CHAPTER 28 ORTHOSTATIC HYPOTENSION
Maintenance of upright posture is made possible by rapid cardiovascular adaptation that ensures blood flow to vital organs, including the brain, and depends primarily on an intact autonomic nervous system. When this system fails, as occurs in neurological disorders affecting autonomic neurons, orthostatic hypotension results. Severely affected patients are completely disabled, unable to stand for more than few seconds before syncope ensues.
PATHOPHYSIOLOGY
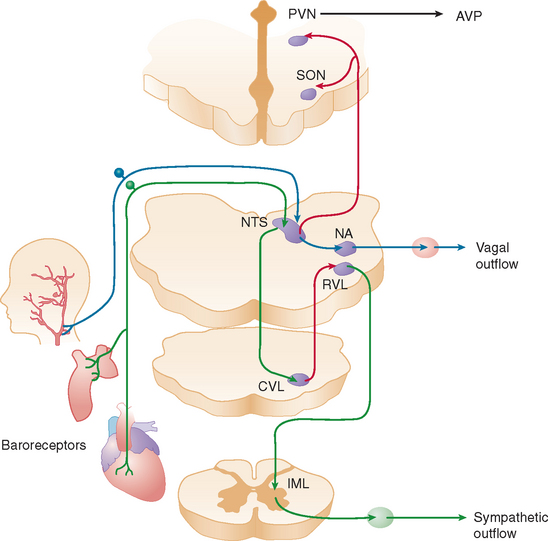
When a normal individual stands, about 700 mL of blood pools in the legs and lower abdominal veins. Venous return decreases, resulting in a transient decline in cardiac output. The reduction in central blood volume and arterial pressure is sensed by pressure-sensitive cardiopulmonary volume receptors and arterial baroreceptors. This leads to baroreflex-mediated sympathetic activation with increases in stroke volume and peripheral vasoconstriction and parasympathetic withdrawal with increase in heart rate (Fig. 28-1). These rapid hemodynamic changes prevent blood pressure from falling; their failure causes orthostatic hypotension.

When arterial blood pressure falls below a critical level, cerebral blood flow also decreases. When systolic blood pressure is around 50 mm Hg at brain level (which corresponds to a systolic blood pressure of approximately 70 mm Hg at cardiac level when a person is standing) (Fig. 28-2), the autoregulatory capacity of cerebral blood flow reaches maximum vasodilation and is unable to compensate for a further fall in blood pressure.
In addition to these almost instantaneous changes in vascular tone and heart rate directly mediated by autonomic innervation, other longer term mechanisms that contribute to the maintenance of upright blood pressure are also influenced by the baroreflex. These mechanisms are impaired in patients with autonomic failure and contribute to their orthostatic hypotension. Increased sympathetic renal nerve activity induces tubular sodium reabsorption directly1 and by stimulating the secretion of renin from the juxtaglomerular apparatus.2 Renin and converting enzyme convert circulating angiotensinogen into angiotensin II, which is a vasoconstrictor and secretagogue of aldosterone from the adrenal medulla. Aldosterone retains sodium in the kidney, increasing extracellular fluid volume. Patients with autonomic failure have low renin and low aldosterone levels. In addition, unloading of thoracic baroreceptors releases the antidiuretic hormone from the neurohypophysis into the systemic circulation.3,4 Acting on specific receptors in vascular smooth muscle cells,5 vasopressin produces vasoconstriction, and in the kidney it causes water retention and expands extracellular fluid volume.6 Baroreflex-mediated vasopressin release is blunted in some patients with autonomic failure.
Two other circulating vasoactive peptides, atrial natriuretic factor (ANF) and endothelin, are involved in the regulation of blood pressure and extracellular fluid volume, and their secretion may also be controlled, at least in part, by autonomic reflexes. ANF is secreted from atrial myocites7 when atrial pressure increases. ANF produces natriuresis, relaxation of vascular smooth muscle, and inhibition of renin and aldosterone secretions.8–10 When right atrial pressure decreases, as during the upright posture, circulating levels of ANF quickly fall, contributing to vasoconstriction and expansion of extracellular fluid volume. Whether ANF is released by the direct effect of pressure on the cardiocytes or by a centrally mediated autonomic reflex is still unclear. Kaufmann and colleagues found that the response of circulating ANF to changes in atrial pressure is preserved in patients with baroreflex impairment, which suggests that a local intracardiac reflex regulates the secretion of the peptide.11 Endothelin, a powerful vasoconstrictor synthesized by endothelial cells,12 has an important role in the local control of the circulation.13 In addition, however, endothelin is synthesized by neurons in the paraventricular and supraoptic nuclei of the hypothalamus14 and is co-released with vasopressin from the neurohypophysis into the bloodstream when thoracic baroreceptors are unloaded.15 The physiological function of the endothelin released into plasma during baroreflex activation remains to be defined, but it is likely that circulating endothelin contributes to the vasoconstriction that maintains blood pressure in the upright posture.15
DIAGNOSIS AND EVALUATION OF ORTHOSTATIC HYPOTENSION
Orthostatic hypotension produces a characteristic clinical history. Symptoms of lightheadedness and tunnel vision occur on standing, never while the person is lying down, and are always relieved immediately on sitting or lying down, because cerebral blood flow is passively restored. Failure to meet these criteria suggests other causes of impaired consciousness (e.g., hypoglycemia, seizures, cardiac arrhythmias).16 In patients with diabetes or other peripheral neuropathies, proprioceptive abnormalities lead to feeling of unsteadiness that patients refer as dizziness, which may wrongly suggest orthostatic hypotension. In addition to the classic lightheadedness with blurred vision and syncope, symptoms of chronic orthostatic hypotension may include vague generalized weakness, fatigue, cognitive impairment, and pain in the shoulders and back of the neck (coat hanger pain).
Many patients with chronic autonomic failure can tolerate very low orthostatic blood pressures with few symptoms of cerebral hypoperfusion, perhaps because their cerebral autoregulatory capacity is well preserved.17 However, physical exercise, prolonged standing, the postprandial state, or mild volume depletion exacerbates orthostatic hypotension and invariably triggers symptoms of cerebral hypoperfusion, including presyncope or syncope.
The diagnosis of orthostatic hypotension relies on simple measurements of blood pressure and heart rate after the patient has lain down for 5 to 10 minutes and after 1 to 3 minutes of standing. Orthostatic hypotension is arbitrarily defined as a decrease in systolic blood pressure of at least 20 mm Hg and in diastolic blood pressure of at least 10 mm Hg within 3 minutes of standing, in association with symptoms of cerebral hypoperfusion.18 On occasion, the diagnosis of orthostatic hypotension may require repeated measurements of blood pressure throughout the day. In patients with autonomic impairment, the severity of orthostatic hypotension is worse early in the morning or about 30 minutes after a meal compared to the rest of the day.
Concomitant measurements of heart rate are crucial for adequate interpretation of results. Side effects of medications are arguably the most common causes of orthostatic hypotension. The most common culprits are tricyclic antidepressants, diuretics, nitrates, and α blockers used to treat benign prostatic hypertrophy. In these cases, there is usually a compensatory increase in heart rate in association with orthostatic hypotension. In contrast, the presence of symptomatic orthostatic hypotension without an adequate compensatory increase in heart rate is sufficient clinical indication of autonomic failure (Fig. 28-3). The diagnosis can be easily confirmed with simple measurements of heart rate and blood pressure (Table 28-1). No single test completely differentiates patients with autonomic failure from age-matched control subjects, but together these tests provide a reliable indicator of the presence and severity of cardiovascular autonomic impairment.
TABLE 28-1 Assessment of Autonomic Function: Bedside Physiological Tests
R-R, R wave to R wave; R-Rexp, R-R interval during expiration; R-Rinsp, R-R interval during inspiration.
CAUSES OF AUTONOMIC FAILURE
Primary forms of autonomic failure are rarely the result of congenital deficiency of the enzyme dopamine β-hydroxylase. Affected patients cannot synthesize norepinephrine in sympathetic nerves.19 Most cases of primary autonomic failure are caused by degeneration of autonomic neurons.20 Autonomic failure is a prominent feature of two of the most prevalent neurodegenerative diseases: the Lewy body syndromes and multiple-system atrophy (MSA). The Lewy body syndromes, so named because of characteristic cytoplasmic eosinophilic neuronal inclusions (Lewy body, Lewy neuritis) found in the brain and peripheral autonomic nerves of affected patients, manifest with three different but overlapping phenotypes: pure autonomic failure, with neuronal degeneration restricted mostly to peripheral autonomic neurons and autonomic failure as the sole clinical finding; Parkinson’s disease, with prominent degeneration of the substantia nigra and other brainstem nuclei, in addition to peripheral autonomic neurons, and clinical findings dominated by motor abnormalities with varying degrees of autonomic failure; and dementia with Lewy bodies, a disorder with extensive cortical involvement, in addition to degeneration of brainstem nuclei and peripheral autonomic neurons and, clinically, severe cognitive impairment associated with parkinsonism and autonomic dysfunction.
The second type of neurodegeneration with prominent autonomic failure, MSA, affects neurons in basal ganglia, cortex, and spinal cord, sparing peripheral autonomic neurons, with characteristic glial cytoplasmic inclusions but no Lewy bodies. MSA has two phenotypes: parkinsonian and cerebellar, named MSAp and MSAc according to the predominant motor abnormality. Severe autonomic failure is prominent in both phenotypes.21
The differential diagnosis between primary neurodegenerative autonomic disorders can be difficult. There are three common diagnostic challenges. First, it is often very difficult to distinguish, early on in the disease process, between pure autonomic failure and MSA because patients with MSA can present initially with isolated autonomic failure. It may be impossible to rule out MSA and make a definitive diagnosis of pure autonomic failure during the first 1 to 2 years of disease onset. Follow-up may be required before a definitive diagnosis can be made. MSA has a more rapid progression, with disability resulting from worsening of the movement disorder, which commonly occurs 6 to 8 years after onset of symptoms. Another differential diagnosis challenge is that between MSA and Parkinson’s disease. Patients with otherwise classical Parkinson’s disease can have severe orthostatic hypotension. Patients with MSA rarely have the onset of unilateral resting tremor typical of Parkinson’s disease and, in general, respond poorly to levodopa. Third, studies suggest that autoimmune autonomic neuropathy can resemble pure autonomic failure clinically but with more prominent gastrointestinal abnormalities.
Autonomic failure with an acute or subacute (less than 3 months) clinical onset is suggestive of an autoimmune autonomic neuropathy. In most cases, the precise cause is not known, but in some patients, ganglionic nicotinic acetylcholine receptor (nAChR) autoantibodies are found. Clinical autonomic impairment is more pronounced in patients with high antibody titers, which indicates a pathogenic role for the nAChR antibodies.22 In addition to orthostatic hypotension, patients have dry eyes and dry mouth, severe upper gastrointestinal motility impairment, large pupils that react poorly to light and accommodation, and neurogenic bladder.23
Autoimmune autonomic neuropathy with a subacute onset may also occur as a paraneoplastic syndrome.24 This is commonly associated with anti-Hu antibodies (also known as type 1 antineuronal nuclear antibody), most often in patients with small cell lung cancer but also in those with other malignancies.25 Other antibodies associated with paraneoplastic autonomic neuropathy include Purkinje cell cytoplasmic antibodies type 2 and antibodies to the neuron cytoplasmic protein collapsin response-mediator protein-5. The clinical onset is generally subacute; acute or chronic manifestations are unusual. As in autoimmune autonomic neuropathy, prominent clinical findings are orthostatic hypotension, bowel hypomotility, bladder dysfunction, pupillomotor and sudomotor dysfunction, and xerophthalmia.
MANAGEMENT
In general, a stepwise approach to treatment according to the severity of the symptoms is preferable.26 Table 28-2 shows general management guidelines, but treatment should be individualized. Some recommendations may actually be contraindicated in a given patient. The initial approach is always to remove any of the conditions enumerated in Table 28-2 that may induce or aggravate orthostatic hypotension. The suggestions for management described as follows are relevant to patients with orthostatic hypotension caused by autonomic impairment.
TABLE 28-2 Stepwise Approach to Management of Orthostatic Hypotension
Nonpharmacological Therapy
In patients with persistent symptoms, conservative nonpharmacological therapy is indicated. Patients with autonomic failure are unable to conserve sodium, and liberalization of sodium intake is generally recommended. These patients have exaggerated nocturnal diuresis with relative hypovolemia and worsening of orthostatic hypotension early in the morning. Elevating the head of the bed with 6- to 9-inch blocks can reduce nocturnal diuresis by reducing nocturnal hypertension. This simple maneuver may ameliorate orthostatic hypotension the following morning. During the day, wearing waist-high custom-fitted elastic support stockings exerts pressure on the legs and reduces venous pooling. It is important to avoid wearing support stockings in the supine position because they may contribute to diuresis and supine hypertension. Support stockings, however, are difficult to wear, and compliance is low. Most of the pooling of blood, however, occurs in the abdomen.27 Therefore, patients can try using an abdominal binder as an alternative to support stockings (Table 28-3).28
TABLE 28-3 Pharmacological Agents for the Treatment of Orthostatic Hypotension
| Agent | Dosage | Side Effects |
|---|---|---|
| Sodium chloride | 1g/meal | Nausea, diarrhea |
| Fludrocortisone (Florinef) | 0.1-0.3 mg/day | Hypokalemia, congestive heart failure, supine hypertension |
| Midodrine (ProAmatine) | 5-10 mg* | Scalp itching, goose bumps |
| Yohimbine (Yocon) | 5.4 mg* | Nervousness, tremor |
| Indomethacin (Indocin) | 25 mg | Gastrointestinal discomfort, peptic ulcer |
| L-Dihydroxyphenyl serine (DOPS)† | Supine hypertension |
Please refer to more detailed sources for complete list of side effects and contraindications.
* A dose of these short-acting pressor agents, given before exertion, improves orthostatic symptoms for 2-3 hours. In general, administration of >3 doses/day is discouraged, to avoid side effects and development of tolerance.
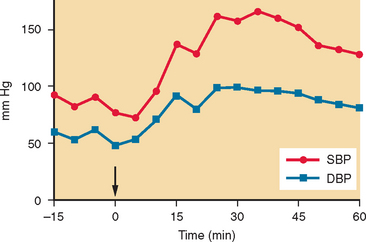
It has been discovered that rapid intake of 500 mL of water results in an acute (within 30 minutes) and transient (lasting about 60 minutes) increase in blood pressure in patients with autonomic failure.29 This pressor effect can be substantial (Fig. 28-4) and can be used to alleviate orthostatic hypotension. It is commonly recommended that patients drink 500 mL of water about 15 minutes before getting out of bed in the morning, when symptoms are at their worst.
Pharmacological Therapy
In addition to nonpharmacological therapy, severely affected patients usually require the use of drugs. The goal of treatment is to minimize symptoms rather than to normalize an upright blood pressure. Therapy is often initiated with fludrocortisone acetate at a low dose (0.1 mg/day) and increased slowly up to 0.3 mg/day if needed.30 As an indication that volume expansion has occurred, a weight gain of 1 to 2 kg and mild ankle edema may be desirable in these patients. However, hypokalemia, supine hypertension, and pulmonary edema may occur, and patients must be monitored carefully. Fludrocortisone is not effective unless it is given in conjunction with increased salt intake (e.g., sodium chloride tablets, 1 g with meals), because its pressor effect is dependent on its ability to enhance renal sodium retention. A common mistake is to increase the dose of fludrocortisone without ensuring that patients have adequate salt supplementation. Fludrocortisone worsens supine hypertension, and its long-term safety in patients suffering from supine hypertension is not known. Patients with autonomic failure who receive fludrocortisone have target organ damage in the form of left ventricular hypertrophy, similar to that in patients with chronic arterial hypertension.31
Most patients with severe autonomic impairment also require short-acting pressor agents also cite Jordan et al., (1998).32 The goal in prescribing these drugs is to provide patients with periods when they can remain upright, rather than to try to keep severely afflicted patients symptom free throughout the day. Most of the agents listed in Table 28-4, if effective in a given patient, increase blood pressure for 2 to 3 hours. In general, these agents are best given before periods of exertion as needed, rather than at fixed (e.g., three-times-a-day) intervals. This approach may reduce the likelihood of side effects and the development of tolerance that reduces their long-term efficacy.33 Patients should also avoid lying down for 4 to 5 hours after taking these drugs, to prevent supine hypertension. These drugs have negligible effects in healthy subjects; the increase in blood pressure seen in patients with autonomic failure is a reflection of their extreme hypersensitivity to most pressor and depressor agents.34 For this reason, treatment should be started at very small doses and should be individualized. This is best done by measuring blood pressure at intervals for 2 to 3 hours after administration of the first dose of each drug. L-Threo-dihydroxyphenyl serine (the biologically active stereoisomer of the amino acid 3,4-dihydroxyphenyl serine) is a precursor of norepinephrine that has shown promise in the treatment of orthostatic hypotension in small clinical trials.35
Treatment of Related Conditions
Autonomic failure can be associated with low-production anemia and inappropriately low serum erythropoietin levels. If other causes of anemia are ruled out, patients can be treated with recombinant erythropoietin (25 to 50 U/kg subcutaneously three times per week). Erythropoietin has been shown to improve upright blood pressure,36,37 and its use may be warranted for this reason alone, rather than as a treatment for anemia.
Many patients may also have supine hypertension resulting from preexisting essential hypertension or as part of autonomic failure.38 In occasional patients, significant hypertension may be present even in the seated position. During the day, supine hypertension is best managed by simply avoiding the supine position. At night, it is necessary for many patients to take vasodilators at bedtime, after which they should be advised against getting up during the night without assistance. Hydralazine hydrochloride (25 to 100 mg) and low doses of nitrates as transdermal preparations (e.g., Nitro-Dur, 0.1 mg/hour, applied at bedtime and removed on arising) or short-acting calcium channel blockers (e.g., nifedipine, 10 mg) are often useful. A stepwise approach to the management of supine hypertension in the setting of orthostatic hypotension is included in Table 28-3 and discussed in detail elsewhere.39
Freeman R. Autonomic peripheral neuropathy. Lancet. 2005;365:1259-1270.
Jordan J, Shannon JR, Biaggioni I, et al. Contrasting actions of pressor agents in severe autonomic failure. Am J Med. 1998;105:116-124.
Jordan J, Shannon JR, Grogan E, et al. A potent pressor response elicited by drinking water. Lancet. 1999;353:723.
Shibao C, Gamboa A, Diedrich A, et al. Management of hypertension in the setting of autonomic failure: a pathophysiological approach. Hypertension. 2005;45:469-476.
Wright RA, Kaufmann H, Perera R, et al. A double-blind, doseresponse study of midodrine in neurogenic orthostatic hypotension. Neurology. 1998;51:120-124.
1 Zambrasky E, DiBona G, Kaloyanides G. Specificity of neural effect on renal tubular sodium reabsorption. Proc Soc Exp Biol Med. 1976;151:543-546.
2 Ganong W, Reid I. Role of the sympathetic nervous system and central alpha and beta adrenergic receptors in regulation of renin secretion. In: Onesti G, editor. Regulation of Blood Pressure by Central Nervous System. New York: Grune & Stratton; 1976:261-273.
3 Segar W, Moore W. The regulation of antidiuretic hormone release in man. Effects of change in position and ambient temperature on blood ADH levels. J Clin Invest. 1968;47:2143-2151.
4 Leimbach WNJr, Schmid PG, Mark AL. Baroreflex control of plasma arginine vasopressin in humans. Am J Physiol. 1984 Oct;247(4 Pt 2):H638-H644.
5 Altura BM, Altura BT. Vascular smooth muscle and neurohypophyseal hormones. Fed Proc. 1977;36:1853-1860.
6 Jard S. Vasopressin receptors. In: Czernichow P, Robinson A, editors. Diabetes Insipidus in Man. Basel: S. Karger; 1985:89-104.
7 de Bold AJ, Borenstein HB, Veress AT, et al. A rapid and potent natriuretic response to intravenous injections of atrial myocardial extract in rats. Life Sci. 1981;28:89-94.
8 Ledsome J, Wilson N, Courneya CA, et al. Release of atrial natriuretic peptide by atrial distension. Can J Physiol Pharmacol. 1985;63:739-742.
9 Garcia J, Thibault G, Cantin M, et al. Effect of a purified atrial natriuretic factor on rat and rabbit vascular strips and vascular beds. Am J Physiol. 1984;247:R34-R39.
10 Atarashi K, Mulrow PJ, Franco-Saenz R, et al. Inhibition of aldosterone production by an atrial extract. Science. 1984;224:992-994.
11 Kaufmann H, Oribe E, Pierotti AR, et al. Atrial natriuretic factor in human autonomic failure. Neurology. 1990;40:1115-1119.
12 Yanagisawa M, Kurihara H, Kimura S. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature. 1988;332:411-415.
13 Vane JR, Anggard EE, Botting RM. Regulatory functions of the vascular endothelium. N Engl J Med. 1990;323:27-36.
14 Yoshizawa T, Shinmi O, Giaid A, et al. Endothelin: a novel peptide in the posterior pituitary system. Science. 1990;247:462-464.
15 Kaufmann H, Oribe E, Oliver JA. Plasma endothelin during upright tilt: relevance for orthostatic hypotension? Lancet. 1991;338:1542-1545.
16 Kaufmann H. Syncope. A neurologist’s viewpoint. Cardiol Clin. 1997;15:177-194.
17 Horowitz DR, Kaufmann H. Autoregulatory cerebral vasodilation occurs during orthostatic hypotension in patients with primary autonomic failure. Clin Auton Res. 2001;11:363-367.
18 Kaufmann H. Consensus statement on the definition of orthostatic hypotension, pure autonomic failure and multiple system atrophy. Clin Auton Res. 1996;6(2):125-126.
19 Robertson D, Haile V, Perry SE, et al. Dopamine betahydroxylase deficiency. A genetic disorder of cardiovascular regulation. Hypertension. 1991;18:1-8.
20 Kaufmann H, Biaggioni I. Autonomic failure in neurodegenerative disorders. Semin Neurol. 2003;23:351-363.
21 Gilman S, Low P, Quinn N, et al. Consensus statement on the diagnosis of multiple system atrophy. American Autonomic Society and American Academy of Neurology. Clin Auton Res. 1998;8(6):359-362.
22 Vernino S, Adamski J, Kryzer TJ, et al. Neuronal nicotinic ACh receptor antibody in subacute autonomic neuropathy and cancer-related syndromes. Neurology. 1998;50:1806-1813.
23 Klein CM, Vernino S, Lennon VA, et al. The spectrum of autoimmune autonomic neuropathies. Ann Neurol. 2003;53:752-758.
24 Freeman R. Autonomic peripheral neuropathy. Lancet. 2005;365:1259-1270.
25 Winkler AS, Dean A, Hu M, et al. Phenotypic and neuropathologic heterogeneity of anti-Hu antibodyrelated paraneoplastic syndrome presenting with progressive dysautonomia: report of two cases. Clin Auton Res. 2001;11(2):115-118.
26 Kaufmann H. Treatment of patients with orthostatic hypotension and syncope. Clin Neuropharmacol. 2002;25:133-141.
27 Diedrich A, Biaggioni I. Segmental orthostatic fluid shifts. Clin Auton Res. 2004;14(3):146-147.
28 Smit AA, Wieling W, Fujimura J, et al. Use of lower abdominal compression to combat orthostatic hypotension in patients with autonomic dysfunction. Clin Auton Res. 2004;14(3):167-175.
29 Jordan J, Shannon JR, Grogan E, et al. A potent pressor response elicited by drinking water [Letter]. Lancet. 1999;353:723.
30 Hickler R. Successful treatment of orthostatic hypotension with 9-alpha fluorohydrocortisone. N Eng J Med. 1959;261:788-791.
31 Vagaonescu TD, Saadia D, Tuhrim S, et al. Hypertensive cardiovascular damage in patients with primary autonomic failure. Lancet. 2000;355:725-726.
32 Kaufmann H, Brannan T, Krakoff L, et al. Treatment of orthostatic hypotension due to autonomic failure with a peripheral alpha-adrenergic agonist (midodrine). Neurology. 1988;38:951-956.
33 Wright RA, Kaufmann HC, Perera R, et al. A double-blind, doseresponse study of midodrine in neurogenic orthostatic hypotension. Neurology. 1998;51:120-124.
34 Jordan J, Shannon JR, Biaggioni I, et al. Contrasting actions of pressor agents in severe autonomic failure. Am J Med. 1998;105:116-124.
35 Kaufmann H, Saadia D, Voustianiouk A, et al. Norepinephrine precursor therapy in neurogenic orthostatic hypotension. Circulation. 2003;108:724-728.
36 Biaggioni I, Robertson D, Krantz S, et al. The anemia of primary autonomic failure and its reversal with recombinant erythropoietin. Ann Intern Med. 1994;121:181-186.
37 Perera R, Isola L, Kaufmann H. Erythropoietin improves orthostatic hypotension in primary autonomic failure. Neurology. 1994;44(Suppl 2):A363.
38 Shannon JR, Jordan J, Diedrich A, et al. Sympathetically mediated hypertension in autonomic failure. Circulation. 2000;101:2710-2715.
39 Shibao C, Gamboa A, Diedrich A, et al. Management of hypertension in the setting of autonomic failure: a pathophysiological approach. Hypertension. 2005;45:469-476.
40 Hainsworth R. Pathophysiology of syncope. Clin Auton Res. 2004;14(Suppl 1):18-24.