Chapter 120 Organ System Considerations that Affect Anesthetic Management
Cardiovascular Performance
Changes with Development
Cardiac assessment of the PICU patient requires a knowledge of normal cardiovascular growth and development and an understanding of the influence of disease and the effects of anesthesia on cardiovascular function. The myocardium of the neonate is less compliant than that of the adult and the neonate’s cardiac output depends primarily on the heart rate. Cardiac output increases substantially with age, whereas cardiac index (cardiac output divided by body surface area) ranges from 2.5 to 4.2 L/min/m2 throughout life. Sympathetic innervation of the neonatal heart is not fully developed, and myocardial catecholamine stores are limited.1 The response of the neonatal heart to vasoactive drugs is attenuated and the capacity for peripheral vasoconstriction during hypovolemia is reduced, probably because of the immature baroreceptor and α-adrenergic receptor systems.2 Systemic vascular resistance is normally 800 to 1200 dynes • sec/cm–5 at birth and reaches the adult value of 1600 by 1 to 2 years of age. Systolic and diastolic blood pressures increase nonlinearly with age.
Effects of Disease
Disease states can affect cardiovascular performance by their effects on the pulmonary and peripheral vasculatures and by their direct effects on the myocardium. Changes in myocardial compliance and filling pressures can profoundly influence myocardial performance. In addition, factors associated with disease states such as hypoxia, hypercarbia, acidosis, and hypothermia can affect pulmonary and systemic vascular resistances and thereby further modify myocardial function. The effects of changes in preload (i.e., the effects of fluid challenges) on the patient’s blood pressure, cardiac output, and stroke volume and the clinical effects of changes in afterload on heart rate and myocardial contractility all influence the anesthesiologist’s care of critically ill children.
Effects of Anesthetic Agents
Anesthetic agents affect myocardial performance. During induction of anesthesia, potent inhaled agents such as halothane and isoflurane are associated with higher incidences of bradycardia, hypotension, and cardiac arrest in infants and children than in adults.3 These cardiovascular depressant effects appear to be more pronounced in infants than in older children. Diaz and Lockard4 found that more than 70% of healthy newborns had greater than 30% decrease in systolic blood pressure during induction with halothane, whereas Friesen and Lichtor3 noted that infants 1 to 6 months of age had a 40% decrease in mean atrial pressure and a 30% decrease in heart rate when they received a halothane and nitrous oxide anesthetic. In a study of healthy children, 1.5 to 12 years of age, undergoing halothane anesthesia, Barash et al.5 noted that systolic blood pressure and heart rate decreased in a dose-dependent manner. Similar hemodynamic effects are noted in infants anesthetized with isoflurane.6
Because it can be problematic to insert invasive monitors in unsedated, awake children, much of the information about the potent inhaled anesthetic agents and their effects on the determinants of cardiac output is derived from animal studies. In a neonatal piglet model, Boudreaux et al.7 noted that the major adverse effect of halothane was its negative inotropic effect and not its negative chronotropic or unloading activity. In similarly designed studies, Schieber et al.8 observed that, although isoflurane reduced contractility and decreased blood pressure and systemic vascular resistance more than did equipotent concentrations of halothane, cardiac index was better preserved in the isoflurane-anesthetized animals. Thus when compared with halothane, the direct myocardial depressant effect of isoflurane is offset by its effect on the peripheral vasculature, which results in afterload reduction.
Desflurane and sevoflurane are two new potent inhalational anesthetic agents. Because of their low blood solubility, they afford patients rapid induction and rapid awakening.9–14 Desflurane has a blood gas solubility coefficient (0.42) that is similar to nitrous oxide in children. However, desflurane’s pungent airway properties result in a high incidence of laryngospasm, coughing, and hypoxemia that limit its utility as an induction agent in nonintubated children.14 The cardiovascular profile of desflurane is age dependent.12 Compared with awake values, arterial blood pressure decreased in children anesthetized with 1 minimal alveolar concentration (MAC) of desflurane by approximately 30%, whereas the heart rate decreased or remained the same. Thus at 1 MAC, desflurane, like isoflurane and halothane, appears to attenuate the baroresponse in children. Weiskopf et al.15 demonstrated that rapid increases in desflurane from 0.55 to 1.66 MAC in adults can transiently increase arterial blood pressure and heart rate. This cardiovascular excitation is associated with an increase in sympathetic and renin-angiotensin system activity.
Information on the hemodynamics of sevoflurane in children suggests that sevoflurane (blood gas coefficient of 0.68) appears to produce the same hemodynamic effects as isoflurane.10 In adults anesthetized with sevoflurane or isoflurane, administration of exogenous epinephrine had similar dysrhythmogenic properties.16 In a study of children, using echocardiograms, in which sevoflurane and halothane were compared at equal MAC, Holzman et al.17 noted sevoflurane had fewer myocardial depressant effects than sevoflurane.
The synthetic opioids may offer more hemodynamic stability than the inhaled anesthetic agents.18–20 Robinson and Gregory21 were the first to report the safety and efficacy of high-dose fentanyl anesthesia in children, in a study of premature infants undergoing patent ductus arteriosus ligation. In subsequent reports on pediatric patients, Hickey and Hansen22 documented the safety of opioid anesthesia in children with complex congenital heart disease. Although these investigators noted that high doses of fentanyl (50 and 75 μg/kg) decreased heart rate and mean arterial pressure (MAP) by 7% and 9%, respectively, in patients with bidirectional shunts, opioids had a salutary effect on pulmonary vascular resistance, increasing transcutaneous oxygenation by 45%.
Because of the prolonged respiratory and sedative effect associated with moderate- and high-dose administration of fentanyl and its congeners, shorter-acting opioids may offer the advantage of more predictable control with a similar cardiovascular profile than the longer-acting opioids. Remifentanil, a new synthetic opioid agonist with an ultrashort half-life, has been introduced into clinical practice. Remifentanil is metabolized by plasma and tissue esterases. It is independent of organ elimination. Consequently, its kinetic parameters do not change with the duration of infusion. This flat, context-sensitive half-time coupled with its ultrashort half-life (7 to 10 minutes) allow better drug effect predictability. Pharmacokinetic studies in children demonstrate faster clearances and larger volumes of distribution in neonates compared with older infants and children. In vitro studies by Olgatree et al.23 have demonstrated that remifentanil has no significant direct negative inotropic effects on the myocardium and that β-adrenergic stimulation of the heart remains intact. Comparative studies of remifentanil to inhaled anesthetic agents in children undergoing pyloromyotomy surgery suggest that the short half-life of remifentanil may be beneficial with regard to postoperative respiratory changes.24,25 Because of remifentanil’s ultrashort half-life and its nonaccumulation with prolonged infusion, it may be a beneficial agent for the short-term (less than 12 hours) sedation of infants and children in the PICU setting. However, the rapid development of tolerance and cost issues will likely preclude its use for longer periods of time.
Dexmedetomidine, a sedative analgesic that has become increasingly popular, like clonidine is an α2-adrenergic agonist and is highly selective with a α2/α1 ratio of 1600:1. Stimulation of these receptors in the central nervous system (CNS) and the spinal cord produces sedation, anxiolysis, analgesia, deceased MAC of inhalational anesthetic agents (increased sensitivity), decreased renin and vasopressin levels with increased diuresis, decreased sympathetic tone with decreased heart rate and blood pressure.26 Dexmedetomidine has a rapid distribution phase (6 minutes) and an elimination half life of 2 hours.27 Petroz et al.28 demonstrated that children, 2 to 12 years of age, have similar pharmacokinetics to adults. Rodarte et al.29 studied the pharmacokinetics in infants 1 to 24 months of age. He concluded that infants have a faster clearance of dexmedetomidine than adults (27 mL/kg/min versus 13 mL/kg/min). A loading dose of 1 μg/kg and continuous infusions of 0.2 to 0.7 μg/kg/hr have been used in various clinical scenarios to produce sedation-analgesia. The most common adverse events are hypotension and bradycardia, whichare enhanced in the presence of cardiac comorbidities or when dexmedetomidine is used with other medications that have negative chronotropic effects (propofol, succinylcholine, digoxin, pyridostigmine).30–32 When a loading dose of dexmedetomidine is administered, there is a biphasic response. The initial response is an increase in blood pressure with a decrease in heart rate, followed by a decrease in blood pressure and a further decrease in heart rate.33 This biphasic effect is thought to be due to dexmedetomidine’s initial ability to stimulate peripheral postsynaptic α2b-adrenergic receptors resulting in vasoconstriction, followed by the more intense central CNS effects on α2a-adrenergic receptors causing sympatholysis. Bloor et al.33 administered boluses of 0.25, 0.5, 1.0, and 2.0 μg/kg to healthy volunteers and noted a decrease in mean arterial blood pressure (MAP) respectively of 14%, 16%, 23%, and 27%. Cardiac output decreased 20% following a loading dose of 1 μg/kg in the first minute and returned to 90% of baseline after 60 minutes. When a loading dose of 2 μg/kg was administered, cardiac output decreased by 60% and returned to 85% of baseline after 1 hour. Venn et al.34 studied the effects of dexmedetomidine in 66 patients with comorbidities who were in the ICU, mechanically ventilated, and who received a loading dose of 1 μg/kg followed by a continuous infusion of 0.2 to 0.7 μg/kg/hr. Hypotension and bradycardia (≥30% decrease from baseline) was observed in 18 of the 66 patients. Reports of bradycardia and sinus arrest have also been noted by Ingersoll-Weng.31 Khan et al.35 reported similar effects of dexmedetomidine during anesthesia with isoflurane. The majority of the effects occurred with end-tidal isoflurane levels greater than or equal to 1%. Animal studies and studies on isolated human papillary muscle have demonstrated no direct negative inotropic effects on myocardial contractility.36 The sympatholytic effects of dexmedetomidine on pediatric patients have been evidenced by Muktar et al.37 Thirty infants and children undergoing CPB were randomized to receive dexmedetomidine (1 μg/kg load followed by a continuous infusion of 0.5 μg/kg/hr) or placebo. Plasma cortisol norepinephrine, epinephrine, and glucose levels were significantly less in the dexmedetomidine group.
Propofol is a sedative hypnotic that is widely used as an induction agent in anesthesia. It is also used as a continuous infusion for prolonged sedation in the ICU. Its popularity in the ICU setting comes from its rapid clearance that allows quick awakening for rapid weaning or neurologic evaluation. Propofol is suspended in a lipid emulsion, which when administered by a continuous infusion results in a significant lipid load. Induction doses for anesthesia vary from 2 to 3 mg/kg, while ICU sedation doses vary from 50 to 250 μg/kg/min. Induction doses of propofol can cause a 10% to 15% decrease of MAP as well as bradycardia, especially when coadministered with other vagotonic drugs. Propofol has a modest negative inotropic effect, due to antagonism of β-adrenergic receptors and calcium channels.38 Since 1992, there have been increasing reports of a fatal adverse reaction that has been termed the Propofol Infusion Syndrome (PRIS).39 To date, 61 cases (32 pediatric and 29 adult) have been reported, and there have been 20 pediatric and 18 adult deaths.40 PRIS is characterized by severe intractable bradycardia that leads to cardiac failure, severe metabolic acidosis, hyperlipidemia, rhabdomyolysis with consequent hyperkalemia, and renal failure.41 Prolonged propofol infusions (more than 48 hours) and infusion rates greater than 4 mg/kg/hr have been linked to PRIS. Priming factors such as critical illness (respiratory failure and traumatic brain injury) and triggering factors such as catecholamine and steroid infusion are associated with the syndrome.44 The underlying mechanism of PRIS is not well understood. It is speculated that high doses of propofol inhibit the mitochondrial respiratory chain. As a result, there is a decreased ATP production and a decreased mitochondrial lipid metabolism, with an accumulation of toxic long fatty acid chains that are arrhythmogenic.45–47 Management of PRIS is very difficult and consists of prompt recognition and interruption of the infusion. Aggressive cardiac resuscitation must be initiated early on with high dose inotropes, fluid administration, and the use of pacing devices. The early administration of hemodialysis and hemofiltration together with ECMO has improved survival.48–50
Anemia and Transfusion
Concerns about transfusion-related disease transmission have forced clinicians to reassess transfusion criteria. Rothstein51 previously recommended that in patients younger than 3 months of age, hemoglobin concentration should be greater than 10 g/dL, whereas in children older than 3 months, a hemoglobin concentration of 9 g/dL was adequate. Slogoff52 concluded that in normovolemic adults, a hematocrit of 20% (hemoglobin 7 g/dL) is adequate. Carson et al.53 retrospectively reviewed mortality in 125 adult surgical patients who refused blood transfusion for religious reasons. Mortality correlated inversely with preoperative hemoglobin level and directly with operative blood loss. No operative deaths occurred among patients with a preoperative hemoglobin level above 8 g/dl and an operative blood loss of less than 500 mL.
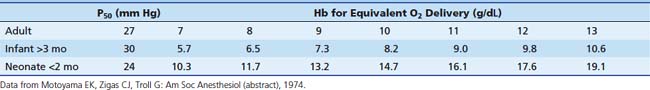
Minimum acceptable hemoglobin (and approximate hematocrit) levels can be inferred from anticipated tissue oxygen delivery (Table 120-1). A hemoglobin concentration of 7 to 8 g/dL (hematocrit 21% to 24%) is reasonable for adults and children based on the previous discussion. Minimum values chosen for infants should be more conservative (i.e., ensure greater oxygen delivery to tissues) than those for older patients. This takes into consideration the higher oxygen consumption of infants and provides them with a larger margin of safety against hypoxic injury. Infants younger than 2 months probably require a hemoglobin level of 13 g/dL (hematocrit of 40%); infants older than 3 months require a hemoglobin level of 7 g/dL (hematocrit of 21%). Infants aged 2 to 3 months are in a transitional phase, and minimum hemoglobin levels are more difficult to predict. These values are intended as guidelines only; each patient must be considered individually.
Patients with chronic anemia increase oxygen delivery by increasing cardiac output. Although the potent inhaled anesthetic agents decrease myocardial function and cardiac output and thereby decrease oxygen delivery, these side effects are offset by the decrease in oxygen consumption that occurs in anesthetized patients. The need to transfuse blood in the perioperative period frequently is determined by the patient’s underlying hemodynamic stability, the type of surgery anticipated, and the known risks of administering blood products. In general, unless the procedure is minor, blood should be available for all ICU patients. Because ICU patients frequently are monitored with invasive catheters, serial measurements of hematocrit or hemoglobin usually can be obtained during the operative procedure to determine whether transfusion is warranted.
Respiratory Failure
When respiratory failure is present, the anesthesiologist must be aware of its precipitating factors. Acute respiratory distress syndrome (ARDS) is a clinical syndrome of respiratory distress, poor pulmonary compliance, and hypoxemia that usually occurs after a nonpulmonary condition such as shock, trauma, sepsis, or an unexplained condition leading to pulmonary parenchymal disease and respiratory failure. Direct mortality from respiratory failure is approximately 10% to 20%; overall mortality may be as high as 65%. ARDS was first described as a clinical entity occurring in pediatric patients in 1980.54 Recognized factors predisposing pediatric patients to the development of ARDS include severe infection, cardiac arrest, shock, and aspiration. Although the cause of this condition is uncertain, the fundamental defect in the lung is injury to the pulmonary capillary endothelial cells, leading to interstitial and, ultimately, alveolar pulmonary edema. This pulmonary vascular leakage results in hypoxemia, decreased pulmonary compliance, increased pulmonary vascular resistance, and venoarterial shunting.
The mainstays of treatment are endotracheal intubation, continuous positive-pressure ventilation with positive end-expiratory pressure (PEEP), and supplemental oxygen. Invasive monitoring by arterial, central venous, and pulmonary arterial cannulation may be necessary to optimize preload and cardiac output in the presence of high PEEP and high transpulmonary pressures. The patient’s requirements for oxygen, PEEP, and pulmonary toilet must be understood preoperatively. Patients with poor pulmonary compliance challenge the ability to maintain adequate intraoperative ventilation. Preoperative knowledge of the interaction of PEEP with the adequacy of ventilation and cardiovascular function is an important concern of the anesthesiologist. Anesthetic agents can modify not only cardiovascular function, but also respiratory function, by their effect on hypoxic pulmonary vasoconstriction. Patients with ARDS may rely in part on regional hypoxic pulmonary vasoconstriction to minimize intrapulmonary shunting. Shifting of pulmonary blood flow away from atelectatic lung regions has been well described. The administration of vasoactive drugs and potent inhaled anesthetic agents inhibits hypoxic pulmonary vasoconstriction and therefore may exacerbate hypoxemia by increasing intrapulmonary shunting.55,56
Neurologic Injury
The leading form of pediatric trauma is traumatic brain injury (TBI), which accounts for 36% of traumatic deaths in the USA.57,58 Eighty percent of trauma patients have central nervous system injury, and in 60% of these patients, the CNS injury is the most severe. It is estimated that 185 of every 100,000 children experience head trauma requiring hospitalization.58,59 Poor outcomes have been associated with early hypotension, low Glasgow Coma Scale score, altered cerebral blood flow (CBF), hyperglycemia, and deranged autoregulation.59 Neurologic injury may be the primary insult resulting from trauma. However, secondary cerebral injury may result from the development of intracranial hypertension and resultant cerebral ischemia. Recognition and control of elevated intracranial pressure (ICP) are key elements to preventing neurologic deterioration and patient morbidity. Secondary traumatic brain injury can be characterized by an array of biochemical, cellular, and molecular events that are associated with ischemia, excitotoxicity, energy failure, and cell death cascades, all of which result in cerebral swelling, axonal injury, inflammation, and regeneration.60
Intracranial Pressure
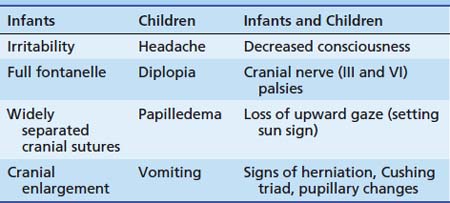
The signs of intracranial hypertension in infants and children are listed in Table 120-2. The presence of these signs and symptoms, in conjunction with the baseline findings on neurologic examination, may dramatically alter the patient’s anesthetic care. The anesthesiologist must be aware of ongoing interventions to control ICP. Control of cerebral perfusion pressure is essential for maintaining neurologic function in pathologic states. Cerebral perfusion pressure is expressed as the difference between MAP and the highest value obtained out of the following: central venous pressure, intrathoracic pressure, or ICP. Thus in the presence of intracranial hypertension (ICP ≥15 to 20 mm Hg), higher systemic blood pressures must be achieved to prevent cerebral ischemia. Intracranial compliance depends on the volumes of the intracranial contents, cerebral tissue, blood, and extracellular fluid. The most dynamic of these is the blood compartment. Changes in cerebral blood volume are mediated primarily through changes in cerebral vascular resistance. Cerebral vascular resistance is affected by both intracranial and extracerebral factors.
Table 120–2 Signs of Intracranial Hypertension in Infants and Children
| Infants | Children | Infants and Children |
|---|---|---|
| Irritability | Headache | Decreased consciousness |
| Full fontanelle | Diplopia | Cranial nerve (III and VI) palsies |
| Widely separated cranial sutures | Papilledema | Loss of upward gaze (setting sun sign) |
| Cranial enlargement | Vomiting | Signs of herniation, Cushing triad, pupillary changes |
Regulation of Cerebral Blood Flow
The normal brain receives 15% of cardiac output. Kennedy and Sokoloff61 determined that healthy children have a high CBF (100 mL/100 gm/min), this value decreases to adult values of 50 mL/100 gm/min during the teenage years. Cerebral blood flow regulation depends primarily on the local chemical and metabolic milieu, particularly the concentrations of hydrogen ion, adenosine, and prostanoids. Production of these compounds correlates with normal cerebral activity and metabolic rate. Neurogenic and myogenic components also have minor roles in regulating cerebral vascular resistance.
Extracerebral factors that can change cerebral vascular resistance and CBF include the arterial partial pressures of carbon dioxide (PaCO2) and oxygen (PaO2), MAP, and various drugs (Figure 120-1). Cerebral circulation is very sensitive to variations of PaCO2, and has been studied using transcranial Doppler ultrasound.62 CBF varies linearly by 2% to 4% for every variation of 1 mm Hg of PaCO2. Data from anesthetized children show that CO2 vasoreactivity is higher than in adults (13.8% vs. 10.3% change).63 Low blood pressure decreases cerebral vascular vasoreactivity.63 As PaCO2 is rapidly lowered toward 20 mm Hg, there is marked cerebral vasoconstriction and reduction in both CBF and ICP. Theoretically, an acute decrease in PaCO2 below 20 mm Hg may be detrimental by reducing CBF enough to cause cerebral ischemia. The salutary effects of acute hyperventilation on ICP are diminished over time because acute changes in cerebrospinal fluid pH are normalized in approximately 6 hours. Arterial oxygenation within the normal clinical range has little effect on cerebral vascular resistance. However, PaO2 less than 50 mm Hg results in cerebral vasodilation and increased CBF. Hyperoxia in excess of 300 mm Hg may produce cerebral vasoconstriction.
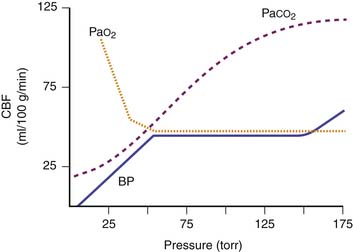
Cerebral autoregulation is a homeostatic process by which the brain maintains a constant CBF over a MAP range from 60 to 150 mm Hg in adults. At an MAP less than 60 mm Hg, symptoms of cerebral ischemia may appear. If the upper limit of MAP for autoregulation is exceeded, the resultant increase in CBF may cause cerebral edema. The autoregulatory curve may shift in the presence of chronic hypertension, intracranial tumors, head trauma, or shock states. This renders the brain more susceptible to ischemic effects.63 The range of MAP over which autoregulation of CBF occurs in infants and children likely shifts in tandem with age-related changes in normal systemic blood pressures and cerebral perfusion pressures. Raju et al.64 suggest that an infant’s mean cerebral perfusion pressure is approximately equal to its gestational age in weeks and that this estimate holds true for growing preterm infants up to 5 weeks after birth.
Effects of Anesthetics on Cerebral Blood Flow
In general, the potent inhaled anesthetic agents impair autoregulation and may cause hypotension and increased CBF (Figure 120-2). Consequently, they must be used cautiously, if at all, in patients with evidence of traumatic brain injury. Halothane and enflurane at 1 MAC abolish cerebral autoregulation. Isoflurane impairs autoregulation less than halothane.65 The effects of anesthetic agents on CBF and cerebral metabolism are summarized in Figure 120-3. In general, the perfect agent would decrease both CBF and cerebral metabolic rate of oxygen (CMRO2). The potent inhaled agents (halothane, enflurane, and isoflurane) “uncouple” the normal relationship between CBF and metabolism and cause marked cerebral vasodilation. Isoflurane is the only inhaled anesthetic agent with which CBF actually may decrease when concomitant hyperventilation to PaCO2 of 20 to 25 mm Hg is used.66 Whereas nitrous oxide alone is known to cause mild cerebral vasodilation and to increase CMRO2 (probably related to inadequate anesthetic depth), these effects are easily countered by the addition of intravenous sedatives, hypnotics, and narcotics. There are limited data regarding the effects of the two newest inhaled agents, sevoflurane and desflurane, on intracranial dynamics. Although the effects of desflurane on CBF have been studied, there is relatively little information regarding the effects of sevoflurane on CNS physiology in human subjects. Artru et al.67 noted that desflurane increases CSF pressure in animals. Ornstein et al.68 showed that in patients with intracranial mass lesions, desflurane and isoflurane have similar effects on CBF. The electroencephalographic effects of desflurane are similar to those of isoflurane, with prominent burst suppression occurring at 1.24 MAC.69
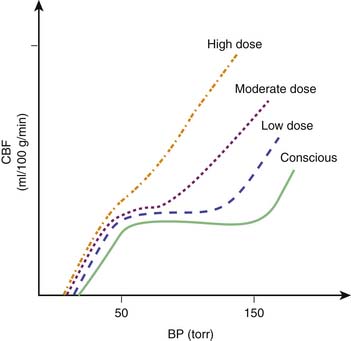
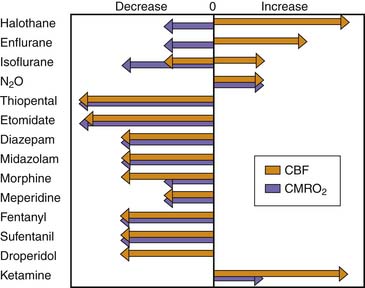
Figure 120–3 Effects of anesthetic agents on cerebral blood flow (CBF) and cerebral metabolic rate of oxygen (CMRO2).
(Modified from Cucchiara RF, Block S, Steinkeler JA: The effects of anesthetic agents on cerebral blood flow and cerebral metabolism: anesthesia for intracranial procedures. In Barash PG, Cullen BF, Stoelters SK, editors: Clinical anesthesia, Philadelphia, 1989, JB Lippincott.)
In volunteers using transcranial Doppler, Bedforth et al.70 noted that at 1 MAC of desflurane, cerebral autoregulation was impaired and that at 1.5 MAC autoregulation was abolished. In a separate study of adult non-neurosurgical patients, Bedforth et al.71 noted that the introduction of desflurane following induction of anesthesia with propofol impaired cerebral autoregulation more than with equi-MAC doses of sevoflurane. In contrast, in studies of static and dynamic compliance of autoregulation, Strebel et al.72 noted that, at 1.5 MAC desflurane and 1.5 MAC sevoflurane, autoregulation was impaired but propofol (200 μg/kg/min) had no effect. Muzzi et al.73 showed that 1 MAC of desflurane in adults with supratentorial lesions increases ICP as opposed to 1 MAC of isoflurane. Desflurane may alter CSF dynamics. Desflurane does not appear to change CSF absorption but does increase CSF production.73
In a study of patients undergoing non-intracranial neurosurgical procedures, Summors et al.74 noted that dynamic cerebral autoregulation using transcranial Doppler ultrasonography is better preserved during 1.5 MAC sevoflurane than 1.5 MAC isoflurane anesthesia. Others noted similar findings with sevoflurane and isoflurane. In patients studied at 0.5 and 1.5 MAC, the dose-dependent vasodilatory effect was less for sevoflurane.75,76 Nishiyama et al.77 assessed comparative cerebral vasodilatory responsiveness to CO2 during either sevoflurane or isoflurane anesthesia. Nishiyama et al.71 noted in a group of adult patients that changes in CBF caused by changes in CO2 are greater during isoflurane anesthesia. However, attempts to decrease ICP by decreasing CO2 were more successful with isoflurane than with sevoflurane. In addition to cerebral vascular autoregulation, sevoflurane use is associated with epileptiform activity. In pediatric patients with and without a preexisting history of epilepsy, induction by sevoflurane of tonic, clonic, and silent seizures during the induction and maintenance of anesthesia is reported.78–80
For the anesthesiologist, knowledge of the patient’s ICP and cranial compliance and an understanding of the pathophysiology of the CNS lesion are important for planning appropriate anesthetic management.81 In experimental animals, Statler et al.81 demonstrated that isoflurane had more neuroprotective effect than fentanyl in rats that had undergone controlled cortical impact lesions. Hendrich et al.82 quantified CBF using labeled magnetic resonance imaging in normal rats anesthetized with fentanyl, isoflurane, or pentobarbital. In this study, CBF values were found to be approximately 2.5 to 3 times lower in most regions analyzed during anesthesia with either fentanyl (with N2O/O2) or pentobarbital versus isoflurane (with N2O/O2). In addition, these investigators noted that CBF was heterogeneous in rats anesthetized with isoflurane (with N2O/O2) but relatively homogenous in rats anesthetized with either fentanyl (with N2O/O2) or pentobarbital. In previous human studies with opioid anesthesia, opioids maintained static cerebral autoregulation. Engelhard et al.83 subsequently demonstrated that remifentanil, a mu-opioid agonist with unique pharmacokinetic properties, when combined with low-dose propofol maintains both static and dynamic compliance of cerebrovascular autoregulation. Propofol also has been used to alter CNS dynamics.84 Positron emission tomography suggests that propofol produces global metabolic depression.85,86 Like thiopental, propofol decreased CMRO2, CBF, and ICP. Cerebral responsiveness to arterial CO2 appears to be preserved during propofol anesthesia.87,88
Hepatic Dysfunction
An assessment of liver function is important for two aspects of anesthetic management. The liver is a major site of drug metabolism and is involved in homeostasis of the coagulation system. Anesthesia and surgery may exacerbate liver dysfunction in patients with preexisting liver disease and, in some instances, may cause life-threatening hepatic failure. Although the cause of hepatic dysfunction in surgical patients is unknown, it may be related to decreases in hepatic blood flow. Consequently, anesthetic drugs and techniques that may decrease hepatic blood flow should be avoided if possible.
Effects of Anesthetics on Hepatic Blood Flow
Blood flow in the splanchnic circulation can be altered by numerous factors. Mechanical ventilation, by altering the normal relationship between splanchnic venous outflow and venous outflow from the kidney and lower extremities, may affect splanchnic blood flow. The changes associated with mechanical ventilation can be further exacerbated by increasing tidal volume and applying PEEP.89 Carbon dioxide concentration also influences splanchnic blood flow.90 Various anesthetic agents may have different effects on hepatic blood flow.91 Gelman et al.92 showed that hepatic oxygen supply is better maintained during isoflurane than during halothane anesthesia. In addition, in hypoxic rat models, the incidence of hepatic centrilobular necrosis was least with isoflurane compared with halothane, nitrous oxide, and fentanyl. Although anesthetic agents can decrease hepatic blood flow, Gelman et al.92 demonstrated that the decrease in hepatic blood flow is small compared with the decrease associated with the surgical stress of a laparotomy. Surgical procedures involving traction and manipulation of the abdominal viscera are associated with the release of various vasoactive compounds that may further alter splanchnic blood flow. In sevoflurane-anesthetized dogs, Frank et al.93 demonstrated that hepatic arterial blood flow was maintained, portal blood flow was decreased, and total hepatic blood flow was unchanged. In desflurane-anesthetized dogs, hepatic artery blood flow was maintained, portal blood flow was decreased, and total hepatic blood flow was reduced.94
Effects of Liver Disease on Pharmacokinetics
In addition to the effects of anesthetic agents on the liver, hepatic disease affects the pharmacology of anesthetic agents. Little is known about the pharmacologic properties of anesthetic agents in pediatric patients with liver disease.95,96 Although the liver is the major site of drug biotransformation, the effects of hepatic dysfunction on drug elimination and disposition are inconsistent.97 The degree of liver dysfunction and a drug’s ability to bind to plasma proteins are important variables in determining drug kinetics in patients with liver disease. For drugs with a high hepatic extraction ratio, hepatic clearance is sensitive to changes in hepatic blood flow, whereas for drugs with a low hepatic extraction ratio, hepatic drug clearance is a function of intrinsic hepatic enzyme activity and protein binding.97 Thus the reported inconsistent effect of liver disease on drug pharmacology may be a function of the heterogeneous pathophysiology of liver disease with respect to hepatocellular function, protein binding, and hepatic blood flow. Table 120-3 compares opioid pharmacokinetics in patients with and without liver disease. Note that the effects of hepatic disease are variable with respect to the pharmacokinetics of opioids. Remifentanil, a μ-opioid agonist that undergoes plasma and tissue esterase metabolism, is unaffected, with respect to its pharmacokinetic profile, by liver disease.98,99
The pharmacokinetics of neuromuscular blocking agents (NMBAs) in adults have been well studied. Of the nondepolarizing NMBAs, pancuronium, vecuronium, and atracurium are significantly metabolized. Of these agents, only pancuronium and vecuronium pharmacokinetic profiles are altered by liver disease. In a study of children with moderate-to-severe liver dysfunction, Brandom et al.100 showed that liver failure has no effect on atracurium pharmacokinetics. Mivacurium is a benzylisoquinolium diester metabolized by plasma cholinesterase. It is an NMBA of short duration. Greene et al.101 noted that the severity of disease did not correlate with the duration of mivacurium blockade in pediatric patients with liver disease compared with controls. However, mivacurium’s initial recovery and overall recovery from neuromuscular blockade did correlate inversely with the concentration of plasma cholinesterase.
Cisatracurium is a stereoisomer of atracurium. In patients with normal hepatic and renal function, the duration of action of cisatracurium is similar to that of atracurium. DeWolf et al.102 noted that although the kinetic profile of cisatracurium differed in patients with liver failure compared with control patients, the duration of clinical effectiveness was similar for the two groups of patients. In patients with hepatic failure, clearance of rocuronium is reduced, and the volume of distribution is increased. Consequently, the elimination half-life is prolonged.
The Role of the Liver in Coagulation
Thromboelastography (TEG) has been in use for more than 60 years.103 Developed during the Second World War, it has had a revival with the advent of transplantation and the ability to perform more complex surgeries. Moreover, new technology has made this point-of-care test easier to execute, more reliable, and reproducible.104,105 TEG is a global dynamic assessment of hemostasis that can yield information on several different components of coagulation within 60 minutes. Its use for the guidance of blood component therapy in liver transplant has been demonstrated.106,107 TEG measures the static viscoelastic properties of whole blood. As a clot begins to form in the sampling cup, the viscoelastic properties of the sample increase and an increase in amplitude is registered on the tracing. At the end of the test a characteristic trace with two arms is created.108 Various measurements are derived from the tracing (Figure 120-4).109 The R-time is the time in minutes from the start of the sample run until the first traces of clot mesh are detected (defined as an increase in amplitude of 2 mm). R-time generally reflects factor levels. The K-time is the time elapsed between the amplitudes of 2 and 20 mm on the TEG tracing and reflects velocity of clot formation. The α-angle is the slope drawn from r to k. The angle reflects fibrinogen activity. The maximum amplitude (MA) is the greatest vertical amplitude. It reflects clot strength and indirectly assesses platelet number and function and fibrin formation. The clot lysis index (A60) is the amplitude 60 minutes after the MA is achieved. It represents fibrinolytic activity. Alternatively, lysis 30 (Ly30) can be used, which assesses fibrinolysis at a shorter time interval.
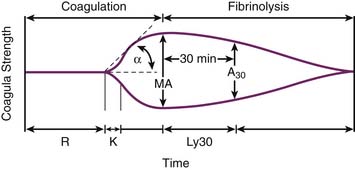
Andrew et al.110 demonstrated that the coagulation system of newborns is immature and undergoes a maturational process in the first year of life, during which time factor levels reach near adult levels. Clinical practice, however, shows that this does not expose neonates and infants to increased bleeding during surgery. Paradoxically, TEG tracings of healthy newborns show that neonates initiate and develop clots faster than adults.111 R-time is significantly shorter despite a prolonged aPTT when compared to adults. MA values are also increased when compared to adults. An explanation of this can be due to the presence of adult levels of factors VIII and XIII, vonWillebrand factor, and platelets, already at birth, and lower levels of inhibitors, especially antithrombin III. This imbalance could promote faster activation and a more resistant clot.111
Thromboelastography has facilitated solid organ transplantation, and much of our understanding of the relation of TEG with the coagulation cascade has come from patients undergoing liver transplantation. Patients during liver transplants are frequently in a hypocoagulable state, with an increased risk for perioperative bleeding. The reasons for this hypocoagulable state included decreased synthesis of coagulation proteins, decreased platelet number and function, and increased fibrinolysis secondary to decreased hepatic clearance of TPA.112 TEG tracings can reflect this state with prolonged r-time, decreased α-angle and decreased MA. During the anhepatic phase (when the liver is removed), fibrinolysis increases and there is further deterioration of the MA and the α-angle. The A60 is also decreased. The TEG tracing shows signs of hyperfibrinolysis. With the absence of the liver, there is no hepatic clearance of TPA and, in addition, low levels exist of the plasmin inhibitor α2-antiplasmin.113 When the donor liver is reperfused (reperfusion phase), a reperfusion coagulopathy can further affect hemostasis. Increased amounts of plasminogen activators released by the damaged endothelium of the donor liver and the presence of exogenous heparin in the donor liver are the cause. TEG tracings can show extreme levels of hyperfibrinolysis, and the r-time can be further prolonged. TEG analysis with heparinase sample cups demonstrates the contribution of heparin to the coagulopathy by a partial correction of the tracing when compared to the native without heparinase.114
In 1985, Kang et al.107 devised a protocol for blood component therapy guided by TEG tracings. This protocol dictated that an r-time of ≥15 min needed 2 units of FFP, an MA of ≤40 mm needed 10 units of platelets, and when the α-angle was ≤45° or there was no improvement with the above blood component therapy, 6 units of cryoprecipitate was given. When blood requirements of this group were compared to a historical control group, they found that the TEG-monitored received 33% less blood. In addition, TEG can be used as an in vitro test to predetermine the effectiveness of the antifibrinolytic agents tranexamic acid and epsilon aminocaproic acid (EACA).115,116
Recombinant activated factor VII (rFVIIa, NovoSeven) is an analogue of the naturally occurring activated Factor VII and is a powerful hemostatic agent. It is approved by the FDA for use in patients with congenital or acquired hemophilia with inhibitors who are actively bleeding or for prophylaxis prior to invasive surgery or procedures.117,118 rFVIIa directly enhances thrombin generation, locally at the site of endothelial damage where tissue factor is exposed, and indirectly on the activated platelet surface, creating the thrombin burst generating enormous amounts of fibrin mesh in the proximity of the damaged vasculature.119,120 Since its marketing in the United States in 1999, there has been an exponential increase of publications regarding the off-label use for life-threatening hemorrhages of various origins in nonhemophiliac patients, giving rFVIIa a potential role as a universal hemostatic agent.121 The lack of randomized trials for off-label use of rFVIIa in children makes it difficult to evaluate its efficacy and safety. At present, the only randomized trials are in children with hemophilia where a standardized treatment regimen of 90 μg/kg every 2 hours was administered until hemostasis was obtained.118 Its fast reconstitution and low volume leads to rapid administration without risk of fluid overload. There is, however, intersubject variability, and its short half-life in children (1.32 hours vs. 2.72 hours in adults)122 increases costs, because higher doses are necessary to achieve the same plasma levels as in adults.123 There are several case series describing the use of rFVIIa in children with severe liver failure.124–126 Varying doses from 40 to 100 μg/kg were used as a last effort to correct severe bleeding after failure of conventional blood component therapy. Pediatric liver transplants have a higher incidence of vascular graft thrombosis because of the smaller caliber of the vascular grafts, and it is feared that the administration of rFVIIa can increase the incidence of vascular graft thrombosis. Kalicinski et al.127 reviewed 89 consecutive patients, aged 1 to 20 years, who underwent cadaveric liver transplant. Twenty-eight of these patients had a high risk of intraoperative blood loss and received rFVIIa prior to transplant. When compared to the 61 patients that had no increased intraoperative risk of blood loss, they noted no difference in blood loss, operating time, reexploration, and hospital stay. Interestingly no thromboembolic events were observed in the group that received rFVIIa, while in the 61 patients that did not receive rFVIIa, three had hepatic artery thrombosis, two had portal vein thrombosis, and one had hepatic vein thrombosis. The authors suggest that despite the use of rFVIIa, the overall hemostatic balance remains modestly hypocoagulable.127 Currently, there are no laboratory tests that can predict the clinical response to rFVIIa. Most clinicians are using the prothrombin time to monitor its hemostatic effect, although it does not reflect in vivo responses.128 Hendricks et al. reported monitoring the effects of rFVIIa by TEG analysis.129 Both onset of clot formation and amplitude and clot stability improved after administration of rFVIIa.
Fluid and Electrolyte Disturbances
The acidosis associated with renal failure would not be expected to interfere with reversal of neuromuscular blockade. Miller and Roderick66 demonstrated that metabolic alkalosis, but not metabolic acidosis, may prevent neostigmine-induced antagonism of pancuronium. The mechanism proposed depends on simultaneous decreases in calcium and potassium levels. Whether the hypocalcemia associated with renal failure clinically affects reversal of NMBAs remains to be demonstrated. In the event of prolonged neuromuscular blockade, dialysis should easily remove both NMBAs and anticholinesterase agents used for reversal of blockade. Both classes of drugs are highly ionized compounds.
Renal Drug Metabolism
The kidneys also function in drug elimination. The effects of renal failure on drug disposition and elimination are of particular interest to the anesthesiologist. Nondepolarizing NMBAs are the major class of anesthetic agents excreted primarily by the kidneys. Drugs that are almost exclusively eliminated by the kidney (e.g., gallamine and metocurine) should be avoided in the presence of renal failure. Pancuronium accumulates despite its partial hepatic metabolism and biliary excretion and may demonstrate as much as a fivefold increase in elimination half-life in anephric patients. The intermediate-duration NMBAs vecuronium and atracurium should be considered for use in patients with renal failure.130,131 Although vecuronium depends primarily on hepatobiliary excretion with only 20% primarily excreted by the kidney, its active hepatic metabolites are dependent on renal excretion and may accumulate with repeated dosing in patients with renal dysfunction. Therefore it may result in prolonged blockade, depending on the degree of renal failure or insufficiency. Atracurium elimination occurs by Hoffmann elimination and ester hydrolysis in the plasma. Because these processes are independent of renal and hepatic function, the elimination half-life of atracurium is constant regardless of failure of these organs.100,132 Atracurium metabolism produces laudanosine, which is associated with seizures in laboratory animals at high plasma concentrations.133 Data in human subjects indicate that the risk of such seizures probably is not clinically relevant. ICU patients receiving atracurium infusions for up to 219 hours had maximum plasma concentrations well below the threshold for canine seizures and showed no evidence of cerebral excitation.134 The effects of renal failure on this scenario are not documented. Mivacurium is a bisquarternary benzylisoquinolinium diester, nondepolarizing NMBA that is metabolized by plasma cholinesterase. It is a mixture of three optical isomers. Mivacurium at a dose of 0.15 mg/kg prolonged neuromuscular blockade by a factor of 1.5 in patients with renal disease compared to patients with normal renal function.135
Cisatracurium is one of the stereoisomers of atracurium and is thought to be three times more potent than atracurium but with a similar duration of action. Like atracurium, cisatracurium is metabolized by Hoffmann elimination, and the drug does not accumulate. Its duration of action is not prolonged in patients with renal failure.136 Rocuronium is a monoquaternary steroidal NMBA of rapid-to-intermediate onset of action and intermediate duration. The clinical duration of rocuronium is similar to that of vecuronium. It has a rapid onset of action, but in patients with renal disease, the onset of action is decreased by 30% to 40% and its duration of action is prolonged and variable.137,138
Opioids are metabolized primarily by the liver. Thus renal failure should not have a major effect on the primary clearance of opioids, although the metabolism of some opioids (meperidine and morphine) results in pharmacologically active metabolites that are water soluble and undergo renal elimination. As such, the elimination and hence the clinical effects of these agents may be prolonged, especially after repeated dosing or continuous infusions in patients with renal dysfunction. The pharmacokinetics of fentanyl, sufentanil, and alfentanil in children appear to be minimally affected by chronic renal failure.139,140 Morphine glucuronide accumulates at higher plasma concentration in patients with chronic renal failure.141,142 Normeperidine, the major metabolite of meperidine, may accumulate and cause prolonged respiratory depression and seizures.
Of the inhaled anesthetic agents, isoflurane is probably the safest for patients with renal failure. It is not metabolized to fluoride ion or associated with potential renal toxicity, as are enflurane and methoxyflurane. It also is not associated with bromide ion production, as is halothane. Although bromide is not a renal toxin, it may account for persistent sedation. The metabolic breakdown of desflurane is insignificant. Less than 1% of the absorbed dose is metabolized. Sevoflurane, however, undergoes metabolic breakdown into organic and inorganic compounds. Inorganic fluoride is a potential cause of nephrotoxicity with the prolonged administration of sevoflurane. Studies with methoxyflurane show that plasma inorganic fluoride levels greater than 50 μmol/L are associated with nephrotoxicity. However, plasma levels may not accurately predict nephrotoxicity. Kharasch et al.143 demonstrated that inhalational anesthetic agents are metabolized within the kidney by the cytochrome P-450 system and that various anesthetic agents are metabolized differently within the kidney. Consequently, it appears that intrarenally produced fluoride. and not the plasma fluoride level. may explain the relationship of nephrotoxicity to anesthetic metabolism. These data suggest that agents that do not undergo primarily metabolism in the kidney (sevoflurane) are less likely to result in nephrotoxicity than older agents that were metabolized by the kidneys (methoxyflurane). The newer agents, sevoflurane and desflurane have been studied in patients with and without renal disease. Desflurane is a polyfluorinated ethyl ether that does not undergo in vivo metabolism. Studies in both animals and humans suggest that desflurane has no hepatic or renal toxic effect; however, it does interact with dry carbon dioxide absorbants and produces carbon monoxide.144 Sevoflurane is a polyfluorinated methylisopropyl ether that undergoes in vivo degradation to difluorovinyl products and fluoride ion. In addition, sevoflurane undergoes degradation by the CO2 absorber system to form fluoromethyl-1-1-difluoro-1-(trifluoromethyl) vinyl ether, also known as compound A. Both compound A and free fluoride ions have potential nephrotoxic effect. However, clinical studies in patients with normal renal function and those with abnormal renal function demonstrate that changes in kidney function following sevoflurane anesthesia are similar to changes that occur after desflurane, isoflurane, or enflurane anesthesia.145–151
Remifentanil is an opioid agonist that is metabolized by plasma and tissue esterases. The metabolic product of remifentanil metabolism has a potency of 1/3400 of its parent compound. It is independent of organ elimination, and its extremely rapid metabolism does not allow for drug accumulation. Ross et al.152 reported the pharmacokinetic properties of remifentanil in children. Its ultrashort half-life (less than 7 minutes) allows patients a rapid recovery from anesthesia. Because it is independent of organ elimination, the pharmacokinetic profile of remifentanil is unchanged in either renal or hepatic insufficiency.153 Remifentanil also demonstrates a flat, context-sensitive half-time. Thus the length of time remifentanil is infused does not affect the time to patient recovery when infusion is discontinued.154,155
In addition to the fluid and electrolyte problems and pharmacologic concerns posed by renal failure, the presence or development of renal failure per se may affect perioperative outcome. In adults, perioperative renal failure is associated with mortality rates as high as 70%. Similar data from children are lacking. However, in a study of pediatric patients undergoing liver transplantation, the 1-year survival rate in patients with renal failure was 53% compared with 81% for patients in whom renal failure did not develop.156
Intravenous Alimentation
ICU patients frequently receive intravenous hyperalimentation for nutritional support. Intravenous hyperalimentation regimens should be adjusted in anticipation of surgery. Surgical trauma results in accelerated protein catabolism. Administration of glucose to traumatized patients does not have the protein-sparing effect observed during starvation. The capacity of postoperative patients to handle exogenous glucose loads is impaired as a result of the neuroendocrine response to injury.157 Hyperglycemic, hyperosmolar, nonketotic coma (HHNKC) is reported as a cause of delayed awakening after anesthesia. In two reports, the patients had diabetes mellitus, and they returned to the baseline level of consciousness within 1 to 3 postoperative days after appropriate therapy. Bedford158 reported the fatal development of HHNKC postoperatively in a nondiabetic patient who received high-dose steroid therapy for thrombotic thrombocytopenic purpura.
References are available online at http://www.expertconsult.com.
1. Kharasch E.D., Hankins D.C., Thummel K.E. Human kidney methoxyflurane and sevoflurane metabolism. Anesthesiology. 1995;82:689.
2. Rogers S.N., Benumof J.L. Halothane and isoflurane do not decrease PaO2 during one-lung ventilation in intravenously anesthetized patients. Anesth Analg. 1985;64:946.
3. Friesen R.H., Lichtor J.L. Cardiovascular depression during halothane anesthesia in infants: a study of three induction techniques. Anesth Analg. 1982;61:42.
4. Diaz J.H., Lockard C.H. Is halothane really safe in infancy? Anesthesiology. 1979;51:S313.
5. Barash P.G., Glanz S., Katz J.D., et al. Ventricular function in children during halothane anesthesia: an echocardiographic evaluation. Anesthesiology. 1978;49:79.
6. Friesen R.H., Lichtor J.L. Cardiovascular effects of inhalation induction with isoflurane in infants. Anesth Analg. 1983;62:411.
7. Boudreaux J.P., Schieber R.A., Cook D.R. Hemodynamic effects of halothane in the newborn piglet. Anesth Analg. 1984;63:731.
8. Schieber R.A., Namnoum A., Sugden A., et al. Hemodynamic effects of isoflurane in the newborn piglet: comparison with halothane. Anesth Analg. 1986;65:633.
9. Davis P.J., Cohen I.T., McGowan F.X.Jr., et al. Recovery characteristics of desflurane vs. halothane for maintenance of anesthesia in pediatric ambulatory patients. Anesthesiology. 1994;80:298.
10. Sarner J.B., Levine M., Davis P.J., et al. Clinical characteristics of sevoflurane in children: a comparison with halothane. Anesthesiology. 1995;82:38.
11. Taylor R.H., Lerman J. Induction, maintenance, and recovery characteristics of desflurane in infants and children. Can J Anaesth. 1992;39:6.
12. Taylor R.H., Lerman J. Minimum alveolar concentration of desflurane and hemodynamic responses in neonates, infants, and children. Anesthesiology. 1991;75:975.
13. Young W.L. Effects of desflurane on the central nervous system. Anesth Analg. 1992;75:S32.
14. Zwass M.S., Fisher D.M., Welborn L.G., et al. Induction and maintenance characteristics of anesthesia with desflurane and nitrous oxide in infants and children. Anesthesiology. 1992;76:373.
15. Weiskopf R.B., Moore M.A., Eger E.I.II, et al. Rapid increase in desflurane concentration is associated with greater transient cardiovascular stimulation than with rapid increase in isoflurane concentration in humans. Anesthesiology. 1994;80:1035.
16. Navarro R., Weiskopf R.B., Moore M.A., et al. Humans anesthetized with sevoflurane or isoflurane have similar arrhythmic response to epinephrine. Anesthesiology. 1994;80:545.
17. Holzman R.S., van der Velde M.E., Kaus S.J., et al. Sevoflurane depresses myocardial contractility less than halothane during induction of anesthesia in children. Anesthesiology. 1996;85:1260-1267.
18. Davis P.J., Cook D.R., Stiller R.L., et al. Pharmacodynamics and pharmacokinetics of high-dose sufentanil in infants and children undergoing cardiac surgery. Anesth Analg. 1987;66:203.
19. Hickey P.R., Hansen D.D. Fentanyl- and sufentanil-oxygen-pancuronium anesthesia for cardiac surgery in infants. Anesth Analg. 1984;63:117.
20. Moore R.A., Yang S.S., McNicholas K.W., et al. Hemodynamic and anesthetic effects of sufentanil as the sole anesthetic for pediatric cardiovascular surgery. Anesthesiology. 1985;62:725.
21. Robinson S., Gregory G.A. Fentanyl-air-oxygen anesthesia for ligation of patent ductus arteriosus in preterm infants. Anesth Analg. 1981;60:331.
22. Hickey P.R., Hansen D.D. Fentanyl- and sufentanil-oxygen-pancuronium anesthesia for cardiac surgery in infants. Anesth Analg. 1984;63:117.
23. Olgatree M.L., Sprung J., Moravec C.S. Effects of remifentanil on the contractility of failing human heart muscle. J Cardiothorac Vasc Anesth. 2005;19(6):763-767.
24. Davis P.J., Galinkin J., McGowan F.X., et al. A randomized multicenter study of remifentanil compared with halothane in neonates and infants undergoing pyloromyotomy. I. Emergence and recovery profiles. Anesth Analg. 93, 2001. 1380138-1380136
25. Galinkin J.L., Davis P.J., McGowan F.X., et al. A randomized multicenter study of remifentanil compared with halothane in neonates and infants undergoing pyloromyotomy. II. Perioperative breathing patterns in neonates and infants with pyloric stenosis. Anesth Analg. 2001;93:1387-1392.
26. Hall J.E., Uhrich T.D., Barney J.A., et al. Sedative, amnestic, and analgesic properties of small-dose dexmedetomidine infusions. Anesth Analg. 2000;90:699-705.
27. Dyck J.B., Shafer S.L. Dexmedetomidine pharmacokinetics and pharmacodynamics. Anesth Pharm Rev. 1993;1:238-245.
28. Petroz G.C., Sikich N., James M., et al. A phase 1, two center study of the pharmacokinetics and pharmacodynamics of dexmedetomidine in children. Anesthesiology. 2006;105(6):1098-1110.
29. Rodarte A., Diaz S.M., Foley J., et al. The pharmacokinetics of dexmedetomidine in post-surgical pediatric intensive care unit patients: A preliminary study. Pediatr Crit Care Med. 2007;8(5):419-424.
30. Peden C.J., Cloote A.H., Stratford N., et al. The effect of intravenous dexmedetomidine premedication on the dose requirement of propofol to induce loss of consciousness in patients receiving alfentanil. Anaesthesia. 2001;56:408-413.
31. Ingersoll-Weng E., Manecke G.R., Thistlewaite P.A. Dexmedetomidine and cardiac arrest. Anesthesiology. 2004;100:738-739.
32. Berkenbosch J.W., Tobias J.D. Development of bradycardia during sedation with dexmedetomidine in an infant concurrently receiving digoxin. Pediatr Crit Care Med. 2003;4:203-205.
33. Bloor B.C., Ward D.S., Belleville J.P., et al. Effects of intravenous dexmedetomidine in humans, II: Hemodynamic changes. Anesthesiology. 1992;77:1134-1142.
34. Venn R.M., Newman P.J., Grounds R.M. A phase II study to evaluate the efficacy of Dexmedetomidine for the sedation in the medical intensive care unite. Intensive Care Med. 2003;29:201-207.
35. Khan Z.P., Munday I.T., Jones R.M. Effects of dexamedetomidine on isoflurane requirements in health volunteers. 1: Pharmacodynamic and pharmacokinetic interactions. Br J Anaesth. 1999;83:372-380.
36. Housmans P.R. Effects of dexmedetomidine on contractility, relaxation, and intracellular calcium transients of isolated ventricular myocardium. Anesthesiology. 1990;73:919-922.
37. Mukhtar A.M., Obayah E.M., Hassona A.M. The use of dexmedetomidine in pediatric cardiac surgery. Anesth Analg. 2006;103:52-56.
38. Kam P.C.A., Cardone D. Propofol infusion syndrome, Anaesthesia. 2007;62:690-701.
39. Bray R.J. Propofol infusion syndrome in children. Paediatr Anaesth. 1998;8:491-499.
40. Cremer O.L., Moons K.G.M., Bouman E.A.C., et al. Long term propofol infusion and cardiac failure in adult head-injured patients. Lancet. 2001;357:117-118.
41. Cannon M.L., Glazier S.S., Bauman L.A. Metabolic acidosis, rhabdomyolysis and cardiovascular collapse after prolonged propofol infusion. J Neurosurg. 2001;95:1053-1056.
42. Burow B.K., Johnson M.E., Packer D.L. Metabolic acidosis associated with propofol in the absence of other causative factors. Anesthesiology. 2004;101:239-241.
43. Kumar M.A., Urrutia V.C., Thomas C.E. The syndrome of irreversible acidosis after prolonged propofol infusion. Neurocrit Care. 2005;3:257-259.
44. Vasile B., Rasulo F., Candiani A., Latronico N. The pathophysiology of propofol infusion syndrome: a simple name for a complex syndrome. Intensive Care Med. 2003;29:1417-1425.
45. Wolf A., Weir P., Segar P., Stone J., Shield J. Impaired fatty acid oxidation in propofol infusion syndrome. Lancet. 2001;357:606-607.
46. Farag E., DeBoer G., Cohen B.H., et al. Metabolic acidosis due to propofol infusion. Anesthesiology. 2005;102:697-698.
47. Cray S.H., Robinson B.H., Cox P.N. Lactic acidaemia and bradyarrhythmia in a child sedated with propofol. Crit Care Med. 1998;26:2087-2092.
48. Culp K.E., Augoustides J.G., Ochroch A.E., Milas B.L. Clinical management of cardiogenic shock associated with prolonged propofol infusion. Anesth Analg. 2004;99:221-226.
49. Abrahams J.M., Reiter G.T., Acker M.A., et al. Propofol. J Neurosurg. 2002;96:1160-1161.
50. Barclay K., Williams A.J., Major E. Propofol infusion syndrome in children. BMJ. 1992;305:953.
51. Rothstein P. What hemoglobin level is adequate in pediatric anesthesia? Anesth Update. 1978;1:2.
52. Slogoff S. Anesthesia considerations in the anemic patient. Anesth Update. 1979;2:7.
53. Carson J.L., Poses R.M., Spence R.K., et al. Severity of anaemia and operative mortality and morbidity. Lancet. 1988;1:727.
54. Pfennigner J., Gerber A., Tschappeler H., et al. Adult respiratory distress syndrome in children. J Pediatr. 1982;101:352.
55. Domino K.B., Borowec L., Alexander C.M., et al. Influence of isoflurane on hypoxic pulmonary vasoconstriction in dogs. Anesthesiology. 1986;64:423.
56. Marshall C., Lindgren L., Marshall B.E. Effects of halothane, enflurane and isoflurane on hypoxic pulmonary vasoconstriction in rat lungs in vitro. Anesthesiology. 1984;60:304.
57. Ward J.D. Craniocerebral injuries. In: Burtain W.L., editor. Management of pediatric trauma. Philadelphia: WB Saunders; 1995:177-188.
58. Hoyert D.L., Heron M.P., Murphy S.L., Kung H.C. Deaths: final data for 2003. Natl Vital Stat Rep. 2006;54:1-120.
59. Luerrsen T.G., Klauber M.R., Marshall L.F. Outcome from head injury related to patient’s age: a longitudinal prospective study of adult and pediatric head injury. J Neurosurg. 1988;68:409-416.
60. Kochanek P.M., Clark R., Ruppel R., et al. Biochemical, cellular, and molecular mechanisms in the evolution of secondary damage after severe traumatic brain injury in infants and children: lesions learned from the bedside. Pediatr Crit Care Med. 2000;11:4-19.
61. Kennedy C., Sokoloff L. An adaptation of the nitrous oxide method to the study of the cerebral circulation in children; normal values for cerebral blood flow and cerebral metabolic rate in childhood. J Clin Invest. 1957;36:1130-1137.
62. Ng S.C., Poon W.S., Chan M.T. Is transcranial Doppler ultrasonography (TCD) good enough in determining CO2 reactivity and pressure autoregulation in head-injured patients? Acta Neurochir Suppl. 2002;81:125-127.
63. Karsli C., Luginbuehl I., Farrar M., Bissonnette B. Cerebrovascular carbon dioxide reactivity in children anaesthetized with propofol. Paediatr Anaesth. 2003;13:26-31.
64. Raju T.N.K., Doshi U.V., Vidyasagar D. Cerebral perfusion pressure studies in healthy preterm and term newborn infants. J Pediatr. 1982;100:139.
65. Todd M.M., Drummond J.C. A comparison of the cerebrovascular and metabolic effects of halothane and isoflurane in the cat. Anesthesiology. 1984;60:276.
66. Scheller M.S., Todd M.M., Drummond J.C. Isoflurance, halothane, and regional cerebral blood flow at various levels of PaCO2 in rabbits. Anesthesiology. 1986;64:598.
67. Artru A.A., Powers K., Doepfner P. CSF, sagittal sinus, and jugular venous pressures during desflurane or isoflurane anesthesia in dogs. J Neurosurg Anesthesiol. 1994;6:239.
68. Ornstein E., Young W.L., Fleischer L.H., et al. Desflurane and isoflurane have similar effects on cerebral blood flow in patients with intracranial mass lesions. Anesthesiology. 1993;79:498.
69. Rampil I.J., Lockhart S.H., Eger E.I., et al. The electroencephalographic effects of desflurane in humans. Anesthesiology. 1991;74:434.
71. Bedforth N.M., Hardman J.G., Nathanson M.H. Cerebral hemodynamic response to the introduction of desflurane: a comparison with sevoflurane. Anesth Analg. 2000;91:152-155.
72. Strebel S., Lam A.M., Matta B., et al. Dynamic and static cerebral autoregulation during isoflurane, desflurane, and propofol anesthesia. Anesthesiology. 1995;83:66-76.
73. Muzzi D.A., Lasasso T.J., Dietz N.M., et al. The effect of desflurane and isoflurane on cerebrospinal fluid pressure in humans with supratentorial mass lesions. Anesthesiology. 1992;76:720-724.
74. Summors A., Gupta A., Matta B. Dynamic cerebral autoregulation during sevoflurane anesthesia: a comparison with isoflurane. Anesth Analg. 1999;88:341-345.
75. Gupta S., Heath K., Matta B.F. Effect of incremental doses of sevoflurane on cerebral pressure autoregulation in humans. Br J Anesth. 1997;79:469-472.
76. Matta B.F., Heath K.J., Tipping K., et al. Direct cerebrovascular vasodilatory effects of sevoflurane and isoflurane. Anesthesiology. 1999;91:677-680.
77. Nishiyama T., Matusukawa T., Yokoyama T., et al. Cerebrovascular carbon dioxide reactivity during general anesthesia: a comparison between sevoflurane and isoflurane. Anesth Analg. 1999;89:1437-1441.
78. Komatsu H., Taie S., Endo S., et al. Electrical seizures during sevoflurane anesthesia in two pediatric patients with epilepsy. Anesthesiology. 1997;81:1535.
79. Woodforth I.J., Hicks R.G., Crawford M.R., et al. Electroencephalographic evidence of seizure activity under deep sevoflurane anesthesia in a nonepileptic patient. Anesthesiology. 1997;87:1579-1582.
80. Young W.L. Effects of desoflurane on the central nervous system. Anesth Analg. 1992;75:S32.
81. Statler K.D., Kochanek P.M., Dixon C.E., et al. Isoflurane improves long-term neurologic outcome versus fentanyl after traumatic brain injury in rats. J Neurotrauma. 2000;12:1179-1189.
82. Hendrich K.S., Kochanek P.M., Melick J.A., et al. Cerebral perfusion during anesthesia with fentanyl, isoflurane, or pentobarbital in normal rats studied by arterial spin-labeled MRI. Magn Reson Med. 2001;46:202-206.
83. Engelhard K., Werner C., Mollenberg O., Kochs E. Effects of remifentanil/propofol in comparison with isoflurane on dynamic cerebrovascular autoregulation in humans. Acta Anaesthesiol Scand. 2001;45:971-976.
84. Van Hemelrijck J., Fitch W., Mattheuseen M., et al. Effect of propofol on cerebral circulation and autoregulation in the baboon. Anesth Analg. 1990;71:49.
85. Alkire M.T., Haier R.J., Barker S.J., et al. Cerebral metabolism during propofol anesthesia in humans studied with positron emission tomography. Anesthesiology. 1995;82:393.
86. Abrahams J.M., Reiter G.T., Acker M.A., Sinson G.P. Propofol. J Neurosurg. 2002;96:1160-1161.
87. Dam M., Ori C., Pizzolato G., et al. The effects of propofol anesthesia on local cerebral glucose utilization in the rat. Anesthesiology. 1990;73:499.
88. Pinaud M., Lelausque J.-N., Chetanneau A., et al. Effects of propofol on cerebral hemodynamics and metabolism in patients with brain trauma. Anesthesiology. 1990;73:404.
89. Matuschak G.M., Pinsky M.R., Rogers R.M. Effects of positive end-expiratory pressure on hepatic blood flow and performance. J Appl Physiol. 1987;62:1377.
90. Fujita Y., Sakai T., Ohsumi A., et al. Effects of hypocapnia and hypercapnia on splanchnic circulation and hepatic function in the beagle. Anesth Analg. 1989;69:152.
91. Gelman S., Dillard E., Bradley E.L.Jr. Hepatic circulation during surgical stress and anesthesia with halothane, isoflurane, or fentanyl. Anesth Analg. 1987;66:936.
92. Gelman S. General anesthesia and hepatic circulation. Can J Physiol Pharmacol. 1987;65:1762.
93. Frank E.J., Morgan S.E., Coetzee A., et al. The effects of sevoflurane, halothane, enflurane, and isoflurane on hepatic blood flow and oxygenation in chronically instrumented greyhound dogs. Anesthesiology. 1992;76:85.
94. Merin R.G., Bernard J.M., Doursout M.F., et al. Comparison of the effects of isoflurane and desflurane on cardiovascular dynamics and regional blood flow in the chronically instrumented dog. Anesthesiology. 1991;74:568-574.
95. Davis P.J., Cook D.R. Anesthetic problems in pediatric liver transplantation. Transplant Proc. 1989;21:3493.
96. Davis P.J., Stiller R.L., Cook D.R., et al. Effects of cholestatic hepatic disease and chronic renal failure on alfentanil pharmacokinetics in children. Anesth Analg. 1989;68:579.
97. Williams R.L. Drug administration in hepatic disease. N Engl J Med. 1984;309:1616.
98. Dershwitz M., Rosow C.E. The pharmacokinetics and pharmacodynamics of remifentanil in volunteers with severe hepatic or renal dysfunction. J Clin Anesth. 1996;8:88S-90S.
99. Hoke J.F., Shlugman D., Dershwitz M., et al. Pharmacokinetics and pharmacodynamics of remifentanil in persons with renal failure compared with healthy volunteers. Anesthesiology. 1997;87:531-541.
100. Brandom B.W., Stiller R.L., Cook D.R., et al. Pharmacokinetics of atracurium in anaesthetized infants and children. Br J Anaesth. 1986;58:1210.
101. Green D.W., Fisher M., Sockalingham I. Mivacurium compared with succinylcholine in children with liver disease. Br J Anaesth. 1998;81:463-465.
102. deWolf A.M., Freeman J.A., Scott V.L., et al. Pharmacokinetics and pharmacodynamics of cisatracurium in patients with end-stage liver disease undergoing liver transplantation. Br J Anaesth. 1996;76:624-628.
103. Hartert H. Blutgerninnungstudien mit der thromboeslastographic, einen neven untersuchingsver fahren. Klin Wochenschr. –260, 1948;16:257.
104. Luddington R.J. Thrombelastography/thromboelastometry. Clin Lab Haematol. 2005;27:81-90.
105. Mallett S.V., Cox D.J. Thromboelastography. Br J Anesth. 1992;69:307-313.
106. Depotis G.J., Joist J.H., Goodnough L.T. Monitoring of hemostasis in cardiac surgical patients: impact of point-of-care testing on blood loss and transfusion outcomes. Clin Chem. 1997;43(9):1684-1696.
107. Kang Y.G., Martin D.J., Marquez J., et al. Intraoperative changes in blood coagulation and thrombelastographic monitoring in liver transplantation. Anesth Analg. 1985;64:888-896,.
108. Michael T: Ganter. MD, Christoph K, et al: Coagulation monitoring: current techniques and clinical use of viscoelastic point-of-care coagulation devices. Anesth Analg. 2008;106:1366-1375.
109. Salooja N., Perry D.J. Thrombelastography, Blood Coag Fibrinol. 2001;12(5):327-337.
110. Andrew M., Vegh P., Johnston M. Maturation of the hemostatic system during childhood. Blood. 1992;80:1998-2005.
111. Miller B.E., Bailey J.M., Mancuso T.J. Functional maturity of the coagulation system in children: an evaluation using thrombelastography. Anesth Analg. 1997;84:745-748.
112. Dzik W.H., Arkus C.F., Jenkins R.L., Stump D.C. Fibrinolysis during liver transplantation in humans: role of tissue type plasminogen activator. Blood. 1988;71:1090-1095.
113. Porte R.J., Bontempo F.A., Knot E.A. Systemic effects of tissue plasminogen activator-associated fibrinolysis and its relation to thrombin generation in orthotopic liver transplantation. Transplantation. –984, 1989;47:978.
114. Pivalizza. Heparinase and thrombelastography in liver transplantation for a patient with von Willebrand’s disease. Anesthesiology. 1996;84:1236-1239.
115. Kaspar M., Ransay M., Nguyen A.-T. Continuous small-dose tranexamic acid reduces fibrinolysis but not transfusion requirements during orthotopic liver transplantation. Anesth Analg. 1997;85:281-285.
116. McSorelty M.W., Taraporewalla K.J. Does prophylactic epsilon aminocaproic acid improve blood loss and coagulation in liver transplantation. Transplant Proc. 1941;1991:23.
117. Hedner U. Recombinant coagulation factor VIIa: from concept to clinical application in hemophilia treatment in 2000. Semin Thromb Hemost. 2000;26:363-366.
118. Shapiro A.D., Gilchrist G.S., Hoots W.K., et al. Prospective, randomized trial of two doses of rFVIIa (NovoSeven) in haemophilia patients with inhibitors undergoing surgery. Thromb Haemost. 1998;80:773-778.
119. Butenas S., Brummel K.E., Branda R.F., et al. Mechanism of factor VIIa-dependent coagulation in hemophilia blood. Blood. 2002;99:923-930.
120. ten Cate H., Bauer K.A., Levi M., et al. The activation of factor X and prothrombin by recombinant factor VIIa in vivo is mediated by tissue factor. J Clin Invest. 1993;92:1207-1212.
121. Hedner U. Recombinant activated factor VII as a universal hemostatic agent. Blood Coagul Fibrinolysis. 1998;9:S147-S152.
122. Villar A., Aronis S., Morfini M., et al. Pharmacokinetics of activated recombinant coagulation factor VII in children vs. adults with hemophilia A. Haemophilia. 2004;10:352-359.
123. Hedner U., Kristensen H., Berntrop E., et al. Pharmacokinetics of rFVIIa in children. Haemophilia. 1998;4:355.
124. Brown J.B., Emerick K.M., Brown D.L., et al. Recombinant factor VIIa improves coagulopathy caused by liver failure. J Pediatr Gastroenterol Nutr. 2003;37:268-272.
125. Chuansumrit A., Chantarojanasiri T., Isarangkura P., et al. Recombinant factor VIIa in children with acute bleeding resulting from liver failure and DIC. Blood Coagul Fibrinolysis. 2000;11(suppl 1):S101-S105.
126. Kalicinski P., Kaminski A., Drewniak T., et al. Quick correction of hemostasis in two patients with fulminant liver failure undergoing liver transplantation by recombinant activated factor VII. Transplant Proc. 1999;31:378-379.
127. Kalicinski P., Markeiwicz M., Kaminski A., et al. Single pretransplant bolus of rFVIIa ameliorates influence of risk factores for blood loss during orthotopic liver transplantation. Pediatr Transplant. 2005;9:299-304.
128. Hedner U., Erhardtsen E. Potential role for rFVIIa in transfusion medicine. Transfusion. 2002;42:114-124.
129. Hendricks H.G., Meijer K., de Wolf J.T., et al. Effects of rFVIIa on coagulation measured by thromboelastography in liver transplantation. Blood Coagul Fibrinol. 2002;13:309-313.
130. Bell C.F., Hunter J.M., Jones R.S., et al. Use of atracurium and vecuronium in patients with oesophageal varices. Br J Anaesth. 1985;57:160.
131. Bencini A.F., Scaf A.H.J., Sohn Y.J., et al. Disposition and urinary excretion of vecuronium bromide in anesthetized patients with normal renal function or renal failure. Anesth Analg. 1986;65:245.
132. deBros F.M., Lai A., Scott R., et al. Pharmacokinetics and pharmacodynamics of atracurium during isoflurane anesthesia in normal and anephric patients. Anesth Analg. 1986;65:743.
133. Chapple D.J., Miller A.A., Ward J.B., et al. Cardiovascular and neurological effects of laudanosine: studies in mice and rats, and in unconscious and anaesthetized dogs. Br J Anesth. 1987;59:218.
134. Yate P.M., Flynn P.J., Arnold R.W., et al. Clinical experience and plasma laudanosine concentrations during the infusion of atracurium in the intensive therapy unit. Br J Anaesth. 1987;59:211.
135. Cook D.R., Freeman J.A., Lai A.A., et al. Pharmacokinetics of mivacurium in normal patients and in those with hepatic or renal failure. Br J Anaesth. 1992;69:580-585.
136. Boyd A.H., Eastwood N.B., Parker C.J.R., et al. Pharmacodynamics of the 1R cis-1¢R cis isomer of atracurium (51W89) in health and chronic renal failure. Br J Anaesth. 1995;174:400-404.
137. Magorian T., Wood P., Caldwell J., et al. The pharmacokinetics and neuromuscular effects of rocuronium bromide in patients with liver disease. Anesth Analg. 1995;80:754-759.
138. Moore E.W., Hunter J.M. The new neuromuscular blocking agents: do they offer any advantages? Br J Anaesth. 2001;87:912-925.
139. Davis P.J., Stiller R.L., Cook D.R., et al. Pharmacokinetics of sufentanil in adolescent patients with chronic renal failure. Anesth Analg. 1988;67:268.
140. Davis P.J., Stiller R.L., Cook D.R., et al. Effects of cholestatic hepatic disease and chronic renal failure on alfentanil pharmacokinetics in children. Anesth Analg. 1989;68:579.
141. Chauvin M., Sandouk P., Schermann J.M., et al. Morphine pharmacokinetics in renal failure. Anesthesiology. 1987;66:327.
142. Osborne R., Joel S., Grebenik K. The pharmacokinetics of morphine and morphine glucuronides in kidney failure. Clin Pharmacol Ther. 1993;54:158.
143. Kharasch E.D., Hankins D.C., Thummel K.E. Human kidney methoxyflurane and sevoflurane metabolism. Anesthesiology. 1995;82:689.
144. Fang Z.X., Eger E.I.II, Laster M.J., et al. Carbon monoxide production from degradation of desflurane, enflurane, isoflurane, halothane, and sevoflurane by soda lime and Baralyme. Anesth Analg. 1995;89:1187-1193.
145. Bito H., Ikeuchi Y., Ikekda K. Effects of low flow sevoflurane anesthesia on renal function. Anesthesiology. 1997;86:1231-1237.
146. Conzen P.F., Nuscheler M., Melotte A., et al. Renal function and serum fluoride concentrations in patients with stable renal insufficiency after anesthesia with sevoflurane or enflurane. Anesth Analg. 1995;81:569-575.
147. Ebert T.J., Frink E.J., Kharasch E.D. Absence of biochemical evidence for renal and hepatic dysfunction after 8 hours of 1.25 minimum alveolar concentration sevoflurane anesthesia in volunteers. Anesthesiology. 1998;88:601-610.
148. Eger E.I.2nd, Koblin D.D., Bowland T., et al. Nephrotoxicity of sevoflurane versus desflurane anesthesia in volunteers. Anesth Analg. 1997;84:160-168.
149. Higuchi H., Adachi Y., Wada H., et al. The Effects of low-flow sevoflurane and isoflurane anesthesia on renal function in patients with stable moderate renal insufficiency. Anesth Analg. 2001;92:650.
150. Higuchi H., Sumita S., Wada H., et al. Effects of sevoflurane and isoflurane on renal function and on possible markers of nephrotoxicity. Anesthesiology. 1998;89:307-322.
151. Kharasch E.D., Frink E.J.Jr., Zager R., et al. Assessment of low-flow sevoflurane and isoflurane effects on renal function using sensitive markers of tubular toxicity. Anesthesiology. 1997;86:1238-1253.
152. Ross A.K., Davis P.J., Dear G.L., et al. Pharmacokinetics of remifentanil in anesthetized pediatric patients undergoing elective surgery or diagnostic procedures. Anesth Analg. 2001;93:1393-1401.
153. Hoke J.F., Shlugman D., Dershwitz M., et al. Pharmacokinetics and pharmacodynamics of remifentanil in persons with renal failure compared with healthy volunteers. Anesthesiology. 1997;87:531-541.
154. Egan T.D., Lemmens H.J., Fiset P., et al. The pharmacokinetics of the new short-acting opioid remifentanil (GI87084B) in healthy adult male volunteers. Anesthesiology. 1993;79:881-892.
155. Glass P., Hardman D., Kamiyama Y. Preliminary pharmacokinetics and pharmacodynamics of an ultra-short-acting opioids: remifentanil (GI87084B). Anesth Analg. 1993;77:1031-1040.
156. Ellis D., Avner E.D., Starzl T.E. Renal failure in children with hepatic failure undergoing liver transplantation. J Pediatr. 1986;108:393.
157. Elliott M.J., Alberti K.G.M.M.Carbohydrate metabolism: effects of preoperative starvation and trauma Biebuyck J.F., editor. Nutritional aspects of anesthesia. Clinics in anesthesiology, vol 1. Philadelphia: WB Saunders, 1983.
158. Bedford R.F. Hyperosmolar hyperglycemia nonketotic coma following general anesthesia: report of a case. Anesthesiology. 1971;35:652.