Ovarian Germ Cell Tumors
General Features
Germ cell tumors arise from primordial germ cells and account for approximately 30% of all ovarian tumors. Over 95% of them are benign dermoid cysts (mature cystic teratomas) and the remaining 5% are malignant. Malignant germ cell tumors (MGCTs) represent approximately 3% of all ovarian cancers in Western countries and about 20% in Asian and African populations, where surface epithelial cancers are much less common.1
Germ cell tumors replicate in a distorted form various stages of normal embryogenesis and, like the embryo, are capable of developing complex and highly differentiated tissues.2 The malignant potential of ovarian germ cell tumors is inversely related to their degree of differentiation. Some traditional views on the histogenesis of germ cell tumors have been recently challenged. Although germ cell tumors of the ovary are morphologically similar to testicular germ cell tumors, they may not necessarily have an identical origin.3 Testicular germ cell tumors originate from primitive germ cells with a malignant character, whereas ovarian germ cell tumors have a parthenogenetic origin from postmeiotic or meiotic cells.4 For this reason, embryonal carcinoma (EC) occurs more frequently in the testis than the ovary. Nevertheless, an unknown percentage of MGCTs developing in phenotypic females may, in fact, represent testicular germ cell tumors (seminomas, ECs, mixed germ cell tumors), as they may have originated from the malignant germ cell component of gonadoblastomas present in dysgenetic gonads of patients with an unrecognized Y chromosome-containing genotype.5 Rarely, germ cell tumors may arise from pre-existing somatic neoplasms of the female genital tract.6–9 In these cases, the teratoid tumors derive most likely from a pluripotent stem cell population of somatic neoplasms. Recent evidence suggests that germinomas may not be end-stage tumors, as traditionally believed, but some of them could be precursors to germ cell neoplasms capable of further differentiation.10–15 The occasional morphologic overlap between the primitive or immature forms of germ cell tumors, such as germinoma, EC, and yolk sac tumor (YST), supports the view that these tumors constitute a closely related group of neoplasms capable of embryonic or extraembryonic differentiation.10,16 The new concepts of tumor histogenesis have been represented in a tridimensional tetrahedron model of inter-relationships between the different components (Figure 29.1). Nevertheless, the most popular histogenetic scheme17 (shown in Figure 29.2) forms the basis for the current World Health Organization (WHO) classification of ovarian germ cell tumors (Table 29.1).18

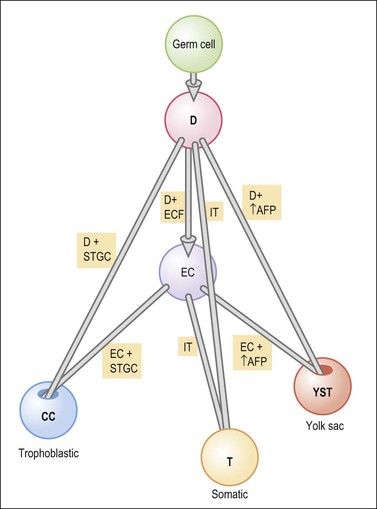
Figure 29.1 Tetrahedron model of germ cell histogenesis. D, dysgerminoma; D+ECF, dysgerminoma with early carcinomatous features; EC, embryonal carcinoma; D+STGC, dysgerminoma with syncytiotrophoblastic giant cells; D+↑AFP, dysgerminoma with elevated α-fetoprotein; IT, immature teratoma; EC+STGC, embryonal carcinoma with syncytiotrophoblastic giant cells; EC+↑AFP, embryonal carcinoma with elevated α-fetoprotein; CC, choriocarcinoma; T, mature teratoma; YST, yolk sac tumor.11 (Modified with permission of Taylor & Francis.)
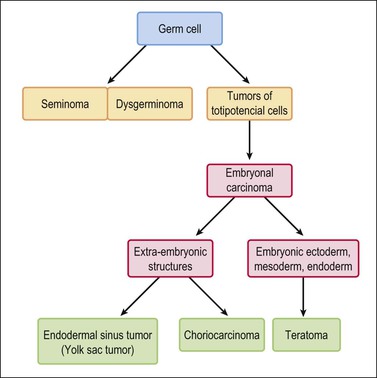
Figure 29.2 Germ cell tumors. Traditional histogenetic scheme.17 (Data from Teilum G. Special tumors of the ovary and testis. Comparative pathology and histological identification. JB Lippincott, Philadelphia, 1976: p. 68)
MGCTs generally occur in younger women (75% in women under 30 years) and account for two-thirds of ovarian cancers in the first two decades.1,19 The tumors are usually large (median size is 16 cm). Bilateral tumors are rare except dysgerminomas (10–20% bilaterality). Abdominal enlargement and pelvic pain are the most common presenting symptoms. Teenagers who present with abdominal masses and who have never menstruated should be evaluated for the possibility of a gonadoblastoma that has undergone malignant progression. Preoperative karyotyping can be helpful to identify underlying chromosomal abnormalities. Human chorionic gonadotropin (hCG) and α-fetoprotein (AFP) levels are useful markers in the diagnosis and in monitoring the postoperative course of these patients. Moreover, recent stem cell research has provided several highly diagnostic pluripotency markers, including transcription factors and cytoplasmic/membranous proteins that are sequentially expressed in MGCTs according to their differentiation stage.20,21
MGCTs are now frequently cured by fertility-sparing surgery and combination chemotherapy including bleomycin, etoposide, and cisplatin.22 For patients with advanced stage disease, maximum cytoreductive surgery appears to be beneficial. About 60–75% of women have stage I disease and 25–30% have stage III disease. For patients with early stage disease, cure rates approach 100%. For those with advanced stage disease, cure rates are reportedly at least 75%.22
The relative frequencies of the various types of MGCTs are shown in Table 29.2. Most MGCTs occur in a pure form, but approximately 15% of cases contain two or more types. The prognosis of patients with a mixed MGCT usually reflects that of its most malignant component. Therefore, it is important to sample these tumors extensively, particularly areas with different gross appearance. One section per every centimeter in tumor diameter is recommended. Most MGCTs are composed of primitive tissues (i.e., dysgerminoma, YST); less frequently, malignant neoplasms of the adult type arise in dermoid cysts, usually in older patients.
Table 29.2
Approximate Frequency of MGCTs of the Ovary
| • Dysgerminoma | 33% |
| • YST | 10% |
| • EC | — |
| • Polyembryoma | — |
| • Choriocarcinoma | <1% |
| • Teratoma | |
| • Immature | 36% |
| • Malignancy in dermoid cyst | 5% |
| • Monodermal | — |
| • Mixed | 15% |
Dysgerminoma
Clinical Features
Dysgerminoma is one of the most common MGCTs of the ovary, but it accounts for only 1–2% of all ovarian cancers. They occur almost exclusively in children and young women. The average patient age is 22 years.23 Most patients present with a rapidly growing abdominal mass that often causes lower abdominal pain or pressure and may simulate pregnancy. Premenarchal patients with a pelvic mass should have their karyotype determined, as approximately 5% of dysgerminomas arise from gonadoblastomas in phenotypic females with abnormal gonads. These patients have pure gonadal dysgenesis (46,XY, bilateral streak gonads) or mixed gonadal dysgenesis (45,X/46,XY, unilateral streak gonad, contralateral testis). The estimated risk of malignancy is 28% by the age of 20 years for the former patients and 19% risk by the same age for the latter. Therefore, prophylactic removal of the gonads is recommended in both groups of patients at an early age.1 Almost all patients with dysgerminoma have elevated serum levels of lactic dehydrogenase (1 and 2 fractions) at presentation.24 In rare cases (3–5%), dysgerminomas may produce hCG and these patients may present with endocrine symptoms that are characteristically estrogenic (menstrual irregularities, isosexual pseudoprecocity, pseudopregnancy), but rarely androgenic.25 Serum levels of CA125, placental-like alkaline phosphatase (PLAP), and neuron-specific enolase (NSE) have been elevated in some cases. Paraneoplastic hypercalcemia has been described in some patients.26 One tumor occurred in a patient with a germline BRCA1 mutation.27 About two-thirds of dysgerminomas are stage IA at diagnosis; higher stage tumors involve the contralateral ovary (20%), pelvic and para-aortic lymph nodes, and/or the pelvic and abdominal peritoneum.
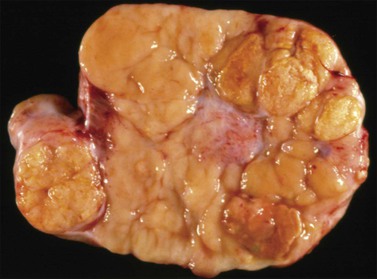
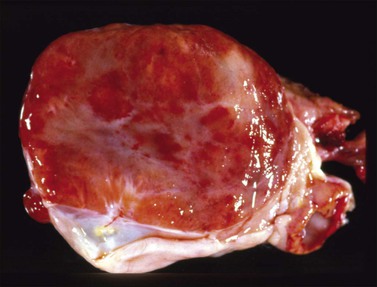
Macroscopic Features
Dysgerminomas are characteristically solid, and are well-encapsulated tumors with an average diameter of 15 cm. On section, they are lobulated, soft, and fleshy, and may appear gray-white or light tan (Figure 29.3). Areas of coagulative necrosis and hemorrhage typically associated with cystic change may be seen. Such areas should be sampled to rule out the presence of other types of MGCT. The presence of calcification suggests an underlying gonadoblastoma. Dysgerminoma is the only MGCT that has a significant rate of bilaterality. Gross involvement of the contralateral ovary is seen in 10% of cases, and in another 10% microscopic foci of tumor are found.1
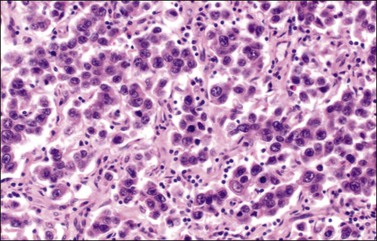
Microscopic Features
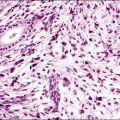
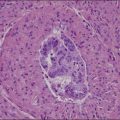
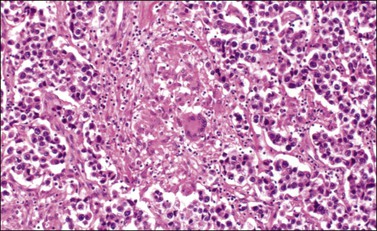
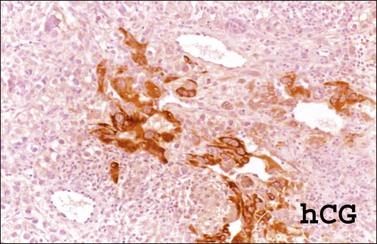
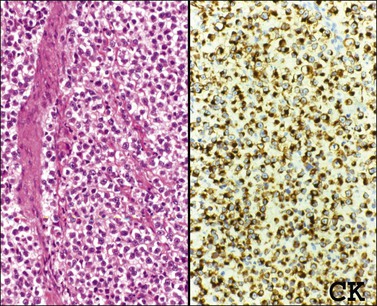
The microscopic appearance of dysgerminoma is identical to that of testicular seminoma and extragonadal germinoma. It is composed of a monotonous population of rounded cells resembling primordial germ cells in a predominantly diffuse or insular arrangement. The cells may also grow as trabeculae, cords, or small nests (Figure 29.4). Lack of intercellular cohesion may result in the formation of pseudoglandular spaces. The tumor cells are polygonal, with discrete cell membranes and abundant eosinophilic to clear, glycogen-rich, cytoplasm. The nuclei are large, central, and rounded, and they contain one or a few prominent nucleoli. Mitotic figures are often numerous. Aggregates of tumor cells are usually separated by thin fibrous septa almost always infiltrated by T-lymphocytes.28 In some tumors, lymphocytes are abundant and may form follicles with germinal centers. In approximately 20% of the cases, epithelioid sarcoid-like granulomas with multinucleated giant cells are present (Figure 29.5).1 About 5% of dysgerminomas contain syncytiotrophoblastic giant cells (SGCs) in the absence of any other non-germinomatous differentiation.25 Such tumors, which have the same prognosis as dysgerminomas in which SGCs are absent, should be sampled extensively to rule out foci of choriocarcinoma or EC. The SGCs have a perivascular location, are immunoreactive for human chorionic gonadotropin (hCG)(Figure 29.6), and are often associated with stromal luteinization. The luteinized stromal cells, which may be admixed with the tumor cells or located at the periphery of the neoplasm,29 are probably responsible for the estrogenic or androgenic symptoms that are found in some patients.
Immunohistochemistry
Dysgerminoma cells show cytoplasmic and membranous immunoreaction for vimentin, PLAP, CD117 (c-kit), and D2-40 (podoplanin); the membranous reaction is characteristic of dysgerminoma.30,31 There is nuclear immunoreaction for the stem cell/primitive germ cell nuclear transcription factors OCT3/4, NANOG, and SALL431–33 (Table 29.3). OCT3/4 is expressed very early during embryogenesis and has an essential role in blastocyst differentiation. However, when the female germ cells enter meiosis the expression of OCT3/4 is downregulated. OCT3/4 regularly shows positivity in dysgerminoma (Figure 29.7) but can also be expressed in EC and in some immature neural elements of ovarian teratoma.
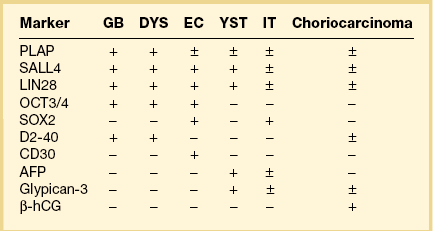
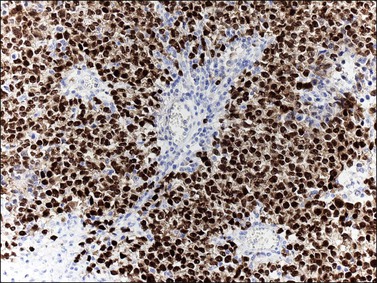
Figure 29.7 Dysgerminoma. OCT3/4-positive nuclear immunoreaction recognizes germ cells in poorly fixed specimen. (Courtesy of Dr. Francisco Nogales.)
SALL4 can also show positivity in EC, YST, and primitive areas of immature teratoma (IT).33 Dysgerminomas may exhibit limited cytoplasmic dot- or rim-like staining for cytokeratin (Figure 29.8) but epithelial membrane antigen (EMA) is negative. The SGCs that are found in a small percentage of dysgerminomas exhibit positive cytoplasmic staining for hCG (Figure 29.6).
Somatic Genetics
Dysgerminomas, like seminomas, show by comparative genomic hybridization frequent (75%) chromosome 12p gain, usually in the form of an isochromosome 12p.34 The c-kit gene encodes a tyrosine kinase receptor (KIT), which is required in normal spermatogenesis and is expressed in seminomas and dysgerminomas. c-kit point mutations localized to exon 17, codon 816 (Asp>Val), involving the phosphotransferase domain, have been identified in 25–50% of ovarian dysgerminomas.35–37 The mutations are in exon 17, not in the exon 11 location, which confers susceptibility to imatinib therapy.
Differential Diagnosis
Dysgerminoma should be distinguished from the solid type of YST, EC, clear cell carcinoma, and large cell lymphoma. The solid YST is almost always associated with other more typical patterns that are not seen in dysgerminomas. YSTs exhibit greater nuclear variation, contain hyaline bodies, immunoreact for AFP and glypican-3, and lack the lymphocytic infiltrate of dysgerminomas. Moreover, in contrast to dysgerminoma, YSTs fail to react for D2-40 and OCT3/4. The extremely rare EC shows larger cells with more ample nuclei that are more hyperchromatic and irregular than those of dysgerminoma. ECs are CD30 and cytokeratin positive, almost always contain SGCs, and lack the stromal infiltrate of lymphocytes. The distinction of dysgerminoma from clear cell carcinoma is discussed in Chapter 27. Large cell lymphomas may resemble dysgerminomas both grossly and microscopically. Lymphomas, however, are bilateral in approximately one-half of the cases, and simultaneous involvement of the ipsilateral tube occurs in 25% of them38 (see Chapter 30). Careful attention to the nuclear features and immunostains for PLAP, D2-40, SALL4, and lymphoid markers facilitate the correct diagnosis. Poorly fixed dysgerminomas may occasionally be misdiagnosed as poorly differentiated carcinomas, as the cellular features of the former tumors are lost.
Treatment and Prognosis
The treatment of patients with dysgerminoma is primarily surgical, including the resection of the ovarian tumor and complete surgical staging. Given that the tumor mainly affects young females, for whom preservation of fertility is important, unilateral adnexectomy with frequent patient follow-up (every 2–3 months for the first 2–3 years) is the treatment of choice. This conservative approach is currently used in young patients even in the presence of metastasis.39 Therefore, biopsy of an apparently normal contralateral ovary is not recommended as it increases the risk of infertility. Dysgerminoma is highly sensitive to chemotherapy, which is usually reserved for the treatment of recurrent disease.23,40 Although these tumors are equally sensitive to radiotherapy, its use has been discontinued in an effort to preserve patient fertility, and cisplatin-based chemotherapy is currently preferred.22 In young patients whose karyotype analysis reveals a Y chromosome, both ovaries should be removed.1 When the patient’s fertility is not a factor, hysterectomy with bilateral salpingo-oophorectomy is recommended; chemotherapy is subsequently administered in the case of higher stage tumors.22 Postoperative work-up includes the serum markers AFP and hCG, chest X-ray, and abdominopelvic CT or MRI. Recurrence in the retained contralateral ovary can occur in 5–15% of cases over the next 2 years.1 About 75% of the recurrences occur within the first postoperative year, and the most common sites are the peritoneal cavity and the retroperitoneal lymph nodes.1
Patients with dysgerminoma have an excellent prognosis. The 5 year survival rate is greater than 95% for patients with stage IA tumor, and 85% for those with advanced stage or recurrent tumor.22 Features associated with recurrence include a tumor diameter greater than 10 cm, and age younger than 20 years.41 Although the designation ‘anaplastic dysgerminoma’ has been applied to tumors with numerous mitoses, there is no evidence that such tumors are associated with a worse prognosis, and therefore this term is not recommended. Most dysgerminomas are nondiploid and ploidy is not a useful prognostic factor.42
Yolk Sac Tumor (Primitive Endodermal Tumor)
Clinical Features
Yolk sac tumors (YSTs) (also referred to as primitive endodermal tumors) account for approximately 10% of MGCTs and are almost as common as dysgerminoma in patients under the age of 20 years.1 They occur at a median age of 18 years and are rare over the age of 40 years.1,43–45 Patients most frequently present with abdominal pain and a large, rapidly growing, pelvic mass. Rupture or torsion of the tumor occurs in about 10% of patients. YSTs consistently produce AFP, which can be demonstrated in the patient’s serum, usually at a level greater than 1000 ng/ml. This substance, which is normally produced in the yolk sac of the developing embryo, may serve as a tumor marker in monitoring the patient during and after therapy. Lower serum levels of AFP may be encountered in other tumors that also occur in young females such as IT and Sertoli–Leydig cell tumors (SLCTs). CA125 and carcinoembryonic antigen (CEA) are also elevated in 100% and 10% of YSTs, respectively. Rare examples of YST have occurred in older patients in association with mucinous or endometrioid tumors; in such cases, a somatic cell rather than a germ cell origin is almost certain.6,8,9
Yolk sac tumor is a highly malignant neoplasm. At laparotomy, evidence of extraovarian spread has been reported in 30–70% of cases.44,45 In an older study,46 subclinical metastases were present in 84% of cases regarded as ‘stage I’ tumors, which most likely reflects incomplete staging. When the tumor spreads beyond the ovary, it involves the omentum and peritoneum, the para-aortic lymph nodes, and the liver.
Macroscopic Features
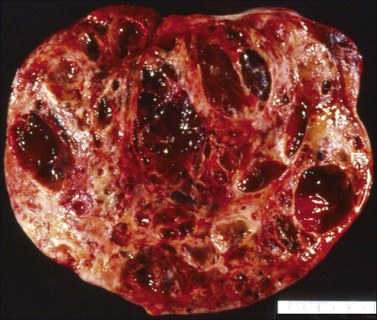
Yolk sac tumors are large, well-encapsulated masses, with an average diameter of 15 cm. The external surface is usually smooth and glistening. The sectioned surface is characteristically solid and cystic and shows soft, gray to yellow tissue with extensive areas of hemorrhage and necrosis (Figure 29.9). When a polyvesicular vitelline component is present, the tumor shows a microcystic appearance.1 Although YSTs are bilateral in less than 5% of patients, the contralateral ovary may contain a dermoid cyst in about 10% of cases.1
Microscopic Features
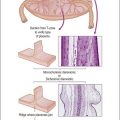
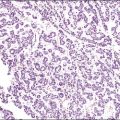
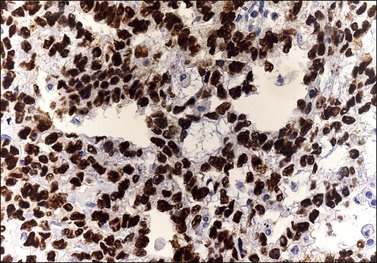
Although YSTs often exhibit a wide variety of microscopic patterns, most tumors have a reticular architecture reflecting extraembryonic differentiation.46,47 They are composed of a network of irregular spaces lined by primitive epithelial cells with glycogen-rich, clear cytoplasm, and large, hyperchromatic nuclei with prominent nucleoli; mitotic figures are numerous (Figure 29.10). The most characteristic feature is the presence of isolated papillary projections with a central blood vessel and peripheral sleeve of embryonic epithelial cells (Schiller–Duval bodies; Figure 29.11). Cross sections of this structure once were erroneously compared with immature glomeruli (Schiller’s mesonephroma).48 In fact, they closely resemble invaginations of yolk sac endoderm, as seen best in the rat placenta, forming the endodermal sinuses of Duval.46 Nevertheless, Schiller–Duval bodies are present in only 20% of cases.47 The term ‘endodermal sinus tumor’ is misleading, since the endodermal sinus is neither a structure present in human embryogenesis nor a constant feature of these neoplasms. Accordingly, the designation primitive endodermal tumor has recently been proposed.49 Another distinctive feature is the presence of brightly eosinophilic, periodic acid–Schiff (PAS)-positive, diastase-resistant hyaline globules (Figure 29.10). Occasionally, multiple small vesicles with eccentric constrictions resembling the yolk sac vesicle of the normal embryo are present, and the tumor is designated a polyvesicular vitelline tumor (Figure 29.12).46,50,51 This pattern recapitulates the subdivision of the primary yolk sac vesicle into a large component lined by flat cells (vestigial primary yolk sac) and a smaller component lined by taller epithelium that simulates the forerunner of the primitive gut and its appendages (secondary yolk sac vesicle) in normal embryogenesis (Figure 29.13). Like the normal yolk sac vesicle, the neoplastic yolk sac may give rise to tumors of embryonal type. These tumors recapitulate primitive gut (glandular YST) and primitive liver (hepatoid YST).52–56
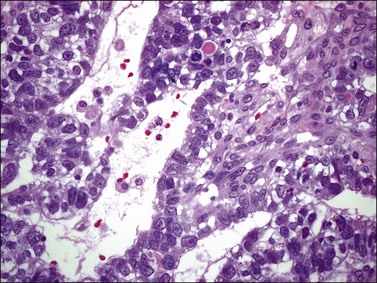
Figure 29.10 Yolk sac tumor. Reticular pattern. Irregular spaces lined by primitive epithelial cells with glycogen-rich, clear cytoplasm, and large, hyperchromatic nuclei with prominent nucleoli. A hyaline body is present (upper center). Mitotic figures are seen.
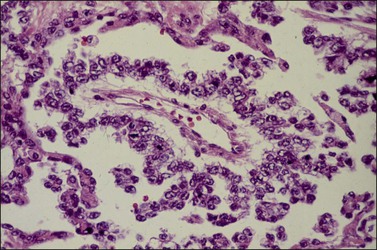
Figure 29.11 Yolk sac tumor. Schiller–Duval (glomeruloid) body. Central blood vessel and peripheral sleeve of embryonic epithelial cells.
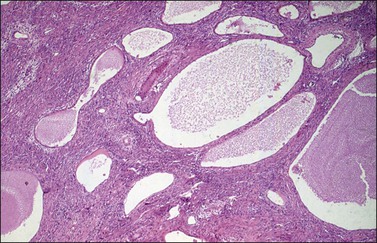
Figure 29.12 Yolk sac tumor, polyvesicular vitelline variant. The vesicles are lined by flattened epithelial cells and some of them exhibit eccentric constrictions.
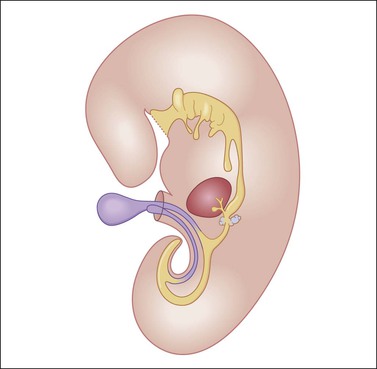
Figure 29.13 Diagram of a median longitudinal section through a 10 mm human embryo (about 6 weeks old). The primitive yolk sac (blue) is in continuity with the midgut. The rudiment of the liver and biliary tract appears attached to the ventral wall of the foregut. (Modified from Prat J, Pathology of the ovary. Saunders, Philadelphia, 2004. With permission.)
Endodermal-type glands may be found in approximately 50% of YSTs, often admixed with reticular and polyvesicular vitelline components.1 In some tumors, a glandular pattern predominates, and may appear as rounded cribriform aggregates of primitive epithelial cells (‘intestinal’ variant; Figure 29.14),52 or glands of ‘endometrioid’ type, which can be mistaken for typical or secretory endometrioid adenocarcinoma (endometrioid-like variant; Figure 29.15);53 foci of carcinoid tumor have been described in one of the latter tumors.57 Occasionally, the mature intestinal component may give rise to a mucinous carcinoid.58
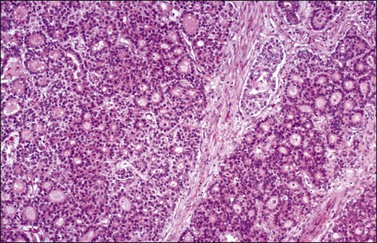
Figure 29.14 Yolk sac tumor, glandular variant, intestinal type. The tumor shows a cribriform pattern.
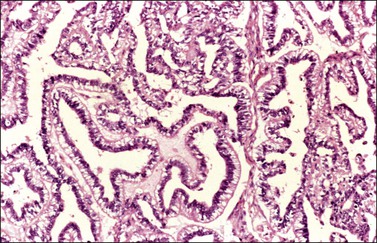
Figure 29.15 Yolk sac tumor, endometrioid-like glandular variant. The tumor glands resemble those of the secretory endometrium.
Although small foci of hepatoid differentiation are found in 16–48% of YSTs,55,56 neoplasms with a predominant hepatoid component are infrequent.54,59 These tumors are characterized by compact masses of large polyhedral cells with abundant eosinophilic cytoplasm, and round central nuclei with prominent single nucleoli, separated by thin fibrous bands, resembling hepatocellular carcinoma (Figure 29.16). Hyaline bodies are usually numerous. In some cases, glandular spaces containing mucin are present. Ultrastructural examination of hepatoid YSTs reveals features similar to hepatocellular carcinoma.54 A ‘parietal’ differentiation characterized by small extracellular accumulations of basement membrane material has been described in over 90% of YSTs.55 Rarely, YSTs may contain SGCs.
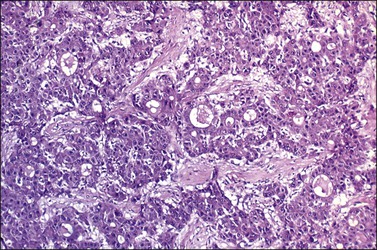
Figure 29.16 Yolk sac tumor, hepatoid variant. The tumor is composed of compact masses and nests of large polyhedral cells with abundant eosinophilic cytoplasm separated by a fibrous stroma. Note the striking resemblance to a hepatocellular carcinoma.
The loose stromal component of YST recapitulates the extraembryonic mesenchyme (magma reticulare),46 and may be the site of origin of the sarcomas that develop in some patients after chemotherapy.60,61
Immunohistochemistry
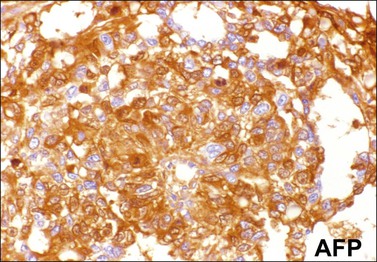
Similar to the primitive gut (secondary yolk sac) and its appendages, YSTs are immunoreactive for AFP (Figure 29.17), glypican-3 (Figure 29.18), SALL4 (Figure 29.19), villin, CDX2. and hepatocyte paraffin antigen 1.33 AFP is expressed often focally either as granular cytoplasmic deposits or delineating intracellular canaliculi. Glypican-3 immunoreaction is usually stronger than that of AFP;62 however, it is not as specific since it is expressed by other germ cell tumors and clear cell carcinoma.63 Nuclear transcription factor SALL4 is regularly expressed in both epithelium and mesenchyme32 and there is cytoplasmic reactivity of RNA-binding protein LIN28.64 Villin is also constantly expressed in membranes and cytoplasms of epithelial cells. The different endodermal elements may be immunoreactive for their corresponding tissue markers, i.e., hepatic components for hepatocyte paraffin antigen 1,65 intestinal elements for CDX2,49 and foregut-derived epithelia for thyroid transcription factor 1.33 However, YSTs are negative for OCT4, SOX2, D2-40, and CD30 (Table 29.3). Combined expression of AFP, glypican-3, villin, SALL4, or LIN28 makes an ideal panel for confirming the diagnosis.49
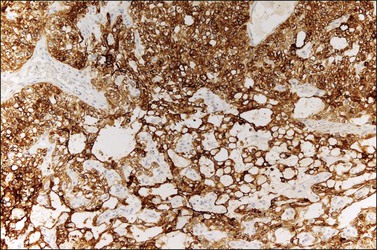
Differential Diagnosis
Because of their various microscopic patterns, YSTs can be confused with many other ovarian tumors, both primary and metastatic. The most common problems in differential diagnosis include clear cell carcinoma and endometrioid adenocarcinoma (see Chapter 27), the retiform variant of SLCT (see Chapter 28), and hepatoid carcinoma (see Chapter 27). YSTs should also be distinguished from other germ cell tumors such as dysgerminoma and the very rare EC. These differential diagnoses are discussed under those headings.
Treatment and Prognosis
The treatment of YST includes surgical exploration, unilateral salpingo-oophorectomy, and frozen section for diagnosis.22,44 Although gross metastases should be removed, a thorough surgical staging is not necessary because all patients need chemotherapy.44 Prior to combination chemotherapy, the prognosis for patients with YSTs was dismal despite apparently adequate surgery. In one large study of patients treated prior to 1975 the 3 year survival was only 13%.47 The development of the vincristine, dactinomycin, and cyclophosphamide (VAC) combination chemotherapy dramatically changed the outlook for patients with YSTs. Currently, cisplatin-based chemotherapy provides still better results. Survival rates have approached 100% for patients with stage I tumors, and 75% for patients with higher stage tumors.22 Serum AFP can be used to monitor the response to treatment and to detect tumor recurrence. Adverse prognostic factors include tumor stage II or higher, gross residual tumor after surgery, and ascitic fluid of more than 100 ml.45 The good long-term outcome in two polyvesicular-vitelline tumor (PVVT) cases supports prior evidence that this form of YST may be more indolent than the conventional forms of the tumor.51
Embryonal Carcinoma
Embryonal carcinoma (EC) is a very rare ovarian tumor that is morphologically identical to EC of the testis. For many years, it was confused with YST, which it resembles.66 From a histogenetic viewpoint, EC has been considered a pluripotent stem cell tumor capable of differentiating along different pathways.67 The patients are young (median age, 12 years), present with an abdominal mass, and consistently have a positive pregnancy test as a result of hCG production by the tumor.66 Premenarchal patients have endocrine manifestations in about half of the cases; usually, they include isosexual pseudoprecocity, irregular bleeding, amenorrhea, or hirsutism. Laparotomy reveals peritoneal spread in 40% of cases.66 Grossly, ECs are large (17 cm), unilateral tumors that exhibit solid and variegated cut surfaces. Foci of hemorrhage and necrosis are common. Histologically, ovarian EC resembles testicular Embryonal carcinoma; it is composed of large primitive cells distributed in solid masses, often exhibiting central necrosis, gland-like spaces, and papillae. The nuclei are round and vesicular. Mitotic figures, including atypical forms, are numerous. Hyaline globules are usually present. SGCs immunoreactive for hCG are almost always present (Figure 29.20).
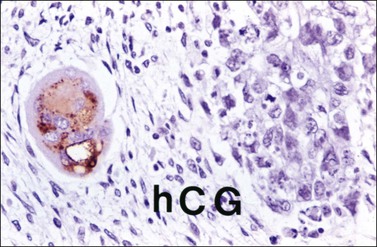
Figure 29.20 Embryonal carcinoma. A syncytiotrophoblastic giant cell shows positive immunostaining for hCG.
Embryonal carcinoma is most often found in the ovary as a component of a mixed germ cell tumor. The tumor cells are immunoreactive for cytokeratin, PLAP, CD30, NSE, AFP, and hCG. The last two substances can be used as tumor markers. Nuclear transcription factors OCT3/4 and SALL4 are positive and SOX2 is variably positive.31,32,68 Immunoreaction for EMA is negative. Most ECs contain an isochromosome, i12p. EC should be distinguished from other MGCTs (dysgerminoma, YST), juvenile granulosa cell tumor (JGCT), and undifferentiated carcinomas in the surface-epithelial category. In contrast to EC, the last two tumors rarely produce AFP and usually lack SGCs. Unilateral salpingo-oophorectomy is the recommended surgical procedure. Although these tumors are highly malignant, cisplatin-based combination chemotherapy has resulted in some long-term survivors.
Polyembryoma
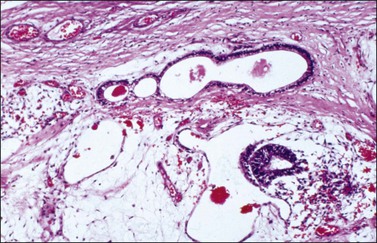
Rare cases of MGCT contain large numbers of embryoid bodies in various stages of development, typically distributed in a primitive mesenchymal stroma.69–72 Only 15 ovarian polyembryomas have been reported in the English literature, most of them found as components of mixed germ cell tumors in children or young women.72 Serum levels of AFP and hCG may be elevated. Grossly, polyembryomas are unilateral large tumors with a microcystic cut surface. Microscopically, the embryoid bodies resemble perfect or imperfect early embryos and contain germ discs, yolk sacs, amniotic cavities, chorionic elements, and extraembryonic mesenchyme in an edematous stroma (Figure 29.21). Well-differentiated enteric glands and mature or immature hepatic tissue, which may secrete bile, are found in some cases. The yolk sac components of the embryoid bodies and the nests of liver cells are immunoreactive for AFP. The behavior of polyembryomas is similar to that of other MGCTs. Conservative surgery and combination chemotherapy is the recommended treatment.
Nongestational Choriocarcinoma
Pure nongestational choriocarcinoma of the ovary is an exceedingly rare and highly malignant tumor that develops before puberty. It accounts for less than 1% of MGCTs.1 More frequently, choriocarcinoma is seen as a component of a mixed MGCT73 (only 45 pure cases have been reported).74 Clinically, these patients present with abdominal enlargement and pain and, occasionally, there is hemoperitoneum simulating a tubal pregnancy.75 The pregnancy test is positive and the elevated serum level of hCG may lead to isosexual pseudoprecocity in children or menstrual abnormalities in older patients.75 Serum β-hCG levels range from hundreds to more than 2,000,000 mIU/ml.74 Grossly, choriocarcinomas are large (4–25 cm), hemorrhagic, and friable tumors. Microscopically, much of the tumor is hemorrhagic and necrotic. The viable areas show cytotrophoblastic and syncytiotrophoblastic cells arranged in a plexiform pattern (Figure 29.22). The former cells are mononucleated with vesicular nuclei, abundant clear cytoplasm, and well-defined borders; the syncytiotrophoblastic cells have abundant vacuolated basophilic or amphophilic cytoplasm and several dark nuclei. In some tumors, the plexiform pattern may not be apparent.1 Intermediate trophoblastic cells are present in some cases. Vascular invasion is frequent. The syncytiotrophoblastic cells are immunoreactive for hCG, cytokeratins, human placental lactogen, PLAP, EMA, CD10, CEA, α-inhibin, and GLP-3.
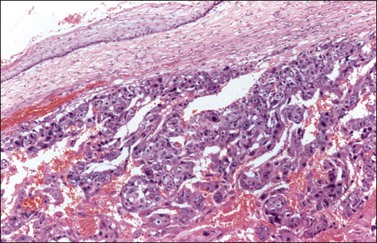
The clinical symptoms and histologic picture of germ cell-derived choriocarcinomas are similar to those of gestational choriocarcinoma; however, the remarkable response to chemotherapy (methotrexate or actinomycin D) associated with the latter tumors does not occur.76 Although gestational choriocarcinomas of the ovary are almost invariably metastatic from uterine or tubal choriocarcinomas, they may occasionally follow an ovarian pregnancy.1,75 The finding of a corpus luteum of pregnancy may be of help in establishing the gestational nature of the tumor. Similarly, the presence of a paternal component by DNA analysis or HLA typing is characteristic of gestational choriocarcinomas.77–79 Rare cases of poorly differentiated carcinomas with choriocarcinomatous differentiation and hCG secretion have been reported.8,80 Unilateral salpingo-oophorectomy followed by combination chemotherapy has resulted in cures or prolonged remissions.
Teratomas
Teratomas are germ cell tumors composed of an array of tissues derived from two or three embryonic layers (ectoderm, mesoderm, and endoderm) in any combination. The great majority of teratomas are benign cystic tumors that contain mature elements and are designated mature cystic teratomas or dermoid cysts. The presence of any immature tissue warrants a diagnosis of IT, which is a potentially malignant neoplasm. Nevertheless, typical dermoid cysts containing very small foci of immature tissue have a benign behavior and should not be reported as grade 1 IT.81 Rarely, teratomas may be predominantly or exclusively composed of endodermal or ectodermal tissues (monodermal teratomas).1 The pathogenesis of teratomas has always excited speculation because of their exotic composition. Cytogenetic analysis, using chromosome-banding techniques, has revealed that ovarian teratomas are parthenogenetic tumors that develop from a single germ cell after its first meiotic division.82 Also, genetic analysis of mature ovarian teratomas has demonstrated genotypic differences between homozygous teratomatous tissue and heterozygous host tissue.83
Mature Teratomas
Clinical Features
Mature teratomas, usually dermoid cysts, represent the most common ovarian neoplasms, accounting for 27–44% of all primary ovarian tumors and over two-thirds of all ovarian tumors in patients under 15 years of age.1,84 Most patients are between 20 and 40 years, but the tumors occur at all ages. Although they usually occur in pure form, dermoid cysts are found grossly within 26% of ITs and in the ovary contralateral to a MGCT in about 5–10% of cases.1,81 Most patients with mature teratomas present the typical signs and symptoms of benign ovarian tumors, but approximately 25% are asymptomatic and the tumors are discovered on routine examination. Radiologically, dermoid cysts are characterized by central areas of very low density enclosed by a ring of increased capsular density.85 Calcified tissues including bone and teeth may be present and facilitate the radiologic diagnosis. Dermoid cysts may undergo torsion or rupture, with acute abdominal symptoms, or hemoperitoneum. Leakage of the cyst contents into the abdominal cavity results in granulomatous peritonitis, which may mimic tuberculosis or metastatic carcinoma.86 Peritoneal melanosis may also occur. Autoimmune hemolytic anemia has been reported in some cases. Mature solid teratomas may occasionally produce mature peritoneal glial implants (grade 0), which have an excellent prognosis.87
Although mature teratomas are benign tumors, malignant transformation may occur in approximately 1–2% of cases, usually in postmenopausal women (mean age, 51–62 years). Almost any component may become malignant, but squamous cell carcinoma accounts for 80% of the cases.1,88
Macroscopic Features
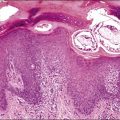
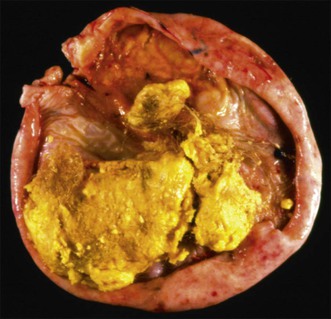
Mature teratomas are almost always cystic tumors and are bilateral in approximately 15% of cases. They range in size from 0.5 to 40 cm (average 15 cm) and have a smooth grayish white external surface. On section, a unilocular or, less frequently, a multilocular cyst lined by skin appears filled with sebaceous material and hair (Figure 29.23). The cyst contents are liquid at body temperature but they solidify at room temperature. A solid nodule composed of fat tissue with teeth or bone usually protrudes into the cyst lumen (Rokitansky’s protuberance). Dermoid cysts may contain a great variety of mature somatic tissues that may be seen grossly, such as brain tissue, bone, cartilage, adipose tissue, mucinous cysts, and thyroid. In rare cases, partially developed organs may be identified. Rarely, mature teratomas are completely solid and differ grossly from ITs by the absence of hemorrhagic and necrotic areas.89 Twenty-two cases of a rare type of mature solid teratoma resembling a malformed human fetus have been reported. This form of teratoma has been designated homunculus or fetiform teratoma.90 Dermoid cysts that are unusually large and adherent to surrounding structures or that contain mural nodules, or extensive necrosis and hemorrhage, are suspicious of harboring a secondary malignant tumor.1
Microscopic Features
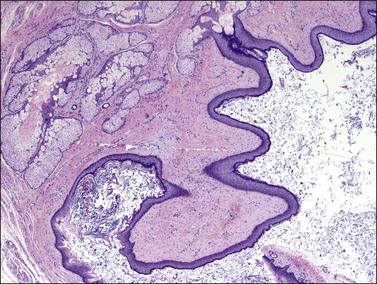
Mature teratomas are composed of adult and sometimes fetal-type tissue derived from two or three embryonic layers. Ectodermal elements predominate in almost all cases, particularly epidermis (Figure 29.24), pilosebaceous structures, teeth, sweat glands, and neural tissue (usually glia).1 In mature solid teratomas, well-differentiated glia may constitute the predominant component. Other ectodermal derivatives, such as cerebrum, cerebellum, choroid plexus, and retina, may also be found. The common presence of cranially derived tissues in one study suggests that the type of differentiation in mature cystic teratomas parallels anterior embryonic plate development.91 Frequent endodermal elements include respiratory and gastrointestinal structures and thyroid tissue. The most common mesodermal derivatives are smooth or striated muscle, adipose tissue, bone, teeth, and cartilage. Less frequently, adrenal, pituitary, pancreatic, renal, thymic, mammary, or prostatic tissue are encountered.92 Although mitotic figures are rare, they may be seen in recognizable fetal tissues.1 The various tissues exhibit an organoid arrangement; for instance, cartilage, mucinous glands, and respiratory epithelium may appear together as in normal bronchus. Rarely, a florid benign vascular proliferation may be found in association with glial tissue and can be misinterpreted as an immature component.93
Benign tumors, such as carcinoid, struma, adrenal adenoma, prolactinoma, glomus tumor, sebaceous adenomas, and nevus, may develop within a dermoid cyst.1,94–97 Six mucinous–intestinal tumors associated with pseudomyxoma peritonei arose in ovarian dermoid cysts.98,99 The great majority of secondary cancers are invasive or, rarely, in situ squamous cell carcinomas (see later). Sebaceous carcinomas, sarcomas, and melanomas are occasionally seen.1,100
Treatment and Prognosis
Dermoid cysts are treated conservatively, particularly in children and young women. Ovarian cystectomy is adequate treatment. With the exception of rare cases in which a secondary malignant tumor develops, dermoid cysts are benign tumors even if they contain microscopic foci of immature neural tissue.81 Similarly, mature solid teratomas, including those associated with mature implants, have a favorable clinical course.87 Patients with squamous cell carcinoma that has disseminated beyond the ovary typically have a poor prognosis.88
Immature Teratomas
Clinical Features
Immature teratomas (ITs) are probably the most common MGCTs of the ovary, representing about 36% of such tumors and 1% of ovarian cancers in general.101–105 Only 3% of ovarian teratomas are immature. They occur predominantly in children and young women, at a median age of 18 years, who have a palpable abdominal mass and complain of pelvic or abdominal pain. In rare cases, ITs are preceded by ipsilateral dermoid cysts that have been removed some years previously.81 Serum AFP can be elevated in patients with pure IT but levels only rarely exceed 1000 ng/ml. Higher levels almost always indicate the presence of a YST component.106 Other tumor markers include hCG, NSE, CEA, and CA125. Extraovarian spread, usually in the form of peritoneal implants and less frequently as lymph node metastases, is found in one-third of patients at the time of surgery.87,102–105 Ruptured or adherent tumors are most commonly associated with peritoneal implants.87
Macroscopic Features
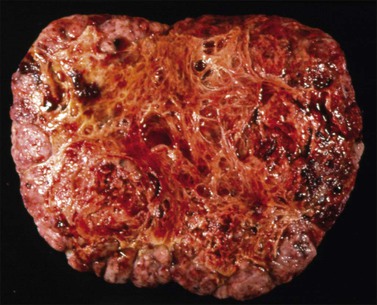
Immature teratomas are typically large (mean size, 18 cm), encapsulated tumors that are ruptured in nearly 50% of cases.102 On section, they are predominantly solid, but small cysts containing mucin or blood are often seen (Figure 29.25). The solid areas, which usually correspond to neural tissue, are soft, fleshy, and gray to pink and frequently show hemorrhage and necrosis. Foci of bone and cartilage may be present. Grossly visible dermoid cysts are found in 26% of cases.81 The tumor is almost always unilateral, but the contralateral ovary contains a dermoid cyst in about 10% of patients.1
Microscopic Features
Most tumors contain a mixture of mature and immature embryonal elements. The embryonic-type tissue varies from microscopic foci to large amounts and is composed predominantly of neuroepithelial rosettes (Figure 29.26) and tubules admixed with areas of hypercellular glia with numerous mitoses. The neuroepithelial rosettes are lined by crowded cells (Figure 29.27) and may be pigmented. Islands of immature cartilage, bone, skeletal muscle, and glandular structures are distributed through a myxoid stroma. Embryonal endodermal elements, such as hepatic tissue and intestinal-type epithelium, may also be found.56 The presence of these elements in grade 3 ITs (see later) with markedly elevated serum AFP levels should alert the pathologist to perform a diligent search for a YST component. In childhood ITs, such a component may develop as well-differentiated glandular or hepatoid YST.106 Two cases of ITs composed predominantly of endodermal elements without neural component have been described (Figure 29.28).107 Rarely, immature renal tissue,108 SGCs, and yolk sac-like tissue may be found.102,109 Vascular proliferation occurs in relationship with neural tissues.93
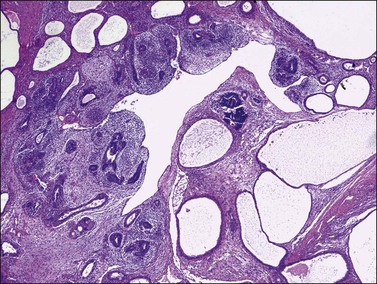
Figure 29.26 Immature teratoma. The tumor shows both immature neuroepithelial and mesenchymal elements.
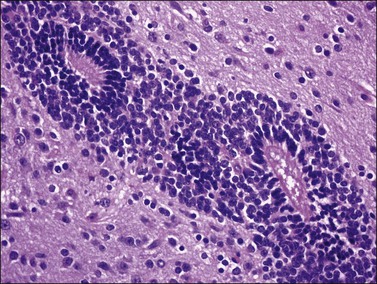
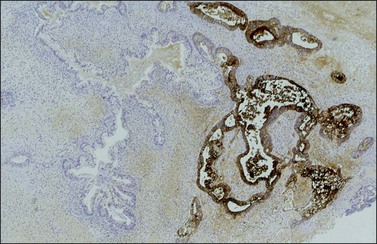
Figure 29.28 Immature endodermal teratoma. The tumor is exclusively composed of endodermal elements without neural component. The intestinal-like glands show positive AFP immunoreaction.
The relative amount of primitive neuroepithelial tissue is an important factor in grading and determining the prognosis of ITs.89 Grading is performed by a subjective, semiquantitative analysis of the relative amount and atypicality of the immature neural tissue (neuroepithelial tubules and neural blastema) present in the tumor. Grade 1 has been applied to tumors with rare foci of immature neural tissue that occupy less than one low-power field (LPF) (40×) in any slide, grade 2 to tumors with moderate amounts of immature neural tissue occupying more than 1 but less than 4 LPF in any slide, and grade 3 to tumors with immature neural tissue occupying 4 or more LPFs in any slide (Table 29.4).102 The grading system is also applied to peritoneal implants (Figure 29.29) and lymph node metastases. A two-grade system (low grade and high grade), which combines grades 2 and 3, has been proposed but has not been formally adopted.109 ITs as well as mature solid teratomas may be associated with mature (grade 0) peritoneal implants (so-called peritoneal gliomatosis).87 The pathogenesis of this phenomenon is unclear and both, direct seeding of immature cells from the primary tumor with subsequent differentiation in the peritoneum87 and peritoneal metaplasia110 from stem cells have been proposed. In some cases, the glial implants appear admixed with endometriosis.111 As mature and immature implants may coexist, generous sampling of the peritoneal lesions is recommended.1 The pelvic and para-aortic lymph nodes may also contain similar glial tissue.112

Immunohistochemistry
SALL4 is positive in both intestinal and neural immature tissues.63 SOX2, however, identifies more specifically immature neural areas, highlighting them, and is helpful for grading of immature teratoma.33 Glypican-3 may also exhibit a patchy neuroepithelium immunoreaction. The neuroectodermal tissues are also immunoreactive for several neural markers, including glial fibrillary acidic protein (GFAP), NSE, S-100 protein, neurofilament protein, synaptophysin, and nerve growth factor receptor. The intestinal-type glands and hepatic tissue stain strongly for AFP.56 SGCs stain positively for hCG.
Somatic Genetics
Immature teratomas seem to originate from premeiotic germ cells.113 By comparative genomic hybridization, ITs exhibit fewer DNA changes than other ovarian germ cell tumors. No gain of 12p or i(12p) has been identified.114 Grades 1–2 ITs are diploid in 90% of cases, whereas most grade 3 tumors are aneuploid.115
Differential Diagnosis
Immature teratoma is distinguished from mature solid teratoma by the presence of immature embryonal tissue. The finding of fetal cartilage and mitotically active cerebrum or cerebellum does not justify the diagnosis of immature teratoma.89 Typical dermoid cysts may contain minor foci of immature neural tissue.81 Malignancy arising in a dermoid cyst occurs most commonly in postmenopausal patients. In contrast to IT, only one element, usually squamous epithelium, is malignant (see later). IT is a frequent component of mixed MGCTs; therefore, the presence of other primitive elements such as dysgerminoma or YST should be excluded by generous sampling. In primitive mixed tumors occurring in children and adolescents, the YST components may be of the glandular (enteric) or hepatoid type and, thus, difficult to distinguish from the embryonal or fetal gastrointestinal glands or hepatocytes that can be found in pure ITs.106 The additional presence of extraembryonal patterns (reticular or polyvesicular vitelline patterns) of YST, as well as the typically elevated serum levels of AFP, facilitate the differential diagnosis.106 The differential diagnosis between immature teratomas and malignant mixed müllerian tumor with heterologous elements and primitive neuroectodermal tumors is discussed in Chapter 27 and in the following sections, respectively.
Treatment and Prognosis
In a young patient whose tumor appears localized to a single ovary (stage IA), a unilateral salpingo-oophorectomy and surgical staging should be done. If preservation of fertility is not an issue, or the contralateral ovary or uterus is involved, a total abdominal hysterectomy and bilateral salpingo-oophorectomy should be carried out. Any peritoneal lesions should be sampled carefully and submitted for histologic evaluation. The primary prognostic factor in patients with immature teratoma is the tumor grade. Patients with stage IA, grade 1 tumors or those with exclusively mature (grade 0) glial implants have an excellent prognosis and are treated by surgery alone. Patients with grade 2 or 3, stage IA, or those with immature implants should receive combination chemotherapy.103–105 The VAC regimen has been replaced by platinum-containing regimens such as cisplatin, etoposide, and bleomycin (BEP). In patients with no residual tumor after surgery, the survival rate is 90–100%.103,116 Prognosis is less favorable for patients with residual gross tumor or recurrent IT. Following chemotherapy, high-grade implants typically disappear, or are replaced by mature, necrotic, or fibrotic tissue.87 Mature implants may continue to grow (‘growing teratoma syndrome’) and may require reoperation. However, on rare occasions, gliomatosis peritonei can induce a florid vascular proliferation that may result in peritoneal hemorrhage and shock.117 Rare cases of glioblastoma multiforme arising from peritoneal gliomatosis have been described.118 Cytogenetically abnormal or aneuploid ITs may be associated with a worse prognosis than karyotypically normal or diploid tumors.119
According to a report from the Pediatric Oncology Group, surgery alone is curative in children and adolescents with IT of any grade, and chemotherapy should only be used for cases with tumor recurrence.106 In such cases, the presence of a YST component rather than the grade of the IT component is the only valid predictor of recurrence.106
Monodermal Teratomas
Struma Ovarii
Clinical Features
Over 20% of dermoid cysts may contain thyroid tissue on microscopic examination, but the term struma ovarii is only applied to teratomas composed either exclusively or predominantly of such tissue. Struma ovarii is the most common form of monodermal teratoma accounting for 3% of all ovarian teratomas.120 Most patients are in their fifth decade.120 Along with a palpable abdominal mass, patients may present with ascites and hydrothorax (‘pseudo-Meigs’ syndrome’), cervical thyroid enlargement, and hyperthyroidism with high pelvic iodine uptake.121
Macroscopic Features
Most tumors are unilateral and range from 0.5 to 10 cm in diameter. On section, the thyroid tissue appears solid, gelatinous, and typically brown or green-brown (Figure 29.30). Usually, struma occurs in a pure form; less frequently, it is associated with a dermoid cyst or is a component of strumal carcinoid. Occasionally, strumas are found in the wall of mucinous cystadenomas or are admixed with Brenner tumor.122 Some cases of struma ovarii appear as unilocular or multilocular cysts containing brown to green gelatinous fluid.123 Rarely, the contralateral ovary contains a dermoid cyst or even another struma.
Microscopic Features
Struma ovarii may appear as normal thyroid tissue or an adenoma of the macrofollicular, microfollicular, embryonal, or mixed type (Figure 29.31). Clear cell, oxyphilic, and solid tubular forms of thyroid adenoma also occur.124 Under polarized light, birefringent calcium oxalate crystals are often found within the colloid, which is immunoreactive for thyroglobulin. Tumor cells also show immunohistochemical expression of TTF-1. They exhibit only mild nuclear atypia and mitotic figures are rare. In cystic strumas, thyroidal differentiation may not be readily apparent. The thin fibrous septa are lined by flat to cuboidal nonspecific epithelial cells and the characteristic thyroid follicles may be sparse in the cyst wall.123
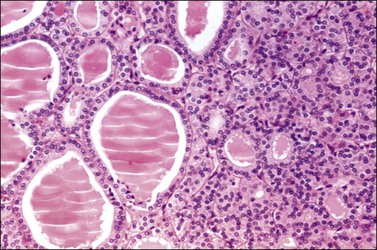
Figure 29.31 Struma ovarii. The tumor resembles a thyroid adenoma with mixed macrofollicular and microfollicular pattern.
The criteria for malignant change in struma ovarii have not been uniformly established. Malignant strumas are rare, and most of them are characterized by a papillary architecture (or a follicular pattern with occasional papillae) and the presence of cells with overlapping ground-glass nuclei.125 Follicular carcinomas are difficult to diagnose since struma has no true capsule but is surrounded only by stroma, and capsular invasion cannot be a criterion for malignancy;126 also, true vascular invasion is exceedingly rare and has not proved to be a valid prognostic parameter.125,126 Poorly differentiated carcinomas are rare.125
Differential Diagnosis
Cystic struma is often mistaken for a serous or mucinous cystic tumor; however, the green to brown color of the cyst contents and, microscopically, the focal presence of thyroid follicles on the cyst wall, and the occasional association with other teratomatous elements in the struma permit its identification.123 Cysts closely resembling thyroid follicles may be found in endometrioid carcinomas, clear cell carcinomas, Sertoli-Leydig cell tumors (SLCTs), and pregnancy luteomas, but other typical morphologic features in such tumors almost always facilitate their distinction. A predominant microfollicular pattern may be misinterpreted as the Call–Exner bodies of a granulosa cell tumor (GCT). Oxyphilic strumas may be mistaken for other oxyphilic tumors, such as steroid cell tumors, and strumas with a solid tubular pattern may simulate Sertoli cell tumors. In these cases, the finding of true thyroid follicles and calcium oxalate crystals and immunoreactivity for thyroglobulin and TTF-1 confirms the diagnosis of struma.124 The rare case of ovarian metastasis from carcinoma of the thyroid gland is usually diagnosed after obtaining additional clinical data.
Treatment and Prognosis
Benign struma ovarii is treated by oophorectomy. Patients with malignant struma should be treated by oophorectomy and removal of as much extraovarian tumor as technically feasible. In some of these patients, extraovarian disease has been treated successfully with 132I.127
Most cases of struma have been clinically benign. Although in the older literature 5–10% of strumas have been considered malignant, only a few cases have been associated with metastasis or recurrence. In the remaining cases, the diagnosis of malignancy has been based exclusively on microscopic criteria, and some tumors originally thought to be malignant are now interpreted as benign strumal carcinoids.128 Clinically malignant struma ovarii can develop from histologically malignant strumas, adenomas of various types, or even benign thyroid tissue. In fact, ovarian tumors with subtle atypical nuclear features, which are suggestive of but not diagnostic for the follicular variant of papillary thyroid carcinoma, still can metastasize.129 No single histologic feature can reliably predict a malignant behavior, but ascites, larger tumor size (over 12 cm), ovarian surface invasion, and extensive adhesions favor a malignant clinical behavior.125,130 Rare cases of benign-appearing struma implants on the peritoneum (so-called peritoneal strumosis) have been associated with indolent clinical course and prolonged survival.131 These cases have also been designated ‘highly differentiated follicular carcinomas.’132 In the largest series reported of malignant struma ovarii, the overall survival rate was 89% at 10 years and 84% at 25 years, indicating the need for routine long-term follow-up.125
Carcinoids
Clinical Features
Ovarian carcinoids are the second most common monodermal teratomas. They may occur in pure form (15%) or, more often, combined with other teratomatous elements (85%) such as a dermoid cyst or a struma ovarii (strumal carcinoid).128 They may also be a component of a mucinous cystic tumor or a Brenner tumor. The pure carcinoids are divided into insular, trabecular, and mucinous types, and resemble carcinoids of the gastrointestinal tract.133–135 Although ovarian carcinoids may occur at any age, most patients are perimenopausal or postmenopausal women who present with nonspecific symptoms, such as abdominal enlargement and pain. The carcinoid syndrome (facial flushing, diarrhea, bronchospasm, hypertension, and edema secondary to carcinoid heart disease) has been described in 30% of cases of insular carcinoid, and less often in trabecular (13%) and strumal (3.2%) carcinoids.136,137 It typically disappears after removal of the tumor.133 Patients with the syndrome usually have tumors larger than 7 cm in diameter. Other clinical findings include chronic constipation, hypoglycemia, and hyperthyroidism associated with strumal carcinoids. Occasionally, androgenic or estrogenic symptoms related to stromal luteinization or peripheral steroid cell proliferation may develop.1
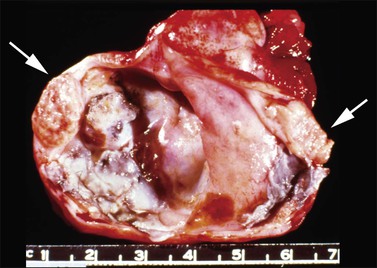
Gross Features
Although ovarian carcinoids are unilateral tumors, the contralateral ovary contains a dermoid cyst, a mucinous tumor, or a Brenner tumor in 15% of cases.1 Carcinoid tumors are firm, yellow-tan, and predominantly solid tumors. Exclusively cystic tumors are rare. Occasionally, the carcinoid may form a nodule that protrudes into the lumen of a dermoid cyst (Figure 29.32) or a mucinous cystic tumor,138 or be admixed with a Brenner tumor or struma (strumal carcinoid). In some cases, the carcinoid and struma can be separately recognized on gross examination.
Microscopic Features
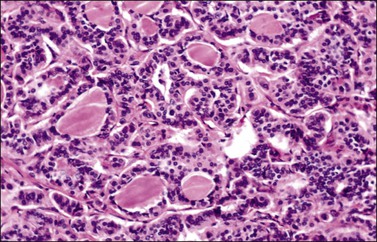
All ovarian carcinoids are composed, at least in part, of round to cuboidal neuroendocrine cells that have uniform round nuclei, coarse chromatin granules, and small nucleoli. Mitotic figures are rare. The cytoplasm is characteristically abundant and varies from clear to eosinophilic. Four histologic types of carcinoid tumor may be found in the ovary. The insular type is the most common, accounting for 26–53% of cases.133,136,137 It resembles midgut carcinoid and is characterized by cellular nests or islands separated by a fibromatous stroma (Figure 29.33). The cellular islands often show small tubular acini lined by columnar cells, particularly at their periphery. The acinar lumens contain eosinophilic secretion, which may be calcified. The peripheral cells of the islands and the acinar cells contain red-brown argentaffin granules. Respiratory or gastrointestinal epithelium may also be present.
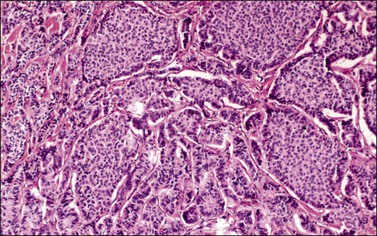
Figure 29.33 Insular carcinoid. Cellular nests containing small round acini are separated by fibrous stroma.
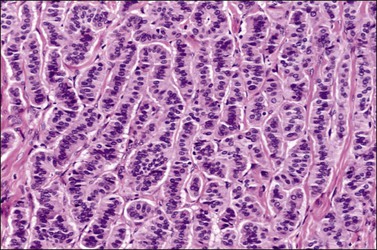
Trabecular carcinoids account for 23–29% of cases.136,137 They are characterized by wavy, parallel cords, ribbons, or trabeculae of cells separated by fibrous stroma and resemble hindgut or foregut carcinoids (Figure 29.34).134 The cords are composed of columnar cells with oblong nuclei oriented perpendicular to the axis of the cord. Typically, intracytoplasmic argyrophilic granules are found at both poles of the nucleus. Associated teratomatous elements are almost always present.
Mucinous (goblet cell) carcinoids constitute only 1.5% of cases and resemble goblet cell carcinoids arising in the appendix.135,137 Unlike the appendiceal tumors, however, the ovarian mucinous carcinoids are often associated with other neoplastic elements. In fact, they are thought to develop from appendiceal tissue within a teratoma. The mucinous carcinoids have been subdivided into three groups: well differentiated, atypical, and carcinomatous.135 In the well-differentiated tumors, small nests or glands composed of goblet cells and cuboidal to columnar cells may be floating within pools of mucin that appear surrounded by a fibrous stroma (Figure 29.35). The nuclei are uniform, small, and round to oval. The neuroendocrine nature of the tumor cells is usually confirmed by immunohistochemistry. Atypical tumors show crowded glands or a cribriform pattern. Tumors with a carcinomatous component contain islands and large masses of tumor cells, crowded glands, and isolated signet-ring cells. These tumors show severe nuclear atypia and numerous mitoses. Combinations of the three histologic subtypes may occur.
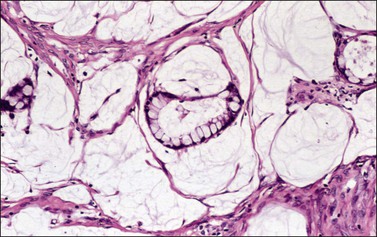
Figure 29.35 Well-differentiated mucinous (goblet cell) carcinoid. Nests composed of cuboidal to columnar goblet cells lie in pools of mucin.
Strumal carcinoid is the second most common type and accounts for 26–44% of cases. It is characterized by coexistence of carcinoid and thyroid tissue.128,136,137 The two components may be contiguous (Figure 29.36) or, more often, intimately admixed; either component may predominate. The carcinoid is trabecular in approximately half of the cases. The thyroid component resembles normal thyroid tissue or follicular adenoma. In areas of intimate mixture, the carcinoid cells characteristically replace the original lining cells of the thyroid follicles (Figure 29.37). Consequently, neuroendocrine granules are found not only within the cells of the trabecular carcinoid, but also within those lining the colloid-filled spaces.128,139 Mucinous glands and cysts or even a mucinous carcinoid component may be present in about 40% of cases.140
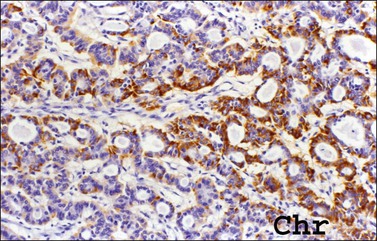
Immunohistochemical Findings
Carcinoids are immunoreactive to neuroendocrine markers such as chromogranin (Figure 29.38), synaptophysin, and Leu-7.141 They are often positive for CDX2. Additionally, various peptide hormones can be detected in about 25% of cases, including serotonin, gastrin, pancreatic polypeptide, vasoactive intestinal peptide, insulin, glucagon, substance P, peptide YY, neurotensin, β-endorphin, ACTH, and somatostatin.142 Insular and trabecular carcinoids are CK7 positive and CK20 negative. In contrast, mucinous (goblet cell) carcinoids show a CK7(−)/CK20 diffuse profile.143 In strumal carcinoids, thyroglobulin, TTF-1, and CK7 are usually expressed in the strumal component but not in the carcinoid component.139,140,144 Calcitonin and amyloid may be present in rare examples of these tumors,128 but immunostains for CEA are negative.
Differential Diagnosis
In the absence of other teratomatous elements, primary ovarian carcinoid may be difficult to distinguish from metastatic carcinoid. Along with a clinical history of carcinoid tumor in the gastrointestinal tract, lung, or elsewhere, evidence favoring metastasis includes bilaterality, multinodularity, peritoneal metastases, and persistence of the carcinoid syndrome or elevated 5-hydroxyindolacetic acid levels in the urine after removal of the ovarian tumor.145 For mucinous (goblet cell) carcinoid, a metastatic appendiceal carcinoma with goblet cell carcinoid-like and signet-ring cell features must be excluded.146 These findings are also helpful in distinguishing ovarian mucinous carcinoid with signet-ring cells from Krukenberg tumor.
Treatment and Prognosis
Ovarian carcinoids are tumors of low malignant potential and less than 5% are complicated by metastasis and death. Most tumors occur in perimenopausal women and, therefore, the standard treatment is hysterectomy and bilateral salpingo-oophorectomy. Unilateral adnexectomy is adequate treatment for young women. Ovarian trabecular carcinoids do not metastasize and are associated with good prognosis. Strumal carcinoid typically follows a benign course and is only rarely associated with metastases.128,139 Most of the malignant tumors have been of the insular or mucinous (carcinomatous) type.135 A few patients have died of progressive carcinoid heart disease.
Neuroectodermal-Type Tumors
Sixty-five neuroectodermal tumors, similar to neoplasms of the central nervous system (CNS), have been described in the ovary.147–153 They included well-differentiated tumors, mainly ependymomas (Figure 29.39), but also astrocytomas and oligodendrogliomas; primitive tumors resembling medulloblastoma, medulloepithelioma, central neurocytoma, and neuroblastoma (Figure 29.40A–D); and anaplastic neoplasms resembling glioblastoma multiforme. Most patients were in their twenties to thirties (range, 6–69 years) and presented with abdominal/pelvic pain, abdominal mass, and fullness.147,151 All tumors but one were unilateral.154 The primitive and anaplastic tumors are typically large (average 14 cm, range 4–29 cm), had a malignant gross appearance, and often extended beyond the ovary.
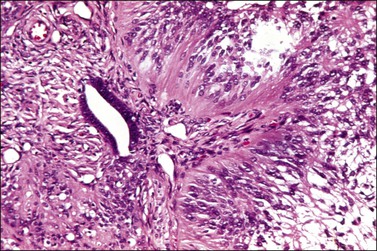
Figure 29.39 Ependymoma. Cells with fibrillary cytoplasmic processes form perivascular pseudorosettes.
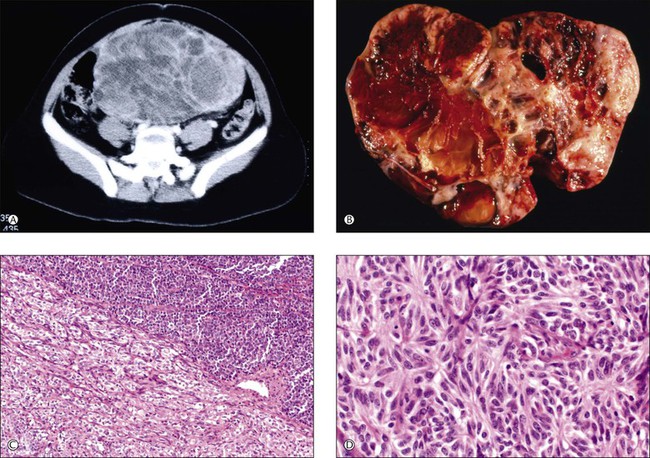
Figure 29.40 Primitive neuroectodermal tumor (PNET). (A) CT scan shows a solid abdominal mass, 18 cm in greatest diameter. (B) The sectioned surface of the tumor is solid and cystic with extensive hemorrhage and necrosis. (C) Typical biphasic pattern composed of large (lower left) and small (upper right) cells. (D) Homer–Wright rosettes.
Given their association with teratomas, most neuroectodermal-type tumors are thought to be of germ cell origin. Tumors not associated with teratomas (particularly ependymomas and primitive neuroectodermal tumors; PNETs) may result from neometaplasia occurring in müllerian-related tissues.155 In the largest series reported, all oligodendrogliomas and most astrocytomas were associated with teratomas but only 1 of 19 ependymomas was so.147,152,153 Also, most primitive tumors (except PNETs) and all glioblastoma multiforme were associated with teratomas.147,151 But even if most cases are essentially germ cell tumors, they should be classified separately from grade 2–3 ITs in which the neuroectodermal elements do not resemble tumors of the CNS.147 Whereas the IT with predominant neuroectodermal differentiation is easily recognized because of the presence of many other teratomatous elements, the pure or almost pure neuroectodermal-type tumor is frequently confused with other types of ovarian tumors such as granulosa cell tumor, small cell carcinoma of the hypercalcemic type, and endometrioid carcinoma. Extensive sampling and immunohistochemical stains for glial fibrillary acidic protein (GFAP) and MIC2 protein (CD99) facilitate the correct diagnosis. Ovarian ependymomas are more likely to express various cytokeratins, and estrogen receptor and progesterone receptor than their CNS counterparts.156 The EWS/FLI-1 chimeric transcript, secondary to t(11;22)(q24;q12) chromosomal translocation, characteristic of the PNET family, has been identified by reverse transcription polymerase chain reaction in a case of ovarian neuroectodermal tumor.157 Clinical stage is the most important prognostic factor. However, ovarian ependymomas are associated with good prognosis even in an advanced stage.148,150 In contrast, the primitive and anaplastic tumors have a poor outcome.147
Carcinoma in Dermoid Cysts
Squamous Cell Carcinoma
Malignant change occurs in 1–2% of dermoid cysts. Any component may be involved, but squamous cell carcinomas account for 80% of cases. Nonepithelial malignant tumors are exceedingly rare.158 The tumors occur at an average age of about 55 years.158–160 Presentation varies from an ordinary dermoid cyst to obvious ovarian cancer. In patients 45 years or older, a diameter larger than 10 cm and elevated CEA and squamous cell carcinoma antigen levels are suspicious of malignancy and imaging may be confirmatory.159
Average size is about 14 cm.159 Tumors are solid or solid and cystic with a polypoid mass showing areas of necrosis. A thickened wall is characteristic. Microscopically, the tumors show papillary, nodular, and infiltrative features.88 Almost all squamous cell carcinomas arise from respiratory or gastrointestinal-type epithelium rather than skin and may show transmural extension and local invasion (Figure 29.41).161 Unlike squamous cell carcinomas associated with other ovarian tumors that are more frequently bilateral, carcinomas arising in dermoid cyst are typically unilateral.88
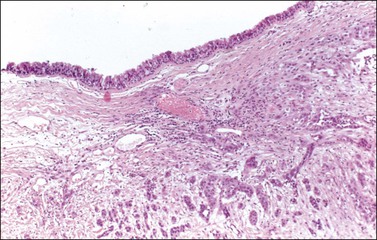
Figure 29.41 Squamous cell carcinoma arising in a dermoid cyst. Invasive squamous cell carcinoma underlies respiratory epithelium lining the cyst.
Outcome is highly dependent on stage and tumors confined to the ovary have a favorable outcome. The overall 5 year survival is 15–52% for all stages161 and 75.7% for stage I tumors.
Adenocarcinoma
Adenocarcinoma is the second most common malignancy arising in dermoid cysts, accounting for 7% of cases.162 Most tumors arise from gastrointestinal and respiratory type epithelium.59,163 Rarely, carcinomas of sweat glands, sebaceous glands, salivary glands, mammary glands,164,165 and carcinosarcoma166 may occur. Patients’ ages range from 17 to 66 years (mean, 39 years) and preoperative elevated serum level of CEA and CA19.9 and imaging are helpful for the preoperative diagnosis.167
Mucinous tumors arising in dermoid cysts show a wide morphologic spectrum, including benign, malignant, and low-grade tumors resembling mucinous appendiceal tumors; some may show goblet cells, signet-ring cells, and carcinoid-like morphology.143,168 Pseudomyxoma ovarii is common and in some cases is associated with pseudomyxoma peritonei. Immunohistochemical stains for CK7 and CK20 are of limited value in the differential diagnosis with a secondary tumor. Search of teratomatous elements in the ovarian mass and careful examination of the intestines, in particular appendix and rectum, are important for establishing the correct diagnosis.
The main prognostic parameters are stage and grade. Mucinous borderline tumors confined to the ovary have a favorable prognosis. Also, patients with adenocarcinomas limited to the ovary may have a favorable outcome.169
Mixed Malignant Germ Cell Tumors
In ~10–20% of malignant germ cell tumors (MGCTs) of the ovary, particularly of a primitive nature, a combination of two or more neoplastic types is found.73 The proportion of each tumor type parallels the incidence of germ cell tumors and influences prognosis. Although any combination is possible, dysgerminoma and YST is the most common; immature teratoma is the third most common component; and EC, choriocarcinoma, and polyembryoma are less frequently encountered.73 In fact, the latter tumors are rarely found in the ovary in a pure form, but rather as components of mixed MGCTs. Mixed MGCTs may secrete either AFP or hCG, or both, or neither, depending on their components. A panel of immunomarkers including OCT3/4, CD30, and glypican-3 is useful for characterizing the tumor component. Proper identification of the various components requires careful gross examination and extensive sampling of the tumor. Contemporary results indicate that tumor stage is the only significant prognostic factor in patients treated with combination chemotherapy.170
Mixed Germ Cell Sex Cord–Stromal Tumor
Gonadoblastoma
Gonadoblastoma is a rare in situ form of MGCT consisting of a mixture of immature sex-cord cells and germ cells that arises almost exclusively in dysgenetic gonads. It occurs mainly in young patients, often before the age of 20 years.171 Most patients are virilized phenotypic females, but nearly all of them are genotypic males, i.e., have a Y chromosome. The underlying gonadal abnormality is almost always 46,XY pure gonadal dysgenesis or mixed gonadal dysgenesis, which is frequently associated with a 45,X/46,XY karyotype (Table 29.5). A gene that increases susceptibility to gonadoblastoma in patients with dysgenetic gonads has been identified in the long arm of the Y chromosome (GBY region). This gene (TSPY1) encodes the testis-specific protein 1, which is thought to have a role in cell cycle regulation.172 Rare patients are phenotypic men with varying degrees of feminization.
Table 29.5
Gonadoblastoma (Phenotype and Karyotype)
| Female—MGD | 90% |
| Male | 10% |
| Y chromosome | 95% |
| • 46,XY | Most frequent |
| • 45,X/46,XY | 1% |
| • Mosaic with Y | |
| 46,XX/45,X0 | 5% |
Grossly, the tumor varies in appearance, from soft and fleshy to firm and completely calcified, depending upon the presence or absence of an associated MGCT, calcification, or both. Whereas pure gonadoblastomas are typically less than 8 cm in size, those with an MGCT are usually larger.1 Gonadoblastomas may arise from indeterminate gonads (60%), dysgenetic testes (20%), or streak gonads (20%);1 the tumors are bilateral in more than 40% of cases.1
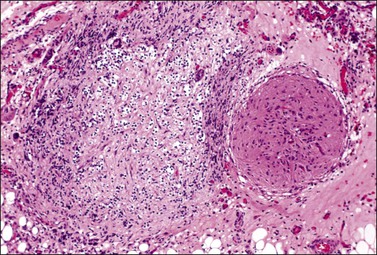
Microscopically, the tumor contains both immature germ cells and sex cord–stromal cells, which resemble granulosa or Sertoli cells, arranged in small islands (Figure 29.42) and intermixed with rounded pink hyaline bodies (basement membrane-type material) (Figure 29.43). Leydig cells or lutein cells are distributed through the intervening stroma in two-thirds of the cases and are responsible for the endocrine manifestations.171 Calcification may be extensive and detected by X-rays. Overgrowth by an MGCT occurs in approximately half the cases.1 The malignant tumor is a germinoma in 80% of cases, and less frequently yolk sac tumor (YST), embryonal carcinoma (EC) choriocarcinoma, or IT.1 MGCTs associated with gonadoblastoma are usually diploid.
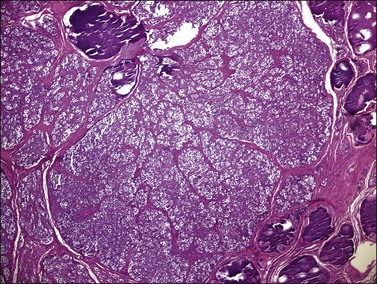
Figure 29.42 Gonadoblastoma. Cellular nests surrounded by connective tissue stroma. Foci of calcification are seen.
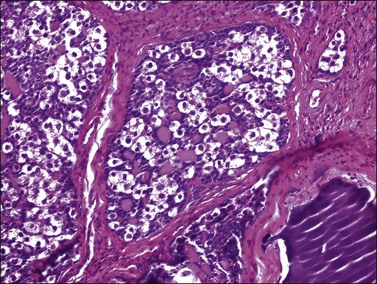
Figure 29.43 Gonadoblastoma. A nest composed of large germ cells admixed with smaller sex cord derivatives. Numerous rounded pink hyaline bodies are surrounded by the smaller cells.
The germ cells in gonadoblastoma have the same immunophenotype as dysgerminoma cells. They show positive membrane and cytoplasmic reactivity for placental alkaline phosphatase (Figure 29.44), CD117 (c-kit), and D2-40 (podoplanin), and they exhibit nuclear reactivity for OCT3/4 and SALL4.32,173 Cytoplasmic and membrane immunoreaction for TSPY is present in the germ cells.174
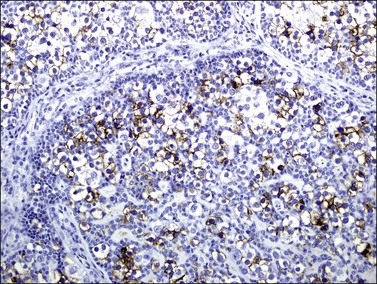
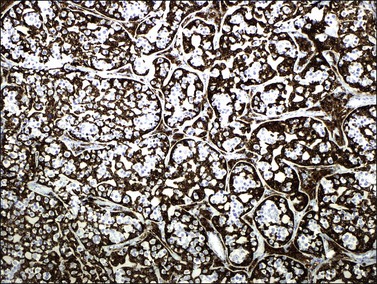
The sex cord–stromal cells show positive cytoplasmic reaction for inhibin (Figure 29.45), vimentin, and cytokeratin, and nuclear staining for WT1 and FOXL2, but they lack reactivity for SOX9.175 This suggests a closer relationship to granulosa cells than to Sertoli cells. The hyaline material in the tumor cell nests is PAS positive and reacts with anti-laminin antibodies. Luteinized cells in the stroma around the gonadoblastoma nests react strongly for inhibin and calretinin.
Gonadoblastoma should be distinguished from pure dysgerminoma and sex cord tumor with annular tubules (SCTAT). In a patient with dysgenetic gonads and a Y chromosome, the finding of calcification within a dysgerminoma should raise the question of its origin in gonadoblastoma. Although SCTAT resembles gonadoblastoma, it lacks a germ cell component.1 Other mixed germ cell sex cord–stromal tumors that lack the characteristic pattern of gonadoblastoma and develop in children with apparently normal gonads and a normal karyotype have been described.176 Pure gonadoblastoma is benign unless it is overgrown by an MGCT; however, because of the frequency of this event, it is regarded as an ‘in situ’ form of MGCT and treated by gonadectomy.1
References
1. Scully, RE, Young, RH, Clement, PB. Tumors of the ovary, maldeveloped gonads, fallopian tube, and broad ligament. In: Atlas of tumor pathology, 3rd series. Fascicle 23. Washington, DC: Armed Forces Institute of Pathology; 1998:239–312.
2. Pierce, GB, Teratocarcinoma: a model for developmental concept of cancer. Current topics in developmental biology Moscona, AA, Monroy, A, eds. Current topics in developmental biology; vol. 2. Academic Press, New York, 1967:223–246.
3. Damjanov, I. Pathobiology of human germ cell neoplasia. Recent Results Cancer Res. 1991; 123:1–19.
4. Ulbright, TM. Germ cell tumors of the gonads: a selective review emphasizing problems in differential diagnosis, newly appreciated, and controversial issues. Mod Pathol. 2005; 18(Suppl 2):S61–S79.
5. Oosterhuis, JW, Stoop, H, Honecker, F, Looijenga, LH. Why human extragonadal germ cell tumours occur in the midline of the body: old concepts, new perspectives [discussion in Int J Androl 2007; 30(4):263–4]. Int J Androl. 2007; 30:256–263.
6. Rutgers, JL, Young, RH, Scully, RE. Ovarian yolk sac tumor arising from endometrioid carcinoma. Hum Pathol. 1987; 18:1296–1299.
7. Mazur, MT, Talbot, WH, Jr., Talerman, A. Endodermal sinus tumor and mucinous cystadenofibroma of the ovary. Occurrence in an 82 year old woman. Cancer. 1988; 62:2011–2015.
8. Oliva, E, Andrada, E, Pezzica, E, Prat, J. Ovarian carcinomas with choriocarcinomatous differentiation. Cancer. 1993; 72:2441–2446.
9. Nogales, FF, Bergeron, C, Carvia, RE, et al. Ovarian endometrioid tumors with yolk sac tumor component, an unusual form of ovarian neoplasm. Analysis of six cases. Am J Surg Pathol. 1996; 20:1056–1066.
10. Walt, H, Arrenbrecht, S, Delozier-Blanchet, CD, et al. A human testicular germ cell tumor with borderline histology between seminoma and embryonal carcinoma secreted beta HCG and AFP only as a xenograft. Cancer. 1986; 58:139–146.
11. Srigley, JR, Mackay, B, Toth, P, Ayala, A. The ultrastructure and histogenesis of male germ cell neoplasia with emphasis on seminoma with early carcinomatous features. Ultrastruct Pathol. 1988; 12:67–86.
12. Damjanov, I. Is seminoma a relative or a precursor of embryonal carcinoma? Lab Invest. 1989; 60:1–3.
13. Oosterhuis, JW, Castedo, SMMJ, Dejong, B, et al. Ploidy of primary germ cell tumors of the testis; pathogenesis and clinical relevance. Lab Invest. 1989; 60:14–21.
14. Czaja, JT, Ulbright, TM. Evidence for the transformation of seminoma to yolk sac tumor, with histogenetic considerations. Am J Clin Pathol. 1992; 97:468–477.
15. Parkash, V, Carcangiu, ML. Transformation of ovarian dysgerminoma to yolk sac tumor: evidence for a histogenetic continuum. Mod Pathol. 1995; 8:881–887.
16. Cardoso de Almeida, PC, Scully, RE. Diffuse embryoma of the testis. A distinctive form of mixed germ cell tumor. Am J Surg Pathol. 1983; 7:633–642.
17. Teilum, G. Special tumors of ovary and testis and related extragonadal lesions. Comparative pathology and histological identification. Philadelphia: JB Lippincott; 1976.
18. Prat, J, Cao, D, Carinelli, S, et al. Tumours of the ovary: Germ cell tumours. In Kurman RJ, Carcangiu ML, Herrington S, Young RH, eds.: World Health Organization (WHO) classification of tumours of female reproductive organs, 4th ed, Lyons, France: IARC Press, 2014.
19. Prat, J. Pathology of the ovary. Philadelphia: Saunders; 2004.
20. Tapia, N, Arauzo-Bravo, MJ, Ko, K, Scholer, HR. Concise review: challenging the pluripotency of human testis-derived ESC-like cells. Stem Cells. 2011; 29(8):1165–1169.
21. Wang, Z, Oron, E, Nelson, B, et al. Distinct lineage specification roles for NANOG, OCT4, and SOX2 in human embryonic stem cells. Cell Stem Cell. 2012; 10:440–454.
22. Gershenson, DM. Management of ovarian germ cell tumors. J Clin Oncol. 2007; 25:2938–2943.
23. Vicus, D, Beiner, ME, Klachook, S, et al. Pure dysgerminoma of the ovary 35 years on: a single institutional experience. Gynecol Oncol. 2010; 17:23–26.
24. Pressley, RH, Muntz, HG, Falkenberry, S, Rice, LW. Serum lactic dehydrogenase as a tumor marker in dysgerminoma. Gynecol Oncol. 1992; 44:281–283.
25. Zaloudek, CJ, Tavassoli, FA, Norris, HJ. Dysgerminoma with syncytiotrophoblastic giant cells. A histologically and clinically distinctive subtype of dysgerminoma. Am J Surg Pathol. 1981; 5:361–367.
26. Radhakrishnan, S, Haq, S, Lofts, F, et al. Ovarian dysgerminoma associated with hypercalcemia. Br J Obstet Gynaecol. 2001; 108:1302–1304.
27. Werness, BA, Ramus, SJ, Whittemore, AS, et al. Primary ovarian dysgerminoma in a patient with a germline BRCA1 mutation. Int J Gynecol Pathol. 2000; 19:390–394.
28. Dietl, J, Horny, HPI, Ruck, P, Kaiserling, E. Dysgerminoma of the ovary: an immunohistochemical study of tumor infiltrating lymphoreticular cells and tumor cells. Cancer. 1993; 71:2562–2568.
29. Rutgers, JL, Scully, RE. Functioning ovarian tumors with peripheral steroid cell proliferation: a report of twenty-four cases. Int J Gynecol Pathol. 1986; 5:319–337.
30. Sever, M, Jones, TD, Roth, LM, et al. Expression of CD117 (c-kit) receptor in dysgerminoma of the ovary: diagnostic and therapeutic implications. Mod Pathol. 2005; 18:1411–1416.
31. Chang, MC, Vargas, SO, Hornick, JL, et al. Embryonic stem cell transcription factors and D2-40 (podoplanin) as diagnostic immunohistochemical markers in ovarian germ cell tumors. Int J Gynecol Pathol. 2009; 28:347–355.
32. Cao, D, Guo, S, Allan, RW, et al. SALL4 is a novel sensitive and specific marker of ovarian primitive germ cell tumors and is particularly useful in distinguishing yolk sac tumor from clear cell carcinoma. Am J Surg Pathol. 2009; 33:894–904.
33. Nogales, FF, Dulcey, I, Preda, O. Issues in gynecologic pathology. Germ cell tumors of the ovary. An update. Arch Pathol Lab Med. 2013; 137:104–112.
34. Cossu-Rocca, P, Zhang, S, Roth, LM, et al. Chromosome 12p abnormalities in dysgerminoma of the ovary: a FISH analysis. Mod Pathol. 2006; 19:611–615.
35. Hoei-Hansen, CE, Kraggerud, SM, Abeler, VM, et al. Ovarian dysgerminomas are characterised by frequent KIT mutations and abundant expression of pluripotency markers. Mol Cancer. 2007; 6:12.
36. Cheng, L, Roth, LM, Zhang, S, et al. KIT gene mutation and amplification in dysgerminoma of the ovary. Cancer. 2011; 117:2096–2103.
37. Hersmus, R, Stoop, H, van de Geijn, GJ, et al. Prevalence of c-KIT mutations in gonadoblastoma and dysgerminomas of patients with disorders of sex development (DSD) and ovarian dysgerminomas. PLoS One. 2012; 7:e43952.
38. Osborne, BM, Robboy, SJ. Lymphomas or leukemia presenting as ovarian tumors. An analysis of 42 cases. Cancer. 1983; 52:1933–1943.
39. Zhang, R, Sun, YC, Zhang, GY, et al. Treatment of malignant ovarian germ cell tumors and preservation of fertility. Eur J Gynaecol Oncol. 2012; 33:489–492.
40. Mangili, G, Sigismondi, C, Lorusso, D, et al. Is surgical restaging indicated in apparent stage IA pure ovarian dysgerminoma? The MITO group retrospective experience. Gynecol Oncol. 2011; 121:280–284.
41. Gershenson, DM, Copeland, JL, Del Junco, G, et al. Second-look laparotomy in the management of malignant germ cell tumors of the ovary. Obstet Gynecol. 1986; 67:789–794.
42. Palmquist, MB, Webb, MJ, Lieber, MM, et al. DNA ploidy of ovarian dysgerminomas: correlation with clinical outcome. Gynecol Oncol. 1992; 44:13–16.
43. Fujita, M, Inoue, M, Tanizawa, O, et al. Retrospective review of 41 patients with endodermal sinus tumor of the ovary. Int J Gynecol Cancer. 1993; 3:329–335.
44. Gershenson, DM, Del Junco, G, Herson, J, Rutledge, FN. Endodermal sinus tumor of the ovary: the M.D. Anderson experience. Obstet Gynecol. 1983; 61:194–202.
45. Kawai, M, Kano, T, Furuhashi, Y, et al. Prognostic factors in yolk sac tumors of the ovary. A clinicopathologic analysis of 29 cases. Cancer. 1991; 67:184–192.
46. Teilum, G. Endodermal sinus tumor of the ovary and testis. Comparative morphogenesis of the so-called mesonephroma ovarii (Schiller) and extraembryonic (yolk sac-allantoic) structures of the rat’s placenta. Cancer. 1959; 12:1092–1105.
47. Kurman, RJ, Norris, HJ. Endodermal sinus tumor of the ovary: a clinicopathologic analysis of 71 cases. Cancer. 1976; 38:2404–2419.
48. Schiller, W. Mesonephroma ovarii. Am J Cancer. 1939; 35:1–21.
49. Nogales, FF, Preda, O, Nicolae, A. Yolk sac tumours revisited: a review of their many faces and names. Histopathology. 2012; 60:1023–1033.
50. Nogales, FF, Jr., Matilla, A, Nogales, O, Galera-Davidson, HL. Yolk sac tumors with pure and mixed polyvesicular vitelline patterns. Hum Pathol. 1978; 9:553–566.
51. Young, RH, Ulbright, TM, Policarpio-Nicolas, ML. Yolk sac tumor with a prominent polyvesicular vitelline pattern: a report of three cases. Am J Surg Pathol. 2013; 37:393–398.
52. Cohen, MB, Friend, DS, Molnar, JJ, Talerman, A. Gonadal endodermal sinus (yolk sac) tumor with pure intestinal differentiation: a new histologic type. Pathol Res Pract. 1987; 182:609–616.
53. Clement, PB, Young, RH, Scully, RE. Endometrioid-like yolk sac tumor of the ovary. A clinicopathological analysis of eight cases. Am J Surg Pathol. 1987; 11:767–778.
54. Prat, J, Bhan, AK, Dickersin, GR, et al. Hepatoid yolk sac tumor of the ovary (endodermal sinus tumor with hepatoid differentiation). A light microscopical, ultrastructural and immunohistochemical study of seven cases. Cancer. 1982; 50:2355–2368.
55. Ulbright, TM, Roth, LM, Brodhecker, CA. Yolk sac differentiation in germ cell tumors. A morphologic study of 50 cases with emphasis on hepatic, enteric, and parietal yolk sac features. Am J Surg Pathol. 1986; 10:151–164.
56. Nakashima, N, Fukatsu, T, Nagasaki, T, et al. The frequency and histology of hepatic tissue in germ cell tumors. Am J Surg Pathol. 1987; 11:682–692.
57. Dickersin, GR, Oliva, E, Young, RH. Endometrioid-like variant of ovarian yolk sac tumor with foci of carcinoid: an ultrastructural study. Ultrastruct Pathol. 1995; 19:421–429.
58. Nogales, FF, Buritica, C, Regauer, S, Gonzalez, T. Mucinous carcinoid as an unusual manifestation of endodermal differentiation in ovarian yolk sac tumors. Am J Surg Pathol. 2005; 29:1247–1251.
59. Devouassoux-Shisheboran, M, Schammel, DP, Tavassoli, FA. Ovarian hepatoid yolk sac tumours: morphological, immunohistochemical and ultrastructural features. Histopathology. 1999; 34:462–469.
60. Michael, H, Ulbright, TM, Brodhecker, CA. The pluripotential nature of the mesenchyme-like component of yolk sac tumor. Arch Pathol Lab Med. 1989; 113:1115–1119.
61. Ulbright, TM, Loehrer, PJ, Roth, LM, et al. The development of non-germ cell malignancies within germ cell tumors. A clinicopathologic study of 11 cases. Cancer. 1984; 54:1824–1833.
62. Zynger, DL, Everton, MJ, Dimov, ND, et al. Expression of glypican 3 in ovarian and extragonadal germ cell tumors. Am J Clin Pathol. 2008; 130:224–230.
63. Rabban, JT, Zaloudek, CJ. A practical approach to immunohistochemical diagnosis of ovarian germ cell tumours and sex cord-stromal tumours. Histopathology. 2013; 62:71–88.
64. Xue, D, Peng, Y, Wang, F, et al. RNA-binding protein LIN28 is a sensitive marker of ovarian primitive germ cell tumours. Histopathology. 2011; 59:452–459.
65. Pitman, MB, Triratanachat, S, Young, RH, Oliva, E. Hepatocyte paraffin 1 antibody does not distinguish primary ovarian tumors with hepatoid differentiation from metastatic hepatocellular carcinoma. Int J Gynecol Pathol. 2004; 23:58–64.
66. Kurman, RJ, Norris, HJ. Embryonal carcinoma of the ovary. A clinicopathologic entity distinct from endodermal sinus tumor resembling embryonal carcinoma of the adult testis. Cancer. 1976; 38:2420–2433.
67. Kleinsmith, LJ, Pierce, GB, Jr. Multipotentiality of single embryonal carcinoma cells. Cancer Res. 1964; 24:1544–1551.
68. Cheng, L, Zhang, S, Talerman, A, Roth, LM. Morphologic, immunohistochemical, and fluorescence in situ hybridization study of ovarian embryonal carcinoma with comparison to solid variant of yolk sac tumor and immature teratoma. Hum Pathol. 2010; 41:716–723.
69. Takeda, A, Ishizuka, T, Goto, T, et al. Polyembryoma of ovary producing alpha-fetoprotein and HCG: immunoperoxidase and electron microscopic study. Cancer. 1982; 14:1878–1889.
70. Nakashima, N, Murakami, S, Fukatsu, T, et al. Characteristics of ‘embryoid body’ in human gonadal germ cell tumors. Hum Pathol. 1988; 19:1144–1154.
71. Prat, J, Matías-Guiu, X, Scully, RE. Hepatic yolk sac differentiation in an ovarian polyembryoma. Surg Pathol. 1989; 2:147–150.
72. Jondle, DM, Shahin, MS, Sorosky, J, Benda, JA. Ovarian mixed germ cell tumor with predominance of polyembryoma. A case report with literature review. Int J Gynecol Pathol. 2002; 21:78–81.
73. Kurman, RJ, Norris, HJ. Malignant mixed germ cell tumor of the ovary. A clinical and pathologic analysis of 30 cases. Obstet Gynecol. 1976; 48:579–589.
74. Lv, L, Yang, K, Wu, H, et al. Pure choriocarcinoma of the ovary: a case report. J Gynecol Oncol. 2011; 22:135–139.
75. Axe, SR, Klein, VR, Woodruff, JD. Choriocarcinoma of the ovary. Obstet Gynecol. 1985; 66:111–114.
76. Jacobs, AJ, Newland, JR, Green, RK. Pure choriocarcinoma of the ovary. Obstet Gynecol Surv. 1982; 37:603–609.
77. Grover, V, Grover, RK, Usha, R, et al. Primary pure choriocarcinoma of the ovary. Gynecol Obstet Invest. 1990; 30:61–63.
78. Fisher, RA, Newlands, ES, Jeffreys, AJ, et al. Gestational and nongestational trophoblastic tumors distinguished by DNA analysis. Cancer. 1992; 69:839–845.
79. Shigematsu, T, Kamura, T, Arima, T, et al. DNA polymorphism analysis of a pure non-gestational choriocarcinoma of the ovary: case report. Eur J Gynaecol Oncol. 2000; 21:153–154.
80. Hafezi-Bakhtiari, S, Morava-Protzner, I, Burnell, MJ, et al. Choriocarcinoma arising in a serous carcinoma of ovary: an example of histopathology driving treatment. J Obstet Gynaecol Can. 2010; 32:698–702.
81. Yanai-Inbar, I, Scully, RE. Relations of ovarian dermoid cysts and immature teratomas: an analysis of 350 cases of immature teratoma and 10 cases of dermoid cyst with microscopic foci of immature tissue. Int J Gynecol Pathol. 1987; 6:203–212.
82. Linder, D, McCaw, BK, Hecht, F. Parthenogenic origin of benign ovarian teratomas. N Engl J Med. 1975; 292:63–66.
83. Vortmeyer, AO, Devouassoux-Shisheboran, M, Li, G, et al. Microdissection-based analysis of mature ovarian teratoma. Am J Pathol. 1999; 154:987–991.
84. Koonings, PP, Campbell, K, Mishell, DR, Jr., Grimes, DA. Relative frequency of primary ovarian neoplasms: a 10-year review. Obstet Gynecol. 1989; 74:921–926.
85. Zakin, D. Radiologic diagnosis of dermoid cysts in the ovary. Obstet Gynecol Surv. 1976; 31:165–184.
86. Comerci, JT, Jr., Licciardi, F, Bergh, PA, et al. Mature cystic teratoma: a clinicopathologic evaluation of 517 cases and review of the literature. Obstet Gynecol. 1994; 84:22–28.
87. Robboy, SJ, Scully, RE. Ovarian teratoma with glial implants on the peritoneum. An analysis of 12 cases. Hum Pathol. 1970; 1:643–653.
88. Pins, MR, Young, RH, Daly, WJ, Scully, RE. Primary squamous cell carcinoma of the ovary. Report of 37 cases. Am J Surg Pathol. 1996; 20:823–833.
89. Thurlbeck, WM, Scully, RE. Solid teratoma of the ovary. A clinicopathological analysis of 9 cases. Cancer. 1960; 13:804–811.
90. Abbott, TM, Hermann, WJ, Jr., Scully, RE. Ovarian fetiform teratoma (homunculus) in a 9-year-old girl. Int J Gynecol Pathol. 1984; 2:392–402.
91. Chen, E, Fletcher, CD, Nucci, MR. Meningothelial proliferations in mature cystic teratoma of the ovary: evidence for the common presence of cranially derived tissues paralleling anterior embryonic plate development. An analysis of 25 consecutive cases. Am J Surg Pathol. 2010; 34:1014–1018.
92. Halabi, M, Oliva, E, Mazal, PR, et al. Prostatic tissue in mature cystic teratoma of the ovary: a report of four cases, including one with features of prostatic adenocarcinoma, and cytogenetic studies. Int J Gynecol Pathol. 2002; 21:261–267.
93. Baker, PM, Rosai, J, Young, RH. Ovarian teratomas with florid benign vascular proliferation: a distinctive finding associated with the neural component of teratomas that may be confused with a vascular neoplasm. Int J Gynecol Pathol. 2002; 21:16–21.
94. Axiotis, CA, Lippes, HA, Merino, MJ, et al. Corticotroph cell pituitary adenoma within an ovarian teratoma. A new cause of Cushing’s syndrome. Am J Surg Pathol. 1987; 11:218–224.
95. Kallenberg, GA, Pesce, CM, Norman, B, et al. Ectopic hyperprolactinemia resulting from an ovarian teratoma. JAMA. 1990; 263:2472–2474.
96. Palmer, PE, Bogojavlensky, S, Bhan, AK, Scully, RE. Prolactinoma in wall of ovarian dermoid cyst with hyperprolactinemia. Obstet Gynecol. 1990; 75:540–543.
97. Silver, SA, Tavassoli, FA. Glomus tumor arising in a mature teratoma of the ovary: report of a case simulating a metastasis from cervical squamous carcinoma. Arch Pathol Lab Med. 2000; 124:1373–1375.
98. Lee, K, Scully, RE. Mucinous tumors of the ovary. A clinicopathologic study of 196 borderline tumors (of intestinal type) and carcinomas. Including an evaluation of 11 cases with ‘pseudomyxoma peritonei.’. Am J Surg Pathol. 2000; 24:1447–1464.
99. Ronnett, BM, Seidman, JD. Mucinous tumors arising in ovarian mature cystic teratomas. Am J Surg Pathol. 2003; 27:650–657.
100. Davis, GL. Malignant melanoma arising in mature ovarian cystic teratoma (dermoid cyst): report of two cases and literature analysis. Int J Gynecol Pathol. 1996; 15:356–362.
101. Smith, HO, Berwick, M, Verschraegen, CF, et al. Incidence and survival rates for female malignant germ cell tumors. Obstet Gynecol. 2006; 107:1075–1085.
102. Norris, HJ, Zirkin, HJ, Benson, WL. Immature (malignant) teratoma of the ovary: a clinical and pathologic study of 58 cases. Cancer. 1976; 37:2359–2372.
103. Gershenson, DM, Del Junco, G, Silva, EG, et al. Immature teratoma of the ovary. Obstet Gynecol. 1986; 68:624–629.
104. Kawai, M, Kano, T, Furuhashi, Y, et al. Immature teratoma of the ovary. Gynecol Oncol. 1991; 40:133–137.
105. Bonazzi, C, Peccatori, F, Colombo, N, et al. Pure ovarian immature teratoma, a unique and curable disease: 10 years’ experience of 32 prospectively treated patients. Obstet Gynecol. 1994; 84:598–604.
106. Heifetz, SA, Cushing, B, Giller, R, et al. Immature teratomas in children: pathologic considerations: a report from the combined Pediatric Oncology Group/Children’s Cancer Group. Am J Surg Pathol. 1998; 22:1115–1124.
107. Nogales, FF, Avila, IR, Concha, A, et al. Immature endodermal teratoma of the ovary: embryologic correlations and immunohistochemistry. Hum Pathol. 1993; 24:364–370.
108. Nogales, FF, Jr., Favara, BE, Major, FJ, et al. Immature teratoma of the ovary with a neural component (“solid” teratoma). A clinicopathologic study of 20 cases. Hum Pathol. 1976; 7:625–642.
109. O’Connor, DM, Norris, HJ. The influence of grade on the outcome of stage I ovarian immature (malignant) teratoma and the reproducibility of grading. Int J Gynecol Pathol. 1994; 13:283–289.
110. Ferguson, AW, Katabuchi, H, Ronnett, BM, Cho, KR. Glial implants in gliomatosis peritonei arise from normal tissue, not from the associated teratoma. Am J Pathol. 2001; 159(1):51–55.
111. Calder, CJ, Light, AM, Rollason, TP. Immature ovarian teratoma with mature peritoneal metastatic deposits showing glial, epithelial, and endometrioid differentiation: a case report and review of the literature. Int J Gynecol Pathol. 1994; 13:279–282.
112. Perrone, T, Steiner, M, Dehner, LP. Nodal gliomatosis and alpha-fetoprotein production: two unusual facets of grade I ovarian teratoma. Arch Pathol Lab Med. 1986; 110:975–977.
113. Zhuang, Z, Devouassoux-Shisheboran, M, Lubensky, IA, et al. Premeiotic origin of teratomas: is meiosis required for differentiation into mature tissues? Cell Cycle. 2005; 4:1683–1687.
114. Kraggerud, SM, Szymanska, J, Abeler, VM, et al. DNA copy number changes in malignant ovarian germ cell tumors. Cancer Res. 2000; 60:3025–3030.
115. Baker, BA, Frickey, L, Yu, IT, et al. DNA content of ovarian immature teratomas and malignant germ cell tumors. Gynecol Oncol. 1998; 71:14–18.
116. Williams, SD, Blessing, JA, Liao, S, et al. Adjuvant therapy of ovarian germ cell tumors with cisplatin, etoposide, and bleomycin: a trial of the Gynecologic Oncology Group. J Clin Oncol. 1994; 12:701–706.
117. Nogales, FF, Aguilar, D. Florid vascular proliferation in grade 0 glial implants from ovarian immature teratoma. Int J Gynecol Pathol. 2002; 21(3):305–307.
118. Dadmanesh, F, Miller, DM, Swenerton, KD, Clement, PB. Gliomatosis peritonei with malignant transformation. Mod Pathol. 1997; 10:597–601.
119. Riopel, MA, Spellerberg, A, Griffin, CA, Perlman, EJ. Genetic analysis of ovarian germ cell tumors by comparative genomic hybridization. Cancer Res. 1998; 58:3105–3110.
120. Woodruff, JD, Rauh, JT, Markley, RL. Ovarian struma. Obstet Gynecol. 1966; 27:194–201.
121. Simkin, PH, Ramirez, LA, Zweizig, SL, et al. Monomorphic teratoma of the ovary: a rare cause of triiodothyronine toxicosis. Thyroid. 1999; 9:949–954.
122. Burg, J, Kommoss, F, Bittinger, F, et al. Mature cystic teratoma of the ovary with struma and benign Brenner tumor: A case report with immunohistochemical characterization. Int J Gynecol Pathol. 2002; 21:74–77.
123. Szyfelbein, WM, Young, RH, Scully, RE. Cystic struma ovarii: a frequently unrecognized tumor. A report of 20 cases. Am J Surg Pathol. 1994; 18:1102–1116.
124. Szyfelbein, WM, Young, RH, Scully, RE. Struma ovarii simulating ovarian tumors of other types: a report of 30 cases. Am J Surg Pathol. 1995; 19:21–29.
125. Robboy, SJ, Shaco-Levy, R, Peng, RY, et al. Malignant struma ovarii: an analysis of 88 cases, including 27 with extraovarian spread. Int J Gynecol Pathol. 2009; 28:405–422.
126. Devaney, K, Synder, R, Norris, HJ, Tavassoli, FA. Proliferative and histologically malignant struma ovarii: a clinicopathologic study of 54 cases. Int J Gynecol Pathol. 1993; 12:333–343.
127. Willemse, PH, Oosterhuis, JW, Aalders, JG, et al. Malignant struma ovarii treated by ovariectomy, thyroidectomy, and 131I administration. Cancer. 1987; 60:178–182.
128. Robboy, SJ, Scully, RE. Strumal carcinoid of the ovary: an analysis of 50 cases of a distinctive tumor composed of thyroid tissue and carcinoid. Cancer. 1980; 46:2019–2034.
129. Garg, K, Soslow, RA, Rivera, M, et al. Histologically bland ‘extremely well differentiated’ thyroid carcinomas arising in struma ovarii recur and metastasize. Int J Gynecol Pathol. 2009; 28:222–230.
130. Shaco-Levy, R, Bean, SM, Bentley, RC, et al. Natural history of biologically malignant struma ovarii: analysis of 27 cases with extraovarian spread. Int J Gynecol Pathol. 2010; 29:212–227.
131. Karseladze, AI, Kulinitch, SI. Peritoneal strumosis. Pathol Res Pract. 1994; 190:1086–1088.
132. Roth, LM, Karseladze, AI. Highly differentiated follicular carcinoma arising from struma ovarii: a report of three cases, a review of the literature, and reassessment of so-called peritoneal strumosis. Int J Gynecol Pathol. 2008; 27:213–222.
133. Robboy, SJ, Norris, HJ, Scully, RE. Insular carcinoid primary in the ovary: a clinicopathologic analysis of 48 cases. Cancer. 1975; 36:404–418.
134. Robboy, SJ, Scully, RE, Norris, HJ. Primary trabecular carcinoid of the ovary. Obstet Gynecol. 1977; 49:202–207.
135. Baker, PM, Oliva, E, Young, RH, et al. Ovarian mucinous carcinoids including some with a carcinomatous component. A report of 17 cases. Am J Surg Pathol. 2001; 25:557–568.
136. Davis, KP, Hartmann, LK, Keeney, GL, Shapiro, H. Primary ovarian carcinoid tumors. Gynecol Oncol. 1996; 61:259–265.
137. Soga, J, Osaka, M, Yakuwa, Y. Carcinoids of the ovary: an analysis of 329 reported cases. J Exp Clin Cancer Res. 2000; 19:271–280.
138. Robboy, SJ. Insular carcinoid of ovary associated with malignant mucinous tumors. Cancer. 1984; 54:2273–2276.
139. Snyder, RR, Tavassoli, FA. Ovarian strumal carcinoid: immunohistochemical, ultrastructural, and clinicopathologic observations. Int J Gynecol Pathol. 1986; 5:187–201.
140. Matias-Guiu, X, Forteza, J, Prat, J. Mixed strumal and mucinous carcinoid tumor of the ovary. Int J Gynecol Pathol. 1995; 14:179–183.
141. Zhao, C, Bratthauer, GL, Barner, R, et al. Comparative analysis of alternative and traditional immunohistochemical markers for the distinction of ovarian Sertoli cell tumor from endometrioid tumors and carcinoid tumor: A study of 160 cases. Am J Surg Pathol. 2007; 31:255–266.
142. Sorrong, B, Falkmer, S, Robboy, SJ, et al. Neurohormonal peptides in ovarian carcinoids: an immunohistochemical study of 81 primary carcinoids and of intraovarian metastases from six mid-gut carcinoids. Cancer. 1982; 49:68–74.
143. Vang, R, Gown, AM, Zhao, C, et al. Ovarian mucinous tumors associated with mature cystic teratomas: morphologic and immunohistochemical analysis identifies a subset of potential teratomatous origin that shares features of lower gastrointestinal tract mucinous tumors more commonly encountered as secondary tumors of the ovary. Am J Surg Pathol. 2007; 31:854–869.
144. Rabban, JT, Lerwill, MF, McCluggage, WG, et al. Primary ovarian carcinoid tumors may express CDX-2: a potential pitfall in distinction from metastatic intestinal carcinoid tumors involving the ovary. Int J Gynecol Pathol. 2009; 28:41–48.
145. Robboy, SJ, Scully, RE, Norris, HJ. Carcinoid metastatic to the ovary. A clinocopathologic analysis of 35 cases. Cancer. 1974; 33:798–811.
146. Hristov, AC, Young, RH, Vang, R, et al. Ovarian metastases of appendiceal tumors with goblet cell carcinoid-like and signet ring cell patterns: a report of 30 cases. Am J Surg Pathol. 2007; 31:1502–1511.
147. Kleinman, GM, Young, RH, Scully, RE. Primary ovarian neuroectodermal tumors: a report of 25 cases. Am J Surg Pathol. 1993; 17:764–778.
148. Kleinman, GM, Young, RH, Scully, RE. Ependymoma of the ovary: report of three cases. Hum Pathol. 1984; 15:632–638.
149. Hirschowitz, L, Ansari, A, Cahill, DJ, et al. Central neurocytoma arising within a mature cystic teratoma of the ovary. Int J Gynecol Pathol. 1997; 16:176–179.
150. Mikami, M, Komura, Y, Sakaiya, N, et al. Primary ependymoma of the ovary, in which long-term oral etoposide (VP-16) was effective in prolonging disease-free survival. Gynecol Oncol. 2001; 83:149–152.
151. MorovicA, Damjanov, I. Neuroectodermal ovarian tumors: a brief overview. Histol Histopathol. 2008; 23:765–771.
152. Stolnicu, S, Furtado, A, Sanches, A, et al. Ovarian ependymomas of extra-axial type or central immunophenotypes. Hum Pathol. 2011; 42:403–408.
153. Ud Din, N, Memon, A, Aftab, K, et al. Oligodendroglioma arising in the glial component of ovarian teratomas: a series of six cases and review of literature. J Clin Pathol. 2012; 65:631–634.
154. Carr, KA, Roberts, JA, Frank, TS. Progesterone receptors in bilateral ovarian ependymoma presenting in pregnancy. Hum Pathol. 1992; 23:962–965.
155. Guerrieri, C, Jarlsfelt, I. Ependymoma of the ovary. A case report with immunohistochemical, ultrastructural, and DNA cytometric findings, as well as histogenetic considerations. Am J Surg Pathol. 1993; 17:623–632.
156. Idowu, MO, Rosenblum, MK, Wei, XJ, et al. Ependymomas of the central nervous system and adult extra-axial ependymomas are morphologically and immunohistochemically distinct—a comparative study with assessment of ovarian carcinomas for expression of glial fibrillary acidic protein. Am J Surg Pathol. 2008; 32:710–718.
157. Kawauchi, S, Fukuda, T, Miyamoto, S, et al. Peripheral primitive neuroectodermal tumor of the ovary confirmed by CD99 immunostaining, karyotypic analysis, and RT-PCR for EWS/FLI-1 chimeric mRNA. Am J Surg Pathol. 1998; 22:1417–1422.
158. Peterson, WF. Malignant degeneration of benign cystic teratomas of the ovary; a collective review of the literature. Obstet Gynecol Surv. 1957; 12:793–830.
159. Dos Santos, L, Mok, E, Iasonos, A. Squamous cell carcinoma arising in mature cystic teratoma of the ovary: A case series and review of the literature. Gynecol Oncol. 2007; 105:321–324.
160. Hackethal, A, Brueggmann, D, Bohlmann, MK, et al. Squamous-cell carcinoma in mature cystic teratoma of the ovary: Systematic review and analysis of published data. Lancet Oncol. 2008; 9:1171–1180.
161. Hirakawa, T, Tsuneyoshi, M, Enjoji, M. Squamous cell carcinoma arising in mature cystic teratoma of the ovary. Clinicopathologic and topographic analysis. Am J Surg Pathol. 1989; 13:397–405.
162. Sumi, T, Ishiko, O, Maeda, K, et al. Adenocarcinoma arising from respiratory ciliated epithelium in mature ovarian cystic teratoma. Arch Gynecol Obstet. 2002; 267:107–109.
163. Song, YJ, Ryu, SY, Choi, SC, et al. Adenocarcinoma arising from the respiratory ciliated epithelium in a benign cystic teratoma of the ovary. Arch Gynecol Obstet.. 2009; 280:659–662.
164. Ribeiro-Silva, A, Chang, D, Bisson, FW, Ré, LO. Clinicopathological and immunohistochemical features of a sebaceous carcinoma arising within a benign dermoid cyst of the ovary. Virchows Arch. 2003; 443:574–578.
165. Boyd, C, Patel, K, O’Sullivan, B, et al. Pulmonary-type adenocarcinoma and signet ring mucinous adenocarcinoma arising in an ovarian dermoid cyst: report of a unique case. Hum Pathol. 2012; 43:2088–2092.
166. Arora, DS, Haldane, S. Carcinosarcoma arising in a dermoid cyst of the ovary. J Clin Pathol. 1996; 49:519–521.
167. Yahata, T, Kawasaki, T, Serikawa, T, et al. Adenocarcinoma arising from respiratory ciliated epithelium in benign cystic teratoma of the ovary: a case report with analyzes of the CT, MRI and pathological findings. J Obstet Gynaecol Res. 2008; 34:408–412.
168. McKenney, JK, Soslow, RA, Longacre, TA. Ovarian mature teratomas with mucinous epithelial neoplasms: morphologic heterogeneity and association with pseudomyxoma peritonei. Am J Surg Pathol. 2008; 32:645–655.
169. Levine, DA, Villella, JA, Poynor, EA, Soslow, RA. Gastrointestinal adenocarcinoma arising in a mature cystic teratoma of the ovary. Gynecol Oncol. 2004; 94:597–599.
170. Gershenson, DM, Del Junco, G, Copeland, LJ, et al. Mixed germ cell tumors of the ovary. Obstet Gynecol. 1984; 64:200–206.
171. Scully, RE. Gonadoblastoma: a review of 74 cases. Cancer. 1970; 25:1340–1356.
172. Salo, P, Kaariainen, H, Petrovic, V, et al. Molecular mapping of the putative gonadoblastoma locus on the Y chromosome. Genes Chromosomes Cancer. 1995; 14:210–214.
173. Cheng, L, Thomas, A, Roth, LM, et al. OCT4: a novel biomarker for dysgerminoma of the ovary. Am J Surg Pathol. 2004; 28:1341–1346.
174. Li, Y, Tabatabai, ZL, Lee, TL, et al. The Y-encoded TSPY protein: a significant marker potentially plays a role in the pathogenesis of testicular germ cell tumors. Hum Pathol. 2007; 38:1470–1481.
175. Hersmus, R, Kalfa, N, de Leeuw, B, et al. FOXL2 and SOX9 as parameters of female and male gonadal differentiation in patients with various forms of disorders of sex development (DSD). J Pathol. 2008; 215:31–38.
176. Talerman, A. A distinctive gonadal neoplasm related to gonadoblastoma. Cancer. 1972; 30:1219–1224.