Chapter 2 Ovarian Hormones: Structure, Biosynthesis, Function, Mechanism of Action, and Laboratory Diagnosis
INTRODUCTION
The main function of the ovaries, maturation and release of oocytes, is accomplished via the production of several steroidal and nonsteroidal hormones that locally modulate a series of complex events. Peripherally, these hormones act on various target organs, including the uterus, vagina, fallopian tubes, mammary glands, adipose tissue, bones, kidneys, and liver, leading to the female phenotype.
The Ovary as an Endocrine Organ
The two-cell theory describes the sequence of events that occurs during ovarian follicular growth and steroidogenesis. According to this theory, LH primarily stimulates thecal cells to produce androstenedione and testosterone, both 19-carbon steroids. In contrast, FSH primarily stimulates granulosa cells to aromatize these 19-carbon steroids into estrogens.1,2
The ovarian production of steroid hormones is regulated both within the ovary, by paracrine (intercellular) and autocrine (intracellular) mechanisms, and by endocrine regulation of FSH secretion by the pituitary. Central to this regulation are several nonsteroidal hormones and factors produced by the ovary.3 This chapter focuses on these aspects of the ovary and discusses the biochemistry, biosynthesis, regulation, and actions of both steroidal and peptide ovarian hormones.
STEROIDOGENESIS AND STEROID HORMONES OF THE OVARY
Steroidogenesis
Steroid hormone formation in the steroid-producing endocrine glands follows the same fundamental pathway and mainly relies on exogenous (or plasma) cholesterol, with the exception of the liver and intestinal mucosa, which are capable of synthesizing cholesterol endogenously from acetyl-coA. The primary source of cholesterol for steroidogenesis in the ovary is derived from the uptake of plasma LDL.4 The rate-limiting step in steroidogenesis is transfer of cholesterol from the cytosol to the inner membrane of the mitochondria.5 This is mediated by an LH-induced mitochondrial enzyme called steroidogenic acute regulatory (StAR) protein.6 The StAR gene is located on chromosome 8p11.2 and codes for a 285-amino acid precursor protein, of which 25 amino acids are cleaved off after transport to the mitochondria.7,8 Nonsense mutations of the StAR gene that result in premature stop codons have been identified as a cause of congenital lipoid adrenal hyperplasia, which is characterized by the presence of intracellular lipid deposits that destroy steroidogenesis.7
Ovary steroid hormones are synthesized in both interstitial and follicular cells. The basic structure of cholesterol is three hexagonal carbon rings and a pentagonal carbon ring to which a side chain is attached (Fig. 2-1). Two important methyl groups are also attached at positions 18 and 19. Progestins and corticosteroids (pregnane series 21-carbon steroids) are produced by partial cleavage of the side chain (i.e., the desmolase reaction). Androgens (androstane series 19-carbon steroids) are produced by the total removal of the side chain. Estrogens (estrane series 18-carbon steroids) are produced by aromatization of one of the three hexagonal carbon rings to a phenolic structure with loss of the 19-methyl group.
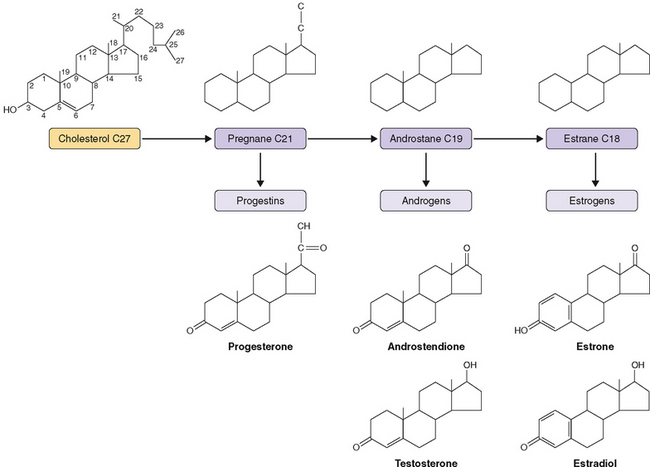
The first step in steroidogenesis is the conversion of cholesterol to pregnenolone via hydroxylation at the carbon 20 and 22 positions, which is followed by cleavage of the side chain (Fig. 2-2). From pregnenolone, steroid hormones are produced by one of two general pathways.5 The pregnenolone (Δ5) pathway produces androgens and estrogens (pregnenolone→17OH-pregnenolone →dehydroepiandrosterone [DHEA]→testosterone→estrogen). The progesterone (Δ4) pathway produces androgens and estrogens (pregnenolone→progesterone→17OH-progesterone→androgen →estrogen). In the adrenal gland, the Δ4 pathway produces mineralocorticoids and glucocorticoids.
The enzymes involved in the intracellular synthesis of steroid hormones include five hydroxylases, two dehydrogenases, a reductase, and an aromatase. The hydroxylases and aromatase belong to the cytochrome P450 (CYP) supergene family (Table 2-1). These enzymes exist on both the mitochondria and endoplasmic reticulum.
Table 2-1 Enzyme Reaction and Cellular Location of Steroidogenic Enzymes
| Enzyme Reaction | Gene (Enzyme) | Cellular Location/Tissue Location |
|---|---|---|
| Cholesterol side chain cleavage | CYP11A (P450scc) | Mitochondria (theca; granulosa) |
| 17α-hydroxylase | CYP17 (P450c17) | ER (theca) |
| 17,20-hydroxylase (lyase) | CYP17 (P450c17) | ER (theca) |
| Aromatase | CYP19 (P450arom) | ER (granulosa) |
| 3β-hydroxysteroid dehydrogenase | 3βHSD | ER (theca; granulosa) |
| 17β-hydroxysteroid dehydrogenase | 17βHSD | ER (granulosa) |
| 21-hydroxylase | CYP21 (P450c21) | ER (adrenal) |
| 11β-hydroxylase | CYP11B1 (P450c11) | Mitochondria (adrenal) |
ER, endoplasmic reticulum
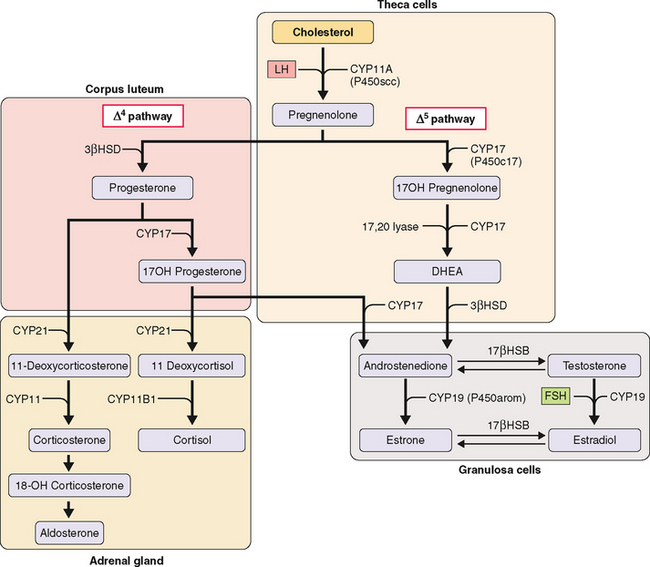
Of these nine enzymes, four key enzymes regulate the main steps of steroidogenesis (see Fig. 2-2): CYP11A (P450scc), a side chain cleavage enzyme that catalyzes the conversion of cholesterol to pregnenolone; 3βHSD, or 3 βa-hydroxysteroid dehydrogenase, which converts pregnenolone to progesterone; CYP17 (P450c17), an hydroxylase that converts pregnenolone to androgens; and CYP19 (P450arom), an aromatase that converts androgens to estrogens. Most reactions are irreversible (denoted by a single arrow in Fig. 2-2). The few reversible reactions (denoted by double arrows) are dependent on cofactor availability (e.g., NADP/NADPH ratio).
The kind of hormone produced depends on the nature of the cell and the presence or absence of the inherent steroidogenic enzymes in the tissue. The adrenal cortex lacks 17βHSD; hence, adrenal androgen production is limited to DHEA and androstenedione. In the testes, LH controls 17βHSD activity and testosterone production. The steroid-producing cells of the ovary (granulosa, theca, corpus luteum) contain the full enzymatic complement for steroid hormone synthesis. In the thecal cells, LH also controls 17βHSD activity and androstenedione production, whereas CYP19 (P450arom) activity in the granulosa cells is controlled by FSH and hence estradiol production. These relationships are the basis for the two-cell, two-gonadotropin system (Fig. 2-3). Aromatization occurs in the endoplasmic reticulum.
In each of the two cell types, the amount of the various enzymes differs depending on the stage of follicle development. CYP11A (P450scc) and 3βHSD are expressed in both thecal and granulosa cells of antral and preovulatory follicles and in the luteinized granulosa and thecal cells of the corpus luteum. In contrast, CYP17 (P450c17) is expressed only in the thecal cells of antral and preovulatory follicles and of the corpus luteum (see Fig. 2-3).
Steroid Hormones of the Ovary
Estrogens
Physiologic Role
Estrogens are essential in the development and maintenance ofthe female phenotype, germ cell maturation, and pregnancy. In addition to their reproductive effects, estrogens also have many other nonreproductive systemic effects, such as bone metabolism/remodeling, nervous system maturation, and endothelial responsiveness.9
At puberty, estrogen stimulates breast development and enlargement and maturation of the uterus, ovaries, and vagina.10,11 Estrogen also works in concert with growth hormone and insulin-like growth factor I (IGF-I) to produce a growth spurt and stimulates the maturation of chondrocytes and osteoblasts, which ultimately leads to epiphyseal fusion.12,13 After midpuberty, estrogen begins to exert a positive feedback on gonadotropin-releasing hormone (GnRH) secretion, leading to the progressive increase of LH and FSH production, culminating in the LH surge, ovulation, and the initiation of the menstrual cycle.
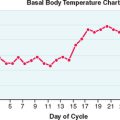
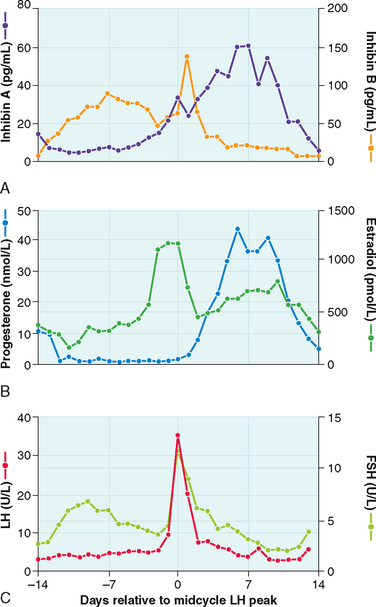
In the adult female, estrogen plays a critical role in maintaining the menstrual cycle.14 The cyclical changes in estradiol, progesterone, and pituitary hormones are illustrated in Figure 2-4. In the early follicular phase of the menstrual cycle, FSH stimulates granulosa cell aromatase activity, resulting in increased follicular concentrations of estrogen. The rising estrogen level further increases the sensitivity of the follicle to FSH and estrogen by increasing the number of estradiol receptors on the granulosa cells. Follicular growth and antral formation is also promoted by estrogen. This sets up a positive feedback cycle, which culminates in one dominant follicle producing an exponential rise in estrogen levels. This exerts a negative feedback on FSH so that falling FSH levels contribute to atresia of other nondominant follicles. The dominant follicle secretes large quantities of estrogen; estradiol levels must be greater than 200 pg/mL for approximately 50 hours before a positive feedback on LH release is achieved.13,15 Once the LH surge is initiated, luteinization of the granulosa cells and progesterone production occurs. In pregnancy, estrogen augments uterine blood flow, although it is not required in itself for the maintenance of pregnancy.16
In the central nervous system, estrogen withdrawal at menopause has been associated with reduced libido, altered mood, and cognitive disturbances. These effects have been attributed to estrogen’s ability to modulate the synthesis, release, and metabolism of many neuropeptides and neurotransmitters.17 Estrogen acts as a serotoninergic agonist by increasing serotonin synthesis in the brain, which may positively influence mood.18 Although prospective observational studies in postmenopausal women have suggested that estrogen replacement therapy might protect against cognitive decline19 and the development of dementia,20 randomized trials of estrogen in the treatment of Alzheimer’s disease have shown no evidence of benefit.21–24
In the skeletal system, estrogen antagonizes the effect of parathyroid hormone by directly inhibiting the function of osteoclasts, which decreases the rate of bone resorption and diminishes bone loss. The Postmenopausal Estrogen/Progestin Interventions (PEPI) trial was a prospective, placebo-controlled trial designed to study the effects of hormone replacement on bone density in postmenopausal women. After 12 months of treatment with estrogen, bone mineral density increased by 1.8% at the hip and by 3% to 5% at the spine.25 The Women’s Health Initiative (WHI) showed that estrogen reduced the risk of both hip and vertebral fractures by 30% to 39%.26
In the cardiovascular system, there is strong evidence that estrogen has a natural vasoprotective role. At a cellular level, estrogen receptors are found on the smooth muscle cells of coronary arteries27 and the endothelial cells of various sites.28 Estrogen causes short-term vasodilation by increasing nitric oxide and prostacyclin release in endothelial cells.29 Several large observational studies, including the Framingham study and the Nurses Health Study, have shown that cardiovascular incidence rates are lower in premenopausal than postmenopausal women.30 There was also a significant association between a younger age at menopause and a higher risk of coronary artery disease.31 These studies led to the conviction that estrogen replacement therapy would consequently prevent the progression of atherosclerosis and coronary heart disease. However, the WHI study and the Heart and Estrogen/progestin Replacement Study (HERS), both large randomized, prospective trials designed to specifically address this issue, have not shown any benefit of estrogen for either the primary or secondary prevention of coronary artery disease, respectively.26,32
Biosynthesis and Metabolism
Estrogens are 18-carbon steroids derived from cholesterol (see Fig. 2-1). The three forms of naturally occurring estrogen include estrone, 17β-estradiol, and estriol. In nonpregnant females, estrone and estradiol are the main biologically active estrogens secreted by the ovary. Estradiol is almost 2 to 5 times more potent than estrone.33 The circulating levels of estradiol are 2 to 4 times higher than those of estrone in premenopausal women. Estradiol concentrations in postmenopausal women are one tenth of those in premenopausal women. Estrone concentrations do not differ with menopausal status; thus, over time, the premenopausal estradiol-to-estrone ratio is reversed.34 In contrast, estriol is not the secretory product of the ovary but is the peripheral metabolite of estrone and estradiol.
The main estrogen in premenopausal women is estradiol, which is produced primarily by the granulosa cells of the ovary. Androstenedione is converted to testosterone via 17βHSD, which is rapidly demethylated at the C-19 position and aromatized to estradiol. Estradiol is also generated to some degree from androstenedione via estrone. Estrone is also a secreted product of the ovary. It constitutes the remaining circulating estrogen (40%) and is mainly derived from the extragonadal peripheral aromatization of adrenal androstenedione.35 Peripheral conversion of androgens to estrogens occurs in skin, muscle, and adipose tissue and in the endometrium.36
In the normal adult female, the production of estradiol varies according to the phase of the menstrual cycle. During the mid luteal phase, for example, the production rate is about 100 to 270 μg/day. In comparison, the production rate for androstenedione is about 3 mg/day, and with its peripheral conversion rate to estrone of about 1.5%, it accounts roughly for about 10% to 30% of estrone production per day. Secondary increases in estrone formation occur in patients with polycystic ovaries or with ovarian cancer characterized by increased androgen production. In such patients, the increased estrogen can disturb the menstrual cycle. In postmenopausal women, the ovarian contribution shrinks, leaving estrone, derived from adrenal androstenedione, as the main source of circulating estrogen.37
In the pregnant woman, the placenta becomes the main source of estrogen in the form of estriol. The placenta is unable to synthesize steroids de novo and depends on circulating precursors from both fetal and maternal steroids. Most of the placental estrogens are derived from fetal androgens (e.g., DHEA sulfate), produced by the fetal adrenal gland.38 Fetal DHEA sulfate is converted to free DHEA by placental sulfatase and then to androstenedione and testosterone before being aromatized to estrone and estradiol. Finally, it is hydroxylated to form estriol.
Estradiol is rapidly converted in the liver to estrone by 17βHSD. Estrone can be further metabolized via three pathways. First, it can be hydroxylated to 16α-hydroxyestrone, which is then converted to estriol. Estriol is further metabolized by sulfation and glucuronidation, and the conjugates are excreted into the bile or urine. Secondly, estrone can be conjugated to form estrone sulfate, which occurs primarily in the liver. Estrone sulfate is biologically inactive and is present in concentrations that are 10-fold to 20-fold higher than concentrations of estrone or estradiol.39 Estrone sulfate can be hydrolyzed by sulfatases present in various tissues to estrone and may serve as a reserve of estrogen in an inactive form. Estrone sulfate may be of some importance in assessing estrogenicity in women and can be detected in serum as well as in urine.40 Thirdly, estrone can also be metabolized by hydroxylation to form 2-hydroxyestrone and 4-hydroxyestrone, which are known as catechol estrogens. These are then converted to the 2-methoxy and 4-methoxy compounds by catechol-O-methyltransferase.
Progesterone
Physiologic Role
During pregnancy, progesterone increases insulin resistance in concert with the production of the other placental counterregulatory hormones, including placental growth hormone, placental lactogens, placental corticotropin-releasing hormone, and cortisol.41
Biosynthesis and Metabolism
Progesterone is part of the group of 21-carbon steroids, which also includes pregnenolone and 17OH-progesterone. Progesterone is responsible for all the progestational effects, whereas pregnenolone is the precursor for all steroid hormones. 17OH-progesterone has little biologic activity. Progesterone and 17OH-progesterone are mainly produced by the corpus luteum in the luteal phase of the menstrual cycle and by the placenta if pregnancy occurs. Circulating levels of progesterone at concentrations greater than 4 to 5 ng/mL (12.7 to 15.9 nmol/L) are indicative of ovulation.42
Androgens
Physiologic Role
In women, androgens originate as 19-carbon steroids from the adrenals and ovaries. The major androgens produced in the ovary, primarily by the thecal cells and to a lesser degree by the ovarian stroma, include DHEA, androstenedione, and a small amount of testosterone. Both DHEA and androstenedione serve as precursors to estrogen synthesis and have little, if any, androgenic activity. However, these biologically inactive androgens are converted by extraglandular metabolism to biologically active androgens such as testosterone and dihydrotestosterone (DHT). Normally, the levels of these potent androgens are low in females and have no significant physiologic function. Excessive production of androgens by the ovary or adrenals has been implicated as the cause of hirsutism and virilization in women.43 In contrast, in the male the androgens, of which testosterone and DHT are the most crucial, are of primary importance.
Biosynthesis and Metabolism
Androgens are 19-carbon steroids derived from cholesterol. The rate-limiting step in androgen synthesis is the conversion of cholesterol to pregnenolone, which is mediated by the action of LH on the ovary and testes. In a normal ovulatory woman, the ovaries secrete approximately 1 to 2 mg of androstenedione, 1 mg of DHEA, and approximately 0.1 mg of testosterone. The majority (≈0.2 mg) of circulating testosterone is derived from peripheral metabolism of DHEA and androstenedione. Overall, testosterone production in women is about 0.3 mg/day; roughly 50% of this is derived from peripheral conversion whereas the remaining 50% is secreted equally by the ovary and the adrenals.44
Transport of Ovarian Steroid Hormones in Plasma
Steroid hormones are not water-soluble and require transport proteins to be carried to their target tissues. The two types of transport proteins are general carrier proteins such as albumin and transthyretin and specific carrier proteins such as thyroxine-binding globulin, sex hormone-binding globulin (SHBG), and transcortin. Both types of proteins are produced in the liver. Less than 2% of ovarian steroid hormones are free in the circulation; the remainder are mostly bound to SHBG and albumin.45,46
Sex hormone-binding globulin, a β-globulin of 95 kDa, is synthesized in the liver. Its gene is localized on the short arm of chromosome 17 (p12-13).47 It is a homodimer composed of two polypeptide chains and has a single binding site for androgens and estrogens. Dimerization is a necessary step in the binding process.48 The bound and free fractions appear to exist in a steady state of equilibrium. The amount of free fraction depends on the concentration of steroid hormone and on the levels and binding affinities of the binding proteins.
Of all the steroid hormones, DHT has the highest affinity for SHBG. Approximately 98% of testosterone circulates bound to SHBG (≈65%) and albumin (≈33%). Estradiol is primarily bound to albumin (≈60%) but also to SHBG (38%); about 2% circulates as the free fraction.49 Progesterone, on the other hand, is mainly bound to albumin (≈80%) but also to transcortin (≈18%). Only approximately 0.6% of progesterone is bound to SHBG and about 2% exists in the free state.
The metabolic clearance of these steroids is inversely related to their binding affinity to SHBG. Thus, conditions that affect levels of SHBG (e.g., pregnancy, oral contraceptives) directly affect the levels of free hormone. Because estrogens increase SHBG synthesis and androgens decrease its synthesis, SHBG levels are twice as high in women compared to men. Several other hormones and other factors are known to influence SHBG levels. Thyroid hormones increase its synthesis and release by the liver.46 Insulin, IGF-I, and prolactin have been shown to inhibit SHBG production in cultured cells.50,51 Furthermore, serum concentrations of SHBG are increased in many disease states, including hyperthyroidism and liver cirrhosis. Certain drugs, including estrogen, tamoxifen, and phenytoin, can also increase serum SHBG concentrations. Carrier protein levels are decreased by hypothyroidism, obesity, and acromegaly and by administration of exogenous androgens, glucocorticoids, and growth hormones.
For many years, only the free fraction of testosterone was regarded as the biologically active component. However, researchers noted that steroid hormones bind with greater affinity to their specific carrier proteins and with much less affinity to albumin. In addition, studies of tissue delivery in vivo showed that the dissociation of albumin-bound testosterone can occur rapidly in a capillary bed so that the active fraction can be larger than the free fraction measured under equilibrium conditions in vitro.52 Thus, unconjugated steroids that are bound to albumin may be treated as free and biologically available.53,54
As mentioned above, SHBG levels can be influenced by numerous disease states. As such, changes in SHBG concentrations can result in large shifts in the free and SHBG-bound fractions. Hence, measurement of SHBG is of great clinical interest because it allows more accurate assessment of free hormones. SHBG is measured by a technique called saturation analysis, in which specific binding capacity of 3H-labeled testosterone is detected.55,56 With modifications, this method can also measure the non-SHBG bound fraction (bioavailable).57 Recently, specific nonisotopic two-site immunoassays for SHBG have become available and are used in most clinical laboratories.
Measurement of Steroid Hormones in Circulation
The technique responsible for the accurate measurement of low concentrations of various steroid hormones and metabolites is competitive inhibition immunoassay or radioimmunoassay (RIA), which was originally described in 1960 by Yalow and Berson.58 However, the development of steroid immunoassays presented several problems. First, they are not immunogenic and have a similar structure—they all have a same cyclopentahaptene nucleus with only minor structural variations—which makes it difficult to generate specific antibodies. Steroids can be made immunogenic via chemical coupling to a carrier protein known as hapten, and antibodies can be raised by immunization with haptens.59 However, the site of the steroid where the protein is conjugated has a significant impact on the specificity of the resulting antibody.59 Antibodies raised to conjugates of BSA coupled at the 19th position show higher specificity than those coupled at the 3rd or 17th positions.60 For accurate clinical interpretation, it is important to know the cross-reactivity data on each antibody that is selected for a given assay. Most commercial assay reagent manufacturers provide cross-reactivity data, but it may not always be reliable and must be evaluated in the clinical laboratories performing the assay.61
Second, the high-affinity binding proteins such as SHBG in the serum compete with the antibody and thus interfere with the measurement of steroid molecules by RIA. This makes direct measurement difficult and necessitates a preassay extraction procedure with organic solvents and often a chromatographic separation of the steroid. Alternatively, the use of certain chemicals, such as 8-anilinonaphthalene sulfonic acid can inhibit the binding of steroids to proteins, which allows the direct measurement of steroid hormones without the extraction step. Direct assays are fast and can be automated. Several automated platforms for measuring estradiol, progesterone, and testosterone are commercially available and are used in most clinical laboratories. These assays, however, have a low sensitivity and, when used to measure very low concentrations, have poor reliability.62 Therefore, they may not be the best choice for clinical applications that require the ability to measure very low hormone concentrations such as estradiol measurement in children and in men (<100 pg/mL),63 testosterone measurement in children and women(<1.5 ng/mL),64 or progesterone measurement during ovarian stimulation (<1 ng/mL).65 To overcome this problem, serum can be extracted with hexane-ethyl acetate (3:2 by volume), dried, and reconstituted in steroid-free serum, which can then be assayed in the automated platform.66
Gas chromatography combined with mass spectrometry (GC-MS) addresses many of the shortcomings of immunoassays and is considered more reliable and accurate than immunoassays. However, this technique requires multiple steps, including solvent extraction, chromatographic fractionation, and chemical dramatization before instrumental analysis, and it is often less sensitive than some immunoassays. This technique has now been superseded by liquid chromatography combined with tamdem mass spectrometry. This newer technique has a higher sensitivity and throughput than GC-MS and is considered a reference methodology.67,68 The technique has been used to simultaneously measure estradiol and estrone in human plasma with no cross-reactivity.69 These methods are recommended as reference methods and can be used to standardize and validate immunoassays, which provide the simplicity and rapid throughput needed for clinical use.
Estrogens
Measurement of estradiol is important in the assessment of female reproductive function. It can be used as an aid in the diagnosis of infertility and oligomenorrhea and to determine menopausal status (Table 2-2). In addition, measurement of estradiol is widely used to monitor ovulation induction and in vitro fertilization protocols.70,71 Estrone levels are of limited clinical value in nonpregnant women because their levels closely parallel those of estradiol, except in the postmenopausal woman, in whom estrone becomes the main form of circulating estrogen. Also, as mentioned above, a specific RIA for the measurement of estrone sulfate has been described and made available commercially. Because estrone sulfate has a large circulating pool, it can serve as a marker of estrogenicity, especially in women on estrogen replacement therapy, in whom estradiol measurements are of little value due to the variable cross-reactivity of conjugated estrogens in the estradiol assays.72,73 In pregnant women, estriol is the main form of estrogen produced, and the amount of estrogen secreted increases from microgram quantities to milligram quantities.
Table 2-2 Assay Techniques and Clinical Applications of Ovarian Steroid Hormones
| Hormone | Assay Techniques | Clinical Application |
|---|---|---|
| Estradiol |
Progesterone
Although GC-MS has been recommended as the reference method for the measurement of progesterone,74 immunoassays using steroid-specific antibodies are again the preferred mode of measurement in most clinical laboratories.75,76 Both RIA and nonisotopic immunoassays for progesterone are available for commercial use. Progesterone measurement is routinely used to detect ovulation and luteal phase defects (see Table 2-2).77 In the follicular phase, progesterone levels are low (<5 nmol/L or 1.5 ng/mL). In the luteal phase, they range between 9 and 79 nmol/L (3 to 25 ng/mL). As noted, progesterone is required to maintain pregnancy, and progesterone measurement in early pregnancy can be valuable in the diagnosis of defects or threatened abortion.78
Androgens
The measurement of androgens, including androstenedione and total testosterone or DHT, can also be accomplished using immunoassays.79–81 The one main drawback of these assays is the cross reactivity with other steroid hormones. Interference with cross-reactive testosterone in the androstenedione assay has been overcome by the use of specific testosterone antiserum to neutralize plasma testosterone.81 Direct measurement of androstenedione and testosterone without extraction is now possible with new commercial assays. However, commercial assays demonstrate high variability, which is greatest in samples from females.82 Total testosterone in hirsute women overlaps significantly with levels seen in normal women, and measurement of free testosterone correlates better with disease.83 Free testosterone has been measured by equilibrium-dialysis, which is a time-consuming and difficult technique for most clinical laboratories to perform. An ultrafiltration technique can be used instead, which depends on MPS-1 centrifugal gel filtration devices and correlates well with the equilibrium-dialysis method83,84 as well as with GC-MS.85 Direct, single-step, nonextraction immunoassay methods using 125I-labeled testosterone analog have been developed commercially and are used in a number of laboratories. The accuracy and validity of this direct assay has been questioned.86,87 Alternatively, an indirect parameter of free testosterone—FAI—can be calculated as a ratio of testosterone to SHBG.88 FAI is a better discriminator of hirsutism than either total testosterone or SHBG levels.87–89 When bound with albumin and transcortin, testosterone dissociates more quickly than when bound with SHBG. This loosely bound teatosterone may be biologically available through dissociation during capillary transit. Cumming and Wall provided evidence for this hypothesis and suggested that this non-SHBG bound testosterone is a marker of hyperandrogenism.57,90
Saliva Measurements
SHBG is either undetectable or minimally present in saliva. Thus, this biologic fluid may reflect the free fraction of plasma steroids. Therefore, measurement of steroid hormones in saliva has attracted considerable attention.91–93 The ease of noninvasive collection combined with the simplicity of measurement makes salivary measurement a promising and attractive alternative to measurement of steroids in plasma. In the future, salivary assays may become useful adjuncts to those performed in plasma.94–97
PEPTIDE HORMONES AND INTRAOVARIAN GROWTH FACTORS OF OVARY
The role of gonadotropins and gonadal steroids in the process of folliculogenesis in the ovary is well recognized. However, multiple other phenomena within the ovary suggest the presence of other intraovarian factors that may fine-tune the effects of gonadotropins and gonadal steroids. For example, the initiation and arrest of meiosis and the different rates of follicular growth leading to the selection of a dominant follicle point toward the existence of an intraovarian modulatory system. The concept of a gonadal factor with endocrine action at the pituitary level can be traced back more than 70 years to Mottram and Cramer.98 The first moiety was identified and named inhibin for its inhibitory effect on the pituitary.99 Since then, there has been an explosion of information regarding multiple and potential intraovarian regulators and their physiology, biochemistry, and biosynthesis as well as the identification of their receptors. Some of these compounds, which have been the subject of intense investigation, include peptide hormones/growth factors, cytokines, and neuropeptides. These factors may act in either an endocrine, autocrine, or paracrine fashion.
Peptide Hormones of the Ovary: Inhibins, Activins, and Follistatins
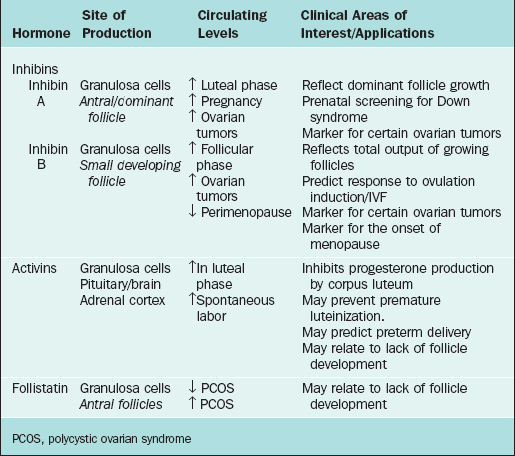
The first water-soluble peptide hormone in testicular extracts that exerted selective inhibitory activity at the pituitary level was described in 1932 and termed inhibin.99 It was not until 1985 (almost 50 years later) that inhibin was isolated and characterized.100,101 This was followed by the identification and characterization of two other related peptide factors (i.e., activin and follistatin).102,103 With the cloning of inhibin α and β subunit genes,104 it was recognized that inhibin and activin belonged to the transforming growth factor-β (TGFβ) superfamily of growth and differentiation factors. Both inhibin and activin are of particular clinical interest and have been extensively reviewed3,105,106 in recent years. They have been found to exert numerous different regulatory functions in a wide variety of both normal and neoplastic cells. Follistatin, a glycoprotein that is structurally distinct but functionally closely linked to inhibin and activin, is also discussed here. The site of production and areas of clinical interest of these peptide hormones are listed in Table 2-3.
Inhibins
The primary sources of inhibin production are the granulosa cells of the ovary and the Sertoli cells of the testis. Inhibin is also produced during pregnancy by the fetus, placenta, decidua, and fetal membranes. The main role of inhibin is to selectively suppress the production of FSH by the pituitary.107,108 This is achieved by modulating FSH biosynthesis through two main mechanisms: by reducing steady-state FSH mRNA in pituitary gonadotropes109 and decreasing the stability of FSH mRNA.110
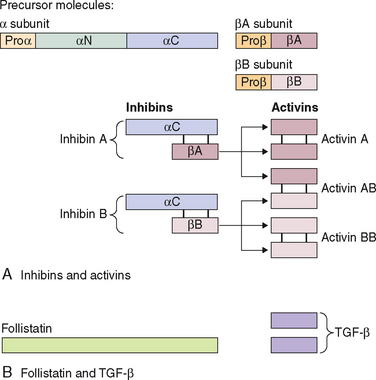
Inhibin is a 32 kDa glycoprotein heterodimer consisting of 2 subunits, α and β, linked by disulfide bonds.111 There is a common α subunit but also two types of β subunits known as βA or βB. Thus, the two isoforms of inhibin are denoted inhibin A and inhibin B. As shown in Figure 2-5, each subunit derives from a separate precursor molecule called prepro-inhibin α (364 amino acid residues), prepro-inhibin βA (424 amino acid residues) and prepro-inhibin βB (407 amino acid residues). These are processed by proteolytic cleavage to yield the mature forms.104 In addition to the fully mature forms (αβ dimers, Mr∼32,000), larger forms of dimeric inhibins with amino terminally extended α and/or β subunits have been identified in follicular fluid, which also possess FSH-suppressing bioactivity.112,113 Furthermore, monomeric forms of both α and β subunits and certain fragments (αN and proαN-αC) generated during subunit processing are present in follicular fluid (see Fig. 2-1) and have intrinsic biologic activities distinct from classical inhibin-like bioactivity.114,115
Because the circulation contains multiple molecular forms of inhibin, it can be difficult to accurately measure inhibin levels using conventional RIAs. Most lack specificity for dimeric inhibin due to the variable cross-reactivity of monomeric forms and of various fragments with the antibody used in the assay.112,116,117 Also, conventional inhibin bioassays based on FSH suppression or release by cultured pituitary cells lack specificity due to the FSH-regulating activities of follistatin and activins. In addition, bioassays lack the sensitivity necessary to measure inhibin levels in the circulation. The development of two-site immunoassays utilizing αβ dimer specific antibodies overcame these problems and allowed specific measurement of the two forms of inhibin dimers (A and B).118 Use of these novel two-site assays allows measurement of inhibin levels throughout the normal menstrual cycle.119
Synthesis of the two isoforms of inhibin differ during the various phases of the menstrual cycle (see Fig. 2-4). Inhibin B levels are highest during the luteal–follicular transition and the early follicular phase, and studies on its presence in follicular fluid and basal granulosa cell secretion suggest that it is secreted by small developing antral follicles.120 In contrast, inhibin A levels in the early and midfollicular phase reflect the sum of FSH- and LH-stimulated inhibin A secretion from all antral follicles. Levels of inhibin A during the late follicular phase mainly reflect secretion from the dominant follicle. Hence, inhibin A and B levels can be used as markers of follicular development. Inhibins have also been investigated as prognostic markers for women undergoing assisted reproductive technologies. In particular, it was suggested that measuring inhibin B levels during the early stages of FSH stimulation for ovulation induction could predict the number of oocytes retrieved and may be useful in monitoring ovarian stimulation for in vitro fertilization. However, because there is a large overlap between normal and subnormal ovarian responses in terms of inhibin B levels, it may be just as effective to obtain Day 3 FSH or perform a clomiphene challenge test.121,122 Women in early perimenopause show significant decrease in inhibin B (no significant change in inhibin A and estradiol), which correlates with a mild increase in FSH levels. This perimenopausal decrease in inhibin B precedes a decrease in inhibin A, suggesting that inhibin B may serve as a sensitive marker for the onset of menopause.123 Studies investigating the role of inhibins in the pathophysiology of polycystic ovary syndrome (PCOS) show conflicting results. Serum inhibin B levels in early follicular phase show significant increase in some studies or no change in others124,125—this awaits future studies to confirm its role in PCOS.
During pregnancy, inhibin A is produced primarily by the fetoplacental unit, whereas inhibin B levels remain low throughout the pregnancy.126 Because there is a twofold elevation of circulating inhibin A levels in the second trimester of Down syndrome pregnancies, measurement of inhibin A is a clinically useful and important test.127 When measurement of inhibin A level is added to alpha-fetoprotein, maternal age, and β-hCG (quad screen), the detection rate for Down syndrome increases from 53% to 75%.128
Inhibin A has also recently been used as a tumor marker for ovarian sex cord tumors. This heterogeneous group of tumors accounts for 7% of all malignant primary ovarian neoplasms. They are composed of granulosa cells, thecal cells, Sertoli cells, Leydig cells, and other nonspecific stromal cells. It is important to distinguish this group of tumors from carcinomas and sarcomas because the former are low-grade tumors with a better prognosis. Inhibin A and its α-subunit have been found to be sensitive immunohistochemical markers of most ovarian sex cord-stromal tumors.129 Inhibin B, however, has been found to be elevated in both sex cord-stromal and epithelial tumors and hence is of limited value in differentiating between the two entities. In addition, low levels of inhibin A in the cyst fluid of epithelial ovarian tumors has recently been reported to be associated with a worse prognosis.130
Inhibin B has been found to be the predominant inhibin secreted in males and is produced by the Sertoli cells of the testes. It also has a negative feedback role on FSH from the pituitary, and its production is regulated by spermatogenesis. Inhibin B levels correlate with sperm count and testicular volume131–133 but cannot distinguish between spermatid arrest and obstructive azoospermia—a condition in which sperm counts are normal.134 As such, it is unlikely to replace testicular biopsy in the evaluation of male infertility.
Activins
Activins are made up of dimers of the inhibin β subunit (βAβA, βAβB, or βBβB) and have a molecular weight of approximately 25 kDa (see Fig. 2-5).135 They are predominantly produced by the granulosa cells of the ovary. Activin/inhibin mRNA and protein have also been detected in extragonadal sources, including the placental trophoblast and decidua, testes, adrenal cortex, brain, spinal cord, and anterior pituitary. This implies that activin has diverse physiologic roles that are not confined to the reproductive system.
Acting either alone or with FSH, activin exerts an autocrine effect on granulosa cells to promote and maintain granulosa cell differentiation. It promotes FSH receptor expression on small undifferentiated granulosa cells136, enhances their response to FSH and LH, and hence increases aromatase activity and estrogen production.137 This may explain how small preantral follicles progress from a gonadotropin-independent to a gonadotropin-dependent stage of development. After the granulosa cells acquire FSH receptors, further growth and differentiation of those cells to a preovulatory stage would be driven by activin acting in concert with FSH. Activin also inhibits both spontaneous and LH/hCG-induced increases in progesterone output by human follicles,137 implying that it plays a role in delaying the onset of premature luteinization.
Activin also has a paracrine effect on thecal steroidogenesis by inhibiting thecal androgen output. It has been proposed that at earlier stages of follicular development, when the androgen requirements are low, thecal androgen synthesis is kept in check due to the relative excess of activin over inhibin and follistatin. However, as dominant follicles approach preovulatory status, increasing granulosa cell expression of inhibin and follistatin upregulates thecal androgen synthesis and ensures that the granulosa cells receive an adequate supply of aromatase substrate for conversion to estradiol. Activin stimulates pituitary FSH production, acting as a functional antagonist to inhibin (Fig. 2-6).102 It achieves this effect by increasing FSH mRNA synthesis as well as by increasing the stability of produced mRNA. Its actions are intimately modulated by intrapituitary concentrations of follistatin, which bind to activin to limit its bioavailability.
Free activin levels as measured by competitive protein binding assay, using follistatin as binding protein, show very little change over the menstrual cycle.138 However, activin levels were elevated throughout the cycle in older versus younger women, suggesting that activin may play an endocrine role in maintaining FSH elevation in reproductive aging.139 Lower levels of activin are detected in PCOS patients with a simultaneous increase in inhibins and follistatin, suggesting that an imbalance in these hormones may contribute to an abnormal LH/FSH ratio.125,140
Follistatin
In the ovary, follistatin is produced by the granulosa cells in antral follicles as well as by luteinized granulosa cells, which are under the positive regulation of FSH. It modulates the function of granulosa cells in favor of luteinization or atresia by neutralizing the effects of activin. It may also directly modulate progesterone metabolism by granulosa cells.141
Follistatin is a single-chain polypeptide (315 amino acids) that functions as the binding protein for activin, thereby neutralizing it. Most of its biology is explained by its antagonism with activin. Follistatin exists in two forms; as full-length follistatin (FS 315) in the circulation and as processed follistatin (FS 288) in follicular fluid142 and the pituitary. It is part of an intrapituitary negative feedback loop where activin promotes FSH biosynthesis and the increased expression of follistatin limits its bioavailability for binding to the activin receptor on target cell membranes. It has been shown that the processed isoform of follistatin (FS 288) binds to cell-surface heparan sulfate proteoglycans with a higher affinity than FS 315.143 Because proteoglycans are anchored to cell membranes, this suggests that this would limit follistatin diffusion from the site of release, leading to high local concentrations of follistatin being maintained. The membrane-anchored follistatin would be able to compete with activin receptors on nearby cells, thus modulating the biologic effects of activin. Once bound to activin, follistatin is able to accelerate the endocytotic internalization and lysosomal degradation of activin by pituitary cells.144
Follistatin levels as measured by sensitive and specific two-site enzyme immunoassay are significantly higher in women with PCOS in comparison to controls. Higher follistatin levels combined with lower activin levels in these patients suggest its role in the lack of pre-ovular follicle development and FSH suppression.125
Growth Factors/Cytokines As Intraovarian Regulators
Certain growth factors have been implicated in cellular communications within the ovary, including insulin-like growth factors IGF-I and IGF-II, epidermal growth factor (EGF), transforming growth factor-α (TGFα), basic fibroblast growth factor (bFGF), cytokines such as interleukins (IL-1 and IL-6), and tumor necrosis factor (TNFα). Growth factors and cytokines that are important as intraovarian regulators are outlined in Table 2-4.
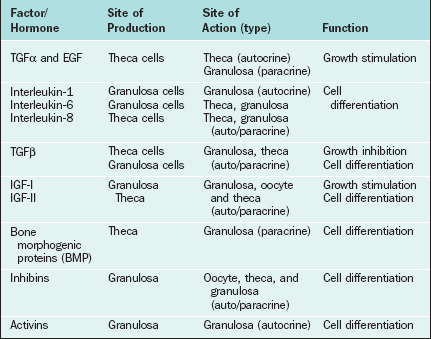
Insulin-like Growth Factors
IGF-I and IGF-II promote cellular mitosis and differentiation in a variety of systems and play an important role in modulating folliculogenesis in an autocrine/paracrine fashion.145–147 Insulin-like growth factors consist of two single-chain polypeptide growth factors that are structurally and functionally similar to proinsulin. The IGF autocrine/paracrine system includes IGFs, their specific receptors in target cells, and a family of IGF-binding proteins that regulate their bioavailability. Both IGF-I and IGF-II are produced in the ovary and augment the effects of the gonadotropins, although the main IGF in human follicles is IGF-II.148 In small antral follicles, both IGF-I and IGF-II are expressed but restricted to thecal cells. However, IGF-I receptor mRNA has been detected only in granulosa cells.146 It serves as an autocrine regulator in thecal cells and as a paracrine regulator in granulosa cells. In dominant follicles, however, no IGF-I mRNA has been detected either in thecal or in granulosa cells, and IGF-II expression is restricted to granulosa cells only. IGF-I receptors are present in granulosa cells only, and IGF-II receptors are expressed in both cell types.146 Thus, in the dominant follicle, IGF-I functions mainly as a paracrine regulator149,150 and IGF-II acts as an autocrine factor. This suggests that IGF-II has an important role in coordinating differential follicular development within the ovary.
Epidermal Growth Factor (EGF), Transforming Growth Factor-α (TGFα), and Basic Fibroblast Growth Factor (bFGF)
A number of other peptide growth factors have been implicated as regulators of follicular development and steroidogenesis (see Table 2-4).151 These include EGF, TGFα, and bFGF. EGF is a single-chain polypeptide of 53 amino acids with three disulfide bonds and has mitogenic effects in a variety of ectodermal and mesodermal tissues. TGFα is a 50-amino acid peptide with 30% to 40% homology with EGF. The EGF receptor is a 170-kDa glycoprotein with tyrosine kinase activity. TGFα binds to EGF receptors with the same affinity as EGF.
The presence of EGF/TGFα and bFGF as well as their receptors in the ovary has been shown both at the protein and mRNA levels. The presence of immunoreactive EGF as well as EGF receptors has been identified in the preovulatory follicles152 and corpus luteum.153,154 Furthermore, studies have shown the presence of TGFα155 and bFGF156 mRNAs in follicular cells; studies have also shown that the TGFα message is upregulated by FSH in vivo. FSH plus TGFα or FSH plus EGF resulted in significantly elevated progesterone and 20α-hydroxyprogesterone levels in granulosa cells in culture.157 The presence of TGFα message in cultured granulosa cells and the fact that it mediates its action via binding to EGF receptor all point to its autocrine role in granulosa cell differentiation, follicle development, and selection. Expression of bFGF and its receptor mRNA has been detected in fetal ovaries and in granulosa cells.158,159 bFGF has been shown to be mitogenic for granulosa cells and to cause an inhibitory action on granulosa cell differentiation and thecal cell steroidogenesis.160,161 It also has potent angiogenic activity.162
Cytokines
Cytokines primarily produced by white blood cells modulate various cellular functions. The cytokines in the ovary are secreted both by the immune cells that are recruited from the circulation in the ovarian stroma and by the thecal and granulosa cells. A number of cytokines have been linked to modulation of ovarian function, including interleukins IL-1 and IL-6 and TNFα. Both IL-1 and IL-6 have been found in significant amounts in follicular fluid.163,164 Granulosa cells accounted for the majority of immunostaining for IL-1 and IL-6 in follicular aspirates, which suggests that these cytokines are produced in granulosa cells164,165 and that they affect granulosa function.166 During folliculogenesis, IL-I promotes proliferation and suppresses differentiation. In the ovulatory process, it promotes ovulation by increasing production of chemokines, steroids, ecosanoids, and vasoactive substances.167 IL-6 demonstrates inhibitory effects on both estradiol and progesterone secretion by FSH-stimulated granulosa cells.168 Elevated IL-6 levels during genital infections may provide a possible link to reproductive dysfunction. TNFα expression has also been detected in granulosa cells of human antral and atretic follicles by immunohistochemistry.169 Also, in vitro treatment with TNFα enhanced steroidogenesis in both healthy and atretic follicles,170 suggesting that TNFα has a paracrine and/or autocrine role. Nonetheless, the physiologic implications of these actions remain unclear and require further investigation.
Neuropeptides
Some evidence suggests that an independent ovarian-central nervous system axis exists.171,172 Electrical stimulation of the hypothalamus in hypophysectomized and adrenalectomized rats produced a change in ovarian steroidal synthesis that was independent of changes in ovarian blood flow.173 In addition, murine thecal cells can produce androgens under adrenergic stimulation.174 Adrenergic innervation of the ovary acts primarily on the thecal-interstitial cells through β2 receptors, synergizing with the effects of gonadotropins in the production of ovarian androgens.174 This may in turn play a role in the regulation of estrogen production by granulosa cells, thereby influencing follicle recruitment and selection.
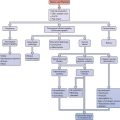
REGULATION OF OVARIAN HORMONES
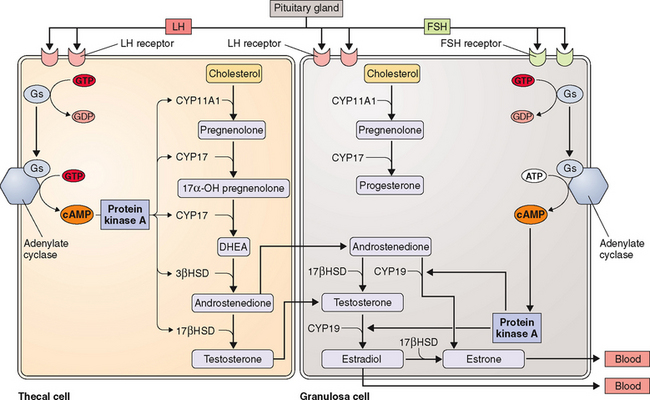
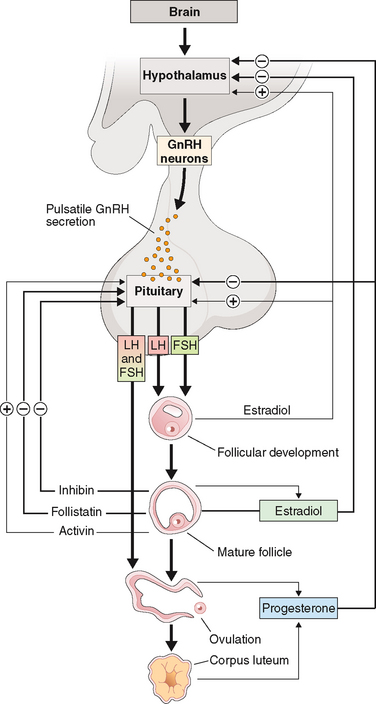
The hypothalamic-pituitary axis plays a key role in the regulation of hormonal synthesis by the ovaries. The hypothalamus is connected to the pituitary gland via a portal vascular system that allows transport of hypothalamic releasing factors from the brain to the pituitary (see Fig. 2-6). The hypothalamus, being the coordinating center, provides precise signals via pusatile release of GnRH to the gonadotrophs, which in turn secrete LH and FSH. Any interruption in this connection results in low gonadotropin levels, leading to failure of ovarian hormone secretion.
Hypothalamic Regulation
Gonadotropin-releasing Hormone
The gonadotropin-releasing activity in hypothalamic extracts was first demonstrated in the late 1950s. In 1971, almost 15 years later, GnRH was first isolated and characterized from a hypothalamic extract. This was followed by its synthesis and its clinical availability. However, early clinical studies proved disappointing until the nature of its pulsatile secretion was recognized. In their classic experiment in the rhesus monkey, Knobil and colleagues showed that normal LH and FSH release required the pulsatile infusion of GnRH at approximately 60-minute intervals175 Furthermore, a change in pulse frequency (lesser or greater) or continuous infusion resulted in failure of LH and FSH secretion and release (Fig. 2-7).176 These observations were confirmed in humans. Pulsatile GnRH infusion reproduces appropriate patterns of hormonal changes, resulting in ovulation and fertility in women with hypothalamic amenorrhea.
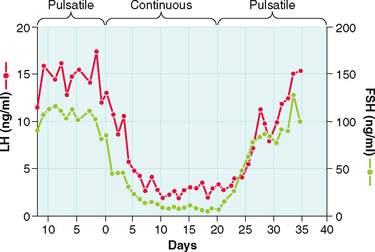
Physiologic Role of GnRH
GnRH is produced by secretory neurons located in the arcuate nucleus of the medial basal hypothalamus and the preoptic area of the anterior hypothalamus. The nerve terminals are found in the lateral portions of the external layer of the median eminence adjacent to the pituitary stalk.177 GnRH has an intrinsically pulsatile pattern of secretion, which is under the control of a hypothalamic pulse generator in the arcuate nucleus.178,179 The frequency and amplitude of the pulsatile rhythm of GnRH secretion are crucial in regulating gonadotropin secretion and hence gonadal activity.180,181 Physiologic frequency (approximately hourly pulses) tends to upregulate GnRH receptors, enhancing pituitary responsiveness to subsequent stimulation by GnRH. This leads to a “self-priming” effect, whereby LH levels have been shown to increase sequentially with sequential GnRH pulses. A longer frequency causes anovulation and amenorrhea; a shorter frequency or constant exposure to GnRH downregulates the GnRH receptors, inducing refractory gonadotropin responses.176,182–184
The “pulse generator” is subject to modification by two main inputs: (1) hormone-mediated signals and (2) neural signals. Hormone signals include negative and positive feedback from the gonadal steroids (e.g., estrogen and progesterone) as well as gonadal protein hormones. Neural signals may come from a wide variety of sources and are mediated by neurotransmitters, including acetylcholine, catecholamines, serotonin, opioids, and γ-aminobutyric acid.185 Norepinephrine is believed to stimulate GnRH release whereas opioids exert inhibitory effects. Dopamine can produce both inhibitory and excitatory GnRH responses, depending on the physiologic state.186
Biochemistry and Biosynthesis
GnRH is a linear decapeptide, derived from the post-translational processing of a large precursor molecule, prepro-GnRH. The prepro-GnRH molecule consists of 92 amino acids in a tripartite structure (Fig. 2-8). It begins with a signal peptide of 23 amino acids, which is followed by the decapeptide and then a Gly-Lys-Arg sequence needed for proteolytic processing and C-terminal amidation of GnRH molecules. The last 56 amino acid residues are collectively known as the GnRH-associated peptide (GAP), which may have prolactin-inhibiting activity.187,188 Knowledge of GnRH structure led to the development of many clinically important long-acting GnRH agonists, including buserelin, leuprolide, and nafareline (see Fig. 2-8).
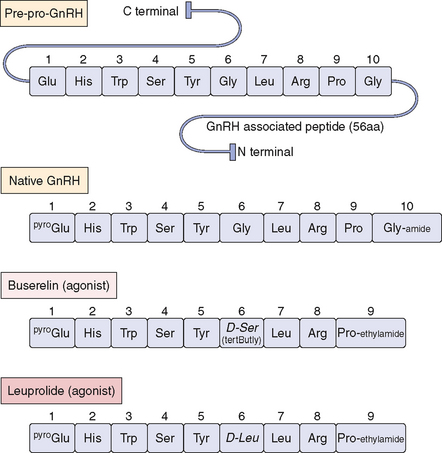
Gonadotropin-releasing hormone is encoded from a single gene on the short arm of chromosome 8p21-p11. The human gene contains 4 exons; exon 2 encodes pro-GnRH, exon 3 and part of exon 2 and 4 encode the GAP protein, and exon 4 encodes a long 3′ untranslated region. Molecular processing occurs primarily in the nucleus of the cell body (soma). After transcription, the mRNA is transported to the cytoplasm where translation takes place and it is converted into the decapeptide. GnRH and its cleavage products, GAP and pro-GnRH, are then transported to the nerve terminals where they are secreted in tandem into the portal circulation.187,189,190
Measurement
GnRH has a short half-life (2–4 min) due to its degradation by peptidases in the hypothalamus and pituitary gland. These peptidases cleave the molecule at the Gly-Leu bond and at position 10. Due to the large dilutional effect, peripheral GnRH serum levels are too low for pulse characteristics to be determined reliably in humans. However, the simultaneous measurement of circulating levels of LH in hypophysial-portal and peripheral blood samples in other species has been shown to correlate closely with GnRH release.191,192 As such, frequent measurements of LH pulses can be used as an accurate indicator of GnRH secretion patterns. FSH also correlates with GnRH secretion, but is less useful due to its long half-life.
Regulation by Pituitary Hormones
As mentioned above, GnRH action on gonadotrophs stimulates gonadotropin production and release in a pulsatile fashion. In addition, LH and FSH release from the pituitary is also affected in both a positive and negative manner by estrogen and progesterone as well as by the protein hormones secretion by the ovaries (see Fig. 2-6). The positive effect of estrogen and the negative effect of progesterone on the gonadotropins depend on the level of steroid hormone and the duration of exposure to the gonadotrophs. On the other hand, both LH and FSH are required for ovarian estrogen synthesis and the level of estrogen production depends on the time of exposure and the level of gonadotropins.193,194 Nonetheless, a disordered signal from the pituitary gland may result in infrequent ovulation (oligo-ovulation) or absent ovulation (anovulation).
Regulation by Luteinizing Hormone and Follicle-stimulating Hormone
Physiologic Role
As discussed above, LH regulates ovarian steroidogenesis. The surge of LH in the middle of the menstrual cycle is also responsible for inducing ovulation. The surge occurs as a result of a dramatic rise in estradiol produced by the preovulatory follicle, which produces a positive feedback on LH. The midcycle surge stimulates the resumption of meiosis and the completion of reduction division in the oocyte with release of the first polar body. Proteolytic enzymes and prostaglandins are increased in response to LH, leading to a release of the oocyte from the ovary.195 Finally, the continued secretion of LH after ovulation converts the remaining follicular cells in the ovary to the corpus luteum and stimulates the corpus luteum to produce progesterone by enhancing the conversion of cholesterol to pregnenolone.
Follicle-stimulating hormone regulates ovarian estrogen synthesis by binding to the FSH receptor on the surface of the granulosa cell and is required for follicular maturation and growth.196,197 This results in elevated cyclic adenosine monophosphate (cAMP) levels and the induction of aromatase, which converts androstenedione from the neighboring thecal cells to estrone. FSH also induces expression of 17βHSD type 1, which converts estrone to estradiol. Increased secretion of estradiol leads to further proliferation of granulosa cells and follicular growth and an increase in the number of estradiol receptors.196,198,199 In the mature follicle, FSH and estradiol increase the LH receptors’ expression in granulosa cells, making these cells responsive to LH and augmenting progesterone secretion. Progesterone then increases FSH release in midcycle.
Biochemistry and Biosynthesis
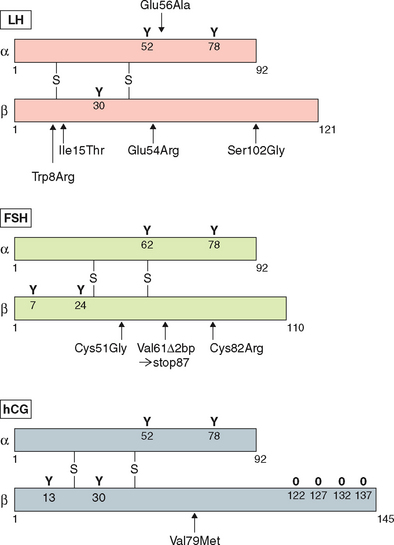
The anterior pituitary produces three glycoprotein hormones: LH, FSH, and thyrotropin-stimulating hormone (TSH), all of which share a similar biochemical structure. Each consists of a heterodimer of two noncovalently linked protein subunits, αβ. Each subunit is cysteine-rich and contains multiple disulfide linkages (Fig. 2-9). There are also multiple carbohydrate moieties on both subunits that are important in the metabolism and biologic activity of the hormones.200,201 The α subunit is common to all three hormones, whereas the β subunit is unique.
α Subunit
The human α subunit gene is located on chromosome 6p21.1-23 and is composed of 4 exons. The first exon is noncoding. The gene encodes a 14-kDa polypeptide that consists of a 24-amino acid signal peptide and the mature α subunit of 92 amino acids with 10 cysteine residues and two N-linked oligosaccharide groups.202 The cysteine residues participate in the intrasubunit disulfide linkages. The α subunit is more abundant than the β subunit, and unassociated, or “free,” α subunits are present in the serum and pituitary. They have little known biologic activity. Hence, only the αβ heterodimer possesses biologic activity.
β Subunit
The β subunits for LH and FSH are encoded by separate genes and are located on different chromosomes. The gene coding for the LHβ subunit consists of three exons and is present in a complex gene cluster on human chromosome 19q13.3.203 The cluster includes six chorionic gonadotropin β (CGβ) genes that are presumably derived from single ancestral LHβ gene by gene duplication.204 Both LHβ and CGβ proteins are structurally and functionally similar. They are approximately 80% homologous in amino acid sequence. Both propeptides contain a 20-amino acid signal sequence. The mature LHβ and CGβ subunits consist of 121 and 145 amino acids, respectively. The major difference between LHβ and CGβ proteins is the presence of a 24-amino acid C-terminal peptide in CGβ, which is heavily glycosylated, with four O-linked carbohydrate moieties (Fig. 2-10).
The gene for the FSHβ subunit is located on the short arm of chromosome 11p13 and, like the gene for LH, it consists of three exons.205 The molecular size of FSH is 33 kDa, consisting of a signal peptide of 18 amino acids and the mature FSHβ protein of 111 amino acids. Like α subunits, both LHβ and FSHβ subunits contain 10 cysteines for disulfide formation. Compared to FSHβ and CGβ subunits, LH does not have a terminal sialic acid on its carbohydrate side chain. This results in a shorter metabolic clearance time for LH, compared to FSH and HCGβ subunits. HCGβ has the highest content of sialic acid and has the longest half-life.206,207 Deglycosylation of gonadotropins has no effect on receptor binding but abolishes signal transduction.208
Mutations in Gonadotropin Genes
Although rare, mutations in gonadotropin genes can result in clinical disorders, which have been described in the literature. In fact, the subject has been recently reviewed extensively.201,209 There are several reports of neutral polymorphisms, but no activating mutations have been reported so far in the α subunit gene.209
Examples of mutations in the LHβ subunit include a single amino acid substitution (Glu to Arg) at codon 54210 that is associated with hypogonadism in homozygous males and with a high incidence of infertility in heterozygotes. In addition two point mutations—Trp to Arg at codon 8 and Ile to Thr at codon 15—have been described in five women with immunologically anomalous LH but with hyperbioactivity.211 However the direct relationship of these mutations with a specific pathogenesis remained elusive. The other variant of LHβ (polymorphism) with substitution of Serine to Glycine at codon 102 in exon 3 has been found to be population-specific in 4% of women in Singapore with menstrual disorders.
Several inactivating mutations have been described in the FSHβ subunit gene (see Fig. 2-9). The first mutation reported was a homozygous 2bp deletion in a codon 61 (Valine) in a woman who presented with delayed puberty, amenorrhea, and infertility.212 The mutation was a nonsense mutation leading to altered amino acid sequence after codon 60, leading to a stop codon at residue 87. The mutated protein was altered and was thus unable to associate with the α subunit and was nonfunctional. The patient was treated with exogenous FSH, leading to follicle maturation and pregnancy. Another case report on FSH mutation with a similar phenotype was found to be heterozygote for two FSH mutations: the first was a nonsense mutation at codon 61 and the second was a substitution of Cysteine to Glycine at codon 51. The loss of the cysteine residue was likely responsible for the altered conformation change and loss of function leading to the phenotype.213 An inactivating mutation leading to the substitution of Cysteine to Arginine residue at codon 82 was described in an infertile man.214
Measurement of LH and FSH
It has been difficult to develop highly specific immunoassays for the gonadotropins due to the high homology of the glycoprotein hormones as well as the need to distinguish between free α subunits and intact hormones. Cross-reactivity with the α subunit has made it difficult to accurately measure LH and FSH with RIAs that use polyclonal antibodies.215–217 In addition, there is microheterogeneity in both pituitary and circulating gonadotropins due to the degree of glycosylation. Variation in oligosaccharide content and structure also causes charge microheterogeneity, resulting in serum isoforms that separate over the pH range of 6.5 to 10 on electrofocusing.218 Although the clinical significance of this microheterogeneity of circulating isoforms remains unknown, it affects immunoreactivity as detected by various antibodies, all of which lead to variability among different immunoassays. Microheterogeneity also affects the biologic activity and may be primarily responsible for the discrepant results generated by immunoassays versus bioassays.
In recent years, two-site directed immunoradiometric (IRMA) or immunochemiluminometric (ICMA) assays have been developed and are based on the use of two monoclonal antibodies. These have helped overcome most limitations of RIAs.219 These assays are automated and show exquisite sensitivity approaching 0.1 mIU/mL. Furthermore, these assays are not affected by the presence of free α subunits and correlate better with bioassays. The high sensitivity of these assays also allows detection of low levels seen in early puberty.
Luteinizing hormone bioassays utilize dispersed mouse or rat Leydig cells or a Leydig cell tumor cell line (MA-10) in culture220,221 and measure testosterone production in vitro. These assays measure the biologic activity of circulating LH under physiologic conditions. This is important because the biologic activity of LH changes with alterations in glycosylation and the tertiary structure of the molecule. The combination of bioassay and RIA permits the calculation of bioactive/immunoreactive ratios, which can provide a useful index of qualitative changes of the LH molecule.219,222 Although the bioactive and immunoactive LH profiles are generally well correlated during physiologic changes, significant discrepancies can occur in some pathologic states. For example, inactivating mutations in the LHβ gene result in elevated levels of immunoreactive LH but a marked loss of bioactivity. In vitro bioassays for FSH activity using rat granulosa cells or Sertoli cells measure production of cAMP or aromatase activity in response to FSH. The sensitivity of these assays is about 2.5 mIU/mL. Alhough in vitro bioassays have been valuable in elucidating the physiology, they remain cumbersome and time consuming and are not practical for routine clinical use.
Measurement of LH and FSH is useful in the diagnosis of gonadal function disorders (Table 2-5). Elevated levels of FSH generally indicate ovarian failure but may be seen in some patients with viable ovarian follicles.223 Although rare, high gonadotropin levels associated with gonadotropin-secreting pituitary tumors or ectopic gonadotropin-producing tumors. In an amenorrheic patient, an elevated LH level with normal FSH and LH-to-FSH ratio typically (but not invariably) of greater than 2 is suggestive of PCOS. Low levels of these hormones are indicative of pituitary or hypothalamic dysfunction and occur together with low serum estradiol levels. For further assessment of pituitary reserve, provocative GnRH testing is required. LH and FSH responses to intravenous injection of 100 μg GnRH are measured at 20 and 60 minutes. Lack of response may suggest the likely diagnosis of hypogonadotropic hypogonadism; however, its sensitivity and specificity are low in patients receiving exogenous sex steroids.224
Table 2-5 Role of Pituitary Hormones in Assessment of Female Infertility
| Hormone | Hormone Levels | Interpretation |
|---|---|---|
| Prolactin | ↑Prolactin | Evaluate for prolactinoma after excluding hypothyroidism, pregnancy, macroprolactinemia as cause. |
| LH and FSH | ↓ LH, ↓ FSH | Hypothalamic or pituitary disease |
| ↑ LH, ↑ FSH | Premature ovarian failure | |
| ↑ LH, ↓ or normal FSH | Polycystic ovary syndrome |
Prolactin
Biochemistry and Biosynthesis
It is produced by lactotrophs in the pituitary, which make up almost 50% of the total pituitary cell population. Its production is under tonic inhibition by dopamine, produced by the tuberoinfundibular cells, and the hypothalamic tuberohypophyseal dopaminergic system. Prolactin is extremely heterogeneous and exists in at least four different molecular forms225–227: (1) little prolactin, molecular weight (MW) 23 kDa, a nonglycosylated monomeric hormone with high receptor binding and bioactivity; (2) G, or glycosylated prolactin, MW 25 kDa, which has reduced immunoreactivity; (3) big prolactin, MW 50 kDa, consisting of a mixture of both dimeric and trimeric forms of G prolactin; and (4) big-big prolactin, MW 100 kDa, consisting of G prolactin covalently coupled with an immunoglobulin, also known as macroprolactin. The big and big-big forms have lower receptor-binding affinity, but may be converted to little prolactin by reduction of the disulfide bonds. As such, discrepancies can exist between measured prolactin levels and the clinical effects.
Measurement
Prolactin levels are measured by use of IRMA and ICMA. These methods give excellent reproducibility, sensitivity, and assay efficiency; however, they vary in their abilty to react with biologically inactive macroprolactin. Hence, a measured serum immunoreactive prolactin level often does not correlate with expected clinical effects. A polyethylene glycol precipitation method should be used to detect the macroprolactinemia.228 The other caveat is that these samples are usually assayed at a single dilution. As such, extremely high levels of prolactin (≈1000 ng/mL), such as those seen in macroprolactinomas, may saturate both capture and localizing antibodies, leading to a falsely low value in some one-step sandwich immunoassays. This has been known as the hook effect. Thus, in patients with macroadenomas, a 1:100 serial dilution should be performed if the assay is prone to this hook effect.
MECHANISMS OF ACTION OF HORMONES
Just on the basis of the location of the hormone receptor (i.e., intracellular/nuclear or cell surface), two distinct mechanisms of hormone actions can be classified. These mechanisms further differ by the nature of the signal transduction pathway or second messenger responsible for mediating hormone action (Table 2-6). Examples of nuclear receptors include steroid hormones that are lipophilic and pass through the cell membrane to interact with receptors located either within the cytoplasm or the nucleus. This in turn affects gene transcription within the nuclear compartment. Polypeptide hormones (i.e., LH, FSH, hCG, GnRH, inhibins, and activins) and growth factors that are hydrophilic interact with cell-surface receptors that are located on the plasma membrane. They trigger a plethora of signaling activity in the membrane and cytoplasmic compartments as well as exerting parallel effects on the transcriptional apparatus in the nuclear compartment. These cell surface receptors can be further classified based on the second messenger into four major subgroups, as listed in Table 2-6.
Table 2-6 Classification of Receptors for Steroid and Peptide Hormones
| Hormones that Bind to Intracellular/Nuclear Receptors | Hormones that Bind to Cell Surface Receptors |
|---|---|
Steroid Hormone Action
Nuclear Receptors Superfamily
Steroid hormone nuclear receptors (estrogen receptor [ER], progesterone receptor, and androgen receptors) are ligand-inducible transcription factors that regulate the expression of target genes involved in reproduction and metabolism. They belong to the superfamily of nuclear hormone receptors and share many structural and functional features.229 Other members of the superfamily include receptors for glucocorticoids, mineralocorticoids, thyroid hormones, 1,25-dihydroxy vitamin D3, retinoic acid, and an ever-increasing number of orphan receptors, which show structural similarity but for which ligands are not known.
Within this nuclear receptor superfamily, three main groups have been identified based on the differences in their functional and recognition characteristics230: type 1, or steroid receptor subclass; type 2, or thyroid/retinoid/vitamin D3 receptor subclass; and a third subclass of orphan receptors.
Type 1 (“Steroid” or “Classical”) Receptor Subfamily
This includes the ER as well as the progesterone, androgen, glucocorticoid, and mineralocorticoid receptors. These receptors cannot bind to DNA in the absence of ligand and thus remain functionally silent. They exist as cytoplasmic/nuclear, multimeric complexes that are in association with heat shock proteins (e.g., HSP90, HSP70, and HSP56). The association of the ligand with the receptor and dissociation of the heat shock proteins are required for activation of the receptor.
Type 2 Receptor Subfamily
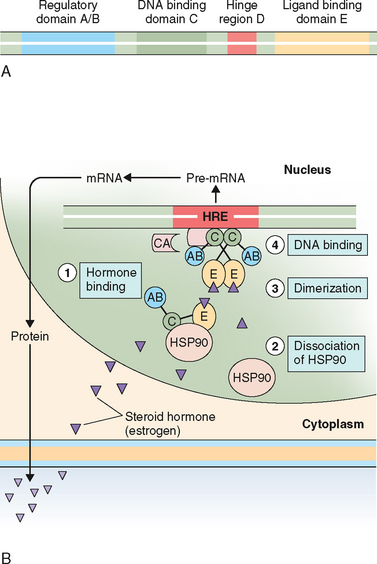
Most of the early knowledge about steroid hormone mechanism of action has been derived from in vitro and in vivo binding studies that used radiolabeled estradiol as a ligand.231 The role of ERs in the regulation of breast cancer growth has been well studied over the past four decades; it was the first steroid receptor to be discovered in the early 1960s. This led to the subsequent elucidation of a general pathway for steroid hormone action. In general, nuclear receptors share a common protein architecture consisting of five functional domains232 as illustrated in Figure 2-10A.
Cellular Mechanism of Action of Steroid Hormones
The steroid hormone nuclear receptors are known as ligand-dependent transcription factors. Binding with their ligands is a necessary step for their function as transcriptional regulators. Unliganded receptors may be localized to either the cytoplasm (e.g., glucocorticoid receptor) or the nucleus (e.g., estrogen, progesterone, and thyroid hormone receptors). For most steroid hormones the unliganded receptors exist in the cell nucleus as large molecular weight oligomers (≈300K; 7-10S sedimentation rate)233 and can be isolated in cytosolic fraction from cells or tissues disrupted in hypotonic media. The oligomers are formed by noncovalent association of a monomeric receptor protein with a dimer of heat shock protein (HSP90, HSP70, or HSP56).234
The general features of the mechanism of action of these hormones are depicted in Figure 2-10B. Steroid hormones that freely diffuse through the cell membrane bind to the specific receptors in the nucleus. Ligand binding to receptor initiates the receptor transformation, or so-called activation process. During this process the receptor undergoes conformational changes that primarily occur as a result of its dissociation from HSP, which exposes the DNA-binding site. Nuclear translocation and dimerization of the activated receptor then occurs. Most evidence suggests that this process is thermodynamically irreversible. The hormone receptor complex then binds to a specific region of DNA, the HRE, which is located upstream of the gene. The first HRE was identified for the glucocorticoid receptor. Later the HREs for the progesterone, androgen, estrogen, and mineralocorticoid receptors were shown to be similar to that of the glucocorticoid receptor.235–237 The steroid HREs in the target genes are a palindromic (inverted-repeat) DNA sequence of 15 base pairs (Table 2-7). This interaction leads to the recruitment of a host of ancillary factors known as coregulators (coactivators or corepressors), creating a transcriptionally permissive or nonpermissive environment at the promoter, as well as communicating with other general transcription factors and RNA polymerase II. Coactivators function as adaptors in a signal transduction pathway. The binding of these coregulators modulates the resulting transcription (i.e., the activation and inactivation of specific genes). Hormone antagonists, for example, induce a different conformation in the TAF-2 that hinders the coactivator-binding site and recruits a corepressor instead, and inhibits gene expression. The availability of these coregulators in different tissues plays an important role in defining the biologic response to both steroid hormone agonists and antagonists.238
Table 2-7 Sequence of DNA Recognition Elements for Steroid Hormone Receptors
| Steroid Hormone Receptor | Element | DNA Recognition Sequence |
|---|---|---|
| Estrogen receptor | ERE |


Sequences read 5′-3′ direction indicated by arrows. S indicates spacer nucleotides (A, G, C, or T).
The Estrogen Receptor
Structure and Function
The structure of ER (now known as ERα) was reported in 1986.239 It consists of five components or domains that are divided into six regions, referred to as A-F (Fig. 2-11), instead of the five regions seen in most steroid receptors. The F region is a C-terminal segment of 42 amino acids that influences the conformational changes that occur after estrogen/antiestrogen binding. Thus, it modulates the level of transcriptional activities, most likely by affecting the interaction with coregulator proteins. It has a molecular weight of 66,000 and contains 595 amino acids. ER mRNA is 6.8 kilobases and contains 8 exons derived from a gene located on the long arm of chromosome 6. More recently a second form of ER has been discovered and named ERβ; it is encoded by a gene located on chromosome 14240 and is in close proximity to the genes that are related to Alzheimer’s disease.241
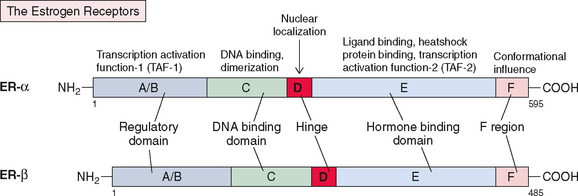
The two receptors show a high degree of homology in the DNA-binding domain (97%) and ligand-binding domain (59%) but less so in hinge (30%), regulatory (17%), and F regions (17.9%)241,242 (see Fig. 2-11). Hence, the binding characteristics of these two receptors are similar, although they differ significantly in their ability to activate gene transcription by regulatory domain TAF-1, which is minimal or absent in ERβ. Both ERα and ERβ are required for normal ovarian function, as shown by specific receptor knockout studies in mice.16 ERα is primarily responsible for estrogenic effects in other tissues, including the uterus.
The two receptors (ERα and ERβ) are differentially expressed in different tissues, leading to the differences in the response to the same hormone subtypes.243,244 The α receptors are predominantly expressed in breast cancer tissue, ovarian stroma, and endometrium. ERβ receptors, on the other hand, are expressed in several nonclassic target tissues, including the kidney, intestinal mucosa, lung, bone, brain, endothelial cells, and the prostate gland. 17β-estradiol and estrone have a higher affinity for α receptors and thus exert their effects predominantly on target tissue with α receptor expression. Phytoestrogens such as genistein and coumestrol, on the other hand, bind predominantly to β receptors245 and would be expected to exert their effects on target tissues expressing these receptors.
The conformational change of the ligand-binding domain also differs in both α and β estrogen receptors, depending on which ligand has been bound to the receptor.244 This distinct conformational change is the major factor that determines the receptor’s ability to interact with coactivators or corepressors. For example, estradiol activates transcription when it binds to ERα but inhibits transcription when bound to ERβ. Raloxifene and tamoxifen, on the other hand, inhibit transcription when forming complexes with ERα and activate transcription when bound to ERβ.
The differential expression of target tissue adaptor proteins and phosphorylation also affect gene transcription. A higher concentration of coactivators or corepressors in the target tissue can affect the cellular response of that tissue to the same ligand. Phosphorylation of the receptor by protein kinases increases the transcriptional activity of the receptor. For example, growth factors such as epidermal growth factor and IGF-I can stimulate protein kinase phosphorylation, activating the estrogen receptor—even in the absence of estrogen.
Somatic mutations in ERα are described and may be associated with certain disease states. A nonsense mutation (premature stop codon) in ERα has been described in a patient with decreased bone mineral density, increased bone turnover, and incomplete closure of bone epiphyses, which demonstrates the role of the ERα in bone growth and homeostasis.246 ER mutations have also been detected in patients with breast cancer. Such mutations include exon 5 deletion within the ligand-binding domain, leading to a constitutively active receptor, and exon 7 deletion, displaying dominant negative activity and inhibition of ER function.247
Antiestrogens and ER
Compounds with antiestrogenic activity can be classified into two categories: those with pure antiestrogenic activity and those with both agonist and antagonist properties. Tamoxifen—an antiestrogen that is used both as a chemopreventive agent and as a hormonal therapeutic agent for breast cancer—inhibits ER action.248
Paradoxically, tamoxifen acts as an estrogen in uterine tissue, and this tissue-specific estrogenic effect is the reason why prolonged tamoxifen therapy increases the risk of uterine cancer.249 Raloxifene, a related benzothiophene analog, retains its antiestrogenic effect in breast and uterine tissue. Both tamoxifen and raloxifene have estrogen-like effects on nonreproductive tissues such as bone and heart and lung tissue.250
Tamoxifen acts by competing with estrogen for receptor binding. Because estrogen-binding affinity is of higher magnitude than tamoxifen, severalfold higher concentrations of tamoxifen are required to inhibit estrogen action. The agonistic or antagonistic effect of tamoxifen is determined by the presence of different promoter elements in the specific cell type.251 Estrogen binding to receptors activates both transcription domains (i.e., TAF-1 and TAF-2). Tamoxifen’s agonistic activity is due to activation of TAF-1, and its antagonistic activity is due to its ability to inhibit the estrogen-dependent activation of TAF-2. The ligand-binding sites for estrogen and antiestrogen are not identical, and tamoxifen binding on receptors induces conformational changes that alter interaction with estrogen-associated proteins and modulates transcriptional activity.251,252 Tamoxifen also activates ER-mediated induction of promoters that are regulated by the TAF-1 site, which explains why it has estrogenic effects on the endometrium—a tissue with significant TAF-1 transcription function. In other cell types, such as those in the breast, TAF-1 has weak transcriptional activity. Hence, antiestrogens have no effect on TAF-1 mediated transcription.253 Raloxifene, on the other hand, may activate estrogen-responsive genes through a response element that is distinct from HRE.254
Pure antiestrogens are derivatives of estradiol that have a long hydrophobic side chain at position 7. Examples of pure antiestrogens include ICI 164,384 and ICI 182,780 (Fulvestrant). Binding of pure antiestrogens may sterically interfere with the dimerization process and thus inhibit DNA binding. Furthermore, these compounds increase the rate of receptor degradation and also inhibit ER-mediated transcription by preferential binding to corepressors, which contributes to their antiestrogenic activity.255,256
Progesterone Receptor
Structure and Function
As with the estrogen receptor, there are two major forms of progesterone receptors— PR-A and PR-B—which are derived from the same gene. PR-A and PR-B are identical except that PR-B contains an additional 164-amino acid sequence at the N-terminal end, which is referred to as the B-upstream segment (BUS) (Fig. 2-12). PR-A has a molecular weight of 94 kDa and contains 768 amino acids; PR-B is 114 kDa with 933 amino acids. The two forms derive from two distinct estrogen-regulated promoters.257 The transcription function domain (TAF-1) in the progesterone receptor is located in the 91-amino acid segment of the regulatory region, and TAF-2 is located in the hormone-binding domain. In PR-B, the BUS contains a third activation domain, TAF-3, which can synergize the actions of other TAFs or autonomously activate transcription.258 TAF-3 recruits and allows a separate subset of coactivators to bind with PR-B that do not interact efficiently with PR-A. Thus, PR-A and PR-B display different transactivation properties that are both cell specific and target gene promoter specific.259 The two isoforms PR-A and PR-B have distinct cellular localization and in the absence of ligand binding, PR-A is predominantly localized in the nucleus and PR-B is present in the cytoplasm.260
The role of progesterone receptor isoforms is not yet fully elucidated. Selective ablation of PR-A expression in mice resulted in severe abnormalities in ovarian and uterine function that led to infertility but did not affect progesterone responses in the mammary gland or thymus.261 In contrast PR-B ablation does not affect ovarian, uterine, and thymic responses to progesterone and manifests as reduced mammary ductal morphogenesis. Thus, PR-A is essential for female fertility; in the absence of PR-A, PR-B functions in a tissue-specific manner and mediates some of the progesterone receptor actions in the mammary gland.261 However, transgenic mice carring an extra copy of the PR-A gene have abnormal mammary gland development, indicating that overexpression of PR-A may have physiologic significance.
The relative levels of the two isoforms differ in the endometrium during the menstrual cycle.262 In the uterus, progesterone action downregulates cell-cycle arrest proteins but upregulates growth factors and their receptors and other regulators263 and is also essential for the initiation and maintenance of pregnancy. Progesterone antagonist RU-486 (mifepristone) initially was synthesized as a glucocorticoid receptor antagonist264 but later was found to display marked antiprogesterone activity.265 It binds to the glucocorticoid recptor with threefold higher affinity than dexamethasone and to the progesterone receptor with a fivefold higher affinity than natural progesterone.266 Unlike progesterone, the RU-486–progesterone receptor complex inhibits transcription because it has a slightly different conformational change in the TAF-2 domain.267 If implantation occurs, it downregulates progesterone-induced genes and results in decidual necrosis and detachment of the conception products.251,266
Androgen Receptor
Structure and Function
The androgen receptor gene was cloned in 1988 and was localized on the human X chromosome between the centromere and q13.268 Like the progesterone receptor, the androgen receptor also exists in two forms: a full-length B form and a shorter A form (molecular weights ≈110 kDa and 87 kDa); both are encoded by the same gene.269 The 87-kDa isoform (AR-A) contains the intact C-terminus but lacks 188 amino acid residues in the N-terminus of the 110-kDa isoform (AR-B) (Fig. 2-13). The ratio of AR-B to AR-A in genital skin fibroblasts from healthy subjects is 10:1. It is not known whether there are functional differences between these isoforms.269 The DNA-binding domain or TAF-2 of the androgen receptor is similar to the TAF-2 regions of other steroid hormone receptors (progesterone, estrogen, glucocorticoid, and mineralocorticoid receptors) but is related most closely to the progesterone receptor.270 Progesterone shows cross-reactivity with androgen receptors, to a degree that becomes clinically relevant only at pharmacologic doses.
A large number of androgen receptor mutations that can alter receptor function have also been described271,272; for example inactivating point mutations in the hormone-binding domain of the androgen receptor can generate different phenotypes that lead to partial to complete androgen insensitivity. A point mutation at residue 689 that substitutes Proline to Histidine may alter the conformation of the ligand-binding domain, which reduces the androgen receptor’s affinity for DHT and abolishes its ability to transactivate the HRE.273 A Serine to Proline substitution at residue 865 eliminates androgen binding and transactivation; thus it also causes complete androgen insensitivity.274 At residue 807, a substitution of Threonine for Methionine causes partial androgen insensitivity by reducing—but not abrogating—the androgen binding to the receptor. However, a Valine or Arginine substitution at the same site totally abrogates androgen binding and causes complete androgen insensitivity syndrome.275
Nongenomic Actions of Steroids
Some steroid hormone actions are independent of their classic genomic actions, as mediated via nuclear receptors—these are called nongenomic actions.276 These effects are rapid, occur within seconds, and are not affected by inhibitors of gene transcription such as dactinomycin or inhibitors of protein synthesis such as cycloheximide. These rapid actions include sodium and calcium ion transport and some neural and cardiovascular effects.277 The messenger and effector system vary with the steroid and the cell type. Thus far, studies have suggested that specific binding sites or receptors are present on the cell membrane and that steroid binding triggers rapid changes in the electrolyte transport system. For example, estrogens have been shown to modulate some cardiovascular effects by inducing Ca2+ flux and vasodilation in the coronary artery.278 Furthermore, steroids can activate second-messenger pathways, generating second messengers capable of altering gene transcription independently of their classical receptor-mediated gene transcription.279,280 Thus, steroid hormones have nongenomic effects as well as genomic effects that are mediated by both classical steroid receptor-mediated pathways and second-messenger pathways.
Tropic Hormone Action
Tropic hormones (i.e., gonadotropins and GnRH) are primarily hydrophilic and depend on their interactions with receptors scattered on the plasma membrane at the cell surface. These receptors, in turn, can be classified into four groups based on their structural homology and type of intracellular messenger involved in activating the signal transduction pathway (see Table 2-6).
G Protein-Coupled Receptors
Gonadotropin Receptors
The gonadotropin receptors are primarily expressed in the gonadal tissues and demonstrate a unique expression pattern that determines their cell-specific roles. LH receptors are present in ovarian thecal and luteal cells and in testicular Leydig cells. FSH receptors are found in ovarian granulosa cells and in testicular Sertoli cells. Furthermore, FSH induces LH receptors in the granulosa cells that are expressed in mature follicles. These receptors are essential in mediating the gonadotropin actions in steroidogenesis and in gonadal growth and differentiation. Their structure, function, and regulation as well as molecular biology have been extensively covered in recent reviews.281,282
Structure and Function
Genes for both LH and FSH receptors are located on chromosome 2p21. The LH receptor gene is about 70 kb and consists of 11 exons and 10 intervening introns.281 The FSH receptor gene is about 54 kb and contains 10 exons and 9 intervening introns.282 As shown in Figure 2-14, the structural similarities between the two receptors are remarkable with the exception of an additional exon in the LH receptor. The long exon 11 in LH receptor and exon 10 in FSH receptor encodes for the seven-transmembrane domains and the intracellular tail as well as the C-terminal end of the hinge region of ectodomain. The remaining 9 exons in the FSH receptor and 10 exons in the LH receptor encode for the entire ectodomain. Both receptors have multiple splice sites, leading to transcription of multiple mRNA splice variants and expression in the ovary and testis.281
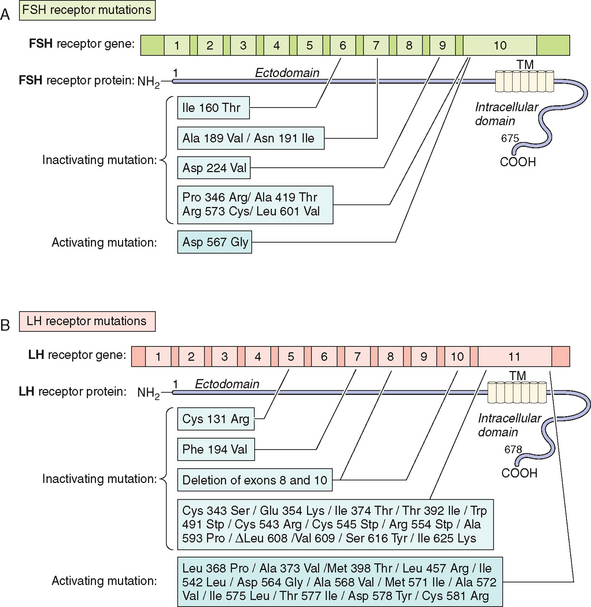
The LH receptor protein contains a 24-amino acid signal peptide (17 in FSH receptor) and the mature protein consists of 675 amino acid residues (678 in FSH receptor) (see Fig. 2-14). The molecular mass is approximately 85 to 90 kDa for both glycosylated LH and FSH receptor proteins. The ectodomains of these receptors are composed of several leucine-rich repeats (LRRs) of about 24 amino acid residues each (9 in LH receptor and 10 in FSH receptor) that form a half donut-shaped structure and are essential for the binding of gonadotropins. The ectodomain is connected to the transmembrane region by the hinge region with conserved sequences. The transmembrane region consists of seven helical structures and is followed by the cytoplasmic C-terminal tail, both of which are important for interaction with intracellular proteins. The binding of LH and FSH to their specific receptors leads to receptor activation and downstream signaling.
Mechanism of Action
The intracellular messenger for LH and FSH is cAMP, which is derived from adenosine triphosphate (ATP) through the action of the enzyme adenylate cyclase. The basic steps involved in the receptor-mediated action of gonadotropins are illustrated in Figure 2-15. The regulation of adenylate cyclase is mediated by a GTP-dependent regulatory G-protein, each of which is composed of three subunits—α, β, and γ—and is associated with the receptor in an inactive GDP-bound form. Gonadotropic binding to their respective receptors induces a conformational change in the receptor and activation of the G-protein complex that leads to release of GDP and binding of the α subunit to GTP.283 Subsequently, the GTP-bound form of the α subunit disassociates from the receptor as well as from the stable β/γ dimer and activates the adenylate cyclase. This in turn leads to an increase in the levels of intracellular cAMP, which in turn activates protein kinase A (PKA) by binding to the inhibitory regulatory subunit of PKA and causing it to dissociate from the complex. Activated PKA phosphorylates many cellular substrates, including several nuclear transcription factors (e.g., cAMP response binding protein [CREB]).281 The binding of CREB to the cAMP response element activates many genes. The fact that PKA activation does not account for all the actions of gonadotropins and that LH can stimulate steroid hormone synthesis without significant changes in cAMP indicated that another pathway may be activated. There is now evidence that the phosphatidylinositol 3,4,5-triphosphate (IP3) pathway is also activated by the LH receptor284 as well as by the FSH receptor,285 although it is not clear whether the IP3 pathway activates the same or different responses in the target cells. The third signaling pathway, the MAPK pathway, has also been shown to be activated by the LH receptor286 (see Fig. 2-15).
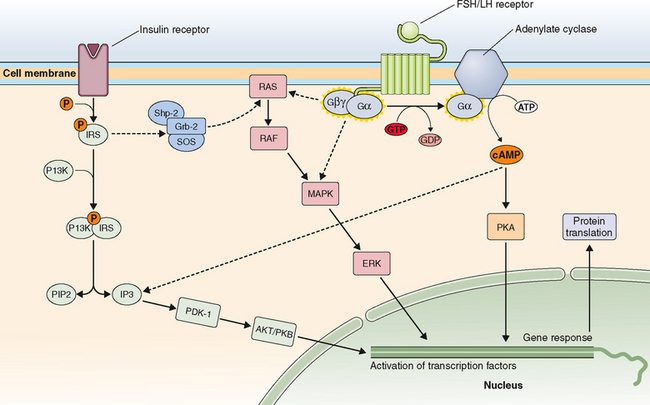
Continuous stimulation of receptors can lead to a decrease in response, a phenomenon known as downregulation. For example, pulsatile LH secretion maintains LH receptors and steroidogenesis in the gonads. However, persistent endogenous elevation of LH or hCG levels downregulates LH receptors and leads to desensitization to the hormonal signal.287 This involves receptor phosphorylation, which uncouples the receptor from the G protein, ending the response. The LH/hCG receptor undergoes desensitization in response to LH or hCG by the phosphorylation of the C-terminal cytoplasmic tail of the receptor.288 Another mechanism of downregulation is the uncoupling of the regulatory and catalytic subunits of the adenylate cyclase enzymes. For example, LH stimulates steroidogenesis by coupling the stimulatory to the catalytic units of adenylate cyclase. Prostaglandin F2α, on the other hand, inhibits the action of LH by an inhibitory regulatory unit that uncouples the catalytic unit to interfere with gonadotropin action.
Mutations in Gonadotropin Receptors
Both activating and inactivating gene mutations have been described in LH and FSH receptors and have been the subject of recent review.209 Interestingly, the activating mutations in the LH receptor gene result in altered testosterone production, and these mutations are known to affect the phenotype of men only, whereas inactivating mutations affect sexual differentiation and fertility in both men and women.
To date, a total of 15 activating mutations have been identified in the LH receptor, all localized in the transmembrane region or in the intracellular loops (see Fig. 2-14A). Transmembrane 6 and the third intracellular loop are recognized as mutational hot spots, and 10 of 14 mutations are localized to these regions. These gain-of-function mutations lead to male-limited precocious puberty characterized by Leydig cell hyperplasia, premature puberty, and onset of spermatogenesis in boys as young as age 3 years.289,290 These patients have blood testosterone levels in the pubertal range, with low or undetectable LH levels. A novel activating LH receptor mutation associated with Leydig cell tumors and nonfamilial precocious puberty has also been described.291
An equal number of inactivating mutations in LHR that cause partial to complete inactivation of receptor have been described.209 These inactivating mutations in LH receptors cause Leydig cell hypoplasia in men and amenorrhea in women. In contrast, loss-of-function mutations are scattered throughout the LH receptor protein. Depending on the mutation, the severity of the male phenotype varies from primary hypogonadism to male pseudohermaphroditism with sexual ambiguity.
Conversely, activating mutations of the FSH receptor are rare. Only one mutation has been identified this far (located in the third intracellular domain of exon 10) in a hypophysectomized patient who was fertile despite the lack of FSH and LH production.292 Likewise, fewer inactivating mutations have been identified in the FSH receptor (see Fig. 2-14B). Most of these are located in the extracellular domain and show reduced ligand binding and signal transduction. They are implicated in hypergonadotropic ovarian dysgenesis in females and variable degrees of spermatogenic failure in males. Recently, two heterozygous missense mutations in the exon 10 encoding the transmembrane region have been described, and both were associated with the familial spontaneous ovarian hyperstimulation syndrome. Although there were no significant differences in the cAMP responses between the wild-type and mutated receptor, the mutated receptor gained sensitivity to hCG.293,294 Low-affinity binding of hCG to the FSH receptor most likely triggered signal transduction that led to hyperstimulation of ovaries.
Gonadotropin-releasing Hormone (GnRH) Receptor
GnRH binds to specific membrane receptors in pituitary gonadotrophs and stimulates LH and FSH secretion (Fig. 2-16). Besides gonadotrophs, GnRH receptors have also been expressed on the gonads, placenta, and brain295–297 Specific GnRH receptors are also characterized in immortalized αT3-1 gonadotroph cells as well as in GnRH-secreting GT1 hypothalamic cells.296,298 In the ovary, GnRH receptors are expressed in granulosa and luteal cells. In the testis, these receptors are expressed in Leydig cells but not Sertoli cells.299 In cultured granulosa cells, the receptor activation stimulates progesterone and prostaglandin synthesis, oocyte maturation, and ovulation300,301 but inhibits FSH-induced steroidogenesis, follicular development, and maturation as well as inhibin secretion.302–304 In luteal cells, it inhibits LH receptor expression and thus LH action.305
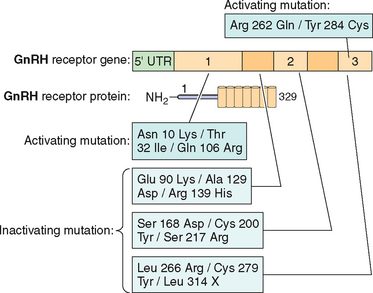
The structure function and intracellular signaling pathways involved in GnRH action have been extensively reviewed in recent years.306–308 The GnRH receptor gene, located on chromosome 4q21.2, consists of three exons309 and encodes a 327-amino acid protein with an approximate molecular weight of 50 to 60 kDa. The receptor has the characteristic features of GPCRs (Fig. 2-17). It consists of an aminoterminal domain, followed by seven transmembrane domains that are connected by three extracellular and three intracellular loops. Unlike other GPCRs, the GnRH receptor lacks the characteristic C-terminal cytoplasmic domain. GnRH binds to the transmembrane domain and the extracellular loop in the hairpin structure and requires partial entry of its N- and C-terminal regions into the transmembrane core of the receptor.310
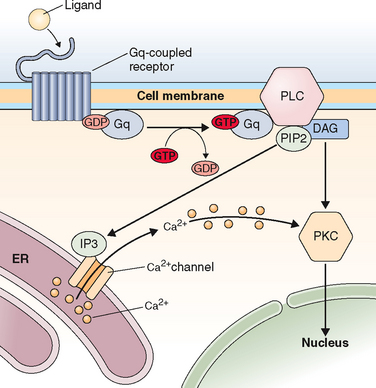
The nature of the intracellular signaling mechanism by the GnRH receptor has been primarily studied in a gonadotroph-derived αT3-1 cell line, as depicted in Figure 2-18. The effector system in the GnRH receptor is the phospholipase C β (PLCβ) and the second messengers are IP3 and 1,2 diacylglycerol (DAG). Its mechanism of action is dependent on calcium. Activation of the receptor by ligand binding initiates a series of steps that lead to transduction of signals. The first step is G protein (Gq/11)-mediated activation of enzyme PLCβ, leading to hydrolysis of phosphoinositides (PIP2) and resulting in the production of IP3 and DAG. The IP3 interacts with a receptor on the endoplasmic reticulum to promote the oscillatory or biphasic release of Ca2+ from intracellular stores, which is known to be an important trigger for gonadotropin secretion. The increased Ca2+ and DAG in turn activate a series of protein kinase C subspecies (PKC). PKC then induce various downstream signal transduction cascades, including the extracellular signal regulated kinase and Jun N-terminal kinase signaling pathways.
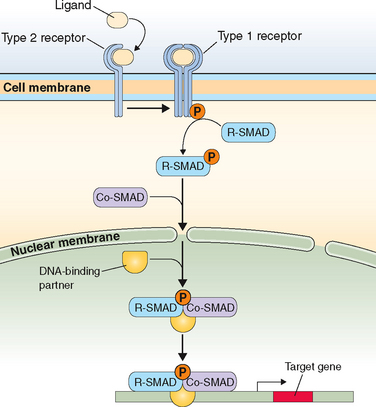
Mutations in GnRH Receptor Gene
A functional GnRH receptor is a prerequisite for normal LH and FSH production. Hence, it is critical for normal pubertal development and reproduction. Several inactivating mutations have been identified in the GnRH receptor gene that are associated with variable phenotypes ranging from modest to complete insensitivity to GnRH. These mutations are present in a number of patients with idiopathic hypogonadotropic hypogonadism (IHH) with an autosomal recessive pattern of inheritance as well as in some patients with sporadic IHH.311 Most identified mutations are heterozygous missense mutations and are scattered throughout the GnRH receptor gene. A total of 14 mutations have been identified to date308 and are listed in Figure 2-16. In vitro studies have shown that some of these mutations make the receptor totally nonfunctional; others have some ability to elicit a response to GnRH.
Peptide Hormone/Growth Factor Action
Pituitary gonadotropin signaling plays a key role in follicular growth, ovulation, and luteinization. However, it is increasingly recognized that their actions are also dependent on their interaction with peptide/growth factor signaling pathways, including IGFs, EGF, and members of the TGFβ family (see Fig. 2-15). Understanding these pathways provides insight into how these factors interact and complement the FSH/LH pathway to control follicular growth.
Family of Receptors with Tyrosine Kinase Activity
All tyrosine kinase receptors have a similar structure: an extracellular domain for ligand binding, a single transmembrane domain, and a cytoplasmic domain. Ligand specificity is determined by the unique amino acid sequences making up the extracellular domain, which determines the three-dimensional conformation of the receptor. The transmembrane domains are heterogeneous, whereas the cytoplasmic domains are fairly homologous. They respond to ligand binding by undergoing conformational changes and autophosphorylation. The structure of the IGF-I receptor is strikingly similar to that of the insulin receptor, with two transmembrane domains linked by disulfide bridges, formed by two α and β subunits.312 The IGF-I receptor gene is located on chromosome 15 at bands q25-26 and contains 21 exons.313
The steps involved in signal transduction by IGF-I/insulin have been extensively reviewed in recent years314,315 and are illustrated in Figure 2-15. The association of the ligand (e.g., insulin) with the receptor’s extracellular domain triggers receptor dimerization. This results in the phosphorylation of tyrosine residues on both the receptor and nonreceptor substrates. The phosphorylation of receptor tyrosine residues occurs at specific locations, which causes these sites to associate with a variety of accessory proteins that have independent signaling capabilities. These accessory proteins include phospholipase Cγ, PI3 kinase (PI3K), GAP, and growth factor receptor-bound protein-2 (GRB2). These interactions are mediated by the presence of highly conserved type 2 src homology domains (SH2) in each accessory molecule, thus named based on their sequence homology to the src proto-oncogene. Each SH2 domain is specific for the amino acids surrounding the phosphotyrosine residues in the receptor molecule. PI3K also produces a second messenger, such as IP3, which in turn activates kinase AKT, also known as protein kinase B (PKB). Proteins phosphorylated by PKB promote cell survival.
Although these associations may trigger immediate signaling events, other accessory proteins (e.g., GRB2) may serve to construct the scaffolding for a more complex signaling apparatus, such as that seen in the RAS-RAF-MEK pathway, This pathway recruits multiple other proteins, resulting in the activation of nuclear transcription and protein synthesis (see Fig. 2-15).
Growth hormone and prolactin fall into another group of receptors that also have a single transmembrane-spanning segment and a short cytoplasmic tail. However unlike IGF-I/insulin receptors they do not possess intrinsic tyrosine kinase activity but interact with other soluble transducer molecules (e.g., Janus kinase 2) that do have tyrosine kinase activity.
Mechanism of Action of Activins and Inhibins
Activins and inhibins are members of the TGFβ superfamily and use a common general mechanism for signal transduction through serine/threonine-specific protein kinases rather than tyrosine kinases. The mechanisms involved in signal transduction by the serine/threonine kinases has been extensively reviewed in recent years.316,317 The activin receptor was first to be cloned and was later followed by other receptors.318,319 They are glycoproteins of approximately 55 kDa and consist of a 500-amino acid sequence. Two types of activin membrane receptors have been identified: type I (ActR-I) and type II receptors (ActR-II). Each contains an intracytoplasmic serine/threonine kinase domain. The steps involved in the mechanism of action of activin are depicted in Figure 2-18. Activin directly interacts with and binds to the relatively short extracellular region of ActR-II when expressed alone or in concert with ActR-I. It can also bind to other TGFβ family members (e.g., bone morphogenic proteins [BMPs] 2, 4, and 7) in concert with BMP type I receptor, which suggests that these receptors have the ability to cross-talk. Activin binding brings together two type II receptors and two type I receptors, which form a receptor complex. One of the receptor kinases phosphorylates and activates the other, which in turn phosphorylates their substrates—the SMAD proteins [the term SMAD is derived from the combination of names of two genes, the C. elegans gene called Sma and the Drosiphila gene Mad]. SMADs are a novel family of signal transducers that can be divided into three groups that include receptor-regulated SMADs (R-SMAD) and a single common SMAD (C-SMAD or SMAD4). The third group includes inhibitory SMADS that antagonize signaling. In activin/TGFβ signaling, the activated type I receptor phosphorylates ligand-specific R-SMADs (SMAD2 and SMAD3), allowing these proteins to associate with SMAD4. The complex then translocates to the nucleus as transcription cofactor. In the nucleus, the action of SMAD complex is modulated by a variety of transcription factors (DNA-binding proteins) at target DNA promoters, which leads to gene transcription.
Inhibin is a heterodimer formed between an inhibin α chain and an activin β chain, and its biologic activity is opposite that of activin. A separate receptor for inhibin has not been identified. The mechanisms by which inhibin antagonizes activin actions are not clearly understood. It has been shown that inhibin competes with activin for binding to the activin receptors but is unable to trigger signaling.320 This may be one of the mechanisms by which inhibins antagonize activin actions. Other actions of inhibin may be mediated by as-yet unidentified inhibin receptors.
SUMMARY
1 McNatty KP, et al. The production of progesterone, androgens and oestrogens by human granulosa cells in vitro and in vivo. J Steroid Biochem. 1979;11:775-779.
2 Hsueh AJ, et al. Hormonal regulation of the differentiation of cultured ovarian granulosa cells. Endocr Rev. 1984;5:76-127.
3 Knight PG. Roles of inhibins, activins, and follistatin in the female reproductive system. Front Neuroendocrinol. 1996;17:476-509.
4 Gwynne JT, Strauss JF3rd. The role of lipoproteins in steroidogenesis and cholesterol metabolism in steroidogenic glands. Endocr Rev. 1982;3:299-329.
5 O’Malley BW. Steroid hormones: Metabolism and mechanism of action. In: Yen SSC, Jaffe RB, Barbieri RL, editors. Reproductive Endocrinology. Philadelphia: WB Saunders; 1999:125-130.
6 Clark BJ, et al. The purification, cloning, and expression of a novel luteinizing hormone-induced mitochondrial protein in MA-10 mouse Leydig tumor cells. Characterization of the steroidogenic acute regulatory protein (StAR). J Biol Chem. 1994;269:28314-28322.
7 Lin D, et al. Role of steroidogenic acute regulatory protein in adrenal and gonadal steroidogenesis. Science. 1995;267:1828-1831.
8 Sugawara T, et al. Human steroidogenic acute regulatory protein: Functional activity in COS-1 cells, tissue-specific expression, and mapping of the structural gene to 8p11.2 and a pseudogene to chromosome 13. Proc Natl Acad Sci USA. 1995;92:4778-4782.
9 Yasui T, et al. Biological effects of hormone replacement therapy in relation to serum estradiol levels. Horm Res. 2001;56:38-44.
10 Winter JS, et al. Gonadotrophins and steroid hormones in the blood and urine of prepubertal girls and other primates. Clin Endocrinol Metab. 1978;7:513-530.
11 Marshall WA, Tanner JM. Variations in pattern of pubertal changes in girls. Arch Dis Child. 1969;44:291-303.
12 Mansfield M.J., et al. Changes in growth and serum growth hormone and plasma somatomedin-C levels during suppression of gonadal sex steroid secretion in girls with central precocious puberty. J Clin Endocrinol Metab. 1988;66:3-9.
13 Boepple PA, et al. Impact of sex steroids and their suppression on skeletal growth and maturation. Am J Physiol. 1988;255:E559-E566.
14 Drummond AE, Findlay JK. The role of estrogen in folliculogenesis. Mol Cell Endocrinol. 1999;151:57-64.
15 Young JR, Jaffe RB. Strength–duration characteristics of estrogen effects on gonadotropin response to gonadotropin-releasing hormone in women. II. Effects of varying concentrations of estradiol. J Clin Endocrinol Metab. 1976;42:432-442.
16 Couse JF, Korach KS. Estrogen receptor null mice: What have we learned and where will they lead us? Endocr Rev. 1999;20:358-417.
17 Panay N, Studd JWW. Oestrogen and behaviour. In: Genazzani AR, Purdy RH, editors. The Brain: Source and Target for Sex Steroid Hormones. UK: The Parthenon Publishing Group; 1996:257-276.
18 Joffe H, Cohen LS. Estrogen, serotonin, and mood disturbance: Where is the therapeutic bridge? Biol Psychiatry. 1998;44:798-811.
19 Carlson MC, et al. Hormone replacement therapy and reduced cognitive decline in older women: the Cache County Study. Neurology. 2001;57:2210-2216.
20 Tang MX, et al. Effect of oestrogen during menopause on risk and age at onset of Alzheimer’s disease. Lancet. 1996;348:429-432.
21 Henderson VW, et al. Estrogen for Alzheimer’s disease in women: Randomized, double-blind, placebo-controlled trial. Neurology. 2000;54:295-301.
22 Shumaker SA, et al. The Women’s Health Initiative Memory Study (WHIMS): A trial of the effect of estrogen therapy in preventing and slowing the progression of dementia. Control Clin Trials. 1998;19:604-621.
23 Mulnard RA, et alfor the Alzheimer’s Disease Cooperative Study. Estrogen replacement therapy for treatment of mild to moderate Alzheimer disease: A randomized controlled trial. JAMA. 2000;283:1007-1015.
24 Shumaker SA, et alfor the Women’s Health Initiative Memory Study. Conjugated equine estrogens and incidence of probable dementia and mild cognitive impairment in postmenopausal women. JAMA. 2004;291:2947-2958.
25 The Writing Group for the PEPI. Effects of hormone therapy on bone mineral density: Results from the postmenopausal estrogen/progestin interventions (PEPI) trial. JAMA. 1996;276:1389-1396.
26 Anderson G.L., et al. Effects of conjugated equine estrogen in postmenopausal women with hysterectomy: The Women’s Health Initiative randomized controlled trial. JAMA. 2004;291:1701-1712.
27 Karas RH, Patterson BL, Mendelsohn ME. Human vascular smooth muscle cells contain functional estrogen receptor. Circulation. 1994;89:1943-1950.
28 Venkov CD, Rankin AB, Vaughan DE. Identification of authentic estrogen receptor in cultured endothelial cells. A potential mechanism for steroid hormone regulation of endothelial function. Circulation. 1996;94:727-733.
29 Kim HP, et al. Nongenomic stimulation of nitric oxide release by estrogen is mediated by estrogen receptor α localized in caveolae. Biochem Biophys Res Commun. 1999;263:257-262.
30 Kannel WB, et al. Menopause and risk of cardiovascular disease: The Framingham study. Ann Intern Med. 1976;85:447-452.
31 Hu FB, et al. Age at natural menopause and risk of cardiovascular disease. Arch Intern Med. 1999;159:1061-1066.
32 Hulley S, et alfor the Heart and Estrogen/progestin Replacement Study (HERS) Research Group. Randomized trial of estrogen plus progestin for secondary prevention of coronary heart disease in postmenopausal women. JAMA. 1998;280:605-613.
33 Le Guevel R, Pakdel F. Assessment of oestrogenic potency of chemicals used as growth promoter by in vitro methods. Hum Reprod. 2001;16:1030-1036.
34 O’Connell MB. Pharmacokinetic and pharmacologic variation between different estrogen products. J Clin Pharmacol. 1995;35(9 Suppl):18S-24S.
35 Thompson EA, Siiteri PK. Studies on the aromatization of C-19 androgens. Ann NY Acad Sci. 1973;212:378-391.
36 Kirschner MA, et al. Androgen–estrogen metabolism in women with upper body versus lower body obesity. J Clin Endocrinol Metab. 1990;70:473-479.
37 Grodin JM, Siiteri PK, MacDonald PC. Source of estrogen production in postmenopausal women. J Clin Endocrinol Metab. 1973;36:207-214.
38 Siiteri PK, MacDonald PC. Placental estrogen biosynthesis during human pregnancy. J Clin Endocrinol Metab. 1966;26:751-761.
39 Ruder HJ, Loriaux L, Lipsett MB. Estrone sulfate: Production rate and metabolism in man. J Clin Invest. 1972;51:1020-1033.
40 Loriaux DL, et al. Estrone sulfate, estrone, estradiol and estriol plasma levels in human pregnancy. J Clin Endocrinol Metab. 1972;35:887-891.
41 Barbieri R. Endocrine disorders in pregnancy. In: Yen SC, Jaffe RB, Barbieri RL, editors. Reproductive Endocrinology. Philadelphia: WB Saunders; 1999:785-811.
42 Goldfien A. Ovaries. In: Greenspan FS, editor. Basic and Clinical Endocrinology. Philadelphia: McGraw-Hill; 2001:453-508.
43 Franks S. Polycystic ovary syndrome. NEJM. 1995;333:853-861.
44 Abraham GE. Ovarian and adrenal contribution to peripheral androgens during the menstrual cycle. J Clin Endocrinol Metab. 1974;39:340-346.
45 Anderson DC. Sex-hormone-binding globulin. Clin Endocrinol (Oxf). 1974;3:69-96.
46 Rosner W, Smith R. Isolation of human testosterone-estradiol-binding globulin. Methods Enzymol. 1975;36:109-120.
47 Berube D, et al. Localization of the human sex hormone-binding globulin gene (SHBG) to the short arm of chromosome 17 (17p12-p13). Cytogenet Cell Genet. 1990;54:65-67.
48 Hammond GL, Bocchinfuso WP. Sex hormone-binding globulin: Gene organization and structure/function analyses. Horm Res. 1996;45:197-201.
49 Dunn JF, Nisula BC, Rodbard D. Transport of steroid hormones: Binding of 21 endogenous steroids to both testosterone-binding globulin and corticosteroid-binding globulin in human plasma. J Clin Endocrinol Metab. 1981;53:58-68.
50 Plymate SR, et al. Inhibition of sex hormone-binding globulin production in the human hepatoma (Hep G2) cell line by insulin and prolactin. J Clin Endocrinol Metab. 1988;67:460-464.
51 Plymate SR, et al. Regulation of sex hormone binding globulin (SHBG) production in Hep G2 cells by insulin. Steroids. 1988;52:339-340.
52 Pardridge WM. Serum bioavailability of sex steroid hormones. Clin Endocrinol Metab. 1986;15:259-278.
53 Manni A, et al. Bioavailability of albumin-bound testosterone. J Clin Endocrinol Metab. 1985;61:705-710.
54 Englebienne P. The serum steroid transport proteins: Biochemistry and clinical significance. Mol Aspects Med. 1984;7:313-396.
55 Rosenfield RL. Plasma testosterone binding globulin and indexes of the concentration of unbound plasma androgens in normal and hirsute subjects. J Clin Endocrinol Metab. 1971;32:717-728.
56 Rudd BT, Duignan NM, London DR. A rapid method for the measurement of sex hormone-binding globulin capacity of sera. Clin Chim Acta. 1974;55:165-178.
57 Cumming DC, Wall SR. Non-sex hormone-binding globulin-bound testosterone as a marker for hyperandrogenism. J Clin Endocrinol Metab. 1985;61:873-876.
58 Yalow RS, Berson SA. Immunoassay of endogenous plasma insulin in man. J Clin Invest. 1960;39:1157-1175.
59 Niswender GD. Hapten-radioimmunoassay for steroid hormones. In: Peron PG, editor. Immunologic Methods in Steroid Determination. New York: Appelton-Century-Crofts; 1970:149-173.
60 Rao PN, et al. Synthesis of new steroid haptens for radioimmunoassay—part V. 19-O-carboxymethyl ether derivative of testosterone. A highly specific antiserum for immunoassay of testosterone from both male and female plasma without chromatography. J Steroid Biochem. 1978;9:539-545.
61 Gupta MK. Interference by luteal phase progesterone in a commercial kit for measurement of 17-α hydroxyprogesterone. Clin Chem. 1985;31:1246-1247.
62 Taieb J, et al. Limitations of steroid determination by direct immunoassay. Clin Chem. 2002;48:583-585.
63 Diver MJ, Nisbet JA. Warning on plasma oestradiol measurement. Lancet. 1987;2:1097.
64 Fuqua JS, et al. Assay of plasma testosterone during the first six months of life: Importance of chromatographic purification of steroids. Clin Chem. 1995;41:1146-1149.
65 Fanchin R, et al. Physiopathology of premature progesterone elevation. Fertil Steril. 1995;64:796-801.
66 Dighe AS, Sluss PM. Improved detection of serum estradiol after sample extraction procedure. Clin Chem. 2004;50:764-766.
67 Marquet P. Progress of liquid chromatography-mass spectrometry in clinical and forensic toxicology. Ther Drug Monit. 2002;24:255-276.
68 Griffiths WJ, et al. Electrospray and tandem mass spectrometry in biochemistry. Biochem J. 2001;355:545-561.
69 Nelson RE, et al. Liquid chromatography-tandem mass spectrometry assay for simultaneous measurement of estradiol and estrone in human plasma. Clin Chem. 2004;50:373-384.
70 Valbuena D, et al. Ovarian stimulation and endometrial receptivity. Hum Reprod. 1999;14(Suppl 2):107-111.
71 Kligman I, Rosenwaks Z. Differentiating clinical profiles: Predicting good responders, poor responders, and hyperresponders. Fertil Steril. 2001;76:1185-1190.
72 Wright K, et al. A specific radioimmunoassay for estrone sulfate in plasma and urine without hydrolysis. J Clin Endocrinol Metab. 1978;47:1092-1098.
73 Ranadive GN, et al. Rapid, convenient radioimmunoassay of estrone sulfate. Clin Chem. 1998;44:244-249.
74 Gaskell SJ, Brownsey BG, Groom GV. Analyses for progesterone in serum by gas chromatography/mass spectrometry: Target data for external quality assessment of routine assays. Clin Chem. 1984;30:1696-1700.
75 Kubasik NP, Hallauer GD, Brodows RG. Evaluation of a direct solid-phase radioimmunoassay for progesterone, useful for monitoring luteal function. Clin Chem. 1984;30:284-286.
76 De Boever J, et al. Solid-phase chemiluminescence immunoassay for progesterone in unextracted serum. Clin Chem. 1984;30:1637-1641.
77 Radwanska E, Hammond J, Smith P. Single midluteal progesterone assay in the management of ovulatory infertility. J Reprod Med. 1981;26:85-89.
78 Radwanska E, Frankenberg J, Allen EI. Plasma progesterone levels in normal and abnormal early human pregnancy. Fertil Steril. 1978;30:398-402.
79 Baxendale PM, Jacobs HS, James VH. Plasma and salivary androstenedione and dihydrotestosterone in women with hyperandrogenism. Clin Endocrinol (Oxf). 1983;18:447-457.
80 Hummer L, Nielsen MD, Christiansen C. An easy and reliable radioimmunoassay of serum androstenedione: Age-related normal values in 252 females aged 2 to 70 years. Scand J Clin Lab Invest. 1983;43:301-306.
81 Putz Z, et al. A selective radioimmunoassay of androstenedione in plasma and saliva. J Clin Chem Clin Biochem. 1982;20:761-764.
82 Boots LR, et al. Measurement of total serum testosterone levels using commercially available kits: High degree of between-kit variability. Fertil Steril. 1998;69:286-292.
83 Vlahos I, et al. An improved ultrafiltration method for determining free testosterone in serum. Clin Chem. 1982;28:2286-2291.
84 Gupta MK. Androgen assessment in hirsutism and alopecia. Cleve Clin J Med. 1990;57:292-297.
85 Van Uytfanghe K, et al. Evaluation of a candidate reference measurement procedure for serum free testosterone based on ultrafiltration and isotope dilution-gas chromatography-mass spectrometry. Clin Chem. 2004;50:2101-2110.
86 Winters SJ, Kelley DE, Goodpaster B. The analog free testosterone assay: Are the results in men clinically useful? Clin Chem. 1998;44:2178-2182.
87 Vermeulen A, Verdonck L, Kaufman JM. A critical evaluation of simple methods for the estimation of free testosterone in serum. J Clin Endocrinol Metab. 1999;84:3666-3672.
88 Mathur RS, et al. Plasma androgens and sex hormone-binding globulin in the evaluation of hirsute females. Fertil Steril. 1981;35:29-35.
89 Carter GD, et al. Investigation of hirsutism: Testosterone is not enough. Ann Clin Biochem. 1983;20:262-263.
90 Davies R, et al. Indirect measurement of bioavailable testosterone with the Bayer Immuno 1 system. Clin Chem. 2002;48:388-390.
91 Riad-Fahmy D, et al. Steroids in saliva for assessing endocrine function. Endocr Rev. 1982;3:367-395.
92 Vining RF, McGinley RA, Symons RG. Hormones in saliva: Mode of entry and consequent implications for clinical interpretation. Clin Chem. 1983;29:1752-1756.
93 Lequin RM, et al. Progesterone in saliva: Pitfalls and consequent implications for accuracy of the determination. Clin Chem. 1986;32:831-834.
94 Vining RF, McGinley R, Rice BV. Saliva estriol measurements: An alternative to the assay of serum unconjugated estriol in assessing feto-placental function. J Clin Endocrinol Metab. 1983;56:454-460.
95 Sufi SB, et al. Multicenter evaluation of assays for estradiol and progesterone in saliva. Clin Chem. 1985;31:101-103.
96 Tallon DF, et al. Direct solid-phase enzyme immunoassay of progesterone in saliva. Clin Chem. 1984;30:1507-1511.
97 Tamate K, et al. Direct colorimetric monoclonal antibody enzyme immunoassay for estradiol-17β in saliva. Clin Chem. 1997;43:1159-1164.
98 Mottram JC. On the general effects of exposure to radium on metabolism and tumour growth in the rat and the special effects on the testis and pituitary. Q J Exp Physiol Cogn Med Sci. 1923;12:209-229.
99 McCullagh D. Dual endocrine activity of testes. Science. 1932;76:19.
100 Rivier J, et al. Purification and partial characterization of inhibin from porcine follicular fluid. Biochem Biophys Res Commun. 1985;133:120-127.
101 Robertson DM, et al. Isolation of a 31 kDa form of inhibin from bovine follicular fluid. Mol Cell Endocrinol. 1986;44:271-277.
102 Ling N, et al. Pituitary FSH is released by a heterodimer of the β-subunits from the two forms of inhibin. Nature. 1986;321:779-782.
103 Ueno N, et al. Isolation and partial characterization of follistatin: A single-chain Mr 35,000 monomeric protein that inhibits the release of follicle-stimulating hormone. Proc Natl Acad Sci USA. 1987;84:8282-8286.
104 Mason AJ, Niall HD, Seeburg PH. Structure of two human ovarian inhibins. Biochem Biophys Res Commun. 1986;135:957-964.
105 Lockwood GM, et al. Circulating inhibins and activin A during GnRH-analogue down-regulation and ovarian hyperstimulation with recombinant FSH for in vitro fertilization-embryo transfer. Clin Endocrinol (Oxf). 1996;45:741-748.
106 Welt C, et al. Activins, inhibins, and follistatins: From endocrinology to signaling. A paradigm for the new millennium. Exp Biol Med (Maywood). 2002;227:724-752.
107 Burger HG. Evidence for a negative feedback role of inhibin in follicle stimulating hormone regulation in women. Hum Reprod. 1993;8(Suppl 2):129-132.
108 de Jong FH, et al. Effects of factors from ovarian follicular fluid and Sertoli cell culture medium on in vivo and in vitro release of pituitary gonadotrophins in the rat: An evaluation of systems for the assay of inhibin. J Reprod Fertil Suppl. 1979;26:47-59.
109 Attardi B, et al. Rapid and profound suppression of messenger ribonucleic acid encoding follicle-stimulating hormone β by inhibin from primate Sertoli cells. Mol Endocrinol. 1989;3:280-287.
110 Attardi B, Winters SJ. Decay of follicle-stimulating hormone-β messenger RNA in the presence of transcriptional inhibitors and/or inhibin, activin, or follistatin. Mol Endocrinol. 1993;7:668-680.
111 Burger HG, et al. Aspects of current and future inhibin research. Reprod Fertil Dev. 1995;7:997-1002.
112 Ireland JL, et al. Alterations in amounts of different forms of inhibin during follicular atresia. Biol Reprod. 1994;50:1265-1276.
113 Sugino K, et al. Purification and characterization of high molecular weight forms of inhibin from bovine follicular fluid. Endocrinology. 1992;130:789-796.
114 Robertson DM, et al. Isolation of inhibin α-subunit precursor proteins from bovine follicular fluid. Endocrinology. 1989;125:2141-2149.
115 Sugino K, et al. Inhibin α-subunit monomer is present in bovine follicular fluid. Biochem Biophys Res Commun. 1989;159:1323-1329.
116 Robertson DM, et al. Inhibin forms in human plasma. J Endocrinol. 1995;144:261-269.
117 Schneyer AL, et al. Immunoreactive inhibin α-subunit in human serum: Implications for radioimmunoassay. J Clin Endocrinol Metab. 1990;70:1208-1212.
118 McConnell DS, et al. Development of a two-site solid-phase immunochemiluminescent assay for measurement of dimeric inhibin-A in human serum and other biological fluids. Clin Chem. 1996;42:1159-1167.
119 Groome NP, et al. Measurement of dimeric inhibin B throughout the human menstrual cycle. J Clin Endocrinol Metab. 1996;81:1401-1405.
120 Welt CK, et al. Differential regulation of inhibin A and inhibin B by luteinizing hormone, follicle-stimulating hormone, and stage of follicle development. J Clin Endocrinol Metab. 2001;86:2531-2537.
121 Corson SL, et al. Inhibin-B as a test of ovarian reserve for infertile women. Hum Reprod. 1999;14:2818-2821.
122 Eldar-Geva T, et al. Relationship between serum inhibin A and B and ovarian follicle development after a daily fixed dose administration of recombinant follicle-stimulating hormone. J Clin Endocrinol Metab. 2000;85:607-613.
123 Burger HG, et al. Serum inhibins A and B fall differentially as FSH rises in perimenopausal women. Clin Endocrinol (Oxf). 1998;48:809-813.
124 Lockwood GM, et al. Mid-follicular phase pulses of inhibin B are absent in polycystic ovarian syndrome and are initiated by successful laparoscopic ovarian diathermy: A possible mechanism regulating emergence of the dominant follicle. J Clin Endocrinol Metab. 1998;83:1730-1735.
125 Norman RJ, et al. Circulating follistatin concentrations are higher and activin concentrations are lower in polycystic ovarian syndrome. Hum Reprod. 2001;16:668-672.
126 Illingworth PJ, et al. Measurement of circulating inhibin forms during the establishment of pregnancy. J Clin Endocrinol Metab. 1996;81:1471-1475.
127 Lambert-Messerlian GM, et al. Second trimester levels of maternal serum inhibin A, total inhibin, α inhibin precursor, and activin in Down’s syndrome pregnancy. J Med Screen. 1996;3:58-62.
128 Aitken DA, et al. Dimeric inhibin A as a marker for Down’s syndrome in early pregnancy. NEJM. 1996;334:1231-1236.
129 Ciris M, et al. Inhibin α and β expression in ovarian stromal tumors and their histological equivalences. Acta Obstet Gynecol Scand. 2004;83:491-496.
130 Chudecka-Glaz A, Rzepka-Gorska I, Kosmowska B. Inhibin A levels in cyst fluid from epithelial ovarian tumors. Acta Obstet Gynecol Scand. 2004;83:501-503.
131 Anderson RA, et al. Inhibin B in seminal plasma: Testicular origin and relationship to spermatogenesis. Hum Reprod. 1998;13:920-926.
132 Nachtigall LB, et al. Inhibin B secretion in males with gonadotropin-releasing hormone (GnRH) deficiency before and during long-term GnRH replacement: Relationship to spontaneous puberty, testicular volume, and prior treatment—a clinical research center study. J Clin Endocrinol Metab. 1996;81:3520-3525.
133 Jensen TK, et al. Inhibin B as a serum marker of spermatogenesis: Correlation to differences in sperm concentration and follicle-stimulating hormone levels. A study of 349 Danish men. J Clin Endocrinol Metab. 1997;82:4059-4063.
134 von Eckardstein S, et al. Serum inhibin B in combination with serum follicle-stimulating hormone (FSH) is a more sensitive marker than serum FSH alone for impaired spermatogenesis in men, but cannot predict the presence of sperm in testicular tissue samples. J Clin Endocrinol Metab. 1999;84:2496-2501.
135 Peng C, et al. Activin and follistatin as local regulators in the human ovary. Biol Signals. 1996;5:81-89.
136 Hasegawa Y, et al. Induction of follicle stimulating hormone receptor by erythroid differentiation factor on rat granulosa cell. Biochem Biophys Res Commun. 1988;156:668-674.
137 Miro F, Smyth CD, Hillier SG. Development-related effects of recombinant activin on steroid synthesis in rat granulosa cells. Endocrinology. 1991;129:3388-3894.
138 Demura R, et al. Human plasma free activin and inhibin levels during the menstrual cycle. J Clin Endocrinol Metab. 1993;76:1080-1082.
139 Santoro N, Adel T, Skurnick JH. Decreased inhibin tone and increased activin A secretion characterize reproductive aging in women. Fertil Steril. 1999;71:658-662.
140 Lockwood GM. The role of inhibin in polycystic ovary syndrome. Hum Fertil (Camb). 2000;3:86-92.
141 Hillier SG, Miro F. Inhibin, activin, and follistatin. Potential roles in ovarian physiology. Ann NY Acad Sci. 1993;687:29-38.
142 Schneyer AL, et al. Follistatin–activin complexes in human serum and follicular fluid differ immunologically and biochemically. Endocrinology. 1996;137:240-247.
143 Sugino H, et al. Follistatin and its role as an activin-binding protein. J Med Invest. 1997;44:1-14.
144 Hashimoto O, et al. A novel role of follistatin, an activin-binding protein, in the inhibition of activin action in rat pituitary cells. Endocytotic degradation of activin and its acceleration by follistatin associated with cell-surface heparan sulfate. J Biol Chem. 1997;272:13835-13842.
145 Adashi EY, et al. Insulin-like growth factors: The ovarian connection. Hum Reprod. 1991;6:1213-1219.
146 Giudice LC. Insulin-like growth factors and ovarian follicular development. Endocr Rev. 1992;13:641-669.
147 Monget P, Bondy C. Importance of the IGF system in early folliculogenesis. Mol Cell Endocrinol. 2000;163:89-93.
148 Geisthovel F, et al. Expression of insulin-like growth factor-II (IGF-II) messenger ribonucleic acid (mRNA), but not IGF-I mRNA, in human preovulatory granulosa cells. Hum Reprod. 1989;4:899-902.
149 el-Roeiy A, et al. Expression of insulin-like growth factor-I (IGF-I) and IGF-II and the IGF-I, IGF-II, and insulin receptor genes and localization of the gene products in the human ovary. J Clin Endocrinol Metab. 1993;77:1411-1418.
150 el-Roeiy A, et al. Expression of the genes encoding the insulin-like growth factors (IGF-I and II), the IGF and insulin receptors, and IGF-binding proteins-1-6 and the localization of their gene products in normal and polycystic ovary syndrome ovaries. J Clin Endocrinol Metab. 1994;78:1488-1496.
151 Carson RS, et al. Growth factors in ovarian function. J Reprod Fertil. 1989;85:735-746.
152 Westergaard LG, Andersen CY. Epidermal growth factor (EGF) in human preovulatory follicles. Hum Reprod. 1989;4:257-260.
153 Khan-Dawood FS. Human corpus luteum: Immunocytochemical localization of epidermal growth factor. Fertil Steril. 1987;47:916-919.
154 Ayyagari RR, Khan-Dawood FS. Human corpus luteum: Presence of epidermal growth factor receptors and binding characteristics. Am J Obstet Gynecol. 1987;156:942-946.
155 Kudlow JE, et al. Ovarian transforming growth factor-α gene expression: Immunohistochemical localization to the theca-interstitial cells. Endocrinology. 1987;121:1577-1579.
156 Lobb DK, et al. Transforming growth factor-alpha in the adult bovine ovary: identification in growing ovarian follicles. Biol Reprod. 1989;40(5):1087-1093.
157 Yeh J, Lee GY, Anderson E. Presence of transforming growth factor-α messenger ribonucleic acid (mRNA) and absence of epidermal growth factor mRNA in rat ovarian granulosa cells, and the effects of these factors on steroidogenesis in vitro. Biol Reprod. 1993;48:1071-1081.
158 Yeh J, Osathanondh R. Expression of messenger ribonucleic acids encoding for basic fibroblast growth factor (FGF) and alternatively spliced FGF receptor in human fetal ovary and uterus. J Clin Endocrinol Metab. 1993;77:1367-1371.
159 Di Blasio AM, et al. Expression of the genes encoding basic fibroblast growth factor and its receptor in human granulosa cells. Mol Cell Endocrinol. 1993;96:R7-R11.
160 McAllister JM, Byrd W, Simpson ER. The effects of growth factors and phorbol esters on steroid biosynthesis in isolated human theca interna and granulosa-lutein cells in long term culture. J Clin Endocrinol Metab. 1994;79:106-112.
161 Adashi EY, et al. Basic fibroblast growth factor as a regulator of ovarian granulosa cell differentiation: A novel nonmitogenic role. Mol Cell Endocrinol. 1988;55:7-14.
162 Koos R. Ovarian angiogenesis. In: Adashi EY, editor. The Ovary. New York: Raven Press; 1993:433-453.
163 Khan SA, et al. Human testis cytosol and ovarian follicular fluid contain high amounts of interleukin-1-like factor(s). Mol Cell Endocrinol. 1988;58:221-230.
164 Machelon V, et al. Interleukin-6 biosynthesis in human preovulatory follicles: Some of its potential roles at ovulation. J Clin Endocrinol Metab. 1994;79:633-642.
165 Piquette GN, et al. Gene regulation of interleukin-1 beta, interleukin-1 receptor type I, and plasminogen activator inhibitor-1 and -2 in human granulosa-luteal cells. Fertil Steril. 1994;62:760-770.
166 Buscher U, et al. Cytokines in the follicular fluid of stimulated and nonstimulated human ovaries: Is ovulation a suppressed inflammatory reaction? Hum Reprod. 1999;14:162-166.
167 Brannstrom M. Potential role of cytokines in ovarian physiology: The case of interleukin-1. In: Leung PCK, editor. The Ovary. 2nd ed. London: Elsevier; 2004:261-271.
168 Salmassi A, et al. Interaction of interleukin-6 on human granulosa cell steroid secretion. J Endocrinol. 2001;170:471-478.
169 Roby KF, et al. Immunological evidence for a human ovarian tumor necrosis factor-α. J Clin Endocrinol Metab. 1990;71:1096-1102.
170 Roby KF, Terranova PF. Effects of tumor necrosis factor-α in vitro on steroidogenesis of healthy and atretic follicles of the rat: Theca as a target. Endocrinology. 1990;126:2711-2718.
171 Marshall JM. Adrenergic innervation of the female reproductive tract: Anatomy, physiology and pharmacology. Ergeb Physiol. 1970;62:6-67.
172 Stefenson A, et al. Comparative study of the autonomic innervation of the mammalian ovary, with particular regard to the follicular system. Cell Tissue Res. 1981;215:47-62.
173 Kawakami M, et al. Involvement of ovarian innervation in steroid secretion. Endocrinology. 1981;109:136-145.
174 Dyer CA, Erickson GF. Norepinephrine amplifies human chorionic gonadotropin-stimulated androgen biosynthesis by ovarian theca-interstitial cells. Endocrinology. 1985;116:1645-1652.
175 Knobil E, et al. Control of the rhesus monkey menstrual cycle: Permissive role of hypothalamic gonadotropin-releasing hormone. Science. 1980;207:1371-1373.
176 Belchetz PE, et al. Hypophysial responses to continuous and intermittent delivery of hypopthalamic gonadotropin-releasing hormone. Science. 1978;202:631-633.
177 Silverman AJ, Jhamandas J, Renaud LP. Localization of luteinizing hormone-releasing hormone (LHRH) neurons that project to the median eminence. J Neurosci. 1987;7:2312-2319.
178 Wetsel WC, et al. Intrinsic pulsatile secretory activity of immortalized luteinizing hormone-releasing hormone-secreting neurons. Proc Natl Acad Sci USA. 1992;89:4149-4153.
179 Rasmussen DD, et al. Pulsatile gonadotropin-releasing hormone release from the human mediobasal hypothalamus in vitro: Opiate receptor-mediated suppression. Neuroendocrinology. 1989;49:150-156.
180 Haisenleder DJ, et al. Influence of gonadotropin-releasing hormone pulse amplitude, frequency, and treatment duration on the regulation of luteinizing hormone (LH) subunit messenger ribonucleic acids and LH secretion. Mol Endocrinol. 1988;2:338-343.
181 Katt JA, et al. The frequency of gonadotropin-releasing hormone stimulation determines the number of pituitary gonadotropin-releasing hormone receptors. Endocrinology. 1985;116:2113-2115.
182 Clayton RN, Catt KJ. Gonadotropin-releasing hormone receptors: Characterization, physiological regulation, and relationship to reproductive function. Endocr Rev. 1981;2:186-209.
183 Yasin M, et al. Gonadotropin-releasing hormone (GnRH) pulse pattern regulates GnRH receptor gene expression: Augmentation by estradiol. Endocrinology. 1995;136:1559-1564.
184 Weiss J, et al. Divergent responses of gonadotropin subunit messenger RNAs to continuous versus pulsatile gonadotropin-releasing hormone in vitro. Mol Endocrinol. 1990;4:557-564.
185 Nazian SJ, et al. Opioid inhibition of adrenergic and dopaminergic but not serotonergic stimulation of luteinizing hormone releasing hormone release from immortalized hypothalamic neurons. Mol Cell Neurosci. 1994;5:642-648.
186 Yen SS, et al. Neuroendocrinology of opioid peptides and their role in the control of gonadotropin and prolactin secretion. Am J Obstet Gynecol. 1985;152:485-493.
187 Seeburg PH, et al. The mammalian GnRH gene and its pivotal role in reproduction. Recent Prog Horm Res. 1987;43:69-98.
188 Seeburg PH, Adelman JP. Characterization of cDNA for precursor of human luteinizing hormone releasing hormone. Nature. 1984;311:666-668.
189 Ronnekleiv OK, et al. Combined immunohistochemistry for gonadotropin-releasing hormone (GnRH) and pro-GnRH, and in situ hybridization for GnRH messenger ribonucleic acid in rat brain. Mol Endocrinol. 1989;3:363-371.
190 Ackland JF, et al. Molecular forms of gonadotropin-releasing hormone associated peptide (GAP): Changes within the rat hypothalamus and release from hypothalamic cells in vitro. Neuroendocrinology. 1988;48:376-386.
191 Levine JE, Ramirez VD. Luteinizing hormone-releasing hormone release during the rat estrous cycle and after ovariectomy, as estimated with push-pull cannulae. Endocrinology. 1982;111:1439-1448.
192 Neill JD, et al. Luteinizing hormone releasing hormone (LHRH) in pituitary stalk blood of rhesus monkeys: Relationship to level of LH release. Endocrinology. 1977;101:430-434.
193 Reiter EO, et al. A role for endogenous estrogen in normal ovarian development in the neonatal rat. Endocrinology. 1972;91:1537-1539.
194 Goldenberg RL, et al. Interaction of FSH and hCG on follicle development in the ovarian augmentation reaction. Endocrinology. 1972;91:533-536.
195 Espey LH. Ovulation. In: Knobil E, Neill JD, editors. The Physiology of Reproduction. New York: Raven Press; 1994:725.
196 Hseuh AJW, Jones PBC, et al. Hormonal regulation of the differentiation of cultured granulosa cells. Endocr Rev. 1984;5:127.
197 Hsueh AJ, et al. Granulosa cells as hormone targets: the role of biologically active follicle-stimulating hormone in reproduction. Recent Prog Horm Res. 1989;45:209-277.
198 Tilly JL, LaPolt PS, Hsueh AJ. Hormonal regulation of follicle-stimulating hormone receptor messenger ribonucleic acid levels in cultured rat granulosa cells. Endocrinology. 1992;130:1296-1302.
199 LaPolt PS, et al. Gonadotropin-induced up- and down-regulation of ovarian follicle-stimulating hormone (FSH) receptor gene expression in immature rats: Effects of pregnant mare’s serum gonadotropin, human chorionic gonadotropin, and recombinant FSH. Endocrinology. 1992;130:1289-1295.
200 Pierce JG, Parsons TF. Glycoprotein hormones: Structure and function. Annu Rev Biochem. 1981;50:465-495.
201 Huhtaniemi I. Functional consequences of mutations and polymorphisms in gonadotropin and gonadotropin receptor genes. In: Leung PCK, editor. The Ovary. 2nd ed. London: Elselvier Academic Press; 2004:55-78.
202 Fiddes JC, Goodman HM. The gene encoding the common α subunit of the four human glycoprotein hormones. J Mol Appl Genet. 1981;1:3-18.
203 Naylor SL, et al. Chromosome assignment of genes encoding the α and β subunits of glycoprotein hormones in man and mouse. Somatic Cell Genet. 1983;9:757-770.
204 Policastro PF, et al. A map of the hCG β-LH β gene cluster. J Biol Chem. 1986;261:5907-5916.
205 Watkins PC, et al. DNA sequence and regional assignment of the human follicle-stimulating hormone β-subunit gene to the short arm of human chromosome 11. DNA. 1987;6:205-212.
206 Ryan RJ, et al. Structure–function relationships of gonadotropins. Recent Prog Horm Res. 1987;43:383-429.
207 Ryan RJ, et al. The glycoprotein hormones: Recent studies of structure–function relationships. Faseb J. 1988;2:2661-2669.
208 Chen HC, et al. Characterization and biological properties of chemically deglycosylated human chorionic gonadotropin. Role of carbohydrate moieties in adenylate cyclase activation. J Biol Chem. 1982;257:14446-14452.
209 Themmen APN, Huhtaniemi IT. Mutations of gonadotropins and gonadotropin receptors: Elucidating the physiology and pathophysiology of pituitary-gonadal function. Endocr Rev. 2000;21:551-583.
210 Weiss J, et al. Hypogonadism caused by a single amino acid substitution in the β subunit of luteinizing hormone. NEJM. 1992;326:179-183.
211 Suganuma N, et al. Effects of the mutations (Trp8→Arg and Ile15→Thr) in human luteinizing hormone (LH) β-subunit on LH bioactivity in vitro and in vivo. Endocrinology. 1996;137:831-838.
212 Matthews CH, et al. Primary amenorrhoea and infertility due to a mutation in the β-subunit of follicle-stimulating hormone. Nat Genet. 1993;5:83-86.
213 Layman LC, et al. Delayed puberty and hypogonadism caused by mutations in the follicle-stimulating hormone beta-subunit gene. NEJM. 1997;337:607-611.
214 Lindstedt G, et al. Follitropin (FSH) deficiency in an infertile male due to FSHβ gene mutation. A syndrome of normal puberty and virilization but underdeveloped testicles with azoospermia, low FSH but high lutropin and normal serum testosterone concentrations. Clin Chem Lab Med. 1998;36:663-665.
215 Midgley ARJr. Radioimmunoassay: A method for human chorionic gonadotropin and human luteinizing hormone. Endocrinology. 1966;79:10-18.
216 Faiman C, Ryan RJ. Radioimmunoassay for human follicle stimulating hormone. J Clin Endocrinol Metab. 1967;27:444-447.
217 Faiman C, Ryan RJ. Radioimmunoassay for human luteinizing hormone. Proc Soc Exp Biol Med. 1967;125:1130-1133.
218 Halvorson LM. Gonadotropic hormones: Biosynthesis, secretion, receptors and action. In: Yen SSC, Barberi RL, editors. Reproductive Endocrinology. Philadelphia: WB Saunders; 1999:89.
219 Jaakkola T, et al. The ratios of serum bioactive/immunoreactive luteinizing hormone and follicle-stimulating hormone in various clinical conditions with increased and decreased gonadotropin secretion: Reevaluation by a highly sensitive immunometric assay. J Clin Endocrinol Metab. 1990;70:1496-1505.
220 Van Damme MP, Robertson DM, Diczfalusy E. An improved in vitro bioassay method for measuring luteinizing hormone (LH) activity using mouse Leydig cell preparations. Acta Endocrinol (Copenh). 1974;77:655-671.
221 Wang C. Bioassays of follicle stimulating hormone. Endocr Rev. 1988;9:374-377.
222 Chappel S. Biological to immunological ratios: Reevaluation of a concept. J Clin Endocrinol Metab. 1990;70:1494-1495.
223 Rebar RW, Connolly HV. Clinical features of young women with hypergonadotropic amenorrhea. Fertil Steril. 1990;53:804-810.
224 Yen SS, et al. Hypothalamic amenorrhea and hypogonadotropinism: Responses to synthetic LRF. J Clin Endocrinol Metab. 1973;36:811-816.
225 Heffner LJ, Gramates LS, Yuan RW. A glycosylated prolactin species is covalently bound to immunoglobulin in human amniotic fluid. Biochem Biophys Res Commun. 1989;165:299-305.
226 Farkouh NH, Packer MG, Frantz AG. Large molecular size prolactin with reduced receptor activity in human serum: High proportion in basal state and reduction after thyrotropin-releasing hormone. J Clin Endocrinol Metab. 1979;48:1026-1032.
227 Markoff E, et al. Glycosylation selectively alters the biological activity of prolactin. Endocrinology. 1988;123:1303-1306.
228 Vieira JG, et al. Extensive experience and validation of polyethylene glycol precipitation as a screening method for macroprolactinemia. Clin Chem. 1998;44:1758-1789.
229 Evans RM. The steroid and thyroid hormone receptor superfamily. Science. 1988;240:889-895.
230 McKenna NJ, Lanz RB, O’Malley BW. Nuclear receptor coregulators: Cellular and molecular biology. Endocr Rev. 1999;20:321-344.
231 Jensen EV, et al. Receptors reconsidered: A 20-year perspective. Recent Prog Horm Res. 1982;38:1-40.
232 Tsai MJ, O’Malley BW. Molecular mechanisms of action of steroid/thyroid receptor superfamily members. Annu Rev Biochem. 1994;63:451-486.
233 King WJ, Greene GL. Monoclonal antibodies localize oestrogen receptor in the nuclei of target cells. Nature. 1984;307:745-747.
234 Bresnick EH, et al. Evidence that the 90-kDa heat shock protein is necessary for the steroid binding conformation of the L cell glucocorticoid receptor. J Biol Chem. 1989;264:4992-4997.
235 Strahle U, Klock G, Schutz G. A DNA sequence of 15 base pairs is sufficient to mediate both glucocorticoid and progesterone induction of gene expression. Proc Natl Acad Sci USA. 1987;84:7855-7871.
236 Klock G, Strahle U, Schutz G. Oestrogen and glucocorticoid responsive elements are closely related but distinct. Nature. 1987;329:734-736.
237 Arriza JL, et al. Cloning of human mineralocorticoid receptor complementary DNA: Structural and functional kinship with the glucocorticoid receptor. Science. 1987;237:268-275.
238 Edwards DP. The role of coactivators and corepressors in the biology and mechanism of action of steroid hormone receptors. J Mammary Gland Biol Neoplasia. 2000;5:307-324.
239 Green S, et al. Cloning of the human oestrogen receptor cDNA. J Steroid Biochem. 1986;24:77-83.
240 Kuiper GG, et al. Cloning of a novel receptor expressed in rat prostate and ovary. Proc Natl Acad Sci USA. 1996;93:5925-5930.
241 Enmark E, et al. Human estrogen receptor β—gene structure, chromosomal localization, and expression pattern. J Clin Endocrinol Metab. 1997;82:4258-4265.
242 Mosselman S, Polman J, Dijkema R. ER β: Identification and characterization of a novel human estrogen receptor. FEBS Lett. 1996;392:49-53.
243 Brzozowski AM, et al. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature. 1997;389:753-758.
244 Pike AC, et al. Structure of the ligand-binding domain of oestrogen receptor β in the presence of a partial agonist and a full antagonist. Embo J. 1999;18:4608-4618.
245 Kuiper GG, et al. Comparison of the ligand binding specificity and transcript tissue distribution of estrogen receptors α and β. Endocrinology. 1997;138:863-870.
246 Smith EP, et al. Estrogen resistance caused by a mutation in the estrogen-receptor gene in a man. NEJM. 1994;331:1056-1061.
247 Fuqua SA, et al. Variant human breast tumor estrogen receptor with constitutive transcriptional activity. Cancer Res. 1991;51:105-109.
248 Grainger DJ, Metcalfe JC. Tamoxifen: Teaching an old drug new tricks? Nat Med. 1996;2:381-385.
249 Kedar RP, et al. Effects of tamoxifen on uterus and ovaries of postmenopausal women in a randomised breast cancer prevention trial. Lancet. 1994;343:1318-1321.
250 Yang NN, et al. Estrogen and raloxifene stimulate transforming growth factor-β 3 gene expression in rat bone: A potential mechanism for estrogen- or raloxifene-mediated bone maintenance. Endocrinology. 1996;137:2075-2084.
251 Berry M, Metzger D, Chambon P. Role of the two activating domains of the oestrogen receptor in the cell-type and promoter-context dependent agonistic activity of the anti-oestrogen 4-hydroxytamoxifen. Embo J. 1990;9:2811-2818.
252 Landel CC, Kushner PJ, Greene GL. The interaction of human estrogen receptor with DNA is modulated by receptor-associated proteins. Mol Endocrinol. 1994;8:1407-1419.
253 Webb P, et al. Tamoxifen activation of the estrogen receptor/AP-1 pathway: Potential origin for the cell-specific estrogen-like effects of antiestrogens. Mol Endocrinol. 1995;9:443-456.
254 Yang NN, et al. Identification of an estrogen response element activated by metabolites of 17β-estradiol and raloxifene. Science. 1996;273:1222-1225.
255 Osborne CK, et al. Double-blind, randomized trial comparing the efficacy and tolerability of fulvestrant versus anastrozole in postmenopausal women with advanced breast cancer progressing on prior endocrine therapy: Results of a North American trial. J Clin Oncol. 2002;20:3386-3395.
256 Bundred N, Howell A. Fulvestrant (Faslodex): Current status in the therapy of breast cancer. Expert Rev Anticancer Ther. 2002;2:151-160.
257 Kastner P, et al. Two distinct estrogen-regulated promoters generate transcripts encoding the two functionally different human progesterone receptor forms A and B. Embo J. 1990;9:1603-1614.
258 Sartorius CA, et al. A third transactivation function (AF3) of human progesterone receptors located in the unique N-terminal segment of the B-isoform. Mol Endocrinol. 1994;8:1347-1360.
259 Giangrande PH, et al. The opposing transcriptional activities of the two isoforms of the human progesterone receptor are due to differential cofactor binding. Mol Cell Biol. 2000;20:3102-3115.
260 Lim CS, et al. Differential localization and activity of the A- and B-forms of the human progesterone receptor using green fluorescent protein chimeras. Mol Endocrinol. 1999;13:366-375.
261 Conneely OM, et al. Reproductive functions of progesterone receptors. Recent Prog Horm Res. 2002;57:339-355.
262 Feil PD, Clarke CL, Satyaswaroop PG. Progestin-mediated changes in progesterone receptor forms in the normal human endometrium. Endocrinology. 1988;123:2506-2513.
263 Ogle TF. Progesterone action in the decidual mesometrium of pregnancy. Steroids. 2002;67:1-14.
264 Moguilewsky M, Philibert D. RU 38486: Potent antiglucocorticoid activity correlated with strong binding to the cytosolic glucocorticoid receptor followed by an impaired activation. J Steroid Biochem. 1984;20:271-276.
265 Baulieu EE. Contragestion and other clinical applications of RU 486, an antiprogesterone at the receptor. Science. 1989;245:1351-1357.
266 Spitz IM, Bardin CW. Mifepristone (RU 486)—a modulator of progestin and glucocorticoid action. NEJM. 1993;329:404-412.
267 Gronemeyer H, et al. Mechanisms of antihormone action. J Steroid Biochem Mol Biol. 1992;41:217-221.
268 Lubahn DB, et al. Cloning of human androgen receptor complementary DNA and localization to the X chromosome. Science. 1988;240:327-330.
269 Wilson CM, McPhaul MJ. A and B forms of the androgen receptor are present in human genital skin fibroblasts. Proc Natl Acad Sci USA. 1994;91:1234-1238.
270 Jenster G, et al. Functional domains of the human androgen receptor. J Steroid Biochem Mol Biol. 1992;41:671-675.
271 Brinkmann AO. Molecular basis of androgen insensitivity. Mol Cell Endocrinol. 2001;179:105-109.
272 Chavez B, et al. Eight novel mutations of the androgen receptor gene in patients with androgen insensitivity syndrome. J Hum Genet. 2001;46:560-565.
273 Rosa S, et al. Complete androgen insensitivity syndrome caused by a novel mutation in the ligand-binding domain of the androgen receptor: Functional characterization. J Clin Endocrinol Metab. 2002;87:4378-4382.
274 Mongan NP, et al. Two de novo mutations in the AR gene cause the complete androgen insensitivity syndrome in a pair of monozygotic twins. J Clin Endocrinol Metab. 2002;87:1057-1061.
275 Ong YC, Kolatkar PR, Yong EL. Androgen receptor mutations causing human androgen insensitivity syndromes show a key role of residue M807 in Helix 8–Helix 10 interactions and in receptor ligand-binding domain stability. Mol Hum Reprod. 2002;8:101-108.
276 Revelli A, Massobrio M, Tesarik J. Nongenomic actions of steroid hormones in reproductive tissues. Endocr Rev. 1998;19:3-17.
277 Revelli A, Tesarik J, Massobrio M. Nongenomic effects of neurosteroids. Gynecol Endocrinol. 1998;12:61-67.
278 Chester AH, et al. Oestrogen relaxes human epicardial coronary arteries through nonendothelium-dependent mechanisms. Coron Artery Dis. 1995;6:417-422.
279 Aronica SM, Kraus WL, Katzenellenbogen BS. Estrogen action via the cAMP signaling pathway: Stimulation of adenylate cyclase and cAMP-regulated gene transcription. Proc Natl Acad Sci USA. 1994;91:8517-8521.
280 Pedram A, et al. Integration of the non-genomic and genomic actions of estrogen. Membrane-initiated signaling by steroid to transcription and cell biology. J Biol Chem. 2002;277:50768-50775.
281 Ascoli M, Fanelli F, Segaloff DL. The lutropin/choriogonadotropin receptor, a 2002 perspective. Endocr Rev. 2002;23:141-174.
282 Simoni M, Gromoll J, Nieschlag E. The follicle-stimulating hormone receptor: Biochemistry, molecular biology, physiology, and pathophysiology. Endocr Rev. 1997;18:739-773.
283 Bourne HR, Sanders DA, McCormick F. The GTPase superfamily: Conserved structure and molecular mechanism. Nature. 1991;349:117-127.
284 Hirsch B, et al. The C-terminal third of the human luteinizing hormone (LH) receptor is important for inositol phosphate release: Analysis using chimeric human LH/follicle-stimulating hormone receptors. Mol Endocrinol. 1996;10:1127-1137.
285 Monaco L, Adamo S, Conti M. Follicle-stimulating hormone modulation of phosphoinositide turnover in the immature rat Sertoli cell in culture. Endocrinology. 1988;123:2032-2039.
286 Hirakawa T, Galet C, Ascoli M. MA-10 cells transfected with the human lutropin/choriogonadotropin receptor (hLHR): A novel experimental paradigm to study the functional properties of the hLHR. Endocrinology. 2002;143:1026-1035.
287 Dufau ML. Endocrine regulation and communicating functions of the Leydig cell. Annu Rev Physiol. 1988;50:483-508.
288 Hipkin RW, Wang Z, Ascoli M. Human chorionic gonadotropin (CG)- and phorbol ester-stimulated phosphorylation of the luteinizing hormone/CG receptor maps to serines 635, 639, 649, and 652 in the C-terminal cytoplasmic tail. Mol Endocrinol. 1995;9:151-158.
289 Laue L, et al. Genetic heterogeneity of constitutively activating mutations of the human luteinizing hormone receptor in familial male-limited precocious puberty. Proc Natl Acad Sci USA. 1995;92:1906-1910.
290 Shenker A, et al. A constitutively activating mutation of the luteinizing hormone receptor in familial male precocious puberty. Nature. 1993;365:652-654.
291 Liu G, et al. Leydig-cell tumors caused by an activating mutation of the gene encoding the luteinizing hormone receptor. NEJM. 1999;341:1731-1736.
292 Gromoll J, Simoni M, Nieschlag E. An activating mutation of the follicle-stimulating hormone receptor autonomously sustains spermatogenesis in a hypophysectomized man. J Clin Endocrinol Metab. 1996;81:1367-1370.
293 Vasseur C, et al. A chorionic gonadotropin-sensitive mutation in the follicle-stimulating hormone receptor as a cause of familial gestational spontaneous ovarian hyperstimulation syndrome. NEJM. 2003;349:753-759.
294 Smits G, et al. Ovarian hyperstimulation syndrome due to a mutation in the follicle-stimulating hormone receptor. NEJM. 2003;349:760-766.
295 Naor Z, Childs GV. Binding and activation of gonadotropin-releasing hormone receptors in pituitary and gonadal cells. Int Rev Cytol. 1986;103:147-187.
296 Krsmanovic LZ, et al. Expression of gonadotropin-releasing hormone receptors and autocrine regulation of neuropeptide release in immortalized hypothalamic neurons. Proc Natl Acad Sci USA. 1993;90:3908-3912.
297 Currie AJ, Fraser HM, Sharpe RM. Human placental receptors for luteinizing hormone releasing hormone. Biochem Biophys Res Commun. 1981;99:332-338.
298 Horn F, et al. Intracellular responses to gonadotropin-releasing hormone in a clonal cell line of the gonadotrope lineage. Mol Endocrinol. 1991;5:347-355.
299 Stojilkovic SS, Reinhart J, Catt KJ. Gonadotropin-releasing hormone receptors: Structure and signal transduction pathways. Endocr Rev. 1994;15:462-499.
300 Clark MR. Stimulation of progesterone and prostaglandin E accumulation by luteinizing hormone-releasing hormone (LHRH) and LHRH analogs in rat granulosa cells. Endocrinology. 1982;110:146-152.
301 Ekholm C, Hillensjo T, Isaksson O. Gonadotropin-releasing hormone agonists stimulate oocyte meiosis and ovulation in hypophysectomized rats. Endocrinology. 1981;108:2022-2024.
302 Hsueh AJ, Erickson GF. Extrapituitary action of gonadotropin-releasing hormone: Direct inhibition ovarian steroidogenesis. Science. 1979;204:854-855.
303 Piquette GN, et al. Regulation of luteinizing hormone receptor messenger ribonucleic acid levels by gonadotropins, growth factors, and gonadotropin-releasing hormone in cultured rat granulosa cells. Endocrinology. 1991;128:2449-2456.
304 LaPolt PS, et al. Regulation of inhibin subunit messenger ribonucleic acid levels by gonadotropins, growth factors, and gonadotropin-releasing hormone in cultured rat granulosa cells. Endocrinology. 1990;127:823-831.
305 Harwood JP, Clayton RN, Catt KJ. Ovarian gonadotropin-releasing hormone receptors. I. Properties and inhibition of luteal cell function. Endocrinology. 1980;107:407-413.
306 Kraus S, Naor Z, Seger R. Intracellular signaling pathways mediated by the gonadotropin-releasing hormone (GnRH) receptor. Arch Med Res. 2001;32:499-509.
307 Poulin B, et al. GnRH signalling pathways and GnRH-induced homologous desensitization in a gonadotrope cell line (αT3-1). Mol Cell Endocrinol. 1998;142:99-117.
308 Millar RP, et al. Gonadotropin-releasing hormone receptors. Endocr Rev. 2004;25:235-275.
309 Chi L, et al. Cloning and characterization of the human GnRH receptor. Mol Cell Endocrinol. 1993;91:R1-R6.
310 Sealfon SC, Weinstein H, Millar RP. Molecular mechanisms of ligand interaction with the gonadotropin-releasing hormone receptor. Endocr Rev. 1997;18:180-205.
311 Karges B, Karges W, de Roux N. Clinical and molecular genetics of the human GnRH receptor. Hum Reprod Update. 2003;9:523-530.
312 Ullrich A, et al. Insulin-like growth factor I receptor primary structure: Comparison with insulin receptor suggests structural determinants that define functional specificity. Embo J. 1986;5:2503-2512.
313 Abbott AM, et al. Insulin-like growth factor I receptor gene structure. J Biol Chem. 1992;267:10759-10763.
314 Richards JS, et al. Novel signaling pathways that control ovarian follicular development, ovulation, and luteinization. Recent Prog Horm Res. 2002;57:195-220.
315 LeRoith D, et al. Molecular and cellular aspects of the insulin-like growth factor I receptor. Endocr Rev. 1995;16:143-163.
316 Massague J. TGF-β signal transduction. Annu Rev Biochem. 1998;67:753-791.
317 Lin SY, et al. Regulation of ovarian function by the TGF-β superfamily and follistatin. Reproduction. 2003;126:133-148.
318 Mathews LS, Vale WW. Expression cloning of an activin receptor, a predicted transmembrane serine kinase. Cell. 1991;65:973-982.
319 ten Dijke P, et al. Activin receptor-like kinases: A novel subclass of cell-surface receptors with predicted serine/threonine kinase activity. Oncogene. 1993;8:2879-2887.
320 Lebrun JJ, Vale WW. Activin and inhibin have antagonistic effects on ligand-dependent heteromerization of the type I and type II activin receptors and human erythroid differentiation. Mol Cell Biol. 1997;17:1682-1691.