlister, W. H. Inherited diseases in bone density in children. Radiol Clin North Am. 1991; 29(1):149–164.
Ihde, L. L., Forrester, D. M., Gottsegen, C. J., et al. Sclerosing bone dysplasias: review and differentiation from other causes of osteosclerosis. Radiographics. 2011; 31(7):1865–1882.
14.10
Paediatric tumours that metastasize to bone
2. Leukaemia – although not truly metastases.
4. Clear cell sarcoma (Wilms’ variant).
7. Ewing’s sarcoma – lung metastases much more common.
8. Osteosarcoma* – lung metastases much more common.
14.11
‘Moth-eaten bone’ in a child
14.12
Periosteal reactions – bilaterally symmetrical in children
1. Normal infants – diaphyseal, not extending to the growth plate, bilaterally symmetrical and a single lamina. Very unusual beyond 4 months of age.
2. Juvenile idiopathic arthritis* – in approximately 25% of cases. Most common in the periarticular regions of the phalanges, metacarpals and metatarsals. When it extends into the diaphysis it will eventually result in enlarged, rectangular tubular bones.
3. Acute leukaemia – associated with prominent metaphyseal bone resorption ± a dense zone of provisional calcification. Osteopenia. Periosteal reaction is due to cortical involvement by tumour cells. Metastatic neuroblastoma can look identical.
4. Rickets* – the presence of uncalcified subperiosteal osteoid mimics a periosteal reaction because the periosteum and ossified cortex are separated.
5. Caffey’s disease – first evident before 5 months of age. Mandible, clavicles and ribs show cortical hyperostosis and a diffuse periosteal reaction. The scapulae and tubular bones are less often affected and tend to be involved asymmetrically.
6. Scurvy* – subperiosteal haemorrhage is most frequent in the femur, tibia and humerus. Periosteal reaction is particularly evident during the healing phase. Age 6 months or older.
7. Prostaglandin E1 therapy – in infants with ductus-dependent congenital heart disease. Severity is related to duration of therapy. Other features include fever, flushing, diarrhoea, skin oedema, pseudowidening of cranial sutures and bone-in-bone appearance.
8. Congenital syphilis – an exuberant periosteal reaction can be due to infiltration by syphilitic granulation tissue or the healing (with callus formation) of osteochondritis. The former is essentially diaphyseal and the latter around the metaphyseal/epiphyseal junction.
14.13
Syndromes and bone dysplasias with multiple fractures as a feature
With reduced bone density
2. Rickets – usually only in presence of severe rachitic change and clear demineralization.
4. Juvenile idiopathic osteoporosis – 2–4 years’ duration, age of onset 2–13 years.
5. Gerodermia osteodysplastica – osteopenia and wormian bones associated with wrinkly skin (cutis laxa) and hip dislocation.
6. Osteoporosis pseudoglioma syndrome – blindness in infancy associated with bony fragility.
7. Mucolipidosis II (I-cell disease) – osteopenia and periosteal ‘cloaking’ in infancy, evolving into dysostosis multiplex.
14.14
Pseudarthrosis in a child
1. Non-union of a fracture – including pathological fracture.
2. Congenital – in the middle to lower third of the tibia ± fibula. 50% present in the first year. Later there may be cupping of the proximal bone end and pointing of the distal bone end.
5. Cleidocranial dysplasia* – congenitally in the femur.
14.15
‘Bone within a bone’ appearance
Brill, P. W., Baker, D. H., Ewing, M. L. Bone within bone in the neonatal spine. Radiology. 1973; 108:363–366.
Frager, D. H., Subbarao, K. The ‘bone within a bone’. JAMA. 1983; 249:77–79.
Matzinger, M. A., Briggs, V. A., Dunlap, H. J., et al. Plain film and CT observations in prostaglandin-induced bone changes. Pediatr Radiol. 1992; 22(4):264–266.
14.16
Irregular or stippled epiphyses
1. Normal – particularly in the distal femur.
2. Avascular necrosis (q.v.) – single, e.g. Perthes’ disease (although 10% are bilateral), or multiple, e.g. sickle-cell anaemia.
3. Congenital hypothyroidism* – not present at birth. Delayed appearance and growth of ossification centres. Appearance varies from slightly granular to fragmentation. The femoral capital epiphysis may be divided into inner and outer halves.
4. Morquio’s syndrome* – irregular ossification of the femoral capital epiphyses results in flattening.
5. Multiple epiphyseal dysplasia – see 14.5.
6. Meyer dysplasia – an epiphyseal dysplasia resembling multiple epiphyseal dysplasia but confined to the femoral heads.
7. Chondrodysplasia punctata (CDP) – punctate calcifications of developing epiphyses in fetus and infant, which resolve in first few years of life, with disturbance of growth of affected bones. All causes affect the peroxisomal metabolic pathway.
(i) Conradi−Hünermann – puncta in long bones, wrists and pelvis. Asymmetric long bone shortening. XD.
(ii) Rhizomelic type – severe and often lethal form. Coronal vertebral clefts and very short humeri. AR.
(iii) Brachytelephalangic type – shortening of distal phalanges with triangular configuration. Puncta mostly in spine. XR.
10. Zellweger syndrome (cerebrohepatorenal syndrome).
11. Fetal alcohol syndrome – mostly calcaneum and lower extremities.
14.17
Solitary radiolucent metaphyseal band
3. Metaphyseal fracture – especially in non-accidental injury*. Depending on the radiographic projection there may be the additional appearance of a ‘corner’ or ‘bucket-handle’ fracture.
5. Leukaemia, lymphoma* or metastatic neuroblastoma.
14.18
Alternating radiolucent and dense metaphyseal bands
1. Growth arrest (Harris or Park’s lines).
3. Rickets* – especially those types that require prolonged treatment such as vitamin D-dependent rickets.
14.19
Solitary dense metaphyseal band
14.21
Fraying of metaphyses
14.22
Cupping of metaphyses
Often associated with fraying.
1. Normal – especially of the distal ulna and proximal fibula of young children. No fraying.
2. Rickets* – with widening of the growth plate and fraying.
3. Trauma – to the growth plate and/or metaphysis. Asymmetrical and localized changes.
4. Bone dysplasias – a sign in a large number, e.g. achondroplasia*, pseudoachondroplasia, metatropic dwarfism, diastrophic dwarfism, the metaphyseal chondrodysplasias and hypophosphatasia*.
5. Scurvy* – usually after fracture.
6. Menke’s disease – Copper deficiency can have similar appearances.
14.23
Erlenmeyer flask deformity
Dysplasias
1. Craniotubular disorders – including Pyle dysplasia, craniometaphyseal dysplasia, craniodiaphyseal dysplasia, progressive diaphyseal dysplasia.
2. Otopalatodigital syndrome type 1, osteodysplasty (Melnick–Needles syndrome) and frontometaphyseal dysplasia – all disorders of filamin A.
3. Osteopetrosis* – in infantile and juvenile forms. Particularly striking in similar disorder of dysosteosclerosis.
Faden, M. A., Krakow, D., Ezgu, F., et al. The Erlenmeyer flask bone deformity in the skeletal dysplasias. Am J Med Genet A. 2009; 149A(6):1334–1345.
Myer, H. S., Cremin, B. J., Beighton, P., et al. Chronic Gaucher’s disease: radiological findings in 17 South African cases. Br J Radiol. 1975; 48:465–469.
14.24
Focal rib lesion (solitary or multiple) in a child
Neoplastic
Secondary more common than primary. Primary malignant more common than benign.
Peripheral PNET including Ewing’s sarcoma (PNET of bone)* and Askin tumour (PNET of chest wall).
14.25
Widening of the symphysis pubis
Congenital
With normal ossification
Cortina, H., Vallcanera, A., Andres, V., et al. The non-ossified pubis. Pediatr Radiol. 1979; 8:87–92.
Muecke, E. C., Currarino, G. Congenital widening of the symphysis pubis. Associated clinical disorders and roentgen anatomy of affected bony pelves. AJR Am J Roentgenol. 1968; 103:179–185.
Patel, K., Chapman, S. Normal symphysis pubis width in children. Clin Radiol. 1993; 47:56–57.
Taybi, H., Lachman, R. S. Radiology of syndromes, metabolic disorders, and skeletal dysplasias, fifth ed. St Louis, MO: Mosby; 2007.
14.27
Abnormal thumbs – congenital
Broad
1. Acrocephalopolysyndactyly (Carpenter type) – two ossification centres for the proximal phalanx in childhood → duplication in adulthood.
2. Acrocephalosyndactyly (Apert type) – partial or complete duplication of the proximal phalanx. Complete syndactyly of digits II–V: ‘mitten hand’ and ‘sock foot’.
3. Diastrophic dysplasia – short, ovoid thumb metacarpal with proximally located thumb.
4. Rubinstein–Taybi syndrome – terminal phalanx + ‘hitch-hiker thumb’.
5. Otopalatodigital syndrome – large cone epiphysis of the distal phalanx.
Short or small
1. Fanconi’s anaemia – ± other radial ray abnormalities. Onset of pancytopenia at 5–10 years of age.
2. Holt–Oram syndrome – finger-like, absent, hypoplastic or triphalangeal thumb + congenital heart disease (ASD, VSD).
4. Cornelia de Lange syndrome – hypoplastic metacarpal.
5. Fetal hydantoin – finger-like thumb with hypoplasia of all the distal phalanges.
14.28
Differential diagnosis of skeletal lesions in non-accidental injury*
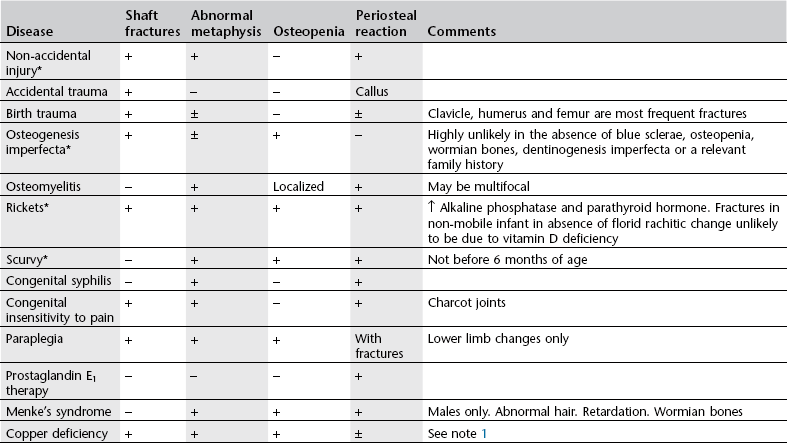
1Copper deficiency. Rare. Unlikely in the absence of at least one risk factor: prematurity, total parenteral nutrition, malabsorption or a low-copper diet. Unlikely in full-term infants less than 6 months of age. Microcytic, hypochromic anaemia. Leukopenia. Normal serum copper and caeruloplasmin does not exclude the diagnosis. Skull fracture never recorded in copper deficiency. Rib fractures only recorded in premature infants.
Bilo, R. A. C., Robben, S. G. F., van Rijn, R. R. Forensic aspects of paediatric fractures. Berlin: Springer; 2009.
Carty, H., Pierce, A. Non-accidental injury: a retrospective analysis of a large cohort. Eur Radiol. 2002; 12(12):2919–2925.
Chapman, S., Hall, C. M. Non-accidental injury or brittle bones. Pediatr Radiol. 1997; 27:106–110.
Kleinman, P. Diagnostic imaging of child abuse, second ed. St Louis, MO: Mosby; 1998.
Offiah, A., Van Rijn, R. R., Perez-Rossello, J. M., et al. Skeletal imaging if child abuse (non-accidental injury). Pediatr Radiol. 2009; 39(5):461–470.
Shaw, J. C. L. Copper deficiency and non-accidental injury. Arch Dis Childhood. 1988; 63:448–455.
14.29
Platyspondyly in childhood
Congenital platyspondyly
1. Thanatophoric dwarfism – inverted ‘U’- or ‘H’-shaped vertebrae with a markedly increased disc space : body height ratio. Telephone handle-shaped long bones.
2. Metatropic dwarfism – flat-appearing vertebral bodies, but large disc spaces mean that overall spinal height is near normal in infancy. As childhood progresses, relative spinal height reduces.
3. Osteogenesis imperfecta* – type IIA.
Acquired platyspondyly
1. Scheuermann’s disease – irregular end-plates and Schmorl’s nodes in the thoracic spine of children and young adults. Disc-space narrowing. May progress to a severe kyphosis.
2. Langerhans’ cell histiocytosis* – the spine is more frequently involved in eosinophilic granuloma and Hand–Schüller–Christian disease than in Letterer–Siwe disease. Most common in young people. The thoracic and lumbosacral spine are the usual sites of disease. Disc spaces are preserved.
3. Osteogenesis imperfecta – multiple spinal compression fractures, resulting in loss of height and spinal deformity among the most serious complications.
4. Sickle-cell anaemia* – characteristic step-like depression in the central part of the end-plate.
14.30
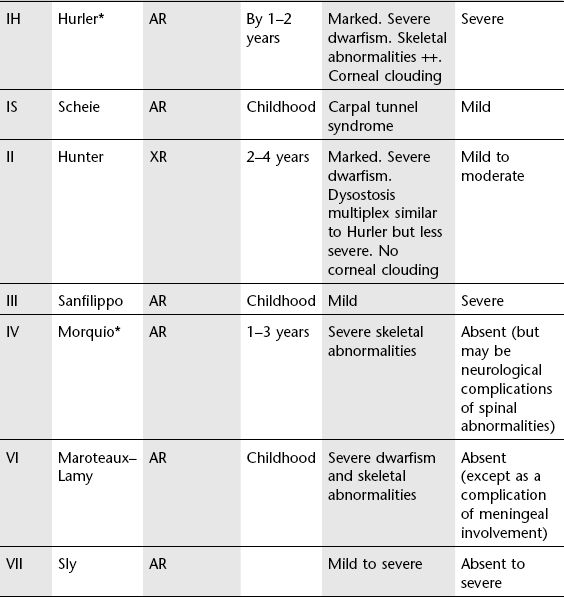
Anterior vertebral body beaks

1. Mucopolysaccharidoses* – with platyspondyly in Morquio’s: this is probably a more useful distinguishing characteristic than the position of the beak, inferior or middle, which is variable.
14.31
Acute upper airway obstruction in a child
1. Laryngotracheobronchitis (croup) – most common 6 months–3 years. Narrowing of the glottic and subglottic airway. Ballooning of hypopharynx on lateral view. ‘Steepling’ of upper airway on frontal view.
2. Acute epiglottitis – the epiglottis is swollen and may be shortened. Other components of the supraglottic region – aryepiglottic folds, arytenoids, uvula and prevertebral soft tissues – are also swollen. The hypopharynx and pyriform sinuses are distended with air.
3. Retropharyngeal abscess – more common < 2 years as retropharyngeral nodes atrophy thereafter. Enlargement of the prevertebral soft tissues which may contain gas or an air–fluid level. Rim enhancement seen following contrast on CT or MRI.
4. Oedema – caused by angio-oedema (allergic, anaphylactic or hereditary), inhalation of noxious gases or trauma. Predominantly laryngeal oedema.
5. Foreign body – more commonly produces a major bronchial occlusion rather than upper airway obstruction.
6. Choanal atresia – most common congenital nasal abnormality bilateral (33%) or unilateral (R > L), bony (90%) or membranous, complete or incomplete. When bilateral and complete, presentation is with severe respiratory distress at birth. Incomplete obstruction is associated with respiratory difficulty during feeding. Diagnosis is by failure to pass a catheter through the nose, and nasopharyngography or CT.
7. Pyriform aperture stenosis – bony overgrowth of the medial nasal process of the maxilla.
8. Retropharyngeal haemorrhage – due to trauma, neck surgery, direct carotid arteriography and bleeding disorders. Widening of the retropharyngeal soft-tissue space.
14.32
Chronic upper airway obstruction in a child
May be associated with overinflation of the lungs.
Nasal
1. Choanal atresia – see 14.31.
2. Nasal angiofibroma – most commonly in adolescent males. Symptoms of nasal obstruction and/or recurrent atraumatic epistaxis. Plain films may show:
Supraglottic
1. Grossly enlarged tonsils and adenoids.
2. Laryngomalacia – presents at or shortly after birth, persists for several months and usually resolves by 2 years. Diagnosis is confirmed by direct laryngoscopy, but fluoroscopy reveals anterior motion of the aryepiglottic folds and distension of the hypopharynx.
3. Micrognathia – in the Pierre Robin syndrome.
4. Cysts – of the epiglottis or aryepiglottic folds. The degree of obstruction depends on the size and location.
Subglottic and tracheal
1. Tracheomalacia – weakness of tracheal wall which may be primary or secondary:
(b) Right arch with left-sided duct/ductal ligament.
(c) Pulmonary artery sling (frequently coexistent intrinsic narrowing).
3. Subglottic haemangioma – the most common subglottic soft-tissue mass in infancy. Occurs before 6 months. 50% have associated cutaneous haemangiomas. Characteristically it produces an asymmetrical narrowing of the subglottic airway.
4. Following prolonged tracheal intubation – may be fixed stenosis or malacia.
5. External compression from other mediastinal structures – e.g. lymphadenopathy.
6. Congenital tracheal stenosis – usually due to presence of complete cartilaginous rings. Associated with pulmonary artery sling.
7. Respiratory papillomatosis – occurs anywhere from the nose to the lungs. Irregular soft-tissue masses which may cavitate around the glottis or in the trachea mostly.
Chess, M. A., Chaturvedi, A., Stanescu, A. L., Blickman, J. G. Emergency pediatric ear, nose, and throat imaging. Semin Ultrasound CT MR. 2012; 33(5):449–462.
Cohen, L. F. Stridor and upper airway obstruction in children. Pediatr Rev. 2000; 21(1):4–5.
John, S. D., Swischuk, L. E. Stridor and upper airway obstruction in infants and children. Radiographics. 1992; 12(4):625–643.
14.33
Neonatal respiratory distress
Pulmonary causes
With no mediastinal shift
1. Hyaline membrane disease (surfactant deficiency disease) – in premature infants. Infants are symptomatic soon after birth but maximum radiographic findings develop at 12–14 hours. Fine granular pattern throughout both lungs, air bronchograms and, later, obscured heart and diaphragmatic outlines. Small lung volume due to diffuse microatelectasis. Often cardiomegaly. May progress to a complete ‘white-out’. Interstitial emphysema, pneumomediastinum and pneumothorax are frequent complications of ventilator therapy. Patchy clearing of infiltrate occurs following surfactant therapy. As oxygenation improves, bidirectional or left-to-right shunting through the ductus arteriosus may lead to pulmonary oedema, cardiomegaly and occasionally pulmonary haemorrhage.
2. Transient tachypnoea of the newborn – prominent interstitial markings and vessels, thickened septa, small effusions and occasionally mild cardiomegaly. May resemble hyaline membrane disease, meconium aspiration or neonatal pneumonia. Resolves within 2–3 days.
3. Meconium aspiration syndrome – predominantly postmature infants. Coarse linear and irregular opacities of uneven size, generalized hyperinflation and focal areas of collapse and emphysema. Spontaneous pneumothorax and effusions in 20%. Pleural effusion in up to two-thirds; never in hyaline membrane disease. No air bronchograms.
4. Pneumonia – in < 1% of newborns. Risk factor – prolonged rupture of membranes. Most commonly group B streptococcus. Segmental or lobal consolidation. Pleural effusions may be large and suggest diagnosis. May resemble hyaline membrane disease or meconium aspiration syndrome, but should be suspected if unevenly distributed.
5. Pulmonary haemorrhage – 75% are less than 2.5 kg. Onset at birth or delayed several days. May occur following surfactant therapy probably due to left-to-right shunting. Resembles meconium aspiration syndrome or hyaline membrane disease.
6. Upper airway obstruction – e.g. choanal atresia and micrognathia.
(a) Neuromuscular abnormalities – often with thin ribs and clavicles.
(b) Skeletal dysplasias – e.g. Jeune’s asphyxiating thoracic dysplasia, thanatophoric dwarfism, osteogenesis imperfecta and metatropic dwarfism.
(c) Pulmonary hypoplasia – e.g. due to fetal renal failure (Potter sequence) or primary (rare).
(d) Major abdominal wall defects (exomphalos/gastroschisis) – short down-sloping ribs with ‘long’ chest.
8. Alveolar capillary dysplasia – often normal radiographic appearances despite severe respiratory distress. Microscopic misalignment of capillaries and pulmonary veins. Universally poor prognosis.
With mediastinal shift away from the abnormal side
1. Diaphragmatic hernia – six times more common on the left side. Multiple lucencies due to gas-containing bowel in the chest. Herniated bowel may appear solid if X-rayed too early but there will still be a paucity of gas in the abdomen.
2. Congenital lobar overinflation – involves the left upper, right upper and right middle lobes (in decreasing order of frequency) with compression of the lung base (cf. pneumothorax which produces symmetrical lung compression). CT is useful, particularly to exclude external compression of a bronchus by an aberrant vessel.
3. Congenital pulmonary airway malformation (previously termed congenital cystic adenomatoid malformation) – translucencies of various shapes and sizes scattered throughout an area of opaque lung with well-defined margins.
4. Pneumothorax – may complicate resuscitation or positive pressure ventilation, or may be spontaneous. Spontaneous pneumothorax is associated with pulmonary hypoplasia, e.g. in Potter sequence. In the supine neonate, pleural air collects anteriorly and may not collapse the lung medially. In the absence of a lung edge, other signs which suggest the presence of a pneumothorax are:
(a) Sharp ipsilateral heart border.
(b) Depression or inversion of the ipsilateral hemidiaphragm.
(c) Sharp ipsilateral parietal pleura in the upper medial part of the hemithorax. If there is tension this may herniate across the superior mediastinum.
(d) Medial deviation of the ipsilateral compressed thymic lobe.
With mediastinal shift towards the abnormal side
1. Atelectasis – most commonly due to incorrect placement of an endotracheal tube down a major bronchus. Much less commonly, primary atelectasis may occur without any other abnormality.
2. Agenesis/aplasia – rare. May be difficult to differentiate from collapse but other congenital defects, especially hemivertebrae, are commonly associated. Agenesis = no bronchus; aplasia = rudimentary bronchus present.
3. Unilateral pulmonary hypoplasia – most commonly due to compression, e.g. by diaphragmatic hernia. May also be associated with vascular anomalies, e.g. absent pulmonary artery, anomalous venous drainage (= scimitar syndrome).
Agrons, G. A., Courtney, S. E., Stocker, J. T., Markowitz, R. I. From the archives of the AFIP: Lung disease in premature neonates: radiologic–pathologic correlation. Radiographics. 2005; 25(4):1047–1073.
Markowitz, R. I., Fellows, K. E. The effects of congenital heart disease on the lungs. Semin Roentgenol. 1998; 33(2):126–135.
Newman, B. Imaging of medical disease of the newborn lung. Radiol Clin North Am. 1999; 37(6):1049–1065.
Schwartz, D. S., Reyes-Mugica, M., Keller, M. S. Imaging of surgical diseases of the newborn chest. Radiol Clin North Am. 1999; 37(6):1067–1078.
14.34
Ring shadows in a child
Neonate
1. Diaphragmatic hernia – unilateral.
2. Interstitial emphysema – secondary to ventilator therapy. May be unilateral or bilateral. Usually transient, but may persist.
3. Congenital pulmonary airway malformation (previously termed congenital cystic adenomatoid malformation) – unilateral.
4. Bronchopulmonary dysplasia – ‘bubbly lung’ appearances with air-trapping.
14.35
Interstitial lung disease unique to childhood
1. Persistent tachypnoea of the newborn – ground-glass opacities and air-trapping. Disorder of pulmonary neuroendocrine cells.
2. Bronchopulmonary dysplasia – patchy atelectasis and air-trapping, interstitial opacities with triangular subpleural opacities.
3. Cellular interstitial pneumonitis of infancy – interstitial infiltrates. Relatively good prognosis.
4. Infantile pulmonary haemosiderosis – recurrent pulmonary haemorrhage leading to fibrotic changes.
5. Chronic pneumonitis of infancy (CPI) – interstitial changes. High mortality.
6. Surfactant protein B deficiency – AR. Similar appearance to hyaline membrane disease but in a full-term newborn infant. May account for some cases of CPI and alveolar proteinosis.
7. Familial desquamative interstitial pneumonitis – worse prognosis than typical desquamative interstitial pneumonitis.
Copley, S. J., Padley, S. P. High-resolution CT of paediatric lung disease. Eur Radiol. 2001; 11:2564–2575.
Koh, D. M., Hansell, D. M. Computed tomography of diffuse interstitial lung disease in children. Clin Radiol. 2000; 55:659–667.
Lynch, D. A., Hay, T., Newell, J. D., Jr., et al. Pediatric diffuse lung disease: diagnosis and classification using high-resolution CT. AJR Am J Roentgenol. 1999; 173(3):713–718.
Owens, C. M. Radiology of diffuse interstitial pulmonary disease in children. Eur Radiol. 2004; 14(Suppl 4):L2–L12.
14.36
The normal thymus
It has the following CT characteristics:
1. Incidence – identifiable in 100% < 30 years of age, decreasing to 17% > 49 years. However, < 10 years of age the distinction from great vessels is very difficult without the use of contrast enhancement.
2. Shape – quadrilateral shape in childhood with, usually, convex, undulating margins. After puberty two separate lobes (ovoid, elliptical, triangular or semilunar) or an arrowhead (triangle). The normal thymus is never multilobular.
3. Size – progressive enlargement during childhood. Maximum absolute size is in the 12–19-year age group but relative to body size it is largest in infancy. Left lobe nearly always larger than right lobe. Becomes narrower with increasing age. Maximum thickness (the perpendicular to the long axis) of one lobe in those > 20 years is 1.3 cm. In those > 40 years there may be linear or oval soft-tissue densities but they are never > 7 mm in size and never alter the lateral contour of the mediastinal fat.
4. Density – homogeneous, isodense or hyperdense when related to chest-wall musculature in childhood. After puberty becoming inhomogeneous and progressively lower in attenuation owing to fatty infiltration. In those > 40 years the majority will have total fatty involution.
14.37
Anterior mediastinal masses in childhood
1. The mediastinum is the most common site of a chest mass in a child.
2. The anterior mediastinum is bounded by the clavicles (superiorly), the diaphragm (interiorly), the sternum (anteriorly), and the anterior surfaces of the heart and great vessels (posteriorly). 45% of paediatric mediastinal masses occur at this site.
Neoplastic
1. Hodgkin’s lymphoma, non-Hodgkin’s lymphoma and leukaemia – the most common cause of an anterior mediastinal mass in children. The majority of neoplastic anterior mediastinal masses are due to Hodgkin’s disease. At presentation, mediastinal lymph nodes are seen in 85% of Hodgkin’s, 50% of non-Hodgkin’s and 5–10% of leukaemics. Comparing mediastinal involvement in Hodgkin’s with non-Hodgkin’s lymphoma:
| Hodgkin’s lymphoma | Non-Hodgkin’s lymphoma |
| Usually > 10 years old | Any age in children |
| Mostly localized. Mediastinal lymphadenopathy (LN) in 85% of those with cervical LN | Disseminated disease in > 75% at presentation |
| Histology usually nodular sclerosing | Histology usually lymphoblastic |
| Displacement of other mediastinal structures rather than compression | Tracheal compression is more likely |
| Paratracheal > hilar > subcarinal LN. Hilar LN without mediastinal LN is rare |
|
| Lung involvement in 10% at diagnosis – direct spread from lymph nodes | Pulmonary involvement is higher |
| Pleural effusion is more common but may be secondary to ascites or lymphatic obstruction |
2. Germ cell tumours – 5–10% of germ cell tumours arise in the mediastinum. Two age peaks: at 2 years and during adolescence. Majority (60%) are teratomas and benign. Endodermal sinus (yolk sac) tumours are more aggressive. Seminomas rare. Tumours may contain calcification (including teeth), fat and cystic/necrotic areas. Radiological appearance does not accurately correlate with histology but large size, marked mass effect and local infiltration suggest an aggressive lesion.
3. Thymoma – 1–2% of mediastinal tumours in childhood. Most occur after 10 years of age. 50% discovered incidentally. Calcification in 10% – linear. Only rarely associated with myasthenia gravis.
14.38
Middle mediastinal masses in childhood
Congenital
1. Foregut duplication cysts – account for 10–20% of paediatric mediastinal masses. The spectrum of abnormalities includes bronchogenic cysts, oesophageal duplication cysts and neurenteric cysts.
(a) Bronchogenic cyst – abnormal lung budding and development of ventral foregut during first trimester. Round or oval, unilocular, homogeneous, water-density mass (usually 0–20 HU, but up to 100 HU due to mucus or milk of calcium contents) with well-defined borders. There may be airway obstruction and secondary infection, both within the cyst and in the surrounding lung. Communication with the tracheobronchial tree, resulting in a cavity, is rare, and may indicate infection. May be located anywhere along tracheobronchial tree, 20% being intrapulmonary:
(i) Paratracheal cysts are attached to the tracheal wall above the carina.
(ii) Carinal cysts are the most common and are attached to the carina ± anterior oesophageal wall.
(iii) Hilar cysts are attached to a lobar bronchus and appear to be intrapulmonary.
(iv) Paraoesophageal cysts may be attached or communicate with the oesophagus but have no communication with the bronchial tree.
(b) Oesophageal duplication cyst – abnormal development of the posterior division of the embryonic foregut. 10–15% of intestinal duplications. Less common than bronchogenic cysts, usually larger and usually upper third of the oesophagus, situated to the right of the midline extending into the posterior mediastinum. May be an incidental finding or produce symptoms related to oesophageal or tracheobronchial tree compression. May contain ectopic gastric mucosa (positive 99mTc-pertechnetate scan) which causes ulceration, haemorrhage or perforation. Communication with the oesophageal lumen is rare.
(c) Neurenteric cyst – failure of separation of gastrointestinal tract from primitive neural crest. Located in the middle or posterior mediastinum, contains neural tissue and maintains a connection with the spinal canal. More commonly right-sided. Vertebral body anomalies (hemivertebra, butterfly vertebra and scoliosis) are usually superior to the cyst.
2. Lymphatic malformation – 5% of lymphatic malformations extend into the mediastinum from the neck. Most present at birth. Cystic with some solid components on all imaging modalities.
14.39
Posterior mediastinal masses in childhood
Neoplastic
1. Ganglion cell tumours – neuroblastoma (most malignant, usually < 5 years; 20% of all paediatric neuroblastoma posterior mediastinal in origin), ganglioneuroblastoma (malignant potential, age 5–10 years) and ganglioneuroma (benign, usually > 10 years). Imaging features of all three types are similar but metastases do not occur with ganglioneuroma. Plain films show a paravertebral soft-tissue mass with calcification in 30%. Thinning of posterior ribs, separation of ribs and enlargement of intervertebral foramina. CT shows calcification in 90%. Both CT and MRI may show extradural extension.
Congenital
Bochdalek hernia – most present at, or shortly after, birth with respiratory distress, but 5% present after the neonatal period. Rarely it may complicate group B streptococcal infection. Bochdalek hernias include:
(a) Persistence of the pleuroperitoneal canal with a posterior lip of diaphragm.
The appearance of herniated liver may provoke thoracentesis and herniated bowel may mimic pneumothorax, pneumatocoeles or cystic adenomatoid malformation.
Berman, L., Stringer, D. A., Ein, S., et al. Childhood diaphragmatic hernias presenting after the neonatal period. Clin Radiol. 1988; 39:237–244.
Donnelly, L. F., Frush, D. P., Zheng, J. -Y., et al. Differentiating normal from abnormal inferior thoracic paravertebral soft tissues on chest radiography in children. AJR Am J Roentgenol. 2000; 175:477–483.
14.40
Solitary pulmonary mass in childhood
Non-neoplastic lesions
1. Pulmonary sequestration – most commonly in medial basal segment of right lower lobe.
2. Intrapulmonary bronchogenic cyst – well-defined rounded lesion that may contain air–fluid level, particularly if previous infection.
3. Granuloma – most commonly following TB.
4. Inflammatory pseudotumour – synonyms include plasma cell granuloma and inflammatory myofibroblastic tumour. Variable size and may be large. Usually peripheral. Calcified in 25%.
Neoplastic lesions
Malignant
1. Solitary metastasis – most commonly Wilms’ tumour and sarcomas. Note that approximately one-third of lung nodules in children with a known primary tumour are NOT metastases, e.g. drug reaction, intrapulmonary lymph node.
2. APUD neuroendocrine tumour – 45% of all primary lung neoplasms in childhood. 80% are carcinoids. Usually malignant, frequently endobronchial, causing lobar collapse or overinflation.
3. Mesenchymal tumour – 25% of all primary lung neoplasms in childhood. Pleuropulmonary blastoma. May be solid (42% 5-year survival), cystic (82% 5-year survival) or mixed. May be very large peripheral and locally invasive mass.
4. Bronchogenic carcinoma – 25% of all primary lung neoplasms in childhood. Most commonly bronchoalveolar cell carcinoma.
14.41
Multiple pulmonary nodules in a child
Benign
1. Miliary TB/other granulomatous infection – miliary pattern of haematogenous spread should be distinguished from ‘tree-in-bud’ pattern of endobronchially disseminated TB.
2. Septic emboli – frequently cavitary.
3. Langerhans’ cell histiocytosis – initial nodular pattern 1–10 mm, developing cavitation or cysts.
4. Wegener’s granulomatosis – may cavitate.
5. Respiratory papillomatosis – represents pulmonary seeding of laryngeal papillomata, occurring in 1% of cases. Nodular and cystic lesions present. Poor prognosis. Risk of malignant transformation.
6. Multiple arteriovenous malformations – two-thirds associated with hereditary haemorrhagic telangiectasia. Multiple in most cases. Usually lower lobes.
14.42
Situs and cardiac malpositions
1. Situs solitus – normal. All structures are concordant.
2. Situs inversus – cardiac apex, aortic arch and stomach are on the right; visceral organs are on the opposite side to normal. Slight increase in the incidence of congenital heart disease. Present in 50% of patients with primary ciliary dyskinesia (the combination is called Kartagener’s syndrome).
3. Situs solitus with dextrocardia – cardiac apex on right with stomach bubble on left. Caused by failure of rotation of the embryonic cardiac loop and > 90% of cases are associated with congenital heart disease, usually cyanotic (corrected TGA, VSD and pulmonary stenosis). Scimitar syndrome is dextrocardia, hypoplastic right lung and partial anomalous pulmonary venous drainage into the inferior cava.
4. Levoversion with abdominal situs inversus – incidence of congenital heart disease 100%.
5. Situs ambiguous with bilateral ‘right-sidedness’: asplenia syndrome – absent spleen, bilateral trilobed lungs, right and left lobes of liver are similar size. Cardiac apex left, right or midline. Complex cardiac anomalies ± small bowel malrotation.
6. Situs ambiguous with bilateral ‘left-sidedness’: polysplenia syndrome – bilateral bilobed lungs, absent hepatic segment of IVC and enlarged azygos and hemiazygos veins. Intracardiac anomalies, but less complex than in bilateral ‘right-sidedness’.
14.43
Neonatal pulmonary venous congestion
1. Prominent interstitial markings.
5. Cardiomegaly – in all except the infradiaphragmatic type of TAPVD.
1st week
1. Overhydration – delayed clamping of the cord and twin–twin transfusion.
2. Asphyxia – the most common cause of cardiomegaly on the first day.
3. Hypoplastic left heart – heart size normal to mild cardiomegaly. Pulmonary vasculature normal or mild oedema. Often a marked discrepancy between the ‘near normality’ of the CXR and severity of clinical symptoms.
14.44
Neonatal cyanosis
With increased pulmonary vascularity
Cyanosis and congestive cardiac failure – either may predominate.

14.45
Cardiovascular involvement in syndromes
| Syndrome | Involvement |
| Cri-du-chat | Variable |
| Down’s* | AV canal, VSD, PDA, ASD and aberrant right subclavian artery |
| Ehlers–Danlos | Mitral valve prolapse, aortic root dilatation, dissecting aortic aneurysm and intracranial aneurysm |
| Ellis–Van Creveld | ASD and common atrium |
| Friedreich’s ataxia | Hypertrophic cardiomyopathy |
| Holt–Oram | ASD and VSD |
| Homocystinuria* | Medial degeneration of the aorta and pulmonary artery causing dilatation. Arterial and venous thromboses |
| Hurler’s/Hunter’s* | Intimal thickening of coronary arteries and valves |
| Kartagener’s | Situs inversus ± septal defects |
| Marfan’s | Cystic medial necrosis of the wall of the aorta, and less commonly the pulmonary artery, leading to dilatation and predisposing to dissection. Aortic and mitral regurgitation |
| Morquio’s* | Late onset of aortic regurgitation |
| Noonan’s | Pulmonary valve stenosis, and branch stenosis of pulmonary arteries, septal defects |
| Osteogenesis imperfecta* | Aortic and mitral regurgitation. Ruptured chordae |
| Rubella | Septal defects, PDA, pulmonary artery branch stenoses and myocardial disease |
| Trisomy 13 | VSD, ASD, PDA and dextroposition |
| Trisomy 18 | VSD, ASD and PDA |
| Tuberous sclerosis* | Cardiomyopathy and rhabdomyoma of the heart |
| Turner’s | Coarctation, aortic and bicuspid aortic valve stenosis |
14.46
Abdominal mass in a child
Non-renal retroperitoneal (23%)
(a) Age – 90% < 5 years; 15–30% < 1 year. Median age 2 years. Accounts for 50% of all neonatal tumours.
(b) Site – adrenal (40%), abdominal sympathetic chain (25%), posterior mediastinal sympathetic chain (15%), neck (5%), pelvis (5%), unknown (10%).
| International Neuroblastoma Risk Group Staging System (INRGSS) | International Neuroblastoma Staging System (INSS) |
| (Preoperative imaging-based system) | (Postsurgical staging system) |
| L1 – localized, not involving vital structures | Stage 1 – localized disease, completely resected |
| L2 – locoregional tumour with 1 or more imaging-defined risk factors | Stage 2A – localized; incomplete gross resection; lymph nodes −ve |
| Stage 2B – localized; complete or incomplete resection; ipsilateral lymph nodes +ve; contralateral lymph nodes −ve | |
| Stage 3 – Unilateral tumour with contralateral +ve lymph nodes or tumour crossing the midline | |
| M – metastatic disease | Stage 4 – metastatic disease |
| MS – < 18 months with skin, liver involvement; better prognosis | 4S – < 12 months with skin, liver involvement |
(i) Ipsilateral tumour extension within two body compartments
– Tumour encasing carotid and/or vertebral artery and/or internal jugular vein.
(iii) Cervicothoracic junction
– Tumour encasing brachial plexus roots.
– Tumour encasing subclavian vessels and/or vertebral and/or carotid artery.
– Tumour encasing the aorta and/or major branches.
– Tumour compressing the trachea and/or principal bronchi.
– Lower mediastinal tumour, infiltrating the costovertebral junction between T9 and T12.
– Tumour infiltrating the porta hepatis and/or the hepatoduodenal ligament.
– Tumour encasing branches of the superior mesenteric artery at the mesenteric root.
– Tumour encasing the origin of the coeliac axis and/or of the superior mesenteric artery.
– Tumour invading one or both renal pedicles.
– Tumour encasing the aorta and/or vena cava.
(vii) Intraspinal tumour extension whatever the location provided that:
– More than one-third of the spinal canal in the axial plane is invaded and/or the perimedullary leptomeningeal spaces are not visible and/or the spinal cord signal is abnormal.
(viii) Infiltration of adjacent organs/structures
(ix) Conditions to be recorded, but not considered imaging-defined risk factors
(d) Clinical presentation – 70% have metastases at presentation and a similar percentage have systemic symptoms. There may be local effects: pain, mass, spinal cord compression, dyspnoea or dysphagia, the effects of metastases (scalp masses, pain, weight loss, anaemia, fatigue, etc.), or other effects due to hormone secretion (opsomyoclonus [cerebellar ataxia and jerky eye movements]; 50% have neuroblastoma), hypertension, diarrhoea (due to vasoactive intestinal peptide), flushing and sweating.
(e) Plain films – calcification in two-thirds, loss of psoas outline, bony metastases, enlargement of intervertebral foramina and, in the chest, abnormal posterior ends of ribs.
(f) US – heterogeneous, echogenic mass.
(g) CT – soft-tissue mass with calcification in nearly all. Encasement rather than displacement of major vessels.
(h) MRI – prolonged T1 and T2 relaxation times. Calcification is not as readily recognized as on CT but MRI is superior for lymph-node metastases, liver metastases and extradural spread of tumour.
(i) Radionuclide scanning – bone scanning (for cortical disease) and MIBG scanning (for medullary disease) are complementary techniques for the demonstration of skeletal metastases. MIBG is superior for follow-up of disease.
Gastrointestinal (18%)
1. Appendix abscess (10%) – particularly spreads to pouch anterior to rectum.
2. Hepatoblastoma – more commonly in right lobe, but 40% in both lobes. 40% calcify. See 14.57.
3. Haemangioma – commonly multiple, involving entire liver. Rarely calcify. ± Associated with congestive heart failure and cutaneous haemangiomas.
4. Choledochal cyst – the classic triad of mass, pain and jaundice is only present in 10%. Dynamic radionuclide scintigraphy with 99mTc-TBIDA is diagnostic. See 14.59.
Balassy, C., Navarro, O. M., Daneman, A. Adrenal masses in children. Radiol Clin North Am. 2011; 49(4):711–727.
Haddad, M. C., Birjawa, G. A., Hemadeh, M. S., et al. The gamut of abdominal and pelvic cystic masses in children. Eur Radiol. 2001; 11:148–166.
Hiorns, M. P., Owens, C. M. Radiology of neuroblastoma in children. Eur Radiol. 2001; 11:2071–2081.
Hoffer, F. A. Magnetic resonance imaging of abdominal masses in the pediatric patient. Semin Ultrasound CT MR. 2005; 26(4):212–223.
14.47
Intestinal obstruction in a neonate
1. It is usually impossible to differentiate small from large bowel.
2. Not all gaseously distended bowel is obstructed. Resuscitation and infants on positive pressure ventilation may lead to significant abdominal distension. A rule of thumb is that bowel that is wider than the width of a lumbar vertebral body is dilated.
3. Ileus is characterized by uniform dilatation of bowel. It is found in sepsis, NEC and electrolyte imbalance. Infants with sepsis and NEC are sick; those with uncomplicated bowel obstruction are usually otherwise well.
4. Bowel obstruction should be considered as ‘high’ (as far as the jejunum) or ‘low’ (for more distal obstructions). The former present with vomiting and are investigated by upper gastrointestinal contrast study, while the lower present with delayed passage of meconium and may require a contrast enema.
High intestinal obstruction
2. Pyloric or prepyloric membrane/antral web – gastric outlet obstruction in the presence of a normal pylorus and the appearance of two duodenal caps. The web may be identified by US.
3. Duodenal atresia/stenosis/web – marked dilatation of the proximal duodenum with the ‘double bubble’ sign, which may also be seen by US of the fetus (50% have a history of polyhydramnios). No gas distally when there is atresia, but a variable amount of gas in the distal bowel when there is stenosis. Duodenal web may produce ‘windsock’ appearance as web balloons into distal duodenum. Bile-stained vomiting in the majority. Associated with annular pancreas (20%), Down’s syndrome (30%), cardiac abnormalities (25%), oesophageal atresia (10%) and other abnormalities of gastrointestinal tract (60%).
4. Preduodenal portal vein – identified on US, CT or MRI. Associated with an intrinsic duodenal obstruction; the vein is not the direct cause of the obstruction.
5. Malrotation and volvulus – sudden onset of bile-stained vomiting. Few radiological signs if the obstruction is recent, intermittent or incomplete. Because of the acute nature of the condition, the duodenum is not dilated. If not recognized, obstruction progresses to bowel ischaemia, infarction and death. A contrast study should demonstrate the normal C-shaped duodenal loop which terminates to the left of the left-sided pedicle at the same level as the duodenal cap. In malrotation without volvulus the duodenojejunal flexure is to the right of and below its normal position. Volvulus with incomplete obstruction is identified by a corkscrew pattern of the jejunum. When there is complete obstruction the distal duodenum terminates as a beak.
6. Congenital fibrous band (of Ladd) – connects caecum to posterolateral abdominal wall and commonly crosses the duodenum. Associated with malrotation and midgut volvulus.
7. Jejunal atresia – 50% of small bowel atresias and 50% are associated with other atretic sites distally (ileum > colon). AXR demonstrates three (‘triple bubble’) or more dilated, air-filled loops. Colon usually normal in calibre.
Low intestinal obstruction
1. Meconium ileus – mottled lucencies (‘soap bubble’ appearance) due to gas trapped in meconium but only few fluid levels (since it is very viscous). Bowel loops of variable calibre. Rapid appearance of fluid levels suggests volvulus. Peritoneal calcification due to perforation occurring in utero is seen in 30%. Secondary microcolon on contrast enema which also shows meconium pellets in the distal ileum. Cystic fibrosis in the majority.
2. Ileal atresia – 50% of small bowel atresia, may be multiple and may coexist with jejunal atresia. Multiple dilated loops with fluid levels. Secondary microcolon.
3. Incarcerated inguinal hernia.
4. Small left colon syndrome/functional immaturity of the left hemicolon – 50% associated with maternal diabetes. Small colon on enema up to level of splenic flexure, sometimes with meconium plugging. Infants should be followed up to exclude Hirschsprung’s disease.
5. Hirschsprung’s disease – multiple dilated loops of bowel. Diagnosis is made by contrast enema which demonstrates normal size, aganglionic distal bowel with a transition zone at the junction with proximal dilated ganglionic bowel, classically reversed rectosigmoid ratio.
6. Meconium plug syndrome – plugged meconium present in distal colon. May be a feature of Hirschsprung’s, small left colon syndrome and a presenting feature of cystic fibrosis (but note this is not the same as meconium ileus).
7. Inspissated milk – presents from 3 days to 6 weeks of age. Dense, amorphous intraluminal masses frequently surrounded by a rim of air, ± mottled lucencies within them. Usually resolves spontaneously.
8. Colonic atresia – 5–15% of intestinal atresias. AXR may be similar to other distal bowel obstructions but some infants show a huge, disproportionately dilated loop (between the atretic segment and a competent ileocaecal valve).
Applegate, K. E., Anderson, J. M., Klatte, E. C. Intestinal malrotation in children: a problem-solving approach to the upper gastrointestinal series. Radiographics. 2006; 5:1485–1500.
Berrocal, T., Torres, I., Gutierrez, J., et al. Congenital anomalies of the upper gastrointestinal tract. Radiographics. 1999; 19:855–872.
Berrocal, T., Lamas, M., Gutierrez, J., et al. Congenital anomalies of the small intestine, colon, and rectum. Radiographics. 1999; 19:1219–1236.
Carty, H., Brereton, R. J. The distended neonate. Clin Radiol. 1983; 34:367–380.
14.48
Intra-abdominal calcifications in the newborn
1. Meconium peritonitis – antenatal bowel perforation results in aseptic peritonitis which rapidly calcifies. Calcification occurs in the peritoneum itself most commonly, but also in the bowel wall and in the scrotum and may be punctate, linear or plaque like. Commonest causes are meconium ileus and ileal atresia, but any cause of bowel obstruction may be associated.
2. Meconium pseudocyst – cyst-like mass with peripheral calcification resulting from walling-off of extruded meconium following perforation.
3. Intraluminal meconium calcification – may occur in association with distal obstruction, particularly meconium ileus and anorectal malformations.
4. Hepatic calcification – neonatal liver calcification occasionally occurs with congenital infections.
Beasley, S. W., de Campo, M. Intraluminal calcification in the newborn: diagnostic and surgical implications. Pediatr Surg. 1986; 1:249.
Lang, I., Daneman, A., Cutz, E., et al. Abdominal calcification in cystic fibrosis with meconium ileus: radiologic–pathologic correlation. Pediatr Radiol. 1997; 27:523–527.
Miller, J. P., Smith, S. D., Sukarochana, K. Neonatal abdominal calcification: is it always meconium peritonitis? J Pediatr Surg. 1988; 23:555.
14.49
Abnormalities of bowel rotation
1. Exomphalos – total failure of the bowel to return to the abdomen from the umbilical cord. Bowel is contained within a sac. To be differentiated from gastroschisis, in which bowel protrudes through a defect in the abdominal wall, classically in a right paraumbilical position.
2. Non-rotation – usually an asymptomatic condition with the small bowel on the right side of the abdomen and the colon on the left side. Small and large bowel lie on either side of the SMA with a common mesentery. CT or transverse US scans show the SMV lying to the left of the SMA (cf. the normal arrangement in which the SMV lies to the right of the SMA).
3. Malrotation – the duodenojejunal flexure lies to the right and caudad to its usual position, which is to the left of the left-sided pedicle on a true AP projection and approximately in the same axial plane as the first part of the duodenum. The caecum is usually more cephalad than normal but is normally sited in 5%. Malrotation is a frequent feature of diaphragmatic hernia and abdominal wall defects. Also associated with visceral heterotaxy. US or CT shows the SMV to the left of the SMA. A normal US does not, however, exclude malrotation (3% false-negative rate): upper gastrointestinal contrast examination remains the gold standard. At risk of volvulus, which is life-threatening.
4. Reverse rotation – rare. Colon lies dorsal to the SMA with jejunum and duodenum anterior to it.
5. Paraduodenal hernia – rare.
6. Cloacal extrophy – rare. No rotation of the bowel, and the ileum and colon open separately onto the extroverted area in the midline below the umbilical cord.
Applegate, K. E., Anderson, J. M., Klatte, E. C. Intestinal malrotation in children: a problem-solving approach to the upper gastrointestinal series. Radiographics. 1992; 26(5):1485–1500.
Dufour, D., Delaet, M. H., Dassonville, M., et al. Midgut malrotation, the reliability of sonographic diagnosis. Pediatr Radiol. 1992; 22:21–23.
Strouse, P. J. Disorders of intestinal rotation and fixation (‘malrotation’). Pediatr Radiol. 2004; 34:837–851.
14.50
Adrenal mass in childhood
Neoplastic
Medullary
2. Ganglioneuroblastoma – one-third adrenal, one-third retroperitoneum, one-third posterior mediastinum, neck or pelvis.
3. Ganglioneuroma – older children, often asymptomatic.
It is not possible to differentiate these entities on imaging alone.
4. Phaeochromocytoma – uncommon. Mean age 11 years, 25% bilateral. Associated with MEN type 2, NF-1 and von Hippel–Lindau disease.
14.51
Primary renal neoplasms in childhood
1. Wilms’ tumour – 8/106 children. 80% present in the first 3 years. Bilateral in 5%. Associated abnormalities: cryptorchidism (3%), hypospadias (2%), hemihypertrophy (2%), sporadic aniridia (1%) (30% of those with aniridia and 10% of those with Beckwith–Wiedemann syndrome [macroglossia, organomegaly, exomphalos ± hemihypertrophy] develop Wilms’ tumour). 90% have favourable histology. Secondaries → lungs and liver. 5% have tumour thrombus in the IVC or right atrium. Hypertension in 25%.
(a) Plain film – bulging flank (75%), loss of renal outline (66%), enlargement of renal outline (33%), displacement of bowel gas (50%), loss of psoas outline (33%), calcification (10%).
(b) US – large, well-defined mass, greater echogenicity than liver. Solid with haemorrhage/necrosis. Lack of IVC narrowing on inspiration suggests occlusion.
(c) CT – large, well-defined, low attenuation, heterogeneous with foci of even lower attenuation due to necrosis. Minimal enhancement compared with the residual rind of functioning renal tissue.
(d) MRI – heterogeneous, low signal (T1W), high signal (T2W). Inhomogeneous enhancement compared with residual renal tissue.
2. Nephroblastomatosis – nephrogenic rests which maintain the potential for malignant induction to Wilms’ tumour. Nephrogenic rests in 40% of unilateral and 99% of bilateral Wilms’ tumours. May be: perilobar (most common, at the lobar surface); intralobar (anywhere in the cortex or medulla, or combined).
(b) CT – low attenuation and non-enhancing (therefore best shown on contrast-enhanced images).
(c) MRI – similar signal to renal cortex. Non-enhancing (therefore best shown on contrast-enhanced images).
3. Congenital mesoblastic nephroma – most common solid renal tumour in the newborn. Mean age at diagnosis is 3.5 months. No recurrence when diagnosed in first 3 months. Indistinguishable from Wilms’ tumour but some demonstrate function.
4. Clear cell sarcoma – 4–6% of childhood renal tumours. Presentation at 3–5 years. Poor prognosis with early secondaries (to bone; usually lytic but may be sclerotic). Never bilateral. No specific imaging features of the primary tumour.
5. Rhabdoid tumour of kidney – 2% of childhood renal tumours. Presentation at 3 months to 4.5 years (50% in first year). Most malignant renal tumour with poorest prognosis. Extrarenal extension or haematogenous secondaries (to brain or bone) often present at diagnosis. Association with midline posterior fossa tumours. Hypercalcaemia sometimes present. Imaging of the primary tumour is similar to Wilms’ tumour; however, areas of necrosis or calcification outlining tumour lobules may suggest rhabdoid tumour.
6. Multilocular cystic nephroma – presents at 3 months to 4 years. Multiple cysts of varying size. Thin septae. Thick septae, nodules or a large solid component suggest Wilms’ tumour with cystic degeneration. Resection is curative and local recurrence is rare. Differential diagnosis is a multicystic dysplastic kidney, but this affects the entire kidney.
(a) US and CT – cystic with thin septae.
(b) MRI – round collections of variable signal intensity suggesting haemorrhage or proteinaceous material.
7. Renal cell carcinoma – rare. Differentiating features from Wilms’ tumour are: older age at presentation (mean 11–12 years), calcification is more common (25%) and more homogeneous, smaller at the time of diagnosis and haematuria is more common. Poorer prognosis compared with Wilms’ tumour. Similar imaging findings. Association with von Hippel–Lindau disease and tuberous sclerosis*.
8. Angiomyolipoma – in 50–80% of patients with tuberous sclerosis*. 50% of patients with angiomyolipomas have tuberous sclerosis. Multiple bilateral tumours, which are usually small.
14.52
Hydronephrosis in a child
1. Pelviureteric junction obstruction – more common on the left side. 20% bilateral. Due to stricture, neuromuscular incoordination or aberrant vessels. Contralateral kidney is dysplastic in 25% of cases and absent in 12%.
2. Bladder outflow obstruction (q.v.) – bilateral upper tract dilatation.
3. Ureterovesical obstruction – more common in males and more common on the left side. May be bilateral.
4. Reflux without obstruction.
5. Associated with urinary tract infection – but no obstruction or reflux? Represents atony.
14.53
Renal mass in the newborn and young infant
1. Hydronephrosis (q.v.) – unilateral or bilateral. The most common cause of an abdominal mass in the first 6 months of life.
2. Multicystic dysplastic kidney (MCDK) – unilateral, but 30% have an abnormal contralateral kidney (mostly pelviureteric junction obstruction). Non-functioning, multilobulated kidney. Rarely, nephrographic crescents and late pooling of contrast medium in cysts are observed. Curvilinear calcification is characteristic but only seen occasionally. US reveals multiple cysts of unequal size. The commonest renal mass in the first year of life.
3. Polycystic kidneys (see Polycystic disease*) – bilateral. Highly echogenic and large on US with autosomal recessive polycystic kidney disease.
4. Renal vein thrombosis (q.v.) – unilateral or bilateral.
14.54
Bladder outflow obstruction in a child
1. Distended bladder with incomplete emptying or reduced bladder volume with trabeculation if long-standing obstruction.
Causes (from proximal to distal)
1. Vesical diverticulum – posteriorly behind the bladder base. It fills during micturition and compresses the bladder neck and proximal urethra. More common in males.
2. Bladder neck obstruction – probably not a distinct entity and only occurs as part of other problems such as ectopic ureterocoele and rhabdomyosarcoma.
3. Ectopic ureterocoele – 80% are associated with the upper moiety of a duplex kidney. 15% are bilateral. More common in females. Opens into the urethra, bladder neck or vestibule. May be largely outside the bladder and the bladder base may be elevated. ‘Drooping lily’ appearance of lower moiety. May prolapse into the urethra.
4. Posterior urethral valves – posterior urethra is dilated and the distal urethra is small. Almost exclusively males.
5. Urethral stricture – post-traumatic strictures are most commonly at the penoscrotal junction and follow previous instrumentation or catheterization.
6. Cowper’s syringocoele – a dilatation of Cowper’s gland ducts. Filling of Cowper’s ducts may be a normal finding. When dilated, occasionally presents with haematuria, infection or urethral obstruction.
7. Anterior urethral diverticulum – a saccular wide-necked, ventral expansion of the anterior urethra, usually at the penoscrotal junction. The proximal lip of the diverticulum may show as an arcuate filling defect and during micturition the diverticulum expands with urine and obstructs the urethra.
10. Meatal stenosis – usually a clinical diagnosis, but may be detected on micturating cystourethrogram: voiding images should include the meatus.
14.55
Vesicoureteric reflux
Congenital = primary reflux
1. Simple congenital reflux – due to incompetence of vesicoureteric junction secondary to abnormal tunnelling of distal ureter through bladder. 10% of normal Caucasian babies and 30% of children with a first episode of UTI. Usually disappears in 80%. Medium- to high-grade VUR can lead to renal damage in association with UTI.
2. Reflux associated with duplex kidneys – usually occurs into lower-moiety ureter, which has a normal position but abnormal tunnelling. Reflux may also occur into a ureterocoele if this everts during filling or voiding.
Acquired = secondary reflux
4. Urethral obstruction – most commonly posterior urethral valves: see 14.54. More commonly on left side.
5. Prune-belly syndrome – almost exclusively males. High mortality. Bilateral hydronephrosis and hydroureters with a distended bladder are associated with undescended testes, hypoplasia of the anterior abdominal wall and urethral obstruction.
Riccabona, M., Avni, F. E., Blickman, J. G., et al. Imaging recommendations in paediatric uroradiology: minutes of the ESPR workgroup session on urinary tract infection, fetal hydronephrosis, urinary tract ultrasonography and voiding cystourethrography, Barcelona, Spain, June 2007. Pediatr Radiol. 2008; 38(2):138–145.
14.57
Hepatic tumours in children
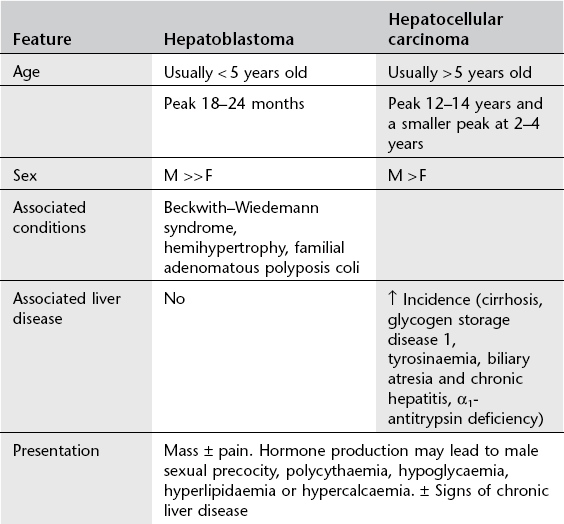
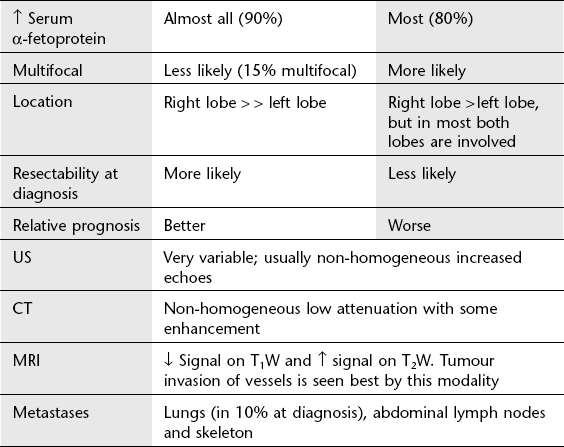
Benign (one-third)
1. Haemangioendothelioma (infantile hepatic haemangioma) – most common benign tumour. Often present in the newborn period with hepatomegaly and congestive cardiac failure. ± Skin haemangiomas (50%) ± consumptive coagulopathy (thrombocytopenia). Unifocal or multifocal, well-defined or diffuse. Typical pattern of enhancement on CT with early rim enhancement and variable delayed ‘filling-in’ of the centre of the tumour over next 30 minutes. On MRI the lesions have a non-specific hypointense T1W and hyperintense T2W appearance with variable areas of T1W hypointensity corresponding to fibrosis and haemosiderin deposition. 99mTc-labelled red cells will accumulate in this tumour. In the neonate, this and cavernous haemangioma may be considered together.
2. Mesenchymal hamartoma – second most common benign tumour. Up to 2 years of age. May be (multi)cystic or stromal, with a ‘Swiss cheese’ appearance. Solid components may enhance.
3. Adenoma – uncommon in paediatric population. Solitary or multiple, occurring spontaneously or complicating glycogen storage disease, Fanconi’s anaemia treated with anabolic steroids, and teenagers on the oral contraceptive pill. Hypodense on CT. Variable appearance on MRI and US. Classically peak arterial enhancement with rapid washout.
14.58
Fetal or neonatal liver calcification
Peritoneal
1. Meconium peritonitis –commonest cause of neonatal abdominal calcification. US reveals intra-abdominal solid or cystic masses with calcified walls.
2. Plastic peritonitis due to ruptured hydrometrocolpos – similar appearance to meconium peritonitis but US may demonstrate a dilated, fluid-filled uterus and vagina.
Parenchymal
1. Congenital infections – TORCH complex (toxoplasmosis, rubella, cytomegalovirus, herpes simplex) and varicella. Randomly scattered nodular calcification. Often calcification elsewhere and other congenital abnormalities.
2. Tumours – haemangioma, hamartoma, hepatoblastoma, teratoma and metastatic neuroblastoma. Complex mass on US.
14.59
Jaundice in infancy
Anatomical abnormalities
1. Biliary atresia – 1 in 15,000 live births. Three types: I (fCBD atresia), extremely rare; II (intrahepatic), uncommon; III (extrahepatic), which is subdivided into subtype 1 (66%) with a bile duct remnant at the porta hepatis and subtype 2 (34%) with no bile duct. Subtype 2 is associated with multiple congenital abnormalities (polysplenia, intestinal malrotation, azygos continuation of the IVC, situs inversus and preduodenal portal vein).
(a) A normal-sized gallbladder that contracts following a fatty meal excludes the diagnosis.
(b) Absence of, or a small or irregular gallbladder or thin-walled gallbladder (gallbladder ‘ghost’ triad), favours the diagnosis, but a normal gallbladder may be seen in 10% of cases.
(c) Liver echogenicity is normal or heterogeneously increased.
(d) A triangular or tubular echogenic structure (due to fibrous tissue) at the porta hepatis is highly specific for extrahepatic biliary atresia (triangular cord sign).
(e) A prominent hepatic artery may also support the diagnosis, but cannot be used in isolation to make the diagnosis.
Normal uptake by hepatocytes but no excretion into the bowel suggests the diagnosis but is not diagnostic since α1-antitrypsin may show similar appearances. Operative cholangiography is indicated.
2. Choledochal cyst – may present in the neonatal period or at a later age. Classification is:
I (80–90%) Fusiform or focal dilatation of the common bile duct ± common hepatic duct.
II (2%) Diverticulum of the common bile duct.
III (2–5%) Outpouching of the common bile duct in the wall of the second part of the duodenum – a choledochocoele.
IVa. Dilatation of the common bile duct and focal dilatations of the intrahepatic ducts.
IVb. Focal dilatations of the common bile duct.
V Focal dilatations of the intrahepatic bile ducts (Caroli’s disease).
3. Alagille syndrome – AD with variable expressivity. Dysmorphic facies, eye abnormalities, cardiovascular abnormalities, especially peripheral pulmonary stenosis or hypoplasia, hypoplasia of intrahepatic bile ducts, butterfly vertebrae, radioulnar synostosis.
14.60
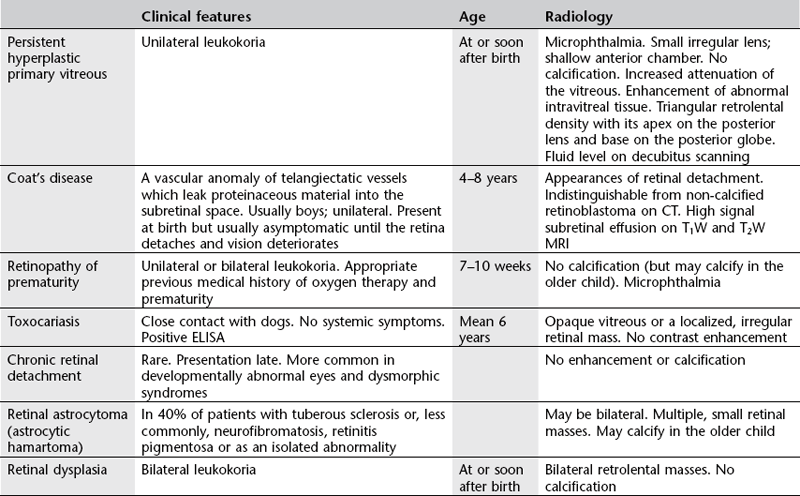
Differential diagnosis of retinoblastoma
Brennan, R. C., Wilson, M. W., Kaste, S., Helton, K. J., McCarville, M. B. US and MRI of pediatric ocular masses with histopathological correlation. Pediatr Radiol. 2012; 42(6):738–749.
Chung, E. M., Specht, C. S., Schroeder, J. W. From the archives of the AFIP: pediatric orbit tumors and tumorlike lesions: neuroepithelial lesions of the ocular globe and optic nerve. Radiographics. 2007; 27(4):1159–1186.
14.61
Prevertebral soft-tissue mass on the lateral cervical X-ray
14.62
Neck masses in infants and children
US is a valuable first imaging modality. MRI is generally preferred to CT.
14.63
Causes of stroke in children and young adults
1. Emboli – cyanotic heart disease (secondary to right-to-left intracardiac shunt), cardiomyopathies, mitral valve prolapse, Osler–Weber–Rendu (secondary to pulmonary arteriovenous malformations).
2. Arterial dissection – trauma, spontaneous, fibromuscular dysplasia (also vessel stenoses and saccular dilatations, intracranial aneurysms), Marfan’s syndrome, Ehlers–Danlos syndrome and homocystinuria (see 12.4).
3. Venous thrombosis – pregnancy, postpartum, oral contraceptive pill, skull base/intracranial sepsis, inflammatory bowel disease, SLE*, Behçet’s disease and malignancy (see 12.3).
4. Infection – purulent meningitis may cause arterial and venous strokes. Viral infection is a well-recognized cause of arterial stroke due to a ‘vasculitis’ that usually involves the proximal MCA (infarction of basal ganglia with sparing of the cortical territories).
5. Trauma – arterial dissection and hypoxia.
6. Drugs – cocaine, amfetamines.
7. Blood disorders – sickle-cell anaemia*, polycythaemia, protein C and S deficiency.
8. Migraine – usually posterior circulation.
9. Vasculopathy, vasculitis – neurofibromatosis*, fibromuscular dysplasia, Kawasaki’s, SLE*, sarcoidosis*.
14.64
Large head in infancy
14.65
Wide cranial sutures
14.66
Hyperechoic lesions in the basal ganglia on cranial ultrasound of neonates and infants
Single punctate, multiple punctate or stripe-like densities.
14.67
Multiple wormian bones
14.68
Craniosynostosis
Primary craniosynostosis
1. Sagittal synostosis – elongated narrow ‘boat-shaped’ skull (scaphocephaly/dolichocephaly).
2. Unilateral coronal synostosis – oblique appearance of the craniofacial structures with harlequin orbit (frontal plagiocephaly).
3. Bilateral coronal synostosis – ‘short head’, often seen with synostosis of other sutures (brachycephaly).
4. Metopic synostosis – triangular-shaped head (trigonocephaly).
5. Unilateral lambdoid synostosis – occipital plagiocephaly.
6. Bilateral lambdoid synostosis – occipital plagiocephaly with flattened occiput. Beware postural flattening of the occiput due to infants being placed to sleep on their backs to prevent sudden infant death syndrome. There is no sutural fusion in these cases.
7. Cloverleaf skull (kleeblattschädel) – synostosis of multiple paired sutures produces a ‘trilobular skull’.
14.69
Cystic lesions on cranial ultrasound in neonates and infants
Infratentorial cysts
1. Mega cisterna magna – occurs in 1%. As isolated finding probably a normal variant.
2. Dandy–Walker malformation – cystic dilatation of posterior fossa in communication with fourth ventricle, with associated vermian hypoplasia.
3. Arachnoid cyst – one-quarter occur in posterior fossa, most commonly retrocerebellar.
Supratentorial cysts
1. Subependymal cysts – located in subependymal region around caudothalamic notch. Most commonly represent previous germinal matrix haemorrhage. May be congenital, probably reflecting germinolysis, particularly in association with CMV infection.
2. Choroid plexus cysts – usually located within body of choroid plexus. Weak markers of aneuploidy, particularly if large and bilateral. No clinical significance if detected after birth.
3. Cystic periventricular leukomalacia – white matter necrosis in preterm infant. Hyperechoic lesions dorsal and lateral to external angles of lateral ventricles, developing into cysts in severe cases.
4. Porencephalic cyst – an area of cystic encephalomalacia filled with CSF, commonly following haemorrhage or infection, with a communication with the ventricular system.
5. Arachnoid cyst – most commonly in the sylvian fissure, and usually incidental. Suprasellar cysts more frequently symptomatic.
6. Vein of Galen malformation – not a cyst, but may appear so on US scan. Colour Doppler flow confirms.
14.70
Disorders of neuronal migration
1. Agyria–pachygyria – poorly formed gyri and sulci, the former being more severe. Focal pachygyria may be the cause of focal epilepsy. Polymicrogyria (see below) may coexist with pachygyria. Extreme cases with a smooth brain may be termed lissencephaly. Complete lissencephaly ≡ agyria. Several distinct forms are recognized.
(a) Type I lissencephaly – small brain with few gyri; smooth, thickened four-layer cortex resembling that of a 13-week fetus with diminished white matter and shallow vertical sylvian fissures (‘figure-of-eight’ appearance on axial images). ± Agenesis of the corpus callosum. Severe mental retardation, diplegia, seizures, microcephaly and limited survival. Some infants have specific dysmorphic features: Miller–Dieker syndrome and Norman–Roberts syndrome. Pachygyria may also be observed in Zellweger syndrome and prenatal CMV infection.
(b) Type II lissencephaly (Walker–Warburg syndrome) – smooth cortex, cerebellar hypoplasia and vermian aplasia and hydrocephalus (in 75%) due to cisternal obstruction by abnormal meninges or aqueduct stenosis.
2. Polymicrogyria – the neurons reach the cortex but are distributed abnormally. Macroscopically the surface of the brain appears as multiple small bumps. Localized abnormalities are more common than generalized and often involve arterial territories, especially the middle cerebral artery. The most common location is around the sylvian fissure. The cortex is isointense to grey matter but in 20% of cases the underlying white matter has high signal on T2W. Linear flow voids due to anomalous venous drainage may be present. Polymicrogyrias may be present in the vicinity of a porencephalic cyst, and may be associated with heterotopic grey matter or agenesis of the corpus callosum or with evidence of fetal infection such as intracranial calcification. Symptoms and signs depend on the size, site and presence of associated abnormalities. The majority have mental retardation, seizures and neurological signs.
3. Schizencephaly – clefts which extend through the full thickness of the cerebral mantle from ventricle to subarachnoid space. The cleft is lined by heterotopic grey matter and microgyrias, indicating that it existed prior to the end of neuronal migration. Unilateral or bilateral (usually asymmetrical) and usually near the sylvian fissure. May be associated with absence of the septum pellucidum or, less commonly, dysgenesis of the corpus callosum. There are variable clinical manifestations, from profound retardation to isolated partial seizures.
4. Heterotopic grey matter – collections of neurons in a subependymal location, i.e. at the site of the germinal matrix or arrested within the white matter on their way to the cortex. Isointense to normal grey matter on all imaging sequences. Nodules or bands and may have mass effect. Frequently a part of complex malformation syndromes or, when isolated, may be responsible for focal seizures which are amenable to surgical treatment. Small heterotopias are probably asymptomatic.
5. Cortical dysplasia – focal disorganization of the cerebral cortex. A single enlarged gyrus resembling focal pachygyria. Usual presentation is with partial epilepsy.
14.71
Supratentorial tumours in childhood
1. Hemispheric astrocytoma – solid, solid with a necrotic centre, or cystic with a mural nodule. Usually large at presentation and can involve the basal ganglia and thalami. Most are low grade. Enhancement with contrast medium does not correlate with histological grade. Associated with NF-1.
2. Craniopharyngioma – more than half of all craniopharyngiomas occur in children (8–14 years). Cystic/solid partially calcified suprasellar mass presenting with headache, visual disturbance and endocrine abnormalities (see 12.37).
3. Optic pathway glioma – low grade, but infiltrating pilocytic astrocytomas associated with NF-1. Solid enhancing tumours that extend along the length of the anterior optic pathways and may invade adjacent structures (e.g. hypothalamus) and extend posteriorly into the optic tracts and radiations.
4. Giant cell subependymal astrocytoma – slow-growing partially cystic, partially calcified tumour occurring in tuberous sclerosis. Located at the foramen of Monro and presents with obstructive hydrocephalus.
5. Germ cell tumours – germinomas, teratoma (see 12.39).
6. Primitive neuroectodermal tumour (PNET) – large heterogeneous hemispheric mass presenting in neonates and small infants. Necrosis, haemorrhage and enhancement are common.
7. Dysembryoplastic neuroepithelial tumour (DNT) – benign cortical tumour often presenting with seizures. Cortical (temporal) mass, usually small, that may demonstrate internal cyst formation and calcification.
8. Ganglioglioma – well-circumscribed peripheral tumour that often presents with seizures. Cystic tumour with mural nodule ± calcification.
9. Choroid plexus papilloma – presents in young children with hydrocephalus. Most occur in the atrium of the lateral ventricle (fourth ventricle in adults) and appear as a well-circumscribed multilobulated avidly enhancing intraventricular mass ± calcification. Invasion of brain suggests choroid plexus carcinoma.
10. Ependymoma – often in the frontal lobe adjacent to the frontal horn, but not usually within the ventricular system.
14.72
Infratentorial tumours in childhood
1. Cerebellar astrocytoma – 20–25% of posterior fossa tumours. Vermis (50%) or hemispheres (20%) or both sites (30%) ± extension into the cavity of the fourth ventricle. Calcification in 20%.
CT/MRI – large lesion displacing the fourth ventricle → obstructive hydrocephalus. 80% are juvenile pilocytic astrocytomas with an excellent prognosis. Tumour can be cystic, solid or solid with central necrosis. 50% of all tumours are a cyst with an isodense enhancing mural nodule. The cyst contents have slightly increased CSF attenuation on CT. 40–45% are solid with central necrosis. The solid component is isodense to hypodense to white matter on CT, low signal on T1W MRI and high signal on T2W MRI. The solid component enhances on CT and MRI. Larger tumour at diagnosis than the solid type. The solid type accounts for 10%.
2. Medulloblastoma – 30–40% of posterior fossa tumours. Short history. 80% located in the vermis; 30% extend into the brainstem.
(a) CT – moderately well-defined, ovoid or spherical mass; slightly increased surrounding cerebellum; rim of oedema. Usually uniform enhancement; non-enhancement rarely. Calcification (in 10%) is usually small, homogeneous and eccentric. Dystrophic calcification occurs after radiotherapy. Small cystic or necrotic areas are unusual.
(b) MRI – low signal on T1W; heterogeneous iso- to low signal on T2W. Variable enhancement.
(a) Seeding of the subarachnoid space.
(b) Retrograde ventricular extension.
(c) Extracranial metastases to bone, lymph nodes or soft tissues.
Recurrence of tumour is demonstrated by:
(a) Enhancement at the site of the lesion.
(b) Enhancement of the subarachnoid space (basal cisterns, sylvian, fissures, sulci and ependymal surfaces of ventricles).
3. Ependymoma – most commonly in the floor of the fourth ventricle. 8–15% of posterior fossa tumours. Usually a long clinical history.
(a) CT – typically, an isodense to hyperdense fourth ventricular mass with punctate calcifications, small cysts and heterogeneous or homogeneous enhancement. Calcification within a fourth ventricular mass or adjacent to the fourth ventricle = ependymoma.
(b) MRI – homogeneous or heterogeneous. Slightly hypointense on T1W and isointense to grey matter on T2W. Tumour extension through the foramen of Magendie, foramen magnum (behind the spinal cord) and foramen of Luschka (into the cerebellopontine angle) are important clues to the diagnosis.
4. Brainstem glioma – 20–30% of posterior fossa tumours. Insidious onset because of the location and tendency to infiltrate cranial nerve nuclei and long tracts without producing CSF obstruction until late. Four subgroups: medullary, pontine, mesencephalic and those associated with NF-1. Tumours may also be diffuse (> 50–75% of the brainstem in the axial plane) or focal (< 50%). Calcification rare.
(a) Medullary – least common. Young children. May be differentiated into focal dorsally exophytic and diffuse forms (with significantly worse prognosis). Low attenuation (CT), low signal (T1W) and high signal (T2W).
(b) Pontine – most common. Diffuse tumours are low attenuation (CT), low signal (T1W) and high signal (T2W). Flattening of the floor of the fourth ventricle. Contrast enhancement is rare. Focal tumours are very uncommon but do exhibit heterogeneous enhancement.
(c) Mesencephalic – focal tumours are more common than diffuse. Symptoms depend on the exact location of the mass.
(d) Associated with NF-1 – most commonly in the medulla. Similar imaging appearances to those without NF-1, but patients may be asymptomatic and progression is slower.
14.73
Intraventricular mass in children
Lateral ventricles
[/level-membership-for-radiology-category][not-level-membership-for-radiology-category]<
[/not-level-membership-for-radiology-category]
