CHAPTER 195 Optic Pathway Hypothalamic Gliomas
Optic pathway hypothalamic gliomas (OPHGs) are included as one of the representative childhood supratentorial tumors. They represent 1% of all central nervous system (CNS) tumors and 3% to 5% of all pediatric brain tumors. OPHGs tend to affect infants and young children, who can have various clinical symptoms. Sixty-five percent of patients with OPHGs are younger than 5 years at diagnosis. They occur with equal frequency in males and females. OPHGs cause symptoms as a result of the anatomic structures involved, such as the optic pathway (including the optic nerve, chiasm, tract, and radiation), the hypothalamus-pituitary axis, the limbic system, the third ventricle, and the circle of Willis. When these structures are compromised by the glioma or by the treatments applied, children may suffer from visual, endocrine, cognitive, and psychobehavioral dysfunction. A mass in the third ventricle produces obstructive hydrocephalus, which may be the primary cause of symptoms. The majority of pediatric low-grade astrocytomas in the hypothalamic/chiasmatic region are typical pilocytic astrocytomas (PAs).1,2 There may be other gliomas such as fibrillary astrocytoma and ganglioglioma, but anaplastic astrocytoma and primitive neuroectodermal tumor are uncommon. PAs have a typical histologic pattern consisting of a biphasic pattern of Rosenthal’s fibers and eosinophilic granular bodies. These tumors are indolent in nature, and an aggressive course is rare. Grossly, they appear partly cystic and have a grayish homogeneous pattern with areas distinct from normal brain tissue. Cerebrospinal fluid (CSF) dissemination is uncommon, and malignant transformation is rare in PAs. A subgroup of PA, pilomyxoid astrocytoma (PMA), was described in 1999.3 PMA typically arises in the hypothalamus or optic chiasm of an infant or young child, with the mean age at initial evaluation being 10 to 18 months of age. PMA has histologic features that are quite unique and different from those of PA. The prominent histologic features of PMA are monotonous, bipolar spindle cells with an angiocentric arrangement within a strikingly myxoid background.3,4 PMA has more aggressive behavior than PA. A local recurrence rate after surgery of 55% to 76% has been reported.3–5 Dissemination through CSF has been demonstrated in 11% to 14% of patients with PMA. Chikai and colleagues speculated that PMA was an infantile form of PA. They suspected that a subset of PMA in the optic pathway/hypothalamus originates from the optic chiasm, possibly derived from radial glia existing in the embryonic optic chiasm.6
These tumors are often associated with neurofibromatosis type 1 (NF1). NF1 is predominantly autosomal dominant, but about 50% of cases result from new mutations. In patients with NF1, the frequency of OPHGs ranges from 6.6% to 20%,7–9 whereas in children with OPHGs, NF1 is diagnosed in about 27% to 58%.1,2 Recent guidelines by the Optic Pathway Glioma Task Force on Neurofibromatosis of the National Institutes of Health recommend a complete ophthalmologic examination at the diagnosis of NF1, with annual examinations until 6 years of age and longer interval thereafter because children younger than 6 years have the greatest risk for the development of symptomatic OPHG.10 However, more recent literature has revealed that OPHG in children with NF1 can be diagnosed after the age of 6 and may progress until the age of 12. These authors recommend more frequent observation than previously proposed.11,12
Clinical Findings
The initial signs and symptoms are dependent on the location and direction of tumor growth and the structures involved. The predominant clinical manifestations are visual disturbances, hydrocephalus, and endocrine dysfunction; the most common initial symptoms are visual complaints. Children may exhibit a decrease in visual acuity or, less often, visual field deficits. If the tumor is located in the intraorbital space, proptosis with deviation of the affected eye may occur. Suprasellar gliomas extending into the third ventricle often cause obstructive hydrocephalus. Of such gliomas, tumors of hypothalamic origin may not cause visual symptoms. Hydrocephalus is manifested as macrocephaly and failure to thrive in infancy. In older patients, the symptoms associated with hydrocephalus are headaches, emesis, lethargy, and decrease in school performance. Hypothalamic-pituitary axis involvement by the tumor leads to partial hypopituitarism, and about 50% of children have measurable endocrine abnormalities.2 Common endocrine dysfunctions are growth hormone deficiency and precocious puberty. Precocious puberty is more common in children with NF1 and OPHG and occurs in about 40% of cases.9 Diabetes insipidus is rare in children with suprasellar gliomas.
Age can play a factor in the type of initial signs seen. Infants often have macrocephaly, hydrocephalus, failure to thrive, or diencephalic syndrome. Younger children may have hydrocephalus, failure to thrive, visual disturbance, or precocious puberty. Older children can have hydrocephalus and visual disturbances as well. Diencephalon syndrome, originally described by Russel, is characterized by a suprasellar glioma in infancy that is manifested as lack of weight gain and emaciation with little subcutaneous fat tissue and alertness. These infants have normal oral intake and height and may exhibit nystagmus or head bobbing. Cappelli and coauthors reported that 6 of 69 children with optic pathway tumors had diencephalon syndrome,2 whereas 6 of 14 children with OPHG diagnosed when younger than 3 years exhibited diencephalon syndrome according to Silva and associates.13 CNS tumor is a rare cause of failure to thrive in childhood, but clinicians should keep it in mind when considering the differential diagnosis. The diagnosis of OPHG is often delayed, and these tumors can be quite large at discovery.14
Neuroimaging
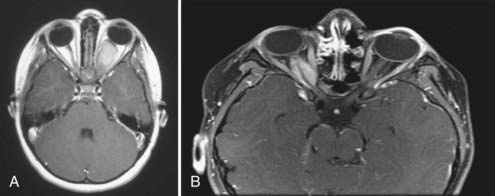
Although CT is useful, magnetic resonance imaging (MRI) is the imaging modality of choice for characterizing an OPHG. Benign gliomas are usually hypointense or isointense on T1-weighted images and hyperintense on T2-weighted images. These tumors generally have various degrees of enhancement ranging from none to strong enhancement with contrast agents. Cyst formation in or around the solid tumor may be present. Intraorbital optic nerve gliomas show fusiform enlargement of the nerve, as well as an elongated length, which results in a tortuous course and often kinking appearance (Fig. 195-1A). The rare optic nerve sheath meningioma in childhood may mimic optic nerve gliomas (Fig. 195-1B).
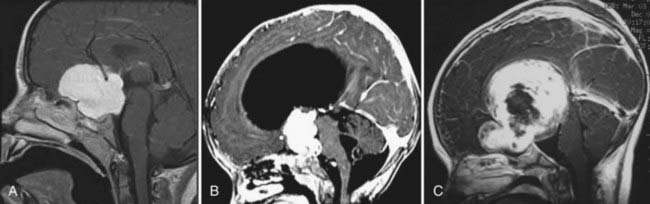
Small chiasmal tumors show expansion of the chiasm with little or no enhancement, which is often seen in children with NF1. Large chiasmal gliomas can exhibit exophytic extension into the suprasellar and parasellar locations and further dorsally into the third ventricle. MRI of these globular suprasellar gliomas shows that the chiasm and hypothalamus are often indistinguishable because of the lack of an anatomic border between these structures (Fig. 195-2A to C). These globular suprasellar gliomas often enhance after intravenous infusion of a contrast agent. They may be associated with cyst formation within or around solid tumor. Another distinct subgroup consists of a diffuse optic pathway glioma. It affects the entire optic pathway, including the optic nerve, chiasm, and tract and the lateral geniculate body. On rare occasion, the optic radiations are also involved, which is more common in children with NF1.15 A diffuse optic pathway glioma affects these structures bilaterally and is characterized by the “mustache” sign. In the children with NF1, it is common to find nonenhancing hyperintense lesions on T2-weighted images in the basal ganglia, brainstem, and cerebellum.16
Treatment
When the patient has symptoms or the tumor is progressing, or both, treatment is warranted. Although radical tumor resection is ideal, because of the anatomic structures present around the tumor, aggressive surgery would compromise visual, neurological, and endocrine function, and total resection is therefore not possible without complications. In one study involving surgical resection of optic pathway gliomas, the progression-free survival rate at 5 years was 52.4%,17 so approximately half of the patients had no recurrence for 5 years.
Surgical Treatment
Optic Nerve Glioma
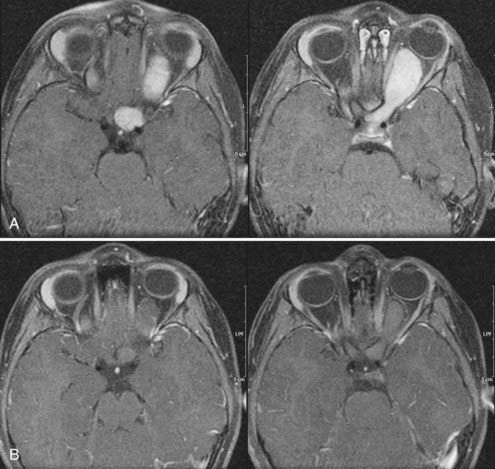
In the past, one of the indications for surgery on optic nerve gliomas was to prevent posterior extension to the chiasm and beyond. However, orbital optic nerve gliomas rarely extend posteriorly after diagnosis.18 Only 5% of optic nerve gliomas recur in the chiasm after “complete” intraorbital excision.19 The present therapeutic recommendation for symptomatic or progressive optic nerve glioma, or both, is chemotherapy (Fig. 195-3A and B). Children with no useful vision, severe eye-threatening proptosis, or a painful eye that failed treatment with chemotherapy may benefit from tumor resection. If both eyes are affected, no indication for surgery is present.
Chiasmal Hypothalamic Tumors
Approximately 50% of patients with suprasellar astrocytoma have obstructive hydrocephalus, which can be treated with a shunt. Because of obstruction at the interventricular foramen or anterior third ventricle, fenestration of the septum pellucidum or bilateral shunts are needed. Endoscopic third ventriculostomy is not feasible. Management of these patients can be complex because their ventricular compartments may become loculated as the tumor enlarges. As with other patients with hydrocephalus, they are then at risk for shunt malfunction and infections. On rare occasion in infants with OPHG, CSF may not be absorbed through the peritoneal cavity and abdominal ascites can result.20
Patients with an enlarged optic chiasm and little or no enhancement are not candidates for radical resection, whereas those with large exophytic tumors extending into the third ventricle and parasellar cisterns should undergo debulking. The surgical approach depends on the direction of maximal tumor extension. Gliomas extending anteriorly to a subfrontal location or laterally to a parasellar location are approached with a pterional/subfrontal technique. In contrast, gliomas extending predominantly to the third ventricle (often hydrocephalic) are removed through an anterior transcallosal approach from either a transforaminal or interforniceal route. It may be difficult to choose the surgical approach for large globular suprasellar astrocytomas extending in both directions (see Fig. 195-2C). The circle of Willis constricts the tumor like a waistband, which usually indicates the limiting point for access when approached with either method. For tumor resection, an operative microscope, ultrasonic aspirator, and frameless stereotaxy are indispensable. In either case, one should avoid overzealous resection, which often causes serious postoperative complications.
Chemotherapy
Chemotherapy has frequently been the primary mode of treatment of symptomatic children over the past 2 decades as a result of the significant latent morbidity associated with RT. At present, many clinicians consider chemotherapy to be the first-line treatment because of fewer side effects than occurs with RT. In children younger than 3 years, it is usually the only adjuvant treatment because intellectual and other functions are relatively well preserved. Various chemotherapeutic agents have been used, including dactinomycin and vincristine21,22 and multiagent chemotherapy consisting of 6-thioguanine, procarbazine, dibromodulcitol, lomustine, and vincristine, with which Petronio and coauthors reported a 5-year recurrence-free survival rate of about 60%.23 More recent studies have recommended a platinum-based chemotherapy regimen. The most widely used of such regimens are probably carboplatin alone24–26; cisplatin, etoposide, and vinblastine27; and carboplatin and vincristine.1,13 Responses (from complete response to stable disease) to carboplatin and vincristine chemotherapy were reported in 105 (85%) of 123 children with OPHGs.1
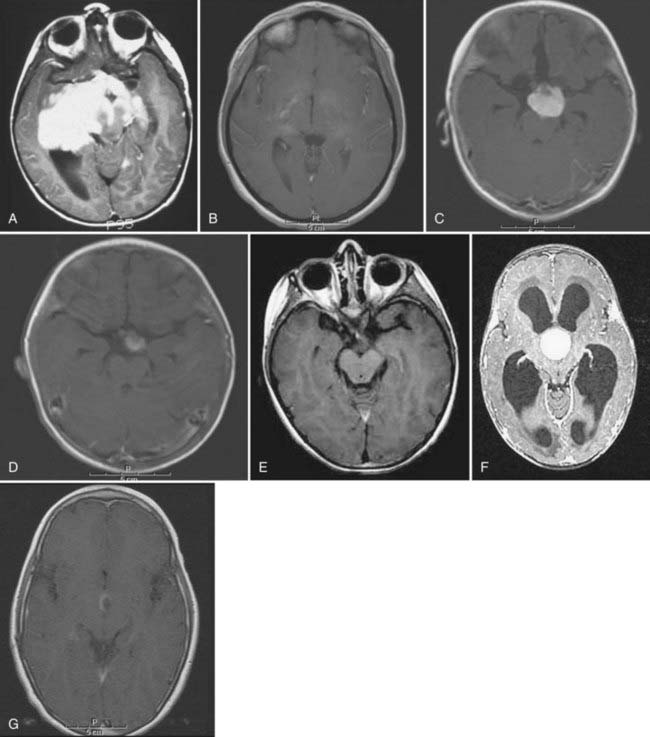
In general, the 5-year recurrence-free survival rate after chemotherapy remains about 60%. Chemotherapy effectively postpones RT, and this delay would reduce the neurological morbidity associated with RT. Children with diencephalon syndrome also respond and improve after chemotherapy.13,28 After completion of chemotherapy, the efficacy of tumor control is sustained for a number of years, but in approximately 60% of children, the OPHG eventually relapses, particularly in younger children. However, in other groups of patients no further therapy is required because of sustained responses to chemotherapy (Fig. 195-4A to G). Patients with NF1 have indolent disease.22
Radiation Therapy
In the prechemotherapy era, 10-year overall and recurrence-free survival rates after RT for OPHGs were reported to be 83% and 65.5%, respectively.2 Older children and those with progressive disease during chemotherapy or with relapse after the completion of chemotherapy are treated with RT. In addition to external beam radiation, other forms of RT have emerged over the past decade, including stereotactic radiosurgery and proton beam therapy. Standard treatment can now be modified and targeted through conformal beam or intensity-modulated RT. RT may be considered in children older than 5 years with progressive disease.
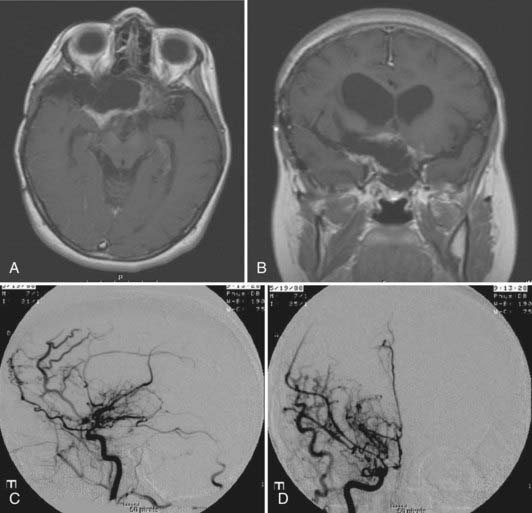
RT probably provides better long-term tumor control. With RT, progression-free survival rates at 5 years were 90%.2 However, one should be aware of radiation-induced side effects, including a second tumor, cerebrovascular complications, neuroendocrine disturbances, optic neuritis, and neurocognitive dysfunction. Second tumors and malignant transformation of benign gliomas may occur after RT. There is a significantly increased risk for a second tumor in patients with NF1 who were treated with RT for optic pathway gliomas.29,30 Malignant transformation of childhood PA is rare, and the majority are related to previous exposure to radiation. Radiation-induced cerebrovascular complications can also occur; Cappelli and coworkers reported that optic pathway gliomas developed in 9 (17%) of 54 children during a median interval of 2.5 years (range, 6 months to 6 years).2 This incidence is expected to be greater if the follow-up is longer. Children with NF1 are much more susceptible to cerebrovascular complications, five times more than those without NF1. They are often symptomatic, and angiography shows stenosis of a major artery or moyamoya disease, or both (Fig. 195-5). The incidence of postirradiation vascular occlusive disorder is particularly high with suprasellar gliomas because the radiation effects are compounded by mechanical compression by persistent tumor around the carotid artery and its major branches. Some may already exhibit arterial occlusion at the time of diagnosis. We would advise that baseline CTA or MRA be performed at the time of diagnosis for all globular suprasellar gliomas. In nearly half of treated children, adverse effects on endocrine function develop and require hormonal replacement therapy. Irradiation of a large field of the brain during the developmental stage causes neurocognitive dysfunction. Delay in administering RT would help, and limiting the field of RT with use of an image-guided technique would also decrease radiation exposure to the surrounding normal brain tissue.
Outcome
The most consistent negative prognostic indicators are young age, posterior tumor location, and PMA subtypes. In addition, optic pathway gliomas associated with NF1 tend to fare better than those without NF1. Survival decreases as the tumor involves structures more posterior to the chiasm—the hypothalamus and optic tracts and radiations. The overall outcome in patients with optic pathway gliomas is generally favorable. However, the prognosis in infants with globular suprasellar gliomas or diffuse optic pathway gliomas is particularly unfavorable.1,19 Patients with a purely optic nerve glioma have survival rates close to 100%.19,31 The long-term visual prognosis is usually stable for the intact side, whereas the affected eye tends to deteriorate over time. Although chemotherapy may reduce the size and degree of contrast enhancement and stabilize visual function, the affected optic nerve does not necessarily regain or show improvement in visual function.18
Patients with PMA have a less favorable prognosis. According to Komotar and colleagues, PMA occurs in a significantly younger population than PA does (mean age at diagnosis of 18 and 58 months, respectively; P < .01).4 Patients with PMA had a slightly higher rate of local recurrence than did patients with PA (76% and 50%, respectively). Furthermore, the PMA group had a substantial rate of CSF dissemination (14%), whereas none of the tumors in the PA group exhibited such spread. Patients with PMA had significantly shorter recurrence-free times than did patients with typical PA (mean progression-free survival times of 26 and 147 months, respectively) and significantly shorter overall survival times than did patients with typical PA (mean overall survival times of 63 and 213 months, respectively). Furthermore, 33% of patients with PMA died as a result of their disease as compared with 17% of patients with PA.
The overall outcome of children with OPHGs is generally favorable, with a 10-year survival rate of 74% to 96% after chemotherapy with or without surgery.2 Chemotherapy has improved the quality of life of young children because RT is delayed or eliminated altogether. In longer follow-up of up to 20 years, the survival rate decreases to 50%. The major cause of the death is tumor progression and involvement of the hypothalamus, which results in multiple electrolyte imbalances, hormone deficits, and hydrocephalus. Complications associated with RT include malignant transformation of the tumor, vasculopathy, and hypothalamic dysfunction.
Ahn Y, Cho BK, Kim SK, et al. Optic pathway glioma: outcome and prognostic factors in a surgical series. Childs Nerv Syst. 2006;22:1136-1142.
Alvord ECJr, Lofton S. Gliomas of the optic nerve or chiasm. Outcome by patients’ age, tumor site, and treatment. J Neurosurg. 1988;68:85-98.
Aquino VM, Fort DW, Kamen BA. Carboplatin for the treatment of children with newly diagnosed optic chiasm glioma: a phase II study. J Neurooncol. 1999;41:255-259.
Astrup J. Natural history and clinical management of optic pathway gliomas. Br J Neurosurg. 2003;17:327-335.
Cappelli C, Grill J, Raquin M, et al. Long term follow up of 69 patients treated for optic pathway tumour before the chemotherapy era. Arch Dis Child. 1998;79:334-338.
Chikai K, Ohnishi A, Kato T, et al. Clinico-pathological features of pilomyxoid astrocytoma of the optic pathway. Acta Neuropathol. 2004;108:109-114.
Fernandez C, Figarella-Branger D, Girard N, et al. Pilocytic astrocytomas in children: prognostic factors—a retrospective study of 80 cases. Neurosurgery. 2003;53:544-553.
Gnekow AK, Kortmann RD, Pietsch T, et al. Low grade chiasmatic-hypothalamic gliomas—carboplatin and vincristine chemotherapy effectively defers radiotherapy within a comprehensive treatment strategy—report from the multicenter treatment study for children and adolescents with a low grade gliomas—HIT-LGG 1996—of the Society of Pediatric Oncology and Hematology. Klin Padiatr. 2004;216:331-342.
Hsu T-R, Wong T-T, Chang F-C, et al. Responsiveness of progressive optic pathway tumors to cisplatin-based chemotherapy in children. Childs Nerv Syst. 2008;24:1457-1461.
Huber J, Sovinz P, Lackner H, et al. Diencephalic syndrome: a frequently delayed diagnosis in failure to thrive. Klin Padiatr. 2007;219:91-94.
Janns AJ, Grundy R, Cnaan A, et al. Optic pathway and hypothalamic/chiasmatic gliomas in children younger than age 5 years with 6-year follow-up. Cancer. 1995;15:1051-1059.
King A, Listernick R, Charrow J, et al. Optic pathway gliomas in neurofibromatosis type 1—the effect of presenting symptoms on occurrence. Am J Med Genet. 2003;122:95-99.
Komotar RJ, Burger PC, Carson BS, et al. Pilocytic and pilomyxoid hypothalamic/chiasmatic astrocytomas. Neurosurgery. 2004;54:72-79.
Lama G, Esposito SM, Grassia C, et al. Neurofibromatosis type 1 and optic pathway glioma. A long-term follow-up. Minerva Pediatr. 2007;59:13-21.
Listernick R, Charrow J, Tomita T, et al. Carboplatin therapy for optic pathway tumors in children with neurofibromatosis type 1. J Neurooncol. 1999;45:185-190.
Listernick R, Frener RE, Piersall L, et al. Late-onset optic pathway tumors in children with neurofibromatosis 1. Neurology. 2004;63:1944-1946.
Listernick R, Louis D, Packer R, et al. Optic pathway gliomas in children with neurofibromatosis 1: consensus statement from NF 1 Optic Pathway Glioma Task Force. Ann Neurol. 1997;41:142-148.
Liu GT, Brodsky MC, Phillips PC, et al. Optic radiation involvement in optic pathway gliomas in neurofibromatosis. Am J Ophthalmol. 2004;137:407-414.
Mahoney DHJr, Cohen ME, Friedman HS, et al. Carboplatin is effective therapy for young children with progressive optic pathway tumors: a Pediatric Oncology Group phase II study. Neuro Oncol. 2000;2:213-220.
Norfray JF, Darling C, Byrd S, et al. Short TE proton MRS and NF-1 intracranial lesions. J Comput Assist Tomogr. 1999;23:994-1003.
Olavarria G, Reitman AJ, Goldman S, et al. Post-shunt ascites in infants with optic chiasmal hypothalamic astrocytoma: role of ventricular gallbladder shunt. Childs Nerv Syst. 2005;21:382-384.
Packer RJ, Sutton LN, Bilaniuk LT, et al. Treatment of chiasmatic/hypothalamic gliomas of childhood with chemotherapy: an update. Ann Neurol. 1988;23:79-85.
Petronio J, Edwards MSB, Prados M, et al. Management of chiasmal and hypothalamic gliomas of infancy and childhood with chemotherapy. J Nneurosurg. 1991;74:701-708.
Poussaint TY, Barnes PD, Nichols K, et al. Diencephalic syndrome: clinical features and imaging findings. AJNR Am J Neuroradiol. 1997;18:1499-1505.
Sharif S, Ferner R, Birch JM, et al. Second primary tumors in neurofibromatosis 1 patients treated for optic glioma: substantial risks after radiotherapy. J Clin Oncol. 2006;24:2570-2575.
Silva MM, Goldman S, Keating G, et al. Optic pathway hypothalamic gliomas in children under three years of age: the role of chemotherapy. Pediatr Neurosurg. 2000;33:151-158.
Singhal S, Birch JM, Kerr B, et al. Neurofibromatosis type 1 and sporadic optic gliomas. Arch Dis Child. 2002;87:65-70.
Sylvester CL, Drohan LA, Sergott RC. Optic-nerve gliomas, chiasmal gliomas and neurofibromatosis type 1. Curr Opin Ophthalmol. 2006;17:7-11.
Thiagalingan S, Flaherty M, Billson F, et al. Neurofibromatosis type 1 and optic pathway gliomas: follow up of 54 patients. Ophthalmology. 2004;111:568-577.
Tihan T, Fisher PG, Kepner JL, et al. Pediatric astrocytomas with monomorphous pilomyxoid features and a less favorable outcome. J Neuropathol Exp Neurol. 1999;58:1061-1068.
Zeid JL, Charrow J, Sandu M, et al. Orbital optic nerve gliomas in children with neurofibromatosis type 1. J AAPOS. 2006;10:534-539.
1 Gnekow AK, Kortmann RD, Pietsch T, et al. Low grade chiasmatic-hypothalamic gliomas—carboplatin and vincristine chemotherapy effectively defers radiotherapy within a comprehensive treatment strategy—report from the multicenter treatment study for children and adolescents with a low grade gliomas—HIT-LGG 1996—of the Society of Pediatric Oncology and Hematology. Klin Padiatr. 2004;216:331-342.
2 Cappelli C, Grill J, Raquin M, et al. Long term follow up of 69 patients treated for optic pathway tumour before the chemotherapy era. Arch Dis Child. 1998;79:334-338.
3 Tihan T, Fisher PG, Kepner JL, et al. Pediatric astrocytomas with monomorphous pilomyxoid features and a less favorable outcome. J Neuropathol Exp Neurol. 1999;58:1061-1068.
4 Komotar RJ, Burger PC, Carson BS, et al. Pilocytic and pilomyxoid hypothalamic/chiasmatic astrocytomas. Neurosurgery. 2004;54:72-79.
5 Fernandez C, Figarella-Branger D, Girard N, et al. Pilocytic astrocytomas in children: prognostic factors—a retrospective study of 80 cases. Neurosurgery. 2003;53:544-553.
6 Chikai K, Ohnishi A, Kato T, et al. Clinico-pathological features of pilomyxoid astrocytoma of the optic pathway. Acta Neuropathol. 2004;108:109-114.
7 Listernick R, Frener RE, Piersall L, et al. Late-onset optic pathway tumors in children with neurofibromatosis 1. Neurology. 2004;63:1944-1946.
8 Lama G, Esposito SM, Grassia C, et al. Neurofibromatosis type 1 and optic pathway glioma. A long-term follow-up. Minerva Pediatr. 2007;59:13-21.
9 King A, Listernick R, Charrow J, et al. Optic pathway gliomas in neurofibromatosis type 1—the effect of presenting symptoms on occurrence. Am J Med Genet. 2003;122:95-99.
10 Listernick R, Louis D, Packer R, et al. Optic pathway gliomas in children with neurofibromatosis 1: consensus statement from NF 1 Optic Pathway Glioma Task Force. Ann Neurol. 1997;41:142-148.
11 Sylvester CL, Drohan LA, Sergott RC. Optic-nerve gliomas, chiasmal gliomas and neurofibromatosis type 1. Curr Opin Ophthalmol. 2006;17:7-11.
12 Thiagalingan S, Flaherty M, Billson F, et al. Neurofibromatosis type 1 and optic pathway gliomas: follow up of 54 patients. Ophthalmology. 2004;111:568-577.
13 Silva MM, Goldman S, Keating G, et al. Optic pathway hypothalamic gliomas in children under three years of age: the role of chemotherapy. Pediatr Neurosurg. 2000;33:151-158.
14 Poussaint TY, Barnes PD, Nichols K, et al. Diencephalic syndrome: clinical features and imaging findings. AJNR Am J Neuroradiol. 1997;18:1499-1505.
15 Liu GT, Brodsky MC, Phillips PC, et al. Optic radiation involvement in optic pathway gliomas in neurofibromatosis. Am J Ophthalmol. 2004;137:407-414.
16 Norfray JF, Darling C, Byrd S, et al. Short TE proton MRS and NF-1 intracranial lesions. J Comput Assist Tomogr. 1999;23:994-1003.
17 Ahn Y, Cho BK, Kim SK, et al. Optic pathway glioma: outcome and prognostic factors in a surgical series. Childs Nerv Syst. 2006;22:1136-1142.
18 Zeid JL, Charrow J, Sandu M, et al. Orbital optic nerve gliomas in children with neurofibromatosis type 1. J AAPOS. 2006;10:534-539.
19 Alvord ECJr, Lofton S. Gliomas of the optic nerve or chiasm. Outcome by patients’ age, tumor site, and treatment. J Neurosurg. 1988;68:85-98.
20 Olavarria G, Reitman AJ, Goldman S, et al. Post-shunt ascites in infants with optic chiasmal hypothalamic astrocytoma: role of ventricular gallbladder shunt. Childs Nerv Syst. 2005;21:382-384.
21 Packer RJ, Sutton LN, Bilaniuk LT, et al. Treatment of chiasmatic/hypothalamic gliomas of childhood with chemotherapy: an update. Ann Neurol. 1988;23:79-85.
22 Janns AJ, Grundy R, Cnaan A, et al. Optic pathway and hypothalamic/chiasmatic gliomas in children younger than age 5 years with 6-year follow-up. Cancer. 1995;15:1051-1059.
23 Petronio J, Edwards MSB, Prados M, et al. Management of chiasmal and hypothalamic gliomas of infancy and childhood with chemotherapy. J Neurosurg. 1991;74:701-708.
24 Mahoney DHJr, Cohen ME, Friedman HS, et al. Carboplatin is effective therapy for young children with progressive optic pathway tumors: a Pediatric Oncology Group phase II study. Neuro Oncol. 2000;2:213-220.
25 Listernick R, Charrow J, Tomita T, et al. Carboplatin therapy for optic pathway tumors in children with neurofibromatosis type 1. J Neurooncol. 1999;45:185-190.
26 Aquino VM, Fort DW, Kamen BA. Carboplatin for the treatment of children with newly diagnosed optic chiasm glioma: a phase II study. J Neurooncol. 1999;41:255-259.
27 Hsu T-R, Wong T-T, Chang F-C, et al. Responsiveness of progressive optic pathway tumors to cisplatin-based chemotherapy in children. Childs Nerv Syst. 2008;24:1457-1461.
28 Huber J, Sovinz P, Lackner H, et al. Diencephalic syndrome: a frequently delayed diagnosis in failure to thrive. Klin Padiatr. 2007;219:91-94.
29 Sharif S, Ferner R, Birch JM, et al. Second primary tumors in neurofibromatosis 1 patients treated for optic glioma: substantial risks after radiotherapy. J Clin Oncol. 2006;24:2570-2575.
30 Singhal S, Birch JM, Kerr B, et al. Neurofibromatosis type 1 and sporadic optic gliomas. Arch Dis Child. 2002;87:65-70.
31 Astrup J. Natural history and clinical management of optic pathway gliomas. Br J Neurosurg. 2003;17:327-335.






