CHAPTER 76 Other Inherited Metabolic Disorders of the Liver
Inborn errors of metabolism encompass a vast variety of disorders with myriad presentations including liver disease and are characterized by complex pathophysiology. Metabolic liver diseases may manifest as acute, life-threatening illnesses in the neonatal period or as chronic liver disease in adolescence or adulthood, with progression to liver failure, cirrhosis, or hepatocellular carcinoma. In the 2007 report of the Scientific Registry of Transplant Recipients in the United States, 3% of all liver transplantations in the United States were performed because of complications resulting from metabolic liver disease.1 When the pediatric population alone is analyzed, this percentage is substantially higher. Nationwide, more than 20% of the liver transplantations performed over the five-year period from 1995 to 2000 were for complications of metabolic liver disease.2 Liver transplantation has become a life-saving measure for many patients with metabolic liver diseases. New nontransplant treatment options have become available that may, in certain cases, obviate the need for liver transplantation and thereby help alleviate the shortage of donor organs.3,4
CLINICAL FEATURES OF METABOLIC LIVER DISEASE
The diverse presenting symptoms of metabolic liver disease are listed in Table 76-1. Certain metabolic liver diseases in young patients may mimic other illnesses, such as acute infections and intoxications. By contrast, the older patient with metabolic liver disease may present with symptoms of chronic disease. Because metabolic diseases can resemble multiple other disorders, a high index of suspicion is required for correct diagnosis.
| Symptoms | Coma |
| Developmental delay | |
| Growth failure | |
| Hyperammonemic symptoms | |
| Hypoglycemic symptoms | |
| Neurologic or motor skill deterioration | |
| Recurrent vomiting | |
| Seizures | |
| Signs | Ascites |
| Abdominal distention | |
| Cardiac dysfunction | |
| Cataracts | |
| Dysmorphic features | |
| Hepatomegaly | |
| Hypotonia | |
| Jaundice | |
| Short stature | |
| Splenomegaly | |
| Unusual odors | |
| Other Findings | Acidosis |
| Cholestasis | |
| Coagulopathy | |
| Fulminant hepatic failure | |
| Ketosis | |
| Rickets |
An infant presenting with cholestasis should undergo an evaluation for metabolic liver disease. (The approach to the patient with jaundice is discussed in detail in Chapter 20.) Any patient with progressive neuromuscular disease, developmental delay, or regression of developmental milestones also requires evaluation. Metabolic liver disease should be an immediate consideration in patients with elevated serum aminotransferase levels, hepatomegaly, acidosis, hypoglycemia, ascites, bleeding diathesis, hyperammonemia, coma, recurrent vomiting, or poor weight gain.
A detailed history can often raise the possibility of metabolic liver disease and help direct the investigation. A family history of consanguinity, multiple miscarriages, or early infant deaths may suggest a metabolic derangement. Close relatives with undiagnosed liver disease, progressive neurologic or muscle disease, or undiagnosed developmental delays should also raise suspicion. A carefully obtained dietary history is also important in dissecting the nature of the illness; introduction of certain foods may correlate with the onset of symptoms, as in patients with urea cycle defects, galactosemia, or fructosemia. A history of specific dietary aversions may also be revealing.5
Recommended initial screening tests, which should direct further diagnostic evaluation, are listed in Table 76-2. Because patients with metabolic liver disease often present with acute and recurrent symptoms, it is of utmost importance that the physician obtain the diagnostic studies as soon as possible and at the time the patient is experiencing symptoms. The laboratory values for many of these illnesses may normalize between acute episodes. In enigmatic cases, serum and urine samples should be obtained during the acute illness and saved (frozen) for definitive studies, if available. A liver biopsy can also be a valuable diagnostic tool. If a metabolic liver disease is suspected, in addition to obtaining specimens for standard histology, a frozen specimen should be saved and a sample prepared for later electron microscopic study to look at the subcellular organelles, which may exhibit characteristic changes in some disorders.
Table 76-2 Screening Laboratory Studies for Metabolic Liver Disease*
| Serum | Albumin |
| Alkaline phosphatase | |
| Amino acids | |
| Aminotransferases | |
| Ammonia | |
| Anion gap calculation | |
| Coagulation profile | |
| Electrolytes | |
| Ferritin | |
| Fractionated bilirubin | |
| Gamma glutamyl transpeptidase | |
| Glucose | |
| Lactate† | |
| Peripheral blood smear | |
| Pyruvate† | |
| Uric acid† | |
| Urine | Organic acids |
| Orotic acid | |
| Reducing substances |
* Specimens of serum and urine obtained during acute episodes should be saved for later studies.
α1-ANTITRYPSIN DEFICIENCY
Deficiency of α1-antitrypsin (α1-AT) is transmitted in an autosomal recessive fashion and leads to an increased risk of lung and liver disease. This deficiency is one of the most common genetic diseases in the world and the second most common metabolic disease affecting the liver, the most common being hereditary hemochromatosis (see Chapter 74).6,7 The following discussion focuses on the effects of α1-AT deficiency on the liver.
Pathophysiology
The prototypical member of the serpin family of protease inhibitors, α1-AT binds with and promotes the degradation of serine proteases in the serum and tissues. The most important of these serine proteases is neutrophil elastase, which is inhibited by α1-AT through formation of a tight 1 : 1 α1-AT-to-elastase complex. Loss of serum α1-AT activity, as occurs in the most common form of α1-AT deficiency, leads to uninhibited neutrophil elastase activity and is one of the primary mechanism for the premature development of pulmonary emphysema in affected patients.8 Another proposed mechanism derives from the observation that α1-ATZ polymers are proinflammatory toward neutrophils and are found in lung lavage fluid of patients with the protease inhibitor (Pi) ZZ phenotype of α1-AT deficiency (see later) and emphysema; moreover, instillation of α1-ATZ polymers in murine lung leads to an influx of neutrophils to the lung.8,9
Allelic α1-AT mutant variants produce Pi gene products that can be distinguished from the normal product by electrophoretic methods; the normal allelic representation is designated PiM. The PiZ variant produces a mutant α1-ATZ protein. Homozygosity at the PiZ allele is the most common and classic pathologic form of α1-AT deficiency and is capable of leading to liver and lung disease. The α1-ATZ molecule represents a single amino acid replacement of glutamine with a lysine residue as a result of a mutation at position 342 of the α1-AT (SERPINA1) gene. More than 100 naturally occurring variants of α1-AT have been described. Although most of these variants are either of no clinical significance or are extremely rare, two additional variants, PiSiiyama and PiMmalton, have been reported to be associated with liver injury and even cirrhosis.8,10–12
α1-AT is produced almost exclusively in the rough endoplasmic reticulum (ER) of hepatocytes and is subsequently targeted to the secretory pathway via the Golgi apparatus. Structural misfolding and polymerization of the mutant α1-ATZ protein causes its aberrant retention in the ER, failure of progression through the secretory pathway, and diminished intracellular degradation. In persons with the phenotype PiZZ, serum α1-AT activity levels are reduced to less than 15% of normal; this loss of function accounts for the development of pulmonary disease. Studies suggest that the rate of intracellular degradation may itself be genetically determined and may influence the expression of disease; α1-ATZ appears to be degraded more slowly in the ER of PiZZ patients who are susceptible to liver disease than in PiZZ patients who are not susceptible to liver disease.13
The exact mechanism for α1-ATZ-induced liver injury is not known. Studies of transgenic mice that express the human ATZ gene suggest a gain-of-function mechanism by which retention in the ER and accumulation in hepatocytes of mutant α1-ATZ is responsible for hepatotoxicity.14 Multiple intracellular signaling pathways, including caspase activation, ER stress responses, and the autophagic response, are activated by the retention of malformed proteins in the ER.15 Autophagy is an intracellular degradative pathway that targets proteins and organelles for destruction during development as well as at times of stress or nutrient deprivation. One hypothesis under investigation is that increased stimulation of autophagy by accumulation of α1-ATZ protein leads to ongoing mitochondrial injury, cellular apoptosis, and a chronic cycle of hepatocellular death and regeneration that may, over time, lead to liver injury and cirrhosis.7,16 Other genetic and environmental modifiers of the ER “quality control” process for handling mutant α1-ATZ protein are undoubtedly involved and account for the wide variation in clinical phenotype observed in the hepatic presentation of PiZZ patients.7
Clinical Features
Although the prevalence of the classic α1-AT deficiency allele, PiZ, is highest in populations derived from northern European ancestry, many racial subgroups are affected worldwide, and millions of persons have combinations of deficiency alleles (i.e., PiSS, PiSZ, or PiZZ).8,17 In the United States, the overall prevalence of deficiency allele combinations is approximately 1 in 490 (i.e., 1 in 1058 for PiSS, 1 in 1124 for PiSZ, and 1 in 4775 for PiZZ).18 Mounting evidence suggests that heterozygous α1-AT deficiency states can contribute to the development of cirrhosis and chronic liver failure in adults through mechanisms similar to those encountered with the PiZZ phenotype.10–1219 In addition, the heterozygous α1-AT deficiency state may contribute to worsening of chronic liver disease caused by hepatitis C viral infection or nonalcoholic fatty liver disease in adults, as well as cholestatic liver diseases in children.7,19–22
In the most unbiased study to date, reported by Sveger,23 on the epidemiology of liver disease in patients with α1-AT deficiency, 200,000 Swedish infants were screened for α1-AT deficiency, 184 infants were found to have abnormal allelic forms of α1-AT (127 PiZZ, 2 PiZnull, 54 PiSZ, and 1 PiSnull), and 6 (5 PiZZ and 1 PiSZ) died in early childhood, although only 2 from cirrhosis.23 Although about 10% of newborns with α1-AT deficiency (PiZZ) present with cholestasis and as many as 50% continue to have elevated serum aminotransferase levels at age three months, most are clinically asymptomatic.23,24 Liver disease does not develop in patients with null α1-AT phenotypes, whereas early-onset emphysema develops in all of them.25 Therefore, the prognosis for patients with liver disease manifesting in infancy as a result of α1-AT deficiency (PiZZ) is highly variable. Even those children in whom cirrhosis develops can have a highly variable progression to end-stage liver disease (ESLD), which infrequently leads to liver transplantation.26 Moreover, siblings with PiZZ have a variable degree of liver involvement; in a study reported by Hinds and colleagues, five of seven children with PiZZ requiring liver transplantation had siblings with PiZZ who had no persistent liver involvement.27 This finding suggests that environmental or additional genetic factors must be involved in determining the severity of liver disease in α1-AT deficiency; this area is under active scientific investigation.28
Of 150 patients with α1-AT deficiency from Sveger’s original study23 who subsequently underwent evaluation at age 16 and 18 years, none had clinical signs of liver disease. Elevated serum aminotransferase or gamma glutamyl transpeptidase (GGTP) levels were found in fewer than 20% of patients with a PiZZ phenotype and in fewer than 15% of those with a PiSZ phenotype.24 By the third decade of life, analysis of this same cohort of affected persons showed that 6% of PiZZ and 9% of PiSZ patients had a marginal increase in serum alanine aminotransferase levels.29
Even though liver disease is often (but not always) mild during infancy and childhood, patients with α1-AT deficiency have an increased risk for development of cirrhosis during adulthood; 42% of all PiZZ patients have histologic evidence of cirrhosis at autopsy.30,31 Moreover, homozygous α1-AT deficiency raises the risk for development of hepatocellular carcinoma, especially in men older than 50 years.31 The diagnosis of α1-AT deficiency should be considered in any patient presenting with noninfectious chronic hepatitis, hepatosplenomegaly, cirrhosis, portal hypertension, or hepatocellular carcinoma.
Diagnosis
α1-AT is considered a hepatic acute-phase reactant, and its release may be stimulated by stress, injury, pregnancy, or neoplasia. Because these factors can influence α1-AT production, even in patients with the PiZZ phenotype, the diagnosis of α1-AT deficiency should be based on phenotype analysis and not solely on the serum α1-AT level.32 A liver biopsy specimen, although not universally recommended, should confirm the diagnosis. Commercial tests are available to detect the most common mutant alleles by polymerase chain reaction analysis of genomic DNA. In addition, a re-sequencing molecular array chip is available for rapid sequencing of the entire SERPINA1 gene to allow identification of rare mutant alleles.33
Screening guidelines to diagnose α1-AT deficiency in asymptomatic persons have been proposed in an effort to reduce the risk of emphysema by counseling patients to avoid smoking.34 Adults with chronic lung disease and siblings of affected patients with lung or liver disease should be targeted for screening, and appropriate education and genetic counseling should be offered to patients with α1-AT deficiency identified by screening.
Treatment
The initial treatment of α1-AT deficiency is with symptomatic care. Breast-feeding until the end of the first year of life has been suggested to decrease the manifestations of cholestatic disease, as may the use of ursodeoxycholic acid.35 The importance of providing fat-soluble vitamins, when indicated, adequate nutrition, and counseling to avoid smoking and second-hand smoke cannot be overemphasized. The role of neonatal screening for α1-AT deficiency is still not settled, although the effect on smoking practices in patients diagnosed at an early age appears to be positive. If effective therapy for liver disease caused by α1-AT deficiency becomes available, neonatal screening may become more useful for preventing the need for liver transplantation.
Although progression to ESLD is uncommon, α1-AT deficiency is the most common metabolic liver disease for which liver transplantation is performed. Besides replacing the injured organ, transplantation corrects the metabolic defect, thereby preventing further progression of systemic disease. Between 1995 and 2004, 161 children and 406 adults underwent liver transplantation for ESLD associated with α1-AT deficiency in the United States; of these, 4.4% of the children and fewer than 1% of the adults were African Americans. Five-year patient survival rates following liver transplantation for the pediatric and adult patients with α1-AT deficiency were 83% and 90% respectively.36 Liver transplantation with grafts from donors with unrecognized α1-AT deficiency appears to have a comparable outcome to transplants using grafts without unrecognized liver disease.37
Replacement therapy with purified α1-AT is the only treatment option approved by the U.S. Food and Drug Administration (FDA) for lung disease associated with α1-AT deficiency. Patients who received replacement therapy in several studies, including a small randomized placebo-controlled trial, have had a slower rate of decline in lung tissue and function compared with control groups, although clinical efficacy has yet to be conclusively demonstrated.38 This therapy would not be expected to benefit α1-AT deficiency–associated liver disease, which results from malprocessed α1-ATZ, not loss of α1-AT protein function.
Other mechanistically based treatment options aimed at influencing the stability or secretion rates of α1-ATZ within the hepatocyte ER are being investigated. Chemical chaperones such as phenylbutyric acid (PBA) markedly increase secretion of α1-ATZ in experimental in vitro and in vivo models of α1-AT deficiency.39 A small pilot study investigated the potential benefits of PBA in the treatment of children with α1-AT–deficient liver disease. Unfortunately, no statistically significant increase in serum α1-AT levels occurred in PBA-treated patients, many of whom experienced unacceptable side effects.40 Another pharmacologic approach in the early stages of investigation involves the design of small molecules to inhibit α1-ATZ protein polymerization and increase clearance of intracellular aggregates.41
α1-AT deficiency is one of many diseases for which reconstitution of the normal genotype through gene therapy is being studied. Long-term expression of human α1-AT in murine liver, at therapeutic levels, has been achieved after hydrodynamics-based intravenous injection of nonviral deoxyribonucleic acid (DNA) constructs.42 Also, delivery of a vector carrying a ribozyme designed to target human α1-ATZ messenger ribonucleic acid (mRNA) in the portal vein of transgenic mice that carry the human PiZ allele led to a 50% reduction in α1-ATZ mRNA levels. In a separate experiment, administration of a vector carrying the normal PiM allele, which is resistant to ribozyme cleavage, led to long-term expression of human α1-AT protein in both liver and serum up to one year following injection.43 These results are promising first steps toward the goal of achieving successful gene replacement therapy for α1-AT deficiency.
GLYCOGEN STORAGE DISEASES
More than 10 distinct inborn disorders of glycogen metabolism have been described in the literature, but only 3 are associated with significant liver disease–glycogen storage disease (GSD) types I, III, and IV.44,45 Other GSDs may cause hepatomegaly or liver histologic changes but generally do not cause clinically important liver disease. The overall incidence of GSD types I, III, and IV is estimated to be between 1 in 50,000 and 1 in 100,000 population.
Glycogen metabolism occurs in many tissues, but the areas of clinical importance are the muscle and liver. The body uses glycogen as a storage system for glucose and as a ready reserve for times when systemic glucose is required (see Chapter 72). Glycogen is composed of long-chain glucose molecules arranged in a linear 1,4 linkage. From 8% to 10% of the glucose molecules are attached in a 1,6 linkage to form branching chains, which permit efficient storage of glucose while minimizing the impact on intracellular osmolality. The substrates for glycogen synthesis, glucose-6-phosphate (Glu-6-P) and glucose-1-phosphate (Glu-1-P), are derived from several pathways, including fructose and galactose metabolic cycles as well as gluconeogenesis and glycogenolysis (Fig. 76-1).
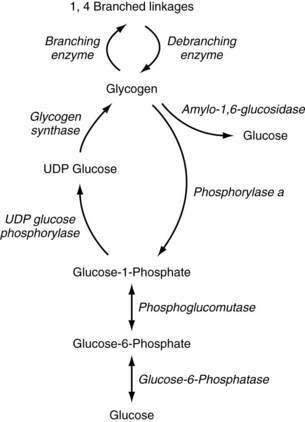
Figure 76-1. Pathway of glycogen synthesis and glycogenolysis. Enzymes are shown in italics. UDP, uridine diphosphate.
Through the action of uridine diphosphate glucose (UDPG) pyrophosphorylase and glycogen synthase, Glu-1-P is metabolized to UDPG and glycogen, sequentially. The 1,4 linkages can be converted to 1,6 linkages by the actions of branching enzymes. Amylo-1,6-glucosidase is a debranching enzyme that can release 8% to 10% of the glucose stored in glycogen. The remaining glucose is released as Glu-1-P through the action of phosphorylase a and is converted to Glu-6-P by phosphoglucomutase. Phosphorylase exists in an active (a) and an inactive form (b); protein kinase is responsible for the conversion of phosphorylase b to a. Protein kinase is stimulated by epinephrine, glucagon, and fasting, and thereby increases glycogenolysis. High levels of glucose influence the conversion of phosphorylase a back to phosphorylase b, thereby decreasing glycogenolysis. Glycogen synthase also exists in active (a) and inactive (b) forms. Phosphorylase a inhibits the conversion to glycogen synthase a, thereby reducing glycogen synthesis. High levels of glycogen favor the formation of glycogen synthase b.45
TYPE I
GSD type I, the most common inborn error of glycogen metabolism, results from deficiency of a two-component enzyme system involved in the transport of Glu-6-P from the cytosol into the ER by Glu-6-P translocase and subsequent cleavage of Glu-6-P by glucose-6-phosphatase (Glu-6-Pase), located on the luminal side of the ER. Clinical and molecular genetic observations have disclosed two subtypes of GSD type I, Ia and Ib, that account for virtually all cases.46 The clinical phenotype with respect to liver disease is similar in the two forms; however, patients with GSD type Ib often have intermittent severe neutropenia and neutrophil dysfunction, making them prone to recurrent episodes of severe bacterial infections and Crohn’s-like intestinal disease.47,48
Clinical Features
Most patients with GSD type I present in infancy with symptoms of metabolic derangement, such as lethargy, seizures, or coma as a result of profound hypoglycemia or metabolic acidosis, a protruding abdomen caused by hepatomegaly, muscular hypotonia, and delayed psychomotor development.49
Physical signs invariably include hepatomegaly, usually with a normal-sized spleen. Patients in whom the disease is poorly controlled for a long time exhibit short stature and growth failure and may be prone to adiposity. Delayed bone age and reduced postpubertal bone mineral density are common.50 Xanthomas can appear after puberty and localize to the elbows, knees, buttocks, or nasal septum, the last leading to epistaxis. Patients with GSD type I are susceptible to a wide spectrum of brain damage that may result in epilepsy, hearing loss, and abnormal neuroradiologic findings, most likely as a result of recurrent episodes of hypoglycemia.51
Other metabolic derangements can be seen. Lactic acid levels can reach four to eight times normal; the accompanying metabolic acidosis may manifest as muscle weakness, hyperventilation, malaise, headache, or recurrent fever. Serum adenosine triphosphate (ATP) and phosphorus levels are low, secondary to an increase in purine synthesis and the inability to release phosphorus from Glu-6-P. Hyperuricemia is common and may lead to gout, arthritis, or progressive nephropathy. Nephromegaly secondary to increased glycogen deposition is common, and with advancing age, progressive renal disease, hypertension, and renal failure requiring dialysis and transplantation may develop.49 Because of hypoglycemia, patients have chronically high serum levels of glucagon with depressed levels of insulin. Hypertriglyceridemia and hypercholesterolemia are present in both GSD Ia and GSD Ib (but more prominently in GSD Ia) and may account for the greater frequency of xanthoma formation.52
In addition to the features already noted, patients with GSD type Ib often have severe intermittent neutropenia and neutrophil dysfunction as well as high platelet counts. Crohn’s-like inflammatory bowel disease often occurs in patients with GSD type Ib at the time of severe neutropenia, and patients are prone to the development of severe bacterial infections, with abscess formation at numerous locations throughout the body.47 No correlation has been found between the severity of bacterial infections or degree of neutropenia and the molecular defect in Glu-6-P transport activity in humans with GSD type Ib, suggesting that other as yet unknown factors, such as modifying genes, define the ultimate disease phenotype.53
Hepatic Involvement
Hepatomegaly in GSD type I results from increased glycogen storage in the liver as well as a large degree of fatty infiltration; the latter likely develops because of a wide array of perturbations in lipid metabolism, including increased free fatty acid flux into the liver.52 Patients demonstrate mild elevations in serum aminotransferase levels but generally do not develop cirrhosis or liver failure.
Hepatic adenomas develop in 22% to 75% of patients, as early as three years of age but most commonly in the second decade of life, and tend to increase in both size and number as the patient ages (see Chapter 94).54 Adenomas in rare instances can transform to hepatocellular carcinoma; unfortunately, serum α-fetoprotein and carcinoembryonic antigen levels and features of the lesions on hepatic imaging are not predictive of malignant transformation.54–56 In the past, reports suggested that the risk of hepatic adenoma was increased in patients who were nonadherent with treatment of GSD type I, but more recent reports have found no differences in the risk of adenoma formation between adherent and nonadherent patients.57 In some patients, hepatic adenomas have been demonstrated to regress and disappear after adequate nutritional therapy, but in general, the course is unpredictable, especially in nonadherent patients.54,55 Because of the unreliability of radiographic imaging and serum marker levels in predicting malignant transformation in this patient population, the better approach—resection of an adenoma or liver transplantation—is uncertain.58
Diagnosis
The hepatic glycogen content is elevated in patients with GSD type I, and the most accurate diagnostic measure is direct analysis of enzyme activity performed on fresh, rather than frozen, liver tissue. Analysis of fresh liver tissue is important to avoid disrupting microsomal Glu-6-Pase activity.59 Fasting serum glucose and lactate levels, a positive glucagon response test result, and the response to fructose or galactose administration (patients with GSD type I do not show the expected rise in serum glucose concentration after administration of glucagon or either sugar) often provide supportive evidence but may not yield a definitive diagnosis. Intermittent severe neutropenia is noted in most patients with GSD type Ib.47,53 DNA analysis-based approaches that integrate biochemical features and the presence or absence of persistent neutropenia have been proposed and may provide a diagnostic alternative to liver biopsy.60
Treatment
Patients with undiagnosed or undertreated GSD type I are at increased risk of death, usually from hypoglycemic comas, seizures, metabolic acidosis, or, in those with GSD type Ib, sepsis from neutropenia.49 Rarely, hepatocellular carcinoma is a cause of death. Management centers on preventing the acute metabolic derangements and potential long-term complications and enabling the patient to attain normal psychological development and a good quality of life.59,61
Consensus guidelines for the management of GSD type I have been proposed.59,61 Biomedical targets for good metabolic control include a preprandial blood glucose level higher than 63 to 77 mg/dL (3.5 to 4.3 mmol/L), urine lactate-to-creatinine ratio higher than 0.06, high-normal serum uric acid level, venous blood base excess higher than −5 mmol/L, bicarbonate level higher than 20 mmol/L, serum triglyceride level lower than 6 mmol/L, and body mass index (BMI) between 0 and 2 standard deviations from normal. In addition, for GSD type Ib, demonstrating a normal fecal α1-AT level is desirable (see Chapter 28).59,62 Because optimal glycemic control is not always possible and the risk of severe hypoglycemia is high if delivery of glucose is interrupted inadvertently, serum lactate levels should be kept at the high end of normal because lactate is an alternative fuel for the brain.
Nutritional supplementation has become the mainstay of therapy for GSD type I. Frequent, high-carbohydrate, daytime feedings, such as uncooked cornstarch, or continuous nighttime drip feedings, or both, allow the steady release of glucose and lead to improved metabolic control and normalized growth and development.52,62 A biochemical target is to maintain the serum glucose level above 70 mg/dL (3.9 mmol/L). Uncooked cornstarch in a dose of 2 g/kg every six hours (6 to 8 mg of glucose/kg/minute) has been suggested; however, alternative regimens have been implemented successfully.
Prophylaxis with antibiotics (e.g., trimethoprim-sulfamethoxazole) is recommended for patients with GSD type 1b and severe neutropenia or recurrent bacterial infections.61 Granulocyte colony stimulating factor (GCSF) has been used with success in patients with GSD type 1b to improve hematologic parameters and neutrophil function and reduce the morbidity associated with severe bacterial infections.63 Splenomegaly may worsen with GCSF therapy, and bone marrow aspiration before and during GCSF therapy may be prudent, given rare occurrences of acute myelogenous leukemia (AML) in patients with GSD type 1b.61 Both GCSF and inflammatory bowel disease raise the risk of osteopenia, and monitoring of bone density is advised.
Adenoviral-mediated gene replacement therapy of recombinant Glu-6-Pase in a canine model of GSD type Ia deficiency, which has all of the major features of GSD type 1a in humans, has led to encouraging results and may be an option in humans in the future.64 An alternative approach, human hepatocyte transplantation, has been performed in a single patient with GSD type 1a with near-resolution of hypoglycemic episodes; however, hypoglycemia subsequently recurred, likely because of the lack of ongoing immunosuppression to reduce the likelihood of recipient rejection of transplanted donor hepatocytes.65 Hepatocyte transplantation, with the use of standard post-transplantation immunosuppression, has been performed successfully in an 18-year-old male adolescent with GSD type 1b, with euglycemia maintained up to 2 years post-transplantation.66
Liver transplantation has corrected the metabolic error in patients with GSD type I and permitted catch-up growth, even in patients in the third decade of life.67,68 Neutrophil counts and function, however, are only variably improved after liver transplantation in patients with GSD type 1b.67
TYPE III
GDE is encoded by a single gene and possesses two independent catalytic activities, an amylo-1,6-glucosidase and oligo-1,4→1,4 glucan transferase. Both of these activities are deficient in the two main clinical subtypes of GSD type III, types IIIa and IIIb. Differential expression of four major GDE mRNA isoforms in liver and muscle tissue distinguishes the two types: type IIIa affects liver and muscle and accounts for 80% of patients, and type IIIb affects the liver only and accounts for 15% of patients. Although subtype-specific mutations in the GDE gene are increasingly identified, the molecular basis for this differential tissue-specific expression of GDE is unknown, and no clear genotype-phenotype correlations have been identified.44,69 Rare isolated loss of one of the two GDE activities has been observed (i.e., glucosidase activity in type IIIc and transferase activity in type IIId).
Clinical Features
Patients with GSD type III may also display progressive muscle weakness, which worsens with activity, and muscle wasting. Nephromegaly is not seen, but cardiomegaly may be present. The diagnosis can be made by direct enzyme analysis of muscle or liver tissue or peripheral leukocytes; mutation analysis of the GDE gene will be increasingly important for diagnosis in the future.45,70 Hepatic adenomas develop in approximately 25% of patients, and isolated reports of cirrhosis leading to hepatocellular carcinoma have been reported.55,71
TYPE IV
Deficiency of the branching enzyme is seen in GSD type IV, a rare syndrome also known as amylopectinosis. Glycogen and amylopectin accumulate in hepatocytes, leading to hepatomegaly, abdominal distention, and failure to thrive, most commonly during infancy. Signs of liver disease, when present, predominate later in the course of the disease. Several variable forms of GSD type IV have been observed—a severe congenital form that manifests as fetal hydrops, neonatal hypotonia, or fetal death72; a childhood subtype that manifests as cardiomyopathy and abnormal neuromuscular development; and other milder, nonprogressive presentations of hepatic disease that do not lead to cirrhosis and are not associated with skeletal muscle or neurologic involvement.73 Genotype-phenotype analyses of the branching enzyme gene have revealed a high degree of molecular heterogeneity without clear clinical associations.73
Hypoglycemia is relatively uncommon, and responses to fructose and galactose challenges are normal. Serum lactate and pyruvate levels are normal, and aminotransferase levels are only moderately elevated until more severe liver involvement becomes apparent. Progressive macronodular cirrhosis is present with an abundance of PAS-positive deposits (amylopectin) in hepatocytes (Fig. 76-2). Cirrhosis may progress to liver failure, and adenomas and hepatocellular carcinoma may develop rarely.74
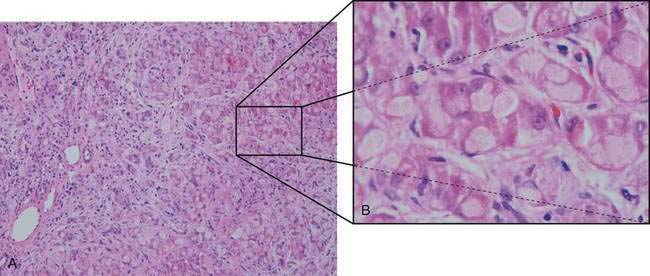
The diagnosis of GSD type IV can be made by direct enzyme analysis of liver tissue or fibroblasts. Most patients die within the first three years of life if the disease is untreated. Diets high in protein and low in carbohydrate have been associated with improved growth but have had little effect on liver involvement. Liver transplantation has been used successfully and results in correction of the metabolic error and normal growth; however, persistence of amylopectin deposits in the heart (with progressive cardiomyopathy) and leukocytes of affected patients has been described.75
CONGENITAL DISORDERS OF GLYCOSYLATION
Congenital disorders of glycosylation (CDGs) comprise a group of inherited defects in the enzymes that synthesize the glycan moiety of glycoproteins or the macromolecules that affect intracellular trafficking and functioning of glycoproteins.76–78 More than 20 CDGs involving both asparagine (N)- and serine/threonine (O)-linked protein glycosylation have been reported to date, and many of these disorders lead to dysfunction of the liver, intestine, or both.76–78 Protein glycosylation is complex and involves multiple enzymatic steps and subcellular compartments.77,78 Secretory glycoproteins with altered carbohydrate moieties in CDGs include coagulation factors, albumin and other binding proteins, growth hormone, apolipoproteins, insulin, and thyroxine-binding globulin.79 Because protein glycosylation occurs in all cells, it is not surprising that patients with a CDG exhibit multisystemic abnormalities, often dominated by central nervous system manifestations.
Two main groups of protein N-glycosylation disorders, groups I and II, have been delineated on the basis of characteristic isoelectric focusing patterns of serum transferrin, a marker protein for this group of disorders.77 Group I disorders (of which there are 12 types) involve aberrant processing of lipid-linked oligosaccharides before transfer to protein targeted for glycosylation and include the three most common and best-characterized types of CDG, types Ia, Ib, and Ic. Clinical features in common among these three disorders are protein-losing enteropathy, coagulopathy (both procoagulant and anticoagulant states), feeding difficulties, and hepatomegaly.77,80
CDG type Ia is caused by defects in phosphomannomutase (PMM), an enzyme that converts mannose-6-phosphate to mannose-1-phosphate (Fig. 76-3). Almost 60 mutations have been found in the encoding PMM2 gene; most patients are compound heterozygotes for mutations that likely preserve some residual PMM enzymatic activity, suggesting that complete loss of PMM activity is incompatible with life; indeed, targeted disruption of the PMM2 gene in the mouse leads to early embryonic lethality.81,82 Patients typically have severe neurologic abnormalities, dysmorphisms (inverted nipples, abnormal fat distribution, and esotropia), and congenital hepatic fibrosis and steatosis in addition to the features described earlier.80,83
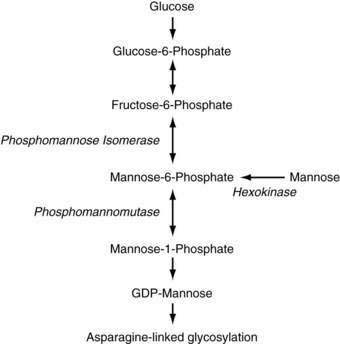
Figure 76-3. Pathway of mannose metabolism. Enzymes are shown in italics. GDP, guanosine diphosphate.
Patients with CDG type Ib have a defect in phosphomannose isomerase (PMI), which converts fructose-6-phosphate to mannose-6-phosphate. In addition to intractable diarrhea, protein-losing enteropathy, and congenital hepatic fibrosis, recurrent episodes of hyperinsulinemic hypoglycemia and cyclic vomiting can occur in patients with CDG type Ib. Neurologic symptoms are usually absent and dysmorphisms are less common than in CDG type 1a.76,80,84 Many patients with CDG type Ib have been treated effectively with dietary mannose, making CDG type Ib the only treatable form of CDG to date, although liver fibrosis can still develop despite improvement in clinical symptoms.78,85,86
Transient hepatomegaly, without congenital hepatic fibrosis, has been noted in a patient with CDG type Ic; otherwise, the clinical features of CDG type Ic are similar to but milder than those of CDG type Ia.80 Patients with CDG type Ih also have protein-losing enteropathy with chronic diarrhea, hepatic fibrosis, and variable degrees of hepatic dysfunction.87 Children with untyped cases of CDG, labeled CDG-X until the genes and protein products can be identified, have been found and have isolated cryptogenic chronic liver disease, mild coagulopathy, and mild portal fibrosis and focal steatosis on liver biopsy specimens.88 In some cases, numerous myelinosomes have been identified in hepatocytes by electron microscopy.
Group II CDGs (six types) involve defects that affect the processing of N-linked glycoproteins.77,78 Most types result from defects in enzymes involved in the trimming of protein-bound oligosaccharides and the subsequent addition of terminal sugars. Patients have marked dysmorphic features and severe developmental retardation. Two infants with hepatosplenomegaly, progressive jaundice, severe epilepsy, recurrent infections, and heart failure have been shown to have mutations in a subunit of the conserved oligomeric Golgi (COG) complex that result in disruption of glycosylated protein intracellular trafficking.89 Another CDG type II disorder with liver involvement has been reported in an infant with recurrent infections, chronic diarrhea, progressive cirrhosis, and normal hepatic excretory and synthetic function.89 The biochemical and clinical features of the remaining types of CDG are less well characterized.
Isoelectrofocusing of apolipoprotein C-III, which carries a single O-linked glycan moiety, has been proposed as a screening method for the O-glycan biosynthesis defects (six types) encountered thus far.76,90 Hepatic dysfunction is usually mild in CDG and usually does not lead to symptoms. Mild hepatic steatosis and fibrosis typically are seen on light microscopy; on electron microscopy, lysosomal vacuoles, termed myelosomes, with concentric electron-dense membranes and variable electron-lucent and electron-dense material, are noted.91 Patients uncommonly can progress to liver failure, with micronodular cirrhosis noted at autopsy.
Any patient with unexplained congenital hepatic fibrosis, protein-losing enteropathy, or a procoagulant tendency should be evaluated for the possibility of CDG (especially type Ib). An initial screening of serum transferrin with isoelectric focusing should be performed, followed by confirmatory enzymatic analysis in fibroblasts, leukocytes, or liver tissue. If the diagnosis of CDG type Ib is confirmed, oral mannose therapy should be initiated.86
PORPHYRIAS
Pathophysiology
The metabolic pathways of heme synthesis are essentially the same in the two tissues in which heme synthesis primarily occurs, the liver (15% to 20%) and the bone marrow (75% to 80%), although synthetic control may be different in the two tissues. The rate-limiting step in hepatic heme synthesis begins with the conversion of glycine and succinyl coenzyme A (CoA) to 5-aminolevulinic acid (ALA) by the action of ALA synthase (Fig. 76-4). ALA synthase activity is decreased by the end-product of the pathway, heme, and is increased by substances that induce the hepatic cytochrome P450 pathway. Six additional enzymatic steps convert ALA to protoporphyrin IX (see Fig. 76-4). In the final step of the pathway, protoporphyrin IX is coupled to ferrous iron by ferrochelatase to create heme. Enzyme deficiencies arising from any of these eight steps of the heme synthetic pathway lead to the clinically apparent diseases known as the porphyrias.
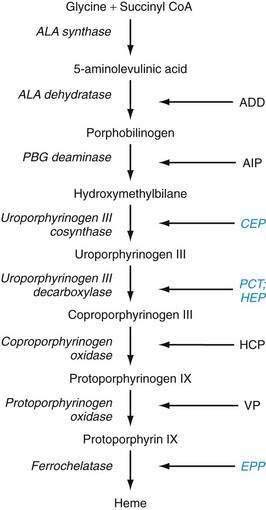
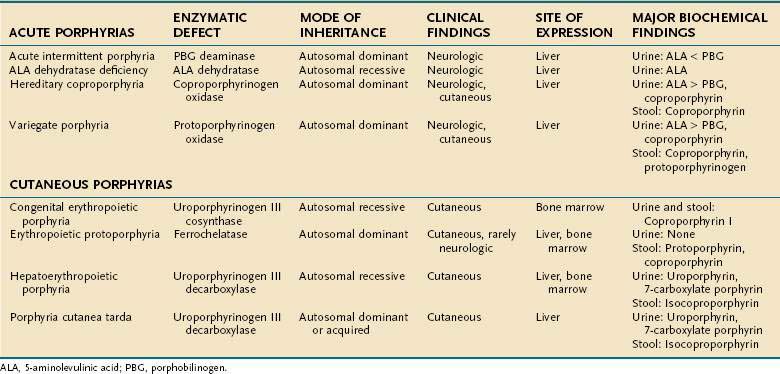
Porphyrias are commonly classified according to clinical features into two main groups, acute porphyrias, which are characterized by dramatic and potentially life-threatening neurologic symptoms, and cutaneous porphyrias, which typically cause few or no neurologic symptoms but instead give rise to a variety of severe skin lesions (Table 76-3). In five of the porphyrias, the liver is the major site of expression; in two others, both the liver and bone marrow are involved; and in one, only the bone marrow contributes.
Acute Porphyrias
The symptoms and signs of the acute neurovisceral attacks that occur in the four acute porphyrias vary considerably. Abdominal pain is present in more than 90% of patients, followed in frequency by tachycardia and dark urine in about 80% of patients. Neuropsychiatric features include hysteria, depression, psychosis, confusion, hallucinations, seizures, and coma, although little evidence suggests that chronic psychiatric illness occurs. Other features are constipation, extremity pain, paresthesias, nausea, vomiting, urinary retention, hypertension, peripheral sensory deficits (often in a “bathing trunk” distribution), and weakness leading to ascending paralysis or quadriplegia. These neurologic attacks appear to be related to the overproduction of ALA and porphobilinogen (PBG), which leads to higher serum and tissue levels of these neurotoxic products.92
Acute episodes are about five times as common in women as in men and may be precipitated by many factors, most commonly drugs, alcohol ingestion, and smoking. Steroids, sex hormones, and medications that stimulate the hepatic cytochrome P450 system, perhaps by increasing the requirements for heme production, are often identified as precipitants.93 Other inciting factors are fasting, infections, and pregnancy; some women report greater problems during the luteal phase of their menstrual cycles.92,94 The disease is clinically latent in 65% to 80% of patients.
ALA dehydratase deficiency is a rare syndrome with autosomal recessive transmission in which the enzyme activity is less than 3%. The enzyme activity is 50% of normal in carriers, who are asymptomatic. Affected patients have severe, recurrent neurologic attacks that may be life threatening. They excrete large amounts of ALA in their urine. Liver transplantation was reported to result in complete resolution of symptoms in one patient with ALA dehydratase deficiency.95
The three remaining acute porphyrias—acute intermittent porphyria (AIP), hereditary coproporphyria (HCP), and variegate porphyria (VP)—result from partial deficiency of the enzymes PBG deaminase, coproporphyrinogen oxidase, and protoporphyrinogen oxidase, respectively. All three disorders are inherited in an autosomal dominant fashion with variable expression. AIP is the most common of the three conditions, occurring in 5 to 10 per 100,000 people, and manifests primarily as derangements in the autonomic nervous system or as a psychiatric disorder.96 VP is more common in South Africa than elsewhere. Although HCP and VP give rise to neurologic symptoms similar to those of AIP, cutaneous lesions also occur in HCP and predominate in VP.97
Cutaneous Porphyrias
The cutaneous porphyrias differ from the acute porphyrias in that affected patients exhibit few or no neurologic symptoms. In these illnesses, excess porphyrins or porphyrinogens are deposited in the upper dermal capillary walls, where these photoreactive compounds cause tissue damage that manifests as cutaneous vesicles and bullae in areas exposed to light or excessive mechanical manipulation. Scarring, infection, pigment changes, and hypertrichosis can follow and even lead to severe mutilation.98
Porphyria cutanea tarda (PCT), the most common of the porphyrias, typically involves a 50% reduction in activity of the enzyme uroporphyrinogen decarboxylase. Patients usually present after 20 years of age. Two types of PCT are recognized. Type I PCT affects 80% of patients and is a sporadic (acquired) form with the enzyme deficiency restricted to the liver. Type II, which affects the other 20% of patients, is familial and inherited in an autosomal dominant fashion with incomplete penetrance; the enzyme deficiency occurs in all tissues.99 Symptoms develop in fewer than 10% of patients with type II PCT. Type I PCT is associated strongly with high alcohol intake, estrogen therapy, and systemic illnesses, including systemic lupus erythematosus, diabetes mellitus, chronic kidney disease, and the acquired immunodeficiency syndrome. For unclear reasons, concomitant hepatitis C infection is strongly associated with expression of PCT (see Chapter 79).100,101 The frequency of mutations of the HFE gene, which causes hereditary hemochromatosis, is increased in patients with types I and II PCT, and these mutations are thus susceptibility factors for clinical expression of the PCT phenotype (see Chapter 74).100,102,103 This association is consistent with pathologic findings in liver biopsy specimens from patients with PCT, of whom 80% have siderosis, 15% have cirrhosis, and most have evidence of iron overload. A patient in whom hepatocellular carcinoma developed has been reported to have unsuspected underlying PCT and hereditary hemochromatosis with bridging fibrosis.104,105 Patients usually do not show signs of overt clinical liver disease, apart from elevated serum aminotransferase levels.
Hepatoerythropoietic porphyria (HEP) is a rare form of porphyria with a pathogenesis similar to that of PCT. HEP results from homozygous uroporphyrinogen decarboxylase deficiency, yielding less than 10% of normal enzyme activity. The cutaneous lesions, which resemble those of PCT, are typically severe and mutilating. The disease usually manifests in the first year of life, and as the patient ages, the dermatologic manifestations may subside, but liver disease, characterized by a nonspecific hepatitis, worsens. Mutation analysis of the uroporphyrinogen decarboxylase gene has revealed numerous mutations that are usually unique to an individual family; no clear genotype-phenotype correlations have been observed.99
Erythropoietic protoporphyria (EPP) is caused by partial deficiency of the enzyme ferrochelatase (FECH), the final step in the heme synthetic pathway. EPP is the second most common type of porphyria and is inherited in an autosomal dominant manner with variable penetrance. Patients with EPP and asymptomatic carriers exhibit an FECH enzyme activity of 30% to 40% and 50%, respectively, even though both groups inherit a defective FECH allele. The mechanism for the variable clinical expression is explained by co-inheritance of a “low-expressing” normal FECH allele in symptomatic patients with EPP and a normal FECH allele in asymptomatic patients.106,107
Clinical liver disease, which develops in 5% to 10% of patients with EPP, results from progressive hepatic accumulation of protoporphyrin.108 Liver disease typically occurs after age 30 but has been described in children. The liver appears black and nodular, with hepatocellular necrosis, portal inflammation, cholestasis, and extensive deposits of dark brown pigment in hepatocytes, Kupffer cells, and biliary structures; birefringence of pigment deposits is seen on polarization microscopy.109 Of 57 patients with EPP followed for more than 20 years in one study,110 50% had normal serum aminotransferase levels and liver histologic findings. Of the remaining patients, cirrhosis occurred in seven and liver failure developed in two. Liver disease is not believed to be secondary to alcohol consumption, viral infections, or external toxins, although these insults can worsen liver function.111 Genetic heterogeneity in the FECH gene has been noted in multiple studies and is seen in patients who need liver transplantation.112
Diagnosis
The approach to the diagnosis of the porphyrias is summarized in Table 76-3. Clinical features alone are usually not specific enough to confirm a diagnosis or distinguish among the various forms of porphyrias, and biochemical test results must be interpreted correctly for accurate diagnosis and management. The diagnosis of porphyria should be considered in patients with recurrent bouts of severe abdominal pain, dark urine, constipation, and neuropsychiatric disturbances or in patients with typical dermatologic findings. To differentiate among the different porphyrias, urine and stool samples should be obtained for porphyrin studies and a urine specimen collected for quantitative ALA and PBG determinations.
In AIP, excretion of PGB and ALA (PGB more than ALA) in dark urine is common during porphyric attacks, but the levels may be normal during asymptomatic periods and in prepubertal patients.113 Patients with HCP and VP excrete high levels of ALA and PBG in the urine; in contrast to those with AIP, these patients excrete more ALA than PGB. A rapid “spot” urine test to detect urinary PGB is recommended to diagnose the acute porphyrias, except for the rare patient with ALA dehydratase deficiency.92 Fecal coproporphyrins are increased in both HCP and VP, whereas only in VP is the amount of fecal protoporphyrin also increased.
Hepatic Involvement
Hepatic involvement in porphyria is variable; in general, patients with acute porphyria may have elevations of serum aminotransferase and bile acid levels, with further increases during acute episodes. Liver biopsy specimens may show steatosis and iron deposition. Although these changes are minor, patients with acute porphyria are at increased risk for development of hepatocellular carcinoma.114
PCT and HEP are more commonly associated with hepatic complications, including liver enlargement with fatty infiltration, inflammation, and granulomatous changes. Siderosis and fibrosis may lead to cirrhosis and liver failure. The risk of hepatocellular carcinoma is increased only slightly in patients with these disorders.105 The patterns of liver injury in CEP are similar to those in PCT and HEP.115
Treatment
The overall survival of patients with acute porphyria is good. Treatment is based on avoidance of drugs and other precipitating factors.93 Generous fluid and glucose administration is recommended during acute attacks and can elicit the “glucose effect” that diminishes ALA synthase activity. Intravenous administration of hematin, a congener of heme, has been shown to decrease the drive for heme synthesis and its excessive by-products. Hematin can also have a dramatic effect on neurologic symptoms, especially if given early in an attack, with clinical improvement often occurring within one to two days.92 Women in whom symptoms are affected by the phases of their menstrual cycle can show improvement while taking oral contraceptives. Liver transplantation has been attempted for several of the porphyrias, with mixed results.92,116,117 Because of the increased frequency of hepatocellular carcinoma, patients with AIP should undergo standard surveillance for this tumor (see Chapter 94).92
Because of the wavelengths of light absorbed by the porphyrins, patients affected by porphyria are at risk from exposure to sunlight as well as to household and fluorescent lights. Patients must use special sunscreen lotions that block rays in the 400- to 410-nm range. Skin trauma should be minimized as much as possible; early treatment of skin infections can decrease scarring. Special screens may be especially useful for protection against indoor lighting. Some patients have incurred severe or lethal internal burns during surgery, including liver transplantation; thus, appropriate precautions must be taken.98
Treatment of PCT initially consists of removal of any offending agent. Historically, treatment has included phlebotomy to decrease iron overload and hepatic siderosis. This approach may provide relief of cutaneous symptoms in four to six months. Chloroquine complexes with uroporphyrin and facilitates its excretion, but caution must be used during therapy with chloroquine because the drug is potentially hepatotoxic.98 The efficacy of chloroquine has been variable in patients with PCT who are homozygous for mutations in the HFE gene; for these patients, phlebotomy should be first-line therapy.118,119 Treatment strategies for HEP are similar to, but have not been as successful as, those for PCT.
Blood transfusions and administration of hematin, charcoal, and cholestyramine all have led to clinical improvement in patients with EPP, but long-term resolution has not been demonstrated. One report of a 63-year-old man with severe cholestatic liver disease caused by EPP showed complete biochemical and histologic resolution of cholestasis with the use of blood transfusions, hematin, cholestyramine, and ursodeoxycholic acid. Subsequently, the patient underwent bone marrow transplantation, with apparently complete resolution of EPP.120 Liver transplantation has been accomplished in patients with EPP and ESLD, with mixed results; the erythropoietic defect persists.117,121 A retrospective review of all 20 patients with EPP who have undergone liver transplantation in the United States revealed unique perioperative complications, including light-induced tissue damage in four patients and neuropathy in six, as well as recurrent EPP-associated liver disease in 65% of patients who survived more than two months. Overall patient and graft survival rates were statistically similar to those for all other patients transplanted in the United States during the same period.117 Therefore, liver transplantation must be considered symptomatic therapy, except in patients with fulminant hepatic failure, given the high risk of recurrent disease in the graft and the added risk of intraoperative photodynamic injury to internal organs.117,122 Bone marrow transplantation after liver transplantation has been performed successfully in a child with EPP-induced ESLD.123
Splenectomy, which lengthens the lifespan of circulating red blood cells and decreases the erythropoietic drive, has been shown to be effective in many patients with CEP. Frequent blood transfusions or hematin infusions inhibit the stimulus for heme production, thereby diminishing or eliminating the cutaneous manifestations of the disease.124 Bone marrow transplantation has proved curative in severely affected patients.124,125
TYROSINEMIA
A disease with autosomal recessive transmission, HT1 has a worldwide incidence of about 1 in 100,000. The incidence is much greater in northern Europe (1 per 8000) and in the Saguenay-Lac-St. Jean region of Quebec, Canada (1 per 1846), where a founder effect has been documented.126
Pathophysiology
The pathway for tyrosine metabolism is shown in Figure 76-5. The enzymatic defect in patients with tyrosinemia has been identified in fumarylacetate hydrolase (FAH), the final step in the degradation process. More than 30 mutations in FAH have been found in patients with HT1, but no clear correlation between FAH genotype and HT1 phenotype has been found.127 FAH deficiency leads to accumulation of the upstream metabolites fumarylacetoacetate (FAA) and maleylacetoacetate, which are then converted to the toxic intermediates succinylacetoacetone (SAA) and succinylacetone (SA). FAA has been shown to deplete blood and liver of glutathione, the consequence of which may be augmentation of the mutagenic potential of FAA.127,128 SA inhibits renal glucose and amino acid transport and the degradation of ALA to PBG, probably via direct modification of amino acids in enzyme active sites. It also inhibits DNA ligase activity in fibroblasts isolated from patients with HT1.127,129 Over time, the combined effects of high levels of FAA and SA on the integrity of DNA and cellular repair mechanisms may account for increased chromosomal breakage in fibroblasts isolated from patients with HT1, as well as an increased risk of hepatocellular carcinoma.127,130
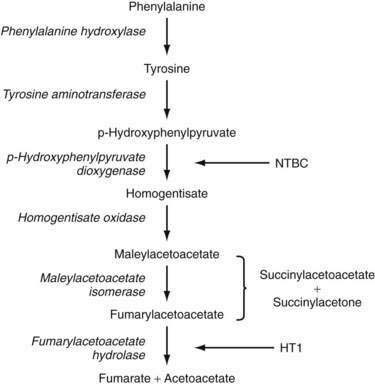
Clinical and Pathologic Features
Patients with HTI present either acutely with liver failure or with chronic liver disease, with or without hepatocellular carcinoma. In the acute form of HTI, patients present with liver disease in the first six months of life; symptoms include those associated with hepatic synthetic dysfunction, such as hypoglycemia, ascites, jaundice, and bleeding diathesis, as well as anorexia, vomiting, and irritability.131 Laboratory studies show elevations of serum aminotransferase, GGTP, and bilirubin levels and decreased levels of coagulation factors. Serum tyrosine, methionine, and α-fetoprotein levels are elevated substantially. Analysis of the urine reveals phosphaturia, glucosuria, hyperaminoaciduria, renal acidosis, and elevated excretion of ALA and phenolic acids.127 The acute form is usually fatal within the first two years of life. In a multicenter study, van Spronsen and associates131 showed that 77% of patients with tyrosinemia presented before the age of six months. The one- and two-year survival rates were 38% and 29%, respectively, if patients presented between zero and two months, and 74% and 74%, respectively, if they presented between two and six months. Survival for both time intervals rose to 96% if the first symptoms appeared after age six months. The cause of death was usually recurrent bleeding and liver failure (35 of 47 deaths); however, hepatocellular carcinoma (7 of 47) and neurologic crisis (3 of 47) accounted for some deaths.
Patients with the chronic form of HTI classically have symptoms that are similar to but milder than those of the acute presentation; sometimes, serum aminotransferase levels as well as plasma tyrosine and methionine levels can be within the normal range.127 These patients usually present after one year of age with hepatomegaly, rickets, nephromegaly, hypertension, and growth retardation. They also are likely to have neurologic problems and hepatocellular carcinoma.
The pathologic changes differ in the acute and chronic forms of the disease. In the acute form, the liver may appear enlarged with a pale nodular pattern or may be shrunken, firm, and brown. Micronodular cirrhosis, fibrotic septa, bile duct proliferation and plugging, steatosis, pseudoacinar and nodular formations, and giant cell transformation may be found on histologic examination. Varying amounts of FAH enzyme activity have been found in liver tissue from patients with HTI as a result of spontaneous reversion of FAH gene mutations. Patients with the chronic form of the disease have a higher level of reversion and a lower frequency of liver dysplasia.132 In an analysis of mutations in the FAH gene in members of 13 unrelated families with HT1, no mutation type predominated in the affected families, and no correlation between genotype and phenotype was observed.133
In the chronic form of tyrosinemia, the liver appears enlarged, coarse, and nodular. In histologic specimens, micronodular and macronodular cirrhosis may be present, as may steatosis, fibrotic septa, and a mild lymphoplasmacytic infiltrate. Cholestasis is less pronounced than in the acute form of HT1. Large- or small-cell dysplasia may be present, reflecting premalignant changes. Because of the nodular changes, identification of progression to hepatocellular carcinoma can be difficult. Because the serum α-fetoprotein value is elevated before hepatocellular carcinoma develops, measurement of α-fetoprotein is not helpful in the diagnosis of hepatocellular carcinoma in this setting. Visualization of both low- and high-attenuation hepatic nodules on computed tomography (CT) is thought to be highly suggestive of hepatocellular carcinoma.134
Renal involvement is nearly universal in patients with tyrosinemia. Findings include a decreased glomerular filtration rate, proximal renal tubular dysfunction, nephromegaly, phosphaturia (which is responsible for the development of rickets), glucosuria, and aminoaciduria. The toxic metabolites SA and SAA are thought to have a direct effect on kidney function. Some patients progress to renal failure and need renal transplantation.135,136 One third of patients develop a cardiomyopathy, most commonly interventricular septal hypertrophy, that is reversible with either medical or surgical management of the disease.137
The neurologic manifestations may be the most concerning feature in older patients with tyrosinemia. More than 40% of patients experience porphyria-like symptoms.138 In a study of 20 children with HTI and 104 neurologic crises, the most common symptoms were pain (96%), hypertonia (76%), vomiting and ileus (69%), weakness (29%), and diarrhea (12%). Eight of the 104 patients required mechanical ventilation.138 A neurologic crisis has been considered to be a frequent cause of death, but the onset of a neurologic crisis may not be associated with worsening liver disease. Blockage of the degradation of ALA by SA is thought to be responsible for the neurotoxicity.
Diagnosis
The diagnosis of tyrosinemia should be suspected in any child with neonatal liver disease or a bleeding diathesis or in any child older than one year with undiagnosed liver disease or rickets. The diagnosis is suggested by increased tyrosine, methionine, phenylalanine, and α-fetoprotein levels. Elevated serum and urine SA and urine ALA levels are regarded as pathognomonic for tyrosinemia. The diagnosis can be confirmed with an assay for FAH in lymphocytes, erythrocytes, skin fibroblasts, or liver tissue.127
Prenatal diagnosis can be performed by determining SA levels in amniotic fluid or measuring FAH activity in chorionic villus biopsy specimens. If the specific gene mutation in a family is known, early genetic diagnosis can be made from chorionic villus biopsy specimens as well.127 Current newborn screening programs measure tyrosine levels on dried blood specimens; unfortunately, neonatal hypertyrosinemia is not specific for a diagnosis of HT1, and the tyrosine levels in newborns with HT1 may overlap substantially with tyrosine levels found in unaffected newborns. Improved newborn screening methodologies have been developed that measure SA in addition to amino acid levels in dried blood specimens; however, these methods are not yet widely employed.139,140
Treatment
Historically, the treatment of tyrosinemia has been dietary management, with restriction of tyrosine and phenylalanine. Dietary restriction has been shown to reverse renal damage and improve metabolic bone disease, although the liver disease progresses. An adequate intake of these amino acids is needed to ensure normal growth and development.127 Few studies of the long-term outcome in tyrosinemia treated with strict dietary management alone are available.
Liver transplantation has become a mainstay of therapy for patients with tyrosinemia. The transplant corrects the phenotype and normalizes FAH activity and liver function. Additionally, the biochemical profiles normalize and kidney disease abates, with improvement in glomerular filtration rate, tubular acidosis, and hypercalcemia in most patients.141,142 Abnormal renal size and architecture persist after liver transplantation,143 and many patients continue to excrete SA despite normal serum values.142 One report has suggested that patients with tyrosinemia who have undergone liver transplantation and receive tacrolimus-based immunosuppression have a high rate of post-transplant neurologic complications.144
In 1992, Lindstedt and associates145 published data on the treatment of tyrosinemia with the herbicide 2-(2-nitro-4-trifluoro-methylbenzoyl)-1,3-cyclohexanedione (NTBC). Later, Holme and Lindstedt146 published the results of a large long-term study of 220 patients with HT1 who were treated with this agent for up to seven years. NTBC is a potent inhibitor of 4-hydroxyphenylpyruvate dioxygenase, one of the initial steps in tyrosine metabolism. Blocking the degradation of tyrosine to its downstream toxic metabolites (i.e., FAA, SA, and SAA) was postulated to lead to improved hepatic function. Treated patients exhibit improved liver synthetic function, as reflected by a shortening of the prothrombin time, as well as decreased serum aminotransferase levels and a reduction in liver parenchymal heterogeneity and nodules on CT. In addition, serum α-fetoprotein and ALA levels are diminished and renal tubular dysfunction reverses.147 Therefore, elevated α-fetoprotein levels in a patient receiving NTBC therapy should raise concern about the patient’s nonadherence to therapy or the development of hepatocellular cancer.127,148 Long-term results show continued improvement in all parameters noted in the earlier report as well as a lower risk for the development of hepatocellular carcinoma in patients who start therapy and were free of hepatocellular carcinoma before the age of two years.146 No patient withdrew from the study because of adverse side effects of the drug. Transient thrombocytopenia and neutropenia as well as ocular symptoms suggestive of corneal irritation have been noted rarely. Cognitive impairment resulting in learning problems may be a long-term complication of chronic use of NTBC in this patient population, possibly from the effects of chronic hypertyrosinemia.149
In another study from Quebec,150 only 4 of 35 patients treated with NTBC underwent liver transplantation; 1 patient received a transplant because of concern about the heterogeneous texture of the liver (suggestive of cirrhosis) shown by ultrasonography coupled with a persistent moderate elevation in the serum α-fetoprotein level. At resection, this child was found to have a small nodule with hepatocellular dysplasia. The 31 remaining patients were monitored while receiving NTBC therapy for up to three years, and none experienced neurologic crises or deterioration of liver disease.150
Therefore, therapy with NTBC significantly improves the clinical course of patients treated at an early age. For those in whom therapy is initiated at a later age, NTBC offers a palliative benefit, although the risk of hepatocellular carcinoma is still high in this group of patients. Because the strategy of treating patients with HT1 with NTBC is relatively new, greater experience is required to enable assessment of the relative costs and long-term outcome and recognition of any possible adverse effects of long-term NTBC therapy.149 Early diagnosis, achieved by inclusion of HT1 in neonatal screening programs, may allow the prompt initiation of effective therapy with NTBC and avoidance of liver transplantation.
UREA CYCLE DEFECTS
Although the syndromes related to the urea cycle defects (UCDs) are not associated with serious liver injury, the basic genetic defect is located within the liver, and the manifestations can mimic those of other metabolic liver diseases. The urea cycle consists of five enzymes that, through several steps, process ammonia derived from amino acid metabolism to urea. Genetic defects in each of these enzymes have been reported, and their overall incidence has been estimated to be 1 in 20,000 to 1 in 30,000.151
Pathophysiology
The steps of the urea cycle are illustrated in Figure 76-6. Carbamyl phosphate synthetase (CPS) I forms carbamyl phosphate from ammonium and bicarbonate. This step requires N-acetyl glutamate as a cofactor, which is synthesized from N-acetyl CoA and glutamic acid by N-acetyl glutamate synthetase. Ornithine transcarbamylase (OTC) combines carbamyl phosphate with ornithine to form citrulline. A second nitrogen enters the cycle as aspartate, which combines with citrulline by the action of argininosuccinate synthetase (AS) to form argininosuccinate. Argininosuccinate is converted to arginine and fumarate by argininosuccinase, or argininosuccinate lyase (AL). Arginase then catalyzes the breakdown of arginine to urea and ornithine in the final step of the pathway. Several amino acid transporters, such as citrin, an aspartate/glutamate carrier protein that supplies aspartate to the urea cycle, are involved in shuttling metabolites into the urea cycle.152
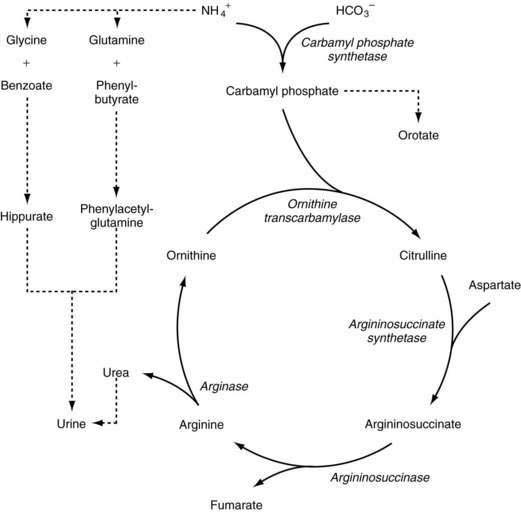
Enzymatic defects have been identified in all five steps involved in the urea cycle. Deficiency of four of these enzymes is transmitted through autosomal recessive inheritance, whereas OTC deficiency is transmitted as an X-linked trait. More than 340 different mutations in the OTC gene give rise to OTC deficiency, the most common urea cycle defect.153 Numerous defects in the other enzymes or amino acid transporters of the cycle (e.g., N-acetylglutamate synthetase or citrin) have been characterized as well.154–156 Moreover, numerous mRNA instability mutations have been found in patients with CPS I deficiency.157
Clinical Features
The spectra of clinical presentations in patients with any of the UCDs are virtually identical; in the neonatal period, these disorders usually manifest as acute life-threatening events. Contrary to the commonly held belief that UCDs are primarily disorders of the newborn period, a large report of the clinical presentation and outcome data of 260 patients with UCDs revealed that two thirds of patients present at greater than 30 days of age, even when female OTC carriers are excluded from the analysis.158 Another large cross-sectional study of 183 patients with UCDs had similar findings.159 Late-onset adult presentations also have been described. Affected infants appear normal for the first 24 to 72 hours until they are exposed to their first feeding, which provides the initial protein load that fosters ammonia production. Symptoms include irritability, poor feeding, vomiting, lethargy, hypotonia, seizures, coma, and hyperventilation, all secondary to hyperammonemia.158 Initially, neonates are often mistakenly thought to have sepsis, despite the absence of perinatal risk factors, and diagnostic laboratory testing is delayed.160 Plasma ammonia levels should be obtained whenever an evaluation for sepsis is initiated in a neonate; levels may exceed 2000 µmol/L (3400 mg/dL), normal levels being 50 µmol/L (85 mg/dL) or less.
For all age groups, overall survival decreases as the peak plasma ammonia level rises for a given episode of hyperammonemia with survival rates of 98% and 47% for peak ammonia levels of less than 200 µmol/L and greater than 1000 µmol/L, respectively.161 Newborns have a survival rate of 73% after their presenting episode of hyperammonemia, whereas patients older than 30 days of age have a survival rate of 98%. Male patients with OTC deficiency have a survival rate of 91% following an episode of hyperammonemia, a rate significantly less than those (93% to 98%) for all other forms of UCDs. Blood gas analysis shows respiratory alkalosis secondary to the hyperventilation caused by the effects of ammonia on the central nervous system. Blood urea nitrogen levels are typically low but can be elevated during times of dehydration or hypoperfusion. Serum levels of liver enzymes are usually normal or minimally elevated. Severe hepatomegaly can occur in early-onset forms of AL deficiency.151
As stated earlier, one third of patients with UCDs present in the neonatal period; the remainder are diagnosed at variable times from infancy to adulthood.158,159 OTC deficiency is the most common UCD (55%), followed by argininosuccinic aciduria (AL deficiency, 16%) and citrullinemia (AS deficiency, 14%).159 Male patients with OTC deficiency have been diagnosed as late as 40 years of age with varied phenotypic presentations.162 As many as 10% of female carriers of OTC deficiency can have symptoms, which may be severe and fatal, although most female carriers have no symptoms or report only nausea after high-protein meals.163 Late-onset CPS deficiency has also been described,164 and the adult form of AS deficiency is relatively common in Japan.155
Symptoms and signs of late-onset UCDs, especially OTC and CPS deficiencies, include episodic irritability, lethargy, or vomiting; self-induced avoidance of protein such as milk, eggs, and meats; and short stature or growth delays. Neurologic symptoms, which can also be episodic, include ataxia, developmental delays, behavioral abnormalities, combativeness, biting, confusion, hallucinations, headaches, dizziness, visual impairment, diplopia, anorexia, and seizures.158 Acute hyperammonemic episodes can resemble Reye’s syndrome (see Chapter 86). Such episodes can be precipitated by high-protein meals, viral or bacterial infections, medications, trauma, or surgery. Infants may present after being weaned from breast milk to infant formulas, which have a higher protein content. Patients with OTC and CPS deficiencies have been reported to present in the postpartum period with acute decompensation and death.165,166
Citrin deficiency is caused by mutations in the SLC25A13 gene and is associated with both adult-onset type 2 citrullinemia and neonatal intrahepatic cholestasis resulting from citrin deficiency (NICCD), a syndrome that primarily affects newborns in East Asia.155,167,168 A case of citrin deficiency in a white infant of European descent who presented with poor weight gain and a bleeding diathesis, but without cholestasis, has been reported, expanding the clinical spectrum of presentations of citrin deficiency.169 NICCD is associated with hyperaminoacidemia (e.g., elevated citrulline, methionine, and tyrosine levels) and cholestasis.155 Hypergalactosemia and elevated acylcarnitine levels also may be observed.170 Hepatic steatosis may be seen in the liver histologically.171 In most patients with NICCD, all biochemical abnormalities resolve spontaneously or with minimal dietary restrictions (e.g., the use of lactose-free formulas); however, several affected infants have required liver transplantation before age one year.167 Therefore, jaundiced infants with multiple abnormal newborn metabolic screen results (e.g., elevation of blood phenylalanine, methionine, or galactose levels) must be observed closely because of the risk for development of ESLD caused by NICCD; a chubby face outside of the realm of normal may be a diagnostic clue.172 The diagnosis can be made by sequence analysis of the SLC25A13 gene or by failure to detect citrin protein in peripheral lymphocytes.168,173
Diagnosis
A high index of suspicion is required for prompt diagnosis of UCDs. Symptoms can mimic those of other acute neonatal problems, such as infections, seizures, and pulmonary or cardiac disease.158,174 Later presentations can mimic other behavioral, psychiatric, or developmental disorders. The first clue may be an elevated serum ammonia level with normal serum aminotransferase levels and without metabolic acidosis. Therefore, if a UCD is considered, the following laboratory measurements should be obtained: serum ammonia, arterial blood gases, urine organic acids, serum amino acids, and urinary orotic acid; Table 76-4 reviews the expected laboratory results.
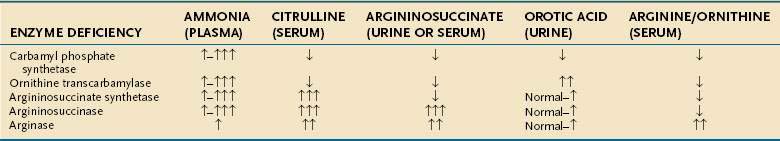
Urinary organic acid profiles are typically normal in UCDs. The plasma amino acid profiles are distinctive, with abnormal levels of arginine, ornithine, and citrulline. Citrulline levels are barely detectable in OTC or CPS deficiencies but markedly raised in AS and AL deficiencies. AS deficiency can be distinguished from AL deficiency by the finding of argininosuccinic acid in the plasma and urine of patients with AL deficiency. OTC deficiency is differentiated from CPS deficiency by the excessive urinary excretion of orotic acid in OTC deficiency. Direct enzyme analysis can be performed and can be useful in patients who have a partial deficiency or who present in adulthood. Prenatal enzyme and genetic linkage analysis can be carried out in family members of known carriers.175 Early neonatal diagnosis leads to improved survival. An N-carbamoyl-glutamic acid test has been advocated for all patients presenting with a suspected UCD while further testing is carried out; patients with N-acetylglutamate synthase deficiency, one of the less common UCDs, have a significant drop in plasma ammonia levels within eight hours of receiving an oral dose of N-carbamoyl glutamic acid, a structural analog of N-acetyl glutamic acid.176,177 An allopurinol loading test, which leads to excretion of orotic acid in amounts that are 10- to 20-fold greater than normal in heterozygote female carriers of OTC deficiency, is nonspecific, and its results must be interpreted with caution; the result can be positive in some patients with mitochondrial disease or defects in pyrimidine metabolism.178 Liver biopsy specimens typically show minimal fatty infiltration; fibrosis or cirrhosis are uncommon findings but have been reported.151,179
Treatment
All external protein intake should be discontinued in infants with UCDs presenting acutely. Serum ammonia levels should be restored to normal. The use of oral lactulose to lower the nitrogen load has not been studied in this patient population. Given the extremely high ammonia levels often encountered, continuous arteriovenous hemodialysis or hemofiltration is often required, but exchange transfusions and peritoneal dialysis are ineffective. Alternative pathways for waste nitrogen disposal should be employed, specifically intravenous administration of sodium benzoate and sodium phenylacetate; however, sodium benzoate should be used with caution in patients with cirrhosis because a paradoxical rise in blood ammonia levels has been observed.180 Arginine, carnitine, and long-chain fatty acids are usually present in low levels in these patients and should be supplemented.181,182 Once the patient stabilizes, low levels of dietary protein, 0.5 to 1.0 gm/kg, may be introduced, with progressive increases as tolerated to provide sufficient protein for growth and tissue repair while minimizing urea production. Long-term therapy and protein restriction are then tailored to the patient; patients with a severe disorder may need essential amino acids to supplement their protein intake.182 Oral phenylbutyrate can be substituted for phenylacetate to improve palatability.
The outcome for patients who present with hyperammonemic coma and a delayed diagnosis is poor.161 The level of ammonia at the time of the first hyperammonemic episode is a rough guide to the eventual neurodevelopmental outcome.183 The sooner the hyperammonemia is treated and the correct diagnosis is made, the better the long-term survival, although for patients who survive the neonatal period, the median survival without liver transplantation is less than four years and is associated with severe developmental delay and neurologic morbidity.160 With optimal dietary and medical management, patients may still have repeated hyperammonemic crises, often during intercurrent viral infections. Symptomatic female OTC heterozygotes also benefit from therapy, which leads to fewer hyperammonemic episodes and a reduced risk of further cognitive decline.184
Patients with a UCD and a deterioration or lack of improvement despite therapy have undergone either total or auxiliary liver transplantation successfully, with normalization of enzyme activity and ammonia levels, the ability to tolerate a normal diet, and five-year survival rates of 90%.185,186 Liver transplantation, if considered, should be done before neurologic damage is permanent because the patient’s neurologic status does not improve after transplantation. The metabolic condition of the patients normalizes completely, and neurologic status does not worsen after transplantation.164 In addition, hepatocyte transplantation has been successful in improving metabolic function in patients with UCDs and has been applied successfully as a bridge to auxiliary liver transplantation.186,187 For patients without severe neurologic compromise before liver transplantation, this therapeutic approach is worthwhile. In addition, the annual cost of care for this group of patients is likely to be reduced dramatically after liver transplantation.
ARGINASE DEFICIENCY
At least two forms of arginase activity occur in humans: Arginase I (AI) predominates in the liver and red blood cells, and arginase II (AII) is found predominantly in kidney and prostate. Arginase deficiency involving AI is the least common of the UCDs. Hyperammonemia is unusual in affected persons, but hyperammonemic coma and death have been reported.188 The clinical disorder is distinct from the other UCDs. It is characterized by indolent deterioration of the cerebral cortex and pyramidal tracts, leading to progressive dementia and psychomotor retardation, spastic diplegia progressing to quadriplegia, seizures, and growth failure. The syndrome is often confused with cerebral palsy.189
Laboratory studies may reveal elevated blood arginine values, mild hyperammonemia, and a mild increase in urine orotic acid excretion. Varying amounts of urea are still produced in these patients secondary to the compensatory elevated expression of AII in the kidneys that ameliorates the clinical disorder. The diagnosis is confirmed by enzymatic analysis, which can be performed prenatally on cord blood samples. Treatment consists of protein restriction and, when needed, medical therapy with sodium phenylbutyrate.189
BILE ACID SYNTHESIS AND TRANSPORT DEFECTS
The pathways for bile acid synthesis and transport within the hepatobiliary system are complex, involving several enzymes and transport processes located in multiple subcellular fractions of the hepatocyte (see Chapter 64). With technologic advances in molecular biology and mass spectrometry, several different inborn errors in bile acid synthesis and transport have been identified as causes of clinical disease. The definition and classification of these disorders have improved, particularly in the clinically heterogeneous subset of cases that comprise progressive familial intrahepatic cholestasis (PFIC) syndromes. For some of these disorders, this progress has led to dramatic advances in often life-saving therapy.
The diagnosis of PFIC is imprecise; the accepted criteria are (1) the presence of chronic, unremitting intrahepatic cholestasis, (2) exclusion of identifiable metabolic or anatomic disorders, and (3) characteristic clinical, biochemical, and histologic features.190 Other symptoms and signs are severe pruritus, hepatomegaly, wheezing and cough, short stature, delayed sexual development, fat-soluble vitamin deficiency, and cholelithiasis. Affected persons exhibit severe and progressive intrahepatic cholestasis, usually manifesting within the first few months of life and often proceeding to cirrhosis and ESLD by the second decade of life. Patients with PFIC syndromes have been found to have defects in bile acid synthetic and transport processes.
Some patients believed previously to have idiopathic neonatal hepatitis or an undiagnosed familial hepatitis syndrome may now be diagnosed accurately as having a form of PFIC. An estimated 1% to 2.5% of patients with idiopathic cholestasis may have defects in bile acid metabolism and transport.191 Table 76-5 lists the known errors of primary and secondary bile acid synthesis and transport.
Table 76-5 Inborn Errors of Bile Acid Synthesis and Transport*
| Defective Bile Acid Synthesis | |
| Primary defects | Cerebrotendinous xanthomatosis (C27-steroid-27-hydroxylase deficiency) |
| 3β-hydroxy-ΔC27-steroid dehydrogenase/isomerase (HSD3B7) deficiency | |
| Δ4-3-oxosteroid 5β-reductase (AKR1D1) deficiency | |
| C24-steroid-7α-hydroxylase (CYP7B1) deficiency | |
| α-Methylacyl-CoA racemase (AMACR) deficiency | |
| Secondary defects (due to organelle damage) | Peroxisomal biogenesis disorders (PBDs) |
| Rhizomelic chondrodysplasia punctata | |
| Zellweger spectrum | |
| Zellweger’s syndrome | |
| Neonatal adrenoleukodystrophy | |
| Infantile Refsum’s disease | |
| Other | |
| Hyperpipecolic acidemia | |
| Leber’s congenital amaurosis | |
| Disorders with loss of single peroxisomal function | |
| Acatalasemia | |
| Acyl-CoA oxidase deficiency | |
| Adult Refsum’s disease | |
| D-bifunctional protein deficiency | |
| Hyperoxaluria type I | |
| Sterol carrier protein X deficiency | |
| Thiolase deficiency (pseudo-Zellweger’s syndrome) | |
| X-linked adrenoleukodystrophy | |
| Contiguous gene syndrome | |
| Generalized hepatic synthetic dysfunction | |
| Fulminant hepatic failure (multiple causes) | |
| Neonatal iron storage disease | |
| Tyrosinemia | |
| Defective Bile Acid or Phospholipid Transport | |
| FIC1 (FIC1) deficiency: progressive familial intrahepatic cholestasis (PFIC) type 1: | |
| Byler’s disease | |
| Benign recurrent intrahepatic cholestasis (BRIC) syndrome | |
| Greenland familial cholestasis | |
| BSEP (ABCB11) deficiency: PFIC type 2 | |
| MDR3 (ABCB4) deficiency: PFIC type 3 | |
| Others | |
| Alagille (Jagged) syndrome (see Chapter 62) | |
| Cholestasis-lymphedema syndrome (Aagenaes syndrome) | |
| North American Indian childhood cirrhosis (Cirhin) | |
* Corresponding genes are shown in italics.
Modified from Balistreri WF. Inborn errors of bile acid biosynthesis: Clinical and therapeutic aspects. In: Hofmann AF, Paumgartner G, Stiehl A, et al, editors. Bile Acids in Gastroenterology, Basic and Clinical Advances. London: Kluwer Academic Publishers; 1995. p 333; Jonas MM, Perez-Atayde AR, editors. Liver disease in infancy and childhood. In: Schiff ER, Sorrell MF, Maddrey WC, editors. Schiff’s Diseases of the Liver, 9th ed. Philadelphia: Lippincott Williams & Wilkins; 2003. p 1459.
BILE ACID SYNTHESIS DEFECTS
Bile acid synthetic pathways are discussed in detail in Chapter 64. Only the most common enzyme deficiencies are described here; all can be diagnosed through mass spectrometry of the urine or serum. General principles of therapy rely on the hypothesis that inborn errors of bile acid biosynthesis lead to underproduction of normal trophic and choleretic primary bile acids and overproduction of hepatotoxic primitive bile acid metabolites.192 Bile acids found in patients with inborn errors also act as cholestatic agents by inhibiting canalicular ATP-dependent bile acid transport (the rate-limiting step in the overall process of bile acid transport across the canalicular membrane).193
Cerebrotendinous xanthomatosis (CTX), C27-steroid-27-hydroxylase deficiency, is characterized by bilateral juvenile cataracts and chronic diarrhea, followed by progressive neurologic dysfunction, hypercholesterolemia, atherosclerosis, and deposition of cholesterol and cholestanol in tissues.192,194,195 CTX should be treated with chenodeoxycholic acid. In one series,196 five children with CTX showed dramatic improvement in biochemical and electroencephalographic abnormalities and resolution of diarrhea with this therapy. In addition, no further delay in motor development was noted, and three patients showed an improved intelligence quotient.196
Patients with 3β-hydroxy-C27-steroid dehydrogenase/isomerase (3β-HSD) deficiency may present with pruritus, jaundice, hepatomegaly, steatorrhea, and fat-soluble vitamin deficiencies. Deficiency of Δ4-3-oxosteroid 5β-reductase also leads to neonatal cholestasis but rapidly progresses to synthetic dysfunction and liver failure. Both conditions have been treated with a primary bile acid (i.e., cholic acid) and ursodeoxycholic acid supplementation.191,197 Cholic acid bypasses the enzymatic block and provides negative feedback to earlier steps in the synthetic pathways, and ursodeoxycholic acid displaces toxic bile acid metabolites and serves as a hepatobiliary cytoprotectant. Other known disorders result from C24-steroid-7α-hydroxylase, α-methylacyl-CoA racemase, and amidation deficiencies.191,192
Many peroxisomopathies have been described; these disorders are associated with multiple clinical abnormalities and up to 50 wide-ranging biochemical abnormalities.198 They are diagnosed through a combination of specialized tests, such as that for very-long-chain fatty acids (VLCFAs) and ultrastructural analysis of tissue biopsy specimens.199 Peroxisomes are responsible for beta-oxidation in the final steps of bile acid synthesis to yield the primary bile acids cholic acid and chenodeoxycholic acid. The disorders can be divided into three groups: peroxisome biogenesis disorders (PBDs), which cause multiple abnormalities; disorders of single proteins, which result in limited dysfunction; and a contiguous gene syndrome (see Table 76-5).198 PBDs, which are grouped because they share similar clinical and biochemical features, include rhizomelic chondrodysplasia punctata (RCDP), which is characterized by severe rhizomelic shortening of the limbs, severe skeletal abnormalities, cataracts, and facial abnormalities, and the three Zellweger spectrum disorders, which include Zellweger’s syndrome (ZS), with the most severe clinical abnormalities, neonatal adrenoleukodystrophy, and infantile Refsum’s disease, with the mildest features.198
ZS is a primary disorder of peroxisome biogenesis. The multiple features of ZS include distinctive dysmorphic features (hypertelorism, large anterior fontanelle, deformed earlobes), neonatal hypotonia, impaired hearing, retinopathy, cataracts, seizures, and skeletal changes. Hepatomegaly is common, and the progressive liver disease that develops in patients with ZS is similar to that identified in other errors of bile acid synthesis.200
Peroxisome biogenesis involves more than 13 PEX genes and requires the targeting and importation of cytosolic proteins into the peroxisomal membrane and matrix. Importation of proteins fated for the peroxisomal matrix requires guidance from one of two peroxisome-targeting signals, PTS1 and PTS2. Patients with Zellweger spectrum disorders display defects in the importation of proteins that use PTS1 and PTS2, whereas patients with RCDP have a defect in the importation of proteins using PTS2.198
The most common disorder of peroxisomes, X-linked adrenoleukodystrophy (X-ALD), is included in the second grouping of peroxisomopathies and results from a defect in the peroxisomal adrenoleukodystrophy protein (ALDP), which is a member of the ATP-binding cassette (ABC) superfamily of membrane transporters. Several other single peroxisomal enzyme deficiencies exist, as noted in Table 76-5. The last group, contiguous gene syndrome, involves a large deletion spanning chromosome Xq28 and affecting multiple genes involved in peroxisome function.198,201
Historically, the treatment of ZS has been supportive, with most patients not surviving the first year of life. The goals of medical therapy have been to improve nutrition and growth, control central nervous system symptoms, and limit progression of liver disease. Treatment with primary bile acids has been shown to improve biochemical studies and histology, increase growth, and improve neurologic symptoms.202 Adrenal hormone replacement therapy is necessary for patients with X-ALD; the only known effective treatment for the cerebral form of this condition is hematopoietic stem cell transplantation, which if performed early, leads to good neurologic outcomes.203,204 In an uncontrolled study, the use of docosahexaenoic acid (DHA) led to improved myelination in patients with peroxisomal disorders; currently, however, the use of DHA should be limited to controlled clinical trials.205
BILE ACID TRANSPORT DEFECTS
FIC1 disease is caused by mutations in the ATP8B1 gene (initially named the FIC1 gene) that encodes the FIC1 protein, a P-type adenosine triphosphatase involved in ATP-dependent aminophospholipid transport. FIC1 protein is expressed in many organs in addition to the liver, including the intestine and pancreas. Severe mutations in FIC1 lead to progressive early-onset FIC1 disease (PFIC type 1), whereas mutations in FIC1 that are predicted to affect protein structure minimally are more commonly seen in patients with BRIC type 1.206,207 Patients with Greenland familial cholestasis also have distinct defects in the FIC1 gene, as do other kindreds of Dominican and Saudi descent.207,208
In PFIC type 1, serum GGTP and cholesterol levels are normal or mildly elevated, and levels of bile acids are elevated in the serum and low in the bile. Serum aminotransferase and bilirubin levels are mildly elevated as well. Impaired bile acid transport in the intestine may account for the striking malabsorption and diarrhea manifested by some patients with PFIC type 1; these intestinal clinical features do not resolve after liver transplantation. Histology of liver tissue from patients with PFIC type 1 disease typically shows a bland canalicular cholestasis, with varying degrees of hepatocellular ballooning and giant cell transformation; portal fibrosis and eventually cirrhosis may be seen later in the course of the disease. The liver histology of patients with BRIC is normal between episodes. On electron microscopic evaluation of liver tissue from patients with PFIC type 1, characteristic coarse, granular bile deposits are seen in the canaliculus (“Byler’s bile”).208
BSEP disease is caused by a wide spectrum of mutations in the ABCB11 gene, which encodes an ABC protein that serves as the canalicular BSEP.206 Patients with the progressive form of BSEP disease (PFIC type 2) present with high serum bile acid levels but low or low-normal serum GGTP levels, and they usually have intense pruritus, jaundice, poor weight gain, and hepatosplenomegaly.208,209 In addition, a genetically distinct form of BRIC (type 2) is associated with mutations in ABCB11; interestingly, patients with BRIC type 2 commonly have cholelithiasis and the absence of extrahepatic manifestations.206,210 Early in the course of PFIC type 2, a nonspecific giant cell hepatitis is found on routine histology of the liver, and on electron microscopy, amorphous bile deposits are seen in the canaliculi. For unclear reasons, patients with PFIC type 2 appear to have an increased risk of developing malignancies of the hepatobiliary system, such as hepatoblastoma, hepatocellular carcinoma, and cholangiocarcinoma, in contrast to patients with other forms of progressive intrahepatic cholestasis.208,211–213
MDR3 disease is caused by mutations in the ABCB4 gene that encodes the MDR3 glycoprotein, an ABC phosphatidylcholine translocase located on the canalicular membrane.206,214 Patients with MDR3 disease present with several disease phenotypes as well, including PFIC type 3, intrahepatic and gallbladder lithiasis, intrahepatic cholestasis of pregnancy, and adult-onset ductopenic cholestatic liver disease.215–217 Moreover, mutations in MDR3 may serve as disease modifiers for primary sclerosing cholangitis, primary biliary cirrhosis, and drug-induced cholestasis.218 MDR3 deficiency is thought to lead to decreased excretion of cytoprotective biliary phospholipids, leaving an increased pool of cytotoxic biliary bile salts that gives rise to subsequent bile duct damage and proliferation and release of GGTP into the serum. Patients with PFIC type 3 present with high serum levels of GGTP and bile acids as well as bile ductular proliferation on routine microscopy. Some female patients with familial intrahepatic cholestasis of pregnancy have been shown to be heterozygous carriers of a mutation in MDR3; the mutations likely lead to a genetic predisposition that requires the coexistence of other nongenetic factors for full expression of the disease.215,219
Other chronic intrahepatic cholestatic diseases such as North American Indian childhood cirrhosis (NAICC), cholestasis-lymphedema syndrome (Aagenaes syndrome),220 neonatal icthyosis and sclerosing cholangitis syndrome, and arthrogryposis-renal dysfunction—cholestasis (ARC) syndrome—have disease causing loci that are genetically distinct from those that cause FIC1, BSEP, or MDR3 diseases.221 Further molecular genetic characterization of these disorders has identified a single-point mutation in the cirhin gene, which encodes a nucleolar protein of unknown function, in patients with NAICC.222 Moreover, mutations in the claudin-1 and VPS33B (vacuolar protein sorting 33B) genes have been identified in patients with neonatal icthyosis and sclerosing cholangitis and in ARC syndromes, respectively; both of these genes encode proteins that are important in membrane fusion events.223,224
Medical treatment of patients with disorders of bile acid transport as a group, with phenobarbital, cholestyramine, opioid antagonists, rifampin, and phototherapy, has been largely ineffective or highly variably effective and of short duration of benefit at best. Therapy with ursodeoxycholic acid may be effective in reducing pruritus and improving liver biochemical test levels in up to 50% of patients with PFIC, regardless of serum GGTP levels.186 Surgical approaches such as ileal exclusion and partial external biliary diversion have provided satisfactory symptomatic relief to some patients by decreasing the bile acid pool and pruritus, especially in patients with PFIC type 1 and 2.225–227 Long-term marked improvement in pruritus and growth has been reported in six pediatric patients treated with biliary diversion.225 If all else fails, liver transplantation leads to good overall outcomes, with normalization of bile acid synthesis and growth, even in patients who receive a living donor organ from a potentially heterozygous parent.228,229
CYSTIC FIBROSIS
Cystic fibrosis (CF) is discussed in detail in Chapter 57; here, a brief discussion of the hepatic complications seen in this multisystemic disorder is presented. Liver disease can be the presenting symptom of CF in the newborn, and CF-associated liver disease has been seen with meconium ileus syndrome. Other risk factors for CF-associated liver disease include male sex, pancreatic insufficiency, and a CF transmembrane regulator genotype associated with severe disease.230–232 Although CF has been identified in fewer than 2% of patients with neonatal cholestasis, the diagnosis should be considered in any infant who presents with neonatal cholestasis.
CF-associated liver disease may become more common as the mean age of survival for patients with CF rises; however, liver involvement is not universal and seems to peak during the adolescent years. Up to 30% of patients may have clinical or symptomatic liver disease after the neonatal period.230
Hepatobiliary diseases noted in patients with CF can be grouped into three categories (Table 76-6). The pathognomonic lesion of CF, focal biliary cirrhosis (FBC), presumably results from defective function of the CF transmembrane regulator protein, which is expressed in bile duct cells. Obstruction of small bile ducts leads to chronic inflammatory changes, bile duct proliferation, and portal fibrosis. At autopsy, FBC has been identified in 25% to 30% of patients older than one year of age.233 Progression to multilobular biliary cirrhosis occurs in approximately 5% to 10% of patients with CF and leads to symptoms associated with portal hypertension, such as splenomegaly and variceal bleeding.230,231 Hepatic steatosis also develops in roughly one half of patients but does not appear to correlate with outcome. Biliary abnormalities range from microgallbladder, which is largely asymptomatic and is found in up to 20% of patients, to cholelithiasis and cholangiocarcinoma.231,234 Interestingly, the presence of liver disease does not correlate with the severity of pulmonary disease.230,232
Table 76-6 Spectrum of Hepatobiliary Disease in Patients with Cystic Fibrosis
| Lesions specific to cystic fibrosis | Hepatic |
| Focal biliary cirrhosis with inspissation | |
| Multilobular biliary cirrhosis with inspissation | |
| Biliary | |
| Microgallbladder | |
| Mucocele | |
| Mucous hyperplasia of the gallbladder | |
| Lesions secondary to extrahepatic disease | Hepatic lesions associated with cardiopulmonary disease |
| Centrilobular necrosis | |
| Cirrhosis | |
| Pancreatic lesions | |
| Fibrosis (leading to bile duct compression/stricture) | |
| Lesions that occur with a higher frequency in patients with cystic fibrosis | Hepatic |
| Drug hepatotoxicity | |
| Fatty liver | |
| Neonatal cholestasis | |
| Viral hepatitis | |
| Biliary | |
| Biliary sludge | |
| Cholangiocarcinoma | |
| Cholelithiasis | |
| Sclerosing cholangitis |
Modified from Balistreri WF. Liver disease in infancy and childhood. In: Schiff ER, Sorrell MF, Maddrey WC, editors. Schiff’s Diseases of the Liver, 9th ed. Philadelphia: Lippincott-Raven; 1999. p 1379.
The variable occurrence and clinical course of liver disease in patients with CF may be related to other genetic factors. For example, elevated concentrations of endogenous ursodeoxycholic acid have been documented in patients with CF without liver disease, thus raising the possibility that ursodeoxycholic acid may protect against liver injury in these patients.235
The diagnosis of liver disease in patients with CF can be difficult because the presenting signs are subtle. Hepatomegaly, which is present in approximately 30% of patients, has been shown to correlate well with the presence of cirrhosis and is often the first finding of liver disease. Liver biochemical test levels may remain relatively normal despite histologic evidence of cirrhosis. Needle biopsy of the liver can be helpful; however, because of the focal distribution of histologic abnormalities, sampling error may occur. Ultrasonography can detect the presence of biliary tree abnormalities as well as heterogeneous or nodular liver parenchyma; however, a normal study does not preclude significant hepatic fibrosis.236,237
Treatment with ursodeoxycholic acid improves liver biochemical test levels in patients with CF; however, conclusive evidence that the drug halts the progression to cirrhosis is not yet available.238 Because patients with CF rarely have true hepatocellular dysfunction, the role of liver transplantation in this patient population remains controversial, particularly given the scarcity of donor livers. Nevertheless, liver transplantation has been performed successfully in patients with CF who have portal hypertension and stable pulmonary function, with post-transplant outcomes and improvement in nutritional status comparable to those for patients without CF.239,240 A portosystemic shunt can be an effective treatment for a patient with CF and portal hypertension, with long-term outcomes comparable to that for patients with CF who undergo liver transplantation.241
MITOCHONDRIAL LIVER DISEASES
A growing number of liver diseases have been attributed to defects in mitochondrial function. In addition to defects in mitochondrial enzymes involved in the urea cycle or energy metabolism, several mitochondrial hepatopathies involve respiratory chain/oxidative phosphorylation/electron transport defects or alterations in mitochondrial DNA (mtDNA) levels. The mitochondrial genome is especially vulnerable to oxidative injury not only because of its spatial relationship to the respiratory chain, but also because of its lack of protective histones and an adequate excision and recombination repair system. Mitochondrial DNA is inherited almost entirely from the maternal ovum; therefore, many primary mitochondrial deficiencies are inherited in a dominant fashion. Many nuclear genes, however, such as DNA polymerase-γ (POLG), thymidine kinase 2 (TK2), deoxyguanosine kinase (DGUOK), SCO1, BCS1L, and MPV17, encode proteins critical to maintaining proper amounts of mitochondrial DNA and to allowing normal mitochondrial respiratory function. Most mitochondrial diseases with primary involvement of the liver are caused by nuclear rather than mitochondrial gene mutations.242
Mitochondrial respiratory chain disorders can affect 1 in 20,000 children, with liver involvement occurring in 10% to 20% of patients.242,243 Striking heterogeneity of clinical features, ranging from single-organ involvement to multisystem disease, can lead to a delayed or missed diagnosis and can confound therapeutic decision making, for example, with respect to the advisability of liver transplantation.242 The diagnosis of a mitochondrial respiratory chain defect should be considered in a patient with liver disease who has unexplained neuromuscular symptoms, including a seizure disorder; involvement of seemingly unrelated organ systems; a rapidly progressive course; or a chronic course that proves to be a diagnostic dilemma.242 In about 80% of patients, symptoms appear early in life, before age two. The plasma lactate level and the ratio of lactate to pyruvate are often, but not universally, elevated, especially with insidious presentations.242
Neonatal liver failure has been reported in association with cytochrome c oxidase deficiency caused by mutations in the SCO1 or BCS1L genes. The key features of these disorders are lactic acidemia and an elevated ratio of plasma lactate to pyruvate levels. Infants with Alpers-Huttenlocher syndrome (progressive neuronal degeneration in childhood with liver disease ascribed to mitochondrial dysfunction) experience vomiting, hypotonia, seizures, and liver failure, often beginning by age six months. Frequently, the liver disease is unsuspected clinically and becomes evident late in the course of the disease; Alpers-Huttenlocher syndrome has been shown to be caused by mutations in POLG.244 Alternatively, in mtDNA depletion syndrome (caused by mutations in the POLG, DGUOK, or MPV17 genes), hypoglycemia, acidosis, and liver failure develop early in infancy, and neurologic abnormalities are less prominent.242,245
Navaho neurohepatopathy has been shown to be caused by mtDNA depletion and a defect in the MPV17 gene product, which is involved in mtDNA maintenance and regulation of oxidative phosphorylation.242,246 Other multisystemic mitochondrial diseases with liver involvement are Pearson’s marrow-pancreas syndrome (caused by large deletions of mtDNA segments) and chronic diarrhea and intestinal pseudo-obstruction with liver involvement.242
Liver biopsy specimens in mitochondrial disorders typically show macrovesicular and microvesicular steatosis, with increased density and occasional swelling of mitochondria on electron microscopy. Immunohistochemical techniques are being used more frequently (e.g., to diagnose cytochrome c oxidase deficiency). Cholestasis may be present, and conditions associated with chronic liver disease can show micronodular cirrhosis. Lactic acidemia may be constant, intermittent, or absent in mitochondrial disorders.247 Direct measurement of the enzymatic activity of the respiratory chain electron transport protein complexes can be performed on frozen tissue from the organ that expresses the clinical disease, although skin fibroblasts and lymphocytes may also be used. Few academic centers around the world can perform the assays for mitochondrial respiration (polarographic studies) or mtDNA analysis at this time.
No known effective therapy has been developed for mitochondrial respiratory chain disorders that alters the course of disease. Several strategies have been proposed to delay the progression of such disorders, including the use of antioxidants such as vitamin E or ascorbic acid; electron acceptors and cofactors, such as coenzyme Q10, thiamine, or riboflavin; and agents proposed to work by other mechanisms, such as carnitine, creatine, or succinate supplementation. The use of these agents, however, is clearly experimental at this time.242 Liver transplantation is generally contraindicated in these patients, given their dire outcome and high frequency of severe extrahepatic organ involvement.242,248
Alissa FT, Jaffe R, Shneider BL. Update on progressive familial intrahepatic cholestasis. J Pediatr Gastroenterol Nutr. 2008;46:241-52. (Ref 208.)
Anderson KE, Bloomer JR, Bonkovsky HL, et al. Recommendations for the diagnosis and treatment of the acute porphyrias. Ann Intern Med. 2005;142:439-50. (Ref 92.)
Campbell KM, Arya G, Ryckman FC, et al. High prevalence of alpha-1-antitrypsin heterozygosity in children with chronic liver disease. J Pediatr Gastroenterol Nutr. 2007;44:99-103. (Ref 20.)
Desmond CP, Wilson J, Bailey M, et al. The benign course of liver disease in adults with cystic fibrosis and the effect of ursodeoxycholic acid. Liver Int. 2007;27:1402-8. (Ref 238.)
Eklund EA, Sun L, Westphal V, et al. Congenital disorder of glycosylation (CDG)-Ih patient with a severe hepato-intestinal phenotype and evolving central nervous system pathology. J Pediatr. 2005;147:847-50. (Ref 87.)
Jaeken J, Matthijs G. Congenital disorders of glycosylation: A rapidly expanding disease family. Annu Rev Genomics Hum Genet. 2007;8:261-78. (Ref 76.)
Lee WS, Sokol RJ. Liver disease in mitochondrial disorders. Semin Liver Dis. 2007;27:259-73. (Ref 242.)
Lindstedt S, Holme E, Lock EA, et al. Treatment of hereditary tyrosinaemia type I by inhibition of 4-hydroxyphenylpyruvate dioxygenase. Lancet. 1992;340:813-17. (Ref 145.)
Liu C, Aronow BJ, Jegga AG, et al. Novel resequencing chip customized to diagnose mutations in patients with inherited syndromes of intrahepatic cholestasis. Gastroenterology. 2007;132:119-26. (Ref 33.)
Shimozawa N. Molecular and clinical aspects of peroxisomal diseases. J Inherit Metab Dis. 2007;30:193-7. (Ref 198.)
Summar ML, Dobbelaere D, Brusilow S, Lee B. Diagnosis, symptoms, frequency and mortality of 260 patients with urea cycle disorders from a 21-year, multicentre study of acute hyperammonaemic episodes. Acta Paediatr. 2008;97:1420-5. (Ref 158.)
Sundaram SS, Bove KE, Lovell MA, Sokol RJ. Mechanisms of disease: Inborn errors of bile acid synthesis. Nat Clin Pract Gastroenterol Hepatol. 2008;5:456-68. (Ref 192.)
Sveger T. Liver disease in alpha1-antitrypsin deficiency detected by screening of 200,000 infants. N Engl J Med. 1976;294:1316-21. (Ref 23.)
Trauner M, Fickert P, Wagner M. MDR3 (ABCB4) defects: A paradigm for the genetics of adult cholestatic syndromes. Semin Liver Dis. 2007;27:77-98. (Ref 215.)
Tuchman M, Lee B, Lichter-Konecki U, et al. Cross-sectional multicenter study of patients with urea cycle disorders in the United States. Mol Genet Metab. 2008;94:397-402. (Ref 159.)
1. The U.S. Organ Procurement and Transplantation Network and The Scientific Registry of Transplant Recipients. 2007 OPTN/STRT Annual Report: Transplant Data 1997-2006. http://www.optn.org/AR2007, 2007. accessed on January 29, 2009
2. Arya G, Balistreri WF. Pediatric liver disease in the United States: Epidemiology and impact. J Gastroenterol Hepatol. 2002;17:521-5.
3. Balistreri WF. Nontransplant options for the treatment of metabolic liver disease: Saving livers while saving lives. Hepatology. 1994;19:782-7.
4. Teckman J, Perlmutter DH. Conceptual advances in the pathogenesis and treatment of childhood metabolic liver disease. Gastroenterology. 1995;108:1263-79.
5. Balistreri WF. Liver disease in infancy and childhood. In: Schiff ER, editor. Schiff’s Diseases of the Liver. 8th ed. Philadelphia: Lippincott-Raven; 1999:1357-512.
6. Powell LW, Leggett BA, Crawford DHG. Hemochromatosis and other iron storage disorders. In: Schiff ER, editor. Schiff’s Diseases of the Liver. Philadelphia: Lippincott-Raven; 1999:1107-30.
7. Teckman JH. Alpha1-antitrypsin deficiency in childhood. Semin Liver Dis. 2007;27:274-81.
8. Lomas DA. The selective advantage of alpha1-antitrypsin deficiency. Am J Respir Crit Care Med. 2006;173:1072-7.
9. Mahadeva R, Atkinson C, Li Z, et al. Polymers of Z alpha1-antitrypsin co-localize with neutrophils in emphysematous alveoli and are chemotactic in vivo. Am J Pathol. 2005;166:377-86.
10. Mahadeva R, Chang WS, Dafforn TR, et al. Heteropolymerization of S, I, and Z alpha1-antitrypsin and liver cirrhosis. J Clin Invest. 1999;103:999-1006.
11. Canva V, Piotte S, Aubert JP, et al. Heterozygous M3Mmalton alpha1-antitrypsin deficiency associated with end-stage liver disease: Case report and review. Clin Chem. 2001;47:1490-6.
12. Janciauskiene S, Eriksson S, Callea F, et al. Differential detection of PAS-positive inclusions formed by the Z, Siiyama, and Mmalton variants of alpha1-antitrypsin. Hepatology. 2004;40:1203-10.
13. Wu Y, Whitman I, Molmenti E, et al. A lag in intracellular degradation of mutant alpha 1-antitrypsin correlates with the liver disease phenotype in homozygous PiZZ alpha 1-antitrypsin deficiency. Proc Natl Acad Sci U S A. 1994;91:9014-18.
14. Dycaico MJ, Grant SG, Felts K, et al. Neonatal hepatitis induced by alpha 1-antitrypsin: A transgenic mouse model. Science. 1988;242:1409-12.
15. Lawless MW, Greene CM, Mulgrew A, et al. Activation of endoplasmic reticulum-specific stress responses associated with the conformational disease Z alpha 1-antitrypsin deficiency. J Immunol. 2004;172:5722-6.
16. Teckman JH, An JK, Blomenkamp K, et al. Mitochondrial autophagy and injury in the liver in alpha 1-antitrypsin deficiency. Am J Physiol Gastrointest Liver Physiol. 2004;286:G851-62.
17. de Serres FJ. Worldwide racial and ethnic distribution of alpha1-antitrypsin deficiency: Summary of an analysis of published genetic epidemiologic surveys. Chest. 2002;122:1818-29.
18. de Serres FJ, Blanco I, Fernandez-Bustillo E. Genetic epidemiology of alpha-1 antitrypsin deficiency in North America and Australia/New Zealand: Australia, Canada, New Zealand and the United States of America. Clin Genet. 2003;64:382-97.
19. Graziadei IW, Joseph JJ, Wiesner RH, et al. Increased risk of chronic liver failure in adults with heterozygous alpha1-antitrypsin deficiency. Hepatology. 1998;28:1058-63.
20. Campbell KM, Arya G, Ryckman FC, et al. High prevalence of alpha-1-antitrypsin heterozygosity in children with chronic liver disease. J Pediatr Gastroenterol Nutr. 2007;44:99-103.
21. Regev A, Guaqueta C, Molina EG, et al. Does the heterozygous state of alpha-1 antitrypsin deficiency have a role in chronic liver diseases? Interim results of a large case-control study. J Pediatr Gastroenterol Nutr. 2006;43(Suppl 1):S30-5.
22. Ferrarotti I, Baccheschi J, Zorzetto M, et al. Prevalence and phenotype of subjects carrying rare variants in the Italian registry for alpha1-antitrypsin deficiency. J Med Genet. 2005;42:282-7.
23. Sveger T. Liver disease in alpha1-antitrypsin deficiency detected by screening of 200,000 infants. N Engl J Med. 1976;294:1316-21.
24. Sveger T, Eriksson S. The liver in adolescents with alpha 1-antitrypsin deficiency. Hepatology. 1995;22:514-17.
25. Lee JH, Brantly M. Molecular mechanisms of alpha1-antitrypsin null alleles. Respir Med. 2000;94(Suppl C):S7-11.
26. Volpert D, Molleston JP, Perlmutter DH. Alpha1-antitrypsin deficiency-associated liver disease progresses slowly in some children. J Pediatr Gastroenterol Nutr. 2000;31:258-63.
27. Hinds R, Hadchouel A, Shanmugham NP, et al. Variable degree of liver involvement in siblings with PiZZ alpha-1-antitrypsin deficiency-related liver disease. J Pediatr Gastroenterol Nutr. 2006;43:136-8.
28. Chappell S, Hadzic N, Stockley R, et al. A polymorphism of the alpha1-antitrypsin gene represents a risk factor for liver disease. Hepatology. 2008;47:127-32.
29. Piitulainen E, Carlson J, Ohlsson K, Sveger T. Alpha1-antitrypsin deficiency in 26-year-old subjects: Lung, liver, and protease/protease inhibitor studies. Chest. 2005;128:2076-81.
30. Elzouki AN, Eriksson S. Risk of hepatobiliary disease in adults with severe alpha 1-antitrypsin deficiency (PiZZ): Is chronic viral hepatitis B or C an additional risk factor for cirrhosis and hepatocellular carcinoma? Eur J Gastroenterol Hepatol. 1996;8:989-94.
31. Eriksson S, Carlson J, Velez R. Risk of cirrhosis and primary liver cancer in alpha 1-antitrypsin deficiency. N Engl J Med. 1986;314:736-9.
32. Lang T, Muhlbauer M, Strobelt M, et al. Alpha-1-antitrypsin deficiency in children: Liver disease is not reflected by low serum levels of alpha-1-antitrypsin—a study on 48 pediatric patients. Eur J Med Res. 2005;10:509-14.
33. Liu C, Aronow BJ, Jegga AG, et al. Novel resequencing chip customized to diagnose mutations in patients with inherited syndromes of intrahepatic cholestasis. Gastroenterology. 2007;132:119-26.
34. Rachelefsky G, Hogarth DK. Issues in the diagnosis of alpha 1-antitrypsin deficiency. J Allergy Clin Immunol. 2008;121:833-8.
35. Deutsch J, Becker H, Aubock L. Histopathological features of liver disease in alpha 1-antitrypsin deficiency. Acta Paediatr Suppl. 1994;393:8-12.
36. Kemmer N, Kaiser T, Zacharias V, Neff GW. Alpha-1-antitrypsin deficiency: Outcomes after liver transplantation. Transplant Proc. 2008;40:1492-4.
37. Pungpapong S, Krishna M, Abraham SC, et al. Clinicopathologic findings and outcomes of liver transplantation using grafts from donors with unrecognized and unusual diseases. Liver Transpl. 2006;12:310-15.
38. Russi EW. Alpha-1 antitrypsin: Now available, but do we need it? Swiss Med Wkly. 2008;138:191-6.
39. Burrows JA, Willis LK, Perlmutter DH. Chemical chaperones mediate increased secretion of mutant alpha 1-antitrypsin (alpha 1-AT) Z: A potential pharmacological strategy for prevention of liver injury and emphysema in alpha 1-AT deficiency. Proc Natl Acad Sci U S A. 2000;97:1796-801.
40. Teckman JH. Lack of effect of oral 4-phenylbutyrate on serum alpha-1-antitrypsin in patients with alpha-1-antitrypsin deficiency: A preliminary study. J Pediatr Gastroenterol Nutr. 2004;39:34-7.
41. Mallya M, Phillips RL, Saldanha SA, et al. Small molecules block the polymerization of Z alpha1-antitrypsin and increase the clearance of intracellular aggregates. J Med Chem. 2007;50:5357-63.
42. Alino SF, Crespo A, Dasi F. Long-term therapeutic levels of human alpha-1 antitrypsin in plasma after hydrodynamic injection of nonviral DNA. Gene Ther. 2003;10:1672-9.
43. Duan YY, Wu J, Zhu JL, et al. Gene therapy for human alpha1-antitrypsin deficiency in an animal model using SV40-derived vectors. Gastroenterology. 2004;127:1222-32.
44. Wolfsdorf JI, Weinstein DA. Glycogen storage diseases. Rev Endocr Metab Disord. 2003;4:95-102.
45. Ghishan FK, Zawaideh M. Inborn errors of carbohydrate metabolism. In: Suchy FJ, Sokol RJ, Balistreri WF, editors. Liver Disease in Children. 3rd ed. New York: Cambridge University Press; 2007:595-625.
46. Matern D, Seydewitz HH, Bali D, et al. Glycogen storage disease type I: Diagnosis and phenotype/genotype correlation. Eur J Pediatr. 2002;161(Suppl 1):S10-19.
47. Visser G, Rake JP, Fernandes J, et al. Neutropenia, neutrophil dysfunction, and inflammatory bowel disease in glycogen storage disease type Ib: Results of the European Study on Glycogen Storage Disease type I. J Pediatr. 2000;137:187-91.
48. Melis D, Parenti G, Della Casa R, et al. Crohn’s-like ileo-colitis in patients affected by glycogen storage disease Ib: Two years’ follow-up of patients with a wide spectrum of gastrointestinal signs. Acta Paediatr. 2003;92:1415-21.
49. Rake JP, Visser G, Labrune P, et al. Glycogen storage disease type I: Diagnosis, management, clinical course and outcome. Results of the European Study on Glycogen Storage Disease Type I (ESGSD I). Eur J Pediatr. 2002;161(Suppl 1):S20-34.
50. Rake JP, Visser G, Huismans D, et al. Bone mineral density in children, adolescents and adults with glycogen storage disease type Ia: A cross-sectional and longitudinal study. J Inherit Metab Dis. 2003;26:371-84.
51. Melis D, Parenti G, Della Casa R, et al. Brain damage in glycogen storage disease type I. J Pediatr. 2004;144:637-42.
52. Bandsma RH, Smit GP, Kuipers F. Disturbed lipid metabolism in glycogen storage disease type 1. Eur J Pediatr. 2002;161(Suppl 1):S65-9.
53. Melis D, Fulceri R, Parenti G, et al. Genotype/phenotype correlation in glycogen storage disease type 1b: A multicentre study and review of the literature. Eur J Pediatr. 2005;164:501-8.
54. Lee PJ. Glycogen storage disease type I: Pathophysiology of liver adenomas. Eur J Pediatr. 2002;161(Suppl 1):S46-9.
55. Labrune P, Trioche P, Duvaltier I, et al. Hepatocellular adenomas in glycogen storage disease type I and III: A series of 43 patients and review of the literature. J Pediatr Gastroenterol Nutr. 1997;24:276-9.
56. Franco LM, Krishnamurthy V, Bali D, et al. Hepatocellular carcinoma in glycogen storage disease type Ia: A case series. J Inherit Metab Dis. 2005;28:153-62.
57. Di Rocco M, Calevo MG, Taro M, et al. Hepatocellular adenoma and metabolic balance in patients with type Ia glycogen storage disease. Mol Genet Metab. 2008;93:398-402.
58. Reddy SK, Kishnani PS, Sullivan JA, et al. Resection of hepatocellular adenoma in patients with glycogen storage disease type Ia. J Hepatol. 2007;47:658-63.
59. Rake JP, Visser G, Labrune P, et al. Guidelines for management of glycogen storage disease type I—European Study on Glycogen Storage Disease Type I (ESGSD I). Eur J Pediatr. 2002;161(Suppl 1):S112-19.
60. Rake JP, ten Berge AM, Visser G, et al. Glycogen storage disease type Ia: Recent experience with mutation analysis, a summary of mutations reported in the literature and a newly developed diagnostic flow chart. Eur J Pediatr. 2000;159:322-30.
61. Visser G, Rake JP, Labrune P, et al. Consensus guidelines for management of glycogen storage disease type 1b—European Study on Glycogen Storage Disease Type 1. Eur J Pediatr. 2002;161(Suppl 1):S120-3.
62. Daublin G, Schwahn B, Wendel U. Type I glycogen storage disease: Favourable outcome on a strict management regimen avoiding increased lactate production during childhood and adolescence. Eur J Pediatr. 2002;161(Suppl 1):S40-5.
63. Visser G, Rake JP, Labrune P, et al. Granulocyte colony-stimulating factor in glycogen storage disease type 1b. Results of the European Study on Glycogen Storage Disease Type 1. Eur J Pediatr. 2002;161(Suppl 1):S83-7.
64. Beaty RM, Jackson M, Peterson D, et al. Delivery of glucose-6-phosphatase in a canine model for glycogen storage disease, type Ia, with adeno-associated virus (AAV) vectors. Gene Ther. 2002;9:1015-22.
65. Muraca M, Burlina AB. Liver and liver cell transplantation for glycogen storage disease type IA. Acta Gastroenterol Belg. 2005;68:469-72.
66. Lee KW, Lee JH, Shin SW, et al. Hepatocyte transplantation for glycogen storage disease type Ib. Cell Transplant. 2007;16:629-37.
67. Davis MK, Weinstein DA. Liver transplantation in children with glycogen storage disease: Controversies and evaluation of the risk/benefit of this procedure. Pediatr Transplant. 2008;12:137-45.
68. Iyer SG, Chen CL, Wang CC, et al. Long-term results of living donor liver transplantation for glycogen storage disorders in children. Liver Transpl. 2007;13:848-52.
69. Lucchiari S, Pagliarani S, Salani S, et al. Hepatic and neuromuscular forms of glycogenosis type III: Nine mutations in AGL. Hum Mutat. 2006;27:600-1.
70. Horinishi A, Okubo M, Tang NL, et al. Mutational and haplotype analysis of AGL in patients with glycogen storage disease type III. J Hum Genet. 2002;47:55-9.
71. Demo E, Frush D, Gottfried M, et al. Glycogen storage disease type III-hepatocellular carcinoma: A long-term complication? J Hepatol. 2007;46:492-8.
72. Maruyama K, Suzuki T, Koizumi T, et al. Congenital form of glycogen storage disease type IV: A case report and a review of the literature. Pediatr Int. 2004;46:474-7.
73. Moses SW, Parvari R. The variable presentations of glycogen storage disease type IV: A review of clinical, enzymatic and molecular studies. Curr Mol Med. 2002;2:177-88.
74. de Moor RA, Schweizer JJ, van Hoek B, et al. Hepatocellular carcinoma in glycogen storage disease type IV. Arch Dis Child. 2000;82:479-80.
75. Rosenthal P, Podesta L, Grier R, et al. Failure of liver transplantation to diminish cardiac deposits of amylopectin and leukocyte inclusions in type IV glycogen storage disease. Liver Transpl Surg. 1995;1:373-6.
76. Jaeken J, Matthijs G. Congenital disorders of glycosylation: A rapidly expanding disease family. Annu Rev Genomics Hum Genet. 2007;8:261-78.
77. Freeze HH, Aebi M. Altered glycan structures: The molecular basis of congenital disorders of glycosylation. Curr Opin Struct Biol. 2005;15:490-8.
78. Leroy JG. Congenital disorders of N-glycosylation including diseases associated with O– as well as N-glycosylation defects. Pediatr Res. 2006;60:643-56.
79. Miller BS, Freeze HH. New disorders in carbohydrate metabolism: Congenital disorders of glycosylation and their impact on the endocrine system. Rev Endocr Metab Disord. 2003;4:103-13.
80. Damen G, de Klerk H, Huijmans J, et al. Gastrointestinal and other clinical manifestations in 17 children with congenital disorders of glycosylation type Ia, Ib, and Ic. J Pediatr Gastroenterol Nutr. 2004;38:282-7.
81. Kjaergaard S, Skovby F, Schwartz M. Absence of homozygosity for predominant mutations in PMM2 in Danish patients with carbohydrate-deficient glycoprotein syndrome type 1. Eur J Hum Genet. 1998;6:331-6.
82. Thiel C, Lubke T, Matthijs G, et al. Targeted disruption of the mouse phosphomannomutase 2 gene causes early embryonic lethality. Mol Cell Biol. 2006;26:5615-20.
83. Kjaergaard S, Schwartz M, Skovby F. Congenital disorder of glycosylation type Ia (CDG-Ia): Phenotypic spectrum of the R141H/F119L genotype. Arch Dis Child. 2001;85:236-9.
84. Vuillaumier-Barrot S, Le Bizec C, de Lonlay P, et al. Protein losing enteropathy-hepatic fibrosis syndrome in Saguenay-Lac St-Jean, Quebec is a congenital disorder of glycosylation type Ib. J Med Genet. 2002;39:849-51.
85. Mention K, Lacaille F, Valayannopoulos V, et al. Development of liver disease despite mannose treatment in two patients with CDG-Ib. Mol Genet Metab. 2008;93:40-3.
86. Westphal V, Kjaergaard S, Davis JA, et al. Genetic and metabolic analysis of the first adult with congenital disorder of glycosylation type Ib: Long-term outcome and effects of mannose supplementation. Mol Genet Metab. 2001;73:77-85.
87. Eklund EA, Sun L, Westphal V, et al. Congenital disorder of glycosylation (CDG)-Ih patient with a severe hepato-intestinal phenotype and evolving central nervous system pathology. J Pediatr. 2005;147:847-50.
88. Mandato C, Brive L, Miura Y, et al. Cryptogenic liver disease in four children: A novel congenital disorder of glycosylation. Pediatr Res. 2006;59:293-8.
89. Wu X, Steet RA, Bohorov O, et al. Mutation of the COG complex subunit gene COG7 causes a lethal congenital disorder. Nat Med. 2004;10:518-23.
90. Wopereis S, Grunewald S, Morava E, et al. Apolipoprotein C-III isofocusing in the diagnosis of genetic defects in O-glycan biosynthesis. Clin Chem. 2003;49:1839-45.
91. Iancu TC, Mahajnah M, Manov I, et al. The liver in congenital disorders of glycosylation: Ultrastructural features. Ultrastruct Pathol. 2007;31:189-97.
92. Anderson KE, Bloomer JR, Bonkovsky HL, et al. Recommendations for the diagnosis and treatment of the acute porphyrias. Ann Intern Med. 2005;142:439-50.
93. Thunell S, Pomp E, Brun A. Guide to drug porphyrogenicity prediction and drug prescription in the acute porphyrias. Br J Clin Pharmacol. 2007;64:668-79.
94. Kauppinen R, Mustajoki P. Prognosis of acute porphyria: Occurrence of acute attacks, precipitating factors, and associated diseases. Medicine (Baltimore). 1992;71:1-13.
95. Thunell S, Henrichson A, Floderus Y, et al. Liver transplantation in a boy with acute porphyria due to aminolaevulinate dehydratase deficiency. Eur J Clin Chem Clin Biochem. 1992;30:599-606.
96. Crimlisk HL. The little imitator—porphyria: A neuropsychiatric disorder. J Neurol Neurosurg Psychiatry. 1997;62:319-28.
97. Elder GH, Hift RJ, Meissner PN. The acute porphyrias. Lancet. 1997;349:1613-17.
98. Murphy GM. The cutaneous porphyrias: A review. The British Photodermatology Group. Br J Dermatol. 1999;140:573-81.
99. Poblete-Gutierrez P, Mendez M, Wiederholt T, et al. The molecular basis of porphyria cutanea tarda in Chile: Identification and functional characterization of mutations in the uroporphyrinogen decarboxylase gene. Exp Dermatol. 2004;13:372-9.
100. Nagy Z, Koszo F, Par A, et al. Hemochromatosis (HFE) gene mutations and hepatitis C virus infection as risk factors for porphyria cutanea tarda in Hungarian patients. Liver Int. 2004;24:16-20.
101. Gisbert JP, Garcia-Buey L, Pajares JM, Moreno-Otero R. Prevalence of hepatitis C virus infection in porphyria cutanea tarda: Systematic review and meta-analysis. J Hepatol. 2003;39:620-7.
102. Brady JJ, Jackson HA, Roberts AG, et al. Co-inheritance of mutations in the uroporphyrinogen decarboxylase and hemochromatosis genes accelerates the onset of porphyria cutanea tarda. J Invest Dermatol. 2000;115:868-74.
103. Harper P, Floderus Y, Holmstrom P, et al. Enrichment of HFE mutations in Swedish patients with familial and sporadic form of porphyria cutanea tarda. J Intern Med. 2004;255:684-8.
104. Elder GH. Porphyria cutanea tarda. Semin Liver Dis. 1998;18:67-75.
105. Mogl MT, Pascher A, Presser SJ, et al. An unhappy triad: Hemochromatosis, porphyria cutanea tarda and hepatocellular carcinoma—a case report. World J Gastroenterol. 2007;13:1998-2001.
106. Bloomer J, Wang Y, Singhal A, Risheg H. Molecular studies of liver disease in erythropoietic protoporphyria. J Clin Gastroenterol. 2005;39:S167-75.
107. Gouya L, Puy H, Robreau AM, et al. The penetrance of dominant erythropoietic protoporphyria is modulated by expression of wildtype FECH. Nat Genet. 2002;30:27-8.
108. Sarkany RP, Cox TM. Autosomal recessive erythropoietic protoporphyria: A syndrome of severe photosensitivity and liver failure. Qjm. 1995;88:541-9.
109. Anstey AV, Hift RJ. Liver disease in erythropoietic protoporphyria: Insights and implications for management. Gut. 2007;56:1009-18.
110. Doss MO, Frank M. Hepatobiliary implications and complications in protoporphyria, a 20-year study. Clin Biochem. 1989;22:223-9.
111. Jensen JD, Resnick SD. Porphyria in childhood. Semin Dermatol. 1995;14:33-9.
112. Bloomer J, Bruzzone C, Zhu L, et al. Molecular defects in ferrochelatase in patients with protoporphyria requiring liver transplantation. J Clin Invest. 1998;102:107-14.
113. Young JW, Conte ET. Porphyrias and porphyrins. Int J Dermatol. 1991;30:399-406.
114. Andant C, Puy H, Faivre J, Deybach JC. Acute hepatic porphyrias and primary liver cancer. N Engl J Med. 1998;338:1853-4.
115. Bloomer JR, Brenner DA. The Porphyrias. In: Schiff ER, Sorrell MF, Maddrey WC, editors. Schiff’s Diseases of the Liver. 9th ed. Philadelphia: Lippincott-Raven; 2003:1231-60.
116. Seth AK, Badminton MN, Mirza D, et al. Liver transplantation for porphyria: Who, when, and how? Liver Transpl. 2007;13:1219-27.
117. McGuire BM, Bonkovsky HL, Carithers RLJr, et al. Liver transplantation for erythropoietic protoporphyria liver disease. Liver Transpl. 2005;11:1590-6.
118. Stolzel U, Kostler E, Schuppan D, et al. Hemochromatosis (HFE) gene mutations and response to chloroquine in porphyria cutanea tarda. Arch Dermatol. 2003;139:309-13.
119. Rossmann-Ringdahl I, Olsson R. Porphyria cutanea tarda: Effects and risk factors for hepatotoxicity from high-dose chloroquine treatment. Acta Derm Venereol. 2007;87:401-5.
120. Wahlin S, Aschan J, Bjornstedt M, et al. Curative bone marrow transplantation in erythropoietic protoporphyria after reversal of severe cholestasis. J Hepatol. 2007;46:174-9.
121. Cox TM, Alexander GJ, Sarkany RP. Protoporphyria. Semin Liver Dis. 1998;18:85-93.
122. Wahlin S, Srikanthan N, Hamre B, et al. Protection from phototoxic injury during surgery and endoscopy in erythropoietic protoporphyria. Liver Transpl. 2008;14:1340-6.
123. Rand EB, Bunin N, Cochran W, et al. Sequential liver and bone marrow transplantation for treatment of erythropoietic protoporphyria. Pediatrics. 2006;118:e1896-9.
124. Fritsch C, Lang K, Bolsen K, et al. Congenital erythropoietic porphyria. Skin Pharmacol Appl Skin Physiol. 1998;11:347-57.
125. Thomas C, Ged C, Nordmann Y, et al. Correction of congenital erythropoietic porphyria by bone marrow transplantation. J Pediatr. 1996;129:453-6.
126. De Braekeleer M, Larochelle J. Genetic epidemiology of hereditary tyrosinemia in Quebec and in Saguenay-Lac-St-Jean. Am J Hum Genet. 1990;47:302-7.
127. Ashorn M, Pitkanen S, Salo MK, Heikinheimo M. Current strategies for the treatment of hereditary tyrosinemia type I. Paediatr Drugs. 2006;8:47-54.
128. Jorquera R, Tanguay RM. The mutagenicity of the tyrosine metabolite, fumarylacetoacetate, is enhanced by glutathione depletion. Biochem Biophys Res Commun. 1997;232:42-8.
129. Prieto-Alamo MJ, Laval F. Deficient DNA-ligase activity in the metabolic disease tyrosinemia type I. Proc Natl Acad Sci U S A. 1998;95:12614-18.
130. Gilbert-Barness E, Barness LA, Meisner LF. Chromosomal instability in hereditary tyrosinemia type I. Pediatr Pathol. 1990;10:243-52.
131. van Spronsen FJ, Thomasse Y, Smit GP, et al. Hereditary tyrosinemia type I: A new clinical classification with difference in prognosis on dietary treatment. Hepatology. 1994;20:1187-91.
132. Demers SI, Russo P, Lettre F, Tanguay RM. Frequent mutation reversion inversely correlates with clinical severity in a genetic liver disease, hereditary tyrosinemia. Hum Pathol. 2003;34:1313-20.
133. Ploos van Amstel JK, Bergman AJ, van Beurden EA, et al. Hereditary tyrosinemia type 1: Novel missense, nonsense and splice consensus mutations in the human fumarylacetoacetate hydrolase gene; variability of the genotype-phenotype relationship. Hum Genet. 1996;97:51-9.
134. Macvicar D, Dicks-Mireaux C, Leonard JV, Wight DG. Hepatic imaging with computed tomography of chronic tyrosinaemia type 1. Br J Radiol. 1990;63:605-8.
135. Kvittingen EA, Talseth T, Halvorsen S, et al. Renal failure in adult patients with hereditary tyrosinaemia type I. J Inherit Metab Dis. 1991;14:53-62.
136. Paradis K, Weber A, Seidman EG, et al. Liver transplantation for hereditary tyrosinemia: The Quebec experience. Am J Hum Genet. 1990;47:338-42.
137. Arora N, Stumper O, Wright J, et al. Cardiomyopathy in tyrosinaemia type I is common but usually benign. J Inherit Metab Dis. 2006;29:54-7.
138. Mitchell G, Larochelle J, Lambert M, et al. Neurologic crises in hereditary tyrosinemia. N Engl J Med. 1990;322:432-7.
139. Turgeon C, Magera MJ, Allard P, et al. Combined newborn screening for succinylacetone, amino acids, and acylcarnitines in dried blood spots. Clin Chem. 2008;54:657-64.
140. la Marca G, Malvagia S, Pasquini E, et al. The inclusion of succinylacetone as marker for tyrosinemia type I in expanded newborn screening programs. Rapid Commun Mass Spectrom. 2008;22:812-18.
141. Herzog D, Martin S, Turpin S, Alvarez F. Normal glomerular filtration rate in long-term follow-up of children after orthotopic liver transplantation. Transplantation. 2006;81:672-7.
142. Pierik LJ, van Spronsen FJ, Bijleveld CM, van Dael CM. Renal function in tyrosinaemia type I after liver transplantation: A long-term follow-up. J Inherit Metab Dis. 2005;28:871-6.
143. Forget S, Patriquin HB, Dubois J, et al. The kidney in children with tyrosinemia: Sonographic, CT and biochemical findings. Pediatr Radiol. 1999;29:104-8.
144. Emiroglu R, Ayvaz I, Moray G, et al. Tacrolimus-related neurologic and renal complications in liver transplantation: A single-center experience. Transplant Proc. 2006;38:619-21.
145. Lindstedt S, Holme E, Lock EA, et al. Treatment of hereditary tyrosinaemia type I by inhibition of 4-hydroxyphenylpyruvate dioxygenase. Lancet. 1992;340:813-7.
146. Holme E, Lindstedt S. Tyrosinaemia type I and NTBC (2-(2-nitro-4-trifluoromethylbenzoyl)-1,3-cyclohexanedione). J Inherit Metab Dis. 1998;21:507-17.
147. Santra S, Preece MA, Hulton SA, McKiernan PJ. Renal tubular function in children with tyrosinaemia type I treated with nitisinone. J Inherit Metab Dis. 2008;31:399-402.
148. Koelink CJ, van Hasselt P, van der Ploeg A, et al. Tyrosinemia type I treated by NTBC: How does AFP predict liver cancer? Mol Genet Metab. 2006;89:310-15.
149. Masurel-Paulet A, Poggi-Bach J, Rolland MO, et al. NTBC treatment in tyrosinaemia type I: Long-term outcome in French patients. J Inherit Metab Dis. 2008;31:81-7.
150. Mitchell GA, Grompe M, Lambert M. Hypertyrosinemia. In: Scriver CR, Sly WS, Childs B, et al, editors. The Metabolic and Molecular Bases of Inherited Disease. 8th ed. McGraw-Hill; 2000:1777-805.
151. Brusilow SW, Horwich AL. Urea cycle enzymes, 8th ed. Scriver CR, Beaudet AL, Sly WS, Valle D, editors, Metabolic and Molecular Basis of Inherited Disease, Volume 2, 2001. McGraw-Hill, New York: 1909-63
152. Palmieri L, Pardo B, Lasorsa FM, et al. Citrin and aralar1 are Ca(2+)-stimulated aspartate/glutamate transporters in mitochondria. Embo J. 2001;20:5060-9.
153. Yamaguchi S, Brailey LL, Morizono H, et al. Mutations and polymorphisms in the human ornithine transcarbamylase (OTC) gene. Hum Mutat. 2006;27:626-32.
154. Haberle J, Schmidt E, Pauli S, et al. Mutation analysis in patients with N-acetylglutamate synthase deficiency. Hum Mutat. 2003;21:593-7.
155. Saheki T, Kobayashi K, Iijima M, et al. Adult-onset type II citrullinemia and idiopathic neonatal hepatitis caused by citrin deficiency: Involvement of the aspartate glutamate carrier for urea synthesis and maintenance of the urea cycle. Mol Genet Metab. 2004;81(Suppl 1):S20-6.
156. Yefimenko I, Fresquet V, Marco-Marin C, et al. Understanding carbamoyl phosphate synthetase deficiency: Impact of clinical mutations on enzyme functionality. J Mol Biol. 2005;349:127-41.
157. Eeds AM, Hall LD, Yadav M, et al. The frequent observation of evidence for nonsense-mediated decay in RNA from patients with carbamyl phosphate synthetase I deficiency. Mol Genet Metab. 2006;89:80-6.
158. Summar ML, Dobbelaere D, Brusilow S, Lee B. Diagnosis, symptoms, frequency and mortality of 260 patients with urea cycle disorders from a 21-year, multicentre study of acute hyperammonaemic episodes. Acta Paediatr. 2008;97:1420-5.
159. Tuchman M, Lee B, Lichter-Konecki U, et al. Cross-sectional multicenter study of patients with urea cycle disorders in the United States. Mol Genet Metab. 2008;94:397-402.
160. Maestri NE, Clissold D, Brusilow SW. Neonatal onset ornithine transcarbamylase deficiency: A retrospective analysis. J Pediatr. 1999;134:268-72.
161. Enns GM, Berry SA, Berry GT, et al. Survival after treatment with phenylacetate and benzoate for urea-cycle disorders. N Engl J Med. 2007;356:2282-92.
162. Finkelstein JE, Hauser ER, Leonard CO, Brusilow SW. Late-onset ornithine transcarbamylase deficiency in male patients. J Pediatr. 1990;117:897-902.
163. Maestri NE, Lord C, Glynn M, et al. The phenotype of ostensibly healthy women who are carriers for ornithine transcarbamylase deficiency. Medicine (Baltimore). 1998;77:389-97.
164. McBride KL, Miller G, Carter S, et al. Developmental outcomes with early orthotopic liver transplantation for infants with neonatal-onset urea cycle defects and a female patient with late-onset ornithine transcarbamylase deficiency. Pediatrics. 2004;114:e523-6.
165. Wong LJ, Craigen WJ, O’Brien WE. Postpartum coma and death due to carbamoyl-phosphate synthetase I deficiency. Ann Intern Med. 1994;120:216-17.
166. Cordero DR, Baker J, Dorinzi D, Toffle R. Ornithine transcarbamylase deficiency in pregnancy. J Inherit Metab Dis. 2005;28:237-40.
167. Ohura T, Kobayashi K, Tazawa Y, et al. Clinical pictures of 75 patients with neonatal intrahepatic cholestasis caused by citrin deficiency (NICCD). J Inherit Metab Dis. 2007;30:139-44.
168. Tabata A, Sheng JS, Ushikai M, et al. Identification of 13 novel mutations including a retrotransposal insertion in SLC25A13 gene and frequency of 30 mutations found in patients with citrin deficiency. J Hum Genet. 2008;53:534-45.
169. Dimmock D, Kobayashi K, Iijima M, et al. Citrin deficiency: A novel cause of failure to thrive that responds to a high-protein, low-carbohydrate diet. Pediatrics. 2007;119:e773-7.
170. Lee NC, Chien YH, Kobayashi K, et al. Time course of acylcarnitine elevation in neonatal intrahepatic cholestasis caused by citrin deficiency. J Inherit Metab Dis. 2006;29:551-5.
171. Yeh JN, Jeng YM, Chen HL, et al. Hepatic steatosis and neonatal intrahepatic cholestasis caused by citrin deficiency (NICCD) in Taiwanese infants. J Pediatr. 2006;148:642-6.
172. Chen HW, Chen HL, Ni YH, et al. Chubby face and the biochemical parameters for the early diagnosis of neonatal intrahepatic cholestasis caused by citrin deficiency. J Pediatr Gastroenterol Nutr. 2008;47:187-92.
173. Tokuhara D, Iijima M, Tamamori A, et al. Novel diagnostic approach to citrin deficiency: Analysis of citrin protein in lymphocytes. Mol Genet Metab. 2007;90:30-6.
174. Choi JH, Kim H, Yoo HW. Two cases of citrullinaemia presenting with stroke. J Inherit Metab Dis. 2006;29:182-3.
175. King LS, Singh RH, Rhead WJ, et al. Genetic counseling issues in urea cycle disorders. Crit Care Clin. 2005;21:S37-44.
176. Guffon N, Schiff M, Cheillan D, et al. Neonatal hyperammonemia: The N-carbamoyl-L-glutamic acid test. J Pediatr. 2005;147:260-2.
177. Caldovic L, Morizono H, Tuchman M. Mutations and polymorphisms in the human N-acetylglutamate synthase (NAGS) gene. Hum Mutat. 2007;28:754-9.
178. Grunewald S, Fairbanks L, Genet S, et al. How reliable is the allopurinol load in detecting carriers for ornithine transcarbamylase deficiency? J Inherit Metab Dis. 2004;27:179-86.
179. Gucer S, Asan E, Atilla P, et al. Early cirrhosis in a patient with type I citrullinaemia (CTLN1). J Inherit Metab Dis. 2004;27:541-2.
180. Efrati C, Masini A, Merli M, et al. Effect of sodium benzoate on blood ammonia response to oral glutamine challenge in cirrhotic patients: A note of caution. Am J Gastroenterol. 2000;95:3574-8.
181. Sanjurjo P, Ruiz JI, Montejo M. Inborn errors of metabolism with a protein-restricted diet: Effect on polyunsaturated fatty acids. J Inherit Metab Dis. 1997;20:783-9.
182. Singh RH, Rhead WJ, Smith W, et al. Nutritional management of urea cycle disorders. Crit Care Clin. 2005;21:S27-35.
183. Uchino T, Endo F, Matsuda I. Neurodevelopmental outcome of long-term therapy of urea cycle disorders in Japan. J Inherit Metab Dis. 1998;21(Suppl 1):151-9.
184. Maestri NE, Brusilow SW, Clissold DB, Bassett SS. Long-term treatment of girls with ornithine transcarbamylase deficiency. N Engl J Med. 1996;335:855-9.
185. Morioka D, Kasahara M, Takada Y, et al. Current role of liver transplantation for the treatment of urea cycle disorders: A review of the worldwide English literature and 13 cases at Kyoto University. Liver Transpl. 2005;11:1332-42.
186. Puppi J, Tan N, Mitry RR, et al. Hepatocyte transplantation followed by auxiliary liver transplantation—a novel treatment for ornithine transcarbamylase deficiency. Am J Transplant. 2008;8:452-7.
187. Horslen SP, McCowan TC, Goertzen TC, et al. Isolated hepatocyte transplantation in an infant with a severe urea cycle disorder. Pediatrics. 2003;111:1262-7.
188. Picker JD, Puga AC, Levy HL, et al. Arginase deficiency with lethal neonatal expression: Evidence for the glutamine hypothesis of cerebral edema. J Pediatr. 2003;142:349-52.
189. Crombez EA, Cederbaum SD. Hyperargininemia due to liver arginase deficiency. Mol Genet Metab. 2005;84:243-51.
190. Bezerra JA, Balistreri WF. Intrahepatic cholestasis: Order out of chaos. Gastroenterology. 1999;117:1496-8.
191. Setchell KD, O’Connell NC. Disorders of bile acid synthesis and metabolism: A metabolic basis for liver disease. In: Suchy FJ, Sokol RJ, Balistreri WF, editors. Liver Disease in Children. 3rd ed. New York: Cambridge University Press; 2007:736-66.
192. Sundaram SS, Bove KE, Lovell MA, Sokol RJ. Mechanisms of disease: Inborn errors of bile acid synthesis. Nat Clin Pract Gastroenterol Hepatol. 2008;5:456-68.
193. Stieger B, Zhang J, O’Neill B, et al. Differential interaction of bile acids from patients with inborn errors of bile acid synthesis with hepatocellular bile acid transporters. Eur J Biochem. 1997;244:39-44.
194. Clayton PT, Verrips A, Sistermans E, et al. Mutations in the sterol 27-hydroxylase gene (CYP27A) cause hepatitis of infancy as well as cerebrotendinous xanthomatosis. J Inherit Metab Dis. 2002;25:501-13.
195. Dotti MT, Rufa A, Federico A. Cerebrotendinous xanthomatosis: Heterogeneity of clinical phenotype with evidence of previously undescribed ophthalmological findings. J Inherit Metab Dis. 2001;24:696-706.
196. van Heijst AF, Verrips A, Wevers RA, et al. Treatment and follow-up of children with cerebrotendinous xanthomatosis. Eur J Pediatr. 1998;157:313-16.
197. Lemonde HA, Custard EJ, Bouquet J, et al. Mutations in SRD5B1 (AKR1D1), the gene encoding delta(4)-3-oxosteroid 5beta-reductase, in hepatitis and liver failure in infancy. Gut. 2003;52:1494-9.
198. Shimozawa N. Molecular and clinical aspects of peroxisomal diseases. J Inherit Metab Dis. 2007;30:193-7.
199. Takemoto Y, Suzuki Y, Horibe R, et al. Gas chromatography/mass spectrometry analysis of very long chain fatty acids, docosahexaenoic acid, phytanic acid and plasmalogen for the screening of peroxisomal disorders. Brain Dev. 2003;25:481-7.
200. FitzPatrick DR. Zellweger syndrome and associated phenotypes. J Med Genet. 1996;33:863-8.
201. Corzo D, Gibson W, Johnson K, et al. Contiguous deletion of the X-linked adrenoleukodystrophy gene (ABCD1) and DXS1357E: A novel neonatal phenotype similar to peroxisomal biogenesis disorders. Am J Hum Genet. 2002;70:1520-31.
202. Setchell KD, Bragetti P, Zimmer-Nechemias L, et al. Oral bile acid treatment and the patient with Zellweger syndrome. Hepatology. 1992;15:198-207.
203. Resnick IB, Abdul Hai A, Shapira MY, et al. Treatment of X-linked childhood cerebral adrenoleukodystrophy by the use of an allogeneic stem cell transplantation with reduced intensity conditioning regimen. Clin Transplant. 2005;19:840-7.
204. Moser HW. Therapy of X-linked adrenoleukodystrophy. NeuroRx. 2006;3:246-53.
205. Martinez M, Vazquez E. MRI evidence that docosahexaenoic acid ethyl ester improves myelination in generalized peroxisomal disorders. Neurology. 1998;51:26-32.
206. Pauli-Magnus C, Stieger B, Meier Y, et al. Enterohepatic transport of bile salts and genetics of cholestasis. J Hepatol. 2005;43:342-57.
207. Klomp LW, Vargas JC, van Mil SW, et al. Characterization of mutations in ATP8B1 associated with hereditary cholestasis. Hepatology. 2004;40:27-38.
208. Alissa FT, Jaffe R, Shneider BL. Update on progressive familial intrahepatic cholestasis. J Pediatr Gastroenterol Nutr. 2008;46:241-52.
209. Strautnieks SS, Bull LN, Knisely AS, et al. A gene encoding a liver-specific ABC transporter is mutated in progressive familial intrahepatic cholestasis. Nat Genet. 1998;20:233-8.
210. van Mil SW, van der Woerd WL, van der Brugge G, et al. Benign recurrent intrahepatic cholestasis type 2 is caused by mutations in ABCB11. Gastroenterology. 2004;127:379-84.
211. Knisely AS, Strautnieks SS, Meier Y, et al. Hepatocellular carcinoma in ten children under five years of age with bile salt export pump deficiency. Hepatology. 2006;44:478-86.
212. Richter A, Grabhorn E, Schulz A, et al. Hepatoblastoma in a child with progressive familial intrahepatic cholestasis. Pediatr Transplant. 2005;9:805-8.
213. Scheimann AO, Strautnieks SS, Knisely AS, et al. Mutations in bile salt export pump (ABCB11) in two children with progressive familial intrahepatic cholestasis and cholangiocarcinoma. J Pediatr. 2007;150:556-9.
214. de Vree JM, Jacquemin E, Sturm E, et al. Mutations in the MDR3 gene cause progressive familial intrahepatic cholestasis. Proc Natl Acad Sci U S A. 1998;95:282-7.
215. Trauner M, Fickert P, Wagner M. MDR3 (ABCB4) defects: A paradigm for the genetics of adult cholestatic syndromes. Semin Liver Dis. 2007;27:77-98.
216. Jacquemin E, De Vree JM, Cresteil D, et al. The wide spectrum of multidrug resistance 3 deficiency: From neonatal cholestasis to cirrhosis of adulthood. Gastroenterology. 2001;120:1448-58.
217. Gotthardt D, Runz H, Keitel V, et al. A mutation in the canalicular phospholipid transporter gene, ABCB4, is associated with cholestasis, ductopenia, and cirrhosis in adults. Hepatology. 2008;48:1157-66.
218. Pauli-Magnus C, Kerb R, Fattinger K, et al. BSEP and MDR3 haplotype structure in healthy Caucasians, primary biliary cirrhosis and primary sclerosing cholangitis. Hepatology. 2004;39:779-91.
219. Schneider G, Paus TC, Kullak-Ublick GA, et al. Linkage between a new splicing site mutation in the MDR3 alias ABCB4 gene and intrahepatic cholestasis of pregnancy. Hepatology. 2007;45:150-8.
220. Drivdal M, Trydal T, Hagve TA, et al. Prognosis, with evaluation of general biochemistry, of liver disease in lymphoedema cholestasis syndrome 1 (LCS1/Aagenaes syndrome). Scand J Gastroenterol. 2006;41:465-71.
221. Carlton VE, Pawlikowska L, Bull LN. Molecular basis of intrahepatic cholestasis. Ann Med. 2004;36:606-17.
222. Chagnon P, Michaud J, Mitchell G, et al. A missense mutation (R565W) in cirhin (FLJ14728) in North American Indian childhood cirrhosis. Am J Hum Genet. 2002;71:1443-9.
223. Hadj-Rabia S, Baala L, Vabres P, et al. Claudin-1 gene mutations in neonatal sclerosing cholangitis associated with ichthyosis: A tight junction disease. Gastroenterology. 2004;127:1386-90.
224. Gissen P, Johnson CA, Morgan NV, et al. Mutations in VPS33B, encoding a regulator of SNARE-dependent membrane fusion, cause arthrogryposis-renal dysfunction-cholestasis (ARC) syndrome. Nat Genet. 2004;36:400-4.
225. Ng VL, Ryckman FC, Porta G, et al. Long-term outcome after partial external biliary diversion for intractable pruritus in patients with intrahepatic cholestasis. J Pediatr Gastroenterol Nutr. 2000;30:152-6.
226. Kurbegov AC, Setchell KD, Haas JE, et al. Biliary diversion for progressive familial intrahepatic cholestasis: Improved liver morphology and bile acid profile. Gastroenterology. 2003;125:1227-34.
227. Kalicinski PJ, Ismail H, Jankowska I, et al. Surgical treatment of progressive familial intrahepatic cholestasis: Comparison of partial external biliary diversion and ileal bypass. Eur J Pediatr Surg. 2003;13:307-11.
228. Cutillo L, Najimi M, Smets F, et al. Safety of living-related liver transplantation for progressive familial intrahepatic cholestasis. Pediatr Transplant. 2006;10:570-4.
229. Englert C, Grabhorn E, Richter A, et al. Liver transplantation in children with progressive familial intrahepatic cholestasis. Transplantation. 2007;84:1361-3.
230. Colombo C, Battezzati PM, Crosignani A, et al. Liver disease in cystic fibrosis: A prospective study on incidence, risk factors, and outcome. Hepatology. 2002;36:1374-82.
231. Colombo C, Russo MC, Zazzeron L, Romano G. Liver disease in cystic fibrosis. J Pediatr Gastroenterol Nutr. 2006;43(Suppl 1):S49-55.
232. Lamireau T, Monnereau S, Martin S, et al. Epidemiology of liver disease in cystic fibrosis: A longitudinal study. J Hepatol. 2004;41:920-5.
233. Oppenheimer EH, Esterly JR. Hepatic changes in young infants with cystic fibrosis: Possible relation to focal biliary cirrhosis. J Pediatr. 1975;86:683-9.
234. Perdue DG, Cass OW, Milla C, et al. Hepatolithiasis and cholangiocarcinoma in cystic fibrosis: A case series and review of the literature. Dig Dis Sci. 2007;52:2638-42.
235. Smith JL, Lewindon PJ, Hoskins AC, et al. Endogenous ursodeoxycholic acid and cholic acid in liver disease due to cystic fibrosis. Hepatology. 2004;39:1673-82.
236. Akata D, Akhan O. Liver manifestations of cystic fibrosis. Eur J Radiol. 2007;61:11-17.
237. Mueller-Abt PR, Frawley KJ, Greer RM, Lewindon PJ. Comparison of ultrasound and biopsy findings in children with cystic fibrosis related liver disease. J Cyst Fibros. 2008;7:215-21.
238. Desmond CP, Wilson J, Bailey M, et al. The benign course of liver disease in adults with cystic fibrosis and the effect of ursodeoxycholic acid. Liver Int. 2007;27:1402-8.
239. Colombo C, Costantini D, Rocchi A, et al. Effects of liver transplantation on the nutritional status of patients with cystic fibrosis. Transpl Int. 2005;18:246-55.
240. Nash KL, Collier JD, French J, et al. Cystic fibrosis liver disease: To transplant or not to transplant? Am J Transplant. 2008;8:162-9.
241. Gooding I, Dondos V, Gyi KM, et al. Variceal hemorrhage and cystic fibrosis: Outcomes and implications for liver transplantation. Liver Transpl. 2005;11:1522-6.
242. Lee WS, Sokol RJ. Liver disease in mitochondrial disorders. Semin Liver Dis. 2007;27:259-73.
243. Skladal D, Halliday J, Thorburn DR. Minimum birth prevalence of mitochondrial respiratory chain disorders in children. Brain. 2003;126:1905-12.
244. Naviaux RK, Nguyen KV. POLG mutations associated with Alpers’ syndrome and mitochondrial DNA depletion. Ann Neurol. 2004;55:706-12.
245. Mandel H, Szargel R, Labay V, et al. The deoxyguanosine kinase gene is mutated in individuals with depleted hepatocerebral mitochondrial DNA. Nat Genet. 2001;29:337-41.
246. Karadimas CL, Vu TH, Holve SA, et al. Navajo neurohepatopathy is caused by a mutation in the MPV17 gene. Am J Hum Genet. 2006;79:544-8.
247. Robinson BH. Lactic acidemia and mitochondrial disease. Mol Genet Metab. 2006;89:3-13.
248. Dimmock DP, Dunn JK, Feigenbaum A, et al. Abnormal neurological features predict poor survival and should preclude liver transplantation in patients with deoxyguanosine kinase deficiency. Liver Transpl. 2008;14:1480-5.