CHAPTER 146 Osseous Tumors
Non-Neoplastic Lesions
Fibrous Dysplasia
FD is an aberration in normal bone development that results from a defect in osteoblastic differentiation and maturation originating in a mesenchymal precursor.1,2 The disease is characterized by foci of abnormal fibro-osseous proliferation that can affect any area of the calvaria and occurs in three distinct clinical patterns: (1) monostotic, the most common form with single bone involvement; (2) polyostotic, with multiple bone involvement; and (3) as part of McCune-Albright syndrome, a rare variant of the polyostotic form with pigmentation and endocrinologic abnormalities.2–5 The disease commonly occurs in the first 3 decades of life, particularly in late childhood and early adolescence.
FD is considered an abnormal overgrowth of bone. Recent evidence has shown that activating mutations of G proteins in osteoblastic cells result in increased activation of adenylate cyclase, thereby leading to overproduction of cyclic adenosine monophosphate (cAMP) and culminating in increased cell proliferation and aberrant cell differentiation.6–8 Interleukin-6 may also increase intracellular cAMP, which can result in osteoclast proliferation and thus contribute to the bone lesions seen in FD.9 Microscopically, FD characteristically appears as woven bone interspersed between areas of fibrous tissue (Fig. 146-1).
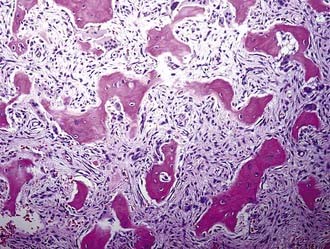
FIGURE 146-1 Histology of fibrous dysplasia (hematoxylin-eosin, ×100). Woven bone is interspersed within areas of fibrous tissue.
On skull radiographs and computed tomography (CT), the lesions have a characteristic “ground-glass” appearance. They may appear sclerotic (35% of cases), cystic (25% of cases), or as a mixture of the two (40% of cases).10 In thinner bones of the cranium (e.g., temporal, frontal, and maxillary bones), the bone undergoes rapid expansion that results in lytic, cavitary lesions (Fig. 146-2). Thicker bones of the skull (e.g., sphenoid) tend to undergo a more diffuse sclerotic reaction.2 CT is an effective imaging tool for detection of the disease, although magnetic resonance imaging (MRI) may be a useful adjunct for identifying affected neurovascular structures.
Clinically, the FD lesion progresses as a painless, nonmobile mass, and orbitocranial swelling can reach significant proportions and result in severe cosmetic deformities. Lesions involving the petrous portion of the temporal bone can give rise to conductive hearing loss via stenosis of the external auditory canal. Nasal obstruction may result from FD involvement of the frontal, ethmoid, sphenoid, or maxillary bones. Skull base involvement may result in diplopia, facial pain or numbness, and headaches. Visual disturbance occurs when the optic canal is involved.2
Treatment of FD ranges from observation to aggressive surgical intervention. Medical treatment, primarily with bisphosphonates, has been reported to be successful.11,12 When feasible, surgical resection with cosmetically acceptable reconstruction is advocated.3 In cases of optic canal involvement (particularly with visual loss), early and aggressive surgical treatment is warranted, and some authors advocate prophylactic enlargement of the optic canals.13 Radiotherapy for FD has been strongly discouraged because the incidence of secondary malignancy is high. FD has also been reported to spontaneously degenerate to more malignant subtypes such as osteosarcoma and, less commonly, to fibrosarcoma, chondrosarcoma, or malignant fibrous histiocytoma.14,15
Langerhans Cell Histiocytosis
LCH is characterized by proliferation and accumulation of histiocytes (i.e., Langerhans cells). LCH can involve any bone in the skeletal system, but the skull is most frequently affected. Three clinical syndromes are associated with LCH: (1) eosinophilic granuloma, (2) Hand-Schüller-Christian disease (with a triad that includes skull lesions, exophthalmos, and diabetes insipidus), and (3) Letterer-Siwe disease (disseminated lesions involving multiple visceral organs).16 In general, the disease is more common in children than adults.
LCH results in bony destruction with replacement of the bone marrow by Langerhans cells, eosinophils, neutrophils, and macrophages. Although its etiology is poorly understood, LCH is thought to be a disorder of the immune system.17,18 There is a neoplastic component because the cells are clonal in origin.16
LCH lesions develop in the diploic spaces of the skull. On plain radiographs they appear as punched-out lytic lesions with a well-defined margin. These lytic lesions may contain a fragment of normal bone, referred to as a sequestrum. Multiple lytic lesions are seen in Hand-Schüller-Christian disease. LCH has also been reported to occur in the skull base, including the clivus.19,20 Here, the disease may result in cranial nerve palsies and brainstem dysfunction. CT can also be used for diagnostic purposes and may be better than plain radiographs at showing bony destruction. MRI is useful for assessing any bone marrow or soft tissue involvement (Fig. 146-3).
The disease is more common in children but can strike at any age. Clinically, patients often complain of localized pain. Petrous bone involvement may result in otorrhea or hearing loss. Some LCH lesions are asymptomatic, which can lead to a delay in diagnosis. Eosinophilic granuloma is the most common form of the disease and is manifested as a monostotic lesion in the skull. The most severe form of the disease, Letterer-Siwe disease, occurs in children younger than 2 years, in whom fever, anemia, hepatosplenomegaly, lymphadenopathy, and skin lesions develop.17
The natural history of LCH is variable. Some lesions will regress spontaneously but may recur years later.21 Some isolated bone lesions may be treated by surgical curettage or direct intralesional injection of methylprednisolone.16 In most patients with cranial involvement, systemic chemotherapy may be warranted, and the agents typically used include vinblastine, etoposide (VP-16), prednisone, methotrexate, and 6-mercaptopurine. Local irradiation has also been used. Patients with solitary lesions have a good prognosis. Poor prognostic indicators include onset before 2 years of age, extensive visceral or extraosseous involvement, anemia, and thrombocytopenia.16,21,22
Aneurysmal Bone Cyst
ABCs are benign osteolytic, multicystic expansile lesions of bone that generally develop in the second or third decade of life. ABCs of the skull are rare and account for 2.5% to 6% of skull pathologies.23 The origin of ABCs is unknown, but one hypothesis suggests that an underlying arteriovenous anomaly results in the dilated vascular spaces.24–26 Secondary development of an ABC has also been described in connection with other initial pathologies.23 The histologic characteristics of ABCs include cavernous, pseudovascular channels consisting of connective tissue with giant cells and trabecular bone.4
Clinically, patients have a tender, palpable scalp mass, although pain is not always present. The treatment of choice is complete resection because subtotal resection is associated with a 50% recurrence rate.23,24,27,28 Preoperative angiographic embolization is recommended as a result of the high vascularity of these lesions.
Benign Tumors
Hemangioma
Hemangiomas are benign tumors of blood vessels. In the skull they are of the cavernous type and consist of large dilated blood vessels separated by fibrous tissue. Hemangiomas of the skull are observed in patients of all ages but are most commonly found in the fourth decade of life.29 Hemangiomas account for 0.2% of osseous tumors.30 Their cause is unknown, although they may be associated with antecedent trauma.29 These tumors develop in the diploic spaces with a vascular supply, typically from the middle meningeal artery or the external carotid artery.
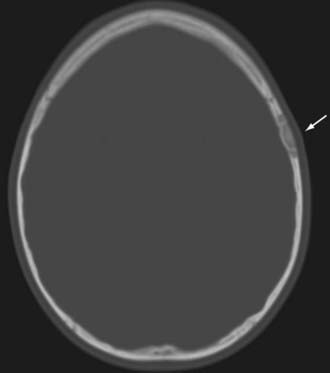
Radiographically, hemangiomas of the skull appear as solitary cystic lesions with a sclerotic rim. Their classic description includes a “honeycomb” or “sunburst” pattern as seen on plain radiographs. On CT, they appear as lytic, expansile, and “bubbly” lesions with a sclerotic rim30,31 (Fig. 146-4). On T1-weighted MRI, hemangiomas generally appear isointense or hypointense. On T2-weighted MRI, they appear hyperintense (Fig. 146-5). Hemangiomas generally show avid contrast enhancement (Fig. 146-6).
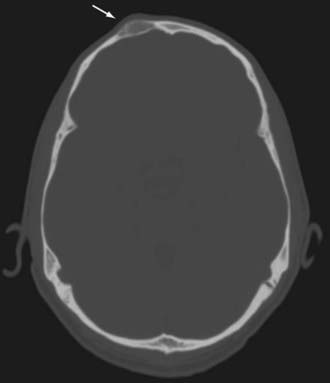
FIGURE 146-4 Axial CT scan showing a right frontal hemangioma (arrow). Note the lytic and expansile nature of the lesion.
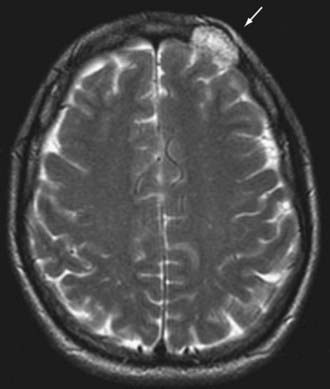
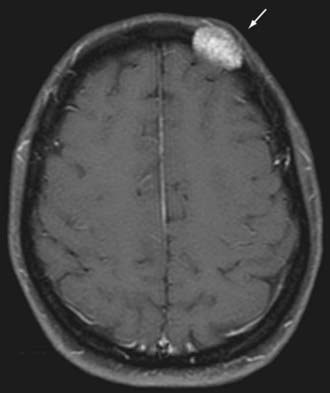
In most patients, hemangiomas are asymptomatic, slow-growing masses, but they may become symptomatic if they compress adjacent structures, such as the meninges. They may also cause isolated skull pain and be palpable masses. The treatment of choice of symptomatic hemangiomas is surgical excision.32
Osseous Meningiomas
Osseous meningiomas (also known as hyperostosing en plaque meningiomas) are primarily a disease of bone. Historically, the nomenclature regarding purely calvarial meningiomas has been confusing. They have been described as occurring in three varieties.33 Tumors that are purely extracalvarial are type I, purely calvarial tumors are type II, and calvarial tumors with extracalvarial extension are type III. Each category is further divided into convexity (C) or skull base (B) subtypes based on their anatomic location. These tumors occur in males and females, with a slight female preponderance, and typically develop in the fifth decade of life.34 Osseous meningiomas represent 1% to 2% of all meningiomas.35
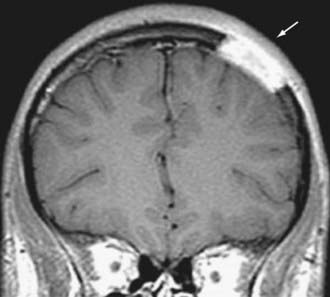
Osseous meningiomas may be osteoblastic or osteolytic, which influences their radiographic appearance. Osteoblastic meningiomas are the most common subtype, and they induce hyperostosis. On plain skull radiographs and CT, these lesions appear hyperdense with areas of calcification and atypical vascular markings.35 On CT, the lesion may have a “ground-glass” appearance similar to that seen in FD (as described earlier).36 Osteolytic meningiomas, which are much rarer, may be manifested as lytic skull lesions. The skull may appear thinned and expanded with disruption of its inner and outer tables. On MRI, both subtypes appear hypointense on T1-weighted images and hyperintense on T2-weighted images. They exhibit avid enhancement after the administration of contrast material (Fig. 146-7).
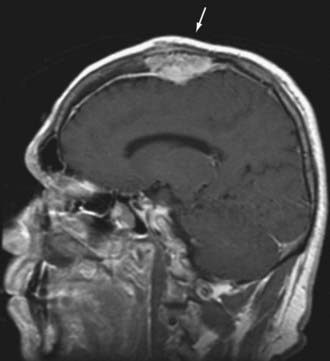
FIGURE 146-7 Sagittal contrast-enhanced magnetic resonance image showing an intraosseous meningioma (arrow).
Histopathologic evaluation of these tumors often shows classic features of meningioma, including psammoma bodies and eosinophilic tumor cells with whorls. The most common histologic type is meningotheliomatous meningioma, although atypical or malignant characteristics may be present.33
These tumors may develop at either the convexity or skull base. They are generally solitary, and symptoms depend on tumor location and the extent of tumor involvement of the calvaria. Most of these tumors are slow growing and painless, but initial symptoms such as neurological deficits, seizures, hearing loss, tinnitus, dizziness, and cranial nerve deficits have been reported.35 Common locations for the variety occurring at the convexity include the periorbital and frontoparietal regions.37 Skull base lesions may also involve the nasal cavity or sinuses. The treatment of choice is wide surgical excision, if possible. Durable reconstruction is also best performed at the time of the initial operation. In the event of subtotal resection of benign meningiomas, observation with serial imaging is an acceptable treatment strategy. However, for atypical or malignant tumors, adjuvant chemotherapy has been advocated.37
Osteoma
Osteomas are benign lesions of abnormally dense bone that is formed in the periosteum. They are most common in the skull and facial bones and usually arise from the outer table.38 Although they are classically described as very common skull lesions, symptomatic lesions are rare, thus making their true incidence difficult to ascertain.39 Three varieties of osteoma have been described: compact, cancellous, and fibrous. The latter two more commonly arise from the inner table (enostotic), and the compact form more commonly arises from the outer table (exostotic).38 They may be intraparenchymal, dural, or skull based or originate from the calvaria. Skull base osteomas are the most common type.
On plain radiographs, the lesion appears as a dense extension from the originating bone. On CT without contrast enhancement, the lesion appears as a very dense hyperostotic lesion without any soft tissue component. Osteomas may be confused with intracranial calcifications. Microscopically, these lesions appear as dense, compact trabeculae of lamellar bone (Fig. 146-8).
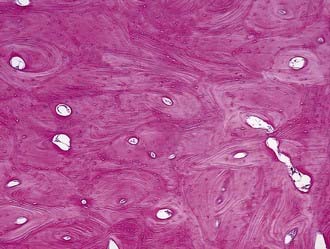
Osteomas are generally asymptomatic, with symptoms occurring as a consequence of their location. Headache is the most frequent symptom, and cosmetic deformity, particularly of the orbit, is also common.39 For symptomatic lesions, surgical removal is the treatment of choice. Care must be taken when resecting osteomas because they may be adherent to the dura.
Giant Cell Tumor
A giant cell tumor is a benign tumor of bone that originates from the nonosteogenic stromal cells in bone marrow.40 It occurs rarely in the skull and accounts for about 5% of all primary bone tumors. In the skull, the mandible and maxilla are the most commonly involved bones, although giant cell tumors have been reported at other sites.41
The most common initial symptom is headache, although tumors involving the skull base may result in cranial nerve deficits.40 Despite being benign, giant cell tumors have a strong propensity for recurrence. They are locally aggressive, and recurrent tumors have been reported to have a higher risk for malignant transformation than do primary giant cell tumors.42 For these reasons, aggressive surgical resection with wide excision and, when possible, en bloc removal of the tumor is advocated. Adjuvant radiation therapy is discouraged because sarcomatous transformation has been reported.43,44
Malignant Tumors
Chordoma
Although considered histologically benign, chordoma is a locally aggressive tumor. Chordomas arise from embryonic remnants of the primitive notochord, a rod-like cord of cells from which the skull base and vertebral column develop. In the skull, which accounts for a third of chordoma cases, these tumors occur in the vicinity of the spheno-occipital bones, particularly the clivus.45 Chordomas have been reported in every age group but are usually seen in adults in the fourth decade of life. There is a strong male preponderance (2 : 1 male-to-female ratio). These tumors are very rare, with an overall incidence of less than 1 in 100,000 individuals annually.46
Grossly, chordomas appear as gelatinous, multilobulated tumors. Two subtypes have been described: typical and chondroid. Typical chordomas are characterized by physaliphorous cells, and the tumor may contain areas of necrosis, hemorrhage, and bone trabeculae (Fig. 146-9). The chondroid variety, seen more frequently in the skull base, has a stromal feature reminiscent of hyaline cartilage with neoplastic cells. There is a resemblance to low-grade chondrosarcoma.
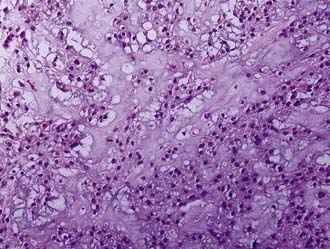
Because chordomas grow slowly, the symptoms and signs of the disease are insidious. The most common symptoms are diplopia and headache as a consequence of clival involvement, with brainstem impingement affecting the sixth cranial nerve. Other sites of involvement include the petrous bone, sellar area, and sinuses.45
Treatment consists of aggressive surgical excision plus radiation therapy for residual tumor because chemotherapeutic options are limited.47 Proton beam radiotherapy is considered the best radiotherapeutic option inasmuch as it can deliver high doses to the tumor while minimizing exposure to surrounding critical structures such as the brainstem.48 With aggressive surgical resection followed by radiation treatment, 5-year survival rates approach 60% to 70%. Recurrences are often treated surgically.
Multiple Myeloma
Multiple myeloma (MM) is a monoclonal neoplastic proliferation of plasma cells originating from bone marrow. Variants of the disease include extramedullary plasmacytoma, nonsecreting myeloma, indolent myeloma, and plasma cell leukemia. There is a predilection for MM to involve the skull, in which it has been reported to occur at multiple locations.49,50 The disease is twice as common in men as in women and usually develops in the fifth through seventh decades of life.
Histopathologically, these tumors can mimic carcinomas, lymphomas, and histiocytic tumors. The key pathologic finding is the identification of plasma cells. Both CT and MRI reveal MM as well-defined destructive lesions.51 On an unenhanced CT scan, MM appears hyperdense, with evidence of bony erosion. On T1- and T2-weighted MRI, MM may appear as a homogeneous, high-intensity mass. The key finding is higher signal intensity on T1-weighted than on T2-weighted images. Contrast enhancement is evident on both CT and MRI sequences.
Treatment of MM is surgical resection if the tumor is in an anatomically suitable location, along with postoperative radiation therapy. For cases not amenable to surgical resection, the treatment of choice is radiation therapy alone. Stereotactic radiosurgery (at a dose of 14 Gy) has been advocated for treatment as well.51
Sarcomas
Angiosarcomas are extremely rare tumors of the skull, with fewer than 20 cases reported in the literature. Most afflicted patients are male, and the most frequently reported location is the frontal bone.52,53 Unlike cutaneous angiosarcomas, skull lesions tend not to ulcerate or form nodules.54 These lesions are usually metastatic, but primary angiosarcomas have been reported.54
Osteosarcomas are more common in children and adolescents than in adults. Although reported in all areas of the skull, they are more common in the calvaria than in the skull base.55 Osteosarcomas are associated with Paget’s disease of bone, which is a focal disorder of bone metabolism and is considered a benign disorder of osteoclast function. Lesions from Paget’s disease can rarely degenerate to osteosarcoma (<1% of cases).56 In the skull, osteosarcomas are also associated with other conditions (such as FD, chronic osteomyelitis, multiple osteochondromatosis, and previous radiotherapy) in 27% to 47% of cases.55 Primary osteosarcomas of the skull are rare, with an incidence of 1.6% to 2% of all osteosarcomas, and it is more common to find metastatic lesions in the skull from osteosarcomas of the extremities.55
Chondrosarcoma is a malignant tumor of cartilage-forming tissue. Although reported at other sites in the skull, the skull base (particularly the clivus) is the most common site of origin.57 These tumors originate from primitive mesenchymal cells or from embryonal rests of the cartilaginous matrix of the skull.58 They occur at an equal frequency between the sexes and are more common in adults, in whom they generally occur between the third and sixth decades of life. These tumors have an incidence of far less than 1 in 100,000.
Imaging of skull sarcomas, whether primary or metastatic, should include both CT and MRI (Figs. 146-10 and 146-11). A CT scan with bone windows is particularly useful in assessing the extent of bony destruction, and MRI is helpful in gauging soft tissue and intracranial involvement. Sarcomas, particularly osteosarcomas, may have a pattern of both osteolytic and osteosclerotic features. Radiating striations may be evident as a result of periosteal elevation.
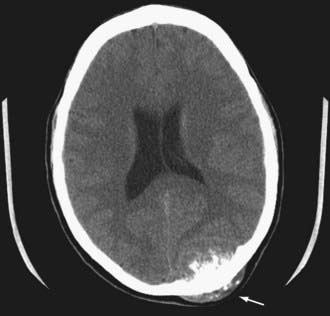
FIGURE 146-10 Axial CT scan showing an osteosarcoma metastatic to the occipital bone of the skull (arrow).
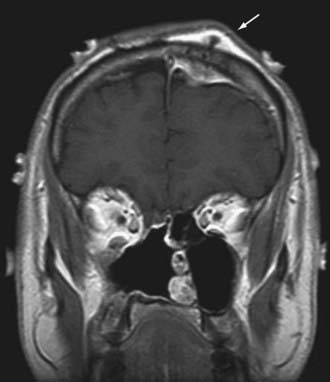
The clinical findings are dependent on location. In the calvaria, many of these tumors will be manifested as palpable masses with or without pain. Skull base lesions, such as chondrosarcoma, more frequently cause headache, visual disturbance, and cranial nerve deficits.58 Treatment varies, depending on tumor location. For calvarial metastases, surgical resection is considered the mainstay of treatment, although for certain histologies (e.g., osteosarcoma), a combination of chemotherapy and surgery is recommended.59 Skull base sarcomas require more extensive surgical strategies given their proximity to critical neurovascular structures.
Developmental Lesions
Epidermoid and Dermoid Cysts
Both epidermoid and dermoid cysts (or tumors) are congenital embryonic lesions derived from ectoderm.60,61 Unlike neoplastic lesions, epidermoid and dermoid cysts have a linear growth pattern and thus grow very slowly.62 They are among the most common skull lesions.61,63 As a consequence of their formation during embryonic development, they are often found at suture lines. Dermoids are more common in the midline, particularly near the fontanelle, and epidermoids are more common laterally, particularly in parietal locations and at the cerebellopontine angle.4,63
Although both epidermoid and dermoid tumors arise from ectoderm, epidermoid cysts generally include only epidermal elements such as keratinaceous debris, whereas dermoid cysts contain dermal elements such as skin, hair, and sebaceous glands. Grossly, epidermoids have a “pearly white” appearance, and when examined histologically, keratinous squamous epithelial cells are seen (Fig. 146-12). Dermoid cysts are characterized by adnexal structures such as sebaceous glands and hair follicles. A sinus tract may be present within either lesion.
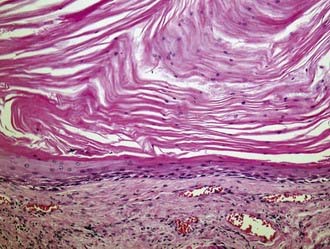
Clinically, these lesions are almost always manifested as painless palpable masses.63 On CT, epidermoid and dermoid cysts have a lytic appearance with marginal calcifications and may erode the skull (Fig. 146-13). The imaging study of choice is MRI, with epidermoid cysts appearing hypointense on T1-weighted images and hyperintense on T2-weighted images (Fig. 146-14). However, they have a very high signal on diffusion-weighted imaging, which is a unique characteristic. Dermoid cysts frequently appear hyperintense on T1-weighted images and hypointense on T2-weighted images because of their high lipid content.
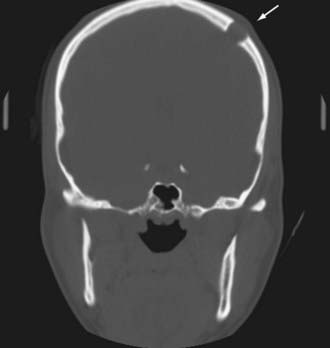
FIGURE 146-13 Coronal CT scan showing a lytic lesion in the left frontal bone (arrow) as a result of an epidermoid cyst.
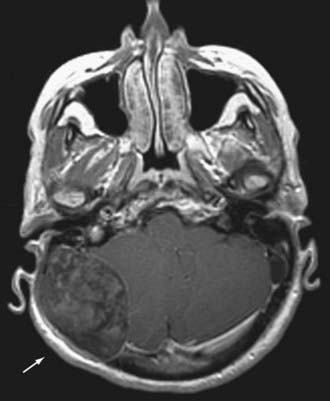
The treatment of choice of either lesion is surgical excision. Surgical removal is curative, and with gross total resection, recurrence is unlikely.61
Azouz EM, Saigal G, Rodriguez MM, et al. Langerhans’ cell histiocytosis: pathology, imaging and treatment of skeletal involvement. Pediatr Radiol. 2005;35:103-115.
Bastug D, Ortiz O, Schochet SS. Hemangiomas in the calvaria: imaging findings. AJR Am J Roentgenol. 1995;164:683-687.
Bonneville F, Savatovsky J, Chiras J. Imaging of cerebellopontine angle lesions: an update. Part 2: intra-axial lesions, skull base lesions that may invade the CPA region, and non-enhancing extra-axial lesions. Eur Radiol. 2007;17:2908-2920.
Cakirer S, Basak M, Celebi I, et al. Aneurysmal bone cyst of the temporal bone. Curr Probl Diagn Radiol. 2003;32:169-175.
Cerase A, Tarantino A, Gozzetti A, et al. Intracranial involvement in plasmacytomas and multiple myeloma: a pictorial essay. Neuroradiology. 2008;50:665-674.
Cho YH, Kim JH, Khang SK, et al. Chordomas and chondrosarcomas of the skull base: comparative analysis of clinical results in 30 patients. Neurosurg Rev. 2008;31:35-43.
Chugh R, Tawbi H, Lucas DR, et al. Chordoma: the nonsarcoma primary bone tumor. Oncologist. 2007;12:1344-1350.
Elder JB, Atkinson R, Zee CS, et al. Primary intraosseous meningioma. Neurosurg Focus. 2007;23(4):E13.
Gibson SE, Prayson RA. Primary skull lesions in the pediatric population: a 25-year experience. Arch Pathol Lab Med. 2007;131:761-766.
Haddad FS, Haddad GF, Zaatari G. Cranial osteomas: their classification and management. Report on a giant osteoma and review of the literature. Surg Neurol. 1997;48:143-147.
Hansen MF, Seton M, Merchant A. Osteosarcoma in Paget’s disease of bone. J Bone Miner Res. 2006;21(suppl 2):P58-63.
Lang FF, Macdonald OK, Fuller GN, et al. Primary extradural meningiomas: a report on nine cases and review of the literature from the era of computerized tomography scanning. J Neurosurg. 2000;93:940-950.
Mascarenhas L, Peteiro A, Ribeiro CA, et al. Skull osteosarcoma: illustrated review. Acta Neurochir (Wien). 2004;146:1235-1239.
Ricalde P, Horswell BB. Craniofacial fibrous dysplasia of the fronto-orbital region: a case series and literature review. J Oral Maxillofac Surg. 2001;59:157-167.
Shah MV, Haines SJ. Pediatric skull, skull base, and meningeal tumors. Neurosurg Clin N Am. 1992;3:893-924.
Singh AD, Chacko AG, Chacko G, et al. Plasma cell tumors of the skull. Surg Neurol. 2005;64:434-438.
Tokgoz N, Oner YA, Kaymaz M, et al. Primary intraosseous meningioma: CT and MRI appearance. AJNR Am J Neuroradiol. 2005;26:2053-2056.
Wein RO, Popat SR, Doerr TD, et al. Plasma cell tumors of the skull base: four case reports and literature review. Skull Base. 2002;12:77-86.
Willatt JM, Quaghebeur G. Calvarial masses of infants and children. A radiological approach. Clin Radiol. 2004;59:474-486.
Yoon SH, Park SH. A study of 77 cases of surgically excised scalp and skull masses in pediatric patients. Childs Nerv Syst. 2008;24:459-465.
1 Papadakis CE, Skoulakis CE, Prokopakis EP, et al. Fibrous dysplasia of the temporal bone: report of a case and a review of its characteristics. Ear Nose Throat J. 2000;79:52-57.
2 Ricalde P, Horswell BB. Craniofacial fibrous dysplasia of the fronto-orbital region: a case series and literature review. J Oral Maxillofac Surg. 2001;59:157-167.
3 Ozek C, Gundogan H, Bilkay U, et al. Craniomaxillofacial fibrous dysplasia. J Craniofac Surg. 2002;13:382-389.
4 Burger PC, Scheithauer BW, Vogel FS. Surgical Pathology of the Nervous System and Its Coverings, ed 4. New York: Churchill Livingstone; 2002.
5 Iseri PK, Efendi H, Demirci A, et al. Fibrous dysplasia of the cranial bones: a case report and review of the literature. Yale J Biol Med. 2005;78:141-145.
6 Riminucci M, Fisher LW, Shenker A, et al. Fibrous dysplasia of bone in the McCune-Albright syndrome: abnormalities in bone formation. Am J Pathol. 1997;151:1587-1600.
7 Marie PJ, de Pollak C, Chanson P, et al. Increased proliferation of osteoblastic cells expressing the activating Gs alpha mutation in monostotic and polyostotic fibrous dysplasia. Am J Pathol. 1997;150:1059-1069.
8 Shenker A, Chanson P, Weinstein LS, et al. Osteoblastic cells derived from isolated lesions of fibrous dysplasia contain activating somatic mutations of the Gs alpha gene. Hum Mol Genet. 1995;4:1675-1676.
9 Yamamoto T, Ozono K, Kasayama S, et al. Increased IL-6-production by cells isolated from the fibrous bone dysplasia tissues in patients with McCune-Albright syndrome. J Clin Invest. 1996;98:30-35.
10 Mendelsohn DB, Hertzanu Y, Cohen M, et al. Computed tomography of craniofacial fibrous dysplasia. J Comput Assist Tomogr. 1984;8:1062-1065.
11 DiMeglio LA. Bisphosphonate therapy for fibrous dysplasia. Pediatr Endocrinol Rev. 2007;4(suppl 4):440-445.
12 Makitie AA, Tornwall J, Makitie O. Bisphosphonate treatment in craniofacial fibrous dysplasia—a case report and review of the literature. Clin Rheumatol. 2008;27:809-812.
13 Chen YR, Noordhoff MS. Treatment of craniomaxillofacial fibrous dysplasia: how early and how extensive? Plast Reconstr Surg. 1990;86:835-842.
14 Ruggieri P, Sim FH, Bond JR, et al. Malignancies in fibrous dysplasia. Cancer. 1994;73:1411-1424.
15 Ruggieri P, Sim FH, Bond JR, et al. Osteosarcoma in a patient with polyostotic fibrous dysplasia and Albright’s syndrome. Orthopedics. 1995;18:71-75.
16 Azouz EM, Saigal G, Rodriguez MM, et al. Langerhans’ cell histiocytosis: pathology, imaging and treatment of skeletal involvement. Pediatr Radiol. 2005;35:103-115.
17 Meyer JS, Harty MP, Mahboubi S, et al. Langerhans cell histiocytosis: presentation and evolution of radiologic findings with clinical correlation. Radiographics. 1995;15:1135-1146.
18 Stull MA, Kransdorf MJ, Devaney KO. Langerhans cell histiocytosis of bone. Radiographics. 1992;12:801-823.
19 Hurley ME, O’Meara A, Fogarty E, et al. Langerhans’ cell histiocytosis of the clivus: case report and literature review. Pediatr Radiol. 2004;34:267-270.
20 Krishna H, Behari S, Pal L, et al. Solitary Langerhans-cell histiocytosis of the clivus and sphenoid sinus with parasellar and petrous extensions: case report and a review of literature. Surg Neurol. 2004;62:447-454.
21 Rao G, Kestle J. Calvarial tumors in children. In: Tonn JC, Westphal M, Rutka JT, et al, editors. Neuro-Oncology of CNS Tumors. Berlin: Springer; 2006:567-573.
22 Shah MV, Haines SJ. Pediatric skull, skull base, and meningeal tumors. Neurosurg Clin N Am. 1992;3:893-924.
23 Cakirer S, Basak M, Celebi I, et al. Aneurysmal bone cyst of the temporal bone. Curr Probl Diagn Radiol. 2003;32:169-175.
24 Chidambaram B, Santosh V, Balasubramaniam V. Aneurysmal bone cyst of the temporal bone. Childs Nerv Syst. 2001;17:411-414.
25 Schonauer C, Tessitore E, Schonauer M. Aneurysmal bone cyst of the frontal bone in a soccer player. Acta Neurochir (Wien). 2000;142:1165-1166.
26 Shah GV, Doctor MR, Shah PS. Aneurysmal bone cyst of the temporal bone: MR findings. AJNR Am J Neuroradiol. 1995;16:763-766.
27 Lippman CR, Jallo GI, Feghali JG, et al. Aneurysmal bone cyst of the temporal bone. Pediatr Neurosurg. 1999;31:219-223.
28 O’Brien DP, Rashad EM, Toland JA, et al. Aneurysmal cyst of the frontal bone: case report and review of the literature. Br J Neurosurg. 1994;8:105-108.
29 Dorner L, Buhl R, Hugo HH, et al. Unusual locations for cavernous hemangiomas: report of two cases and review of the literature. Acta Neurochir (Wien). 2005;147:1091-1096.
30 Nguyen BD, McNaughton D. AJR teaching file: nuclear imaging of a tender skull mass. AJR Am J Roentgenol. 2007;189:S61-S63.
31 Bastug D, Ortiz O, Schochet SS. Hemangiomas in the calvaria: imaging findings. AJR Am J Roentgenol. 1995;164:683-687.
32 Khanam H, Lipper MH, Wolff CL, et al. Calvarial hemangiomas: report of two cases and review of the literature. Surg Neurol. 2001;55:63-67.
33 Lang FF, Macdonald OK, Fuller GN, et al. Primary extradural meningiomas: a report on nine cases and review of the literature from the era of computerized tomography scanning. J Neurosurg. 2000;93:940-950.
34 Tokgoz N, Oner YA, Kaymaz M, et al. Primary intraosseous meningioma: CT and MRI appearance. AJNR Am J Neuroradiol. 2005;26:2053-2056.
35 Elder JB, Atkinson R, Zee CS, et al. Primary intraosseous meningioma. Neurosurg Focus. 2007;23(4):E13.
36 Daffner RH, Yakulis R, Maroon JC. Intraosseous meningioma. Skeletal Radiol. 1998;27:108-111.
37 Crawford TS, Kleinschmidt-DeMasters BK, Lillehei KO. Primary intraosseous meningioma. Case report. J Neurosurg. 1995;83:912-915.
38 Willatt JM, Quaghebeur G. Calvarial masses of infants and children. A radiological approach. Clin Radiol. 2004;59:474-486.
39 Haddad FS, Haddad GF, Zaatari G. Cranial osteomas: their classification and management. Report on a giant osteoma and review of the literature. Surg Neurol. 1997;48:143-147.
40 Wang Y, Honda K, Suzuki S, et al. Giant cell tumor at the lateral skull base. Am J Otolaryngol. 2006;27:64-67.
41 Sethi A, Passey JC, Mrig S, et al. Giant cell tumour (osteoclastoma) of the zygoma: an extremely unusual neoplasm. Acta Otolaryngol. 2006;126:327-329.
42 Goldenberg RR, Campbell CJ, Bonfiglio M. Giant-cell tumor of bone. An analysis of two hundred and eighteen cases. J Bone Joint Surg Am. 1970;52:619-664.
43 Caudell JJ, Ballo MT, Zagars GK, et al. Radiotherapy in the management of giant cell tumor of bone. Int J Radiat Oncol Biol Phys. 2003;57:158-165.
44 Tucker MA, D’Angio GJ, Boice, et al. Bone sarcomas linked to radiotherapy and chemotherapy in children. N Engl J Med. 1987;317:588-593.
45 Erdem E, Angtuaco EC, Van Hemert R, et al. Comprehensive review of intracranial chordoma. Radiographics. 2003;23:995-1009.
46 Chugh R, Tawbi H, Lucas DR, et al. Chordoma: the nonsarcoma primary bone tumor. Oncologist. 2007;12:1344-1350.
47 Muro K, Das S, Raizer JJ. Chordomas of the craniospinal axis: multimodality surgical, radiation and medical management strategies. Expert Rev Neurother. 2007;7:1295-1312.
48 Igaki H, Tokuuye K, Okumura T, et al. Clinical results of proton beam therapy for skull base chordoma. Int J Radiat Oncol Biol Phys. 2004;60:1120-1126.
49 Singh AD, Chacko AG, Chacko G, et al. Plasma cell tumors of the skull. Surg Neurol. 2005;64:434-438.
50 Wein RO, Popat SR, Doerr TD, et al. Plasma cell tumors of the skull base: four case reports and literature review. Skull Base. 2002;12:77-86.
51 Cerase A, Tarantino A, Gozzetti A, et al. Intracranial involvement in plasmacytomas and multiple myeloma: a pictorial essay. Neuroradiology. 2008;50:665-674.
52 Lopes M, Duffau H, Fleuridas G. Primary spheno-orbital angiosarcoma: case report and review of the literature. Neurosurgery. 1999;44:405-407.
53 Shuangshoti S, Chayapum P, Suwanwela N, et al. Unilateral proptosis as a clinical presentation in primary angiosarcoma of skull. Br J Ophthalmol. 1988;72:713-719.
54 Chou YC, Chang YL, Harnod T, et al. Primary angiosarcoma of the cranial vault: a case report and review of the literature. Surg Neurol. 2004;61:575-579.
55 Mascarenhas L, Peteiro A, Ribeiro CA, et al. Skull osteosarcoma: illustrated review. Acta Neurochir (Wien). 2004;146:1235-1239.
56 Hansen MF, Seton M, Merchant A. Osteosarcoma in Paget’s disease of bone. J Bone Miner Res. 2006;21(suppl 2):P58-63.
57 Yagisawa M, Ishitoya J, Tsukuda M, et al. Chondrosarcoma of the temporal bone. Auris Nasus Larynx. 2007;34:527-531.
58 Cho YH, Kim JH, Khang SK, et al. Chordomas and chondrosarcomas of the skull base: comparative analysis of clinical results in 30 patients. Neurosurg Rev. 2008;31:35-43.
59 Smeele LE, Kostense PJ, van der Waal I, et al. Effect of chemotherapy on survival of craniofacial osteosarcoma: a systematic review of 201 patients. J Clin Oncol. 1997;15:363-367.
60 Bonneville F, Savatovsky J, Chiras J. Imaging of cerebellopontine angle lesions: an update. Part 2: intra-axial lesions, skull base lesions that may invade the CPA region, and non-enhancing extra-axial lesions. Eur Radiol. 2007;17:2908-2920.
61 Yoon SH, Park SH. A study of 77 cases of surgically excised scalp and skull masses in pediatric patients. Childs Nerv Syst. 2008;24:459-465.
62 Sirin S, Gonul E, Kahraman S, et al. Imaging of posterior fossa epidermoid tumors. Clin Neurol Neurosurg. 2005;107:461-467.
63 Gibson SE, Prayson RA. Primary skull lesions in the pediatric population: a 25-year experience. Arch Pathol Lab Med. 2007;131:761-766.






