Chapter 41 Overview of Vasculitis
The vasculitides are a group of rare diseases linked by the pathological consequences of vascular inflammation, including bleeding, ischemia, and infarction of downstream organs (Box 41-1). However, the clinical spectrum of these diseases is wide ranging and includes a myriad of clinical and pathological findings. Not all disease phenotypes that occur in the vasculitides are due to true “vasculitis” (i.e., inflammation of vascular structures). Some damage in vasculitis is due to nonvascular inflammation. For example, arthritis, uveitis, and pulmonary nodules are parts of different vasculitides but are not due to interruption of vascular flow. The pathophysiology of vasculitis is covered in Chapter 9 and in individual chapters on Takayasu’s arteritis (TA) (Chapter 42), giant cell arteritis (GCA; Chapter 43), and Kawasaki disease (Chapter 45).
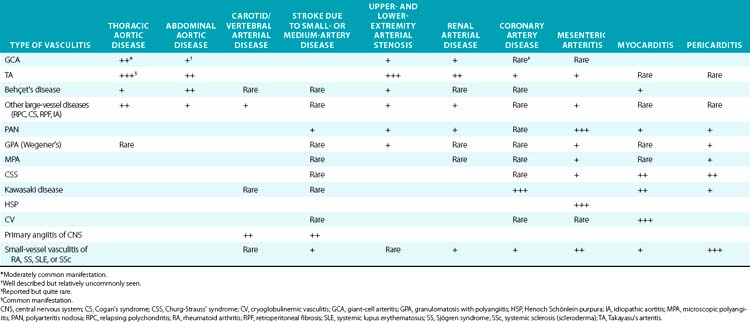
 Box 41-1 Classification of Vasculitis*by Predominant Size of Vessel Involvement
Box 41-1 Classification of Vasculitis*by Predominant Size of Vessel Involvement
This chapter reviews the major types of vasculitis, discusses evaluation of suspected cases of vasculitis, and outlines approaches to treatment and management of these disorders. There is a focus on differentiating inflammatory from noninflammatory disease as it relates to the types of patients physicians specializing in vascular medicine are likely to encounter in a consultative practice (Table 41-1). The newest advances in diagnosis and treatment are also reviewed briefly.
Classification of Vasculitis
Two major classification systems for vasculitis exist: the American College of Rheumatology (ACR) system1 and the Chapel Hill Consensus Conference definitions.2 These systems were not meant to be strictly diagnostic systems, but rather classification systems. These are definitions to apply to established vasculitis and differentiate one vasculitis from another. The main use of these systems has been for clinical trials and other types of clinical research. Nevertheless, these systems have been adapted for use by clinicians as helpful guides to practice. Not all types of vasculitis are included in the ACR or Chapel Hill systems; both are currently undergoing reevaluation and revision.3
Perhaps the simplest method of sorting out the vasculitides, albeit also incomplete and not fully accurate, is to list them according to the size of artery (predominantly but not necessarily exclusively) involved (see Box 41-1). This results in considering small-vessel, medium-vessel, and large-vessel vasculitides. This system, although not applied for clinical trials or even clinically for treatment purposes, is an easy one to use as a first approach to describing the diseases and their major manifestations, and is used to outline the descriptions of the vasculitides in this chapter. However, when specific diseases and results of treatment trials are mentioned, the ACR and Chapel Hill Consensus systems are applied.
Large-Vessel Vasculitis
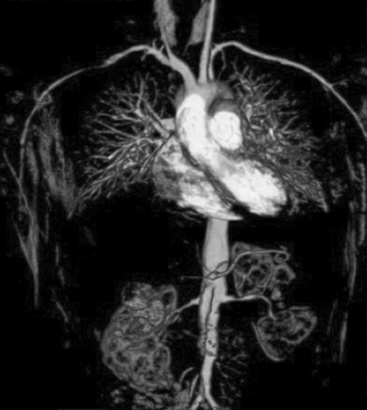
The large-vessel vasculitides are disorders in which the aorta and its main branches are affected, including the subclavian, carotid, vertebral, renal, mesenteric, and iliac arteries4 (Fig. 41-1). Because such vessels are so frequently involved in noninflammatory vascular diseases, and patients with these diseases are frequently encountered by specialists in vascular medicine, these disorders are particularly highlighted in this textbook. Also included are individual chapters on TA (Chapter 42), giant cell (temporal) arteritis (Chapter 43), and Kawasaki disease (Chapter 45). The vasculitides involving large arteries are briefly described in this section, but it is important to realize that many of them also involve smaller-sized vessels.
Giant Cell Arteritis
Giant-cell arteritis, also commonly known as temporal arteritis and described in detail in Chapter 43, is the most common of the idiopathic vasculitides.4,5 Giant-cell arteritis affects men and women aged 50 and older but is especially prevalent after age 70. Many vascular and systemic manifestations are seen in this disease. Vascular disease occurs in the aorta and its branches, with predilection for the branches of the carotid arteries, especially the ophthalmic artery, with resulting headaches, jaw claudication, and visual impairment. Rapid-onset irreversible monocular blindness is the most feared complication, but stroke, limb ischemia, and aortic disease can occur, the latter more common than generally appreciated, especially several years after the initial presentation. Common systemic manifestations include fever, anemia, proximal arthralgias (polymyalgia rheumatica), and fatigue. Diagnosis is often established on finding arteritis on temporal artery biopsy, but this is not required for a diagnosis. Elevated acute phase reactants are seen in 90% of cases. Treatment with high-dose glucocorticoids is highly effective but often results in significant drug-related morbidity.
Takayasu’s Arteritis
Takayasu’s arteritis, described in detail in Chapter 42, is a vasculitis that involves the aorta and all its major branches and the pulmonary arteries, including but not limited to the brachiocephalic, carotid, vertebral, subclavian, renal, femoral, and coronary arteries. This disease often results in stenoses, occlusions, and ischemic damage to end organs and limbs.4,6 Stroke, myocardial infarction (MI), limb claudication, and severe renovascular hypertension are all complications well known to occur in this disease. It is mostly seen in women and usually first presents clinically in the second or third decade, but it can occur at older ages. Many patients have associated systemic symptoms of fever, arthralgias, and malaise. The disease has a waxing and waning course, and delay in diagnosis is common. Treatment involves glucocorticoids in almost all patients and often the addition of immunosuppressive medications. Surgical bypass procedures may be necessary in some cases.
Behçet’s Disease
Behçet’s disease is a systemic inflammatory disease with multiple mucocutaneous manifestations, especially including genital and oral ulcers and often severe sight-threatening inflammatory eye disease.7 Arthritis, gastrointestinal disease (including mucosal lesions), epididymitis, and secondary amyloidosis can also occur. Although its prevalence is markedly increased in countries in the Eastern Mediterranean, Middle East, and East Asia and descendents of people from these regions, Behçet’s disease is found in populations worldwide.
Relapsing Polychondritis
Relapsing polychondritis is a rare connective tissue disease that predominantly affects the cartilaginous structures of the eyes, ears, nose, and subglottis/trachea, but may also affect a wide variety of other organ systems and is associated with vasculitis, especially of large vessels.8 The cardinal feature of polychondritis is auriculitis, inflammation of the outer ear, usually sparing the noncartilaginous lobe. Auriculitis, which is also a feature of GPA and CSS but virtually of no other diseases, is readily treated with glucocorticoids and can result in disfigurement if allowed to go untreated. Other common manifestations include inflammatory eye disease that can lead to blindness, destruction of nasal cartilage leading to internal derangement and external disfigurement, sensorineural hearing loss and vertigo, arthritis, and subglottic inflammation with resulting stenosis, a life-threatening condition. Each of these features can also be seen in GPA, although auriculitis is rare in this disease, and relapsing polychondritis is not associated with parenchymal pulmonary manifestations.
Cogan’s Syndrome
Cogan’s syndrome is a rare disorder characterized by inflammatory eye and inner ear/vestibular disease that can also involve inflammatory vasculitis.9 It is a disease of young adults, usually first affecting patients before age 40, although both children and older patients have also been affected.
Idiopathic Aortitis
Aortitis may be found in the absence of any other manifestations of a systemic inflammatory disease.10–12 These cases often come to the attention of vascular medicine specialists when patients undergoing surgical repair of aortic aneurysms and dissections are found to have inflammation consistent with aortitis on pathological specimens. Autopsies and studies of large numbers of surgical specimens have demonstrated that noninfectious aortitis occurs in 4% to 15% of cases. Although on detailed investigation, many of these patients are retrospectively found to have had evidence of GCA, TA, relapsing polychondritis, GPA, or another definable vasculitis, it is common among these cases to find no evidence of more systemic inflammatory disease. The majority of cases of so-called idiopathic aortitis involve thoracic lesions, in contrast to the overall predominance of abdominal aortic lesions for noninflammatory disease.
The approach to treatment of idiopathic aortitis is unclear; many patients never develop other findings of vasculitis. However, new aneurysms and significant vascular disease do occur in some cases.11 Comprehensive evaluation of evidence of systemic disease is necessary and should include a detailed physical examination, diagnostic imaging, laboratory studies, and other approaches outlined later in this chapter. Appropriate treatment should be given if inflammatory disease other than that seen in the surgical specimen is found, but not all patients require glucocorticoids, especially in the postoperative period. Furthermore, regular follow-up of such patients by a specialist knowledgeable about vasculitis is imperative because lesions may develop subtly and only years after the initial pathological diagnosis is made.
Medium-Vessel Vasculitis
Polyarteritis Nodosa
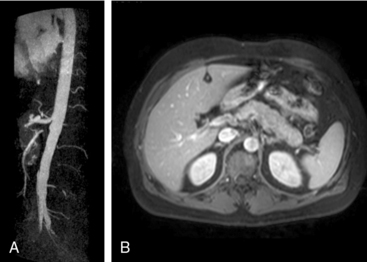
Polyarteritis nodosa (PAN) is among the “purer” vasculitides in that most of its manifestations are due to true vascular inflammation.13 With the identification of other types of vasculitis, the spectrum of what is now diagnosed as PAN has narrowed over the past 50 years. Although characterized as a medium-vessel disease, PAN may also involve small vessels such as those in the skin. Polyarteritis nodosa frequently involves inflammation leading to multiple small aneurysms that often appear angiographically as a “string of beads.” Ischemia and infarction of kidneys, intestines, and skin are common in PAN, with arthralgias, myalgias, and fevers also frequently seen. Diagnosis is based on angiographic appearance (Fig. 41-2) or tissue pathology, often from surgical specimens such as a resected ischemic bowel segment. Interestingly, PAN in one subset of patients is associated with either hepatitis B or hepatitis C infections.13,14 Importantly, there is a difference between hepatitis C–associated PAN and hepatitis C–associated cryoglobulinemic vasculitis (CV, see later section). Cardiac manifestations of PAN are due to coronary arteritis or malignant hypertension (secondary to renal artery disease) and include myocardial ischemia, heart failure, and arrhythmias.
Granulomatosis with Polyangiitis (Wegener’s)
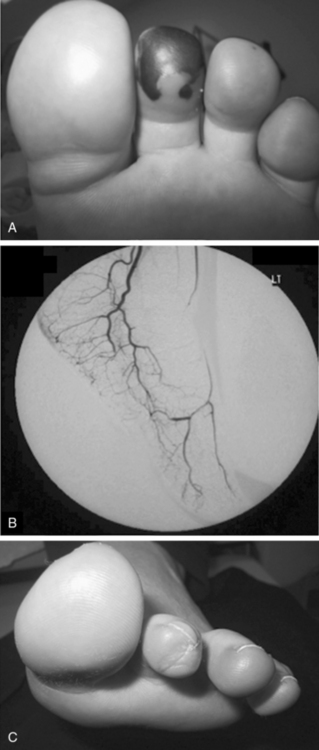
Granulomatosis with polyangiitis is characterized by the triad of inflammation and destruction of tissue in the upper airway and sinuses (Fig. 41-3), lower airway (Fig. 41-4), and kidneys (Fig. 41-5), as well as the development of ANCAs.15,16 Approximately 70% of patients with GPA are positive for ANCA at diagnosis, although some will develop the antibodies later in the course of their illness. Among patients with GPA and glomerulonephritis, more than 90% are positive for ANCA. Although the combination of these features is common in GPA, many patients present with only a subset of these findings. Granulomatosis with polyangiitis also frequently involves many other organ systems. The upper airway lesions include destructive rhinitis, often leading to nasal bridge collapse and the “saddle nose” deformity, sinusitis, and subglottic inflammation that can lead to life-threatening tracheal stenosis. The most severe form of pulmonary disease in GPA is alveolar hemorrhage, and this is a common cause of early death. Other common pulmonary lesions include nodules, with or without cavitation, and tracheobronchitis. Other common features of GPA are retroorbital pseudotumor with resulting proptosis, conductive and sensorineural hearing loss, mononeuritis multiplex, arthritis, and purpura. Peripheral vascular involvement with gangrene is seen in GPA and may be the presenting feature (Fig. 41-6).
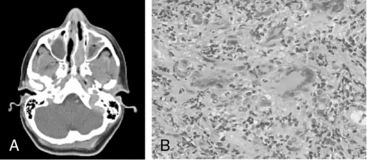
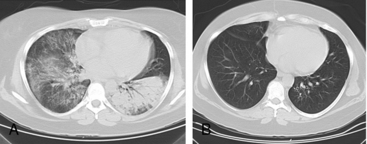
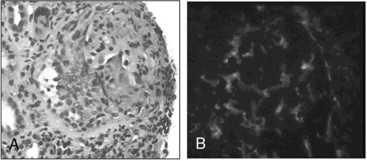
Figure 41-5 (See also Color Plate 41-5.) Renal biopsy in a patient with granulomatosis with polyangiitis (GPA; Wegener’s same patient as in Fig. 41-4) with rapidly progressive glomerulonephritis.
Venous thromboses, including both deep vein thromboses (DVTs) and pulmonary emboli (PEs), occur frequently in GPA and may be associated with active disease.17,18 Although some of the pathology in GPA is indeed granulomatous with histiocytes, piecemeal necrosis, and occasional giant cells and eosinophils, other manifestations of inflammation are also seen in the disease. True vasculitis occurs and includes capillaritis. The renal disease of GPA is identical to other ANCA-positive diseases, and the pathology is that of rapidly progressive glomerulonephritis.
Untreated, GPA most often leads to death or serious damage.19 Glucocorticoids are always used for treatment, but the prognosis of GPA changed considerably when a protocol using cyclophosphamide was introduced in the 1970s at the National Institutes of Health (NIH). The morbidity and mortality of GPA was markedly improved by cytotoxic therapy: 1-year mortality changed from more than 80% to less than 20%.16,19,20 However, serious side effects are common with the use of cyclophosphamide, and the rate of recurrent disease in GPA after therapy is above 50%. In recent years, new treatment protocols have been tested in open and controlled trials that incorporate less toxic immunosuppressive drugs, including methotrexate and azathioprine.21–23 Two multicentered randomized controlled trials (RCTs) have demonstrated that treatment with rituximab, a monoclonal antibody directed against the CD20 receptor on B cells, was equivalent to cyclophosphamide for induction of remission in ANCA-associated vasculitis (GPA and MPA).24,25 In 2011, the U.S. Food and Drug Administration (FDA) approved rituximab for the treatment of GPA and MPA, and it has quickly become an established alternative to cyclophosphamide.
Microscopic Polyangiitis
With the publication of the Chapel Hill Consensus Conference classification system, recognition of MPA as a separate entity gained acceptance.2,13 Microscopic polyangiitis is a mostly small- to medium-vessel ANCA-associated vasculitis with manifestations that strongly overlap with GPA. Its key features include glomerulonephritis, alveolar hemorrhage, skin lesions, and mononeuritis multiplex, but many other organ systems may be involved as well. Unlike GPA, the pathology of MPA is nongranulomatous and does not involve the type of nonvascular disease seen in GPA or CSS. The glomerulonephritis of MPA is identical to that seen in GPA. Most patients with MPA are positive for ANCA, and the predominant ANCA antigen specificity is myeloperoxidase. Cardiac manifestations of MPA are uncommon, but peripheral artery disease (PAD) and gangrene are seen and may be confused with noninflammatory vascular disease. Microscopic polyangiitis should be differentiated from classic PAN. Polyarteritis nodosa is more of a medium-vessel disease and does not include glomerulonephritis or pulmonary capillaritis. Microscopic polyangiitis does not produce the microaneurysms seen in PAN. Treatment of MPA is essentially identical to that for GPA.
Churg-Strauss’ Syndrome
Churg-Strauss’ syndrome, also known as allergic granulomatous angiitis, is a rare disease characterized by the triad of asthma, pulmonary infiltrates, and hypereosinophilia.26,27 Churg-Strauss’ syndrome can, however, involve almost all the clinical features seen in GPA, including the presence of ANCA in some cases (30%-40%). As with GPA, much of the pathology seen in CSS is due to inflammation that is not “vasculitis” per se but is every bit as damaging as vascular inflammation. Tissue eosinophilia, although seen in other types of vasculitis, is particularly common in CSS and often striking. More than 90% of patients have asthma, often severe; the hypereosinophilia may be a marker of disease activity for some patients but is not always present. Pulmonary manifestations include dense infiltrates that rapidly clear with glucocorticoid therapy. Additionally, neuropathies—especially mononeuritis multiplex and gastrointestinal ischemia—are common features and quite damaging. Diagnosis is based on the combination of clinical findings, hypereosinophilia, and pathology specimens that often show granulomatous and eosinophilic inflammation.
Kawasaki Disease
Kawasaki disease, a vasculitis of young children involving medium and small arteries, is a leading cause of acquired CAD in children.28 The disease manifests as a systemic illness with high fevers, conjunctival injection, erythematous oropharyngeal lesions, erythematous rashes and skin desquamation, lymphadenopathy, and other signs and symptoms. Cardiac involvement is frequent in Kawasaki disease and can result in long-term morbidity. Myocarditis and pericarditis are common and can be serious, but coronary artery aneurysms are the most feared aspect of the disease. Both panarteritis and granulomas can be seen in the vessels, with subsequent scarring and aneurysm formation. Treatment includes aspirin and intravenous immunoglobulin (IVIG); such regimens have been shown to markedly reduce the incidence of coronary complications. Kawasaki disease is described in detail in Chapter 45.
Small-Vessel Vasculitis
Henoch-Schönlein Purpura
Henoch-Schönlein purpura is a small-vessel vasculitis that classically involves the clinical triad of inflammatory arthritis, ischemic abdominal pain, and purpura, although not all cases exhibit all three manifestations.29 The most feared manifestation of HSP is glomerulonephritis, which can lead to renal failure. Cardiac disease is not a feature of HSP, but hypertension from renal insufficiency can be severe. The lesions in HSP often involve leukocytoclasia and immunoglobulin (Ig)A deposition. An elevated serum IgA level is commonly seen in patients with HSP.
Cryoglobulinemic Vasculitis
Cryoglobulinemic vasculitis occurs when cryoglobulins, any of various types of Igs that precipitate from serum at temperatures below body temperature, induce an immune complex–mediated inflammatory process in any organ.30 Several types of cryoglobulins can occur, and cryoglobulinemia is subclassified based on the mix of IgG and IgM antibodies that make up the cryoglobulin portion of serum (the “cryocrit”), and whether the excess cryoproteins are polyclonal or monoclonal.
Primary Angiitis of the Central Nervous System
Primary angiitis of the central nervous system (PACNS) is a quite rare necrotizing angiitis limited to the central nervous system (CNS).31,32 PACNS is frequently associated with subacute nonfocal neurological deficits and chronic meningitis, although strokes and hemorrhage can also be seen.
Diagnosis of PACNS necessitates first suspecting this rare disease. Conventional angiography may be helpful in identifying other entities, such as aneurysms and emboli, but is often not diagnostic for vasculitis for several reasons (Fig. 41-7). First, in older patients the endothelial changes of atherosclerosis may mimic those of vasculitis. Second, vasospasm can be confused with stenosis from either atherosclerosis or inflammation. Finally, the resolution of conventional angiography is such that small arteries are not well visualized, and thus many cases of vasculitis may be missed by this technique. Leptomeningeal biopsies or larger tissue samples from affected brain areas are often necessary to demonstrate PACNS and provide the level of evidence required to institute therapy. The histopathology is that of vasculitis, but granulomas and giant cells are not always seen; there may be no inflammation in the vasospastic variant. Tests of cerebrospinal fluid are often normal in patients with PACNS, but are important in evaluating patients for other conditions, especially infection or malignancy.
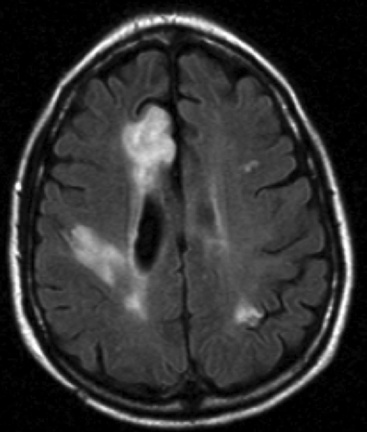
It is imperative to differentiate PACNS from the increasingly recognized set of reversible cerebral vasoconstrictive syndromes (RCVS) previously referred to as benign PACNS33; RCVS is not a vasculitis. It is characterized by acute onset of severe headache (“thunderclap headaches”) and a focal neurological event. RCVS is treated with aggressive vasodilators, including calcium channel blockers, and strict avoidance of smoking and vasoconstricting drugs and substances, such as caffeine, cocaine, sympathomimetics, and serotonin receptor agonists (e.g., sumatriptan).
Drug-Induced Vasculitis
Many drugs or other toxins have been implicated as causing inflammatory vasculitis involving vessels of all sizes, especially small arteries. A full list of drugs considered to be causative for vasculitis and details regarding the clinical syndromes of drug-induced vasculitis are available in recent reviews.34,35 There is an interesting subset of patients with ANCA-associated vasculitis whose disease is caused by exposure to certain medications.36
Evaluation and Diagnosis of Possible Vasculitis
Physical Examination
A full physical examination is required whenever a patient is evaluated for potential vasculitis, and several examination findings should always prompt consideration of vasculitis in any patient. Blood pressure should be measured in both arms for discrepancies. Obtaining pressures in the legs may be appropriate if lower-extremity stenoses are suspected. Hypertension may result from renal artery stenoses (RAS) from vasculitis, and similar physiology occurs with some tight suprarenal aortic stenoses. A full examination of bilateral pulses should include radial, ulnar, brachial, carotid, femoral, popliteal, posterior tibial, and dorsalis pedis pulses. Bruits should be listened for over the aorta and the carotid, femoral, axillary, subclavian, and renal arteries. Presence of blood pressure discrepancies, absent pulses, or arterial bruits are each highly specific for major arterial disease but are not highly sensitive for major arterial lesions in large-vessel vasculitis.37
Laboratory Studies
Antineutrophil cytoplasmic autoantibodies testing
The discovery of ANCAs and their association with GPA, microscopic polyangiitis, CSS, and renal-only pauci-immune glomerulonephritis was extremely important in the evolution of diagnostic testing for vasculitis.38 ANCA testing, when performed properly, is highly specific for these syndromes, and in the correct clinical setting may be the last piece of data necessary to establish a diagnosis, even in the absence of a tissue biopsy. Additionally, the finding of a positive test for ANCA in a patient with already established vasculitis essentially narrows the diagnosis to one of four ANCA-associated diseases.
Specificity of properly performed ANCA testing is better than 90% and may be closer to 99% in certain laboratories.38,39 Sensitivity of ANCA testing varies with the type of disease and clinical manifestations. Although more than 90% of patients with GPA who have renal involvement are ANCA positive, this rate drops to about 70% for patients without renal disease. Most patients with MPA and renal-limited pauci-immune glomerulonephritis are ANCA positive, but the rate of ANCA positivity among patients with CSS varies in the literature from 40% to 80%. Thus, although ANCA is highly specific for vasculitis when present, a negative test by no means excludes the diagnosis.
Diagnostic Vascular Imaging
Radiographic assessment of vascular structures has long been an important tool for diagnosing patients with vasculitis. This is especially true when medium and large vessels are involved because they are much more likely to be visualized by the techniques available.40 Although small- and even medium-sized arteries can often be seen on diagnostic biopsies or surgical specimens, large vessels are not usually amenable to tissue biopsy. Thus diagnostic imaging is crucial to assessment and management of patients with large-vessel vasculitis. The two great challenges inherent in interpretation of imaging of large vessels are (1) differentiating inflammatory disease from atherosclerotic disease and (2) trying to determine whether vascular lesions represent “active” disease. In recent years, interest in large-vessel vascular imaging has greatly increased as investigators and clinicians working in vasculitis strive to properly incorporate advances in various radiological modalities into practice. Imaging of organs and tissues other than arteries themselves is of obvious benefit for specific syndromes to help understand the extent of disease, facilitate choice of tissue biopsy, and rule out other pathology.
Treatment of Vasculitis
The goals of treatment for inflammatory vasculitis are to stop the active inflammation and prevent permanent damage. Unlike many other systemic inflammatory diseases, true clinical remission is not only possible in many cases of vasculitis but should be the goal of treatment. Thus, protocols are increasingly being referred to as involving either “remission induction” or “remission maintenance” treatments. The mainstays of therapy for vasculitis remain glucocorticoids and various immunosuppressive drugs. Treatment protocols are tailored to the specific type of vasculitis and the extent of disease. Clinical trial data are increasingly available to guide treatment for GPA, microscopic polyangiitis, Kawasaki disease, and GCA, whereas other diseases rely on either case series for guidance or extrapolated data from studies in related, but not identical, vasculitides. Box 41-2 outlines the treatments used for patients with inflammatory vasculitis.
 Box 41-2
Box 41-2Medical Therapies for Vasculitis
Medical management of patients with vasculitis should only be directed by physicians familiar with both the use of chronic immunosuppressive agents and the clinical presentation and management of vasculitis. The acute and chronic toxicities of these drugs should not be underestimated and can result in significant morbidity. Treatment protocols are beyond the scope of this chapter, but the most commonly used medications for vasculitis are briefly outlined. Many different agents have been used for vasculitis (see Box 41-2).
Cyclophosphamide is widely considered the most effective agent for inducing and maintaining remission in various types of vasculitis.16 The introduction of cyclophosphamide-based therapy changed the prognosis of many of these diseases. Although cyclophosphamide is extremely effective, its multiple toxicities are severe and include cytopenias, especially neutropenia with associated infections, gonadal failure, teratogenicity, hemorrhagic cystitis, transitional cell carcinoma of the bladder, myelodysplasia, mucositis, hair loss, and others. Controversy exists about the best route of administration for cyclophosphamide, orally or intravenously (IV).
Multiple alternative agents have been tested and proposed to limit use of cyclophosphamide. Methotrexate, taken orally or intramuscularly (IM) weekly, has demonstrated efficacy as both a remission-induction agent and remission-maintenance agent for GPA and is used for both purposes for multiple vasculitides.41 The toxicities of methotrexate include cytopenias, gastrointestinal upset, oral ulcers, teratogenicity, and hepatic disease. Azathioprine is another commonly used agent for remission maintenance in vasculitis.21 Azathioprine can cause cytopenias, infections, nausea, mucositis, hair loss, pancreatitis, and other problems, but like methotrexate, is usually well tolerated even for extended periods. Mycophenolate mofetil is also used for vasculitis. Cyclosporine has long been used in Behçet’s disease and occasionally in other vasculitides.
Many biological agents (“biologics”) have been and continue to be studied as treatment for vasculitides23 (see Box 41-2). This rapidly expanding group of agents that inhibit specific targets within the immune system are having a remarkable impact on the care of patients with a wide variety of autoimmune and systemic inflammatory diseases. The studies of rituximab for GPA and MPA demonstrated efficacy of the new agent.24,25 However, despite initial enthusiasm from open-label studies, randomized trials studying the use of inhibitors of TNF-α did not demonstrate efficacy of this class of drug for treatment of either GPA42 or GCA.43 This experience highlights the need for properly conducted randomized clinical trials in vasculitis. Studies testing the efficacy of several other biologics for various forms of vasculitis are either currently in process or in the planning stages.
Miscellaneous Issues in the Treatment of Vasculitis
Gonadal function and pregnancy-related problems
Treatment of vasculitis often involves drugs that adversely affect patients’ gonadal function or are problematic during pregnancy, or both.44 Cyclophosphamide can cause both male and female sterility and is a highly teratogenic agent. Methotrexate is teratogenic and an abortifacient drug. Glucocorticoids cause significant maternal and some fetal problems. Several of the other treatments in Box 41-2 are either directly contraindicated during pregnancy, or their safety during pregnancy has not been established.
Accelerated atherosclerosis
Although there is little firm evidence yet, concern is growing that patients with inflammatory vasculitis are at increased risk of accelerated atherosclerosis and coronary artery disease, as is seen in patients with other chronic inflammatory diseases such as rheumatoid arthritis and SLE.45 The etiology of the atherosclerosis is likely multifactorial but includes glucocorticoid usage, lipid disorders associated with disease and treatment regimens, and other treatment-related problems, such as nephrotic syndrome and diabetes mellitus. The impact of chronic inflammation itself, however, may be the most important factor in the development of atherosclerosis. Ongoing research is directed at the interaction between inflammation and atherogenesis. Whether or not chronic therapy with statins or other agents that act via lipid or inflammatory pathways is appropriate has yet to be proved in clinical studies.
1 Bloch D.A., Michel B.A., Hunder G.G., et al. The American College of Rheumatology 1990 criteria for the classification of vasculitis. Patients and methods. Arthritis Rheum. 1990;33:1068–1073.
2 Jennette J.C., Falk R.J., Andrassy K., et al. Nomenclature of systemic vasculitides. Proposal of an international consensus conference. Arthritis Rheum. 1994;37:187–192.
3 Watts R.A., Suppiah R., Merkel P.A., et al. Systemic vasculitis–is it time to reclassify? Rheumatology (Oxford). 2011;50:643–645.
4 Kissin G.Y., Merkel P.A. Large-vessel vasculitis. In: Coffman J.D., Eberhardt R.T. Peripheral arterial disease: diagnosis and treatment. Totawa, NJ: Humana Press; 2002:319.
5 Weyand C.M., Goronzy J.J. Giant-cell arteritis and polymyalgia rheumatica. Ann Intern Med. 2003;139:505–515.
6 Kerr G.S., Hallahan C.W., Giordano J., et al. Takayasu arteritis. Ann Intern Med. 1994;120:919–929.
7 Sakane T., Takeno M., Suzuki N., et al. Behcet’s disease. N Engl J Med. 1999;341:1284–1291.
8 Sridharan S.T. Relapsing polychondritis. In: Hoffman G.S., Weyand C.M. Inflammatory diseases of blood vessels. New York: Marcel Dekker; 2001:675.
9 St Clair E.W., McCallum R.M. Cogan’s syndrome. Curr Opin Rheumatol. 1999;11:47–52.
10 Liang K.P., Chowdhary V.R., Michet C.J., et al. Noninfectious ascending aortitis: a case series of 64 patients. J Rheumatol. 2009;36:2290–2297.
11 Merkel P.A. Noninfectious ascending aortitis: staying ahead of the curve. J Rheumatol. 2009;36:2137–2140.
12 Rojo-Leyva F., Ratliff N.B., Cosgrove D.M.3rd, et al. Study of 52 patients with idiopathic aortitis from a cohort of 1,204 surgical cases. Arthritis Rheum. 2000;43:901–907.
13 Guillevin L. Polyarteritis nodosa and microscopic polyangiitis. In: Ball G.V., Bridges L. Vasculitis. New York: Oxford University Press; 2002:300.
14 Guillevin L., Lhote F., Cohen P., et al. Polyarteritis nodosa related to hepatitis B virus. A prospective study with long-term observation of 41 patients. Medicine (Baltimore). 1995;74:238–253.
15 Hoffman G.S., Gross W.L. Wegener’s granulomatosis: clinical aspects. In: Hoffman G.S., Weyand C.M. Inflammatory diseases of blood vessels. New York: Marcel Dekker; 2001:381.
16 Hoffman G.S., Kerr G.S., Leavitt R.Y., et al. Wegener’s granulomatosis: an analysis of 158 patients. Ann Intern Med. 1992;116:488–498.
17 Allenbach Y., Seror R., Pagnoux C., et al. High frequency of venous thromboembolic events in Churg-Strauss syndrome, Wegener’s granulomatosis and microscopic polyangiitis but not polyarteritis nodosa: a systematic retrospective study on 1130 patients. Ann Rheum Dis. 2009;68:564–567.
18 Merkel P.A., Lo G.H., Holbrook J.T., et al. Brief communication: high incidence of venous thrombotic events among patients with Wegener granulomatosis: the Wegener’s Clinical Occurrence of Thrombosis (WeCLOT) Study. Ann Intern Med. 2005;142:620–626.
19 Walton E.W. Giant-cell granuloma of the respiratory tract (Wegener’s granulomatosis). BMJ. 1958;2:265–270.
20 Reinhold-Keller E., Beuge N., Latza U., et al. An interdisciplinary approach to the care of patients with Wegener’s granulomatosis: long-term outcome in 155 patients. Arthritis Rheum. 2000;43:1021–1032.
21 Jayne D., Rasmussen N., Andrassy K., et al. A randomized trial of maintenance therapy for vasculitis associated with antineutrophil cytoplasmic autoantibodies. N Engl J Med. 2003;349:36–44.
22 Langford C.A. Treatment of ANCA-associated vasculitis. N Engl J Med. 2003;349:3–4.
23 Langford C.A., Sneller M.C. Biologic therapies in the vasculitides. Curr Opin Rheumatol. 2003;15:3–10.
24 Jones R.B., Tervaert J.W., Hauser T., et al. Rituximab versus cyclophosphamide in ANCA-associated renal vasculitis. N Engl J Med. 2010;363:211–220.
25 Stone J.H., Merkel P.A., Spiera R., et al. Rituximab versus cyclophosphamide for ANCA-associated vasculitis. N Engl J Med. 2010;363:221–232.
26 Guillevin L., Lhote F., Cohen P. Churg-Strauss syndrome: clinical aspects. In: Hoffman G.S., Weyand C.M. Inflammatory diseases of blood vessels. New York: Marcel Dekker; 2001:399.
27 Guillevin L., Cohen P., Gayraud M., et al. Churg-Strauss syndrome. Clinical study and long-term follow-up of 96 patients. Medicine (Baltimore). 1999;78:26–37.
28 Barron K.S. Kawasaki disease: etiology, pathogenesis, and treatment. Cleve Clin J Med. 2002;69(Suppl 2):SII69–SII78.
29 Saulsbury F.T. Henoch-Schonlein purpura. Curr Opin Rheumatol. 2001;13:35–40.
30 Cacoub P., Costedoat-Chalumeau N., Lidove O., et al. Cryoglobulinemia vasculitis. Curr Opin Rheumatol. 2002;14:29–35.
31 Calabrese L.H., Duna G.F., Lie J.T. Vasculitis in the central nervous system. Arthritis Rheum. 1997;40:1189–1201.
32 Hajj-Ali R.A., Singhal A.B., Benseler S., et al. Primary angiitis of the CNS. Lancet Neurol. 2011;10:561–572.
33 Calabrese L.H., Dodick D.W., Schwedt T.J., et al. Narrative review: reversible cerebral vasoconstriction syndromes. Ann Intern Med. 2007;146:34–44.
34 Merkel P.A. Drug-induced vasculitis. Rheum Dis Clin North Am. 2001;27:849–862.
35 Merkel P.A. Drug-induced vasculitis. In: Hoffman G.S., Weyand C.M. Inflammatory diseases of blood vessels. New York: Marcel Dekker; 2001:727.
36 Choi H.K., Merkel P.A., Walker A.M., et al. Drug-associated antineutrophil cytoplasmic antibody-positive vasculitis: prevalence among patients with high titers of antimyeloperoxidase antibodies. Arthritis Rheum. 2000;43:405–413.
37 Grayson P.C., Tomasson G., Cuthbertson D., et al. Association of vascular physical examination findings and arteriographic lesions in large vessel vasculitis. J Rheumatol. 2012;39:303–309.
38 Niles J.L. Antineutrophil cytoplasmic antibodies in the classification of vasculitis. Annu Rev Med. 1996;47:303–313.
39 Merkel P.A., Polisson R.P., Chang Y., et al. Prevalence of antineutrophil cytoplasmic antibodies in a large inception cohort of patients with connective tissue disease. Ann Intern Med. 1997;126:866–873.
40 Kissin E.Y., Merkel P.A. Diagnostic imaging in Takayasu arteritis. Curr Opin Rheumatol. 2004;16:31–37.
41 Langford C.A., Sneller M.C., Hoffman G.S. Methotrexate use in systemic vasculitis. Rheum Dis Clin North Am. 1997;23:841–853.
42 WGET Research Group. Etanercept plus standard therapy for Wegener’s granulomatosis. N Engl J Med. 2005;352:351–361.
43 Hoffman G.S., Cid M.C., Rendt-Zagar K.E., et al. Infliximab for maintenance of glucocorticosteroid-induced remission of giant cell arteritis: a randomized trial. Ann Intern Med. 2007;146:621–630.
44 Langford C.A., Kerr G.S. Pregnancy in vasculitis. Curr Opin Rheumatol. 2002;14:36–41.
45 Hahn B.H. Systemic lupus erythematosus and accelerated atherosclerosis. N Engl J Med. 2003;349:2379–2380.