Chapter 450 Other Microcytic Anemias
Sideroblastic anemias result from acquired and hereditary disorders of heme synthesis. The anemias are characterized by hypochromic microcytic red blood cells (RBCs) mixed with normal RBCs, thus giving an overall picture of a dimorphic population of erythrocytes, and the complete blood cell count indicates an extremely high RBC distribution width (RDW). The serum iron concentration usually is elevated, and the transferrin saturation of iron is increased. In all cases of sideroblastic anemia, regardless of the specific cause, impaired heme synthesis leads to retention of iron within the mitochondria. Morphologically, this is seen in marrow nucleated RBCs with iron granules (aggregates of iron in mitochondria) that have a perinuclear distribution. These unusual cells, known as ringed sideroblasts (see  Fig. 450-1 on the Nelson Textbook of Pediatrics website at www.expertconsult.com), are found only in pathologic states and are distinct from the sideroblasts (RBC precursors that contain diffuse cytoplasmic ferritin granules) in the marrow of normal subjects. Sideroblastic anemias most commonly occur in adulthood, and these acquired disorders can be idiopathic or secondary to drugs, alcohol, or myelodysplastic disorders. A few sideroblastic anemias are seen in children.
Fig. 450-1 on the Nelson Textbook of Pediatrics website at www.expertconsult.com), are found only in pathologic states and are distinct from the sideroblasts (RBC precursors that contain diffuse cytoplasmic ferritin granules) in the marrow of normal subjects. Sideroblastic anemias most commonly occur in adulthood, and these acquired disorders can be idiopathic or secondary to drugs, alcohol, or myelodysplastic disorders. A few sideroblastic anemias are seen in children.
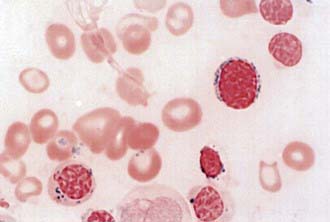
Figure 450-1 Ring sideroblast in myelodysplastic syndrome (refractory anemia with ring sideroblasts)—iron stain.
(From Ryan DH, Cohen HJ: Bone marrow examination. In Hoffman R, Benz EJ Jr, Shattil SJ, et al, editors: Hematology, ed 4, Philadelphia, 2005, Churchill Livingstone.)
Congenital Sideroblastic Anemia
A unique variant of congenital sideroblastic anemia is Pearson syndrome, characterized by the early onset of transfusion-dependent anemia, neutropenia, and thrombocytopenia. In addition to the usual marrow abnormalities of sideroblastic anemia, children with this syndrome also have vacuolization of RBC and myeloid precursors. In contrast to other sideroblastic anemias, which are microcytic, this is a macrocytic anemia and consequently it sometimes is confused with Diamond-Blackfan anemia (Chapter 442).
Rare Types of Hypochromic Microcytic Anemia
Several patients have had refractory hypochromic anemia associated with lymphatic tumors or lymphoid hyperplasia. Correction of the anemia followed removal of the abnormal lymphatic tissue in these patients (Chapters 483 and 500).
Alcindor T, Bridges KR. Sideroblastic anaemias. Br J Haematol. 2002;116:733-743.
Ayas M, Al-Jefri A, Mustafa MM, et al. Congenital sideroblastic anaemia successfully treated using allogeneic stem cell transplantation. Br J Haematol. 2001;113:938.
Chalco JP, Huicho L, Alamo C, et al. Accuracy of clinical pallor in the diagnosis of anemia in children: a meta-analysis. BMC Pediatrics. 2005;5:46.
Fleming MD. The genetics of inherited sideroblastic anemias. Semin Hematol. 2002;39:270-281.
Guernsey DL, Jiang H, Campagna DR, et al. Mutations in mitochondrial carrier family gene SLC25A38 cause nonsyndromic autosomal recessive congenital sideroblastic anemia. Nat Genet. 2009;41:651-656.
Moy RJ. Prevalence, consequences and prevention of childhood nutritional iron deficiency: a child public health perspective. Clin Lab Haematol. 2006;28:291-298.
Panagiotou JP, Douros K. Clinicolaboratory findings and treatment of iron-deficiency anemia in childhood. Pediatr Hematol Oncol. 2004;21:521-534.
Sandoval C, Jayabose S, Eden AN. Trends in diagnosis and management of iron deficiency during infancy and early childhood. Hematol Oncol Clin North Am. 2004;18:1423-1438. x
Walter T. Effect of iron-deficiency anemia on cognitive skills and neuromaturation in infancy and childhood. Food Nutr Bull. 2003;24(4 Suppl):S104-S110.





