Chapter 48 Diseases of Pulmonary Circulation
Early referral to expert centers is crucial to patient survival.
Developmental Pulmonary Vascular Anatomy
Embryology
Distinctions can be recognized between the proximal and distal pulmonary vasculature both in terms of their embryonic origins and the morphogenetic processes by which they develop. The sixth branchial arch is the embryonic origin of the proximal pulmonary vasculature, and it develops by the process of vasculogenesis. Vasculogenesis is the differentiation and segregation of angioblasts within the mesenchyme, which forms early vascular channels (arteries, veins, and lymphatics), depending on local influences from the epithelium and the mesenchyme. The lung mesenchyme is the embryonic origin of the distal vasculature, which develops by angiogenesis. Angiogenesis is the formation of new vessels from preexisting vascular channels by proliferation and migration of endothelial cells at the tips, which form multiple capillary sprouts. Abnormal maturation or maturational arrest in pulmonary arterial development is reflected in functional derangement that can appear in the newborn period. Persistent pulmonary hypertension of the newborn (PPHN) has been reported in association with pulmonary arterial maturational arrest at week 5 of gestation.1 Normal growth and development of the pulmonary circulation in utero is critical for achieving successful transition to postnatal life.
Multiple factors influence development and growth of pulmonary vasculature in utero, including growth factors. Growth factors such as vascular endothelial growth factor2 and fibroblast growth factor3 appear to be responsible for orderly growth and branching morphogenesis of blood vessels. Quantity, timing, and location are critical determinants of the net effects of these agents. The mechanical stress that endothelial cells must withstand may be the predominant force responsible for development of large vessels after onset of circulation.4
Vascular Smooth Muscle
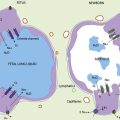
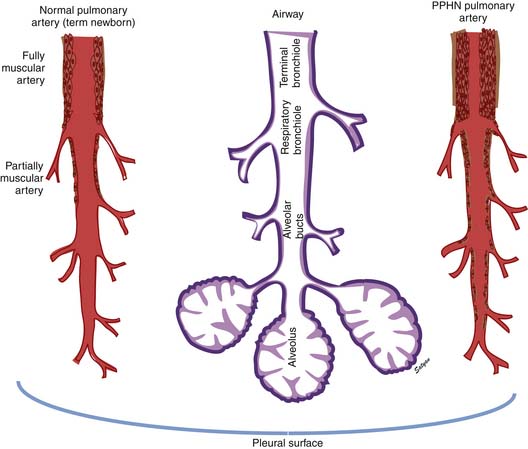
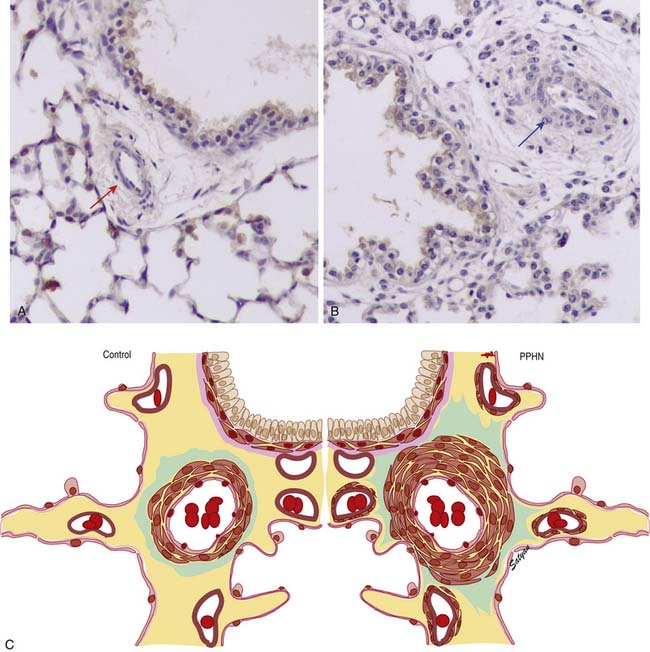
In the normal fetal and term lung, fully muscularized thick-walled preacinar arteries extend to the level of terminal bronchioles, whereas the intra-acinar arteries (i.e., those accompanying respiratory bronchioles) are partially muscular (surrounded by a spiral of muscle) or nonmuscular. Arteries at alveolar ducts and alveolar walls are nonmuscular. Preacinar arteries in the fetus late in gestation and in the newborn have thicker coats of smooth muscle relative to the arterial diameter than do similar arteries in adults, although the fetus actually has less distal extension of smooth muscle in smaller arteries than do older children or adults.5 Figure 48-1 shows the diagrammatic representation of the extension of arterial smooth muscle within the acinus in a normal term newborn and in infants with severe PPHN.6 An increase in intrauterine vascular smooth muscle, resulting from peripheral extension of smooth muscle into vessels that do not normally contain muscle layers, may contribute to the pathophysiology of PPHN7 (see Figure 48-1). Increased muscularization of the pulmonary arteries has been described in infants with severe meconium aspiration syndrome (MAS) with PPHN.8 Neonates who die of PPHN may have a striking distal extension of smooth muscle in the intra-acinar region, thickening of the media and adventitia, and excessive accumulation of the matrix protein in the pulmonary vessels.9 The increase in vascular smooth muscle and its peripheral extension can occur either prenatally or postnatally. In utero ductal ligation 1 to 2 weeks prior to delivery results in severe PPHN at birth associated with distal extension of vascular smooth muscle in newborn lambs10 (Figure 48-2) similar to the changes seen in human infants dying with severe PPHN.
Developmental Pulmonary Vascular Physiology
Hemodynamic Features of Fetal Circulation
Oxygenated blood (PaO2 approximately 30 to 40 mm Hg) returning from the placenta in the umbilical vein11 splits in the liver with slightly more than half passing through the ductus venosus to the inferior vena cava (IVC). The oxygenated blood streams along the medial aspect of the IVC as it enters the right atrium (Figure 48-3). Approximately two thirds of the IVC flow is directed toward the foramen ovale by the eustachian valve and the septum primum and enters the left atrium. The remaining third of the IVC flow mixes with the blood from the superior vena cava and enters the right ventricle. The majority of the right ventricular (RV) output enters the ductus arteriosus and the descending aorta. A small portion enters the lungs via the pulmonary arteries.12 The ratio of blood flow to the pulmonary arteries to the flow that traverses the ductus arteriosus is determined by the fetal pulmonary vascular resistance (PVR).
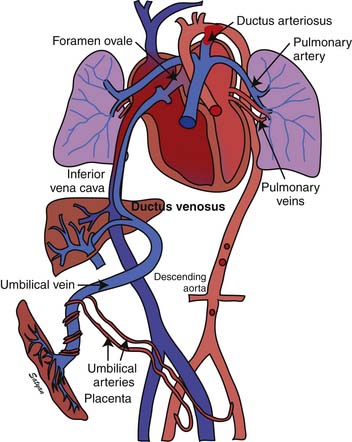
Figure 48–3 Fetal circulation showing the shunting of blood at the ductus venosus, foramen ovale, and ductus arteriosus.
(Copyright Satyan Lakshminrusimha.)
The fetal pulmonary circulation fulfills a unique function. Close to term, fetal lungs in the lamb receive about 5% to 10% of combined ventricular output to meet the metabolic demands of an actively growing organ. More recent data in human fetuses using Doppler echocardiography suggest that approximately 25% of combined ventricular output may enter the lungs near term.13 The presence of large fetal shunts, the foramen ovale and the ductus arteriosus (see Figure 48-3), allows the fetal lung to regulate the amount of blood flow it receives by active vasoconstriction. The distribution of combined ventricular output, measured in fetal lambs ranging from 60 to 150 days gestational age, indicates that the lungs receive only about 3.5% of total output at 0.4 term, but the fraction increases to almost 8% at term.14 This remarkable increase in fetal pulmonary perfusion presumably is related to growth of the fetal lung. The lung increases in weight by sixfold from midgestation to term, and the number of pulmonary vessels increases more than tenfold.15,16 Thus the tremendous increase in cross-sectional area of pulmonary vasculature permits pulmonary blood flow (Qp) to increase throughout gestation. However, Qp remains relatively constant when corrected for wet lung weight with advancing gestation.17 A gradual increase in pulmonary arterial pressure (PAP), accompanied by a relatively constant flow per unit lung tissue, results in increasing fetal PVR with advancing gestation.17 A major part of this increase comes from elevated pulmonary vascular tone associated with low PO2 in the fetal lung.18,19 Maintaining high PVR in utero is important because the function of gas exchange is performed not by the lungs but by the placenta.
Regulation of Pulmonary Vascular Tone in Utero
Low oxygen tension and various mediators play a crucial role in maintaining elevated fetal PVR.20 Vasoconstriction in response to low oxygen tension contributes to high PVR in the fetal lamb as it approaches term.18,19 Decreasing oxygen tension in fetuses at 103 to 104 days of gestation does not increase PVR, but it doubles resistance in fetuses at 132 to 138 days. Conversely, increasing oxygen tension does not change PVR before 100 days of gestation, but it decreases resistance markedly and increases blood flow to normal newborn levels at 135 days of gestation.17
Arachidonic acid metabolites are some of the most powerful vasoconstrictors known. Prostaglandin (PG) F2α and thromboxane A2 (TXA2) are synthesized by the fetal lung via the cyclooxygenase pathway21 and are pulmonary vasoconstrictors in the fetal and newborn lungs. However, TXA2 does not appear to be responsible for maintenance of high PVR in the fetus.22 Blocking PG or thromboxane synthesis does not decrease fetal PVR but prevents the decrease in PVR in response to rhythmic distension of the lung in fetal lambs.23 The cytochrome P450 metabolism of arachidonic acid results in the formation of epoxyeicosatrienoic acids, dihydroxyeicosatetraenoic acids, and 20-hydroxyeicosatetraenoic (HETE) acids. Of these compounds, HETE acids have been shown to constrict pulmonary circulation in newborn piglets.24 However, inhibition of 20-HETE did not reduce basal PVR in fetal lambs.25 Thus it does not appear that cyclooxygenase and CYP450 metabolites of arachidonic acid cause the high vascular tone of the fetal lung.
Leukotrienes are formed from arachidonic acid through the 5-lipooxygenase pathways. Lipoxygenase activity has been demonstrated in human fetal lung as early as 12 to 18 weeks of gestation.26 Leukotriene inhibition has been shown to decrease PVR in fetal lambs by 45%.27 It also reverses hypoxic pulmonary vasoconstriction in newborn lambs but not in newborn piglets.28,29 The specific role of leukotrienes in maintaining high PVR in immature fetuses is unclear. It is possible that they play a role in pathologic states such as hypoxia or inflammation.
Endothelins (ETs) are 21-residue peptides30,31 whose role in regulating vascular tone and vasomotor responses has been studied intensively in the past decade. Three distinct ET isoforms have been described: endothelin-1 (ET-1), endothelin-2 (ET-2), and endothelin-3 (ET-3), cleaved from ET precursors big ET-1, big ET-2, and big ET-3, respectively, by an ET-converting enzyme. ET-1 synthesized by vascular endothelial cells is a potent vasoconstrictor,32 and its effects in both animal and human studies vary with the tone of the pulmonary vessels, dose of ET-1, and the maturation of vessels.33,34 Of the ETs, ET-1 is the best characterized, and its actions of fetal pulmonary circulation are best studied. Both ET-1 and ET-2 have been shown to dilate the fetal (normally high tone) pulmonary vasculature and constrict the bed when the tone is reduced by ventilation.35 Thus it appears that the response of the pulmonary vasculature to ETs is tone-dependent. In contrast, infusion of big ET-1, the precursor of ET-1, into fetal lambs causes sustained pulmonary vasoconstriction,36 suggesting that this might be the predominant effect of endogenous ET-1.37 Currently at least two receptor subtypes, ETA and ETB, are thought to mediate responses to ETs (Figure 48-4). The ETB receptor plays a role in vasodilation and the ETA receptor plays a role in vasoconstriction. Selective blockade of the ETA receptor causes fetal pulmonary vasodilation.38–40 Some investigators suggest a significant role for an endothelial ETB receptor in vasodilation and a smooth muscle ETB receptor and ETA receptor in vasoconstriction.41,42 Vasoconstriction induced by ET-1 is mediated by calcium,43 whereas the vasodilator properties are mediated by endothelium-derived nitric oxide (NO).42,44 ET-1 may play a role in the change that occurs in the pulmonary vasculature at birth. It is possible that prenatally, endogenous ET-1 primarily stimulates ETA receptors to cause vasoconstriction and that ETB receptors are less active in fetal life.38 However, ETB receptors mediate the vasodilator responses to ET-1 in the fetus, and there is a suggestion that an abundance of ETB receptors may be of physiologic importance in decreasing PVR at birth.40
Transitional Circulation
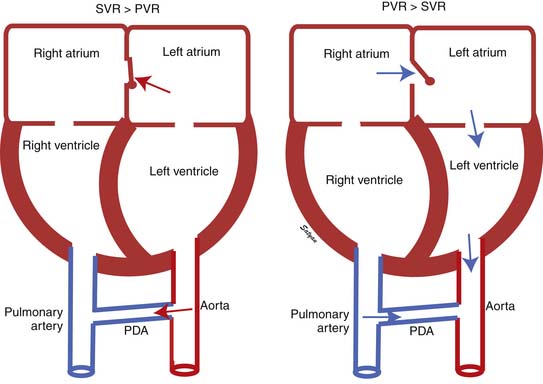
The first stage of transitional circulation is essentially a fetal pulmonary circulation that is characterized by high pressure and low flow because of both passive and active elevation of PVR (Figure 48-5, A). The passive resistance most likely is related to compression of pulmonary capillaries by fetal lung liquid, but there is also a high degree of active vasomotor tone resulting from various mediators and hypoxic stimuli. Thus PVR exceeds systemic vascular resistance (SVR), resulting in right atrial and ventricular pressures exceeding left atrial and ventricular pressures. High PVR results in right-to-left shunting of blood across the foramen ovale, and most of the blood ejected by the right ventricle flows across the ductus arteriosus into the descending aorta. Persistence of elevated PVR after birth without the benefit of placental oxygenation results in the profound hypoxemia that characterizes PPHN.
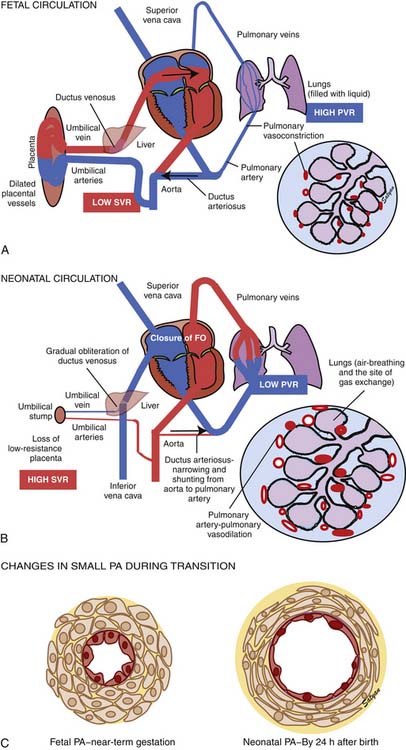
The second stage of normal transition is accomplished when the fluid-filled fetal lungs are distended with air during the first breath (Figure 48-5, B).20 A rapid decrease in PVR occurs with mechanical distension of the pulmonary vascular bed. The entry of air into the alveoli improves oxygenation of the pulmonary vascular bed, further decreasing PVR.45 At birth PVR decreases dramatically, which leads to an eightfold increase in Qp. The increase in Qp raises left atrial pressures above right atrial pressures, closing the foramen ovale. SVR increases at birth, in part because of removal of the low resistance bed of the placenta. As PVR becomes less than systemic, flow across the ductus reverses. Within the first 5 minutes after birth, oxygen-induced vasodilation and lung expansion decrease PVR to approximately half of systemic resistance. Over the first few hours after birth, the ductus arteriosus closes, largely in response to the increase in oxygen tension. At this point the normal postnatal circulatory pattern is established.
The third stage of the transitional circulation occurs for 12 to 24 hours after birth and accounts for the greatest reduction in PVR. In the final phase of neonatal pulmonary vascular transition, further decline in PVR is accompanied by rapid structural remodeling of the entire pulmonary bed from the main pulmonary arteries to the capillaries.46 During this remodeling, changes in the shape and geometric orientation of endothelial and smooth muscle cells cause luminal enlargement. Maturation of smooth muscle function, thinning of endothelial cells (Figure 48-5, C), and more gradual changes in elastic and connective tissue occur during the next few weeks.
Factors Responsible for Decrease in Pulmonary Vascular Resistance at Birth
The onset of ventilation with rhythmic inflation of the lungs at birth with a resultant increase in oxygen tension in the lungs leads to a decrease in PVR. Each of these stimuli has been shown to decrease vascular resistance and increase blood flow in the lungs of fetal lambs.45 Increasing oxygen tension alone by using a hyperbaric chamber decreased PVR and increased Qp by tenfold in mature fetal lambs.17 Inflation of lungs with gas may decrease resistance, at least in part by mechanical effects. Oxygen may dilate in part by direct effects on vascular endothelial and smooth muscles.20,47
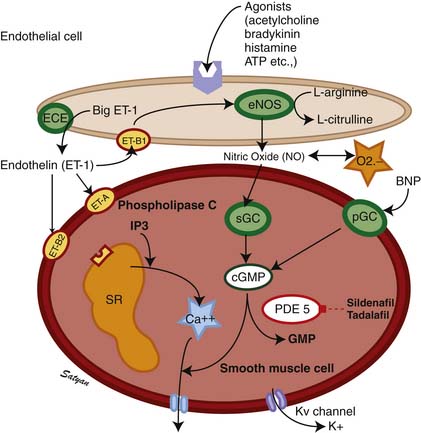
The drop in PVR soon after birth is accompanied by production of prostacyclin (PGI2) and NO. Arachidonic acid metabolites such as PGs, generated through the cyclooxygenase pathway, are potent pulmonary vasodilators in the fetus. PGI2 synthesized by endothelial cells appears to relax smooth muscle by producing cyclic adenosine monophosphate (cAMP). PGI2 and its metabolites are more potent vasodilators than PGE2. Even though blockade of PG synthesis, either by indomethacin48,49 or meclofenamate,23 blunts the decrease in PVR, it does not completely disrupt the transition to gas exchange. In addition, cyclooxygenase through the PGE2 pathway plays an important role in maintaining patency of the ductus arteriosus. PPHN has been observed in infants of mothers receiving aspirin or nonsteroidal antiinflammatory drugs (NSAIDs) that inhibit cyclooxygenase activity.50 In this situation, the inhibitor acts through prenatal constriction of the ductus arteriosus or by decreasing PGI2 synthesis at birth.51 The cAMP signal transduction pathway is shown in Figure 48-6.
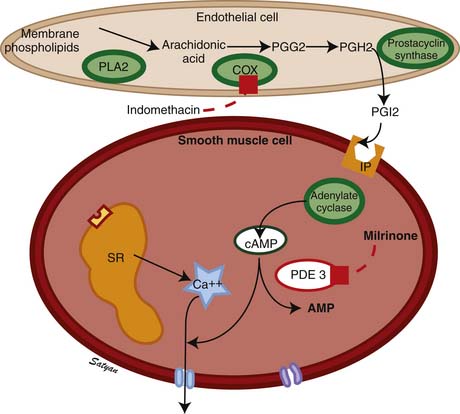
Acetylcholine,52 bradykinin,53 and histamine54 are fetal pulmonary vasodilators, which in many species act by an endothelium-dependent mechanism (see Figure 48-4). They stimulate the production of NO by vascular endothelium. NO activates soluble guanylate cyclase to produce the second messenger cyclic guanosine monophosphate (cGMP). cGMP induces relaxation of vascular smooth muscle through activation of a cGMP-dependent protein kinase that produces a lowering of cytosolic ionic calcium, in part through activation of potassium channels.55 There is strong evidence that NO is an important mediator of the decrease in PVR at birth. NO is a potent dilator of the fetal pulmonary circulation.56 The dilation of the fetal pulmonary circulation caused by an increase in oxygen tension is mediated in large part by endogenous synthesis of NO.57 In late gestation lambs, prolonged administration of nitric oxide synthase (NOS) inhibitors, which blocks endogenous NO synthesis, does not affect basal PVR but markedly blunts the decrease in PVR observed at birth.58
Evidence suggests that many other mediators can act as pulmonary vasodilators during fetal life and at birth. The purines adenosine triphosphate and adenosine are potent pulmonary vasodilators in the fetal lamb that may also be involved at birth.59–63 Natriuretic peptides such as atrial natriuretic peptide, B-type natriuretic peptide, and C-type natriuretic peptide dilate fetal pulmonary vasculature by increase cGMP through particulate guanylate cyclase.64 Arachidonic acid metabolites such as epoxyeicosatrienoic acids, which are generated through the cytochrome P450 pathway, are potent pulmonary vasodilators.65 Because pulmonary vasodilation at birth is a vital step in establishing postnatal life, it is logical that there would be sufficient redundant vasodilators to compensate for failure or inadequacy of any single pathway.37
Persistent Pulmonary Hypertension of the Newborn
PPHN is a serious clinical condition that can result from diverse etiologies and is characterized by failure of the pulmonary circulation to adapt to extrauterine life.
Pathophysiology
Elevated pulmonary to systemic vascular resistance ratio (PVR/SVR) resulting from either vasoconstriction, structural remodeling of the pulmonary vasculature, intravascular obstruction, or lung hypoplasia (Figure 48-7) characterizes PPHN. This leads to right-to-left shunting of blood across the foramen ovale and ductus arteriosus, resulting in hypoxemia. Numerous disease states with diverse etiologies can result in a similar final pathophysiology. About 10% of cases with PPHN are idiopathic, with no associated pulmonary airspace pathology. However, PPHN is usually associated with other acute respiratory conditions, such as MAS, respiratory distress syndrome (RDS), pneumonia, or congenital diaphragmatic hernia (CDH) (Figure 48-8). Hypoxemia in these conditions can be due to ventilation/perfusion (V/Q) mismatch and intrapulmonary, as well as extrapulmonary, right-to-left shunting of blood. In some newborns with hypoxic respiratory failure, a single mechanism predominates (e.g., extrapulmonary right-to-left shunting in idiopathic PPHN). However, more commonly, several of these mechanisms contribute to hypoxemia. In MAS, obstruction of the airways by meconium results in decreasing V/Q ratios and increasing intrapulmonary right-to-left shunt. Other segments of the lungs may be overventilated relative to perfusion, causing increased physiologic dead space. The same patient also may have severe PPHN with extrapulmonary right-to-left shunting at the ductus arteriosus and foramen ovale. The PPHN in patients with MAS may result from the alveolar hypoxia, from inflammatory mediators, or from abnormal pulmonary vascular muscularization.
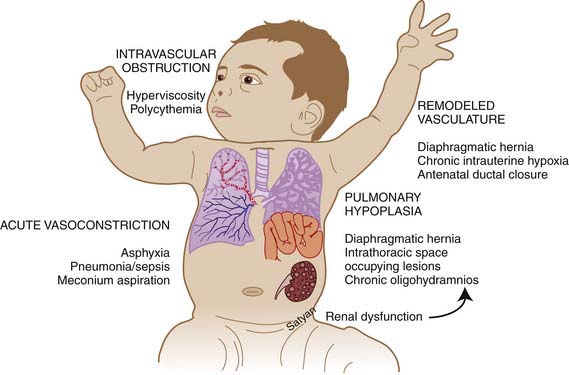
Figure 48–7 Mechanisms of persistent pulmonary hypertension of the newborn.
(Copyright Satyan Lakshminrusimha.)
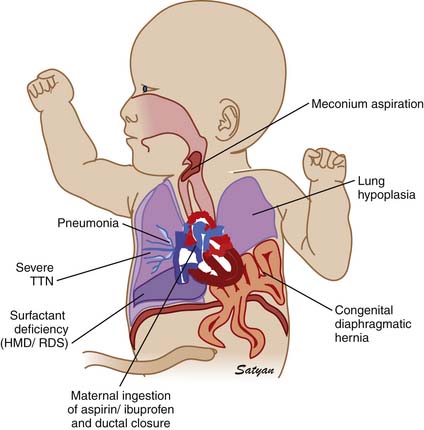
Figure 48–8 Clinically common conditions associated with persistent pulmonary hypertension of the newborn.
(Copyright Satyan Lakshminrusimha.)
Pneumonia or meconium aspiration may release inflammatory mediators that induce vasoconstriction. Vasoconstrictors such as leukotrienes, platelet-activating factor, thromboxanes,66 and ET-167 have been found to be elevated in patients with PPHN. Chronic intrauterine ETA receptor blockade following ductal ligation decreases pulmonary arterial pressure in utero and decreases RV hypertrophy, and distal muscularization of small pulmonary arteries increases the fall in PVR at delivery in newborn lambs with PPHN.39 Thus ET-1 acting through the ETA receptor stimulation might contribute to the pathogenesis and pathophysiology of PPHN. Derangements in the NO pathway of vasodilation also can result in the physiologic characteristics of PPHN. Pulmonary endothelial nitric oxide synthase (eNOS) gene and protein expression and enzyme activity are decreased in fetal lambs with PPHN induced by antenatal ductal ligation.68 In addition, the response to stimulators of eNOS is lost.69 In these lambs with PPHN, the vascular response to NO itself is also diminished,70 whereas the response to cGMP is normal. Thus the decreased responsiveness appears to result from decreased vascular smooth muscle sensitivity to NO at the level of soluble guanylate cyclase. The cGMP pathway of signal transduction is shown in Figure 48-4. Because NO both vasodilates and inhibits vascular smooth muscle growth, diminished eNOS expression may contribute to both abnormal vasoreactivity and excessive muscularization of pulmonary vessels in patients with PPHN.
PH sometimes occurs because of an abnormal pulmonary vascular bed despite the absence of alveolar hypoxia and hypercapnia and of lung inflammation. These infants can be grouped according to the degree of muscularization and the number of pulmonary arteries.71 In infants with hypoplastic lungs, as in those with congenital heart disease (CHD) and oligohydramnios sequence, PPHN may arise primarily as a consequence of a decreased number of vessels causing a decreased cross-sectional area of the pulmonary vascular bed, leading to flow restriction (see Figure 48-7). Patients with alveolar capillary dysplasia may have a similar vascular hypoplasia. These cases may be complicated by increased muscularization of the vessels.
Antenatal exposure to NSAIDs such as aspirin and ibuprofen has been associated with PPHN.50 More recently, an association between the use of selective serotonin reuptake inhibitor class of antidepressants after 20 weeks of gestation and PPHN has been found.72,73 Prenatal exposure to fluoxetine (a selective serotonin reuptake inhibitor) induced fetal PH in rats.74 The exact mechanism of this association is not clear. Polycythemia and hyperviscosity also may increase PVR and contribute to PPHN. Similarly, hypothermia, acidosis, and hypoxemia can aggravate PPHN.
Clinical Presentation
Labile hypoxemia is the hallmark of PPHN (Figure 48-9). A newborn infant who is extremely labile, with frequent desaturation episodes and wide swings in arterial PO2 without changes in ventilator settings, should suggest the possibility of PPHN. Lability also can occur in the face of significant parenchymal disease when V/Q mismatch is severe. Auscultation of the heart in babies with PPHN may reveal a single S2, which can be loud, and a systolic murmur of tricuspid regurgitation. Chest radiograph findings may vary depending on the etiology of PPHN. Hypoxemia out of proportion to the degree of parenchymal disease severity on chest radiography should suggest PPHN. Measurement of preductal and postductal arterial oxygenation can confirm PPHN. A difference in arterial PO2 20 mm Hg or greater or oxygen saturation 10% or greater should be considered significant. Minimal or no difference in oxygen tension does not exclude PPHN because shunting at the atrial level produces no ductal gradient and probably is the most common site of shunting. Thus in clinical practice, the gold standard in defining PPHN rests on the echocardiographic findings of right-to-left shunting of blood at the foramen ovale and/or the ductus arteriosus, as well as estimates of PAP. Doppler measurements of atrial and ductal level shunts provide essential information when managing a newborn with hypoxic respiratory failure.75 For example, left-to-right shunting at the foramen ovale and ductus arteriosus with marked hypoxemia suggests predominant intrapulmonary shunting, and interventions should be directed at optimizing lung inflation. Similarly, presence of right-to-left shunting at the ductal level and left-to-right shunting at the atrial level suggests PPHN with left ventricular dysfunction with some pulmonary venous hypertension (see Table 48-1). This finding may be associated with CHD.76
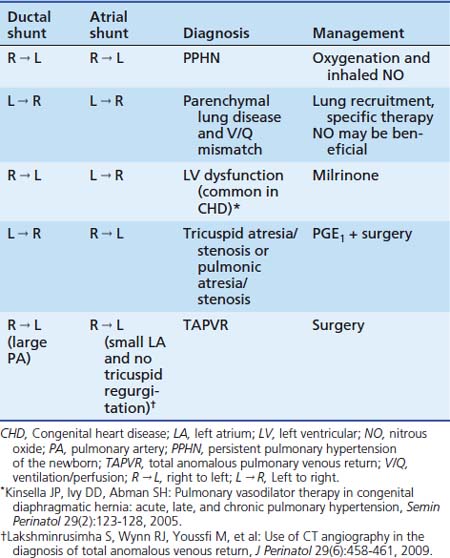
Treatment
General Measures
Understanding the dynamic pathophysiology underlying right-to-left shunting is important to the successful management of PPHN. In patients with PPHN, even mild stress can cause PO2 to plummet within minutes. Vigorous and persistent resuscitative measures are often necessary to promote pulmonary vasorelaxation. Accordingly, neonates and children with PPHN are often very sensitive to activity and agitation. Minimal stimulation and sedation with narcotics such as fentanyl and morphine are commonly used to achieve this goal. However, paralysis should be avoided because it is associated with increased mortality.77 The lability in PaO2 may result from the fact that when pulmonary and systemic arterial pressures are similar, small alterations in the ratio of the two can produce large changes in extrapulmonary shunting (see Figure 48-8).78 Systemic blood pressures should be maintained at a high normal range for age and gestation, because an increased systemic resistance may decrease the degree of right-to-left shunting. Hypotension resulting from hypovolemia should be treated aggressively with volume replacement. If hypotension persists despite volume replacement, inotropic support with dopamine, dobutamine, and epinephrine may be required. All patients with PPHN benefit from a core group of therapies that includes management of hypothermia, hypocalcemia, acidosis, hypoglycemia, and polycythemia. In cases of PPHN resulting from perinatal asphyxia, correcting alveolar hypoxia, hypercarbia, and metabolic acidosis with administration of oxygen, conventional ventilation, and a buffer should restore normal pulmonary vasodilation. Surprisingly, few prospective randomized trials have been conducted of most of the therapeutic modalities advocated for the treatment of PPHN. Older therapies such as hyperventilation and alkalosis were introduced into clinical practice based on animal studies or short-term studies that included very small numbers of patients and used physiologic response rather than patient outcome as the end point. Only the newer therapies of inhaled nitric oxide (iNO) and extracorporeal membrane oxygenation (ECMO) have been rigorously evaluated in controlled clinical trials. In a National Institute of Child Health and Human Development (NICHD) observational study,77 the use of hyperventilation varied among the 12 NICHD centers from 33% to 92%, and alkali infusion ranged from 27% to 93%. Similar variation was seen in the use of inotropic agents (46% to 100%) and intravenous vasodilators (13% to 81%). High-frequency ventilation (HFV) use ranged from 0% to 73%, and ECMO use varied from 0% to 85%. Some of the variation is expected given the wide range of pathophysiology contributing to PPHN. A more likely explanation for the wide variation seen, however, is the overall lack of proven efficacy of many of these therapies.
Hyperventilation and Alkali Infusion
Animal studies documented the sensitivity of the pulmonary vasculature to both hypoxia and acidosis.79 Short-term studies during cardiac catheterization showed reductions in PAP and elevation of oxygen tension following hyperventilation.78,80 Based on these studies, hyperventilation became the mainstay of ventilation of term infants with hypoxic respiratory failure and PPHN. In some patients the benefits of hyperventilation may be outweighed by risks of barotrauma or volutrauma. Moreover, subsequent observations have raised concern of impaired cerebral perfusion and neurosensory deafness at extremes of alkalosis.81,82 Studies of infants with PPHN maintaining normal PCO2 (40 to 60 mm Hg) indicate similar or better outcomes and with less chronic lung disease.83,84 Many neonatologists have moved away from the practice of hyperventilation in neonates with PPHN. Animal studies have shown that the beneficial effects of hyperventilation result from altered pH rather than from changes in PCO2 or minute ventilation.85,86 At one time alkali infusion to maintain alkaline pH to induce pulmonary vasodilation was a standard practice. In the aforementioned NICHD observational study, the group treated with alkali had a greater chance of treatment with ECMO compared with those treated with hyperventilation and an increased rate of supplemental oxygen at 28 days.77 Alkali infusion with bicarbonate increases CO2 production, necessitating higher ventilator support. Lack of patient outcome data with both hyperventilation and alkali infusion and availability of better therapeutic options have led to less use of these outdated management strategies.
Oxygen
Oxygen is a specific and potent pulmonary vasodilator, and increased oxygen tension is an important mediator of reduction in PVR at birth. Alveolar hypoxia and hypoxemia increase PVR and contribute to the pathophysiology of PPHN. Avoiding hypoxemia by mechanical ventilation with high concentrations of oxygen continues to be the mainstay of PPHN management. However, exposure to hyperoxia may result in formation of oxygen free radicals and lead to lung injury. Recent evidence suggests that brief exposure to 100% oxygen in newborn lambs results in increased contractility of pulmonary arteries87 and reduces response to inhaled NO.88,89 The biological half-life of endogenous NO is related to the local concentration of superoxide anions (see Figure 48-4).90 Administration of intratracheal recombinant human superoxide dismutase (an antioxidant that breaks down superoxide anions) results in improved oxygenation in lambs with PPHN.91,92 Based on these studies, it appears that avoiding hyperoxia is as important as avoiding hypoxia in the management of PPHN.
The optimal PaO2 in the management of PPHN is not clear. Wung and colleagues84 have suggested that gentle ventilation with avoidance of hyperoxia and hyperventilation results in good outcome in neonates with respiratory failure. Decreasing PaO2 below 45 to 50 mm Hg results in increased PVR in newborn calves79 and lambs.89 In contrast, maintaining PaO2 greater than 70 to 80 mm Hg does not result in additional decrease in PVR in both control lambs and lambs with PPHN. In animal studies, hypoxemia results in pulmonary vasoconstriction and normoxemia reduces PVR but hyperoxemia does not result in additional pulmonary vasodilation. However, to date, randomized studies comparing different PaO2 targets have not been conducted in infants with PPHN.
Lung Recruitment Strategies
Surfactant
Clinical reports suggest that surfactant therapy improves oxygenation in term infants with RDS, pneumonia, and MAS.93,94 Surfactant administration results in reduced need for ECMO in infants of 36 weeks or greater gestation at birth. Near-term and term infants delivered by elective repeat cesarean section may be one subset of patients who are at risk for deficiency or dysfunction of surfactant and progressive hypoxic respiratory failure.95 If the cause of PPHN is parenchymal lung disease such as RDS, meconium aspiration or pneumonia, exogenous surfactant may be beneficial in improving oxygenation and reducing the need for ECMO.94
High-Frequency Ventilation
Many clinicians use HFV to manage infants with PPHN. Considering the important role of parenchymal lung disease in specific disorders resulting in PPHN, adequate lung inflation and optimal ventilation are as essential as pharmacologic vasodilator therapy. In the case of inhaled vasodilators, optimal inflation and ventilation may be necessary for drug delivery.96 Infants with PPHN from a variety of causes have been successfully treated with HFV.97 High-frequency oscillatory ventilation (HFOV) decreases PaCO2 and increases oxygenation in infants with PPHN. HFOV may improve oxygenation through safer use of higher mean airway pressures to maintain lung volume and prevent atelectasis. Two studies have evaluated the effectiveness of HFV compared with conventional ventilation in rescuing infants with respiratory failure and PPHN from potential ECMO therapy.98,99 Neither mode of ventilation was more effective in preventing ECMO in these infants. In clinical pilot studies using iNO, combination of HFOV and iNO resulted in the greatest improvement in oxygenation in some newborns who had severe PPHN complicated by diffuse parenchymal lung disease and underinflation.100 A randomized controlled trial demonstrated that treatment with HFOV and iNO was often successful in patients who failed to respond to HFOV or iNO alone in severe PPHN, and the differences in responses were related to the specific disease associated with PPHN. Infants with RDS and MAS benefit most from a combination of HFOV and iNO therapy.101,102
Nitric Oxide
In 1996 the Food and Drug Administration (FDA) approved iNO for use in neonates who are 35 weeks’ gestation and older with PPHN. The physiologic rationale for using iNO for treatment of PPHN103,104 is based on its ability to achieve potent and selective pulmonary vasodilation without decreasing systemic vascular tone (Figure 48-9). Once iNO enters the intravascular space, it combines with hemoglobin to form methemoglobin and does not exert a vasodilator effect on the systemic circulation. Inhaled NO also exerts a microselective effect and reduces V/Q mismatch. Being an inhaled vasodilator, NO enters only ventilated alveoli and redirects pulmonary blood by dilating adjacent pulmonary arterioles and reduces V/Q mismatch (Figure 48-10). Studies in newborn lambs have shown that prolonged administration of NO increased survival rates without increasing the incidence of acute lung injury in lambs with PPHN.105
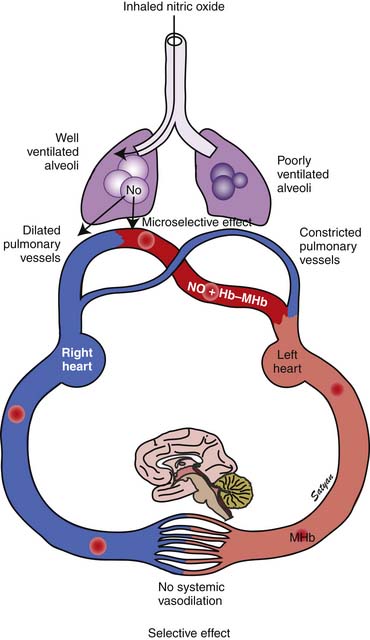
Three randomized trials on the use of iNO in newborns with PPHN and respiratory failure were published in 1997. In a randomized controlled clinical trial of patients with PPHN reported by Roberts and colleagues,106 oxygenation doubled in 58% of treated patients in response to 80 ppm iNO. In addition, twice the proportion of the treated group avoided ECMO compared with the control group. In the Neonatal Inhaled Nitric Oxide Study, 235 infants older than 34 weeks’ gestation who were diagnosed with hypoxic respiratory failure were randomly assigned to receive 20 ppm iNO or were assigned to a control group. Infants whose partial pressure of arterial oxygen increased by 20 mm Hg or less were studied for a response to 80 ppm iNO or control gas. The end point of this trial was death or ECMO. Although the mortality rate was no different in either treatment arm, there was a 40% reduction in the need for ECMO among infants treated with iNO compared with control subjects.107 Only 6% of infants who failed to respond to treatment with 20 ppm of iNO responded to the higher dose in this study. In a study by Kinsella and colleagues,102 205 infants with PPHN were randomly assigned to receive iNO and conventional ventilation or to receive HFOV alone. Those who did not respond to either therapy received the combination of iNO and HFOV. Treatment with HFOV in combination with iNO was successful in some patients who did not respond to one treatment alone. The differences were partly related to the specific disease (RDS and MAS) associated with PPHN.102 Because adequate lung inflation appears to be necessary for optimal response to iNO, lung recruitment strategies with HFOV should augment the response to iNO. Clark and colleagues108 reported a 38% reduction in ECMO and no difference in mortality among 248 infants randomly assigned to receive 20 ppm iNO for 24 hours followed by 5 ppm for no more than 96 hours. A meta-analysis of the results of seven randomized trials of iNO use in newborns with PPHN demonstrated that 58% of hypoxic near-term and term infants responded to iNO within 30 to 60 minutes.109 Mortality was not reduced in any of the NO studies analyzed, but use of ECMO as a rescue therapy in nonresponders was significantly decreased.
Inhaled NO by itself is toxic at higher concentrations. Potential adverse effects include methemoglobinemia, pulmonary edema, and platelet dysfunction. NO reacts with superoxide anion to form peroxynitrite, which causes lipid peroxidation and other oxidative injury to cell membranes. NO2 is even more toxic. Careful monitoring of both NO and NO2 levels during administration is mandatory. Because the optimal dosing and timing of iNO administration remains unclear and the potential toxicities are dose-related, lower doses might afford both safety and efficacy in the management of these infants. Two studies using 2 ppm iNO yielded contradictory results. In one study, 2 ppm iNO diminished the clinical response to 20 ppm.110 In the other study, the initial exposure to a very low dose did not compromise the response to higher doses.111 In a study involving direct measurements of PAP during cardiac catheterization, iNO produced peak improvement in oxygenation at 5 ppm, whereas peak improvement in the pulmonary-to-systemic arterial pressure ratio did not occur until an iNO dose of 20 ppm, which suggests that an initial dose of 20 ppm is optimum for the treatment of PPHN.112 The use of higher doses of iNO is associated with increased methemoglobin levels, especially at 80 ppm but not at 40 or 20 ppm.113 The results of randomized controlled trials support the use of iNO at starting does of 20 ppm in near-term and term infants. In summary, iNO offers substantial benefit to a large proportion of near-term and term newborns with hypoxic respiratory failure who do not respond to ventilatory support, lung recruitment, and oxygen.
The timing of initiation of iNO in patients with hypoxic respiratory failure is not clear. Severity of hypoxemia in neonates is measured by oxygenation index (OI = Mean airway pressure in cm of water × FIO2 × 100/PaO2 [in mm Hg]). An OI of 40 is often used as an indication for ECMO therapy. An OI of 25 is associated with a 50% risk of requiring ECMO or dying.37 Thus the acceptable indication for treatment with iNO include an OI greater than 25 with echocardiographic evidence of PPHN or a higher OI with or without evidence of right-to-left shunt. However, it has been suggested that initiating iNO at a lower OI may be associated with reduced need for ECMO.114 Konduri and colleagues115 randomly assigned neonates who were born at 34 weeks’ gestation or later, required assisted ventilation, and had an OI of 15 or greater and less than 25 to receive early iNO or to receive simulated initiation of iNO (control). Infants who had an increase in OI to 25 or more were given iNO as standard therapy. Arterial oxygen tension increased by more than 20 mm Hg in 73% of infants who received early iNO (n = 150) and in 37% of infants in the control group (n = 149) after study gas initiation. Infants in the control group received standard iNO and deteriorated to an OI score greater tha 40 more often than did infants who were given early iNO. The incidence of death (early iNO group, 6.7% vs. control group, 9.4%), ECMO (10.7% vs. 12.1%), and their combined incidence (16.7% vs. 19.5%) were similar in both groups.115 Follow-up evaluations showed no differences between the 2 groups in the incidence of neurodevelopmental impairment (early iNO group, 27%; control group, 25%) and hearing impairment (early iNO group, 23%; control group, 24%). Mental development index scores were similar in the two groups; however, psychomotor developmental index scores were significantly higher in the control group (early iNO group, 89 ± 17.7; control group, 93.5 ± 18.4).116 These findings suggest that caution must be exercised while initiating iNO at lower OI levels.
Inhaled NO should be weaned gradually to avoid rebound PH.117 A flow diagram showing the protocol for weaning iNO at The Women and Children’s Hospital of Buffalo is shown in Figure 48-11.
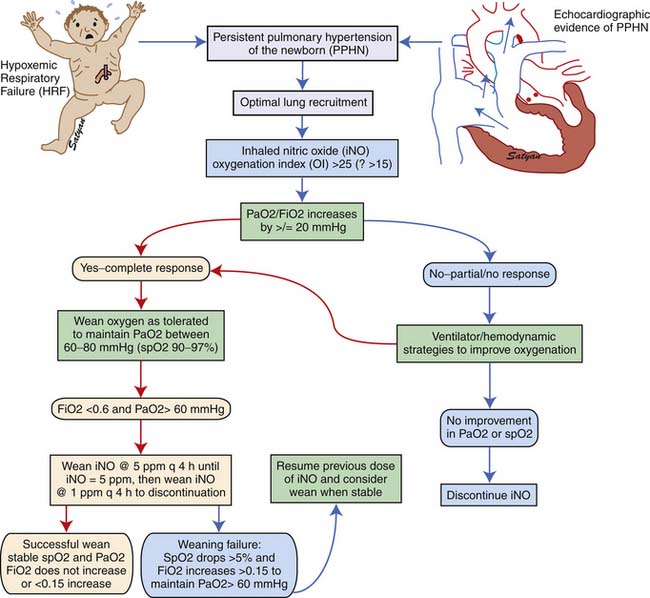
Figure 48–11 Weaning protocol for inhaled nitric oxide in use at The Women and Children’s Hospital of Buffalo, NY.
The outcome of infants treated with iNO collectively supports both the efficacy and safety of this mode of treatment. Reports identify significant medical and neurodevelopmental sequelae of PPHN with or without iNO and point out the necessity for coordinated multidisciplinary follow-up for these infants. The overall rate of neurodevelopmental handicap in infants treated with NO was 46%, with 25% mildly affected and 21% severely affected at age 1 year in one study.118 In another study, mild and severe neurodevelopmental handicaps were 14% and 12% at age 1 year and 9% and 12% at age 2 years.119 In these studies sensorineural hearing loss was present in 6% to 19% of infants with PPHN treated with iNO. No difference was noted between control and treatment groups for these outcomes. Published reports on the use of iNO in ECMO centers have not substantiated early concerns that iNO would adversely affect outcome by delaying ECMO utilization. Inhaled NO treatment may play an important role in stabilizing patients before ECMO is initiated, thus improving the chances of ECMO cannulation without further clinical deterioration. The Committee on the Fetus and Newborn of the American Academy of Pediatrics has suggested that iNO use be limited to tertiary care centers where ECMO is available.120 In non-ECMO centers, a system should be in place to continue iNO during transport even if a response occurs, because not all physiologic responders avoid ECMO. For the same reason, the combination of HFOV and iNO should be used cautiously in non-ECMO centers.
Phosphodiesterase Inhibitors
Vasodilators such as NO and prostacyclin relax vascular smooth muscle by increasing intracellular concentrations of the second messengers cGMP (see Figure 48-4) and cAMP, respectively (see Figure 48-5). However, nearly 40% of patients with PPHN do not demonstrate a sustained improvement in oxygenation with this management. Because iNO is not universally effective, there has been considerable interest in understanding and targeting other biochemical pathways that regulate pulmonary vasoconstriction in PPHN. Inhibition of the cGMP degrading phosphodiesterase (PDE5) by sildenafil and inhibition of the cAMP degrading phosphodiesterase (PDE3) by milrinone offer additional tools to achieve pulmonary vasodilation in patients with PPHN. Two studies have evaluated the use of oral sildenafil in PPHN.121,122 In centers lacking iNO therapy, oral sildenafil (dose range 1 to 3 mg/kg every 6 hours) has been shown to improve oxygenation and reduce mortality. A recent study showed that intravenous (IV) sildenafil was effective in improving oxygenation in patients with PPHN with and without prior exposure to iNO.123 Systemic hypotension was the most common adverse effect. Administration of a loading dose slowly over 3 hours followed by a maintenance dose of sildenafil reduced the risk of systemic hypotension. These data suggest a beneficial effect for oral as well as IV sildenafil in patients with PPHN. Because oral or IV agents are not selective to pulmonary circulation, caution must be exercised about the risk of systemic hypotension with oral sildenafil. IV sildenafil is not available currently in the United States, but the same caution regarding systemic hypotension exists. The use of oral sildenafil for treatment of PH is approved by the FDA for adults but not for children.
Milrinone is the prototype PDE3 inhibitor and is commonly used in adult and pediatric intensive care settings as an inotropic vasodilator. Agents that increase cAMP levels in pulmonary arterial smooth muscle cells (such as milrinone and prostacyclin) provide an alternate pathway of pulmonary vascular relaxation and potentially result in improved oxygenation in patients with poor response to iNO. In newborn lambs with PPHN, milrinone effectively relaxes pulmonary arteries124 and improves oxygenation.125 There is a marked increase in PDE3 activity and reduced cAMP in pulmonary arteries following ventilation of lambs with iNO.126 Milrinone can be effective in PPHN by inhibiting PDE3 and increasing cAMP, causing direct pulmonary vasodilation and a synergistic effect with iNO. Milrinone may also improve cardiac function by positive inotropy (improved contraction), lusitropy (improved relaxation), and reduced ventricular afterload. Bassler and colleagues127 and McNamara and associates128 described 13 patients with PPHN refractory to iNO from Ontario, Canada, who were treated effectively with intravenous milrinone. This therapy is associated with a risk of hypotension and intraventricular hemorrhage and is not currently approved by the FDA.
Extracorporeal Membrane Oxygenation
If the heart and lungs cannot support the newborn, they can be bypassed with ECMO. Several randomized trials have indicated improved survival of infants supported with ECMO. In a large prospective trial conducted in the United Kingdom, 121 infants with severe respiratory failure were randomly assigned to ECMO or conventional management.129 Survival in the patients treated with ECMO was significantly greater than in the control group (68% vs. 41%). Neurologic outcome was similar among survivors of either treatment arm, indicating that ECMO likely did not contribute to morbidity in this group of critically ill infants. ECMO has been shown to be both clinically129 and economically130 justifiable for mature newborn infants with severe respiratory failure. ECMO is not a specific treatment for any disease but rather a method of supportive treatment, in which the patient is kept alive while the lungs and their vasculature recover. As newer treatment modalities including HFOV, surfactant therapy, and NO have become available for treatment of hypoxemic respiratory failure, ECMO use in newborns has decreased considerably.131,132 Because of serious inherent risks, such as systemic and intracranial hemorrhage, for infants with PPHN, the procedure presently is reserved for newborn infants with reversible pulmonary disease in whom alternative therapies have failed. However, ECMO should be initiated before the infant is moribund.
Pulmonary Arterial Hypertension in Children
After the newborn period, pulmonary arterial hypertension (PAH) in children is an uncommon disease characterized by elevated PAP either at rest or during exercise. Progressive obliteration of the pulmonary vascular bed is the hallmark of PAH. Primary pulmonary hypertension (PPH) is a rare disease in children that predominantly affects adolescents and young people in whom no apparent cause for PAH can be identified. In secondary PAH, a coexisting disease can be identified that presumably explains the PAH. As per World Health Organization (WHO) classification,133 the term PAH refers to a disease spectrum with a similar clinical and pulmonary histopathology that includes PPH and PAH, which cannot be distinguished from PPH. This includes patients with PPH, both sporadic and familial, as well as pulmonary arterial hypertension associated with collagen vascular disorders, congenital systemic to pulmonary shunts, human immunodeficiency virus, CHD, portal hypertension, hemoglobinopathies, myeloproliferative disorders, and appetite suppressants (Box 48-1). Familial PAH often results from a mutation in the bone morphogenic protein receptor-2 and is inherited as an autosomal dominant disease with incomplete penetrance. These groups together comprise WHO Group I PAH. Other WHO categories include Group II, PH with left heart disease; Group III, associated with lung disease and/or hypoxemia; Group IV, PH due to chronic thrombotic and/or embolic disease; and Group V, miscellaneous cause of PAH. Despite recent advances leading to early recognition and new therapies, PAH remains a serious life-threatening condition. Most of the abnormalities in PAH have been linked to the recent advances in our understanding of smooth muscle cell physiology, endothelial and platelet dysfunction, vascular remodeling, and genetics to provide insight into its pathophysiology. It is important to establish an accurate diagnosis with respect to etiology of PAH because therapy may vary with etiology. For instance, thromboembolic disease is often amenable to surgery with excellent results from thromboendarterectomy. Similarly, secondary PAH may result from a left-sided heart lesion with pulmonary venous hypertension.
Box 48–1 Revised WHO Classification of PH
From McLaughlin VV, Archer SL, Badesch DB, et al: Expert consensus document on pulmonary hypertension, J Am Coll Cardiol 53(17):1573-1619, 2009.
HIV, Human immunodeficiency virus; PH, pulmonary hypertension; WHO, World Health Organization.
Pathophysiology
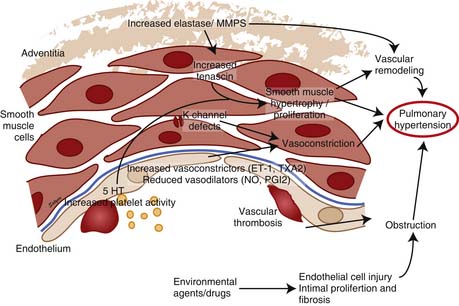
PAH is a syndrome resulting from restricted flow through the pulmonary arterial circulation, which leads to pathologic increases in PVR and ultimately to right heart failure. The mechanisms responsible for the development of PAH are complex and incompletely understood. In persons with PPH, hyperreactive lung vessels in which various stimuli initiate vasoconstriction may explain the subsequent development of characteristic vascular lesions.134 The vascular endothelium is regarded as an important source of locally active mediators that contribute to the control of vasomotor tone. The thromboxane/PGI2 and NO/ET-1 system are two of the important balances that control pulmonary vasomotor tone. Endothelial injury/dysfunction may lead to vascular remodeling and progressively increasing vascular obstruction and loss of vascular luminal cross-section and obliteration (Figure 48-12). Risk factors that are associated with PPH include the use of anorexic drugs, family history of PH, infection with human immunodeficiency virus, cirrhosis, and the use of cocaine or intravenous drugs.135
ET-1 is a potent vasoconstrictor and stimulates pulmonary artery smooth muscle cell (PASMC) proliferation. Plasma levels of ET-1 are increased in patients with PAH and correlate with the severity of PAH and prognosis.136 Of the two ET-1 receptors, ETa receptors, which are expressed mainly in the smooth muscle, mediate vasoconstriction and cell proliferation, whereas ETb receptors, which are expressed mainly in the endothelial cells, are important for clearance of ET-1, release of NO and PGI2, and inhibition of endothelin-converting enzyme-1.30,137 In experimental studies of hypoxia- induced138 and monocrotaline-induced PH,139 chronic ET receptor blockade lowered PAP and the incidence of vascular and pulmonary injury and improved NO-mediated pulmonary vasodilation. Studies with L-ω-nitroarginine methyl ester–induced PH suggest that ET-1 is linked to the dysfunction of the L-arginine/NO pathway140 because ETa selective141 but not combined ET blockade142 improves endothelial function. Thus selective inhibition of ETa receptors improves the endothelial L-arginine/NO pathway that agrees with observations in humans.143 ET-1 expression in pulmonary arteries is increased in patients with primary and secondary PH.144,145 ET-1 increases at high altitudes in mountaineers and correlates with pulmonary pressures and oxygen tension.146 ET increases even more in mountaineers prone to high altitude pulmonary edema.147 Increased local production145 and elevated circulating levels of endothelin148 have been demonstrated in patients with Eisenmenger syndrome,149 PPH,150 and in PAH associated with CHD.151
PGI2 is a powerful vasodilator and an inhibitor of platelet aggregation, whereas TXA2 is a vasoconstrictor and induces platelet aggregation leading to microvascular thrombus formation and hence vascular injury. Because TXA2 and PGI2 have opposing effects on platelet aggregation and pulmonary vascular smooth muscle, an imbalance in their biosynthesis could contribute to the progressive increase in PVR seen in older untreated patients with pulmonary hypertensive CHD.152 Decreased urinary excretion of 2,3-dinor-6-keto prostaglandin F1α, a metabolite of PGI2, occurs in patients with PPH.153 In patients with PAH, the balance between these two molecules is shifted toward TXA2,153 favoring thrombosis, proliferation, and vasoconstriction. A decrease in expression of the enzyme PGI2 synthase (PGI2-S) in the lung may be an important manifestation of pulmonary endothelial dysfunction in patients with severe PH. PGI2-S is decreased in small- and medium-sized pulmonary arteries, and the loss of expression of PGI2-S may represent one of the phenotypic alterations present in the pulmonary endothelial cells in patients with severe PAH.154 Pulmonary PGI2-S overexpression in transgenic mice protects against development of hypoxic PH.155 PGI2-S may play a major role in modifying the pulmonary vascular response to injury. Intratracheal transfer of human PGI2-S gene has been shown to augment pulmonary prostacyclin synthesis, ameliorate monocrotaline (MCT)-induced PH and improve survival in MCT rats.156
Potassium (K+) channels play an important part in smooth muscle cell electrophysiology, which has implications for the development of PAH. The inhibition of voltage-gated K+ (Kv) channels results in an accumulation of positively charged K+ ions within the cell, hence depolarizing the cell and activating the voltage-gated, L-type calcium channel.157 Calcium then enters the cell, activating the contractile apparatus, leading to vasoconstriction, and possibly initiating cell proliferation. Acute hypoxia seems to initiate vasoconstriction in part by inhibiting the Kv channel in PASMC.157 In humans with PPH, Kv1.5 messenger ribonucleic acid (mRNA) levels are reduced in PASMC.158 This down-regulation of Kv1.5 is associated with inhibition of the K+ current, membrane depolarization, and elevation of cytosolic Ca++. Thus decreased expression or function of K+ channels in PASMC in patients with PPH could initiate and/or maintain pulmonary vasoconstriction and play a role in the pathogenesis of PPH.158 The use of anorexic agents like aminorex, fenfluramine, and dexfenfluramine is associated with the development of PPH,135 and these agents are thought to act by blocking the Kv channel.159 Kv2.1 is inhibited by dexfenfluramine, a weight-loss drug that is associated with the development of PAH.159 Anorexigen-induced Kv channel inhibition and membrane depolarization can contribute to pulmonary vasoconstriction.160,161 Fenfluramine reduces the Kv1.5 mRNA levels by 50% in PASMC from normotensive patients,162 suggesting that inhibited gene transcription and expression of Kv channels may play an important role in anorexigen-induced PAH.
Matrix metalloproteinases (MMP) and extracellular matrix are thought to be important in vascular remodeling and hence proliferation of SMCs. Endothelial abnormalities early in the course of PAH may permit extravasation of factors that stimulate SMC production of vascular serine elastase.163,164 This results in the liberation of matrix-bound SMC mitogens, such as basic fibroblast growth factor, and enhances matrix degradation by activating other MMPs. The MMPs stimulate the production of mitogenic cofactor, tenascin, leading to phosphorylation of growth factor receptors and smooth muscle cell (SMC) proliferation. When MMPs are inhibited, tenascin levels fall, leading to apoptosis.163 Direct inhibition of MMP-2 and serine elastases lead to complete regression of experimental PH in rats,165 suggesting that they regulate vascular tone. MMP-2 and MMP-9 can activate platelets,166 and intravascular MMP-2 can enhance the formation of vasoconstrictors and inhibit the action of endogenous vasodilators.167
A number of other less well-studied factors potentially are involved in the pathogenesis of PH. Abnormalities of the thrombomodulin/protein C anticoagulant system with a decrease in soluble thrombomodulin and an increase in fibrinolytic inhibitor plasminogen activator 1 have been noted in patients with PPH.168 Whether hypercoagulability occurs in response to PAH or can actually initiate PAH is unclear, but it likely contributes to disease progression. Serotonin (5-hydroxytryptamine or 5-HT) and serotonin transporter (5-HTT) have been shown to promote development of hypoxic PH by stimulating PASMC growth.169 5-HTT activity may play a role in the pathogenesis of PASMC proliferation in PPH, and a 5-HTT polymorphism may confer susceptibility to PPH.170 Mutations in the 2 genes in the transforming growth factor-β receptor pathway BMPR2 and activin-like kinase 1, have been implicated in the pathogenesis of familial PAH.171,172 Many different BMPR2 mutations occur in familial PAH. These mutations, which lead to loss of function in the SMAD signaling pathway, are prevalent in familial PAH.171 Activin-like kinase-1 mutations, detected in a group of patients with hereditary hemorrhagic telangiectasia and PAH,172 are also thought to result in growth-promoting alterations of SMAD-dependent signaling.
Pulmonary Vascular Histopathology
PAH is a panvasculopathy predominantly affecting small pulmonary arteries and is characterized by a variety of arterial abnormalities, including intimal hyperplasia, medial hypertrophy, adventitial proliferation, thrombosis in situ, varying degrees of inflammation,and plexiform arteriopathy (Figure 48-13). Heath-Edwards introduced the first comprehensive classification of PAH based on the histopathology of pulmonary vasculature173 in children with CHD or idiopathic PH (Table 48-2). Grade I referred to changes seen in patients with CHD associated with PH from birth. Grades II to IV were classified as a result of raised PAP. Grades V and VI often overlap and represent advanced disease. Rabinovitch174 introduced a morphometric classification system in 1978 by studying lung biopsies of 50 patients with CHD (Table 48-3). The presence and severity of pathologic lesions correlated with increased pulmonary blood flow (PBF), elevated PAP, and elevated PVR. The most familiar patterns of pulmonary vascular lesions begin with medial hypertrophy followed by cellular proliferation and concentric laminar intimal fibrosis. Eventually dilatated lesions, fibrinoid necrosis, and plexiform lesions develop, and the disease is referred as Eisenmenger syndrome.175 The progressive vascular lesions that occur in patients with PPH are identical to those that occur with CHD involving systemic to pulmonary shunts.173 Based on these findings, similar therapeutic approaches have been used in children who have PPH and those who have PAH associated with CHD.
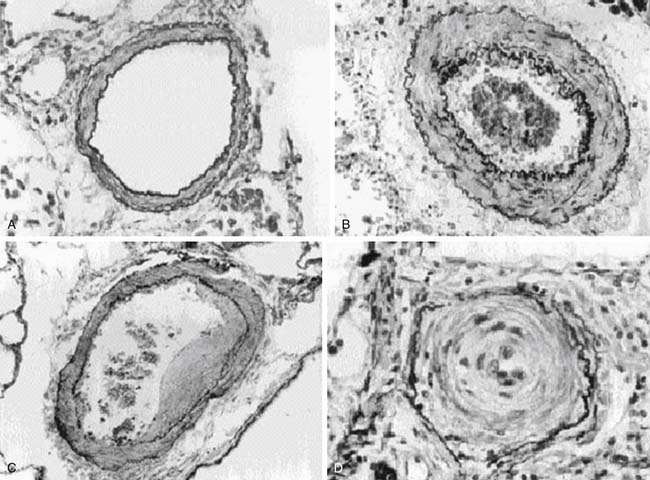
Table 48–2 Heath-Edwards Classification of Pulmonary Vascular Disease
| Grade | Pulmonary Vascular Pathology |
|---|---|
| I | Retention of fetal-type pulmonary vessels |
| II | Medial hypertrophy with cellular intimal proliferation |
| III | Medial hypertrophy; intimal fibrosis; early generalized vascular dilatation in severe instances |
| IV | Progressive generalized arterial dilation and occlusion by intimal fibrosis with the formation of complex dilation lesions (plexiform lesions) |
| V | Chronic dilation with formation of numerous dilation lesions including veinlike branches of hypertrophied arteries, cavernous and angiomatous lesions, and pulmonary hemosiderosis |
| VI | Necrotizing arteritis |
Table 48–3 Morphometric Classification of Pulmonary Vascular Disease
| Grading | Vascular Pathology |
|---|---|
| Grade A | Abnormal extension of smooth muscle along smaller peripheral arteries with intra-acinar extension (arteries accompanying respiratory bronchiole, alveolar duct and alveolar wall) (corresponds to Heath-Edwards grade I); associated with ↑ PBF without evidence of ↑ PAP |
| Grade B | In association with abnormal extension of the muscle, the medial muscular coat of the intra-acinar and preacinar arteries is thicker than normal (corresponds to Heath-Edwards grade I); when mild not associated with PAH; when more than twice normal is associated with PAH |
| Grade C | In association with abnormal extension and increased thickness of the muscular coat, the number of intra-acinar and preacinar arteries are reduced in number (high alveolar-arterial ratio); associated with moderate to severe elevation in PVR |
PAH, Pulmonary arterial hypertension; PAP, pulmonary artery pressure; PBF, pulmonary blood flow; PVR, pulmonary vascular resistance.
In patients with CHD, morphologic criteria175 and pulmonary hemodynamics are both used to determine the stage beyond which surgical correction is no longer indicated because established disease will cause an unacceptable high operative risk or will continue to progress despite surgical repair.176 Abnormalities in muscularization and growth of pulmonary arteries obtained at biopsy during surgical repair from patients with CHD correlated with preoperative hemodynamic data of increased pulmonary blood flow, pressure, and resistance.175 Pulmonary vascular structural changes assessed by morphometry175 (see Table 48-3) and Heath-Edwards classification173 (see Table 48-2) in lung tissue obtained by biopsy at the time of surgical repair correlated with the postoperative hemodynamic findings of PAP and PVR measured 1 day and 1 year after surgery. One year after repair, mean PAP and PVR were normal in all patients whose conditions were surgically corrected before 9 months of age regardless of the severity of pulmonary vascular changes. PAP and PVR were increased in all patients whose conditions were repaired after 2 years of age with grade C morphometric findings and to a severe degree if associated with Heath-Edwards grade III. Thus although Heath-Edwards grade usually can be used to identify patients at risk for PH in the early postoperative period, both the morphometric and Heath-Edwards grades as well as age of the patient at the time of repair can be used to determine whether PAP and PVR eventually return to normal or remain elevated.
Congenital Heart Disease
Pulmonary vascular disease remains a significant cause of morbidity and mortality in children with CHD. Congenital heart lesions resulting in increased PBF or pulmonary venous obstruction produce pulmonary artery smooth muscle hypertrophy and hyperplasia and pulmonary vasoconstriction.178 Unless PH is treated either by medical or surgical means, pulmonary vasoconstriction may persist, progress to vascular obliteration, and produce a high morbidity. Pulmonary hypertensive crisis in the immediate postoperative period is a life-threatening complication in children with certain types of CHD, despite a good surgical repair.179 Even though early surgical repair has been advocated to prevent later pulmonary vascular obstructive disease, it does not abolish the occurrence of PH in the immediate postoperative period.180 Microemboli, platelet aggregation, compliment activation, pulmonary leukosequestration, excess production of vasoactive mediators, atelectasis, and hypoxia during cardiopulmonary bypass all contribute to elevated PVR in the immediate postoperative period. The effects on the pulmonary vasculature may be insidious over several hours, presenting as low cardiac output and right heart failure or more acutely as pulmonary hypertensive crisis. In such situations, the PAP increases to systemic or suprasystemic levels, systemic blood pressure falls, and the arterial oxygen saturation drops. In one large center, half of the postoperative cardiac children who had pulmonary hypertensive crises died during their hospitalization.181 In the newborn, parenchymal lung disorders including pulmonary edema, intrapulmonary shunting, and pneumonia may coexist with heart disease, resulting in hypoxic respiratory failure following cardiopulmonary bypass.
Pulmonary endothelial damage is an early histological event in children with CHD and pulmonary hypertension.182 Reduced pulmonary vasodilation to acetylcholine demonstrates a physiologic impairment of endothelial function in these children.183 Loss of local endothelial NOS activity may be one of the contributing factors in postoperative pulmonary hypertensive crisis. It has been suggested that cardiopulmonary bypass also may contribute to endothelial dysfunction. Arteries with endothelial damage are particularly sensitive to exogenous NO, and response to inhaled NO in the postoperative period suggests that the capacity for smooth muscle relaxation and pulmonary vasodilatation is intact in these children.184 Even though NO-induced vasodilation varies among children with PH and elevated PVR, a decline in selective response seems to parallel the progression of established vascular disease and thus may help in selecting patients to undergo surgery.185 An increase in the number of ETa receptors in lung arteries and lung parenchyma has been noted in patients with increased PVR and low PBF.186 Up-regulation of the ET-1 system may play a role in the pathogenesis of secondary PH associated with CHD in children.186
Clinical Presentation
Often no correlation exists between the time PPH is thought to have started, the age at which it is diagnosed, and the severity of symptoms. The disease seems to progress fairly rapidly, especially in children. Frequent tiredness, dyspnea, dizziness, and fainting spells are the typical early symptoms. Some of the other symptoms include edema of legs, cyanosis, palpitations, and chest pain. Exertional dyspnea, chest pain, and syncope result from the inability to increase cardiac output in the presence of increased oxygen demand. Examination findings compatible with PH in children include jugular venous distention, systolic murmur of tricuspid regurgitation, loud second heart sound at the base of the heart, and a diastolic murmur of pulmonary regurgitation (Graham Steel murmur). In patients with PAH and right heart failure hepatomegaly, ascites and peripheral edema may be seen. Initially the RV hypertrophies to maintain cardiac output at rest, although the ability to increase cardiac output during exercise may be impaired. As pulmonary vascular disease progresses, RV dysfunction ensues, resulting in right heart failure. Although the left side of the heart is not directly affected by pulmonary vascular disease, progressive right heart dilatation can impair left ventricular filling.187 Depending on the degree of physical limitation by symptoms, the functional level of patients is graded from I to IV, with higher grades representing severe disease and worse prognosis. The New York Heart Association (NYHA) classification of dyspnea has been modified by the WHO to categorize PH by the severity of symptoms that, unlike PAP, correlate well with survival (Table 48-4).
Table 48–4 WHO Functional Classification for Pulmonary Hypertension (Modified After NYHA Classification)
| WHO Class | Description |
|---|---|
| Class I | Patients with pulmonary hypertension but without resulting limitation of physical activity; ordinary physical activity does not cause undue dyspnea or fatigue, chest pain, or near syncope |
| Class II | Patients with pulmonary hypertension resulting in slight limitation of physical activity; they are comfortable at rest; ordinary physical activity causes undue dyspnea or fatigue, chest pain, or near syncope |
| Class III | Patients with pulmonary hypertension resulting in marked limitation of physical activity; they are comfortable at rest; less than ordinary activity causes undue dyspnea or fatigue, chest pain, or near syncope |
| Class IV | Patients with pulmonary hypertension with inability to carry out any physical activity without symptoms; these patients manifest signs of right heart failure; dyspnea and/or fatigue may be present even at rest; discomfort is increased by any physical activity |
NYHA, New York Heart Association; WHO, World Health Organization.
Diagnostic Approach
Doppler echocardiography may offer clues to the potential etiologies of PH, particularly left-sided heart lesions. RV systolic pressures greater than 35 to 40 mm Hg in a patient with unexplained dyspnea may warrant further evaluation of PAH. Common echocardiographic findings of PAH include right atrial and rRV enlargement, reduced RV function, tricuspid regurgitation, flattening of intraventricular septum, and underfilling of the left ventricle. In a clinical setting, findings of Doppler echocardiography suggest the diagnosis of PAH, but right heart catheterization (RHC) may eventually be required for an accurate assessment of PVR to confirm the diagnosis. Other noninvasive modalities such as computed tomography (CT) and/or magnetic resonance imaging (MRI) should be explored before subjecting the patient to a catheterization procedure. CT scanning may suggest an etiology of PAH such as a severe airway or parenchymal lung disease. Chronic thromboembolic pulmonary hypertension (CTEPH) is a form of PH and should be sought in all patients with clinically significant PH because it is curable with surgery. CT and V/Q scans help in the diagnosis of CTEPH. Presence of ground-glass opacities, particularly with a centrilobular distribution, septal lines, and adenopathy on a CT scan are suggestive of pulmonary venoocclusive disease in a patient displaying symptoms of PH.188 Pulmonary venoocclusive disease is an unusual form of PH, characterized by elevated PAP with a normal pulmonary capillary wedge pressure and evidence of venous congestion on chest radiograph. Cardiac MRI accurately assesses size and function of the RV with a high degree of reproducibility and hence is used as a prognostication tool in the management of PAH.189 Despite the aforementioned noninvasive investigations, catheterization may be needed to define the hemodynamic profile, to confirm the diagnosis of PAH, to perform vasoreactivity testing on confirmation of diagnosis, and most important, to begin a specific management plan for the treatment of PAH.
The current hemodynamic definition of PAH is a mean PAP greater than 25 mm Hg; a pulmonary capillary wedge pressure, left atrial pressure or left ventricular end-diastolic pressure of 15 mm Hg or less and a PVR greater than 3 Wood units.190 Any patient with suspected PAH requires a careful invasive assessment of pulmonary hemodynamics. Many causes of elevated PA pressure (PH) are not caused by pulmonary vascular disease. Pulmonary hypertension commonly occurs with high transpulmonary flow states such as exercise, anemia, pregnancy, sepsis, portopulmonary syndrome, or thyrotoxicosis. In these states, the pulmonary vascular bed is anatomically normal and the PH resolves when the cardiac output returns to normal levels. The consensus document on PH requires elevated PVR as opposed to simply elevated mean PAP in the setting of a normal left heart filling pressures. PVR is a more robust diagnostic criterion for PAH because it reflects the influence of transpulmonary gradient and cardiac output and is only elevated if the vascular obstruction occurs within the precapillary pulmonary circulation. PVR is a useful measure to apply to patients with increased mean PAP. PVR distinguishes passive PH (i.e., elevated mean PAP and normal PVR) from PPH caused by pulmonary vascular disease (i.e., elevated mean PAP and elevated PVR). By definition, PVR and PAP are both elevated in PAH.133
All patients suspected of having PAH after noninvasive evaluation should undergo RHC prior to initiation of therapy specific to PAH. Although RHC and pulmonary angiography are invasive, they can be performed safely by experienced operators even in patients with severe PH and right heart failure. In a recent review of 7218 RHC procedures collected retrospectively and prospectively, 76 serious events (1.1%) were reported. Most of them were related to venous access (e.g., hematoma or pneumothorax), followed by arrhythmias and hypotensive episodes related to vagal reactions or pulmonary vasoreactivity testing with a overall procedure-related mortality of 0.05% (4/76).191
Acute Vasodilator Testing
Vasoreactivity testing in an essential component in the diagnostic evaluation of PAH, because this has therapeutic implications. Responders are more likely to respond to oral calcium channel blockers (CCB) than are nonresponders and seem to have a better prognosis.192 Acute vasodilator testing (AVT) is usually performed during the same procedure as the diagnostic RHC. AVT is performed using iNO, intravenous PGI2,194 or intravenous adenosine.195 Inhaled NO is the commonly used drug for AVT at doses of 20 to 40 ppm for 5 minutes. Hemodynamic measurements are recorded prior to and after administration of iNO, and the drug is discontinued completely. Based largely on the data from Sitbon and colleagues,192 the European Society of Cardiology and American College of Chest Physicians guidelines propose that the acute response to vasodilator testing be defined as a decrease in mean PAP by at least 10 mm Hg to an absolute level of less than 40 mm Hg without a decrease in cardiac output.196 While these criteria are insufficient to capture all patients who may be responsive to long-term CCB therapy, it will reliably identify those who are least likely to benefit from oral CCB therapy and therefore provide the greatest degree of safety.133 Patients with PAH due to conditions other than idiopathic PAH, such as BMPR2 genotype or anorexigen-induced PAH, have a very low rate of long-term responsiveness to oral CCB therapy. Accordingly, the decision to proceed with AVT in such patients should be individualized. AVT is not indicated and may be harmful in patients with significantly elevated left heart filling pressures.133
Treatment
General Measures
Treatment of PAH has evolved considerably during the past decade, and treatment algorithms have been formulated by the American College of Chest Physicians for adult patients.196 Unfortunately, because of limited data in children with PAH, management decisions often are extrapolated from adult studies. Without treatment, the natural history for children with idiopathic PAH is worse than that for adults; however, with treatment, the outcome appears better in children than in adults. In the absence of studies specifically reporting the clinical response of children to PAH therapy, similar clinical strategies have been suggested for the management of PAH in children. Treatment goals in children are similar to those in adults. They include improvements in patient symptoms such as dyspnea, enhancing exercise capacity measured objectively by an assessment of exercise endurance in older children (such as a 6-mile walk test), improving hemodynamic profile (lowering PAP), preventing the progression of the disease, and improving survival rates.
Treatment of PPH is usually treatment for life. The therapeutic regimen has to be tailored in a given child and adjusted according to the clinical and hemodynamic response. Optimizing the management in these children will improve the quality of life and survival. Most important is the avoidance and treatment of hypoxemia. Patients are advised to avoid heavy physical exercise because this may evoke exertional syncope. Exposure to high altitude may contribute to hypoxic pulmonary vasoconstriction and may not be well tolerated. Because hypoxemia is a potent pulmonary vasoconstrictor, most experts recommend supplemental oxygen to maintain oxygen saturation greater than 90%. A sodium-restricted diet is advised and is particularly important to manage volume status in patients with RV failure. Routine immunizations, such as against influenza and pneumococcal pneumonia, are advised. The hemodynamic fluctuations of pregnancy, labor, and delivery are potentially devastating to patients with PAH with a demonstrated maternal mortality of 30% to 50%.197 Current guidelines recommend that pregnancy be avoided or terminated early in women with PAH.196 It is important to discuss effective methods of birth control with women with PAH who have child-bearing potential, although the preferred method is not clear. It also is advisable to avoid elective major surgeries, especially during periods of progression or bad control of the disease. Conventional treatment of children with PPH consists of an anticoagulant and an oral vasodilator, usually a CCB and supplemental oxygen.198 Anticoagulation with Warfarin has been shown to be beneficial in these patients.199 In a more recent study, anticoagulant therapy had a positive influence on long-term survival and a significantly improved quality of life in patients with PPH, particularly in patients with a history of anorectic drug intake.200
Therapy in secondary PH is directed at the cause of PAH. Closure of the heart defect will resolve the PAH resulting from increased PBF. Management of pulmonary venous hypertension includes controlling left ventricular failure and relieving the cause of venous obstruction (commonly left heart obstructive lesions). Medical management includes administering oxygen, diuretics, and digoxin. Vasodilator therapy is the mainstay in the management of pulmonary vascular obstruction. Even small reductions in RV afterload following vasodilator therapy will produce substantial improvement in RV output in patients with PH. The goal of vasodilator therapy in patients with PAH is to reduce PAP and increase cardiac output without symptomatic systemic hypotension. The response to vasodilators is important in making a decision for surgical correction (lung or heart/lung transplantation). Patients with severe intractable cases of left ventricular dysfunction may need heart transplantation.
Pulmonary Vasodilators
Calcium Channel Blockers
CCBs are the drugs most frequently used in the treatment of PH outside of the neonatal period. The response to CCBs is better in children than in adults with PPH. Acute response to the vasodilator drug during cardiac catheterization will help in determining the desirability of using chronic oral CCBs. A positive response to a vasodilator is taken as a decrease in mean PAP of 20% or more with no fall in cardiac index. Patients who meet criteria for oral CCBs should be followed closely for both safety and efficacy of CCB therapy. If the patient meets the definition of an acute response but does not improve to functional class I or II while receiving CCB therapy, the patient should not be considered a chronic responder and alternative PAH therapy should be considered. Long-acting nifedipine, diltiazem, or amlodipine are the most commonly used CCBs. Verapamil should be avoided because of its potentially negative inotropic effects. The response to acute vasodilator testing is higher in children compared with adults (41% vs. 12% in adults).201 The acute response in children is also age dependent. Oral CCBs help in reducing PAP in about 40% of children with PPH.202 In the remaining 60%, acute vasodilator testing with agents such as iNO or intravenous PGI2 failed to demonstrate responsiveness, and these children have not benefited from chronic oral CCB therapy.202 Chronic CCB therapy improved survival and quality of life in children who acutely respond to vasodilator drug testing.203 A 5-year survival rate for patients treated with chronic oral CCB who respond acutely to vasodilator testing was 97%, versus 35% for those who do not respond acutely.202 Chronic use of CCBs in patients with fixed PVR and unfavorable hemodynamics may worsen right heart failure. Regular, noninvasive monitoring of PAP and cardiac function is as essential part of the management of these patients.
Prostanoids
Epoprostenol
Prostacyclin (sodium epoprostenol) has been studied extensively during the past decade in patients with PPH201,202,204,205 and secondary pulmonary hypertension.206 Chronic administration of prostacyclin is usually well tolerated with minimal decrease in systemic pressures and modest decrease in PAP. Higher doses can lead to high cardiac output states, suggesting that it has important positive inotropic effects.204 In patients who are nonresponders to vasodilator testing and responders who fail to improve while taking CCBs, continuous intravenous infusion of PGI2 improves survival.202 Long-term, continuous IV administration of PGI2 has been shown to improve survival, quality of life, and exercise capacity and to significantly reduce PAP in children with PPH.202,207 Ongoing studies suggest that in some children who were previously believed to have fixed pulmonary vascular disease with irreversible obstruction, their condition may be reversible with long-term IV administration of prostacyclin, perhaps via remodeling of the pulmonary vascular bed.202,208 Although in the late 1980s and early 1990s the aim of chronic prostacyclin therapy was to act as a bridge to lung or heart/lung transplantation,209 continuous prostacyclin now is being used as an alternative therapy to transplantation in selected children.201 Chronic PGI2 infusion improves hemodynamics and quality of life in patients with PH associated with CHD for whom conventional therapy fails.210 Continuous IV PGI2 is the only one of the “specific” therapies currently approved for use in patients with chronic PH. However, drawbacks to this therapy exist. Tolerance develops in some patients, requiring dose escalation, which results in additional adverse events and increased drug costs. More importantly, complications are associated with administration of PGI2, such as risk of serious infections, catheter thrombosis, and rebound PAH due to pump failure with subsequent interruptions in therapy. Given its considerable complexity, epoprostenol use should to limited to centers experienced with its administration and with systematic follow-up of patients.
Treprostinil
The propensity for serious catheter-related infections with epoprostenol administration led to the development of treprostinil, a stable prostacyclin analogue for subcutaneous infusion with a half-life of about 4 hours. Chronic subcutaneous infusion of treprostinil improved indices of dyspnea, signs and symptoms of PH, and hemodynamics and had an acceptable safety profile in patients with PAH.211 Dosage used in this study was 1.25 ng/kg/min to 22.5 ng/kg/min via subcutaneous infusion with use of a micro infusion pump with a catheter placed in the subcutaneous tissue. The FDA approved subcutaneously administered treprostinil in 2002 for use in persons with functional class II, III, and IV PAH. More recently, IV treprostinil has been studied in an open-label, uncontrolled fashion. Even though the drug proved to be of benefit in the treatment of persons with PAH,212 a Centers for Disease Control and Prevention report recently raised concern for an increased risk of bloodstream infections, particularly with gram-negative organisms, in patients receiving IV treprostinil.213 Given the complexity of administration of both IV and subcutaneous treprostinil, administration should be limited to centers experienced with this agent.
Iloprost
Iloprost is a stable prostacyclin analog with a serum half-life of 20 to 25 minutes. Aerosolization of prostacyclin or its stable analog iloprost causes selective pulmonary vasodilatation, increases cardiac output, and improves venous and arterial oxygenation in patients with severe PH.214 Thus it may offer a new strategy for treatment of this disease. Long-term treatment with aerosolized iloprost has been shown to be safe and has sustained effects on exercise capacity and pulmonary hemodynamics in patients with PPH.215 In a placebo-controlled trial, iloprost inhalation (2.5 μg or 5 μg per inhalation; median dose of 30 μg per day) improved pulmonary hemodynamics and quality of life in patients with severe PH.216 Inhaled iloprost represents a significant advance from intravenous PGI2 in the management of PAH. Aerosolized iloprost might be an alternative to iNO for early testing of vascular reactivity and for the postoperative treatment of acute PH in children with CHD.217 Common adverse effects of inhaled iloprost include cough, headache, flushing, and jaw pain. Iloprost was approved by the FDA in 2004 for use in persons with functional class III and IV PAH. Its short half-life and frequent inhalations do not make it an ideal candidate for chronic treatment of PPH in children.
Beraprost
Beraprost is a prostacyclin analog for oral administration. It has been shown that beraprost reduces PVR in patients with PPH218–220 and also may be effective in the treatment of secondary precapillary pulmonary hypertension.221 Oral administration of beraprost sodium also may improve exercise capacity and ventilatory efficiency in patients with both primary and chronic thromboembolic pulmonary hypertension.222 Limited studies suggest the efficacy of oral beraprost in infants and children with PH resulting from congenital heart defects.223
Endothelin Receptor Antagonists
Bosentan
Bosentan (Tracleer), an oral antagonist of both ETA and ETB receptors, is the most widely studied endothelin receptor antagonist in clinical trials. In a placebo-controlled randomized trial, bosentan increased exercise capacity and improved hemodynamics in patients with PH.224 Patients given bosentan had a reduced Borg dyspnea index and an improved WHO functional class indicating exercise tolerance.224 In another large double-blind, placebo-controlled multicenter study, bosentan given for 16 weeks was well tolerated, improved exercise tolerance, and increased time to clinical worsening.225 Similar improvements in exercise capacity, symptoms, and clinical worsening were seen in the larger Bosentan Randomized Trial of Endothelin Antagonist Therapy of Pulmonary Hypertension study.226 Bosentan was well tolerated at a dose of 125 mg twice daily in these adult patients. Studies in children of the physiologic and long-term effects of bosentan appear to be warranted. Potentially serious hepatotoxicity and teratogenicity has been reported with bosentan that requires close monitoring and cautious use. Because of the risk of hepatic toxicity, the FDA requires that liver function tests be performed at least monthly and hematocrit be checked every 3 months for these patients. There is concern that the endothelin antagonists as a class may be capable of causing testicular atrophy and male infertility. Younger males who may consider conceiving should be counseled regarding this possibility prior to taking these drugs.
Selective ETA Receptor Antagonists
Sitaxsentan and ambrisentan are the two commercially available selective ETA receptor antagonists. In patients with PAH who were in NYHA functional class II, III or IV, sitaxsentan improved exercise capacity as assessed by the 6-mile walk test and improved functional class after 12 weeks of treatment.227 The incidence of liver abnormalities was more favorable for the 100-mg dose group (0%) compared with placebo (3%) and the 300-mg group (10%). The Sitaxsentan to Relieve Impaired Exercise-2 study again demonstrated that the 100-mg dose of sitaxsentan improved exercise capacity as assessed by the 6-mile walk test and improved functional class compared with placebo. Although a 50-mg dose of sitaxsentan had a lower incidence of liver enzyme abnormalities, it was not different from the placebo group.228 In a double-blind study, ambrisentan was evaluated at four dosages (1, 2.5, 5, or 10 mg) once daily for 12 weeks. Results of the 6-mile walk test improved over baseline with ambrisentan.229 Two phase III clinical trials of ambrisentan in persons with PAH have been completed. These trials randomly assigned 202 and 192 patients with PAH, respectively, to placebo and ambrisentan. Doses of 5 and 10 mg of ambrisentan were compared with placebo in the Ambrisentan in Pulmonary Arterial Hypertension, Randomized, Double-blind, Efficacy Study (ARIES), and doses of 2.5 and 5 mg of ambrisentan were compared with placebo in ARIES-2.230 After 12 weeks, improvements were noted in the primary end point of the 6-mile walk test in both the studies. Ambrisentan was approved by the FDA in 2007 for patients with PAH with functional class II and III symptoms. As a class, endothelin receptor antagonists have a potential for liver injury and teratogenicity. Monthly monitoring of liver function tests, a monthly pregnancy test in women of child-bearing potential, and a periodic hemoglobin measurement are required. Precautions regarding contraception and testicular atrophy are similar to those for bosentan.
Nitric Oxide
The acute responsiveness to iNO seems to predict the subset of patients who might be responsive to oral CCBs and thus is a relatively safe and easy test to perform during cardiac catheterization.231 Experience with long-term use of iNO in children with PAH is limited. Inhaled NO, however, is expensive and requires a fairly sophisticated delivery and monitoring system. Nonetheless, iNO dilates the pulmonary circulation while avoiding unwanted and dangerous systemic vasodilation and is economical and extremely effective in the management of PH in critically ill patients in intensive care units.232 Inhaled NO has been used to assess the vasodilator capacity of the pulmonary vascular bed in children with CHD and elevated PVR.184,233 Low-dose iNO (2 to 20 ppm) has been effective in the management of postoperative PH following corrective surgery in infants with CHD.234 NO at higher doses (20 to 80 ppm) produced selective pulmonary vasodilatation in children with CHD and pulmonary hypertension.235 Hemodynamic benefit with NO has been shown in newborns with total anomalous pulmonary venous connection and congenital mitral stenosis and in postoperative patients with preexisting left-to-right shunts and other lesions.236 It can be used to help discriminate anatomic obstruction to pulmonary blood flow from pulmonary vasoconstriction, and it may be used in the treatment or prevention of pulmonary hypertensive crisis after cardiopulmonary bypass in neonates.236,237 A trial of iNO may diminish the need for ECMO in patients with cardiopulmonary failure following cardiac surgery.238,239 In children with PH and CHD, both iNO and aerosolized iloprost were equally effective in selectively lowering PVR.217
Phosphodiesterase Inhibitors
Type 5 phosphodiesterase (PDE-5) is primarily responsible for degradation of cGMP to inactive metabolite GMP. PDE-5 appears to be particularly abundant in pulmonary vessels. One way of augmenting the concentration of cGMP in pulmonary vessels is by inhibiting the activity of PDE-5. PDE-5 inhibitors such as sildenafil and tadalafil might therefore be expected to prolong the vasodilating effects of cGMP. Sildenafil is a potent and selective inhibitor of cGMP-specific PDE-5240 and has been shown to cause selective pulmonary vasodilatation in an ovine model of acute pulmonary hypertension.241 Several agents that inhibit PDE-5 are shown to augment the response to NO.242–245 Dipyridamole, a nonselective PDE-5 inhibitor, in combination with iNO, augments the decrease in PVR index and blunts the severity of acute hypoxic pulmonary vasoconstriction in children with PH.246 When administered alone in children with PH, dipyridamole reduces the PVR index primarily through an increase in cardiac index.246 In the Sildenafil Use in Pulmonary Arterial Hypertension study, 278 patients with PAH were randomly assigned to either placebo or sildenafil (20, 40, or 80 mg) orally 3 times daily for 12 weeks.247 Results of the 6-mile walk test increased from baseline in all sildenafil groups. In all sildenafil groups, mean PAP was reduced and functional class improved. The incidence of clinical worsening did not differ significantly between the patients treated with sildenafil versus placebo. Long-term data in 222 patients (available for the 80-mg group) completing 1 year of treatment with sildenafil monotherapy showed sustained improvement from baseline at 1 year in the 6-mile walk test. The FDA-approved dose of sildenafil in patients with PAH is 20 mg administered three times daily for adults. Adverse effects include headache, flushing, dyspepsia, and epistaxis. The question of whether higher doses might confer additional hemodynamic benefit has generated considerable debate, and such doses continue to be studied in clinical trials. Another long-acting PDE-5 inhibitor, tadalafil, is currently undergoing clinical study. Like sildenafil, tadalafil has been approved by the FDA for erectile dysfunction. However, it continues to be investigated in patients with PAH.
Combination Therapy
Combining two agents may be clinically useful. As previously noted, agents that inhibit degradation of cGMP (PDE-5 inhibitors) may act synergistically with iNO. Similarly inhibiting PDE-3, a principal metabolizer of cAMP, might be expected to augment the response to PGI2. Inhaled milrinone, either alone or in combination with inhaled PGI2, selectively dilated the pulmonary vasculature without systemic effects in cardiac surgical patients with PH.248 The goal of combination therapy is to maximize efficacy and to minimize toxicity. The safety and efficacy of combination therapy in persons with PAH is a subject of active investigation. In one recent study, inhaled Iloprost has been studied in patients who remained symptomatic (NYHA functional class III or IV) while undergoing stable bosentan therapy for at least 3 months.249 After 12 weeks, the primary efficacy measure, a postinhalation 6-mile walk test, improved significantly in the Iloprost group compared with the placebo group. Improvements in mean PAP, functional class, and clinical worsening also were found. More recently sildenafil or placebo was evaluated in 267 patients with PAH who remained symptomatic while receiving a stable dose of epoprostenol for at least 3 months.250 Patients treated with sildenafil experienced a placebo-adjusted improvement in the 6-mile walk test, mean PAP, and time to clinical worsening.
Atrial Septostomy
Despite advances in medical treatment for PAH, many patients experience progressive functional decline, largely because of worsening right heart failure. Fortunately, it is uncommon to see patients in the pediatric age group in end-stage PAH compared with adults. In these patients, interventional and surgical options, including atrial septostomy and lung or combined heart and lung transplantation, should be considered. In patients with symptomatic cor-pulmonale secondary to pulmonary vascular disease, atrial septostomy can improve symptoms and may serve as a palliative bridge to heart and/or lung transplantation.251,252 Several case series have reported hemodynamic and clinical improvement following atrial septostomy.251,252 Improved cardiac output appears to be the principal hemodynamic benefit. Improvements in NYHA functional class and the 6-mile walk test also have been reported.253 The success rates for bridging patients to transplantation, with septostomy, ranges from 30% to 40%.251,252 The procedural mortality rate is high, however, with an estimate of 15% based on published series. Currently, atrial septostomy is recommended for patients with severe PAH and intractable right heart failure despite maximal medical therapy, including optimized PAH-specific agents and inotropes. The goals of the procedure are palliation and restoration and maintenance of clinical stability until a transplant can be performed.
Lung and Combined Heart/Lung Transplantation
Before PGI2 became available as the long-term therapy, lung transplantation was the only option for survival for patients with severe PH. Single-lung, double-lung, and heart-lung transplantation have all been advocated as the operation of choice in persons with PPH. Transplantation is offered after maximal therapeutic effort with vasodilators and oxygen has been attempted and when the estimated 2-year survival is less than 50%.254 Most centers now perform bilateral lung or heart-bilateral lung transplantation for persons with PPH. Intravenous PGI2 has been used as a bridge to stabilize the patient until transplantation is accomplished. Of the 70 lung transplants performed in 1997, PH accounted for 7% of infants and 18% of older children requiring transplantation.255 Survival rates have improved considerably in the 1990s, suggesting both technical improvements and earlier referral for transplantation. Recent data from the International Society for Heart and Lung Transplantation (January 1998 to December 2001) indicate an overall survival following lung transplant of 70% to 80% at 1 year and 50% at 3 years in children younger than17 years. Survival in all patients with PPH following lung transplant was 72% at 1 year and 58% at 3 years. In a single-center study, survival following heart-lung transplant in patients with PPH was 72% at 1 year, 67% at 2 years, and 42% at 5 years.256 Because outcomes for infants listed for lung or heart/lung transplantation is similar to that of children, very young age should not be considered a contraindication to lung or heart/lung transplantation.257 Earlier diagnosis and listing may decrease pretransplant mortality.257 Even though the early outcome following lung transplantation has improved considerably, long-term complications, including infections, bronchiolitis obliterans, and complications of immunosupression remain significant problems.258 Aggressive medical therapy combined with early recognition of lung transplant candidates will improve quality of life and overall survival of these children.
Prognosis and Survival in Pulmonary Arterial Hypertension
Most of the studies have been focusing on short time end points, such as 12 weeks’ duration or the 6-mile walk test, and long-term follow-up guidelines for these patients is lacking. Figure 48-14 summarizes the treatment algorithm of PAH with an emphasis on initial diagnosis and initial management of these patients from the expert consensus document for PH.133 Although therapies to manage PAH are evolving rapidly, long-term experience with epoprostenol has suggested the importance of early follow-up, because patients who remain in NYHA functional class III or IV despite therapy have poor outcomes and should be considered for a lung transplant.259
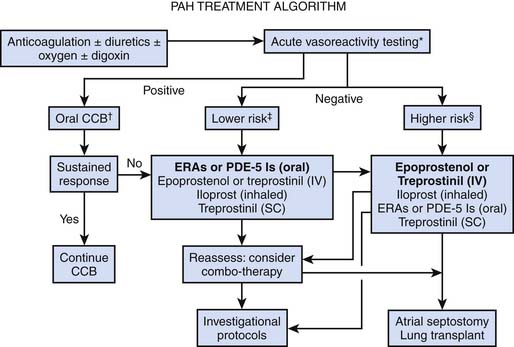
Figure 48–14 Treatment algorithm for pulmonary arterial hypertension.
(From McLaughlin VV, et al: Expert Consensus Document on Pulmonary Hypertension, J Am Coll Cardiol 53(17):1573-1619, 2009; adapted with permission from Elsevier.)
Some of the earlier studies found that NYHA functional class, 6-mile walk, and hemodynamic variables were all related to prognosis and survival. All of these parameters were studied as primary end points in many prospective trials. The National Institutes of Health cohort study showed that among 194 patients who received a diagnosis of idiopathic PAH, the risk of death was higher among patients in NYHA functional class III or IV than among those in functional class I or II. Median survival among patients presenting with NYHA functional class I or II symptoms was nearly 6 years versus 2.5 years for functional class III and 6 months for functional class IV symptoms.260 Other studies have confirmed the importance of functional class as a prognostic variable, even during treatment.261 Patients who improved to functional class I or II with epoprostenol had a better prognosis than did patients who remained in functional class III or IV.261
In the pivotal epoprostenol idiopathic PAH trial, performance in the 6-mile walk test was found to be an independent predictor of survival, leading to use of this test as the primary end point in many trials.262 All large published evaluations implicate hemodynamics as an important predictor of survival.259,261 In the National Institutes of Health registry, three hemodynamic variables that were associated with an increased risk of death included increased mean PAP (>55 mm Hg), increased right atrial pressure (>12 mm Hg), and decreased cardiac index (<2.0 L/min/m2).260 Similarly, responders to vasodilator testing have an excellent prognosis with up to a 95% survival at 5 years.192 In a recent single-center study of 65 children with PAH, the 5-, 10- and 15-year survival rates were 96%, 92%, and 65%, respectively, and no significant correlation between outcome and immediate response to the vasodilators was found.263
While echocardiography has been a pivotal screening test, studies evaluating the prognostic value of echocardiography have been limited to relatively small series. The presence of any degree of pericardial effusion has been proven to be a consistent predictor of mortality.264 More recently, tricuspid annular plane systolic excursion on echocardiography has been shown to be a robust measure of RV function and a powerful predictor of patient survival among those with PH.265 Tricuspid annular plane systolic excursion of less than 1.8 identifies patients with more advanced RV dysfunction in patients with PH. In addition, these patients had reduced survival over a median follow-up of 19 months. Cardiac MRI accurately assesses size and function of the RV with a high degree of reproducibility. RV function is an important prognostic indicator in persons with PAH, and MRI of poor RV function was found to be an independent predictor of mortality and treatment failure.189 Cardiac MRI holds promise as a noninvasive tool to assess for evidence of RV response and remodeling with treatment. Brain natriuretic peptide and NT-proBNP appear to be significantly elevated and are independent predictors of poor prognosis.266
References are available online at http://www.expertconsult.com.
1. Goldstein J.D., et al. Unusual vascular anomalies causing persistent pulmonary hypertension in a newborn. Am J Cardiol. 1979;43(5):962-968.
2. Drake C.J., Littl C.D. Exogenous vascular endothelial growth factor induces malformed and hyperfused vessels during embryonic neovascularization. Proc Natl Acad Sci U S A. 1995;92(17):7657-7661.
3. Klein S., et al. Basic fibroblast growth factor modulates integrin expression in microvascular endothelial cells. Mol Biol Cell. 1993;4(10):973-982.
4. Risau W. Embryonic angiogenesis factors. Pharmacol Ther. 1991;51:371-376.
5. Hislop A., Reid L. Pulmonary arterial development during childhood: branching pattern and structure. Thorax. 1973;28(2):129-135.
6. Reid L.M. Structure and function in pulmonary hypertension. New perceptions. Chest. 1986;89(2):279-288.
7. Murphy J.D., et al. The structural basis of persistent pulmonary hypertension of the newborn infant,. J Pediatr. 1981;98(6):962-967.
8. Perlman E.J., Moore G.W., Hutchins G.M. The pulmonary vasculature in meconium aspiration,. Hum Pathol. 1989;20(7):701-706.
9. Murphy J.D., Vawter G.F., Reid L.M. Pulmonary vascular disease in fatal meconium aspiration. J Pediatr. 1984;104(5):758-762.
10. Wild L.M., Nickerson P.A., Morin F.C.III. Ligating the ductus arteriosus before birth remodels the pulmonary vasculature of the lamb. Pediatr Res. 1989;25(3):251-257.
11. Rudolph A.M. Distribution and regulation of blood flow in the fetal and neonatal lamb. Circ Res. 1985;57(6):811-821.
12. Rudolph A.M. Fetal and neonatal pulmonary circulation. Am Rev Respir Dis. 1977;115(6 pt 2):11-18.
13. Rasanen J., et al. Role of the pulmonary circulation in the distribution of human fetal cardiac output during the second half of pregnancy. Circulation. 1996;94(5):1068-1073.
14. Rudolph A.M., Heymann M.A. Circulatory changes during growth in the fetal lamb. Circ Res. 1970;26(3):289-299.
15. Cassin S., et al. The vascular resistance of the foetal and newly ventilated lung of the lamb,. J Physiol. 1964;171:61-79.
16. Levin D.L., et al. Morphological development of the pulmonary vascular bed in fetal lambs. Circulation. 1976;53(1):144-151.
17. Morin F.C.III, Egan E.A. Pulmonary hemodynamics in fetal lambs during development at normal and increased oxygen tension. J Appl Physiol. 1992;73(1):213-218.
18. Dawes G.S., Mott J.C. The vascular tone of the fetal lung,. J Physiol. 1962;164:465-477.
19. Heymann M.A. Control of the pulmonary circulation in the perinatal period. J Dev Physiol. 1984;6(3):281-290.
20. Lakshminrusimha S., Steinhorn R.H. Pulmonary vascular biology during neonatal transition. Clin Perinatol. 1999;26(3):601-619.
21. Cassin S., et al. Leukotrienes and prostaglandins in fetal lung liquid. J Appl Physiol. 1990;68(5):2214-2222.
22. Clozel M., et al. Thromboxane is not responsible for the high pulmonary vascular resistance in fetal lambs. Pediatr Res. 1985;19(12):1254-1257.
23. Velvis H., Moore P., Heymann M.A. Prostaglandin inhibition prevents the fall in pulmonary vascular resistance as a result of rhythmic distension of the lungs in fetal lambs. Pediatr Res. 1991;30(1):62-68.
24. Fuloria M., et al. 20-hydroxyeicosatetraenoic acid is a vasoconstrictor in the newborn piglet pulmonary microcirculation. Am J Physiol Lung Cell Mol Physiol. 2004;287(2):L360-L365.
25. Parker T.A., et al. Inhibition of 20-HETE abolishes the myogenic response during NOS antagonism in the ovine fetal pulmonary circulation. Am J Physiol Lung Cell Mol Physiol. 2005;289(2):L261-L267.
26. Saeed S.A., Mitchell M.D. Arachidonate lipoxygenase activity in human fetal lung. Eur J Pharmacol. 1982;78(3):389-391.
27. Soifer S.J., et al. Leukotriene end organ antagonists increase pulmonary blood flow in fetal lambs. Am J Physiol. 1985;249(3 pt 2):H570-H576.
28. Leffler C.W., Mitchell J.A., Green R.S. Cardiovascular effects of leukotrienes in neonatal piglets. Role in hypoxic pulmonary vasoconstriction? Circ Res. 1984;55(6):780-787.
29. Schreiber M.D., Heymann M.A., Soifer S.J. Leukotriene inhibition prevents and reverses hypoxic pulmonary vasoconstriction in newborn lambs. Pediatr Res. 1985;19(5):437-441.
30. Yanagisawa M. The endothelin system. A new target for therapeutic intervention. Circulation. 1994;89(3):1320-1322.
31. Yanagisawa M., et al. A novel potent vasoconstrictor peptide produced by vascular endothelial cells,. Nature. 1988;332(6163):411-415.
32. Luscher T.F. Endothelin: systemic arterial and pulmonary effects of a new peptide with potent biologic properties. Am Rev Respir Dis. 1992;146(5 pt 2):S56-S60.
33. Luscher T.F. Endothelium-derived vasoactive factors and regulation of vascular tone in human blood vessels. Lung. 1990;168(Suppl):27-34.
34. Wong J., et al. Developmental effects of endothelin-1 on the pulmonary circulation in sheep. Pediatr Res. 1994;36(3):394-401.
35. Cassin S., et al. Tone-dependent responses to endothelin in the isolated perfused fetal sheep pulmonary circulation in situ. J Appl Physiol. 1991;70(3):1228-1234.
36. Jones O.W.III, Abman S.H. Systemic and pulmonary hemodynamic effects of big endothelin-1 and phosphoramidon in the ovine fetus. Am J Physiol. 1994;266(3 pt 2):R929-R955.
37. Aschner J.L., Fike C.D. New developments in the pathogenesis and management of neonatal pulmonary hypertension. In: Bancalari E., editor. The newborn lung. Philadelphia: Saunders Elsevier; 2008:241-299.
38. Ivy D.D., Kinsella J.P., Abman S.H. Physiologic characterization of endothelin A and B receptor activity in the ovine fetal pulmonary circulation. J Clin Invest. 1994;93(5):2141-2148.
39. Ivy D.D., et al. Prolonged endothelin A receptor blockade attenuates chronic pulmonary hypertension in the ovine fetus. J Clin Invest. 1997;99(6):1179-1186.
40. Wong J., Fineman J.R., Heymann M.A. The role of endothelin and endothelin receptor subtypes in regulation of fetal pulmonary vascular tone. Pediatr Res. 1994;35(6):664-670.
41. Elton T.S., et al. Normobaric hypoxia stimulates endothelin-1 gene expression in the rat. Am J Physiol. 1992;263(6 pt 2):R1260-R1264.
42. Wong J., et al. Endothelin-1 vasoactive responses in lambs with pulmonary hypertension and increased pulmonary blood flow. Am J Physiol. 1995;269(6 pt 2):H1965-H1972.
43. Mann J., Farrukh I.S., Michael J.R. Mechanisms by which endothelin 1 induces pulmonary vasoconstriction in the rabbit. J Appl Physiol. 1991;71(2):410-416.
44. Tod M.L., Cassin S. Endothelin-1-induced pulmonary arterial dilation is reduced by N omega-nitro-L-arginine in fetal lambs. J Appl Physiol. 1992;72(5):1730-1734.
45. Teitel D.F., Iwamoto H.S., Rudolph A.M. Changes in the pulmonary circulation during birth-related events. Pediatr Res. 1990;27(4 pt 1):372-378.
46. Haworth S.G., Hislop A.A. Adaptation of the pulmonary circulation to extra-uterine life in the pig and its relevance to the human infant. Cardiovasc Res. 1981;15(2):108-119.
47. Cornfield D.N., et al. Oxygen causes fetal pulmonary vasodilation through activation of a calcium-dependent potassium channel. Proc Natl Acad Sci U S A. 1996;93(15):8089-8094.
48. Leffler C.W., Tyler T.L., Cassin S. Effect of indomethacin on pulmonary vascular response to ventilation of fetal goats. Am J Physiol. 1978;234(4):H346-H351.
49. Tyler T., et al. The effects of indomethacin on the pulmonary vascular response to hypoxia in the premature and mature newborn goat. Proc Soc Exp Biol Med. 1975;150(3):695-698.
50. Alano M.A., et al. Analysis of nonsteroidal antiinflammatory drugs in meconium and its relation to persistent pulmonary hypertension of the newborn. Pediatrics. 2001;107(3):519-523.
51. Turner G.R., Levin D.L. Prostaglandin synthesis inhibition in persistent pulmonary hypertension of the newborn. Clin Perinatol. 1984;11(3):581-589.
52. Tiktinsky M.H., Cummings J.J., Morin F.C.III. Acetylcholine increases pulmonary blood flow in intact fetuses via endothelium-dependent vasodilation. Am J Physiol. 1992;262(2 pt 2):H406-H410.
53. Frantz E., et al. Bradykinin produces pulmonary vasodilation in fetal lambs: role of prostaglandin production. J Appl Physiol. 1989;67(4):1512-1517.
54. Truog R.D., Accurso F.J., Wilkening R.B. Fetal pulmonary vasodilation by histamine: response to H1 and H2 stimulation. Dev Pharmacol Ther. 1990;14(3):180-186.
55. Archer S.L., et al. Nitric oxide and cGMP cause vasorelaxation by activation of a charybdotoxin-sensitive K channel by cGMP-dependent protein kinase. Proc Natl Acad Sci U S A. 1994;91(16):7583-7587.
56. Abman S.H., et al. Role of endothelium-derived relaxing factor during transition of pulmonary circulation at birth. Am J Physiol. 1990;259(6 pt 2):H1921-H1927.
57. Tiktinsky M.H., Morin F.C.III. Increasing oxygen tension dilates fetal pulmonary circulation via endothelium-derived relaxing factor. Am J Physiol. 1993;265(1 pt 2):H376-H380.
58. Fineman J.R., et al. Chronic nitric oxide inhibition in utero produces persistent pulmonary hypertension in newborn lambs. J Clin Invest. 1994;93(6):2675-2683.
59. Konduri G.G., et al. Adenosine infusion improves oxygenation in term infants with respiratory failure. Pediatrics. 1996;97(3):295-300.
60. Konduri G.G., Gervasio C.T., Theodorou A.A. Role of adenosine triphosphate and adenosine in oxygen-induced pulmonary vasodilation in fetal lambs. Pediatr Res. 1993;33(5):533-539.
61. Konduri G.G., et al. Purine nucleotides contribute to pulmonary vasodilation caused by birth-related stimuli in the ovine fetus. Am J Physiol. 1997;272(5 pt 2):H2377-H2384.
62. Konduri G.G., et al. Adenosine triphosphate and adenosine increase the pulmonary blood flow to postnatal levels in fetal lambs. Pediatr Res. 1992;31(5):451-457.
63. Konduri G.G., et al. Adenosine is a pulmonary vasodilator in newborn lambs. Am Rev Respir Dis. 1992;146(3):670-676.
64. Lakshminrusimha S., et al. C-type natriuretic peptide system in fetal ovine pulmonary vasculature. Am J Physiol Lung Cell Mol Physiol. 2001;281(2):L361-L368.
65. Fuloria M., Smith T.K., Aschner J.L. Role of 5,6-epoxyeicosatrienoic acid in the regulation of newborn piglet pulmonary vascular tone. Am J Physiol Lung Cell Mol Physiol. 2002;283(2):L383-L389.
66. Dobyns E.L., et al. Eicosanoids decrease with successful extracorporeal membrane oxygenation therapy in neonatal pulmonary hypertension. Am J Respir Crit Care Med. 1994;149(4 pt 1):873-880.
67. Langleben D., et al. Endothelin-1 in acute lung injury and the adult respiratory distress syndrome. Am Rev Respir Dis. 1993;148(6 pt 1):1646-1650.
68. Shaul P.W., et al. Pulmonary endothelial NO synthase gene expression is decreased in fetal lambs with pulmonary hypertension. Am J Physiol. 1997;272(5 pt 1):L1005-L1012.
69. McQueston J.A., et al. Chronic pulmonary hypertension in utero impairs endothelium-dependent vasodilation. Am J Physiol. 1995;268(1 pt 2):H288-H294.
70. Steinhorn R.H., Russell J.A., Morin F.C.III. Disruption of cGMP production in pulmonary arteries isolated from fetal lambs with pulmonary hypertension. Am J Physiol. 1995;268(4 pt 2):H1483-H1489.
71. Geggel R.L., Reid L.M. The structural basis of PPHN,. Clin Perinatol. 1984;11(3):525-549.
72. Chambers C.D., et al. Selective serotonin-reuptake inhibitors and risk of persistent pulmonary hypertension of the newborn. N Engl J Med. 2006;354(6):579-587.
73. Kallen B., Olausson P.O. Maternal use of selective serotonin re-uptake inhibitors and persistent pulmonary hypertension of the newborn. Pharmacoepidemiol Drug Saf. 2008;17(8):801-806.
74. Fornaro E., et al. Prenatal exposure to fluoxetine induces fetal pulmonary hypertension in the rat. Am J Respir Crit Care Med. 2007;176(10):1035-1040.
75. Lakshminrusimha S., Wynn R.J., Youssfi M., et al. Use of CT angiography in the diagnosis of total anomalous venous return. J Perinatol. 2009;29(6):458-461.
76. Kinsella J.P., Ivy D.D., Abman S.H. Pulmonary vasodilator therapy in congenital diaphragmatic hernia: acute, late, and chronic pulmonary hypertension. Semin Perinatol. 2005;29(2):123-128.
77. Walsh-Sukys M.C., et al. Persistent pulmonary hypertension of the newborn in the era before nitric oxide: practice variation and outcomes. Pediatrics. 2000;105(1 pt 1):14-20.
78. Drummond W.H., et al. The independent effects of hyperventilation, tolazoline, and dopamine on infants with persistent pulmonary hypertension,. J Pediatr. 1981;98(4):603-611.
79. Rudolph A.M., Yuan S. Response of the pulmonary vasculature to hypoxia and H+ ion concentration changes. J Clin Invest. 1966;45(3):399-411.
80. Peckham G.J., Fox W.W. Physiologic factors affecting pulmonary artery pressure in infants with persistent pulmonary hypertension. J Pediatr. 1978;93(6):1005-1010.
81. Bifano E.M., Pfannenstiel A. Duration of hyperventilation and outcome in infants with persistent pulmonary hypertension. Pediatrics. 1988;81(5):657-661.
82. Hendricks-Munoz K.D., Walton J.P. Hearing loss in infants with persistent fetal circulation. Pediatrics. 1988;81(5):650-656.
83. Dworetz A.R., et al. Survival of infants with persistent pulmonary hypertension without extracorporeal membrane oxygenation. Pediatrics. 1989;84(1):1-6.
84. Wung J.T., et al. Management of infants with severe respiratory failure and persistence of the fetal circulation, without hyperventilation. Pediatrics. 1985;76(4):488-494.
85. Lyrene R.K., et al. Alkalosis attenuates hypoxic pulmonary vasoconstriction in neonatal lambs. Pediatr Res. 1985;19(12):1268-1271.
86. Morin F.I. Hyperventilation, alkalosis, prostaglandins and the pulmonary circulation of the newborn. Appl Physiol. 1986;61:2088-2094.
87. Lakshminrusimha S., et al. Pulmonary arterial contractility in neonatal lambs increases with 100% oxygen resuscitation. Pediatr Res. 2006;59(1):137-141.
88. Lakshminrusimha S., et al. Pulmonary hemodynamics in neonatal lambs resuscitated with 21%, 50%, and 100% oxygen. Pediatr Res. 2007;62(3):313-318.
89. Lakshminrusimha S., et al. Oxygen concentration and pulmonary hemodynamics in newborn lambs with pulmonary hypertension. Pediatr Res. 2009;66(5):539-544.
90. Faraci F.M., Didion S.P. Vascular protection: superoxide dismutase isoforms in the vessel wall. Arterioscler Thromb Vasc Biol. 2004;24(8):1367-1373.
91. Lakshminrusimha S., et al. Superoxide dismutase improves oxygenation and reduces oxidation in neonatal pulmonary hypertension. Am J Respir Crit Care Med. 2006;174(12):1370-1377.
92. Steinhorn R.H., et al. Recombinant human superoxide dismutase enhances the effect of inhaled nitric oxide in persistent pulmonary hypertension. Am J Resp Crit Care Med. 2001;164(5):834-839.
93. Auten R.L., et al. Surfactant treatment of full-term newborns with respiratory failure. Pediatrics. 1991;87(1):101-107.
94. Lotze A., et al. Multicenter study of surfactant (beractant) use in the treatment of term infants with severe respiratory failure. Survanta in Term Infants Study Group. J Pediatr. 1998;132(1):40-47.
95. Keszler M., et al. Severe respiratory failure after elective repeat cesarean delivery: a potentially preventable condition leading to extracorporeal membrane oxygenation. Pediatrics. 1992;89(4 pt 1):670-672.
96. Pabalan M.J., et al. Methemoglobin to cumulative nitric oxide ratio and response to inhaled nitric oxide in PPHN. J Perinatol. 2009;29(10):698-701.
97. Carlo W.A., et al. High-frequency jet ventilation in neonatal pulmonary hypertension. Am J Dis Child. 1989;143(2):233-238.
98. Clark R.H., Yoder B.A., Sell M.S. Prospective, randomized comparison of high-frequency oscillation and conventional ventilation in candidates for extracorporeal membrane oxygenation. J Pediatr. 1994;124(3):447-454.
99. Engle W.A., et al. Controlled prospective randomized comparison of high-frequency jet ventilation and conventional ventilation in neonates with respiratory failure and persistent pulmonary hypertension. J Perinatol. 1997;17(1):3-9.
100. Kinsella J.P., Abman S.H. Clinical approaches to the use of high-frequency oscillatory ventilation in neonatal respiratory failure. J Perinatol. 1996;16(2 pt 2):S52-S55.
101. Kinsella J.P., Abman S.H. High-frequency oscillatory ventilation augments the response to inhaled nitric oxide in persistent pulmonary hypertension of the newborn: Nitric Oxide Study Group. Chest. 1998;114(Suppl 1):100S.
102. Kinsella J.P., et al. Randomized, multicenter trial of inhaled nitric oxide and high-frequency oscillatory ventilation in severe, persistent pulmonary hypertension of the newborn. J Pediatr. 1997;131(1 pt 1):55-62.
103. Kinsella J.P., et al. Low-dose inhalation nitric oxide in persistent pulmonary hypertension of the newborn. Lancet. 1992;340(8823):819-820.
104. Roberts J.D., et al. Inhaled nitric oxide in persistent pulmonary hypertension of the newborn. Lancet. 1992;340(8823):818-819.
105. Zayek M., Cleveland D., Morin F.S.III. Treatment of persistent pulmonary hypertension in the newborn lamb by inhaled nitric oxide. J Pediatr. 1993;122(5 pt 1):743-750.
106. Roberts J.D.Jr., et al. Inhaled nitric oxide and persistent pulmonary hypertension of the newborn. The Inhaled Nitric Oxide Study Group. N Engl J Med. 1997;336(9):605-610.
107. Ninos. Inhaled nitric oxide in full-term and nearly full-term infants with hypoxic respiratory failure. The Neonatal Inhaled Nitric Oxide Study Group [erratum N Engl J Med 337(6):434, 1997],. N Engl J Med. 1997;336(9):597-604.
108. Clark R.H., et al. Low-dose nitric oxide therapy for persistent pulmonary hypertension of the newborn. Clinical Inhaled Nitric Oxide Research Group. N Engl J Med. 2000;342(7):469-474.
109. Finer NN, Barrington KJ: Nitric oxide for respiratory failure in infants born at or near term [update in Cochrane Database Syst Rev (2):CD000399, 2001; PMID: 11405963], Cochrane Database Syst Rev (2):CD000399, 2000.
110. Cornfield D.N., et al. Randomized, controlled trial of low-dose inhaled nitric oxide in the treatment of term and near-term infants with respiratory failure and pulmonary hypertension. Pediatrics. 1999;104(5 pt 1):1089-1094.
111. Finer N.N., et al. Randomized, prospective study of low-dose versus high-dose inhaled nitric oxide in the neonate with hypoxic respiratory failure. Pediatrics. 2001;108(4):949-955.
112. Tworetzky W., et al. Inhaled nitric oxide in neonates with persistent pulmonary hypertension. Lancet. 2001;357(9250):118-120.
113. Davidson D., et al. Inhaled nitric oxide for the early treatment of persistent pulmonary hypertension of the term newborn: a randomized, double-masked, placebo-controlled, dose-response, multicenter study. The I-NO/PPHN Study Group,. Pediatrics. 1998;101(3 pt 1):325-334.
114. Konduri G.G., Kim U.O. Advances in the diagnosis and management of persistent pulmonary hypertension of the newborn. Pediatr Clin North Am. 2009;56(3):579-600.
115. Konduri G.G., et al. A randomized trial of early versus standard inhaled nitric oxide therapy in term and near-term newborn infants with hypoxic respiratory failure,. Pediatrics. 2004;113(3 pt 1):559-564.
116. Konduri G.G., et al. Early inhaled nitric oxide therapy for term and near-term newborn infants with hypoxic respiratory failure: neurodevelopmental follow-up. J Pediatr. 2007;150(3):235-240. 240 e1
117. Aly H., Sahni R., Wung J.T. Weaning strategy with inhaled nitric oxide treatment in persistent pulmonary hypertension of the newborn. Arch Dis Child Fetal Neonatal Educ. 1997;76(2):F118-F122.
118. Lipkin P.H., et al. Neurodevelopmental and medical outcomes of persistent pulmonary hypertension in term newborns treated with nitric oxide [comment]. J Pediatr. 2002;140(3):306-310.
119. Rosenberg A.A., et al. Longitudinal follow-up of a cohort of newborn infants treated with inhaled nitric oxide for persistent pulmonary hypertension. J Pediatr. 1997;131(1 pt 1):70-75.
120. American Academy of Pediatrics. Committee on Fetus and Newborn: Use of inhaled nitric oxide. Pediatrics. 2000;106(2 pt 1):344-345.
121. Baquero H., et al. Oral sildenafil in infants with persistent pulmonary hypertension of the newborn: a pilot randomized blinded study. Pediatrics. 2006;117(4):1077-1083.
122. Vargas-Origel A., Gómez-Rodriguez G., Aldana-Valenzuela C., et al. The use of sildenafil in persistent pulmonary hypertension of the newborn,. Am J Perinatol. 2010;27(3):225-230.
123. Steinhorn R.H., et al. Intravenous sildenafil in the treatment of neonates with persistent pulmonary hypertension. J Pediatr. 2009;155(6):841-847. el
124. Lakshminrusimha S., et al. Milrinone enhances relaxation to prostacyclin and iloprost in pulmonary arteries isolated from lambs with persistent pulmonary hypertension of the newborn. Pediatr Crit Care Med. 2009;10(1):106-112.
125. Rashid N., et al. Effects of prostacyclin and milrinone on pulmonary hemodynamics in newborn lambs with persistent pulmonary hypertension induced by ductal ligation. Pediatr Res. 2006;60(5):624-629.
126. Chen B., Lakshminrusimha S., Czech L., et al. Regulation of phosphodiesterase 3 in the pulmonary arteries during the perinatal period in sheep. Pediatr Res. 2009;46(6):682-687.
127. Bassler D., et al. Neonatal persistent pulmonary hypertension treated with milrinone: four case reports. Biol Neonate. 2006;89(1):1-5.
128. McNamara P.J., et al. Milrinone improves oxygenation in neonates with severe persistent pulmonary hypertension of the newborn. J Crit Care. 2006;21(2):217-222.
129. UK collaborative randomised trial of neonatal extracorporeal membrane oxygenation. UK Collaborative ECMO Trail Group. Lancet. 1996;348(9020):75-82.
130. Roberts T.E. Economic evaluation and randomised controlled trial of extracorporeal membrane oxygenation: UK collaborative trial. The Extracorporeal Membrane Oxygenation Economics Working Group. BMJ. 1998;317(7163):911-915.
131. Hintz S.R., et al. Decreased use of neonatal extracorporeal membrane oxygenation (ECMO): how new treatment modalities have affected ECMO utilization. Pediatrics. 2000;106(6):1339-1343.
132. Kennaugh J.M., et al. Impact of new treatments for neonatal pulmonary hypertension on extracorporeal membrane oxygenation use and outcome. J Perinatol. 1997;17(5):366-369.
133. McLaughlin V.V., et al. ACCF/AHA 2009 expert consensus document on pulmonary hypertension a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association developed in collaboration with the American College of Chest Physicians: American Thoracic Society, Inc: and the Pulmonary Hypertension Association. J Am Coll Cardiol. 2009;53(17):1573-1619.
134. Wagenvoort C.A., Wagenvoort N. Primary pulmonary hypertension: a pathologic study of the lung vessels in 156 clinically diagnosed cases. Circulation. 1970;42:1163-1184.
135. Abenhaim L., et al. Appetite-suppressant drugs and the risk of primary pulmonary hypertension. International Primary Pulmonary Hypertension Study Group. N Engl J Med. 1996;335(9):609-616.
136. Rubens C., et al. Big endothelin-1 and endothelin-1 plasma levels are correlated with the severity of primary pulmonary hypertension. Chest. 2001;120(5):1562-1569.
137. Luscher T.F., Barton M. Endothelins and endothelin receptor antagonists: therapeutic considerations for a novel class of cardiovascular drugs. Circulation. 2000;102(19):2434-2440.
138. Chen S.J., et al. Endothelin-receptor antagonist bosentan prevents and reverses hypoxic pulmonary hypertension in rats. J Appl Physiol. 1995;79(6):2122-2131.
139. Prie S., Stewart D.J., Dupuis J. EndothelinA receptor blockade improves nitric oxide-mediated vasodilation in monocrotaline-induced pulmonary hypertension. Circulation. 1998;97(21):2169-2174.
140. Panza J.A., et al. Effect of increased availability of endothelium-derived nitric oxide precursor on endothelium-dependent vascular relaxation in normal subjects and in patients with essential hypertension. Circulation. 1993;87(5):1475-1481.
141. d’uscio L.V., et al. Blood pressure-independent effects of chronic selective ETa-receptor blockade in L-name-induced hypertension. J Hypertens. 1998;16(Suppl 2):S90.
142. Moreau P., et al. Blood pressure and vascular effects of endothelin blockade in chronic nitric oxide-deficient hypertension. Hypertension. 1997;29(3):763-769.
143. Verhaar M.C., et al. Endothelin-A receptor antagonist-mediated vasodilatation is attenuated by inhibition of nitric oxide synthesis and by endothelin-B receptor blockade. Circulation. 1998;97(8):752-756.
144. Giaid A. Nitric oxide and endothelin-1 in pulmonary hypertension. Chest. 1998;114(Suppl 3):208S-212S.
145. Giaid A., et al. Expression of endothelin-1 in the lungs of patients with pulmonary hypertension. N Engl J Med. 1993;328(24):1732-1739.
146. Goerre S., et al. Endothelin-1 in pulmonary hypertension associated with high-altitude exposure. Circulation. 1995;91(2):359-364.
147. Sartori C., et al. Exaggerated endothelin release in high-altitude pulmonary edema. Circulation. 1999;99(20):2665-2668.
148. Stewart D.J., et al. Increased plasma endothelin-1 in pulmonary hypertension: marker or mediator of disease? Ann Intern Med. 1991;114(6):464-469.
149. Cacoub P., et al. Endothelin-1 in primary pulmonary hypertension and the Eisenmenger syndrome. Am J Cardiol. 1993;71(5):448-450.
150. Yoshibayashi M., et al. Plasma endothelin concentrations in patients with pulmonary hypertension associated with congenital heart defects. Evidence for increased production of endothelin in pulmonary circulation. Circulation. 1991;84(6):2280-2285.
151. Allen S.W., et al. Circulating immunoreactive endothelin-1 in children with pulmonary hypertension. Association with acute hypoxic pulmonary vasoreactivity. Am Rev Respir Dis. 1993;148(2):519-522.
152. Adatia I., et al. Thromboxane A2 and prostacyclin biosynthesis in children and adolescents with pulmonary vascular disease. Circulation. 1993;88(5 pt 1):2117-2122.
153. Christman B.W., et al. An imbalance between the excretion of thromboxane and prostacyclin metabolites in pulmonary hypertension. N Engl J Med. 1992;327(2):70-75.
154. Tuder R.M., et al. Prostacyclin synthase expression is decreased in lungs from patients with severe pulmonary hypertension. Am J Respir Crit Care Med. 1999;159(6):1925-1932.
155. Geraci M.W., et al. Pulmonary prostacyclin synthase overexpression in transgenic mice protects against development of hypoxic pulmonary hypertension. J Clin Invest. 1999;103(11):1509-1515.
156. Nagaya N., et al. Gene transfer of human prostacyclin synthase ameliorates monocrotaline-induced pulmonary hypertension in rats. Circulation. 2000;102(16):2005-2010.
157. Weir E.K., Archer S.L. The mechanism of acute hypoxic pulmonary vasoconstriction: the tale of two channels,. FASEB J. 1995;9(2):183-189.
158. Yuan X.J., et al. Attenuated K+ channel gene transcription in primary pulmonary hypertension. Lancet. 1998;351(9104):726-727.
159. Patel A.J., Lazdunski M., Honore E. Kv2.1/Kv9.3, a novel ATP-dependent delayed-rectifier K+ channel in oxygen-sensitive pulmonary artery myocytes. EMBO J. 1997;16(22):6615-6625.
160. Michelakis E.D., et al. Dexfenfluramine elevates systemic blood pressure by inhibiting potassium currents in vascular smooth muscle cells. J Pharmacol Exp Ther. 1999;291(3):1143-1149.
161. Weir E.K., et al. A role for potassium channels in smooth muscle cells and platelets in the etiology of primary pulmonary hypertension. Chest. 1998;114(Suppl 3):200S-204S.
162. Wang J., et al. Action of fenfluramine on voltage-gated K+ channels in human pulmonary-artery smooth-muscle cells. Lancet. 1998;352(9124):290.
163. Cowan K.N., Jones P.L., Rabinovitch M. Elastase and matrix metalloproteinase inhibitors induce regression, and tenascin-C antisense prevents progression, of vascular disease. J Clin Invest. 2000;105(1):21-34.
164. Rabinovitch M. It all begins with EVE (endogenous vascular elastase). Isr J Med Sci. 1996;32(10):803-808.
165. Cowan K.N., et al. Complete reversal of fatal pulmonary hypertension in rats by a serine elastase inhibitor. Nature Med. 2000;6(6):698-702.
166. Fernandez-Patron C., et al. Differential regulation of platelet aggregation by matrix metalloproteinases-9 and -2. Thromb Haemost. 1999;82(6):1730-1735.
167. Fernandez-Patron C., Radomski M.W., Davidge S.T. Vascular matrix metalloproteinase-2 cleaves big endothelin-1 yielding a novel vasoconstrictor. Circ Res. 1999;85(10):906-911.
168. Welsh C.H., et al. Coagulation and fibrinolytic profiles in patients with severe pulmonary hypertension. Chest. 1996;110(3):710-717.
169. Eddahibi S., et al. Treatment with 5-HT potentiates development of pulmonary hypertension in chronically hypoxic rats. Am J Physiol. 1997;272(3 pt 2):H1173-H1181.
170. Eddahibi S., et al. Serotonin transporter overexpression is responsible for pulmonary artery smooth muscle hyperplasia in primary pulmonary hypertension [comment]. J Clin Invest 108(8). 2001:1141-1150.
171. Lane K.B., et al. Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension. The International PPH Consortium. Nature Genet. 2000;26(1):81-84.
172. Trembath R.C., et al. Clinical and molecular genetic features of pulmonary hypertension in patients with hereditary hemorrhagic telangiectasia. N Engl J Med 345(5). 2001:325-334.
173. Heath D., Edwards J.E. The pathology of hypertensive pulmonary vascular disease: a description of six grades of structural changes in the pulmonary arteries with special reference to congenital cardiac septal defects,. Circulation. 1958;18:533-547.
174. Rabinovitch M., et al. Lung biopsy in congenital heart disease: a morphometric approach to pulmonary vascular disease. Circulation. 1978;58(6):1107-1122.
175. Wood P. The Eisenmenger syndrome or pulmonary hypertension with reversed central shunt. Br Med J. 1958:701-709.
176. Blackstone E.H., et al. Optimal age and results in repair of large ventricular septal defects. J Thorac Cardiovasc Surg. 1976;72(5):661-679.
177. Rabinovitch M., et al. Vascular structure in lung tissue obtained at biopsy correlated with pulmonary hemodynamic findings after repair of congenital heart defects. Circulation. 1984;69(4):655-667.
178. Haworth S.G., Reid L. Structural study of pulmonary circulation and of heart in total anomalous pulmonary venous return in early infancy. Br Heart J. 1977;39(1):80-92.
179. Wheller J., et al. Diagnosis and management of postoperative pulmonary hypertensive crisis. Circulation. 1979;60(7):1640-1644.
180. Hanley F.L., et al. Repair of truncus arteriosus in the neonate. J Thorac Cardiovasc Surg. 1993;105(6):1047-1056.
181. Hopkins R.A., et al. Pulmonary hypertensive crises following surgery for congenital heart defects in young children. Eur J Cardio Thorac Surg. 1991;5(12):628-634.
182. Hall S.M., Haworth S.G. Onset and evolution of pulmonary vascular disease in young children: abnormal postnatal remodelling studied in lung biopsies. J Pathol. 1992;166(2):183-193.
183. Celermajer D.S., Cullen S., Deanfield J.E. Impairment of endothelium-dependent pulmonary artery relaxation in children with congenital heart disease and abnormal pulmonary hemodynamics. Circulation. 1993;87(2):440-446.
184. Wessel D.L., et al. Use of inhaled nitric oxide and acetylcholine in the evaluation of pulmonary hypertension and endothelial function after cardiopulmonary bypass. Circulation. 1993;88(5 pt 1):2128-2138.
185. Berner M., et al. Inhaled nitric oxide to test the vasodilator capacity of the pulmonary vascular bed in children with long-standing pulmonary hypertension and congenital heart disease. Am J Cardiol. 1996;77(7):532-535.
186. Lutz J., et al. Endothelin-1- and endothelin-receptors in lung biopsies of patients with pulmonary hypertension due to congenital heart disease. Clin Chem Lab Med. 1999;37(4):423-428.
187. Rubin L.J. Primary pulmonary hypertension. Chest. 1993;104(1):236-250.
188. Resten A., et al. Pulmonary hypertension: CT of the chest in pulmonary venoocclusive disease. AJR Am J Roentgenol. 2004;183(1):65-70.
189. van Wolferen S.A., et al. Prognostic value of right ventricular mass, volume, and function in idiopathic pulmonary arterial hypertension. Eur Heart J. 2007;28(10):1250-1257.
190. McGoon M., et al. Screening, early detection, and diagnosis of pulmonary arterial hypertension: ACCP evidence-based clinical practice guidelines. Chest. 2004;126(Suppl 1):14S-34S.
191. Hoeper M.M., et al. Complications of right heart catheterization procedures in patients with pulmonary hypertension in experienced centers. J Am Coll Cardiol. 2006;48(12):2546-2552.
192. Sitbon O., et al. Long-term response to calcium channel blockers in idiopathic pulmonary arterial hypertension. Circulation. 2005;111(23):3105-3111.
193. Pepke-Zaba J., et al. Inhaled nitric oxide as a cause of selective pulmonary vasodilatation in pulmonary hypertension. Lancet. 1991;338(8776):1173-1174.
194. Rubin L.J., et al. Prostacyclin-induced acute pulmonary vasodilation in primary pulmonary hypertension. Circulation. 1982;66(2):334-338.
195. Schrader B.J., et al. Comparison of the effects of adenosine and nifedipine in pulmonary hypertension. J Am Coll Cardiol. 1992;19(5):1060-1064.
196. Badesch D.B., et al. Medical therapy for pulmonary arterial hypertension: updated ACCP evidence-based clinical practice guidelines. Chest. 2007;131(6):1917-1928.
197. Weiss B.M., et al. Outcome of pulmonary vascular disease in pregnancy: a systematic overview from 1978 through 1996. J Am Coll Cardiol. 1998;31(7):1650-1657.
198. Bowyer J.J., et al. Effect of long term oxygen treatment at home in children with pulmonary vascular disease. Br Heart J. 1986;55(4):385-390.
199. Fuster V., et al. Primary pulmonary hypertension: natural history and the importance of thrombosis. Circulation. 1984;70(4):580-587.
200. Frank H., et al. The effect of anticoagulant therapy in primary and anorectic drug-induced pulmonary hypertension,. Chest. 1997;112(3):714-721.
201. Barst R.J. Treatment of primary pulmonary hypertension with continuous intravenous prostacyclin. Heart. 1997;77(4):299-301.
202. Barst R.J., Maislin G., Fishman A.P. Vasodilator therapy for primary pulmonary hypertension in children. Circulation. 1999;99(9):1197-1208.
203. Rich S., Kaufmann E., Levy P.S. The effect of high doses of calcium-channel blockers on survival in primary pulmonary hypertension [comment],. N Engl J Med. 1992;327(2):76-81.
204. Rich S., McLaughlin V.V. The effects of chronic prostacyclin therapy on cardiac output and symptoms in primary pulmonary hypertension [comment],. J Am Coll Cardiol. 1999;34(4):1184-1187.
205. Barst R.J., et al. A comparison of continuous intravenous epoprostenol (prostacyclin) with conventional therapy for primary pulmonary hypertension. The Primary Pulmonary Hypertension Study Group [comment]. N Engl J Med. 1996;334(5):296-302.
206. McLaughlin V.V., et al. Compassionate use of continuous prostacyclin in the management of secondary pulmonary hypertension: a case series [comment]. Ann Intern Med. 1999;130(9):740-743.
207. Channick R.N., et al. Pulsed delivery of inhaled nitric oxide to patients with primary pulmonary hypertension: an ambulatory delivery system and initial clinical tests. Chest. 1996;109(6):1545-1549.
208. McLaughlin V.V., et al. Reduction in pulmonary vascular resistance with long-term epoprostenol (prostacyclin) therapy in primary pulmonary hypertension [comment]. N Engl J Med. 1998;338(5):273-277.
209. Barst R.J., et al. Survival in primary pulmonary hypertension with long-term continuous intravenous prostacyclin. Ann Intern Med. 1994;121(6):409-415.
210. Rosenzweig E.B., Kerstein D., Barst R.J. Long-term prostacyclin for pulmonary hypertension with associated congenital heart defects. Circulation. 1999;99(14):1858-1865.
211. Simonneau G., et al. Continuous subcutaneous infusion of treprostinil, a prostacyclin analogue, in patients with pulmonary arterial hypertension: a double-blind, randomized, placebo-controlled trial. Am J Respir Crit Care Med. 2002;165(6):800-804.
212. Gomberg-Maitland M., et al. Transition from intravenous epoprostenol to intravenous treprostinil in pulmonary hypertension. Am J Respir Crit Care Med. 2005;172(12):1586-1589.
213. Bloodstream infections among patients treated with intravenous epoprostenol or intravenous treprostinil for pulmonary arterial hypertension—seven sites, United States, 2003-2006. MMWR Morb Mortal Wkly Rep. 2007;56(8):170-172.
214. Olschewski H., et al. Aerosolized prostacyclin and iloprost in severe pulmonary hypertension. Ann Intern Med. 1996;124(9):820-824.
215. Hoeper M.M., et al. Long-term treatment of primary pulmonary hypertension with aerosolized iloprost, a prostacyclin analogue. N Engl J Med. 2000;342(25):1866-1870.
216. Olschewski H., et al. Inhaled iloprost for severe pulmonary hypertension. N Engl J Med. 2002;347(5):322-329.
217. Rimensberger P.C., et al. Inhaled nitric oxide versus aerosolized iloprost in secondary pulmonary hypertension in children with congenital heart disease: vasodilator capacity and cellular mechanisms. Circulation. 2001;103(4):544-548.
218. Ichida F., et al. Acute effect of oral prostacyclin and inhaled nitric oxide on pulmonary hypertension in children. J Cardiol. 1997;29(4):217-224.
219. Nagaya N., et al. Effect of orally active prostacyclin analogue on survival of outpatients with primary pulmonary hypertension. J Am Coll Cardiol. 1999;34(4):1188-1192.
220. Okano Y., et al. Orally active prostacyclin analogue in primary pulmonary hypertension [erratum Lancet 350(9088):1406, 1997]. Lancet. 1997;349(9062):1365.
221. Ono F., et al. Hemodynamic and hormonal effects of beraprost sodium, an orally active prostacyclin analogue, in patients with secondary precapillary pulmonary hypertension. Circ J. 2003;67(5):375-378.
222. Nagaya N., et al. Oral beraprost sodium improves exercise capacity and ventilatory efficiency in patients with primary or thromboembolic pulmonary hypertension. Heart Br Cardiac Soc. 2002;87(4):340-345.
223. Saji T., et al. Short-term hemodynamic effect of a new oral PGI2 analogue, beraprost, in primary and secondary pulmonary hypertension. Am J Cardiol. 1996;78(2):244-247.
224. Channick R.N., et al. Effects of the dual endothelin-receptor antagonist bosentan in patients with pulmonary hypertension: a randomised placebo-controlled study. Lancet. 2001;358(9288):1119-1123.
225. Rubin L.J., et al. Bosentan therapy for pulmonary arterial hypertension [erratum N Engl J Med 346(16):1258, 2002]. N Engl Med. 2002;346(12):896-903.
226. Rubin L.J., et al. Bosentan therapy for pulmonary arterial hypertension. N Engl J Med. 2002;346(12):896-903.
227. Barst R.J., et al. Sitaxsentan therapy for pulmonary arterial hypertension. Am J Respir Crit Care Med. 2004;169(4):441-447.
228. Barst R.J., et al. Clinical efficacy of sitaxsentan, an endothelin-A receptor antagonist, in patients with pulmonary arterial hypertension: open-label pilot study. Chest. 2002;121(6):1860-1868.
229. Galie N., et al. Ambrisentan therapy for pulmonary arterial hypertension. J Am Coll Cardiol. 2005;46(3):529-535.
230. Galie N., et al. Ambrisentan for the treatment of pulmonary arterial hypertension: results of the ambrisentan in pulmonary arterial hypertension, randomized, double-blind, placebo-controlled, multicenter, efficacy (ARIES) study 1 and 2. Circulation. 2008;117(23):3010-3019.
231. Ricciardi M.J., et al. Inhaled nitric oxide in primary pulmonary hypertension: a safe and effective agent for predicting response to nifedipine [comment]. J Am Coll Cardiol. 1998;32(4):1068-1073.
232. Petros A.J., Turner S.C., Nunn A.J. Cost implications of using inhaled nitric oxide compared with epoprostenol for pulmonary hypertension. J Pharm Technol. 1995;11(4):163-166.
233. Ivy D.D., et al. Atrial natriuretic peptide and nitric oxide in children with pulmonary hypertension after surgical repair of congenital heart disease. Am J Cardiol. 1996;77(1):102-105.
234. Miller O.I., et al. Very-low-dose inhaled nitric oxide: a selective pulmonary vasodilator after operations for congenital heart disease. J Thorac Cardiovasc Surg. 1994;108(3):487-494.
235. Roberts J.D.Jr., et al. Inhaled nitric oxide in congenital heart disease. Circulation. 1993;87(2):447-453.
236. Atz A.M., Wessel D.L. Inhaled nitric oxide in the neonate with cardiac disease. Semin Perinatol. 1997;21(5):441-455.
237. Atz A.M., et al. Inhaled nitric oxide in children with pulmonary hypertension and congenital mitral stenosis. Am J Cardiol. 1996;77(4):316-319.
238. Journois D., et al. Inhaled nitric oxide as a therapy for pulmonary hypertension after operations for congenital heart defects. J Thorac Cardiovasc Surg. 1994;107(4):1129-1135.
239. Goldman A.P., et al. Nitric oxide might reduce the need for extracorporeal support in children with critical postoperative pulmonary hypertension. Ann Thorac Surg. 1996;62(3):750-755.
240. Boolell M., et al. Sildenafil, a novel effective oral therapy for male erectile dysfunction. Br J Urol. 1996;78(2):257-261.
241. Bigatello L.M., et al. Sildenafil can increase the response to inhaled nitric oxide. Anesthesiology. 2000;92(6):1827-1829.
242. Cohen A.H., et al. Inhibition of cyclic 3′-5′-guanosine monophosphate-specific phosphodiesterase selectively vasodilates the pulmonary circulation in chronically hypoxic rats. J Clin Invest. 1996;97(1):172-179.
243. Ichinose F., et al. Prolonged pulmonary vasodilator action of inhaled nitric oxide by Zaprinast in awake lambs. J Appl Physiol. 1995;78(4):1288-1295.
244. Ichinose F., et al. Selective pulmonary vasodilation induced by aerosolized zaprinast. Anesthesiology. 1998;88(2):410-416.
245. Ivy D.D., et al. Dipyridamole attenuates rebound pulmonary hypertension after inhaled nitric oxide withdrawal in postoperative congenital heart disease. J Thorac Cardiovasc Surg. 1998;115(4):875-882.
246. Ziegler J.W., et al. Effects of dipyridamole and inhaled nitric oxide in pediatric patients with pulmonary hypertension. Am J Respir Crit Care Med. 1998;158(5 pt 1):1388-1395.
247. Galie N., et al. Sildenafil citrate therapy for pulmonary arterial hypertension. N Engl J Med. 2005;353(20):2148-2157.
248. Haraldsson S.A., Kieler-Jensen N., Ricksten S.E. The additive pulmonary vasodilatory effects of inhaled prostacyclin and inhaled milrinone in postcardiac surgical patients with pulmonary hypertension. Anesth Analg. 2001;93(6):1439-1445.
249. McLaughlin V.V., et al. Randomized study of adding inhaled iloprost to existing bosentan in pulmonary arterial hypertension. Am J Respir Crit Care Med. 2006;174(11):1257-1263.
250. Simonneau G., et al. Addition of sildenafil to long-term intravenous epoprostenol therapy in patients with pulmonary arterial hypertension: a randomized trial. Ann Intern Med. 2008;149(8):521-530.
251. Reichenberger F., et al. Atrial septostomy in the treatment of severe pulmonary arterial hypertension. Thorax. 2003;58(9):797-800.
252. Rothman A., et al. Atrial septostomy as a bridge to lung transplantation in patients with severe pulmonary hypertension. Am J Cardiol. 1999;84(6):682-686.
253. Sandoval J., et al. Graded balloon dilation atrial septostomy in severe primary pulmonary hypertension. A therapeutic alternative for patients nonresponsive to vasodilator treatment,. J Am Coll Cardiol. 1998;32(2):297-304.
254. Bridges N.D. Lung transplantation in children. Curr Opin Cardiol. 1998;13(1):73-77.
255. Boucek M.M., et al. The Registry of the International Society of Heart and Lung Transplantation: Second Official Pediatric Report—1998. J Heart Lung Transplant. 1998;17(12):1141-1160.
256. Whyte R.I., et al. Heart-lung transplantation for primary pulmonary hypertension. Ann Thorac Surg. 1999;67(4):937-941.
257. Ro P.S., Spray T.L., Bridges N.D. Outcome of infants listed for lung or heart/lung transplantation. J Heart Lung Transplant. 1999;18(12):1232-1237.
258. Gaynor J.W., et al. Update on lung transplantation in children. Curr Opin Pediatr. 1998;10(3):256-261.
259. McLaughlin V.V., Shillington A., Rich S. Survival in primary pulmonary hypertension: the impact of epoprostenol therapy. Circulation. 2002;106(12):1477-1482.
260. D’Alonzo G.E., et al. Survival in patients with primary pulmonary hypertension. Results from a national prospective registry. Ann Intern Med. 1991;115(5):343-349.
261. Sitbon O., et al. Long-term intravenous epoprostenol infusion in primary pulmonary hypertension: prognostic factors and survival. J Am Coll Cardiol. 2002;40(4):780-788.
262. Barst R.J., et al. A comparison of continuous intravenous epoprostenol (prostacyclin) with conventional therapy for primary pulmonary hypertension. The Primary Pulmonary Hypertension Study Group. N Engl J Med. 1996;334(5):296-302.
263. Kim H.W., Kim G.B., Je H.G., et al. Pulmonary arterial hypertension in children: a single center experience. Korean Circ J. 2008;38:644-650.
264. Eysmann S.B., et al. Two-dimensional and Doppler-echocardiographic and cardiac catheterization correlates of survival in primary pulmonary hypertension. Circulation. 1989;80(2):353-360.
265. Forfia P.R., et al. Tricuspid annular displacement predicts survival in pulmonary hypertension. Am J Respir Crit Care Med. 2006;174(9):1034-1041.
266. Fijalkowska A., et al. Serum N-terminal brain natriuretic peptide as a prognostic parameter in patients with pulmonary hypertension. Chest. 2006;129(5):1313-1321.