Chapter 426 Other Congenital Heart and Vascular Malformations
426.1 Anomalies of the Aortic Arch
Vascular Rings
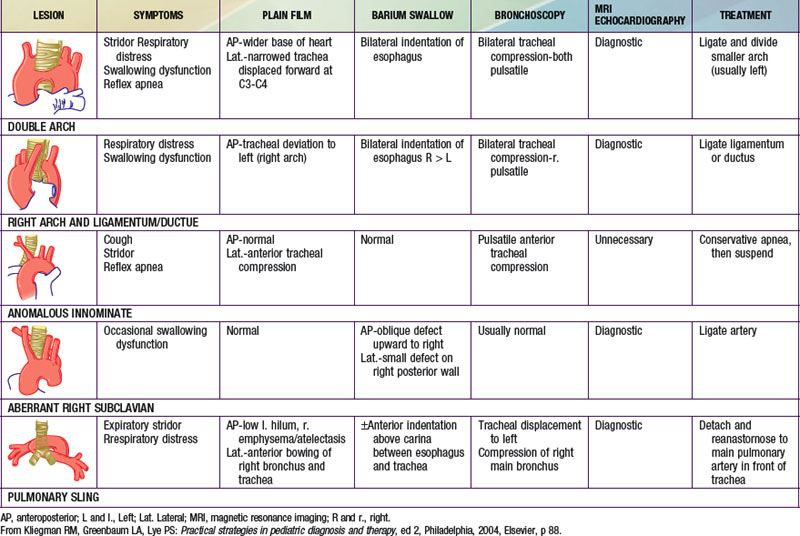
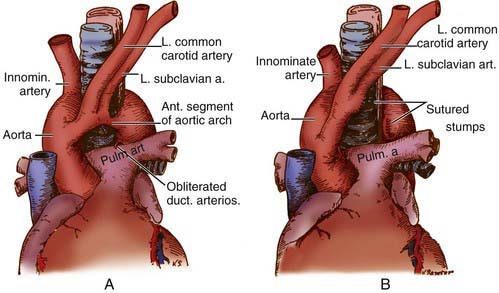
Congenital abnormalities of the aortic arch and its major branches result in the formation of vascular rings around the trachea and esophagus with varying degrees of compression (Table 426-1). The origin of these lesions can best be appreciated by reviewing the embryology of the aortic arch (Fig. 414-1). The most common anomalies include (1) double aortic arch (Fig. 426-1A), (2) right aortic arch with a left ligamentum arteriosum, (3) anomalous innominate artery arising farther to the left on the arch than usual, (4) anomalous left carotid artery arising farther to the right than usual and passing anterior to the trachea, and (5) anomalous left pulmonary artery (vascular sling). In the latter anomaly, the abnormal vessel arises from an elongated main pulmonary artery or from the right pulmonary artery. It courses between and compresses the trachea and the esophagus. Associated congenital heart disease may be present in 5-50% of patients, depending on the vascular anomaly.
Diagnosis
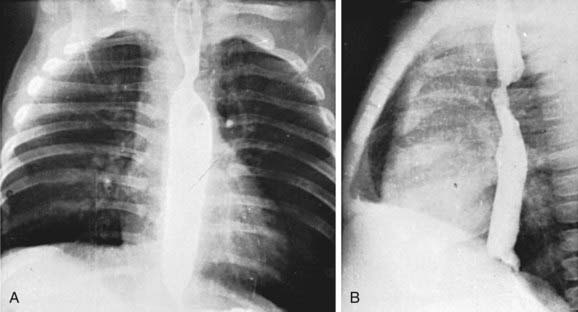
Standard roentgenographic examination is not usually helpful, however, in the past, performing a barium esophagogram was the standard method of diagnosis (Fig. 426-2). Echocardiography in combination with either MRI or CT will usually define the lesion. Cardiac catheterization is reserved for cases with associated anomalies or in rare cases where these other modalities are not diagnostic. Bronchoscopy may be helpful in more severe cases to determine the extent of airway narrowing.
Treatment
Surgery is advised for symptomatic patients who have evidence of tracheal compression. The anterior vessel is usually divided in patients with a double aortic arch (see Fig. 426-1B). Compression produced by a right aortic arch and left ligamentum arteriosum is relieved by division of the latter. Anomalous innominate or carotid arteries cannot be divided; attaching the adventitia of these vessels to the sternum usually relieves the tracheal compression. An anomalous left pulmonary artery is corrected by division at its origin and re-anastomosis to the main pulmonary artery after it has been brought in front of the trachea. Severe tracheomalacia, if present, may require reconstruction of the trachea as well.
Azarow KS, Pearl RH, Hoffman MA, et al. Vascular ring: does magnetic resonance imaging replace angiography? Ann Thorac Surg. 1992;53:882-885.
Bertrand J-M, Chartrand C, Lamarre A, et al. Vascular ring: clinical and physiological assessment of pulmonary function following surgical correction. Pediatr Pulmonol. 1986;2:378-383.
Kussman BD, Geva T, McGowan FX. Cardiovascular causes of airway compression. Paediatr Anaesth. 2004;14:60-74.
Murdison KA, Andrews BA, Chin AJ. Ultrasonographic display of complex vascular rings. J Am Coll Cardiol. 1990;15:1645-1653.
van Son JA, Julsrud PR, Hagler DJ, et al. Imaging strategies for vascular rings. Ann Thorac Surg. 1994;57:604-610.
426.2 Anomalous Origin of the Coronary Arteries
Anomalous Origin of the Left Coronary Artery from the Pulmonary Artery (Alcapa)
Diagnosis
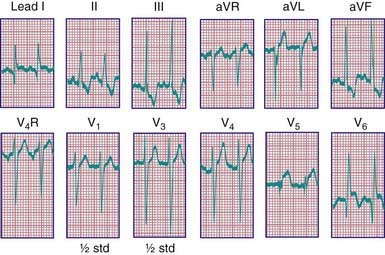
Roentgenographic examination confirms the cardiomegaly. The electrocardiogram resembles the pattern described in lateral wall myocardial infarction in adults. A QR pattern followed by inverted T waves is seen in leads I and aVL. The left ventricular surface leads (V5 and V6) may also show deep Q waves and exhibit elevated ST segments and inverted T waves (Fig. 426-3). Two-dimensional echocardiography can usually suggest the diagnosis; however, echocardiography is not always reliable in diagnosing this condition. On two-dimensional imaging alone, the left coronary artery may appear as though it is arising from the aorta. Color Doppler ultrasound examination has improved the accuracy of diagnosis of this lesion, demonstrating the presence of retrograde flow in the left coronary artery. CT or MRI may be helpful in confirming the origin of the coronary arteries. Cardiac catheterization is diagnostic; aortography shows immediate opacification of the right coronary artery only. This vessel is large and tortuous. After filling of the intercoronary anastomoses, the left coronary artery is opacified, and contrast can be seen to enter the pulmonary artery. Pulmonary arteriography may also opacify the origin of the anomalous left coronary artery. Selective left ventriculography usually demonstrates a dilated left ventricle that empties poorly and mitral regurgitation.
Treatment and Prognosis
Surgical treatment consists of detaching the anomalous coronary artery from the pulmonary artery and anastomosing it to the aorta to establish normal myocardial perfusion. A seriously ill infant with a tiny left coronary artery may present a difficult technical problem. In patients who have already sustained a significant myocardial infarction, cardiac transplantation may be the best option (Chapter 437.1).
Chang RR, Allada V. Electrocardiographic and echocardiographic features that distinguish anomalous origin of the left coronary artery from pulmonary artery from idiopathic dilated cardiomyopathy. Pediatr Cardiol. 2001;22:3-10.
Frommelt PC, Berger S, Pelech AN, et al. Prospective identification of anomalous origin of left coronary artery from the right sinus of Valsalva using transthoracic echocardiography: Importance of color Doppler flow mapping. Pediatr Cardiol. 2001;22:327-332.
Johnsrude CL, Perry JC, Cecchin F, et al. Differentiating anomalous left main coronary artery originating from the pulmonary artery in infants from myocarditis and dilated cardiomyopathy by electrocardiogram. Am J Cardiol. 1995;75:71-74.
Schmidt KG, Cooper MJ, Silverman NH, et al. Pulmonary artery origin of the left coronary artery: diagnosis by two-dimensional echocardiography, pulsed Doppler ultrasound and color flow mapping. J Am Coll Cardiol. 1988;11:396-402.
426.3 Pulmonary Arteriovenous Fistula
Patients who have undergone a Glenn cavopulmonary anastomosis for cyanotic congenital heart disease (Chapter 424.4) are also at risk for the development of pulmonary arteriovenous malformations. In these patients, the arteriovenous malformations are usually multiple and the risk increases over time after the Glenn procedure. These malformations rarely occur after the heart disease is fully palliated by completion of the Fontan operation. This finding suggests that the pulmonary circulation requires an as yet undetermined hepatic factor to suppress the development of arteriovenous malformations. The hallmark of the development of these malformations is a decrease in the patient’s oxygen saturation. The diagnosis can often be made with contrast echocardiography; cardiac catheterization is the definitive test. Completion of the Fontan circuit, so that inferior vena cava blood flow (containing hepatic venous drainage) is routed through the lungs, usually results in improvement or resolution of the malformations.
Feinstein JA, Moore P, Rosenthal DN, et al. Comparison of contrast echocardiography versus cardiac catheterization for detection of pulmonary arteriovenous malformations. Am J Cardiol. 2002;89:281-285.
Freedom RM, Yoo SJ, Perrin D. The biological “scrabble” of pulmonary arteriovenous malformations: considerations in the setting of cavopulmonary surgery. Cardiol Young. 2004;14:417-437.
Marchuk DA. Genetic abnormalities in hereditary hemorrhagic telangiectasia. Curr Opin Hematol. 1998;5:332-338.
Paterson A. Imaging evaluation of congenital lung abnormalities in infants and children. Radiol Clin North Am. 2005;43:303-323.
Srivastava D, Preminger T, Lock JE, et al. Hepatic venous blood and the development of pulmonary arteriovenous malformations in congenital heart disease. Circulation. 1995;92:1217-1222.




