Chapter 69 Oncology

Uveal malignant melanoma: Introduction
Jerry A. Shields and Carol L. Shields
General considerations
Uveal malignant melanoma is the most frequent intraocular malignancy encountered in a practice of ophthalmology1,2.This neoplasm is important because of its potential to cause blindness and death due to systemic metastasis. Hence, clinicians should be familiar with uveal melanoma and make an accurate diagnosis and recommend referral to a subspecialist who manages this neoplasm. Approximately 85% of uveal melanomas arise in the choroid, 10% in the ciliary body, and 5% in the iris. This subchapter briefly discusses demographics, clinical features, differential diagnosis, diagnostic approaches, pathology, management, and prognosis for uveal melanoma.
Demographics
The annual age-adjusted incidence of uveal melanoma is six cases per 1 million population in the United States and Europe. Uveal melanoma is decidedly more common in adult Caucasians and is uncommon in children and dark-skinned individuals. In our experience with 8033 patients with uveal melanoma, the median age at presentation was 59 years (range 3 to 99 years) and the male/female ratio was 50/50%. The melanoma occurred in Caucasians in 98% cases followed by Hispanic (1%), African American (<1%), Asian <1%), Middle Eastern (<1%), Native American (<1%), and Asian Indian (<1%)3. Hence, it is diagnosed more often in Europe and America and infrequently in Africa and Asia. There is no predisposition for gender. Predisposing conditions for uveal melanoma include congenital ocular melanocytosis, Caucasian race, and possibly the dysplastic nevus syndrome and excess exposure to sunlight.
Clinical features
The clinical features of uveal melanoma are covered in detail in a comprehensive textbook and numerous articles on the subject1. The clinical findings vary with the location of the lesion in the uveal tract.
Iris melanoma
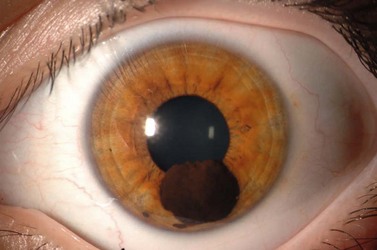
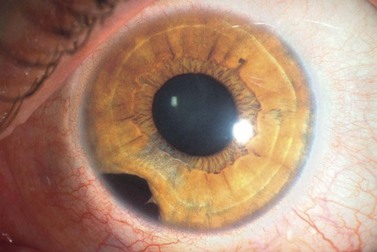
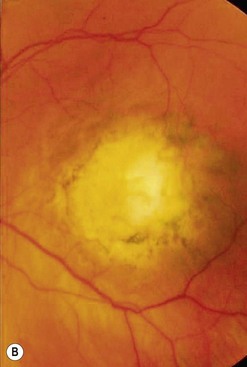
Iris melanoma can be circumscribed (nodular) or diffuse. Circumscribed iris melanoma appears as a variably pigmented, well-defined mass in the iris stroma (Fig. 69.1). More than 80% are located in the inferior half of the iris. It can be totally pigmented, partly pigmented, or clinically amelanotic. The size and shape can vary considerably from case to case. Some are relatively small and almost flat and others are larger and more elevated. Like iris nevus, which is generally smaller, it can cause an irregular pupil and ectropion of the pigment epithelium at the pupillary margin.
The less common diffuse iris melanoma has a tendency to produce acquired hyperchromic heterochromia and secondary glaucoma due to tumor infiltration of the trabecular meshwork1. It can diffusely affect the entire iris or it can appear as irregular geographic patches of pigment. Some patients with diffuse iris melanoma present with unilateral ipsilateral glaucoma and there is a delay in diagnosis while the ‘idiopathic’ or ‘pigmentary’ glaucoma is treated.
Ciliary body melanoma
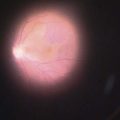
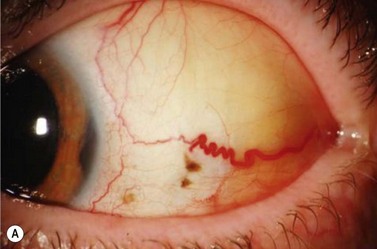
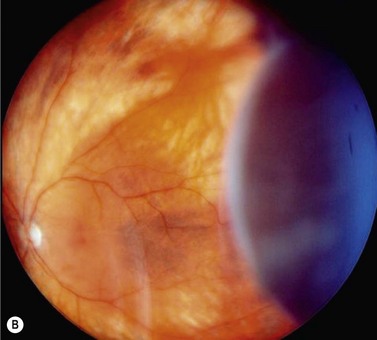
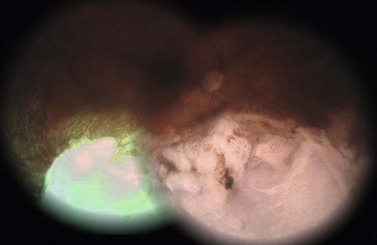
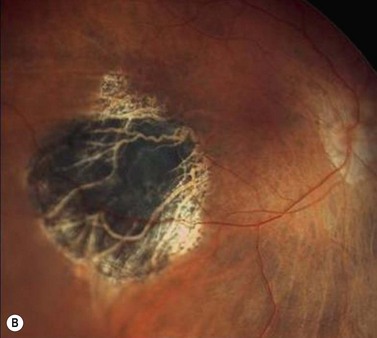
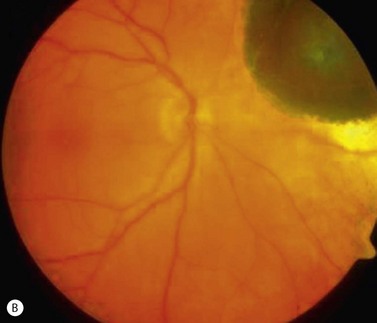
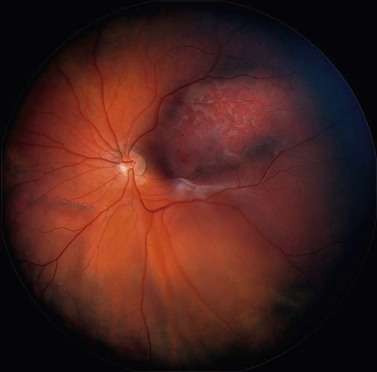
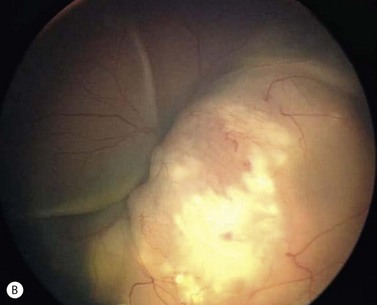
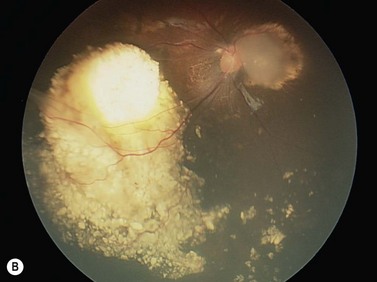
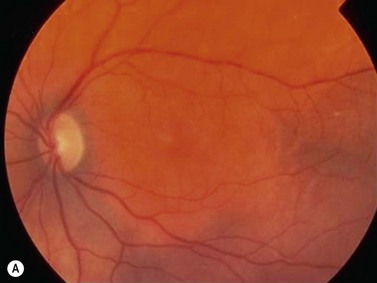
In contrast to iris melanoma, ciliary body melanoma often attains a larger size before it is recognized clinically1 (Fig. 69.2A). However, it is frequently associated with external signs that suggest the underlying diagnosis. The most important is one or more dilated episcleral blood vessels (sentinel vessels) that develop over the base of the tumor. A second sign is an epibulbar pigmented lesion characteristic of transcleral extension of the tumor. When the pupil is dilated widely in such cases, the ciliary body tumor can be visualized as a dome-shaped mass (Fig. 69.2B). Less often, it can assume a circumferential ring growth pattern (ring melanoma). Ciliary body melanoma frequently causes subluxation of the lens and cataract. It can grow posteriorly into the choroid (ciliochoroidal melanoma) and anteriorly into the anterior chamber angle and iris (iridociliary melanoma). It can infiltrate the trabecular meshwork, causing secondary glaucoma.
Choroidal melanoma
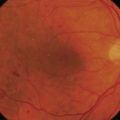
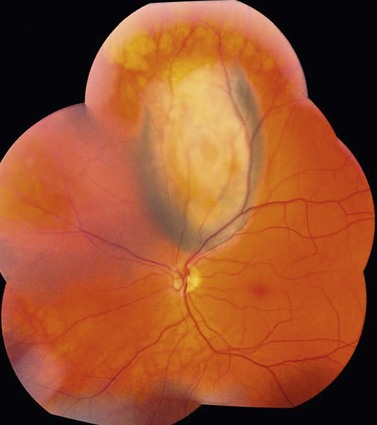
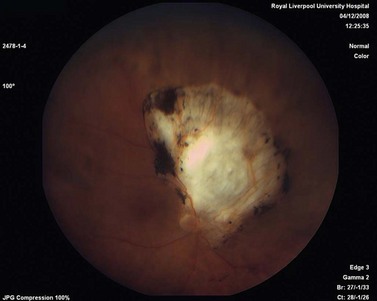
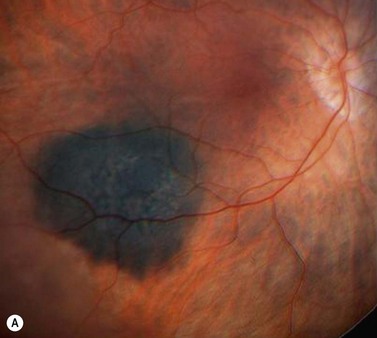
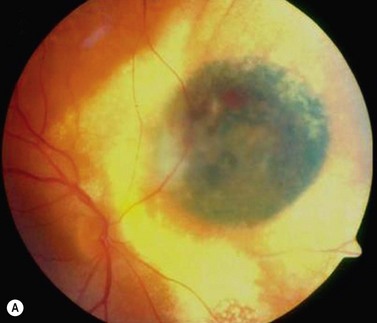
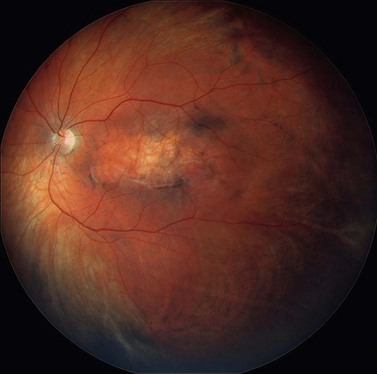
Choroidal melanoma usually presents as a sessile, dome-shaped, or mushroom-shaped mass deep to the sensory retina (Fig. 69.3). It is usually moderately or deeply pigmented but it can be entirely non-pigmented, in which case the diagnosis can be more difficult. A posterior choroidal melanoma can display clumps of overlying orange pigment on its surface at the level of the retinal pigment epithelium. A secondary non-rhegmatogenous retinal detachment frequently occurs. In contrast to a rhegmatogenous detachment, in which the subretinal fluid does not shift, the fluid with melanoma and other tumors shifts with positional changes of the patient’s head. When the melanoma is amelanotic and mushroom shaped, dilated blood vessels in the tumor are visible ophthalmoscopically. When a choroidal melanoma breaks through Bruch’s membrane and assumes such a mushroom shape, it has a tendency to bleed into the subretinal space and vitreous, often obscuring a view of the underlying tumor. Choroidal melanoma can also assume a diffuse growth pattern with only minimal elevation of the tumor1.
Differential diagnosis
The differential diagnosis of uveal melanoma is discussed in more detail elsewhere1. Iris melanoma can resemble iris nevus, epithelioma (adenoma) of the iris pigment epithelium, iris cyst, iridocorneal endothelial syndrome, leiomyoma, and miscellaneous other conditions. Ciliary body melanoma must be differentiated from tumors of the ciliary body pigmented epithelium and non-pigmented epithelium, leiomyoma, cyst, ciliochoroidal effusion, and several other tumors and pseudotumors. Pigmented choroidal melanoma can resemble a large choroidal nevus, subretinal hemorrhage, and a number of other tumors and pseudotumors. Non-pigmented choroidal melanoma must be differentiated from amelanotic choroidal nevus, choroidal metastasis, choroidal hemangioma, granuloma, and other conditions.
Pathology
Most pathologists use the Mclean modification of the Callender classification, in which uveal melanoma is divided into spindle, epithelioid, and mixed cell types1. Most iris melanomas and smaller posterior uveal melanomas are predominantly of spindle cell type, whereas larger melanomas contain a greater proportion of epithelioid cells. Histopathologic criteria for a worse prognosis include more epithelioid cells, greater mitotic activity, greater basal diameter of the tumor, diffuse growth pattern, and extrascleral extension of the melanoma. Genetic factors related to poor prognosis include chromosome 3 monosomy, especially with 8q addition.
Management
Management of uveal melanoma depends on several clinical findings and includes close observation, laser ablation, transpupillary thermotherapy, plaque brachytherapy, charged particle irradiation, local resection, enucleation, and a combination of methods. Plaque radiotherapy and enucleation are the two most commonly employed methods, with the goal being complete eradication of the malignancy. Studies comparing both methods have found similar survival rates4.
Prognosis
Useful features for predicting prognosis for vision and life are mentioned above and described elsewhere. Recently, cytogenetic alterations in uveal melanoma cells have emerged as the most reliable methods of predicting systemic prognosis. Monosomy of chromosome 3 and gain in chromosome 8 (trisomy 8) are most reliable for predicting a worse prognosis5,6. These are also discussed in more detail elsewhere.
Brachytherapy of uveal melanoma
Tara A. McCannel and Bertil Damato
Introduction
Uveal melanoma has traditionally been treated by enucleation, plaque brachytherapy, or external beam radiation with local resection or phototherapy in some cases. Brachytherapy with iodine-125 and enucleation have been evaluated prospectively in a multi-centered fashion by the Collaborative Ocular Melanoma Study (COMS). The COMS did not show mortality rates for medium-sized melanomas to be significantly worse after iodine-125 plaque brachytherapy than after enucleation7. Brachytherapy with iodine-125, which delivers gamma irradiation, is the favored treatment modality in the United States because it has good tissue penetration and its short half-life contributes to its ease of use. Furthermore, the dosimetry can be adjusted for each individual tumor by adjusting the number and distribution of the iodine-125 seeds, which are embedded in the resin lining the underside of the shell. In Europe, ruthenium-106, which mostly emits beta irradiation, is the preferred radioisotope for local treatment of uveal melanoma8,9. Other radioisotopes have been utilized for brachytherapy including cobalt-60, palladium-103, and iridium-192.
Goals of surgery
The goal of brachytherapy is to control local tumor growth by sterilizing the primary tumor while conserving the eye with as much useful vision as possible. It is not known whether brachytherapy or any other form of treatment for uveal melanoma alters patient mortality and if so in whom. Furthermore, treatments for metastatic uveal melanoma are not usually effective10.
Indications for surgery
Uveal melanomas up to 5 mm thick can be treated with a ruthenium-106 plaque, and tumors up to 10 mm thick may be treated with an iodine-125 plaque9. Tumors beyond 20 mm in the largest basal dimension are difficult to treat with episcleral plaque. Brachytherapy is also contraindicated by bulky extraocular extension, unless such tumors can be excised. Optic nerve involvement is not necessarily a contraindication if dosimetry suggests that the entire tumor can be irradiated. Notched iodine-125 plaques can be designed to allow adequate treatment to the nerve11.
Operation techniques
Ruthenium-106 plaque
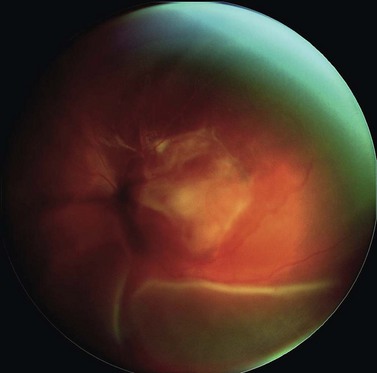
In Liverpool, 15 mm, 20 mm, and 25 mm ruthenium plaques are used for tumors with basal diameters not exceeding 10 mm, 15 mm, and 20 mm respectively. The intended location of the anterior plaque edge is marked on the sclera. A transparent template with four perforations is sutured to the sclera with releasable sutures and its position in relation to the tumor is checked by performing binocular indirect ophthalmoscopy with a right-angled 20-gauge transilluminator passed through the posterior perforation of the template. With tumors extending close to optic disk or fovea, the template is placed with its posterior edge at the posterior tumor margin. Once the template is correctly positioned, a mattress suture is placed in the sclera and left loose. The template is removed and replaced by the radioactive plaque, which is secured by sutures (Fig. 69.4). If trans-scleral tumor biopsy is performed (e.g. for genetic tumor typing), this is done after tumor localization and immediately before inserting the radioactive plaque. If the biopsy is trans-retinal, this is undertaken after radioactive plaque insertion.
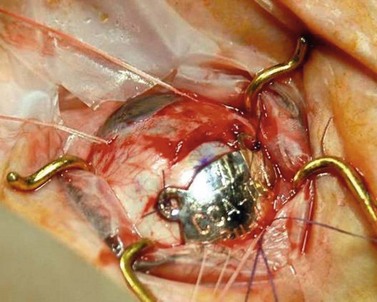
Iodine plaque
At the Jules Stein Eye Institute in Los Angeles, a thinner modified COMS plaque has evolved with dosing and dimensions customized to each individual patient tumor. Plaque placement is similar to that for ruthenium plaques, with transillumination performed to mark the anterior edge of the tumor. Confirmation of plaque placement by indirect ophthalmoscopy is particularly helpful in treating amelanotic uveal melanomas. The author (TM) performs routine intraoperative ultrasonography before permanent tying of the plaque sutures. This has been demonstrated to increase the accuracy of plaque placement and potentially reduce the risk of marginal local recurrence12.
Postoperative complications
Iodine-125 emits relatively low energy photons, which theoretically decreases radiation-related complications. In spite of this favorable profile, iodine-125 brachytherapy can cause keratitis, cataract, neovascular glaucoma, maculopathy, and optic neuropathy13,14.
Assessment of surgery
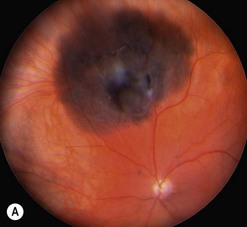
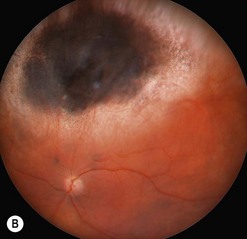
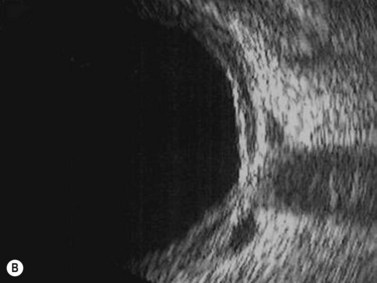
The main quality indicator of brachytherapy is local tumor control. Following brachytherapy, the tumor size may vary within the first 2 months due to local inflammation. Ultrasonography may be performed at the 3-month postoperative visit to confirm a tumor response. Either stabilization of tumor growth or a decrease in tumor height by ultrasonography is considered a successful response to brachytherapy (Fig. 69.5). Rapid shrinkage of the melanoma actually portends a poor prognosis for the patient15.
Proton beam radiotherapy of uveal melanoma
Ann Schalenbourg and Leonidas Zografos
Introduction
External beam radiotherapy has the advantage over brachytherapy in delivering a uniform irradiation to the target volume. Additionally, its indication is less restricted by size, shape, and/or location of the tumor. Teletherapy can be delivered through photons (X-rays), electrons, gamma rays, or accelerated heavy particles (protons, alpha particles). Proton therapy was first applied in Boston in 197516, in Lausanne and the Paul Scherrer Institut (PSI) in 198417,18, and is becoming available in a growing number of centers around the world16–21.
Goals of treatment
As the treatment through which local tumor control is achieved (enucleation or conservative therapy) has no influence on its metastatic risk22, the main goal of protontherapy is to achieve maximal local tumor control while conserving a comfortable eye, and, if possible, useful residual vision.
Indications for proton beam radiotherapy
Some centers use proton therapy for nearly all uveal (iris, ciliary body, and choroidal) melanomas17,20, whereas others reserve this modality for tumors that cannot adequately be treated conservatively by brachytherapy or local resection19,23. In all centers, contraindications include: tumor volume of more than 50% of the eye, large extraocular extension, (sub)total retinal detachment, suspicion of optic nerve invasion and neovascular glaucoma.
Operation techniques for tantalum marker insertion
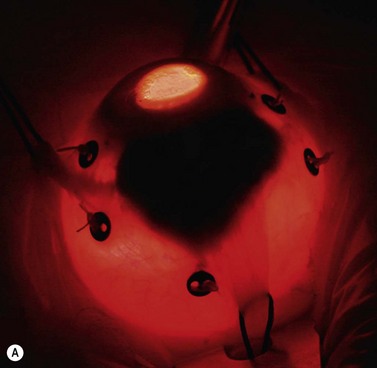
A 180–360° conjunctival peritomy is made. The eye is rotated with muscle slings or, if any rectus muscles have been disinserted, with traction sutures placed in the sclera. The tumor margins are localized by transillumination and marked on the sclera with ink or a pen. Transparent tumor margins are visualized by indentation and indirect ophthalmoscopy. Tantalum markers, four to seven in number, are sutured to the sclera close to the tumor margins (Fig. 69.6A). These buttons are 2.5 mm in diameter, inert, and non-magnetic. Measurements are taken of distances from each marker to: (i) the nearest tumor margin, (ii) the other markers, and (iii) the limbus. The limbus diameter is also recorded. Any disinserted muscles are reinserted and the conjunctiva is closed in the usual fashion.
Simulation
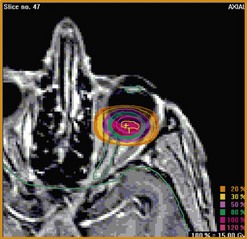
A face-mask and a bite-block are molded for each patient, to immobilize the head precisely in a frame that is secured to the delivery apparatus. The position of the patient’s chair is adjusted so that the tumor is in the target area. Radiographs are taken to localize the tantalum markers for computerized modeling of the tumor within the eye. A brass collimator is prepared for each patient, with an opening adapted to the projected tumor shape (Fig. 69.6B). The irradiation depth can be modulated by interposing absorbers in the proton beam trajectory. The optimal ocular position is determined, so that minimal doses of radiation are delivered to optic disc and other structures.
Proton beam radiotherapy
A total dose of 60–70 Cobalt Gy-equivalent is delivered in four to five fractions16,17. The patient is seated with the head immobilized by the mask and bite-block, gazing at a strategically located target. The eyelids are usually retracted with a speculum. However, when the upper eyelid margin cannot be avoided, treatment is sometimes administered through closed eyelids, the patient fixating with the other eye.
Intraoperative complications
Intraoperative complications are rare, the most evident being retinal perforation when suturing a marker to the sclera. The most common is an over- or underestimation of the tumor margins on transillumination. A penumbra caused by oblique transillumination can cause an overestimation of the tumor extent. Conversely, relative differences in tumor pigmentation as well as the parallax phenomenon – the fact that the posterior tumor margin, to the tangentially placed observer’s eye, is projected about 1 mm anteriorly on the external scleral face – are responsible for an underestimation of the tumor margins24.
Postoperative complications
Long-term complications such as radiation-induced optic neuropathy, maculopathy, and cataract can usually be predicted at the time of treatment planning and therefore occur unavoidably when the tumor extends close to those structures25.
Maculopathy caused by edema or hard exudates can occur even when the macula receives little or no radiation. The risks of persistent retinal detachment and neovascular glaucoma increase with tumor volume18.
Assessment of surgery
Because survival is usually predetermined by the time the ocular tumor is detected and treated22; the main quality indicator is local tumor control (Fig. 69.7). Local tumor recurrence ranges from 1–5% and figures among the lowest of conservative treatment techniques for uveal melanoma17,19–21,26,27. Marginal tumor recurrence can occur if tumor extent is underestimated, which can occur with tumors involving ciliary body. Central tumor recurrences are rare. Conservation of the eye is mainly related to tumor dimensions, the main cause of secondary enucleation being neovascular glaucoma20. Residual vision depends on tumor location and size rather than the type of conservative therapy.
Stereotactic photon beam radiation techniques for uveal melanoma
Martin Zehetmayer and Richard Poetter
Preoperative assessment
During SEBI the patient’s head is immobilized with a stereotactic head frame such as the Leksell head frame used for the GammaKnife28–3034.
For the LINAC, non-invasive head masks are used and these are made from thermoplastic materials31–3335.
Stereotactic radiotherapy (fractionated)
Stereotactic radiotherapy is performed using a LINAC with a specific device and the use of non-invasive head frames, in combination with non-invasive ocular monitoring systems. A robot-mounted LINAC ‘Cyberknife’ has recently been introduced for fractionated and one-fraction stereotactic treatments36,37.
Fractionated treatment with the GammaKnife has been investigated in a study applying two and three fractions for uveal melanoma28. More fractions are not clinically feasible.
With the LINAC, a single beam of radiation is aimed at the tumor from many different directions. Treatment can be delivered during an arc rotation of the LINAC beam around the target isocenter. Round collimators of different sizes and 5–12 arcs of rotation are generally used to cover the target volume. Radiation also can be performed with static conformal fields. Here, the beam shape is adjusted to the target contour by a micro multi-leaf collimator (Fig. 69.8).
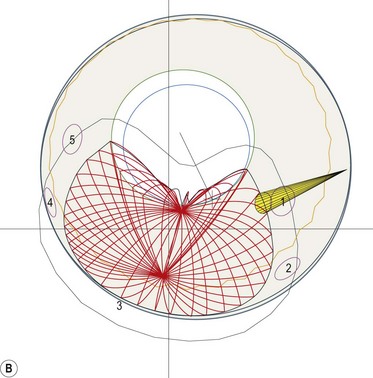
The entire treatment and the total dose (50–70 Gy) are given in four to five fractions of 10–14 Gy over 2 weeks31–3335. Because of the non-invasive and less rigid head and ocular fixation devices, a safety margin of 2.0–2.5 mm in all directions is usually added to the tumor margins.
Stereotactic radiosurgery (one fraction)
Stereotactic radiosurgery is defined as a single delivery of an effective therapeutic radiation dose to a defined limited target. Most patients undergoing radiosurgery have been treated with the Leksell GammaKnife (GK). This comprises 201 cobalt sources placed in a metal hemispheric device that is positioned around the patient’s head so that highly collimated beams of gamma rays converge on the tumor (Fig. 69.9). A total radiation dose of 25–40 Gy is delivered with a safety margin of 1–2 mm29,30,34,38,39.

Anesthesia, akinesia, and ocular fixation
For the GammaKnife and the Cyberknife treatment this is usually accomplished by a retrobulbar injection of an anesthetic to obtain akinesia of the periocular muscles28–30,34,36,37. This technique is often combined with traction sutures, which are passed through two rectus muscles30. For the GammaKnife, an alternative ocular fixation system with a suction-assisted contact lens has been described28.
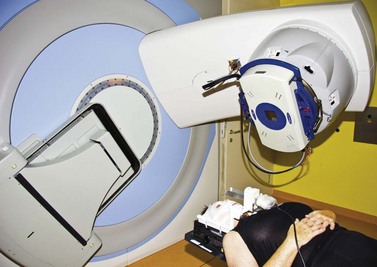
For stereotactic radiotherapy with a LINAC, different approaches have been used to stabilize the eye and thus the tumor. For most patients, a CCTV ocular monitoring system with a head fixation mask has been used31,32,33,35. The patient is asked to fix a defined point or a flashing light. This approach is non-invasive and hence no injection is necessary (Fig. 69.10).
Assessment of treatment-related adverse side effects
Fractionated LINAC SRT is increasingly being used and is similar in principle to the proton therapy approach. At present, most centers use 50–70 Gy total dose delivered in five fractions with 14–10 Gy per fraction31–3335.
Both methods seem to produce similar results with regards to local control; however, the therapeutic window of single-fraction SRS with the GammaKnife seems to be narrower. High single-dose treatment (e.g. 50–80 Gy) was abandoned because of common and severe complications38,39. Single radiation doses below 40 Gy (at the tumor margin) seem to lead to high local tumor control rates with an acceptable incidence of radiogenic side effects29,30,34,39.
Beside the total dose, there is evidence that the amount of irradiated eye and tumor volume influences outcome28.
With Cyberknife SRS, early good results are reported with a 22–18 Gy prescribed marginal dose36.
Local resection of uveal melanoma
Bertil Damato, Heinrich Heimann and Carl Groenewald
Introduction
Local resection procedures include: iridectomy, irido-cyclectomy, trans-scleral choroidectomy with or without cyclectomy, and trans-retinal choroidectomy40. Tumor resection may be primary, or secondary following radiotherapy.
Indications
Primary local resection
Iridectomy is indicated for nodular melanomas involving up to 4 clock hours of iris and not extending to angle. In several centers, this has been replaced by brachytherapy or proton beam radiotherapy40.
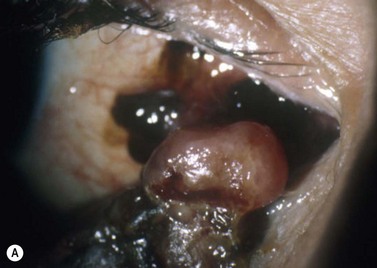
Iridocyclectomy is performed for tumors involving up to 4 clock hours of angle and/or ciliary body and is preferred to radiotherapy when tissue is desired for diagnosis and/or prognostic studies (Fig. 69.11)40.
Trans-scleral choroidectomy and cyclochoroidectomy are performed in few centers and then only if the tumor is considered unsuitable for radiotherapy because the thickness is too great for brachytherapy, and if proton beam radiotherapy or stereotactic radiotherapy is undesirable because of risks such as optic neuropathy or canalicular obstruction40,41. Contraindications to trans-scleral local resection are: diffuse tumor spread, extensive retinal invasion, extraocular spread, involvement of optic nerve or more than 4 clock-hours of the ciliary body, and poor general health precluding hypotensive anesthesia (Fig. 69.12).
Endoresection is indicated for posterior tumors up to 10–13 mm in diameter as a means of avoiding radiation-induced optic neuropathy or maculopathy if the patient is keen to retain good vision and accepts the controversial nature of this surgery (Fig. 69.13).
Operation techniques
Iridectomy
A broad iridectomy is usually required, dealing with the iris defect by iridoplasty, intraocular iris implant, or a cosmetic contact lens40.
Iridocyclectomy
Various methods have been described. Our technique is to constrict the pupil. After performing a 180° conjunctival peritomy, the tumor is localized by transillumination and its margins marked on the sclera with a pen. A lamellar–scleral flap is created, hinged anteriorly and extending into cornea. Deep scleral incisions are made around the tumor with blunt-tipped scissors. The uvea is perforated at the iridociliary junction and resected, either circumferentially or postero-anteriorly, so as to conserve as much of the iris as possible40. Any uncontrollable vitreous bulge is treated by core vitrectomy through a separate sclerotomy or by open-sky vitrectomy. The sclera is closed with non-absorbable sutures, in case histology indicates a need for urgent adjunctive brachytherapy.
Choroidectomy
After 180° conjunctival peritomy and disinsertion of any extraocular muscles in the field, the tumor is localized by transillumination and its margins marked on the sclera40. A polyhedral, lamellar, scleral flap is created, hinged posteriorly. The eye is decompressed by performing limited core vitrectomy, without infusion, using a contact lens and illumination from the operating microscope. Deep scleral incisions are made around the tumor, first laterally, then posteriorly, and finally anteriorly, so as to create a stepped scleral wound edge. The uvea is grasped posterior to the ora with two pairs of notched microforceps and gently stretched to create a small opening for blunt-tipped, spring scissors. These are used to resect the tumor, taking care not to damage the adjacent retina and to preserve as much of the ciliary epithelium as possible. Tumor that has invaded the retina is left in situ, using a Bard–Parker scalpel. The deep scleral lamella is used as a handle to lift the tumor out of the eye. The lamellar scleral flap is closed with interrupted 8-0 nylon sutures. The choroid posterior to the flap is indented with two sponge cells during scleral suturing, to prevent subretinal hematoma formation. Once scleral closure is complete, balanced salt solution is injected into the vitreous cavity. The muscles are re-inserted and the conjunctiva is closed. If there is a retinal break, this is treated by appropriate vitreoretinal surgery and silicone oil tamponade, which are performed after the intraocular injection of balanced salt solution. If adjunctive brachytherapy is required, the plaque is inserted after reconstituting the globe and after any vitreoretinal surgery. The plaque is removed after delivering 100 Gy to a depth of 1–2 mm, leaving disinserted muscles on slings in the mean time.
Endoresection
Total pars plana vitrectomy is performed40,43. The retina is perforated at the tumor apex, avoiding any major retinal blood vessels. The tumor is removed piecemeal, together with a rim of healthy choroid. Fluid–air exchange is performed and any subretinal fluid gravitating into the coloboma is aspirated. Endolaser is applied to the scleral bed to treat any residual tumor, and to the surrounding retina to achieve retinopexy. The peripheral retina is examined with indentation to identify any retinal breaks, which are treated by endolaser photocoagulation. Air–silicone exchange is performed. Cryotherapy is applied to the scleral ports in case of tumor seeding. Adjunctive plaque radiotherapy is administered, either at the end of the procedure or a few weeks later, if histology and cytogenetics indicate high grade malignancy. The silicone oil is removed 12 weeks, when phacoemulsification and lens implantation are also performed.
Intraoperative complications
The main intraoperative complications of trans-scleral local resection include: malposition of scleral flap, buttonholes in superficial or deep sclera, uncontrolled hemorrhage, lens touch, retinal tear, and incomplete tumor resection40. With endoresection, the most common complications are: uncontrolled hemorrhage, incomplete tumor resection, lens touch, and entry-site tears40.
Postoperative complications
Conservation of vision depends on the size and location of the tumor and the avoidance of complications44. Local tumor recurrence is most likely to occur with large, high grade melanomas extending far posteriorly and if adjunctive brachytherapy has not been administered45. Recurrent disease can occur many years after the local resection45. It usually arises at the margin of the coloboma but can develop anywhere in the eye. If not treated quickly, the tumor can extend extraocularly. After endoresection, recurrent tumor can extend trans-sclerally or through the retinotomy to seed around the vitreous cavity46,47.
Rhegmatogenous retinal detachment usually occurs within the first postoperative month and requires urgent surgery, as proliferative vitreoretinopathy tends to develop rapidly48.
Vitreous hemorrhage is rare in the absence of a retinal break.
Metastatic disease is related to tumor size and extent, histological grade, and genetic type49,50. There is no evidence that ocular treatment influences survival51.
Phototherapy of uveal melanoma
Introduction
Phototherapy is useful as primary treatment for small choroidal melanomas, as a means of avoiding radiotherapy52. It is also administered as an adjunct to radiotherapy, to prevent local tumor recurrence53. Secondary phototherapy can induce regression of exudation after radiotherapy, thereby improving vision54.
Fundamental principles
’Trans-pupillary thermotherapy (TTT)’ heats the tumor to 60–65°C by means of a diode laser. This method supersedes photocoagulation, which heats to tumor to temperatures exceeding 65°C52. Photodynamic therapy (PDT) is an investigational method that uses light energy of the appropriate wavelength to activate an agent such as verteporfin with the production of toxic free radicals55,56. Photocoagulation is regarded as obsolete and is not considered in this subchapter57.
Goals of treatment
The goal of treatment is to sterilize the melanoma, with minimal damage to surrounding tissues.
Indications and contraindications
On its own, TTT is associated with a high rate of local tumor recurrence so that it is reserved for small tumors, less than 3 mm thick. TTT therefore tends to be combined with brachytherapy, as part of the primary treatment to reduce any chances of local tumor recurrence (i.e. ‘sandwich therapy’)53,57,58. It is also useful as a secondary procedure, as a treatment for radiation-induced tumor exudation. Contraindications include: media opacities, small pupil, peripheral tumor location and, if used as sole treatment, tumor thickness exceeding 3 mm. TTT tends to be less effective with amelanotic melanomas, which are more likely to respond to PDT than pigmented tumors55. Optic disc involvement increases the risk of local tumor recurrence.
Transpupillary thermotherapy
A contact lens with an anti-reflective coating is used. Overlapping spots of diode laser are administered to the entire tumor surface and a surround of 2–3 mm, with a beam diameter of 3 mm, each application having a duration of 1 minute. The initial power setting is around 400 mW and is increased in 50 mW increments until retinal blanching begins to occur at 45 seconds. At this intensity, it should be possible to treat the tumor without damaging any overlying retinal vessels. If the TTT is administered as a treatment for radiation-induced exudative maculopathy, then only the visible tumor is treated, without safety margins (Figs 69.14 and 69.15)54.
Photodynamic therapy
Several PDT protocols exist. In Liverpool, this therapy is administered as for choroidal neovascular membranes (Fig. 69.16)56.
Treatment of choroidal hemangioma
Introduction
Choroidal hemangiomas are benign, vascular tumors that occur posteriorly, usually involving the retina at the optic disc and/or near the macula. Circumscribed hemangiomas are solitary, whereas diffuse hemangiomas tend to be associated with the Sturge–Weber syndrome (Fig. 69.17). On ophthalmoscopy, circumscribed choroidal hemangiomas appear as round to oval, orange subretinal lesions. Diffuse choroidal hemangiomas tend to cause a widespread reddish thickening of the choroid with indistinct edges, usually involving more than half of the entire choroid. Ultrasonography is routinely performed to measure tumor dimensions and it typically displays a high internal acoustic reflectivity59. Additional ancillary studies may include fluorescein and indocyanine green angiography as well as OCT for the assessment of macular edema60,61. Patients with diffuse choroidal hemangioma and Sturge–Weber syndrome require a systemic and neurological workup62.
Although thought to be present from birth, choroidal hemangiomas may remain asymptomatic or cause symptoms only later in life, usually during the 2nd to 5th decades63. Visual symptoms are caused by exudative retinal detachment and macular edema, which may lead to secondary changes of the RPE, retinal atrophy, and permanent visual loss. The natural history varies considerably. Choroidal hemangiomas may enlarge clinically; this is thought to be caused by venous congestion, and not by multiplication of cells64.
Previously, circumscribed hemangiomas were treated by photocoagulation, but this has been superseded by radiotherapy, which has in turn been largely replaced by photodynamic therapy (PDT)63,65,66. Treatment of diffuse choroidal hemangiomas remains a challenge; most cases may be too extensive for PDT and are more likely to be treated with radiotherapy65.
Photodynamic therapy
Since the introduction of PDT for choroidal hemangiomas, treatment strategies have changed. Initially it was thought that more powerful treatment settings compared to standard parameters for AMD were necessary in order to induce regression in these highly vascularized tumors66. Changes included bolus injection vs. infusion of verteporfin over 10 minutes, increased laser energy, longer exposure time, and re-treatments. However, this may have resulted in over-treatment, as visual loss secondary to chorioretinal atrophy following PDT for choroidal hemangioma has been described and excellent outcomes have now been reported using standard PDT parameters60,67 (Fig. 69.18). There is also a current tendency to reduce the number of re-treatments with PDT; it seems that, in the vast majority of cases, regression can be achieved with only one treatment60.
Radiotherapy
External beam radiotherapy can be delivered using a lens-sparing technique and administering a dose of up to 20 Gy in ten divided fractions65. Proton beam radiotherapy comprising 18–27 Gy in four fractions is preferred by some because it minimizes radiation to healthy tissues65,68. Plaque radiotherapy may be considered for tumors not extending close to the optic disc or fovea, with an apical dose of 48 Gy being recommended69.
Postoperative care
After photodynamic therapy, patients are assessed after about 2 months to determine whether repeated treatment is necessary. This includes the monitoring of clinical signs of evolving atrophy within the hemangioma, measurements of tumor dimensions, and evaluation of associated exudative changes, including OCT for macular edema60. After any form of treatment, patients are reviewed after about 6 months and then intermittently thereafter, if at all.
Postoperative complications
After PDT, the tumor may not respond fully, making it necessary for the treatment to be repeated. However, it has to be kept in mind that a residual thickening of the choroid can sometimes be detected despite regression of the hemangioma. It also may take several months before a significant regression of tumor thickness or associated exudative retinal changes can be seen, and that over-treatment may result in unnecessary visual loss67. Such complications that have been associated with PDT for choroidal hemangioma are choroidal atrophy resulting in visual loss and polypoidal choroidal vasculopathy67,70.
Assessment of treatment
Improvement in vision is likely to occur if the patient presents with mild visual loss and a short history (i.e. less than 3 months). This can currently be achieved in about 70% of treated cases60. Eyes with subfoveal hemangiomas tend to be irreversibly amblyopic as a result of hypermetropia, suggesting that these tumors are congenital. Regression of macular tumors following PDT may also result in atrophy of the overlying RPE and visual loss67. In some advanced cases, particularly in diffuse choroidal hemangioma associated with Sturge–Weber syndrome, the disease cannot be controlled with current treatment methods, resulting in total retinal detachment and intractable neovascular glaucoma (Fig. 69.19).
Treatment of uveal metastasis
Heinrich Heimann and Sarah E. Coupland
Introduction
Uveal metastasis are the most common form of intraocular malignancy, with an estimated annual incidence of 20 000 patients in the USA71. They are usually overshadowed by the underlying disease; however, it is often the ophthalmologist to whom the patient presents.
Clinical features
Although metastasis may occur anywhere in the uvea, they are usually seen in the choroid. In women, the most common primary sites are the breast in 70–80% and the lung in 10% of patients72. In men, by far the commonest primary site is the lung73. Rare sources include the gastrointestinal tract (adenocarcinoma and neuroendocrine carcinoma), thyroid, prostate, kidney, cutaneous melanoma, and lymphoma/leukemia72,74. Metastasis from the lung are usually the initial manifestation of this disease, whereas secondaries from the breast tend to present after treatment of the primary tumor.
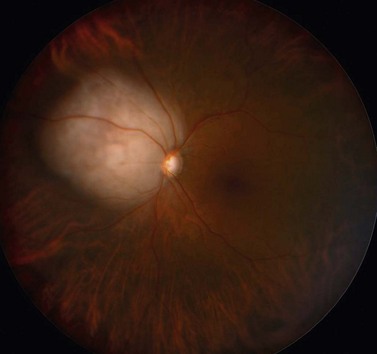
Most patients complain of blurred vision, floaters, and photopsia, as a result of the intraocular mass itself and the associated exudative retinal detachment. Uveal metastasis may, however be, asymptomatic in as many as 11% of all patients75. In many patients, choroidal metastasis are bilateral and multifocal, tending to involve the posterior pole (Fig. 69.20). Those occurring in the iris are rapidly growing uni- or multifocal, non-pigmented tumors with associated inflammation, hyphema, or pseudohypopyon76.
Preoperative assessment
Baseline measurements of tumor dimensions, severity of serous retinal detachment, and visual acuity are required to be able to assess the tumor response to therapy. Tumor biopsy may be indicated to confirm the diagnosis of a suspected metastasis, and, in patients with an unknown primary, to direct systemic investigation according to the differential diagnosis. Informative samples can be obtained using a 25-gauge needle or vitreous cutter via a trans-scleral or trans-retinal approach, albeit at a small risk of tumor seeding or hemorrhage (Fig. 69.21)77,78.
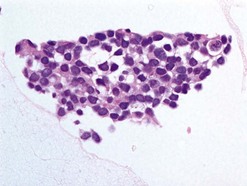
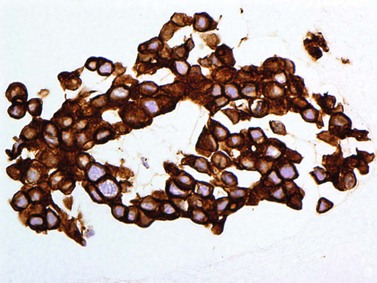
Pathology
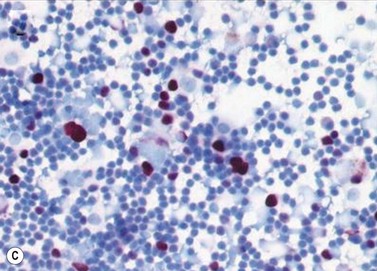
Pathological examination entails: (i) light microscopy of either cytological spin preparations or sections of cell blocks stained with conventional stains, and (ii) staining with immunohistochemical panels selected according to the cytomorphology and differential diagnosis (Fig. 69.22)79. In breast and lung cancers, the cytospins reveal medium to large sized atypical cells on a necrotic or hemorrhagic background. These will be highlighted in pancytokeratin stains, with specific markers being immunoreactive in particular malignancies (e.g. estrogen or progesterone receptors in some breast cancers; prostate-specific antigen (PSA) in prostate cancer, and chromogranin or synaptophysin in neuroendocrine carcinomas)80.
External beam radiotherapy
In focal disease, external beam radiotherapy is recommended: this usually consists of a total dose of 30 Gy, delivered in daily fractions of 3 Gy. A dose of 45–50 Gy in smaller fractions may be administered to patients with a good prognosis (e.g. breast carcinoma). Using this approach, visual acuity can be stabilized or improved in most patients81.
Brachytherapy
Plaque radiotherapy may be useful for solitary choroidal metastasis. This treatment has the advantage of being completed within a week. Brachytherapy may also be useful for persistent tumor after previous external beam radiotherapy82.
Other methods
Photodynamic therapy and transpupillary thermotherapy are reported to achieve local tumor regression in some patients, but these treatments are currently to be considered investigational. In patients with vitreous metastasis from cutaneous melanoma, vitrectomy may provide the required palliation74. Otherwise, the only remaining surgical option is enucleation because the eye is blind and painful as a result of secondary glaucoma.
Postoperative complications
Treatment of choroidal metastasis with radiotherapy and/or systemic chemotherapy is usually effective, preserving vision in most cases. The main complications are radiation retinopathy and cataract, which are more likely to occur in patients with prolonged survival81. The life expectancy of these patients, however, is poor with a median survival of 7 months.
Retinoblastoma
Carol L. Shields and Jerry A. Shields
Introduction
Retinoblastoma is the most common intraocular cancer of childhood83,84. It represents approximately 4% of all pediatric malignancies.
Terminology
Retinoblastoma is basically classified in three different ways: familial or sporadic, bilateral or unilateral, and heritable (germline) or non-heritable (somatic). From a clinical standpoint, the first two classification schemes are most often employed. Thus, a case is classified as unilateral sporadic, bilateral sporadic, unilateral familial, or bilateral familial. About two-thirds of all cases are unilateral and one-third are bilateral. From a genetic standpoint, it is simpler to discuss retinoblastoma with the latter classification of germline or somatic. Bilateral and familial retinoblastoma are caused by a germline mutation and are thus a heritable tumor85,86. Unilateral sporadic retinoblastoma is usually somatic and thus not heritable. However, it is estimated that approximately 15% of children with unilateral sporadic retinoblastoma have a germline mutation. Genetic testing using DNA analysis of the patient’s tumor and peripheral blood can help identify those patients with a germline mutation86,87. Patients with germline mutation retinoblastoma are at risk for related second cancers88,89.
Genetics
The retinoblastoma gene is located on the long arm of chromosome 13 (13q). The 13q deletion syndrome can manifest as several phenotypic abnormalities90. Many patients have minimal or no visible abnormality. The characteristic findings include dysmorphic features such as microcephaly, broad prominent nasal bridge, hypertelorism, microphthalmos, epicanthus, ptosis, protruding upper incisors, micrognathia, short neck with lateral folds, large prominent low set ears, facial asymmetry, imperforate anus, genital malformations, perineal fistula, hypoplastic or absent thumbs, toe abnormalities, and psychomotor and mental retardation.
Clinical features
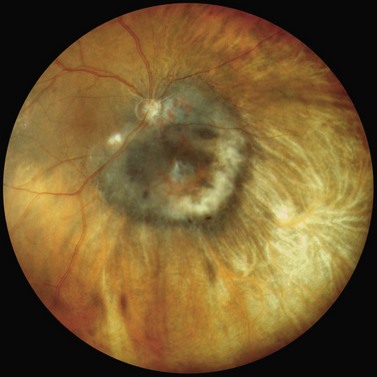
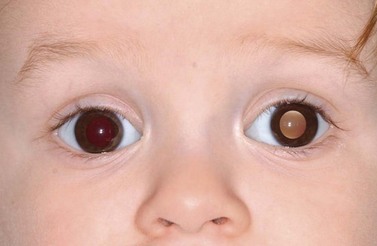
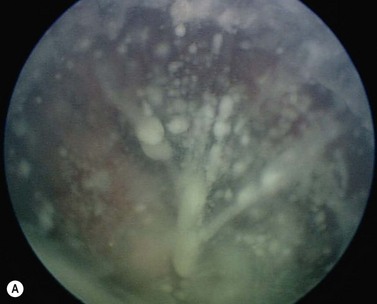
The clinical manifestations of retinoblastoma vary with the stage of the disease at the time of recognition. In its earliest clinical stage, a small retinoblastoma, under 2 mm in basal dimension, appears ophthalmoscopically as a subtle, transparent or slightly translucent lesion in the sensory retina83,84. Slightly larger tumors lead to dilated retinal blood vessels that feed and drain the tumor. Some larger tumors show foci of chalk-like calcification that resemble cottage cheese. A retinoblastoma of any size can produce leukocoria (Fig. 69.23). Larger tumors more often present with leukocoria. This white pupillary reflex is a result of reflection of light from the white mass in the retrolental area. Some normal eyes show leukocoria from the reflection of the normal optic disc through the pupil.
Retinoblastoma growth patterns are subdivided into intraretinal, endophytic, exophytic, and diffuse83,84,91 (Fig. 69.24). Intraretinal tumors are those listed above, limited to the substance of the retina. An endophytic retinoblastoma is one that grows from the retina inward toward the vitreous cavity. Hence, it is characterized by a white hazy mass with obscuration of the retinal blood vessels. Because of its friable nature, an endophytic tumor can seed the vitreous cavity and simulate endophthalmitis or toxocariasis. An exophytic retinoblastoma is one that grows from the retina outward into the subretinal space. Such tumors produce a progressive retinal detachment, with the retina often displaced anteriorly behind a clear lens. An exophytic retinoblastoma can clinically resemble Coats’ disease or other exudative retinal detachment. Occasionally a retinoblastoma can assume a diffuse infiltrating pattern, characterized by a relatively flat infiltration of the retina by tumor cells without an obvious mass91. In such cases, the diagnosis can be more difficult and this pattern can simulate uveitis or endophthalmitis. Less frequently, the presenting feature can be pseudohypopyon due to tumor seeding in the anterior chamber, hyphema secondary to iris neovascularization, vitreous hemorrhage, or signs of orbital cellulitis.
Classification
Several classifications of retinoblastoma have been developed to assist in prediction of globe salvage. The Reese Ellsworth classification was employed from the 1960s to current day. The International Classification of Retinoblastoma (ICRB) has been proposed to simplify the grouping scheme and allow a more practical approach to judging results of chemoreduction92,93 (Table 69.1).
Table 69.1 International Classification of Retinoblastoma (ICRB), long form
| Group | Quick reference | Specific features |
|---|---|---|
| A | Small tumor | Rb ≤3 mm in size* |
| B | Larger tumor Macula Juxtapapillary Subretinal fluid |
Rb >3 mm in size* or |
Rb: retinoblastoma.
* Refers to 3 mm in basal dimension or thickness
Data from Shields CL, Shields JA. Basic understanding of current classification and management of retinoblastoma. Curr Opin Ophthalmol 2006;17:228–34.
Differential diagnosis
Several ocular disorders in infants and children can clinically resemble retinoblastoma94. Despite the classic appearance of retinoblastoma, nearly 50% of patients diagnosed initially with possible retinoblastoma prove, on referral to ocular oncologists, to have simulating conditions and not retinoblastoma, including Coats’ disease, persistent hyperplastic primary vitreous, and ocular toxocariasis (Tables 69.2 and 69.3). Therefore, it is important that the diagnosis of retinoblastoma be established without question, prior to embarking on treatment. Consultation with ocular oncologists experienced with retinoblastoma may be helpful in confirming the clinical diagnosis of retinoblastoma and assisting in designing a management plan for this potentially deadly disease.
Table 69.2 Conditions that closely simulate retinoblastoma in an analysis of 212 pseudoretinoblastomas
| Condition | Percentage |
|---|---|
| Persistent hyperplastic primary vitreous | 28% |
| Coats’ disease | 16% |
| Ocular toxocariasis | 16% |
| Retinopathy of prematurity | 5% |
| Combined hamartoma | 4% |
| Coloboma | 4% |
| Vitreous hemorrhage | 4% |
| Astrocytic hamartoma | 3% |
| Familial exudative vitreoretinopathy | 2% |
| Bilateral retinal vascular hypoplasia with persistent primary vitreous | 2% |
| Rhegmatogenous retinal detachment | 2% |
| X-linked retinoschisis | 2% |
| Medulloepithelioma | 2% |
| Congenital cataract | 2% |
| Retinal capillary hemangioma | 1% |
| Circumscribed choroidal hemangioma | 1% |
| Diffuse choroidal hemangioma | 1% |
| Peripheral uveoretinitis | 1% |
| Toxoplasmic retinitis | 1% |
| Idiopathic endophthalmitis | 1% |
| Norrie’s disease | <1% |
| Incontinentia pigmenti | <1% |
| Morning glory disc anomaly | <1% |
Data from Shields JA, Parsons HM, Shields CL, Shah P. Lesions simulating retinoblastoma. J Pediatr Ophthalmol Strabismus 1991;28:338–40.
Table 69.3 Differentiation of Coats’ disease from retinoblastoma
| Feature | Coats’ disease | Retinoblastoma |
|---|---|---|
| Age of onset (mean) | 5 years | 1.5 years |
| Gender: | ||
| male | 76% | 50% |
| female | 24% | 50% |
| Laterality: | ||
| unilateral | 95% | 60% |
| bilateral | 5% | 40% |
| Family history of disease | 0% | 10% |
| Eye findings | ||
| Anterior chamber | Rare cholesterol crystals | Rare white cells with hypopyon |
| Iris neovascularization | Present in 8% | Present in 17% |
| Cataract | Absent | Absent |
| Vitreous | Clear | White fluffy seeds |
| Retinal vessels | Irregular dilation with telangiectasia Remain visible throughout course Most commonly seen inferotemporally, temporally, and superotemporally |
Tortuous, but regular dilation towards a mass Disappear into tumor Occur in quadrant of tumor |
| Retinal exudation | Present | Absent |
| Retinal mass | Absent | Present |
| Retinal gliosis | Present, often forming a subretinal mass | Absent |
| Retinoschisis | Present in 5% | Absent |
| Subretinal fluid | Present, with golden yellow with cholesterol | Present, with faint white free floating seeds |
| Diagnostic testing | ||
| Ultrasonography | Retinal detachment Subretinal echoes are minimal or none Rare calcification at level of retinal pigment epithelium |
Retinal detachment Subretinal echoes from seeds Calcification within retinal tumor in 90% cases |
| Computed tomography | Retinal detachment | Retinal detachment and calcified retinal mass |
| Magnetic resonance tomography | Retinal detachment | Retinal detachment with enhancement of retinal mass |
Local therapy of retinoblastoma
Alison H. Skalet and Joan O’Brien
Introduction
Therapeutic advances have improved the retinoblastoma survival rate from 5% to 99% in the developed world, with more than 90% of children retaining normal vision in at least one eye95. The therapeutic goals are: (i) tumor eradication, (ii) vision preservation, and (iii) prevention of second tumors in patients with germline RB1 mutations96.
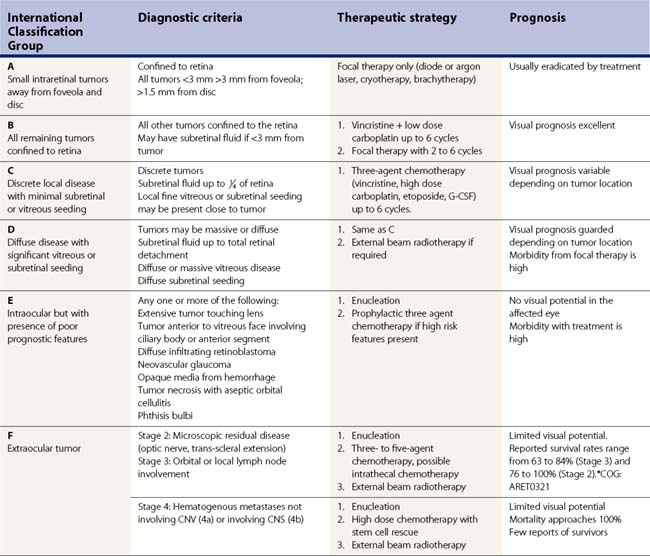
Treatment of individual patients is largely based upon disease staging using the international classification system for intraocular retinoblastoma (Table 69.4)97,98.
Treatment methods
Thermotherapy or laser ablation
Thermotherapy using a diode (810 nm) laser or ablation using an argon green (532 nm) laser is used as primary treatment for Group A eyes or as consolidation treatment for more advanced tumors99. The power is increased until tumor blanching is just visible. Anterior tumors may be treated using scleral depression or a trans-scleral applicator. With both forms of phototherapy, care is taken to avoid cataract and vitreous seeding from excessive energy. Areas of subretinal fluid are avoided to avoid retinal hole formation and retinal detachment.
Trans-scleral cryotherapy
Cryotherapy is used for small, anterior tumors, including tumors with localized vitreous seeds that can be included in the ice-ball100. The triple freeze–thaw technique is applied. A single treatment may be effective with tumors less than 1 mm thick. Cryotherapy is contraindicated with tumors that are diffuse, macular, or juxtapapillary. The main complications include serous retinal detachment, rhegmatogenous retinal detachment, and painful uveitis.
Brachytherapy
Brachytherapy is sometimes selected for equatorial or anterior Group B tumors that are not suitable for laser or cryotherapy alone101. It is useful for tumors up to 15 mm in diameter and 8 mm in thickness not extending close to the optic disc or fovea. The most commonly used radioactive sources are iodine-125 and ruthenium-106. An apical dose of 40 Gy is usually administered. Brachytherapy is contraindicated in eyes with disseminated vitreous seeding. Complications include radiation retinopathy and optic neuropathy, particularly in patients receiving chemotherapy concurrently or previously.
Systemic chemotherapy
The primary goal of systemic chemotherapy in retinoblastoma is to reduce tumor volume to allow focal therapy (chemoreduction) (Fig. 69.25)102. Systemic chemotherapy for retinoblastoma is also indicated when the risk of systemic disease is high. Most chemotherapeutic protocols use two- or three-drug regimens with carboplatin, etoposide, and vincristine; however, inter-institutional variability exists in the exact protocol chosen103,104. The major complications of chemotherapy using the CEV regimen are: (i) renal toxicity, ototoxicity, and myelosuppression with thrombocytopenia from carboplatin; (ii) secondary malignancy caused by etoposide; and (iii) peripheral neuropathy from vincristine.
Local chemotherapy
The objective of local chemotherapy is to achieve higher vitreous drug concentrations while limiting systemic side effects. Two methods of local chemotherapy are being investigated: subTenon’s carboplatin and intra-arterial carboplatin or melphalan96. SubTenon’s injections can cause significant morbidity, including strabismus and optic atrophy. Controlled-release drug formulations may improve the safety of this approach. Cannulation of the ophthalmic artery allows local delivery of chemotherapy at a fraction of the systemic dose, but can potentially cause stroke96. Local chemotherapy delivery modalities require further investigation.
External beam radiation
External beam radiotherapy (EBR) has largely been replaced by chemoreduction. Currently, EBR is administered when local therapies are not possible or have failed. Retinoblastoma is usually treated with 45–50 Gy, delivered in 1.5–2.0 Gy fractions, with the total dose depending on the size of the tumor105.
Enucleation
Enucleation is the primary therapy for Group E disease (see Table 69.4), particularly if the fellow eye is healthy. Removal of the wrong eye is avoided by intraoperative indirect ophthalmoscopy after patching the other eye. Care is taken to avoid scleral perforation, which severely worsens the prognosis. It is essential to obtain an optic nerve stump of at least 10 mm in case of optic nerve invasion. This is achieved by leaving the medial and lateral rectus stumps long, clamping these with mosquito clamps, and exerting gentle traction on the globe. We prefer small, curved enucleation scissors and pass these along the medial orbital wall, cutting the nerve at the orbital apex. The eye and optic nerve should be inspected for extraocular tumor.
Treatment strategies
Group D (’Diffuse, Large’)
Group D eyes have extensive intraocular dissemination that does not meet the criteria outlined below for Group E disease. On the current COG protocol (ARET0231), patients with group D disease receive six cycles of systemic vincristine, etoposide, and high dose carboplatin therapy; local consolidation treatment; and, if necessary, external beam radiotherapy. While prognosis for survival remains 90%, many of these eyes come to enucleation103.
Treatment of vasoproliferative tumor
Heinrich Heimann and Javier Elizalde
Introduction
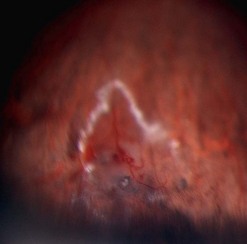
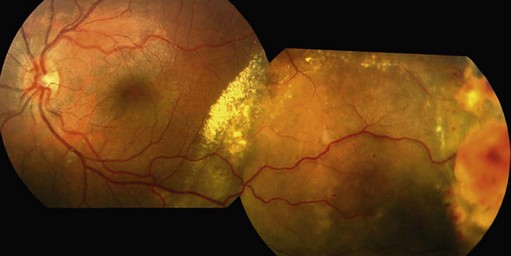
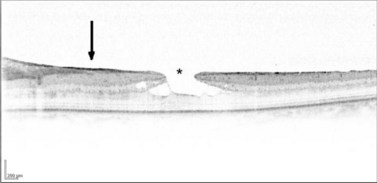
Vasoproliferative tumor is a benign retinal tumor characterized by peripheral location, exudation, and epiretinal membrane formation. It is usually primary but can be secondary to conditions such as uveitis, retinal detachment, Coats’ disease, and retinitis pigmentosa106,107. The typical clinical picture is of an orange or pink retinal mass in the peripheral fundus, associated with retinal vascular changes and exudation (Fig. 69.26). Vasoproliferative tumors can be solitary or multiple and are usually pre-equatorial, mostly in the infero-temporal region (Fig. 69.27). All age groups can be affected, but the disease is most commonly seen between 40 and 60 years of age108. Histologically, the tumors consist of proliferating retinal glial cells, which stain positively for GFAP (glial fibrillary acid protein), and retinal pigment epithelial cells, with a rich capillary network and neovascularizations109,110. Vasoproliferative tumors tend to cause hard exudates, serous retinal detachment, and epiretinal membranes, all of which can affect the macula (Figures 69.26–69.28). On slit-lamp examination, cells in the anterior chamber are almost always present. In addition, teleangiectatic retinal vessels and retinal pigment epithelial proliferations surround the tumor109. In contrast to retinal hemangioblastoma, the feeder vessels are only mildly dilated, if at all. In the advanced stages, the disease can be associated with: epiretinal neovascularizations on the disc and elsewhere, subretinal neovascularization, total retinal detachment, vitreous hemorrhage, and rubeosis iridis106,108–110. The early stages of the disease can be asymptomatic, and the tumor may be diagnosed on routine fundus examination; however, in most cases, patients present with visual loss, metamorphopsia photopsia caused by retinal exudation or floaters, photophobia, or red eye caused by intraocular inflammation. The natural course is thought to be slowly progressive. The disease may advance to total retinal detachment with intractable neovascular glaucoma.
Goals of treatment
The main goals of treatment are to prevent visual loss and preserve the eye. These are achieved by eradicating the tumor, eliminating exudation and macular edema, and, in many patients, treating epiretinal membranes111.
Indications for ocular treatment
Not all vasoproliferative tumors cause visual loss, so that observation may be appropriate in asymptomatic cases112. Treatment is indicated for symptomatic tumors and those threatening to cause visual loss109,111.
Preoperative assessment
The diagnosis is usually established by the typical clinical appearance on indirect ophthalmoscopy. The initial work-up includes echography, which usually demonstrates an inhomogeneous, high internal acoustic reflectivity, and a tumor thickness of 2–6 mm. Measurement of tumor dimensions, photographic documentation of exudates, and OCT measurement of macular edema are required in order to assess the efficacy of any treatment. Fluorescein angiography is rarely necessary; when performed, it usually demonstrates a connection of tumor vessels to mildly dilated retinal feeder vessels and marked epi- and subretinal leakage of the dye from the tumor vasculature109. Systemic investigation is indicated if retinal hemangioblastoma cannot be excluded with certainty.
Ocular treatment
Various forms of treatment are possible and there is no consensus as to which is the best method. Early disease is most commonly treated by laser photocoagulation or cryotherapy107,108. Our approach is to start with photodynamic therapy113, repeating this once or twice if the initial response is inadequate. If this approach is unsuccessful, we then attempt cryotherapy, using a retinal probe with the triple freeze–thaw technique. Persistent disease is treated with brachytherapy109,111,114. Intravitreal injections of anti-VEGF agents can be used to treat secondary macular edema; a positive effect of this treatment on the tumor itself has also been reported115. Vitrectomy with epiretinal membrane peeling may be required in some patients111. In the advanced stages, trans-scleral tumor resections have been performed110,116. Intractable, painful neovascular glaucoma associated with end-stage disease may require enucleation109–111.
Postoperative care
Follow-up is aimed at detecting and treating unresponsive tumors and managing any maculopathy. Epiretinal membranes may develop following initiation of treatment and usually become apparent once exudation has regressed. Some patients may develop bilateral disease or additional tumors in the initially affected eye109,117.
Postoperative complications
Apart from an inadequate response to treatment, the most common complication is maculopathy, caused by either exudation or epiretinal membrane formation. Increasing exudation has been described following cryotherapy of tumors with a thickness of ≥2 mm109. Epitretinal membrane formation is more common following treatment of advanced stages of the disease and after plaque radiotherapy111. In a minority of patients, the disease cannot be controlled with current treatment modalities. This is usually associated with advanced stages of the disease at the initiation of treatment, in particular neovascular glaucoma110,111.
Treatment of retinal capillary angioma
Arun D. Singh and Andrew P. Schachat
Introduction
Retinal capillary angioma (syn. retinal hemangiomblastoma) can occur in isolation or as part of von Hippel–Lindau (VHL) disease118. This syndrome is caused by a mutation on chromosome 3120, which is inherited in an autosomal dominant fashion with incomplete penetrance118,119. Retinal capillary angioma can be classified on the basis of the location within the retina (peripheral and juxtapapillary), morphology (endophytic, exophytic, and sessile), effects on the retina (exudative form and tractional form), and the relationship to VHL disease (with or without VHL disease)118.
Fundamental principles
Treatment of retinal angiomas can be challenging because of the presence of bilateral multiple tumors. The treatment is determined mostly by the size of the tumor, location, activity, and the secondary effects on the retina and vitreous120. As juxtapapillary retinal capillary angioma tends to remain stable, initial observation in an asymptomatic case without evidence of exudation should always be considered121,122.
Preoperative assessment
The fundus findings of retinal capillary angioma are characteristic and diagnosis of retinal capillary angioma can usually be made based solely on ophthalmoscopic examination. The retinal capillary angioma is usually a circumscribed, round retinal lesion with an orange-red color but it can vary in appearance depending upon whether the retinal capillary hemangioma is endophytic, exophytic, or sessile118. Secondary effects such as intraretinal and subretinal exudation are often observed surrounding the angioma, which may extend into the macula producing star exudates.
Of all the ancillary tests, fluorescein angiography is the most informative diagnostic tool because of the vascular nature of the tumor (Fig. 69.29). The angioma fills rapidly with the dye showing early circumscribed hyperfluorescence and delayed leakage. Prominent feeder arteriole and draining vein are also evident. Fluorescein angiography also helps in differentiating the feeder arteriole from the draining vein and is therefore important for treatment planning. Juxtapapillary retinal capillary angioma has similar angiographic features but lacks feeder and draining vessels123. Optical coherent tomography of the macula helps in differentiating tractional maculopathy from exudative maculopathy in association with juxtapapillary tumors.
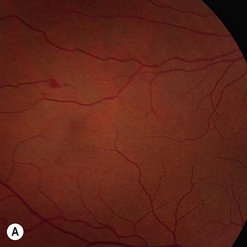
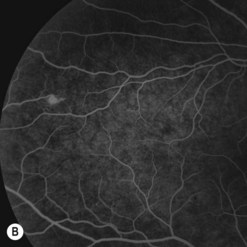
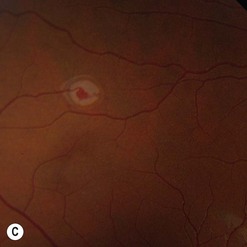
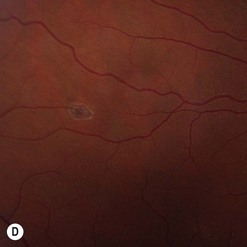
Patients with a solitary tumor and a negative family history should usually undergo systemic evaluation or genetic testing to exclude VHL124. Those with definite or possible VHL should have a systemic evaluation for central nervous system hemangioblastomas, renal carcinomas, pheochromocytoma, deafness, and other abnormalities125.
Ocular treatment
Small posterior retinal angiomas are treated by laser photocoagulation (see Fig. 69.29C)126. Larger tumors up to 2 mm in thickness usually respond to cryotherapy, which may need to be repeated120,127. Large peripheral tumors may respond to brachytherapy128. More recently, photodynamic therapy has been reported to induce occlusion of juxtapapillary and peripheral tumors122,129. Systemic or intravitreal treatments with anti-VEGF agents do not shrink the tumor, although there may be beneficial effects of resolution of the SRF in some cases130. Tumor removal with a vitreous cutter is possible but may be followed by severe proliferative vitreoretinopathy. Vitreoretinal surgery is therefore limited to releasing any retinal traction once regression of the tumor has been achieved.
Postoperative complications
The main complications are incomplete tumor response to treatment and aggravation of any cicatricial effects. The cumulative lifetime risk of significant permanent vision loss is estimated at 60%, and about 20% of patients have vision worse than 20/100 in at least one eye131.
Treatment of ocular lymphoma
Sarah E. Coupland and Bertil Damato
Introduction
The current WHO classification of lymphomas categorizes the various subtypes according to clinical, histomorphological, immunophenotypical and genotypical features132. Intraocular lymphomas can be classified according to whether they are retinal or uveal, and primary or secondary133. Ocular adnexal lymphomas can be clinically staged according to their location and extent, using the tumor, node, metastasis (TNM/AJCC) system134.
Clinical features
Retinal lymphomas
Most retinal lymphomas present after the age of 60 years, usually as chronic uveitis unresponsive to steroid therapy133,135,136. Creamy-white subretinal tumor deposits are usually present (Fig. 69.30A). The large majority are high-grade B-cell lymphomas, which can be subtyped as diffuse large B-cell lymphomas (DLBCL) (Fig. 69.30B). Before or after the appearance of ocular disease, about 70–80% of patients show central nervous system involvement, usually with a fatal outcome.
Uveal lymphomas
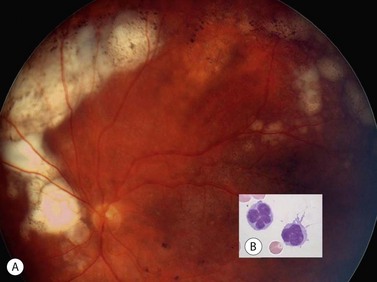
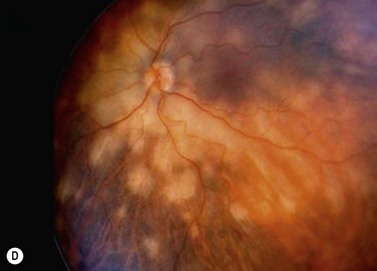
Primary choroidal lymphomas are relatively rare, presenting after the age of 50 years with unilateral loss of vision. Ophthalmoscopy shows a diffuse, pink, choroidal tumor (Fig. 69.31A), which on echography has a low internal acoustic reflectivity. Extraocular extension can occur near the optic nerve and/or sub-conjunctivally near the limbus (Fig. 69.31B). These are mostly low grade, extranodal marginal zone lymphomas (EMZL) with a good survival prognosis (Fig. 69.31C)133. Secondary choroidal lymphomas are usually of the non-Hodgkin’s type, representing an ocular involvement of a systemic lymphoma. They present as multiple, unilateral or bilateral, creamy-white lesions (Fig. 69.31D). The behavior of the ocular deposits usually reflects the malignancy of the underlying systemic disease.
Iridal lymphomas are exceedingly rare, presenting with unilateral pain, redness, and pseudohypopyon. They can be of T- or B-cell type, and tend to ultimately disseminate systemically, resulting in a poor prognosis for survival. Only one case of ciliary body lymphoma has been described and this was of low grade, B-cell type133.
Ocular adnexal lymphomas
These tumors originate in conjunctiva, eyelids, lacrimal gland, and drainage system as well as other orbital tissues (Fig. 69.32A). Two-thirds of these lymphomas are low grade B-cell lymphomas, and can be subtyped as extranodal marginal zone B-cell lymphomas (EMZL) of the MALT type (i.e. mucosa-associated lymphoid tissue) (Fig. 69.32B)137,138 and these occur as localized disease. Although various micro-organisms and chromosomal abnormalities have been linked to the ocular adnexal EMZL, their pathogenesis remains uncertain. High grade B-cell lymphomas, such as DLBCL and mantle cell lymphoma, can occur as primary or secondary tumors in the ocular adnexa, and tend to show more aggressive clinical courses.
Ophthalmic assessment
All patients require full ophthalmic examination. If any form of intraocular lymphoma is suspected, then echography or other ocular imaging is required. In the case of retinal lymphoma, vitreous biopsy should also be performed, ideally also sampling a representative subretinal deposit. Steroid therapy should be discontinued several days before biopsy to increase cellular yield and to avoid false negative results139. For uveal lymphoma, biopsy of the affected choroid, ciliary body, or iris should be performed.
Laboratory techniques
The surgeon should liaise closely with the pathologist, to ensure that the specimen is transported to the laboratory in the correct medium and container and without delay139. A fixative such as buffered formalin is required for tissue specimens, if they are unlikely to reach the laboratory in less than one hour. In contrast, cytological specimens (i.e. vitreous or aqueous taps) should be sent either fresh or in soft fixatives such as Cytolyte or HOPE fixation139. Cytomorphological analysis is performed to determine the grade of malignancy of ocular lymphomas, and to exclude any microorganisms. Immunohistochemistry and/or flow cytometry differentiate reactive from neoplastic lymphocytes and identify the subtype of lymphoma139. The polymerase chain reaction (PCR) directed against the immunoglobulin heavy chain gene or the TCR-receptor determines whether the lymphocytes are monoclonal or polyclonal139. Levels of interleukins 6 and 10 in vitreous help determine whether infiltrates are likely to be inflammatory or neoplastic140.
Treatment
The first line of therapy for most ocular adnexal lymphomas is low dose radiotherapy with or without systemic chemotherapy, depending on the lymphoma stage141. The role of radiotherapy for retinal lymphoma is controversial, as a result of associated morbidities (see below). The current key chemotherapeutic agent in retinal and cerebral lymphoma is methotrexate, administered either intraocularly or intrathecally; possible new regimens could also include high dose cytarabine as this appears to be particularly effective142. Isolated (recurrent) retinal lymphoma may be treated with either intravitreal methotrexate or rituximab143.
Follow-up
Patients with retinal lymphoma are at high risk of developing CNS disease within a few years whether or not there is intraocular tumor recurrence. Ocular treatment is therefore symptomatic, whereas systemic management is usually undertaken by an oncologist with expertise in lymphomas. Although ocular adnexal lymphomas are of low grade type, they undergo high grade transformation in about 10% of cases. Recurrence of ocular adnexal lymphomas in other extranodal sites occurs in one-third of patients, and systemic dissemination in up to 25%139. Long-term follow-up is therefore, required.
Treatment of conjunctival melanoma
Bertil Damato and Sarah E. Coupland
Introduction
Conjunctival melanoma is rare144. Treatment depends on the tumor’s size and location and the presence or absence of diffuse intraepithelial disease. It is believed that local recurrence after initial treatment increases the likelihood of metastatic death.
Invasive melanoma
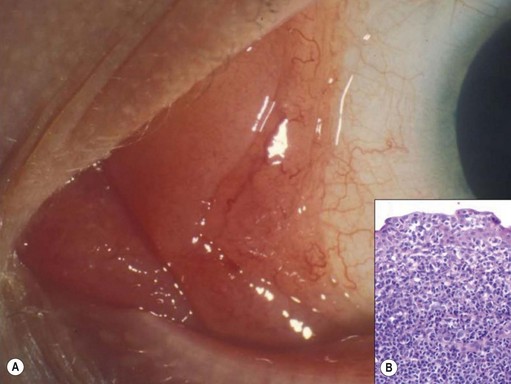
Most invasive conjunctival melanomas arise in the bulbar conjunctiva, particularly at the temporal limbus145. Many are associated with ‘primary acquired melanosis’ (PAM) (see below)145,146.
Metastasis can occur to regional lymph nodes or systemically. Risk factors include: involvement of non-bulbar conjunctiva, particularly caruncle; aggressive tumor histology; and local tumor recurrence147. Ten-year mortality rates of approximately 20–30% are reported144,147.
Primary acquired melanosis/conjunctival melanocytic intraepithelial neoplasia
In our opinion, the term ‘PAM’ is best reserved for clinical use, referring to acquired pigmentation unrelated to any causative ocular or systemic disease (e.g. Addison’s disease)148.
We feel that the histological correlates of PAM should be described more precisely, distinguishing ‘conjunctival hypermelanosis’ (i.e. melanin overproduction) from ‘conjunctival melanocytic intraepithelial neoplasia’ (C-MIN), which can occur without or with cellular atypia148.
In ‘C-MIN’ without cellular atypia, there is an increased number of normal melanocytes along the basal epithelial layer148.
‘C-MIN’ with cellular atypia varies in its grade of malignancy, according to (i) the degree of morphological atypia (determined by cytoplasmic, nuclear, and nucleolar features and the presence/absence of mitoses), (ii) the growth pattern e.g. (pagetoid, isolated nests, confluent nests), and the extent of epithelial involvement (i.e. <50%, 50–90%, or >90% of epithelial thickness)148. We have produced a scoring system to increase the reproducibility between pathologists; preliminary validation studies have demonstrated good reproducibility between observers.
Traditionally, conjunctival melanoma in situ was diagnosed only when full-thickness replacement of the epithelium by atypical melanocytes was present. Recently, the definition of conjunctival melanoma in situ has been modified to include less advanced disease (‘PAM with atypia replacing more than 75% of the conjunctival epithelium, with features of epithelioid changes and/or the presence of intraepithelial cell nests’), and this has been entered as a new stage (pTis) in the 7th edition of the TNM/AJCC staging system150. Applying our C-MIN scoring system, we are confident that a particular score will be identified as representing conjunctival melanoma in situ, and that this will be useful when treating patients and designing clinical trials.
Preoperative assessment
It is essential to examine the entire conjunctiva and palpate the regional lymph nodes. Ocular assessment involves determining the extent of any tumors and pigmented areas in each conjunctival region (i.e. limbus, bulbar conjunctiva, fornix, etc.) recording circumferential spread in clock hours or minutes in each involved region145. Tumor dimensions are also recorded in millimeters. If possible, the tumor thickness is measured with high frequency echography.
The scope of confocal microscopy is still undefined. Some authors recommend sentinel node biopsy for high risk cases149.
Staging can be performed according to the latest edition of the tumor, node, metastasis (TNM/AJCC) clinical staging system150.
Therapeutic techniques
Treatment of invasive melanoma
With a bulbar tumor, a line of diathermy burns is placed around the tumor, about 1–2 mm from the tumor margins, along the intended line of conjunctival incision (Fig. 69.33). The corneal epithelium adjacent to any tumor is devitalized with alcohol and scraped towards the main tumor with a Bard–Parker scalpel. The conjunctiva and the underlying episclera are divided with scissors along the line of the diathermy burns and separated from the sclera with the scalpel. If possible, Bowman’s layer is preserved, to prevent deep corneal invasion. Any deep tumor extensions in sclera are diathermied. The tumor is excised en bloc using a no-touch technique. Hemostasis is strictly maintained by meticulous diathermy. The specimen is placed onto paper and into fixative. Any suspicious areas of residual intra-scleral tumor are treated with diathermy or cryotherapy. Some would perform a lamellar scleral excision151. Using fresh instruments, the conjunctiva is undermined and closed with interrupted sutures. Extensive wounds are allowed to heal by second intention or closed with an amniotic membrane graft.
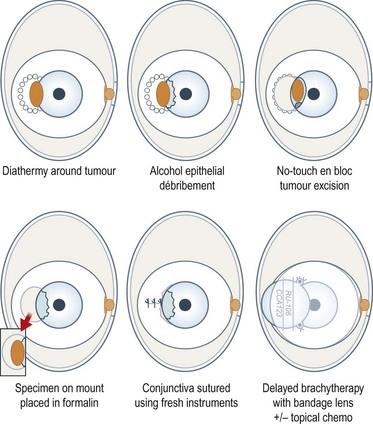
With invasive melanoma, adjunctive brachytherapy can be administered, delivering 100 Gy to a depth of 1 mm using a strontium applicator, ruthenium plaque, or another device, such as an unshielded iodine plaque. If such brachytherapy is not possible, for example with caruncular tumors, then external beam radiotherapy or proton beam radiotherapy can be used (Fig. 69.34)152. We delay adjunctive radiotherapy until the conjunctiva has healed, usually about 2 weeks after the surgical excision. There is no consensus over whether adjunctive radiotherapy should be given to all patients with invasive melanoma or only if histology suggests incomplete surgical clearance.
Cryotherapy is widely used as adjunctive therapy151. Our impression is that it is less reliable than radiotherapy and more likely to cause complications, at least with bulbar tumors.
Complications
Several published series report high rates of local tumor recurrence147,153,154. Presumably, such recurrences have occurred despite measures such as adjunctive cryotherapy and histological assessment of surgical clearance. We therefore administer adjunctive radiotherapy after all excisions for invasive melanoma, rarely if ever seeing any recurrent disease within the irradiated field155. Residual and recurrent melanomas threaten invasion of the eye and orbit as well as seeding and metastasis. There is some evidence that such seeding can be iatrogenic, caused by incisional biopsy, incomplete tumor excision, or carriage of cells by surgical instruments.
Epibulbar cryotherapy can cause uveitis, macular edema, and hypotony155. Adjunctive radiotherapy may delay conjunctival healing and can cause necrosis of any exposed sclera. Excision near extraocular muscles can cause ocular motility problems. Extensive excision of the fornices can result in symblepharon and eyelid malposition155. Topical chemotherapy can cause punctal stenosis and limbal stem cell failure.
Ocular surface squamous neoplasia
Introduction
Ocular surface squamous neoplasia (OSSN) is a spectrum of malignancies of the conjunctiva, limbus, and cornea that includes epithelial dysplasia, carcinoma in situ, and invasive squamous cell carcinoma (SCC).156,157
The most important causes are ultraviolet radiation158, human papilloma virus (HPV), and acquired immunodeficiency syndrome (AIDS)159.
Clinical features and diagnosis
OSSN lesions are usually interpalpebral and mostly at the limbus, although they may be found in or extend to any part of the conjunctiva and cornea. Corneal involvement is usually secondary. OSSN may appear gelatinous, papilliform, or leukoplakic. It may be nodular, especially when invasive, or diffuse, mimicking chronic conjunctivitis156,157. OSSN may be pigmented. Visual acuity is not affected unless the tumor involves central cornea. In most cases the history is of several months.
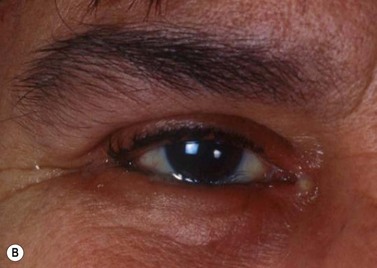
Clinically, it may be difficult to distinguish between conjunctival epithelial dysplasia, carcinoma in situ (Fig. 69.35), invasive SCC (Fig. 69.36), pinguecula, pterygium, and squamous papilloma. The definitive diagnosis is histological.
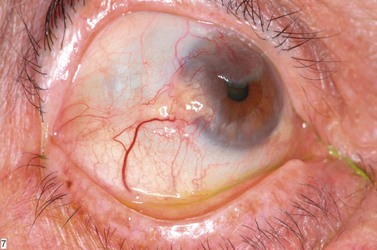
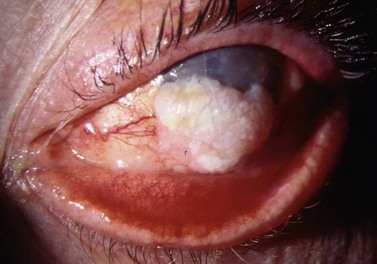
Preoperative diagnostic cytology is useful when planning surgery, helping to avoid excessive excision in benign lesions and prevent incomplete excision of malignant tumors. Exfoliative cytology and impression cytology are popular but only sample superficial conjunctiva, so that invasion cannot be assessed160.
Only histopathologic evaluation of the excised lesion allows differentiation between the three lesions within the spectrum of OSSN156,157. It is important to distinguish non-invasive dysplastic epithelial lesions and carcinoma in situ from invasive SCC, which breaches the basement membrane and invades the subepithelial tissues. Most conjunctival SCC are well differentiated. However, there are three aggressive types of invasive conjunctival SCC: spindle cell squamous carcinoma, mucoepidermoid carcinoma, and adenoid squamous cell carcinoma.
Surgical techniques
Surgical success depends on adequate clearance of the lateral and deep margins. This requires safety margins of 2–3 mm, with some authors even advocating 4–5 mm161. It is important not to touch the affected conjunctiva and to use fresh instruments for closure, to prevent iatrogenic seeding of tumor cells to healthy tissue.
Superficial lesions can be excised with scissors; however, when the sclera is involved, it is necessary to perform a lamellar sclerectomy using a Beaver blade. Frozen sections and modified Mohs’ micrographic techniques have been used by some surgeons, but are not routine. For conjunctival tumors extending posteriorly, the appropriate rectus muscle can be hooked and isolated, to provide traction for better exposure and to avoid inadvertent muscle injury. With limited excisions, the surgical wound can be left without sutures to re-epithelialize. Large defects require suturing using 6/0 or 7/0 absorbable sutures, after undermining the surrounding conjunctiva161.
Corneal involvement is usually limited to the epithelium so that corneal epitheliectomy can be performed without disrupting the Bowman’s layer, which serves as a natural barrier to invasion. Absolute alcohol is applied with a cotton-tipped applicator, to facilitate removal of the epithelium in one piece and to denaturate the neoplastic cells. When the OSSN lesion invades the deeper layers of the cornea, lamellar keratectomy is performed157,161.
Reconstruction of the ocular surface may be needed after large excisions, and the use of autologous conjunctival transplantation and autologous limbal transplants to restore corneal and limbal areas has been described. Others have used amniotic membrane transplantation for reconstruction after excision of large ocular surface neoplasia162.
Adjuvant therapy
After excision alone, recurrence of OSSN is common, with rates averaging 30%. The risk of recurrence is about 5% when the surgical margins are free, increasing to 53–69% when the surgical margins are involved156,157. Treatment with multiple modalities has therefore been recommended.
Fraunfelder and others advocated the use of combined excision and cryotherapy using nitrous oxide and liquid nitrogen163. The cryoprobe can be applied to the surgical margins and to the surgical bed to destroy any tumor cells and obliterate microcirculations. Such adjuvant cryotherapy decreases the recurrence rate to about 0–12.3%. Possible complications include iritis, increased or deceased intraocular pressure, inflammation, corneal edema and scarring, sector iris atrophy, ablation of the peripheral retina, ectropion, and superficial corneal vascularization.
Brachytherapy has been used for many years in treating OSSN, either as monotherapy or in combination with surgical excision. The most commonly used irradiation sources are beta emitters such as strontium-90 and ruthenium-106164. Gamma irradiation has also been administered. Possible complications include post-irradiation inflammation, dry eye, cataract, conjunctival telangiectasis, scarring, symblepharon, scleral ulceration, and corneal perforation.
Since the 1990s topical chemotherapy using mitomycin C, 5-fluorouracil (5FU), and interferon alpha-2b have been used for treating intraepithelial conjunctival and corneal lesions165. These agents have also been administered as adjuvant therapy, either after incomplete excision of conjunctival SCC166 or preoperatively, to decrease the extent of the surgery, eliminating intraepithelial disease. The main adverse reactions to the topical chemotherapy include conjunctival hyperemia, pain, and a burning sensation, all of which are transient.
Treatment of conjunctival–corneal intraepithelial neoplasia
Conjunctival and corneal epithelial dysplasia and carcinoma in situ can be excised when the lesion is small. However, in recent years, good results have been reported with topical chemotherapy using mitomycin C 0.02% or 5FU 1% drops, or intralesional or topical recombinant interferon alpha-2b165. Cryotherapy and brachytherapy are used for extensive or recurrent disease163,164.
1 Shields JA, Shields CL. Uveal melanoma. In: Shields JA, Shields CL, editors. Intraocular Tumors. An Atlas and Textbook. 2nd edn. Philadelphia, Lippincott: Williams & Wilkins; 2008:69-117.
2 Damato B. Uveal melanoma, Butterworth Heinemann, OxfordButterworth-Heinemann. Damato B, editor. Ocular Tumors, 2000:57-93.
3 Shields CL, Furuta M, Thangappan A, et al. Metastasis of uveal melanoma millimeter-by-millimeter in 8033 consecutive eyes. Arch Ophthalmol. 2009;127:989-998.
4 Hawkins BS, Moy CS, Reynolds SM, et al. The COMS randomized trial of iodine-125 brachytherapy for choroidal melanoma, III: initial mortality findings. COMS Report No. 18. Arch Ophthalmol. 2001;119:969-982.
5 Prescher G, Bornfeld N, Hirche H, et al. Prognostic implications of monosomy 3 in uveal melanoma. Lancet. 1996;347:1222-1225.
6 Shields CL, Ganguly A, Materin MA, et al. Chromosome 3 analysis of uveal melanoma using fine needle aspiration biopsy at the time of plaque radiotherapy in 140 consecutive cases. The Deborah Iverson MD Lectureship. Arch Ophthalmol. 2007;125:1017-1024.
7 The COMS randomized trial of iodine-125 brachytherapy for choroidal melanoma: V. Twelve-year mortality rates and prognostic factors: COMS report No. 28. Arch Ophthalmol. 2006;124(12):1684-1693.
8 Damato B, Patel I, Campbell IR, et al. Visual acuity after ruthenium [106] brachytherapy of choroidal melanomas. Int J Radiat Oncol Bio Phys. 2005;63(2):392-400.
9 Damato B, Patel I, Campbell IR, et al. Local tumor control after 106Ru brachytherapy of choroidal melanoma. Int J Radiat Oncol Bio Phys. 2005;63(2):385-391.
10 Augsburger JJ, Correa ZM, Shaikh AH. Effectiveness of treatments for metastatic uveal melanoma. Am J Ophthalmol. 2009;148(1):119-127.
11 Sagoo MS, Shields CL, Mashayekhi A, et al. Plaque radiotherapy for choroidal melanoma encircling the optic disc (circumpapillary choroidal melanoma). Arch Ophthalmol. 2007;125(9):1202-1209.
12 Harbour JW, Murray TG, Byrne SF, et al. Intraoperative echographic localization of iodine 125 episcleral radioactive for posterior uveal melanoma. Retina. 1996;16(2):129-134.
13 Wen JC, McCannel TA. Treatment of radiation retinopathy following plaque brachytherapy for choroidal melanoma. Curr Opin Ophthalmol. 2009;20(3):200-204.
14 Wen JC, Oliver SC, McCannel TA. Ocular complications following I-125 brachytherapy for choroidal melanoma. Eye (Lond). 2009;23(6):1254-1268.
15 Augsburger JJ, Gamel JW, Shields JA, et al. Post-irradiation regression of choroidal melanomas as a risk factor for death from metastatic disease. Ophthalmology. 1987;94(9):1173-1177.
16 Gragoudas ES, Goitein M, Verhey L, et al. Proton beam irradiation: an alternative to enucleation for intraocular melanomas. Ophthalmology. 1980;87:571-581.
17 Egger E, Schalenbourg A, Zografos L, et al. Maximizing local tumor control and survival after proton beam radiotherapy of uveal melanoma. Int J Radiat Oncol Biol Phys. 2001;51:138-147.
18 Egger E, Zografos L, Schalenbourg A, et al. Eye retention after proton beam radiotherapy for uveal melanoma. Int J Radiat Oncol Biol Phys. 2003;55:867-880.
19 Damato B, Kacperek A, Chopra M, et al. Proton beam radiotherapy of choroidal melanoma: the Liverpool–Clatterbridge experience. Int J Radiat Oncol Biol Phys. 2005;62:1405-1411.
20 Dendale R, Lumbroso-Le Rouic L, Noel G, et al. Proton beam radiotherapy for uveal melanoma: results of Curie Institut–Orsy Proton therapy center (ICPO). Int J Radiat Oncol Biol Phys. 2006;65:780-787.
21 Hoecht S, Bechrakis NE, Nausner M, et al. Proton therapy of uveal melanomas in Berlin. 5 years experience at the Hahn–Meitner Institute. Strahlenther Onkol. 2004;180:419-424.
22 Collaborative Ocular Melanoma Study Group. The COMS randomized trial of iodine 125 brachytherapy for choroidal melanoma:V. Twelve year mortality rates and prognostic factors: COMS report No.28. Arch Ophthalmol. 2006;124:1684-1693.
23 Damato B, Kacperek A, Chopra M, et al. Proton beam radiotherapy of iris melanoma. Int J Radiat Oncol Biol Phys. 2005;63:109-115.
24 Zografos L. Examen clinique et modalités de diagnostic des mélanomes de l’uvée. Transillumination (diaphanoscopie. In: Zografos L, editor. Tumeurs Intraoculaires. Paris: Société Française d’Ophtalmologie et Masson; 2002:165-166.
25 Zografos L, Desjardins L, Uffer S, et al. Traitement conservateur des mélanomes. Traitement des mélanomes de la choroïde et du corps ciliaire par irradiation. In: Zografos L, editor. Tumeurs Intraoculaires. Paris: Société Française d’Ophtalmologie et Masson; 2002:248-272.
26 Gragoudas ES, Lane AM, Munzenrider J, et al. Long-term risk of local failure after proton therapy for choroidal/ciliary body melanoma. Trans Am Ophthalmol Soc. 2002;100:43-48.
27 Char DH, Kroll S, Phillips TL, et al. Late radiation failures after iodine 125 brachytherapy for uveal melanoma compared with charged-particle (proton or helium ion) therapy. Ophthalmology. 2002;109:1850-1854.
28 Zehetmayer M, Kitz K, Menapace R, et al. Local tumor control and morbidity after one to three fractions of stereotactic external beam irradiation for uveal melanoma. Radiother Oncol. 2000;55(2):135-144.
29 Mueller AJ, Talies S, Schaller UC, et al. Stereotactic radiosurgery of large uveal melanomas with the gamma-knife. Ophthalmology. 2000;107(7):1381-1387.
30 Langmann G, Pendl G, Müllner K, et al. Gamma knife radiosurgery for uveal melanomas: an 8-year experience. J Neurosurg. 2000;93(Suppl. 3):184-188.
31 Dieckmann K, Georg D, Zehetmayer M, et al. LINAC based stereotactic radiotherapy of uveal melanoma: 4 years clinical experience. Radiother Oncol. 2003;67(2):199-206.
32 Emara K, Weisbrod DJ, Sahgal A, et al. Stereotactic radiotherapy in the treatment of juxtapapillary choroidal melanoma: preliminary results. Int J Radiat Oncol Biol Phys. 2004;59(1):94-100.
33 Muller K, Novak PJCM, de Pan C, et al. Effectiveness of fractionated stereotactic radiotherapy for uveal melanoma. Int J Rad Oncol Biol Phys. 2005;63(1):116-122.
34 Modorati G, Miserocchi E, Galli L, et al. Gamma knife radiosurgery for uveal melanoma. Br J Ophthamol. 2009;93(1):40-44.
35 Somani S, Sahgal A, Krema H, et al. Stereotactic radiotherapy in the treatment of juxtapapillary choroidal melanoma: 2-year follow-up. Can J Ophthalmol. 2009;44(1):61-65.
36 Muacevic A, Nentwich M, Wowra B, et al. Development of a streamlined, non-invasive robotic radiosurgery method for treatment of uveal melanoma. Technol Cancer Res Treat. 2008;7(5):369-374.
37 Zorlu F, Selek U, Kiratli H. Initial results of fractionated CyberKnife radiosurgery for uveal melanoma. J Neurooncol. 2009;94(1):111-117.
38 Haas A, Pinter O, Papaefthymiou G, et al. Incidence of radiation retinopathy after high-dosage single-fraction gamma knife radiosurgery for choroidal melanoma. Ophthalmology. 2002;109(5):909-913.
39 Marchini G, Gerosa M, Piovan E, et al. Gamma Knife stereotactic radiosurgery for uveal melanoma: clinical results after 2 years. Stereotact Funct Neurosurg. 1996;66(Suppl. 1):208-213.
40 Damato B, Groenewald C, Singh AD, Damato BE, Pe’er Jet al, editors. Clinical Ophthalmic Oncology. Philadelphia: Saunders; 2007:259-266.
41 Kivela T, Puusaari I, Damato B. Transscleral resection versus iodine brachytherapy for choroidal malignant melanomas 6 millimeters or more in thickness: a matched case–control study. Ophthalmology. 2003;110:2235-2244.
42 Damato BE, Foulds WS, Schachat AP, Ryan SJ, editors. Retina, 4th edn. St Louis: Mosby; 2006:769-778.
43 Damato B, Groenewald C, McGalliard J, et al. Endoresection of choroidal melanoma. Br J Ophthalmol. 1998;82:213-218.
44 Damato BE, Paul J, Foulds WS. Predictive factors of visual outcome after local resection of choroidal melanoma. Br J Ophthalmol. 1993;77:616-623.
45 Damato BE, Paul J, Foulds WS. Risk factors for residual and recurrent uveal melanoma after trans-scleral local resection. Br J Ophthalmol. 1996;80:102-108.
46 Damato B, Wong D, Green FD, et al. Intrascleral recurrence of uveal melanoma after transretinal ‘endoresection’. Br J Ophthalmol. 2001;85:114-115.
47 Hadden PW, Hiscott PS, Damato BE. Histopathology of eyes enucleated after endoresection of choroidal melanoma. Ophthalmology. 2004;111:154-160.
48 Damato B, Groenewald CP, McGalliard JN, et al. Rhegmatogenous retinal detachment after transscleral local resection of choroidal melanoma. Ophthalmology. 2002;109:2137-2143.
49 Damato BE, Paul J, Foulds WS. Risk factors for metastatic uveal melanoma after trans-scleral local resection. Br J Ophthalmol. 1996;80:109-116.
50 Damato B, Coupland SE. Translating uveal melanoma cytogenetics into clinical care. Arch Ophthalmol. 2009;127:423-429.
51 Damato B. Choroidal melanoma endoresection, dandelions and allegory-based medicine. Br J Ophthalmol. 2008;92:1013-1014.
52 Shields CL, Shields JA, Perez N, et al. Primary transpupillary thermotherapy for small choroidal melanoma in 256 consecutive cases: outcomes and limitations. Ophthalmology. 2002;109:225-234.
53 Shields CL, Cater J, Shields JA, et al. Combined plaque radiotherapy and transpupillary thermotherapy for choroidal melanoma: tumor control and treatment complications in 270 consecutive patients. Arch Ophthalmol. 2002;120:933-940.
54 Damato B, Joussen AM, Gardner TM, Kirchhof B, et al, editors. Retinal Vascular Disease. Berlin: Springer; 2007:582-591.
55 Wachtlin J, Bechrakis NE, Foerster MH. [Photodynamic therapy with verteporfin for uveal melanoma]. Ophthalmologe. 2005;102:241-246.
56 Lang GE, Mennel S, Spital G, et al. [Different indications of photodynamic therapy in ophthalmology]. Klin Monatsbl Augenheilkd. 2009;226:725-739.
57 Foulds WS, Damato BE. Low-energy long-exposure laser therapy in the management of choroidal melanoma. Graefes Arch Clin Exp Ophthalmol. 1986;224:26-31.
58 Bartlema YM, Oosterhuis JA, Journee-de Korver JG, et al. Combined plaque radiotherapy and transpupillary thermotherapy in choroidal melanoma: 5 years’ experience. Br J Ophthalmol. 2003;87:1370-1373.
59 Verbeek AM, Koutentakis P, Deutman AF. Circumscribed choroidal hemangioma diagnosed by ultrasonography. A retrospective analysis of 40 cases. Int Ophthalmol. 1995;19(3):185-189.
60 Boixadera A, Garcia-Arumi J, Martinez-Castillo V, et al. Prospective clinical trial evaluating the efficacy of photodynamic therapy for symptomatic circumscribed choroidal hemangioma. Ophthalmology. 2009;116(1):100-105e1.
61 Schalenbourg A, Piguet B, Zografos L. Indocyanine green angiographic findings in choroidal hemangiomas: A study of 75 cases. Ophthalmologica. 2000;214(4):246-252.
62 Comi AM. Advances in Sturge–Weber syndrome. Curr Opin Neurol. 2006;19(2):124-128.
63 Augsburger JJ, Shields JA, Moffat KP. Circumscribed choroidal hemangiomas: long-term visual prognosis. Retina. 1981;1(1):56-61.
64 Shields JA, Stephens RF, Eagle RCJr, et al. Progressive enlargement of a circumscribed choroidal hemangioma. A clinicopathologic correlation. Arch Ophthalmol. 1992;110(9):1276-1278.
65 Hocht S, Wachtlin J, Bechrakis NE, et al. Proton or photon irradiation for hemangiomas of the choroid? A retrospective comparison. Int J Radiat Oncol Biol Phys. 2006;66(2):345-351.
66 Jurklies B, Bornfeld N. The role of photodynamic therapy in the treatment of symptomatic choroidal hemangioma. Graefes Arch Clin Exp Ophthalmol. 2005;243(5):393-396.
67 Singh AD, Kaiser PK, Sears JE, et al. Photodynamic therapy of circumscribed choroidal haemangioma. Br J Ophthalmol. 2004;88(11):1414-1418.
68 Levy-Gabriel C, Rouic LL, Plancher C, et al. Long-term results of low-dose proton beam therapy for circumscribed choroidal hemangiomas. Retina. 2009;29(2):170-175.
69 Lopez-Caballero C, Saornil MA, de Frutos J, et al. High dose iodine 125 episcleral brachytherapy for circumscribed choroidal hemangioma. Br J Ophthalmol. 2009;94:470-473.
70 Tuncer S, Demirci H, Shields CL, et al. Polypoidal choroidal vasculopathy following photodynamic therapy for choroidal hemangioma. Eur J Ophthalmol. 2009;19(1):159-162.
71 Nelson CC, Hertzberg BS, Klintworth GK. A histopathologic study of 716 unselected eyes in patients with cancer at the time of death. Am J Ophthalmol. 1983;95:788-793.
72 Shields CL, Shields JA, Gross NE, et al. Survey of 520 eyes with uveal metastases. Ophthalmology. 1997;104:1265-1276.
73 Kreusel KM, Bechrakis NE, Wiegel T, et al. Incidence and clinical characteristics of symptomatic choroidal metastasis from lung cancer. Acta Ophthalmol. 2008;86:515-519.
74 Shankar J, Damato BE, Hiscott P. Palliative vitrectomy for intraocular metastasis from cutaneous melanoma. Eye. 2002;16:660-662.
75 Wiegel T, Kreusel KM, Bornfeld N, et al. Frequency of asymptomatic choroidal metastasis in patients with disseminated breast cancer: results of a prospective screening programme. Br J Ophthalmol. 1998;82:1159-1161.
76 Shields JA, Shields CL, Kiratli H, et al. Metastatic tumors to the iris in 40 patients. Am J Ophthalmol. 1995;119:422-430.
77 Bechrakis NE, Foerster MH, Bornfeld N. Biopsy in indeterminate intraocular tumors. Ophthalmology. 2002;109:235-242.
78 Sen J, Groenewald C, Hiscott PS, et al. Transretinal choroidal tumor biopsy with a 25-gauge vitrector. Ophthalmology. 2006;113:1028-1031.
79 Augsburger JJ. Diagnostic biopsy of selected intraocular tumors. Am J Ophthalmol. 2005;140:1094-1095.
80 Trichopoulos N, Augsburger JJ. Neuroendocrine tumours metastatic to the uvea: diagnosis by fine needle aspiration biopsy. Graefes Arch Clin Exp Ophthalmol. 2006;244:524-528.
81 Rudoler SB, Corn BW, Shields CL, et al. External beam irradiation for choroid metastases: identification of factors predisposing to long-term sequelae. Int J Radiat Oncol Biol Phys. 1997;1:251-256.
82 Shields CL, Shields JA, De Potter P, et al. Plaque radiotherapy for the management of uveal metastasis. Arch Ophthalmol.. 1997;115:203-209.
83 Shields JA, Shields CL. Management and prognosis of retinoblastoma. In: Intraocular Tumors. A Text and Atlas. Philadelphia: WB Saunders; 1992:377-392.
84 Shields JA, Shields CL. Retinoblastoma. In: Shields JA, Shields CL, editors. Atlas of Intraocular Tumors. Philadelphia: Lippincott Williams & Wilkins; 2008:293-365.
85 Knudson AG, Meadows AT, Nichols WW, et al. Chromosomal deletion in retinoblastoma. NEJM. 1976;295:1120-1123.
86 Nichols KE, Houseknecht MD, Godmilow L, et al. Sensitive multistep clinical molecular screening of 180 unrelated individuals with retinoblastoma detects 36 novel mutations in the RB1 gene. Hum Mutat. 2005;25:566-574.
87 Schefler AC, Abramson DH. Retinoblastoma: what is new in 2007–2008. Curr Opin Ophthalmol. 2008;19:526-534.
88 Roarty JD, McLean IW, Zimmerman LE. Incidence of second neoplasms in patients with retinoblastoma. Ophthalmology. 1988;95:1583-1587.
89 Kleinerman FA, Tucker MA, Abramson DH, et al. Risk for soft tissue sarcomas by individual subtype in survivors of hereditary retinoblastoma. J Natl Cancer Inst. 2007;99:24-31.
90 Allderdice PW, Davis JG, Miller OJ, et al. The 13q– deletion syndrome. Am J Hum Genet. 1969;21:499-512.
91 Shields CL, Ghassemi F, Tuncer S, et al. Clinical spectrum of diffuse infiltrating retinoblastoma in 34 consecutive eyes. Ophthalmology. 2008;115:2253-2258.
92 Shields CL, Shields JA. Basic understanding of current classification and management of retinoblastoma. Curr Opin Ophthalmol. 2006;17:228-234.
93 Shields CL, Shields JA. Retinoblastoma management: advances in enucleation, intravenous chemoreduction, and intra-arterial chemotherapy. Curr Opin Ophthalmol. 2010;21:203-212.
94 Shields JA, Parsons HM, Shields CL, et al. Lesions simulating retinoblastoma. J Pediatr Ophthalmol Strabismus. 1991;28:338-340.
95 Abramson DH. Retinoblastoma in the 20th century: Past success and future challenges the Weisenfeld lecture. Invest Ophthalmol Vis Sci. 2005;46(8):2683-2691.
96 Lin P, O’Brien JM. Frontiers in the management of retinoblastoma. Am J Ophthalmol. 2009;148(2):192-198.
97 Linn Murphree A. Intraocular retinoblastoma: the case for a new group classification. Ophthalmol Clin North Am. 2005;18(1):41-53. viii
98 Shields CL, Mashayekhi A, Au AK, et al. The International Classification of Retinoblastoma predicts chemoreduction success. Ophthalmology. 2006;113(12):2276-2280.
99 Shields CL, Santos MC, Diniz W, et al. Thermotherapy for retinoblastoma. Arch Ophthalmol. 1999;117(7):885-893.
100 Shields JA, Parsons H, Shields CL, et al. The role of cryotherapy in the management of retinoblastoma. Am J Ophthalmol. 1989;108(3):260-264.
101 Shields CL, Shields JA, Cater J, et al. Plaque radiotherapy for retinoblastoma: long-term tumor control and treatment complications in 208 tumors. Ophthalmology. 2001;108(11):2116-2121.
102 Shields CL, Honavar SG, Meadows AT, et al. Chemoreduction for unilateral retinoblastoma. Arch Ophthalmol. 2002;120(12):1653-1658.
103 Shields JA, Shields CL, Meadows AT. Chemoreduction in the management of retinoblastoma. Am J Ophthalmol. 2005;140(3):505-506.
104 Zage PE, Reitman AJ, Seshadri R, et al. Outcomes of a two-drug chemotherapy regimen for intraocular retinoblastoma. Pediatr Blood Cancer. 2008;50(3):567-572.
105 Hernandez JC, Brady LW, Shields JA, et al. External beam radiation for retinoblastoma: results, patterns of failure, and a proposal for treatment guidelines. Int J Radiat Oncol Biol Phys. 1996;35(1):125-132.
106 Damato B. Vasoproliferative retinal tumour. Br J Ophthalmol. 2006;90(4):399-400.
107 Lafaut BA, Meire FM, Leys AM, et al. Vasoproliferative retinal tumors associated with peripheral chorioretinal scars in presumed congenital toxoplasmosis. Graefes Arch Clin Exp Ophthalmol.. 1999;237(12):1033-1038.
108 Shields CL, Shields JA, Barrett J, et al. Vasoproliferative tumors of the ocular fundus. Classification and clinical manifestations in 103 patients. Arch Ophthalmol. 1995;113(5):615-623.
109 Heimann H, Bornfeld N, Vij O, et al. Vasoproliferative tumours of the retina. Br J Ophthalmol. 2000;84(10):1162-1169.
110 Hiscott P, Mudhar H. Is vasoproliferative tumour (reactive retinal glioangiosis) part of the spectrum of proliferative vitreoretinopathy? Eye (Lond). 2009;23(9):1851-1858.
111 Anastassiou G, Bornfeld N, Schueler AO, et al. Ruthenium-106 plaque brachytherapy for symptomatic vasoproliferative tumours of the retina. Br J Ophthalmol. 2006;90(4):447-450.
112 McCabe CM, Mieler WF. Six-year follow-up of an idiopathic retinal vasoproliferative tumor. Arch Ophthalmol. 1996;114(5):617.
113 Barbezetto IA, Smith RT. Vasoproliferative tumor of the retina treated with PDT. Retina. 2003;23(4):565-567.
114 Cohen VM, Shields CL, Demirci H, et al. Iodine I 125 plaque radiotherapy for vasoproliferative tumors of the retina in 30 eyes. Arch Ophthalmol. 2008;126(9):1245-1251.
115 Kenawy N, Groenwald C, Damato B. Treatment of a vasoproliferative tumour with intravitreal bevacizumab (Avastin). Eye (Lond). 2007;21(6):893-894.
116 Irvine F, O’Donnell N, Kemp E, et al. Retinal vasoproliferative tumors: surgical management and histological findings. Arch Ophthalmol. 2000;118(4):563-569.
117 Wachtlin J, Heimann H, Jandeck C, et al. Bilateral vasoproliferative retinal tumors with identical localization in a pair of monozygotic twins. Arch Ophthalmol. 2002;120(6):860-862.
118 Singh AD, Shields CL, Shields JA. von Hippel–Lindau disease. Surv Ophthalmol. 2001;46:117-142.
119 Latif F, Tory K, Gnarra J, et al. Identification of the von Hippel–Lindau disease tumor suppressor gene. Science. 1993;260:1317-1320.
120 Singh AD, Nouri M, Shields CL, et al. Treatment of retinal capillary hemangioma. Ophthalmology. 2002;109:1799-1806.
121 Oosterhuis JA, Rubinstein K. Hemangioma at the optic disc. Ophthalmologica. 1972;164:362-374.
122 Schmidt-Erfurth UM, Kusserow C, Barbazetto IA, et al. Benefits and complications of photodynamic therapy of papillary capillary hemangiomas. Ophthalmology. 2002;109:1256-1266.
123 Gass JDM, Braunstein R. Sessile and exophytic capillary angiomas of the juxtapapillary retina and optic nerve head. Arch Ophthalmol. 1980;98:1790-1797.
124 Stolle C, Glenn G, Zbar B, et al. Improved detection of germline mutations in the von Hippel–Lindau disease tumor suppressor gene. Hum Mutat. 1998;12:417-423.
125 Maher ER, Yates JR, Harries R, et al. Clinical features and natural history of von Hippel–Lindau disease. Q J Med. 1990;77:1151-1163.
126 Blodi CF, Russell SR, Pulido JS, et al. Direct and feeder vessel photocoagulation of retinal angiomas with dye yellow laser. Ophthalmology. 1990;97:791-797.
127 Welch RB. The recognition and treatment of early angiomatosis retinae and use of cryosurgery as an adjunct to therapy. Trans Am Ophthalmol Soc. 1970;68:367-424.
128 Kreusel KM, Bornfeld N, Lommatzsch A, et al. Ruthenium-106 brachytherapy for peripheral retinal capillary hemangiomas. Ophthalmology. 1998;105:1386-1392.
129 Atebara NH. Retinal capillary hemangioma treated with verteporfin photodynamic therapy. Am J Ophthalmol. 2002;134:788-790.
130 Chew EY. Ocular manifestations of von Hippel–Lindau disease: clinical and genetic investigations. Trans Am Ophthalmol Soc. 2005;103:495-511.
131 Webster AR, Maher ER, Moore AT. Clinical characteristics of ocular angiomatosis in von Hippel–Lindau disease and correlation with germline mutation. Arch Ophthalmol. 1999;117:371-378.
132 Swerdlow SH, Campo E, Harris NL, et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon: IARC; 2008.
133 Coupland SE, Damato B. Understanding intraocular lymphomas. Clin Exp Ophthalmol. 2008;36:564-578.
134 Coupland SE, White V, Rootman J, et al. TNM Staging of Ocular Adnexal Lymphomas. New York: Springer; 2009.
135 Chan CC, Gonzales J. Primary Intraocular Lymphoma. Singapore, London, Hackensack NJ: World Scientific Publishing Co; 2007.
136 Davis JL. Diagnosis of intraocular lymphoma. Ocul Immunol Inflamm. 2004;12:7-16.
137 Coupland SE, Damato B. Lymphomas involving the eye and the ocular adnexa. Curr Opin Ophthalmol. 2006;17:523-531.
138 Ferry JA, Fung CY, Zukerberg L, et al. Lymphoma of the ocular adnexa: A study of 353 cases. Am J Surg Pathol. 2007;31:170-184.
139 Coupland SE. The pathologist’s perspective on vitreous opacities. Eye (Lond). 2008;22:1318-1329.
140 Merle-Beral H, Davi F, Cassoux N, et al. Biological diagnosis of primary intraocular lymphoma. Br J Haematol. 2004;124:469-473.
141 Kennerdell JS, Flores NE, Hartsock RJ. Low-dose radiotherapy for lymphoid lesions of the orbit and ocular adnexa. Ophthal Plast Reconstr Surg. 1999;15:129-133.
142 Ferreri AJ, Reni M, Foppoli M, et al. High-dose cytarabine plus high-dose methotrexate versus high-dose methotrexate alone in patients with primary CNS lymphoma: a randomised phase 2 trial. Lancet. 2009;374(9700):1512-1520. Epub 2009 Sep 18
143 Frenkel S, Hendler K, Siegal T, et al. Intravitreal methotrexate for treating vitreoretinal lymphoma: 10 years of experience. Br J Ophthalmol. 2008;92:231-235.
144 Seregard S. Conjunctival melanoma. Surv Ophthalmol. 1998;42:321-350.
145 Damato B, Coupland SE. Clinical mapping of conjunctival melanomas. Br J Ophthalmol. 2008;92:1545-1549.
146 Shields JA, Shields CL, Mashayekhi A, et al. Primary acquired melanosis of the conjunctiva: risks for progression to melanoma in 311 eyes. The 2006 Lorenz E. Zimmerman lecture. Ophthalmology. 2008;115:511-519.
147 Shields CL, Shields JA, Gunduz K, et al. Conjunctival melanoma: risk factors for recurrence, exenteration, metastasis, and death in 150 consecutive patients. Arch Ophthalmol. 2000;118:1497-1507.
148 Damato B, Coupland SE. Conjunctival melanoma and melanosis: a reappraisal of terminology, classification and staging. Clin Exp Ophthalmol. 2008;36:786-795.
149 Esmaeli B. Regional lymph node assessment for conjunctival melanoma: sentinel lymph node biopsy and positron emission tomography. Br J Ophthalmol. 2008;92:443-445.
150 Damato B, Coupland SE. Management of conjunctival melanoma. Expert Rev Anticancer Ther. 2009;9:1227-1239.
151 Shields JA, Shields CL, De Potter P. Surgical management of conjunctival tumors. The 1994 Lynn B. McMahan Lecture. Arch Ophthalmol. 1997;115:808-815.
152 Zografos L, Uffer S, Bercher L, et al. [Combined surgery, cryocoagulation and radiotherapy for treatment of melanoma of the conjunctiva]. Klin Monatsbl Augenheilkd. 1994;204:385-390.
153 Anastassiou G, Heiligenhaus A, Bechrakis N, et al. Prognostic value of clinical and histopathological parameters in conjunctival melanomas: a retrospective study. Br J Ophthalmol. 2002;86:163-167.
154 Missotten GS, Keijser S, de Keizer RJ, et al. Conjunctival melanoma in the Netherlands: a nationwide study. Invest Ophthalmol Vis Sci. 2005;46:75-82.
155 Damato B, Coupland SE. An audit of conjunctival melanoma treatment in Liverpool. Eye. 2008;23:801-809.
156 Lee GA, Hirst LW. Ocular surface squamous neoplasia. Surv Ophthalmol. 1995;39:429-450.
157 Pe’er J. Ocular surface squamous neoplasia. Ophthalmol Clin N Am. 2005;18:1-13.
158 Ng J, Coroneo MT, Wakefield D, et al. Ultraviolet radiation and the role of matrix metalloproteinases in the pathogenesis of ocular surface squamous neoplasia. Invest Ophthalmol Vis Sci. 2008;49:5295-5306.
159 Verma V, Shen D, Sieving PC, et al. The role of infectious agents in the etiology of ocular adnexal neoplasia. Surv Ophthalmol. 2008;53:312-331.
160 Tananuvat N, Lertprasertsuk N, Mahanupap P, et al. Role of impression cytology in diagnosis of ocular surface neoplasia. Cornea. 2008;27:269-274.
161 Galor A, Jeng BH, Singh AD. Conjunctiva and corneal tumors. Surgical technique. In: Singh AD, Damato BE, Pe’er J, Murphree AL, Perry JD, editors. Clinical Ophthalmic Oncology, Chapter 29. Edinburgh: Saunders/Elsevier, 2007.
162 Espana EM, Prabhasawat P, Grueterich M, et al. Amniotic membrane transplantation for reconstruction after excision of large ocular surface neoplasia. Br J Ophthalmol. 2002;86:640-645.
163 Fraunfelder FT, Wallace TR, Farris HE, et al. The role of cryosurgery in external ocular and periocular disease. Trans Am Acad Ophthalmol Otolaryngol. 1977;83:713-724.
164 Lommatzsch P. Beta-ray treatment of malignant epithelial tumors of the conjunctiva. Am J Ophthalmol. 1976;81:198-206.
165 Stone DU, Butt AL, Chodosh J. Ocular surface squamous neoplasia: a standard of care survey. Cornea. 2005;24:297-300.
166 Frucht-Pery J, Rozenman Y, Pe’er J. Topical Mitomycin-C for partially-excised conjunctival squamous cell carcinoma. Ophthalmology. 2002;109:548-552.