[level-membership-for-pediatrics-category]
Chapter 692 Osteogenesis Imperfecta
Clinical Manifestations
Osteogenesis Imperfecta Type III (Progressive Deforming)
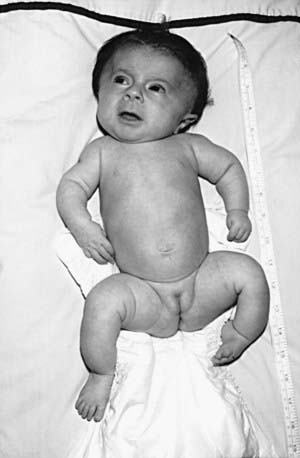
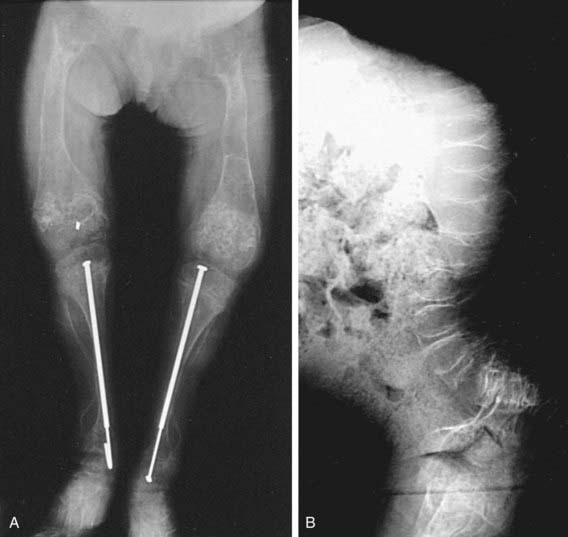
OI type III is the most severe nonlethal form of OI and results in significant physical disability. Birthweight and length are often low normal. Fractures usually occur in utero. There is relative macrocephaly and triangular facies (Fig. 692-1). Postnatally, fractures occur from inconsequential trauma and heal with deformity. Disorganization of the bone matrix results in a “popcorn” appearance at the metaphyses (Fig. 692-2). The rib cage has flaring at the base, and pectal deformity is frequent. Virtually all type III patients have scoliosis and vertebral compression. Growth falls below the curve by the 1st year; all type III patients have extreme short stature. Scleral hue ranges from white to blue.
Complications
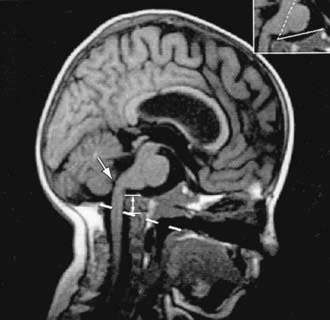
Neurologic complications include basilar invagination, brainstem compression, hydrocephalus, and syringohydromyelia. Most children with OI types III and IV have basilar invagination, but brainstem compression is uncommon. Basilar invagination is best detected with spiral CT of the craniocervical junction (Fig. 692-3).
Antoniazzi F, Bertoldo F, Mottes M, et al. Growth hormone treatment in osteogenesis imperfecta with quantitative defect of type I collagen synthesis. J Pediatr. 1996;129:432-439.
Åström E, Jorulf H, Söderhäll S. Intravenous pamidronate treatment of infants with severe osteogenesis imperfecta. Arch Dis Child. 2007;92:332-338.
Barnes AM, Chang W, Morello R, et al. Deficiency of cartilage-associated protein in recessive lethal osteogenesis imperfecta. N Engl J Med. 2006;355:2757-2764.
Cabral WA, Chang W, Barnes AM, et al. Prolyl 3-hydroxylase 1 deficiency causes a recessive metabolic bone disorder resembling lethal/severe osteogenesis imperfecta. Nat Genet. 2007;39:359-365.
Glorieux FH, Rauch F, Plotkin H, et al. Type V osteogenesis imperfecta: a new form of brittle bone disease. J Bone Miner Res. 2000;15:1650-1658.
Glorieux FH, Rauch F, Ward LM, et al. Alendronate in the treatment of pediatric osteogenesis imperfecta. J Bone Miner Res. 2004;19(Suppl 1):S12.
Glorieux FH, Ward LM, Rauch F, et al. Osteogenesis imperfecta type VI: a form of brittle bone disease with a mineralization defect. J Bone Miner Res. 2002;17:30-38.
Letocha A, Cintas HL, Troendle JF, et al. Controlled trial of pamidronate in children with types III and IV osteogenesis imperfecta confirms vertebral gains but not short-term functional improvement. J Bone Miner Res. 2005;20:977-986.
Marini JC. Should children with osteogenesis imperfecta be treated with bisphosphonates? Nat Clin Prac Endocrinol Metab. 2006;2:14-15.
Marini JC, Hopkins E, Glorieux FH, et al. Positive linear growth and bone responses to growth hormone treatment in children with types III and IV osteogenesis imperfecta. J Bone Miner Res. 2003;18:237-243.
Plotkin H, Rauch F, Bishop NJ. Pamidronate treatment of severe osteogenesis imperfecta in children under 3 years of age. J Clin Endocrinol Metab. 2000;85:1846-1850.
Sakkers R, Kok D, Engelbert R, et al. Skeletal effects and functional outcome with olpadronate in children with osteogenesis imperfecta: a 2-year randomized placebo-controlled study. Lancet. 2004;363:1427-1431.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]
Chapter 692 Osteogenesis Imperfecta
