Chapter 455 Other Membrane Defects
Paroxysmal Nocturnal Hemoglobinuria
Etiology
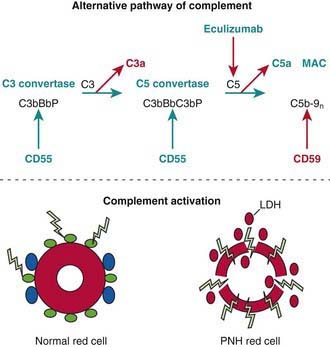
Paroxysmal nocturnal hemoglobinuria (PNH) reflects an abnormality of marrow stem cells that affects each blood cell lineage. The disease is not inherited; it is an acquired disorder of hematopoiesis characterized by a defect in proteins of the cell membrane that renders the red blood cells (RBCs) and other cells susceptible to damage by normal plasma complement proteins (see Fig. 455-1 on the Nelson Textbook of Pediatrics website at  www.expertconsult.com). The deficient membrane-associated proteins include decay-accelerating factor, the C8 binding protein, and other proteins that normally impede complement lysis at various steps. The underlying defect involves the glycolipid anchor that maintains these protective proteins on the cell surface. Various mutations in the PIGA gene that are involved in glycosylphosphatidylinositol biosynthesis have been identified in patients with PNH. More than one PIGA gene mutation often occurs in an individual patient, suggesting multiclonality. Glycosylphosphatidylinositol-deficient cells are found at low frequency in normal persons, suggesting that injury to the normal marrow stem cells provides a selective advantage to the progeny of PNH clones in the genesis of this disease.
www.expertconsult.com). The deficient membrane-associated proteins include decay-accelerating factor, the C8 binding protein, and other proteins that normally impede complement lysis at various steps. The underlying defect involves the glycolipid anchor that maintains these protective proteins on the cell surface. Various mutations in the PIGA gene that are involved in glycosylphosphatidylinositol biosynthesis have been identified in patients with PNH. More than one PIGA gene mutation often occurs in an individual patient, suggesting multiclonality. Glycosylphosphatidylinositol-deficient cells are found at low frequency in normal persons, suggesting that injury to the normal marrow stem cells provides a selective advantage to the progeny of PNH clones in the genesis of this disease.
Acanthocytosis
Acanthocytosis is characterized by RBCs with irregular circumferential pointed projections (see Fig. 452-4E). This morphologic finding is seen with alterations in the cholesterol:phospholipid ratio in some patients with liver disease and in congenital abetalipoproteinemia associated with malabsorption, neuromuscular abnormalities, and retinitis pigmentosa (Chapters 80.3 and 622). It also is associated with the rare X-linked McLeod syndrome, which includes absence of the Kx (Kell) antigen, late-onset myopathy, neurologic abnormalities such as chorea, splenomegaly, and hemolysis with acanthocytosis.
Brodsky RA. Advances in the diagnosis and therapy of paroxysmal nocturnal hemoglobinuria. Blood Rev. 2008;22:65-74.
Gold MM, Shifteh K, Bello JA, et al. Chorea-acanthocytosis: a mimicker of Huntington disease. Case report and review of the literature. Neurologist. 2006;12:327-329.
Hillmen P, Young NS, Shubert J, et al. The complement inhibitor eculizumab in paroxysmal nocturnal hemoglobinuria. N Engl J Med. 2006;355:1233-1243.
Parker C. Eculizumab for paroxysmal nocturnal haemoglobinuria. Lancet. 2009;373:759-767.
Rosse WF, Nishimura J. Clinical manifestations of paroxysmal nocturnal hemoglobinuria: present state and future problems. Int J Hematol. 2003;77:113-120.
Smith LJ. Paroxysmal nocturnal hemoglobinuria. Clin Lab Sci. 2004;17:172-177.
Suenaga K, Kanda Y, Niiya H, et al. Successful application of nonmyeloablative transplantation for paroxysmal nocturnal hemoglobinuria. Exp Hematol. 2001;29:639-642.
Van den Heuvel-Eibrink MM, Bredius RG, te Winkel ML, et al. Childhood paroxysmal nocturnal hemoglobinuria (PHH): a report of 11 cases in the Netherlands. Br J Haematol. 2005;128:571-577.





