[level-membership-for-pathology-category]
Chapter 7 Disorders of metabolism and homeostasis
Some metabolic disorders, congenital or acquired, are specific abnormalities of metabolic pathways, often having considerable clinical effects. Congenital metabolic disorders usually result from inherited enzyme deficiencies.
Other metabolic disorders are characterised by perturbations of the body’s homeostatic mechanisms maintaining the integrity of fluids and tissues. These conditions are almost always acquired and their effects can be diverse.
INBORN ERRORS OF METABOLISM



The concept of inborn errors of metabolism was formulated by Sir Archibald Garrod in 1908 as a result of his studies on a condition called alkaptonuria, a rare inherited deficiency of homogentisic acid oxidase.
Inborn (usually inherited) errors of metabolism are important causes of illness presenting in infancy. Some require prompt treatment to avoid serious complications. Others defy treatment. All deserve accurate diagnosis so that parents can be counselled about the inherited risk to further pregnancies. Inborn metabolic errors are potentially chronic problems, because the primary abnormality is innate rather than due to any external cause that could be eliminated by treatment.
Inborn errors of metabolism are single-gene defects resulting in the absence or deficiency of an enzyme or the synthesis of a defective protein. Single-gene defects occur in about 1% of all births, but the diseases caused by them show geographic variations in incidence; this is exemplified by the high incidence of thalassaemias—due to defects in haemoglobin synthesis (Ch. 23)—in Mediterranean regions. These variations reflect the prevalence of specific abnormal genes in different populations.
Inborn errors of metabolism have four possible consequences:
Accumulation of an intermediate metabolite may have toxic or hormonal effects. However, in some conditions the intermediate metabolite accumulates within the cells in which it has been synthesised, causing them to enlarge and compromising their function or that of neighbouring cells; these conditions are referred to as storage disorders (e.g. Gaucher’s disease). Other inborn metabolic errors lead to the production of a protein with defective function; for example, the substitution of just a single amino acid in a large protein can have considerable adverse effects (e.g. haemoglobinopathies).
The genetic basis of the inheritance of these disorders is discussed in Chapter 3.
Inherited metabolic disorders may be classified according to the principal biochemical defect (e.g. amino acid disorder) or the consequence (e.g. storage disorder).
Disorders of carbohydrate metabolism
The commonest disorder of carbohydrate metabolism with an inherited component in its aetiology is diabetes mellitus (see p. 128). Much less common, but with an autosomal recessive pattern of inheritance and often presenting at an early age, are:
Disorders of amino acid metabolism
Several inherited disorders of amino acid metabolism involve defects of enzymes in the phenylalanine/tyrosine pathway (Fig. 7.1).
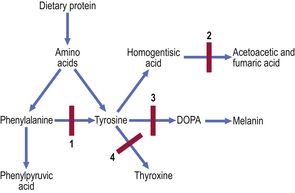
Fig. 7.1 Inborn errors of metabolism in the phenylalanine/tyrosine pathway. 1. Phenylketonuria. Lack of phenylalanine hydroxylase blocks conversion of phenylalanine to tyrosine; phenylalanine and phenylpyruvic acid appear in the urine. 2. Alkaptonuria. Lack of homogentisic acid oxidase causes accumulation of homogentisic acid. 3. Albinism. Lack of the enzyme tyrosinase prevents conversion of tyrosine via DOPA to melanin. 4. Familial hypothyroidism. Deficiency of any one of several enzymes impairs iodination of tyrosine in the formation of thyroid hormone.
Phenylketonuria
This autosomal recessive disorder affects approximately 1 in 10000 infants. It is due to a deficiency of phenylalanine hydroxylase, an enzyme responsible for the conversion of phenylalanine to tyrosine (Fig. 7.1).
In the UK and many other countries the clinical effects of phenylketonuria are now seen only very rarely, because it is detected by screening all newborn infants and treated promptly. Testing is done by analysing a drop of blood, dried on to filter paper, for phenylalanine (Guthrie test). If phenylketonuria is not tested for in this way and the affected infant’s diet contains usual amounts of phenylalanine, then the disorder manifests itself with skin and hair depigmentation, fits and mental retardation. Treatment involves a low phenylalanine diet until the child is at least 8 years old. When affected females themselves become pregnant, the special diet must be resumed to avoid the toxic metabolites damaging the developing fetus.
Alkaptonuria
This rare autosomal recessive deficiency of homogentisic acid oxidase (Fig. 7.1) is a good example of an inborn metabolic error that does not produce serious effects until adult life. The condition is sometimes recognised from the observation that the patient’s urine darkens on standing; the sweat may also be black! Homogentisic acid accumulates in connective tissues, principally cartilage, where the darkening is called ochronosis. This accumulation causes joint damage. The underlying condition cannot be cured; treatment is symptomatic only.
Homocystinuria
Like most inherited disorders of metabolism, homocystinuria is an autosomal recessive disorder. There is a deficiency of cystathionine synthase, an enzyme required for the conversion of homocystine via homocysteine to cystathionine. Homocysteine and methionine, its precursor, accumulate in the blood. Homocystine also accumulates, interfering with the cross-linking of collagen and elastic fibres. The ultimate effect resembles Marfan’s syndrome (see p. 128), but also with mental retardation and fits.
Storage disorders
Inborn metabolic defects result in storage disorders if a deficiency of an enzyme, usually lysosomal, prevents the normal conversion of a macromolecule (e.g. glycogen or gangliosides) into its smaller subunits (e.g. glucose or fatty acids). The macromolecule accumulates within the cells that normally harbour it, swelling their cytoplasm (Fig. 7.2) and causing organ enlargement and deformities. This situation is harmful to the patient because the swelling of cells often impairs their function, or that of their immediate neighbours due to pressure effects, and because of conditions resulting from deficiency of the smaller subunits (e.g. hypoglycaemia in the case of glycogen storage disorders).
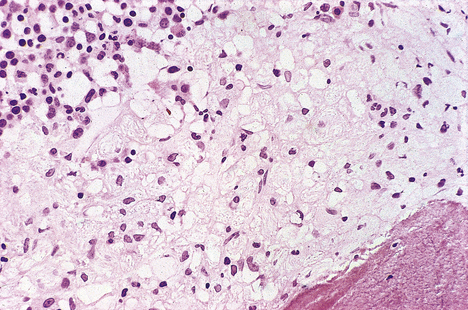
Fig. 7.2 Bone marrow biopsy revealing Gaucher’s disease. Pale foamy macrophages distended with gangliosides have displaced much of the haemopoietic tissue (top left), thereby causing anaemia.
Major categories of these autosomal recessive disorders (Table 7.1) are:
Table 7.1 Examples of inborn errors of metabolism resulting in storage disorders
| Type of disease/examples | Deficiency | Consequences |
|---|---|---|
| Glycogenoses | Debranching enzyme |
McCardle’s syndromeMuscle phosphorylase von Gierke’s diseaseGlucose-6-phosphate dehydrogenase Pompe’s diseaseAcid maltase MucopolysaccharidosesLysosomal hydrolase
Hurler’s syndromeAlpha-l-iduronidase Hunter’s syndromeIduronate sulphate sulphatase SphingolipidosesLysosomal enzyme
Gaucher’s diseaseGlucocerebrosidase Niemann–Pick diseaseSphingomyelinase Tay–Sachs diseaseHexosaminidase A
Disorders of cell membrane transport
Inborn metabolic errors can lead to impairment of the specific transport of substances across cell membranes. Examples include:
Channelopathies
A channelopathy is a disease caused by the dysfunction of a specific ion channel in cell membranes. The ion channel dysfunction may result from:
Cystic fibrosis
Cystic fibrosis, a channelopathy, is the commonest serious inherited metabolic disorder in the UK; it is much commoner in Caucasians than in other races. The autosomal recessive abnormal gene is carried by approximately 1 in 20 Caucasians and the condition affects approximately 1 in 2000 births. The defective gene, in which numerous mutations have been identified, is on chromosome 7 and ultimately results in abnormal water and electrolyte transport across cell membranes.
Cystic fibrosis transmembrane conductance regulator (CFTR)
The commonest abnormality (ΔF508) in the CFTR gene is a deletion resulting in a missing phenylalanine molecule. The defective CFTR is unresponsive to cyclic AMP control, so transport of chloride ions and water across epithelial cell membranes becomes impaired (Fig. 7.3).
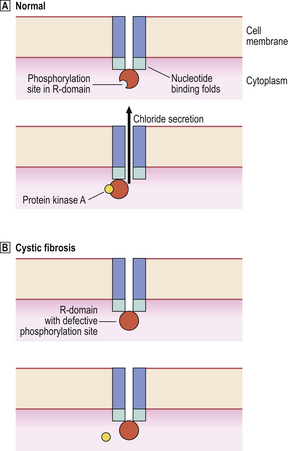
Fig. 7.3 Defective chloride secretion in cystic fibrosis. The normal CFTR is a transmembrane molecule with intracytoplasmic nucleotide binding folds and a phosphorylation site on the R-domain.  In normal cells, interaction of the R-domain with protein kinase A results in opening of the channel and chloride secretion.
In normal cells, interaction of the R-domain with protein kinase A results in opening of the channel and chloride secretion.  In cystic fibrosis, a common defect prevents phosphorylation of the R-domain with the result that chloride secretion is impaired.
In cystic fibrosis, a common defect prevents phosphorylation of the R-domain with the result that chloride secretion is impaired.
Clinicopathological features
Cystic fibrosis is characterised by mucous secretions of abnormally high viscosity. The abnormal mucus plugs exocrine ducts, causing parenchymal damage to the affected organs. The clinical manifestations are:
Porphyrias
The porphyrias, transmitted as autosomal dominant disorders, are due to defective synthesis of haem, an iron–porphyrin complex, the oxygen-carrying moiety of haemoglobin. Haem is synthesised from 5-aminolaevulinic acid. The different types of porphyrin accumulate due to inherited defects in this synthetic pathway (Fig. 7.4).
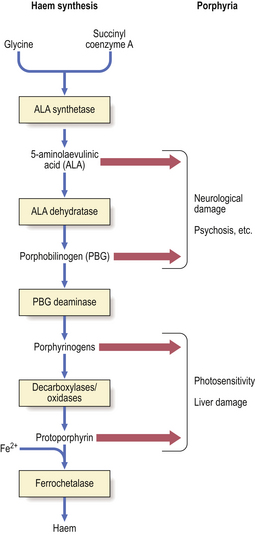
Fig. 7.4 Porphyrias. Enzyme deficiencies in the pathway of synthesis of haem from glycine and succinyl coenzyme A through 5-aminolaevulinic acid result in the accumulation of toxic intermediate metabolites. Removal of product inhibition due to deficient synthesis of haem enhances the formation of intermediate metabolites. Accumulation of 5-aminolaevulinic acid or porphobilinogen tends to be associated with neurological damage and psychiatric symptoms. Accumulation of porphyrinogens, of which there are several types (uro-, copro-, proto-), tends to be associated with photosensitivity.
Clinicopathological features
Accumulation of porphyrins can cause clinical syndromes characterised by:
The pain and psychiatric disturbances are episodic. During the acute attacks, the patient’s urine contains excess 5-aminolaevulinic acid and porphobilinogen. Consequently, the urine may gradually become dark red, brown or even purple (‘porphyria’ is derived from the Greek word ‘porphura’ meaning purple pigment) on exposure to sunlight.
Acute attacks of porphyria can be precipitated by some drugs, alcohol and hormonal changes (e.g. during the menstrual cycle). The most frequently incriminated drugs include barbiturates, sulphonamides, oral contraceptives and anticonvulsants; these should therefore be avoided.
The skin lesions are characterised by severe blistering, exacerbated by light exposure, and subsequent scarring. This photosensitivity is a distressing feature, but it has led to the beneficial use of injected porphyrins in the treatment of tumours by phototherapy with laser light.
Disorders of connective tissue metabolism
Most inherited disorders of connective tissue metabolism affect collagen or elastic tissue. Examples include:
Osteogenesis imperfecta
Osteogenesis imperfecta is a group of disorders in which there is an inborn error of type I collagen synthesis (Ch. 25). Type I collagen is most abundant in bone, so the principal manifestation is skeletal weakness resulting in deformities and a susceptibility to fractures; the other names for this condition are ‘fragilitas ossium’ and ‘brittle bone disease’. The teeth are also affected and the sclerae of the eyes are abnormally thin, causing them to appear blue. It occurs in dominantly and recessively inherited forms with varying degrees of severity.
Marfan’s syndrome
Marfan’s syndrome is a combination of unusually tall stature, long arm span, dislocation of the lenses of the eyes, aortic and mitral valve incompetence, and weakness of the aortic media predisposing to dissecting aneurysms (Ch. 13). The condition results from a defect in the FBN1 gene encoding for fibrillin, a glycoprotein essential for the formation and integrity of elastic fibres.
ACQUIRED METABOLIC DISORDERS
Many diseases result in secondary metabolic abnormalities. In others the metabolic disturbance is the primary event. For example, renal diseases almost always result in metabolic changes that reflect the kidneys’ importance in water and electrolyte homeostasis. In contrast, a disease like gout is often due to a primary metabolic disorder that may secondarily damage the kidneys. This section deals with metabolic abnormalities as both consequences and causes of disease. Acquired metabolic disorders frequently cause systemic problems affecting many organs: for example, diabetes mellitus is associated with microvascular damage in the retinas, nerves, kidneys and other organs; electrolyte imbalance compromises the function of cells in all tissues.
Two of the disorders discussed in this section—diabetes mellitus and gout—are categorised as ‘acquired’ largely because they occur most commonly in adults, but both have a significant genetic component in their aetiology.
Diabetes mellitus




Diabetes mellitus is a group of diseases characterised by impaired glucose homeostasis resulting from a relative or absolute insufficiency of insulin. Insulin insufficiency causes hyperglycaemia and glycosuria. Diabetes is covered in Chapter 17, but a brief account is relevant here.
The aetiology is multifactorial; although the disorder is acquired, there is a significant genetic predisposition. Diabetes mellitus is subclassified into primary and secondary types. Primary diabetes is much more common than diabetes secondary to other diseases.
Primary diabetes mellitus
Primary diabetes mellitus (DM) is subdivided into:
Juvenile-onset diabetes, usually appearing before the age of 20 years, is almost always of IDDM type. There is an inherited predisposition associated with human leukocyte antigens (HLA) DR3 and DR4. The initiating event may be a viral infection of the insulin-producing beta-cells of the islets of Langerhans, precipitating their immune destruction.
Diabetes mellitus developing in adults (over 25 years of age) is most likely to be NIDDM type. It is associated with an acquired resistance to insulin. There is no HLA association, but there is a familial tendency to develop the disease. The affected patients are often, but not always, obese.
Complications of diabetes mellitus
Good control of blood sugar levels reduces the risk of complications. Nevertheless, many diabetics develop complications of their disease. These are covered in more detail in the relevant chapters, but a summary is given here:
To this list should be added hypoglycaemia, which is a frequent and troublesome complication of insulin therapy in IDDM.
There are two possible biochemical explanations for the tissue damage that results from long-term diabetes mellitus:
Gout


Gout is a common disorder resulting from high blood uric acid levels. Uric acid is a breakdown product of the body’s purine (nucleic acid) metabolism (Fig. 7.5), but a small proportion comes from the diet. Most uric acid is excreted by the kidneys. In the blood, most uric acid is in the form of monosodium urate. In patients with gout, the monosodium urate concentration may be very high, forming a supersaturated solution, thus risking urate crystal deposition in tissues causing:
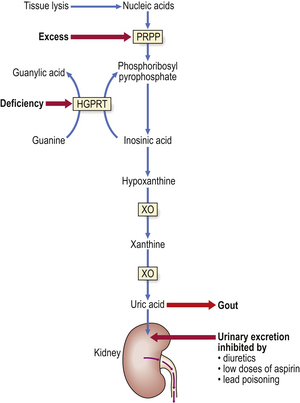
Fig. 7.5 Pathogenesis of gout. The metabolic pathway shows the synthesis of uric acid from nucleic acids. Primary gout can arise from an inherited (X-linked) deficiency of hypoxanthine guanine phosphoribosyl transferase (HGPRT) or excessive activity of 5-phosphoribosyl-1-pyrophosphate (PRPP). Secondary gout results either from increased tissue lysis (e.g. due to tumour chemotherapy) liberating excess nucleic acids or from inhibition of the urinary excretion of uric acid. Xanthine oxidase (XO) is inhibited by allopurinol, an effective long-term remedy for gout.
Gout occurs more commonly in men than in women, and is rare before puberty. A rare form of gout in children—Lesch–Nyhan syndrome—is due to absence of the enzyme HGPRT (hypoxanthine guanine phosphoribosyl transferase) (Fig. 7.5) and is associated with mental deficiency and a bizarre tendency to self-mutilation.
Aetiology
Like diabetes mellitus, the aetiology of gout is multifactorial. There is a genetic component, but the role of other factors justifies the inclusion of gout under the heading of acquired disorders. Aetiological factors include:
Some of these factors may, of course, be interdependent. Accordingly, gout can be subdivided into primary gout, due to some genetic abnormality of purine metabolism, or secondary gout, due to increased liberation of nucleic acids from necrotic tissue or decreased urinary excretion of uric acid.
Clinicopathological features
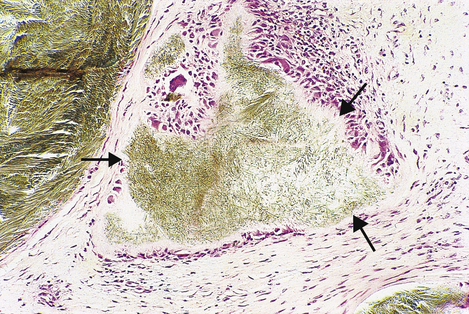
The clinical features of gout are due to urate crystal deposition in various tissues (Fig. 7.6). In joints, a painful acute arthritis results from phagocytosis of the crystals by neutrophil polymorphs, in turn causing release of lysosomal enzymes along with the indigestible crystals, thus accelerating and perpetuating a cyclical inflammatory reaction. The first metatarsophalangeal joint is typically affected.
Water homeostasis




Water and electrolyte homeostasis is tightly controlled by various hormones, including antidiuretic hormone (ADH), aldosterone and atrial natriuretic peptide, acting upon selective reabsorption in the renal tubules (Ch. 21). The process is influenced by the dietary intake of water and electrolytes (in food or drinking in response to thirst or social purposes) and the adjustments necessary to cope with disease or adverse environmental conditions.
Many diseases result in problems of water and electrolyte homeostasis. Disturbances can also occur in post-operative patients receiving fluids and nutrition parenterally. Fortunately, any changes are fairly easy to monitor and control by making adjustments to the fluid and electrolyte intake.
Water is constantly lost from the body—in urine, in faeces, in exhaled gas from the lungs, and from the skin. The replenishment of body water is controlled by a combination of the satisfaction of the sensation of thirst and the regulation of the renal tubular reabsorption of water mediated by ADH.
Water excess
Excessive body water may occur in patients with extensive oedema or if there is inappropriate production of ADH (e.g. as occurs with small-cell lung carcinoma) or if the body sodium concentration increases due to excessive tubular reabsorption (for example, due to an aldosterone-secreting tumour of the adrenal cortex). Water overload can be caused iatrogenically by excessive parenteral infusion of fluids in patients with impaired renal function; this should be avoided by carefully monitoring fluid input and output.
Dehydration
Dehydration results from either excessive water loss or inadequate intake or a combination of both. Inadequate water intake is a common problem in regions of the world affected by drought and famine.
Excessive water loss can be due to:
Dehydration is recognised clinically by a dry mouth, inelastic skin and, in extreme cases, sunken eyes. The blood haematocrit (proportion of the blood volume occupied by cells) will be elevated, causing an increase in whole blood viscosity. This results in a sluggish circulation and consequent impairment of the function of many organs.
The plasma sodium and urea concentrations are typically elevated, reflecting haemoconcentration and impaired renal function.
Oedema and serous effusions



Oedema is an excess of fluid in the intercellular compartment of a tissue. A serous effusion is an excess of fluid in a serous or coelomic cavity (e.g. peritoneal cavity, pleural cavity). The main ingredient of the fluid is always water. Oedema and serous effusions share common mechanisms.
Oedema is recognised clinically by diffuse swelling of the affected tissue. If the oedema is subcutaneous, the affected area shows pitting; i.e. if the skin is indented firmly with the fingers, an impression of the fingers is left transiently on the surface. There is, therefore, usually little difficulty in diagnosing subcutaneous oedema. Oedema of internal tissues may be evident during surgery because they are swollen and, when incised, clear or slightly opalescent fluid oozes from the cut surfaces. Pulmonary oedema gives a characteristic appearance of increased radio-opacity on a plain chest X-ray and can be heard, through a stethoscope, as crepitations on inspiration.
Oedema, irrespective of its cause, has serious consequences in certain organs. For example, pulmonary oedema fluid fills the alveoli and reduces the effective lung volume available for respiration; the patient becomes breathless (dyspnoeic) and, if the oedema is severe, cyanosed. Cerebral oedema is an ominous development because it occurs within the rigid confines of the cranial cavity; compression of the brain against the falx cerebri, the tentorial membranes or the base of the skull leads to herniation of brain tissue, possibly causing irreversible and fatal damage. Cerebral oedema can be diagnosed clinically by finding papilloedema (oedema of the optic disc) on ophthalmoscopy.
Oedema and serous effusions are due to:
Oedema is classified into four pathogenetic categories (Fig. 7.7):
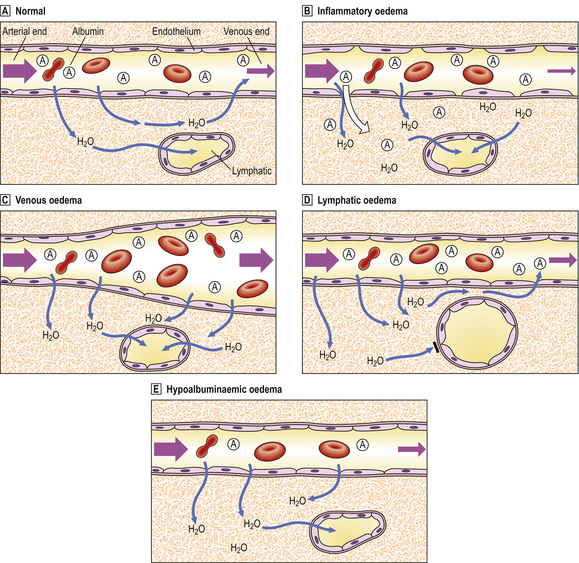
Fig. 7.7 Pathogenesis of oedema.  Normal. Hydrostatic blood pressure forces water out of capillaries at the arterial end, but the plasma oncotic pressure attributable to albumin sucks water back into capillary beds at the venous end. A small amount of water drains from the tissues through lymphatic channels.
Normal. Hydrostatic blood pressure forces water out of capillaries at the arterial end, but the plasma oncotic pressure attributable to albumin sucks water back into capillary beds at the venous end. A small amount of water drains from the tissues through lymphatic channels.  Inflammatory oedema. Gaps between endothelial cells (mostly at venular level) allow water and albumin (and other plasma constituents) to escape. There is increased lymphatic drainage, but this cannot cope with all the water released into the tissues and oedema results.
Inflammatory oedema. Gaps between endothelial cells (mostly at venular level) allow water and albumin (and other plasma constituents) to escape. There is increased lymphatic drainage, but this cannot cope with all the water released into the tissues and oedema results.  Venous oedema. Increased venous pressure (e.g. from heart failure, venous obstruction due to thrombus) causes passive dilatation and congestion of the capillary bed. Increased venous pressure exceeds that of plasma oncotic pressure and so water remains in the tissues.
Venous oedema. Increased venous pressure (e.g. from heart failure, venous obstruction due to thrombus) causes passive dilatation and congestion of the capillary bed. Increased venous pressure exceeds that of plasma oncotic pressure and so water remains in the tissues.  Lymphatic oedema. Lymphatic obstruction (e.g. by tumour deposits, filarial parasites) prevents drainage of water from tissues.
Lymphatic oedema. Lymphatic obstruction (e.g. by tumour deposits, filarial parasites) prevents drainage of water from tissues.  Hypoalbuminaemic oedema. Low plasma albumin concentration reduces the plasma oncotic pressure so that water cannot be sucked back into the capillary bed at the venous end.
Hypoalbuminaemic oedema. Low plasma albumin concentration reduces the plasma oncotic pressure so that water cannot be sucked back into the capillary bed at the venous end.
Inflammatory oedema
Oedema is a feature of acute inflammation (Ch. 10). In acutely inflamed tissues there is increased vascular (mainly venular) permeability due to the separation of endothelial cells under the influence of chemical mediators. Fluid with a high protein content leaks out of the permeable vessels into the inflamed tissue causing it to swell. This is beneficial, because the proteins in the oedema fluid assist in defeating the cause of the inflammation. For example:
In addition to the fluid component, the extravasate contains numerous neutrophil polymorphs.
Tissues affected by inflammatory oedema also have the other features of acute inflammation, such as pain and redness.
Venous oedema
Oedema results from increased intravenous pressure because this pressure opposes the plasma oncotic pressure, largely due to the presence of albumin, that draws fluid back into the circulation at the venous end of capillary beds. Increased intravenous pressure results from either heart failure or impairment of blood flow due to venous obstruction by a thrombus or extrinsic compression. The affected tissues are often intensely congested due to engorgement by venous blood under increased pressure. In heart failure, there is also pulmonary congestion with oedema and so-called passive venous congestion of the liver.
Venous oedema is seen most commonly in dependent parts of the body, notably the legs; indeed, it is not unusual for mild degrees of venous oedema to occur at the ankles and feet of normal people who have sat in aircraft on long flights—immobilisation impairs venous return. The fluid in venous oedema has a low protein content.
Oedema of just one leg is almost always due to venous obstruction by a thrombus. This is a common complication of immobilisation following major surgery or trauma. Bilateral leg oedema, if due to venous causes (there may be other explanations, see below), is more likely to be due to heart failure than venous thrombotic obstruction. In either case it is a serious manifestation prompting immediate attention to the underlying condition.
Lymphatic oedema
Some fluid normally leaves capillary beds and drains into adjacent lymphatic channels to return eventually to the circulation through the thoracic duct. If the lymphatic channels are obstructed, the fluid remains trapped in the tissues and oedema results.
Causes of lymphatic oedema include blockage of lymphatic flow by filarial parasites (Ch. 3) or by tumour metastases (Ch. 11), or as a complication of surgical removal of lymph nodes. Blockage of inguinal lymphatics by filarial parasites frequently causes gross oedema of the legs and, in males, the scrotum; the resulting deformity is called elephantiasis. Blockage of lymphatic drainage from the small intestine, usually because of tumour involvement, causes malabsorption of fats and fat-soluble substances. Blockage of lymphatic drainage at the level of the thoracic duct, or at least close to it, causes chylous effusions in the pleural and peritoneal cavities; the fluid is densely opalescent due to the presence of numerous tiny fat globules (chyle).
Fortunately, oedema due to surgical removal of lymph nodes is now a rare event. It used to be a complication of radical mastectomy for breast cancer, but surgical treatment for this tumour now tends to be more conservative.
Hypoalbuminaemic oedema
A low plasma albumin concentration results in oedema because of the reduction in plasma oncotic pressure; thus, fluid cannot be drawn back into the venous end of capillary beds and it remains in the tissues. Causes of hypo-albuminaemia are:
Hypoalbuminaemia as the cause of oedema can be verified easily by measuring the albumin concentration in serum. The underlying cause is then investigated and, if possible, treated. Infusions of albumin will have a beneficial, but temporary, effect.
Ascites and pleural effusions
Ascites is an excess of fluid in the peritoneal cavity. It is one of the five general causes of a distended abdomen: the complete alliterative list is—fluid, fat, faeces (constipation or obstruction), fetus, flatus (gas in the bowel).
Ascites and pleural effusions may be due to any of the above causes of oedema. However, the increased vascular permeability causing inflammatory oedema and effusions may also be induced by tumours. Thus, tumour cells growing within the cavities or on their serous linings cause excessive leakage of fluid. Serous effusions may be a presenting feature of cancer or they may complicate a previously diagnosed case. The fluid has a high protein content, and cytological examination to look for abnormal cells is often diagnostic.
Serous effusions may be divided into transudates and exudates by their protein content. Transudates have a protein concentration of less than 2g/100ml, whereas the concentration in exudates is higher. Involvement by tumour is the most important cause of an exudate.
Electrolyte homeostasis
Of all the electrolytes in plasma, sodium and potassium are among the most abundant and the most likely to be affected by pathological processes (Table 7.2).
Table 7.2 Common abnormalities of serum electrolytes
| Abnormality | Causes | Consequences |
|---|---|---|
| Hypernatraemia (i.e. high sodium) |
Compensatory increased blood volume OedemaHyponatraemia (i.e. low sodium)
Reduced blood volume HypotensionHyperkalaemia (i.e. high potassium)
Risk of cardiac arrestHypokalaemia (i.e. low potassium)
The abnormalities listed often do not occur in isolation and may be associated with other electrolyte changes.
Sodium and potassium homeostasis
 Sodium may be retained excessively by the body due to inappropriately high levels of mineralocorticoid hormones acting on renal tubular reabsorption
Sodium may be retained excessively by the body due to inappropriately high levels of mineralocorticoid hormones acting on renal tubular reabsorption



Hypernatraemia
Hypernatraemia (high serum sodium) may occur in conditions in which there is excessive mineralocorticoid (such as aldosterone) production acting on renal tubular reabsorption; Conn’s syndrome, due to an adrenal adenoma of the zona glomerulosa cells, is a typical example. The increased total body sodium content may be concealed by a commensurate increase in body water content in an attempt to sustain a normal plasma osmolarity; the serum sodium concentration may therefore underestimate the increase in total body sodium.
Hyponatraemia
Hyponatraemia (low serum sodium) is a logical consequence of impaired renal tubular reabsorption of sodium. This occurs in Addison’s disease of the adrenal glands due to loss of the aldosterone-producing zona glomerulosa cortical cells. Sodium is the electrolyte most likely to be lost selectively in severe sweating in hot climates or during physical exertion such as marathon running; the syndrome of ‘heat exhaustion’ is due mainly to a combination of dehydration and hyponatraemia. Falsely low serum sodium concentrations may be found in hyperlipidaemic states; the sodium concentration in the aqueous phase of the serum is actually normal but the lipid contributes to the total volume of serum assayed.
Hyperkalaemia
Potassium is more abundant within cells than in extracellular fluids, so relatively small changes in plasma concentration can underestimate possibly larger changes in intracellular concentrations. Furthermore, extensive tissue necrosis can liberate large quantities of potassium into the plasma, causing the concentration to reach dangerously high levels. The commonest cause is renal failure causing decreased urinary potassium excretion. Severe hyperkalaemia (>c. 6.5mmol/l) is a serious medical emergency demanding prompt treatment because of the risk of cardiac arrest. Moderate hyperkalaemia is relatively asymptomatic, emphasising the importance of regular biochemical monitoring to avoid sudden fatal complications.
Hypokalaemia
Hypokalaemia (low serum potassium) has many causes (Table 7.2). It is often accompanied by a metabolic alkalosis due to hydrogen ion shift into the intracellular compartment. Clinically, it presents with muscular weakness and cardiac dysrhythmias.
Vomiting and diarrhoea result in combined loss of water, sodium and potassium. Superimposed on this may be alkalosis from vomiting due to loss of hydrogen ions, or acidosis from diarrhoea due to loss of alkaline intestinal secretions.
Calcium homeostasis



Serum calcium levels are regulated by the vitamin D metabolite—1,25-dihydroxyvitamin D—and by parathyroid hormone (PTH). The precise role of calcitonin in humans is uncertain, but it has a serum calcium-lowering effect when administered to patients with hypercalcaemia; however, patients with the calcitonin-producing medullary carcinoma of the thyroid (Ch. 17) do not present with hypocalcaemia.
Hypercalcaemia
Acute hypercalcaemia causes fits, vomiting and polyuria. Persistent hypercalcaemia additionally results in ‘metastatic’ calcification (see p. 141) of tissues and urinary calculi. Causes of hypercalcaemia include:
Primary hyperparathyroidism is most commonly due to an adenoma of the parathyroid glands. The excessive and uncontrolled PTH secretion enhances the absorption of calcium and the osteoclastic erosion of bone, thus releasing calcium.
Hypercalcaemia due to neoplasms of other organs is seen most commonly with breast cancer. In the absence of extensive skeletal metastases, this is attributed to a PTH-like hormone secreted by the tumour cells.
Hypocalcaemia
Hypocalcaemia causes neuromuscular hypersensitivity manifested by tetany. This condition can be corrected rapidly by giving calcium gluconate intravenously. The commonest cause of acute hypocalcaemia is accidental damage to or removal of parathyroid glands during thyroid surgery. Low serum calcium levels resulting from renal disease or intestinal malabsorption are rapidly corrected, in a patient with intact parathyroid glands, by stimulation of PTH secretion. This eventually causes hyperplasia of the parathyroid glands (secondary hyperparathyroidism) and weakening of the skeleton due to excessive osteoclastic resorption under the influence of PTH.
Tetany also results from respiratory alkalosis, often in patients with hysterical hyperventilation who excessively eliminate carbon dioxide, due to a reduction in the ionised calcium concentration as the pH rises.
Acid–base homeostasis




Metabolic pathways are intolerant of pH deviations. The extracellular pH is tightly controlled at an approximate value of 7.4, but the intracellular pH is marginally lower and varies within an even narrower range. Acidic deviation outside the normal plasma pH range is sensed by chemoreceptors at the carotid bifurcations (carotid bodies), in the aortic arch and in the medulla of the brain.
The body has an innate tendency towards acidification due to production of:
This acidic tendency is counteracted by basic (alkaline) buffers (bicarbonate, proteins) in the first instance, but these have limited capacity. Acid–base balance in the plasma is ultimately regulated by:
Acidosis and alkalosis
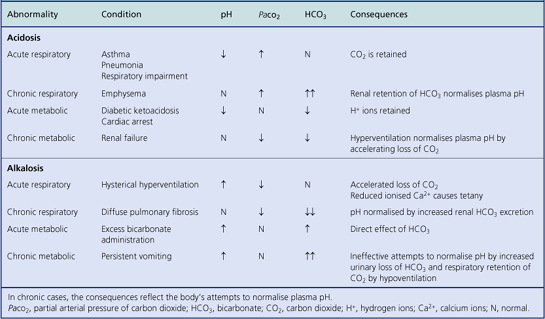
Deviations outside the normal pH range are called acidosis (low pH) and alkalosis (high pH). Either deviation may be further classified as respiratory (due to insufficient or excessive elimination of carbon dioxide from the lungs) or metabolic (due to non-respiratory causes). Thus there are four possible combinations:
The causes of these abnormalities of acid–base balance are shown in Table 7.3. The role of normal respiration and respiratory tract diseases in influencing acid–base balance is discussed in Chapter 14.
Respiratory acidosis
Respiratory acidosis can be corrected by increased renal tubular reabsorption of bicarbonate ions (which are alkaline) or by increased urinary loss of hydrogen ions (which are acidic). By either mechanism, the pH is not corrected as promptly as it can be in metabolic acidosis by immediate stimulation of hyperventilation.
Metabolic acidosis
Metabolic acidosis stimulates hyperventilation, often with deep sighing respiratory excursions (Kussmaul respiration), in order to blow off carbon dioxide and thereby maintain the equilibrium of the bicarbonate/carbonic acid ratio, restoring the pH to neutrality.
METABOLIC CONSEQUENCES OF MALNUTRITION
Malnutrition, a serious medical and socio-economic problem, may be a consequence or a cause of disease. Diseases and conditions commonly complicated by malnutrition include:
This section concentrates on the clinicopathological consequences of malnutrition. Malnutrition may be:
Protein–energy malnutrition




Protein–energy malnutrition results from the frequent combination of insufficient protein, carbohydrate and fat in the diet. Carbohydrate and fat together account for approximately 90% of the energy content of a typical healthy diet. Protein alone cannot replace the necessary energy yield from fats and carbohydrates.
Protein–energy malnutrition frequently co-exists with infections. The infections may exacerbate the deficiency, thus exposing the malnourished state, or they may complicate the deficiency because of impaired body defence mechanisms. In children prolonged malnutrition leads to stunted development due to retardation of linear growth. A shorter period of malnutrition produces body wasting.
Malnutrition in children
Severe malnutrition in children results in two clinical conditions (Fig. 7.8):
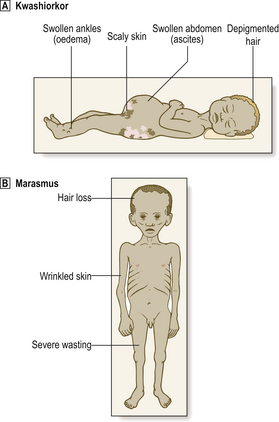
Fig. 7.8 Kwashiorkor and marasmus. Malnutrition in both cases leads to severe wasting.  Wasting is concealed to some extent in kwashiorkor by the oedema and ascites.
Wasting is concealed to some extent in kwashiorkor by the oedema and ascites.  Wasting is obvious in marasmus.
Wasting is obvious in marasmus.
The factors determining which condition will develop in a malnourished child remain uncertain; some cases show features of both conditions. These conditions often co-exist with infections, parasitic infestations and vitamin deficiencies.
Kwashiorkor
Kwashiorkor is characterised by oedema, which may be very extensive and so belie the extreme wasting of the underlying tissues. The skin is scaly and the hair loses its natural colour. The condition often develops when a child is weaned off breast milk, but without the compensation of adequate dietary protein.
The serum albumin is low and this accounts for the oedema due to reduced plasma oncotic pressure. Hypokalaemia and hyponatraemia are common. The liver is enlarged due to severe fatty change; this occurs because the lack of protein thwarts the production of lipoprotein and, therefore, transport of fat from the liver.
Cachexia
Cachexia is a state of severe debilitation associated with profound weight loss. It is seen in malnutrition (marasmus is akin to cachexia), but is most widely associated with the profound weight loss suffered by patients with cancer. When the tumour involves the gastrointestinal tract, the explanation for the cachexia is often obvious. However, weight loss can be a very early manifestation of any cancer and is a particularly common feature of carcinoma of the lung; in this instance, it may be due to factors causing increased protein catabolism as the patient’s food intake may be still within normal limits. Among several factors postulated to be responsible for the increased catabolic state in cachexia is tumour necrosis factor, a peptide secreted by tumour tissue.
Vitamin deficiencies


Deficiencies of vitamins—so named by Casimir Funk (1884–1967) because he believed (mistakenly) that they were all vital amines—produce more specific abnormalities (Table 7.4) than those encountered in protein–energy malnutrition. This is because of their involvement in specific metabolic pathways. Some vitamin deficiencies merit comment here, because either they are relatively frequent or the consequences are profound.
Table 7.4 Vitamin deficiency states
| Vitamin | Dietary sources | Consequence of deficiency |
|---|---|---|
| A | Beta-carotene in carrots, etc. Vitamin A in fish, eggs, liver, margarine | Night blindness, xerophthalmia, mucosal infections |
| B1 (thiamine) | Cereals, milk, eggs, fruit, yeast extract | Beri-beri, neuropathy, cardiac failure, Korsakoff’s psychosis, Wernicke’s encephalopathy |
| B2 (riboflavine) | Cereals, milk, eggs, fruit, liver | Mucosal fissuring |
| B6 (pyridoxine) | Cereals, meat, fish, milk | Confusion, glossitis, neuropathy, sideroblastic anaemia |
| B12 (cobalamin) | Meat, fish, eggs, cheese | Megaloblastic anaemia, subacute combined degeneration of the spinal cord |
| Niacin (nicotinic acid) | Meat, milk, eggs, peas, beans, yeast extract | Pellagra, dermatitis, diarrhoea, dementia |
| Folate | Green vegetables, fruit | Megaloblastic anaemia, mouth ulcers, villous atrophy of small gut |
| C (ascorbic acid) | Citrus fruits, green vegetables | Scurvy, lassitude, swollen bleeding gums, bruising and bleeding |
| D | Milk, fish, eggs, liver | Rickets (in childhood), osteomalacia (in adults) |
| E | Cereals, eggs, vegetable oils | Neuropathy, anaemia |
| K | Vegetables, liver | Blood coagulation defects |
Thiamine (B1) deficiency
Thiamine deficiency impairs glycolytic metabolism and affects the nervous system and the heart. The classic deficiency state is called beri-beri (from the Sinhalese word ‘beri’ meaning weakness). This state is characterised by peripheral neuropathy and, in some cases, cardiac failure.
Alcoholism is a common predisposing cause in countries such as the UK, where it is often associated with an inadequate diet. Alcoholics with thiamine deficiency can develop two central nervous system syndromes:
Folate and vitamin B12 deficiency
Folate and vitamin B12 (cobalamin) are essential for DNA synthesis. Deficiency of either impairs cellular regenera-tion; the effects are seen most severely in haemopoietic tissues, resulting in megaloblastic changes and macrocytic anaemia (Ch. 23). In addition, vitamin B12 deficiency also causes subacute combined degeneration of the spinal cord (Ch. 26).
Vitamin C deficiency
Vitamin C deficiency is now most common in elderly people and in chronic alcoholics, whose diet is often lacking in fresh fruit and vegetables. The vitamin (ascorbic acid) is essential principally for collagen synthesis: it is necessary for the production of chondroitin sulphate and hydroxyproline from proline. Minor degrees of deficiency may be responsible for lassitude and an unusual susceptibility to bruising. Severe deficiency causes scurvy, a condition characterised by swollen, bleeding gums, hyperkeratosis of hair follicles, and petechial skin haemorrhages.
Vitamin D deficiency
Vitamin D is derived either from the diet (milk, fish, etc.) as ergocalciferol (D2) or from the action of ultraviolet light on 7-dehydrocholesterol (D3) to form cholecalciferol in the skin. The intermediate precursors are activated by hydroxylation sequentially in the liver and kidneys to give 1,25-dihydroxy-cholecalciferol, a steroid hormone. Hydroxylation in the kidney is stimulated by parathyroid hormone and hypocalcaemia. An apparent deficiency can therefore result from:
People of races with deeply pigmented skin rely more heavily on dietary vitamin D when they migrate to countries with less sunlight than in their native lands.
Vitamin D is vital for normal calcium homeostasis. Its action resembles that of parathyroid hormone, ultimately causing elevation of the serum calcium concentration. It does so by:
In children, lack of vitamin D impairs mineralisation of the growing skeleton, thus causing rickets. In adults, vitamin D deficiency results in osteomalacia (Ch. 25). However, the pathogenesis of rickets and osteomalacia is identical; the two conditions are different clinical manifestations of vitamin D deficiency occurring at different stages of skeletal development.
Vitamin K deficiency
Vitamin K is essential for the synthesis of blood-clotting factors. It is involved in the carboxylation of glutamic acid residues on factors II, VII, IX and X. The principal dietary sources are vegetables, leguminous plants and liver. Deficiency may result from:
The commonest situation leading to dietary insufficiency is found in neonates on breast milk deficient in vitamin K.
Bruising and an abnormal bleeding tendency are the clinical manifestations of vitamin K deficiency. This occurs not only in the circumstances outlined above, but also in patients with liver failure in whom there is impaired hepatic synthesis of the vitamin K-dependent clotting factors; this can be corrected by giving large doses of vitamin K. It is essential to check the prothrombin time before performing a liver biopsy or any surgery on a patient with suspected liver disease.
OBESITY
Obesity is defined as a body mass index equal to or greater than 30. The body mass index is calculated by dividing the individual’s body weight in kilograms by the square of their height in metres (kg/m2). The cause of obesity is now cosidered to be multifactorial, resulting from an interaction between genetic and environmental factors, and is not regarded in all cases as being simply due to overeating. In a few cases, mutations of the leptin gene or the leptin receptor have been discovered.
Obesity significantly reduces life expectancy by increasing the risk of many serious pathological disorders, including:
For conditions treated surgically, there is also a substantially increased risk of serious post-operative complications, such as deep leg vein thrombosis and wound infections.
The prevalence of obesity is increasing rapidly, particularly in the USA and Europe, where it has been described as an ‘epidemic’ (Fig. 7.9). This has considerable implications for the health of the population and for the future demands on the health care system.
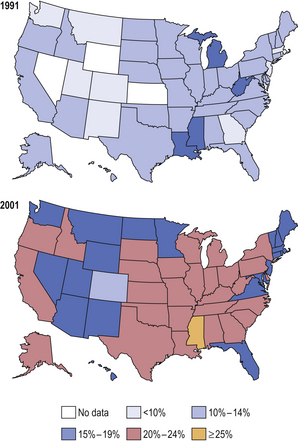
Fig. 7.9 Prevalence of obesity in the USA. Within just 10 years there was a massive increase in the prevalence of obesity (body mass index >30) in the USA. This predisposes affected individuals to an increased risk of serious disorders and a reduced life expectancy. (Data from Mokdad AH, Bowman BA, Ford ES et al 2001 The continuing epidemics of obesity and diabetes in the United States. JAMA 286: 1195–1200.)
Metabolic syndrome
Although the metabolic syndrome (also called syndrome X and insulin resistance syndrome) was first recognised in the early 1900s, marked rises in obesity and type 2 diabetes—common features of the syndrome—have greatly increased its prevalence and importance. In the USA, for example, over 40% of those >60 years of age are affected.
The diagnostic criteria for the metabolic syndrome are still debated, but commonly cited features are:
This cluster of features of the metabolic syndrome has been called the ‘deadly quartet’.
The metabolic syndrome is associated with an increased risk of cardiovascular disease, principally atheroma and its complications, and of type 2 diabetes (in those who have not already developed it as one of the diagnostic criteria for the syndrome). However, it should be noted that several features of the syndrome also independently increase the risk and severity of atheroma, so the precise extent of the morbidity and mortality due to the syndrome itself is difficult to quantify.
TRACE ELEMENTS AND DISEASE
Trace elements are those present at an arbitrarily defined low concentration in a given situation. Some trace elements in humans are of vital importance, despite the meagre quantities found in the human body. Trace elements cause disease when the body levels are higher or lower than normal, depending on the specific biological effects of the element.
Many elements, such as iron, cannot be regarded as trace elements because of their abundance in the body; nevertheless, diseases can result from either a deficiency or an excess (anaemia and haemosiderosis respectively in the case of iron).
Diseases associated with trace element abnormalities are summarised in Table 7.5. Some examples of well-documented associations of disease and trace elements will be summarised.
Table 7.5 Trace elements and disease
| Element | Abnormality | Consequences |
|---|---|---|
| Aluminium | Excess |
CobaltExcessCardiomyopathyCopperExcess
IodineDeficiencyGoitreLeadExcess
MercuryExcessNeuropathySeleniumDeficiency
ZincDeficiency
Aluminium
Aluminium is one of the most abundant elements in the Earth’s crust, but only traces are found in the normal human body. Toxic quantities can enter the body in a variety of ways. Aluminium is present in variable concentrations in water supplies and it is used therapeutically in the form of aluminium hydroxide as an antacid. Aluminium is also used in some cooking utensils, from which it can be leached under acid conditions. Aluminium powder has also been used for the treatment of pneumoconiosis, a chronic lung disorder resulting from the inhalation of toxic or allergenic dusts (Ch. 14).
Aluminium has been incriminated in the development of skeletal abnormalities and encephalopathy in patients on regular haemodialysis for chronic renal failure. In such cases, aluminium has been found deposited on mineralisation fronts in the skeleton, where it may interfere with bone turnover. Dialysis encephalopathy, first reported in 1972, is characterised by progressive dementia, epileptic fits and tremors. In 1976, dialysis encephalopathy was shown to be associated with an abnormally high aluminium concentration in brain tissue obtained from autopsies on affected patients. This finding then led to discovery of a link between aluminium and dialysis bone disease.
Aluminium is often detectable in the brain lesions in Alzheimer’s disease, a relatively common neurodegenerative disorder, but evidence that it is an aetiological factor is very weak.
Copper
Copper is essential for the function of several enzymes (e.g. superoxide dismutase). Copper deficiency appears to be rare. Some people with arthritis claim to derive benefit, undoubtedly psychological, from wearing copper bracelets, although the only observable change is a green discoloration of the underlying skin.
Wilson’s disease is the most important disorder of copper metabolism. This is inherited as an autosomal recessive condition in which copper accumulates in the liver (Fig. 7.10), basal ganglia of the brain, kidneys and eyes. The brown ring of copper deposition around the corneal limbus—the Kayser–Fleischer ring—is absolutely diagnostic. Serum caeruloplasmin levels are usually low. In the liver, the copper accumulation is associated with chronic hepatitis, frequently culminating in cirrhosis (Ch. 16). The neurological changes are seriously disabling. Although Wilson’s disease is rare, it is absolutely vital to consider the diagnosis in any patient presenting with chronic liver disease and neurological signs. d-Penicillamine, a chelating agent, has revolutionised the treatment of Wilson’s disease, but it is to little avail if the liver and brain have already been irreversibly damaged.
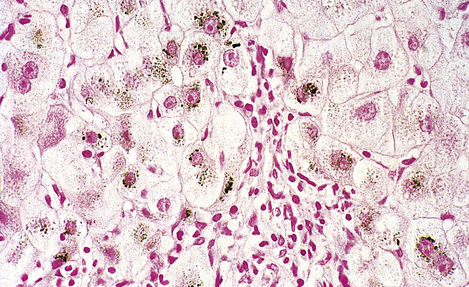
Fig. 7.10 Copper in liver. Liver biopsy, stained for copper (dark granules), showing excessive copper in periportal liver cells. No stainable copper would be present in a normal liver. Copper accumulates in the liver in Wilson’s disease and in patients with chronic obstructive jaundice (e.g. primary biliary cirrhosis).
Iodine
The human body contains only 15–20 mg of iodine, most of which is in the thyroid gland. Iodine is almost unique among elements in having just one known role in the human body: it is essential for the synthesis of thyroxine.
Ingestion of modestly excessive quantities of iodine (as potassium iodide, for example) has no serious adverse consequences. Indeed, large stocks of potassium iodide tablets are kept in the vicinity of nuclear power stations for use in the event of accidental release of radioactive iodine, a cause of thyroid cancer. The potassium iodide competes with the smaller amounts of radioactive iodine for uptake by the thyroid gland.
Iodine deficiency results in goitre (enlargement of the thyroid gland, Ch. 17). Goitre was prevalent in regions where the water and solid food lacked an adequate iodine content, usually in mountainous regions (hence, for example, ‘Derbyshire neck’). Maternal iodine deficiency during pregnancy causes cretinism in neonates, characterised by mental retardation and stunted growth. These problems have been eliminated in many countries by the addition of iodides and iodates to table salt.
Lead
Much effort is being made in many countries to reduce environmental contamination by lead. The human body contains approximately 120mg of lead and the daily intake should not exceed 500 μg. Excessive ingestion or inhalation can result from contaminated food, water or air; the main sources in the UK appear to be old lead piping in water supplies, and tetra-ethyl and tetra-methyl lead added to petrol as anti-knocking agents. Old plumbing is gradually being replaced and the use of unleaded petrol now common.
Toxic effects of lead include central and peripheral nervous system damage, renal damage and sideroblastic anaemia (Ch. 23). It has been alleged that lead exposure may be responsible for mental retardation in children, but it has been difficult to dissociate this from the other consequences of socio-economic deprivation prevalent in the urban environments contaminated with lead.
Mercury
The average human body contains only 13 mg of mercury. The safe daily intake is <50 μg.
Mercury has been used in dental amalgams for filling tooth cavities since 1818. Although doubts have been expressed about its safety, metallic mercury and mercury-containing dental amalgams are insoluble in saliva and are, therefore, not absorbed to an appreciable extent. Dentists must, of course, use mercury cautiously to minimise the risk of cumulative occupational exposure.
Mercury is neurotoxic. Chronic poisoning also results in a characteristic blue line on the gums. Perhaps the best-known (but fictitious) case is that of the Mad Hatter in Alice in Wonderland; hatmakers used mercuric nitrate for making felt out of animal fur! In the 1950s at Minamata, Japan, there was serious water pollution with methyl mercury, causing at least 50 deaths and many more cases of permanent neurological disability.
Despite its known toxicity, mercury has been used therapeutically, though not to any great effect. It was a popular, though ineffectual, remedy for syphilis; this gave rise to the heavenly adage ‘A night with Venus; a lifetime on Mercury’! More recently, pharmaceutical preparations containing mercury were advocated for treating childhood ailments such as measles, teething and diarrhoea. One such preparation containing calomel (mercurous chloride) was sold as a teething powder. Many years later this was suggested—and eventually proven—to be the cause of ‘pink disease’, a distressing condition affecting infants and young children, formerly of unknown aetiology.
TISSUE DEPOSITIONS
Tissues can become altered as a result of deposition of excessive quantities of substances present normally in only small amounts. These include haemosiderin, as in haemochromatosis (Ch. 16), lysosomal storage disorders (see p. 125), and lipofuscin, which accumulates particularly in the liver with ageing (Ch. 12). Pathological calcification and amyloid deposition are detailed below.
Calcification




Although calcium ions are vital for the normal function of all cells, precipitates of calcium salts are normally found only in bones, otoliths and teeth. In disease states, however, tissues can become hardened by deposits of calcium salts; this process is called calcification. Calcification may be:
‘Metastatic’ calcification must not be confused with the process of metastasis of tumours. It is an entirely separate condition. In the context of calcification, ‘metastatic’ only means widespread.
Dystrophic calcification
Calcification is said to be dystrophic if it occurs in tissue already affected by disease. In these cases the serum calcium is normal. The calcification is due to local precipitation of insoluble calcium salts. Common examples are:
The calcified lesions will often be detectable on a plain X-ray as opacities or, if detected at surgery, will feel extremely hard. Dystrophic calcification does not usually have any special consequences for the patient, with the notable exception of calcification of a congenitally bicuspid aortic valve. A bicuspid aortic valve can function quite normally, but when it becomes calcified, a common event in the elderly, the valve cusps become thick and rigid; this causes stenosis, incompetence and, ultimately, cardiac failure (Ch. 13). The biochemical basis of dystrophic calcification is uncertain except in the instance of fat necrosis, a common result of trauma to adipose tissue or of acute pancreatitis (Ch. 16); the liberated fatty acids bind calcium to form insoluble calcium soaps, sometimes causing hypocalcaemia and tetany.
The presence of dystrophic calcification in breast lesions, particularly some carcinomas, is one of the abnormalities looked for by radiologists in the interpretation of mammograms (X-rays of the breasts) when screening for breast cancer.
A few tumours contain minute concentric lamellated calcified bodies. These are called psammoma bodies (‘psammos’ is Greek for sand) and are most commonly found in:
Psammoma bodies assist the histopathologist in correctly identifying the type of tumour, but their pathogenesis is unknown.
‘Metastatic’ calcification
Metastatic calcification is much less common than dystrophic calcification and occurs as a result of hypercalcaemia. Calcification may be widespread and occurs in otherwise normal tissues. Frequent causes are:
In hyperparathyroidism, an adenoma or, less often, a diffuse hyperplasia of the parathyroid glands secretes excess quantities of parathyroid hormone; this liberates calcium from the bone, resulting in hypercalcaemia. In some patients with malignant neoplasms, hypercalcaemia results from either the secretion of a parathyroid hormone-like substance or extensive bone erosion due to skeletal metastases.
In this condition the calcium salts are precipitated on to connective tissue fibres (e.g. collagen, elastin; Fig. 7.11).
Amyloid

 Composed of immunoglobulin light chains, serum amyloid protein A, peptide hormones, prealbumin, etc.
Composed of immunoglobulin light chains, serum amyloid protein A, peptide hormones, prealbumin, etc.



Amyloid (meaning starch-like from the Greek ‘amylon’) is the name given to a group of proteins or glycoproteins that, when deposited in tissues, share the following properties:
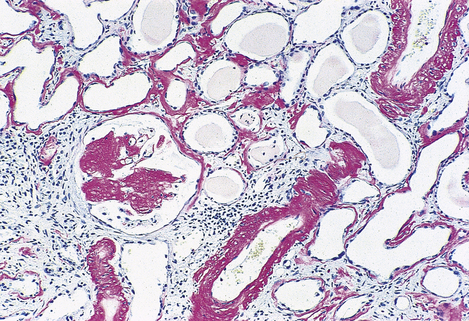
Fig. 7.12 Renal amyloidosis. Renal biopsy stained to show amyloid (red). The amyloid is deposited in the glomeruli, blood vessel walls and tubular basement membranes.
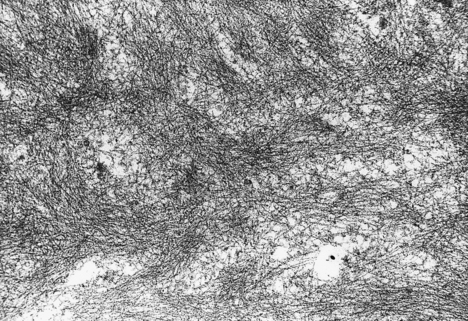
Fig. 7.13 Amyloid ultrastructure. Amyloid substances are characterised by a fibrillar appearance on electron microscopy.
Small asymptomatic deposits of amyloid are not uncommon in the spleen, brain, heart and joints of elderly people.
The beta-pleated molecular configuration is an important feature because the body has no enzymes capable of digesting large molecules in this form, so they remain permanently in the tissues.
Classification
Amyloid can be classified according to:
These are all equally legitimate and complementary methods of classification. The chemical composition often correlates with the clinical classification (Table 7.6); it can, therefore, be helpful diagnostically and lead to the discovery of the aetiology in an individual case.
Table 7.6 Classification of amyloid substances
| Condition | Amyloid substance |
|---|---|
| Myeloma-associated (primary) | AL (immunoglobulin light chains or fragments) |
| Reactive (secondary) | AA (serum amyloid protein A, an acute-phase reactant) |
| Alzheimer’s disease | A-beta (derived from amyloid precursor protein) |
| Haemodialysis-associated | A-beta-2M (beta-2 microglobulin) |
ATTR (transthyretin)Familial Mediterranean feverAAFinnish amyloidosisAGel (gelsolin)Medullary carcinoma of thyroidAMCT (calcitonin)
In addition to those amyloid substances listed, all amyloid deposits also contain amyloid P glycoprotein as a common constituent.
Clinically, however, amyloidosis presents with organ involvement which is either:
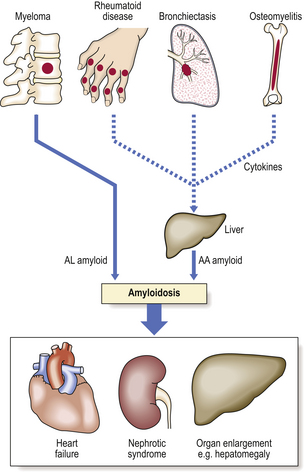
Fig. 7.14 Common causes and consequences of systemic amyloidosis. In primary or myeloma-associated amyloidosis the AL amyloid comprises light chains secreted by neoplastic plasma cells. In reactive or secondary amyloidosis the production of AA amyloid by the liver is stimulated by cytokines secreted by chronic inflammatory cells.
Systemic amyloidosis
In systemic amyloidosis the material is deposited in a wide variety of organs; virtually no organ is exempt. Clinical features suggesting amyloidosis include generalised diffuse organ enlargement (e.g. hepatomegaly, splenomegaly, macroglossia) and evidence of organ dysfunction (e.g. heart failure, proteinuria).
Systemic amyloidosis is further classified according to its aetiology:
Myeloma-associated amyloidosis
The amyloid substance in myeloma-associated amyloidosis is AL amyloid—immunoglobulin light chains.
A myeloma is a plasma cell tumour, often multiple, arising in bone marrow and causing extensive bone erosion. It produces excessive quantities of immunoglobulin of a single class (e.g. IgG) with a uniform light chain (e.g. kappa). The light chain forms the amyloid material. The amyloid is deposited in many organs—heart, liver, kidneys, spleen, etc.—but shows a predilection for the connective tissues within these organs.
In some cases, myeloma-associated amyloidosis is called primary amyloidosis because of the absence of any clinically obvious myeloma. However, invariably there is a clinically occult plasma cell tumour, with little bone erosion to declare itself, but with a monoclonal immunoglobulin band on serum electrophoresis; this is referred to as a benign monoclonal gammopathy.
Amyloidosis is a serious complication of myeloma, exacerbating the ill health of the patient.
Reactive (secondary) amyloidosis
The amyloid substance in reactive or secondary amyloidosis is AA amyloid, derived from serum amyloid protein A.
Serum amyloid protein A, synthesised in the liver, is an acute phase reactant protein, one of several so called because the serum concentrations rise in response to the presence of a variety of diseases.
Reactive amyloidosis, by definition, always has a predisposing cause; this is invariably a chronic inflammatory disorder. Chronic inflammatory disorders frequently predisposing to secondary amyloidosis are:
The amyloid in reactive amyloidosis shows the same tendency to widespread deposition as in myeloma-associated amyloidosis, but it has a predilection for the liver, spleen and kidneys (Fig. 7.14).
Senile amyloidosis
Minute deposits of amyloid, usually derived from serum prealbumin, may be found in the heart and in the walls of blood vessels in many organs of elderly people. However, only in a few cases do they result in significant signs or symptoms.
Haemodialysis-associated amyloidosis
The association of amyloidosis with long-term haemodialysis for chronic renal failure has been recognised only recently. The clinical manifestations include arthropathy and carpal tunnel syndrome. In a few cases there is much more extensive involvement of other organs. The amyloid material deposited in the affected tissues appears to be beta-2 microglobulin.
Localised amyloidosis
Amyloid material is often found in the stroma of tumours producing peptide hormones. It is particularly characteristic of medullary carcinoma of the thyroid, a tumour of the calcitonin-producing interfollicular C-cells. In this instance, the amyloid contains calcitonin molecules arranged in a beta-pleated sheet configuration.
Localised deposits of amyloid may be found, without any obvious predisposing cause, in virtually any organ; this is, however, a rare occurrence. The skin, lungs and urinary tract seem to be the most frequent sites.
Cerebral amyloid is found in Alzheimer’s disease (see Ch. 26) and in the brains of elderly people in:
The amyloid in plaques in Alzheimer’s disease comprises A-beta protein complexed with apolipoprotein E (apoE). The latter occurs in several allelic variants, of which apoE4 is a risk factor for Alzheimer’s disease.
Clinical effects and diagnosis
The clinical manifestations of amyloidosis are:
Amyloidosis may be suspected on clinical examination because of enlargement of various organs, especially the liver and spleen. As the kidneys are often involved and the amyloid is deposited in glomerular basement membranes, altering their filtration properties, the patients often have proteinuria; in severe cases the proteinuria can result in nephrotic syndrome (Ch. 21). The diagnosis is best confirmed by biopsy of the rectal mucosa, commonly involved in cases of systemic amyloidosis; this procedure is relatively safe and painless. The amyloid in the biopsy can be stained histologically using Congo red or Sirius red dyes, or immunohistochemically using specific antibodies. When examined using one fixed and one rotating polarising filter in the light path on either side of the section, the red colour changes to green (dichroism); this simple optical test is quite specific for amyloid. Using special techniques it may be possible to characterise the amyloid substance more precisely to determine its origin and to identify thereby the underlying cause.
Localised amyloid in a tumour is of no clinical consequence other than serving to assist the histopathologist in correctly identifying the tumour as, for example, a medullary carcinoma of the thyroid.
A solitary amyloid deposit is of clinical significance either because it mimics a tumour (e.g. on a plain chest X-ray) or because it compresses a vital structure (e.g. a ureter).
Commonly confused conditions and entities relating to disorders of metabolism and homeostasis
| Commonly confused | Distinction and explanation |
|---|---|
| Cystine, cysteine, homocysteine and homocystinuria | Both cystine and cysteine are sulphur-containing amino acids: one molecule of cystine can be reduced to two molecules of cysteine. Homocysteine is an intermediate in the synthesis of cysteine; high blood levels are found in homocystinuria. |
| Tetany and tetanus | Tetany is muscular spasm induced by hypocalcaemia, either an absolute reduction in serum calcium or, as in respiratory alkalosis (e.g. hysterical hyperventilation), a reduction in the amount of ionised calcium. Tetanus is the disease resulting from infection by Clostridium tetani which produces a toxin causing muscular spasm. |
| Kwashiorkor and marasmus | Marasmus is severe wasting due to protein–energy malnutrition. In kwashiorkor the wasting is somewhat concealed by oedema of the tissues. |
| Dystrophic and ‘metastatic’ calcification | Calcification of diseased tissues (e.g. atheromatous plaques, old tuberculous lesions) is called dystrophic. Calcification of previously normal tissues in a patient with hypercalcaemia is often said to be ‘metastatic’, but this should not be confused with the process of tumour metastasis. |
| Primary and secondary amyloidosis | In primary amyloidosis the amyloid deposits contain immunoglobulin light chains; although there may be no underlying cause (hence ‘primary’), the light chains probably originate from a neoplastic clonal proliferation of plasma cells. In secondary amyloidosis the amyloid comprises serum amyloid protein A produced by the liver in response to cytokines from chronically inflamed tissues. |
Daneman D.. Type 1 diabetes. Lancet. 2006;367:847-858.
Eckel R.H., Grundy S.M., Zimmet P.Z.. The metabolic syndrome. Lancet. 2005;365:1415-1428.
Kass R.S.. The channelopathies: novel insights into molecular and genetic mechanisms of human disease. Journal of Clinical Investigation. 2005;115:1986-1989.
Kellum J.A.. Determinants of blood pH in health and disease. Critical Care. 2000;4:6-14.
Kraut J.A., Madias N.E.. Approach to patients with acid–base disorders. Respiratory Care. 2001;46:392-403.
Labib M.. The investigation and management of obesity. Journal of Clinical Pathology. 2003;56:17-25.
Lenihan J.. The crumbs of creation: trace elements in history, medicine, industry, crime and folklore. Bristol: Adam Hilger; 1988.
Marshall W.J.. Clinical chemistry. Edinburgh: Mosby; 2000.
Nyhan W.L., Barshop B.A., Ozand P.T. Atlas of metabolic diseases. Oxford: Oxford University Press, 2005
Pepys M.B.. Amyloidosis. Annual Review of Medicine. 2006;57:223-241.
Ratjen F., Doring G.. Cystic fibrosis. Lancet. 2003;361:681-689.
Scriver’s Online Metabolic & Molecular Bases of Inherited Disease. http://www.ommbid.com/
Wong L.L., Verbalis J.G.. Systemic diseases associated with disorders of water homeostasis. Endocrinology and Metabolism Clinics of North America. 2002;31:121-140.
[/level-membership-for-pathology-category][not-level-membership-for-pathology-category]
Chapter 7 Disorders of metabolism and homeostasis
Some metabolic disorders, congenital or acquired, are specific abnormalities of metabolic pathways, often having considerable clinical effects. Congenital metabolic disorders usually result from inherited enzyme deficiencies.
Other metabolic disorders are characterised by perturbations of the body’s homeostatic mechanisms maintaining the integrity of fluids and tissues. These conditions are almost always acquired and their effects can be diverse.
INBORN ERRORS OF METABOLISM
The concept of inborn errors of metabolism was formulated by Sir Archibald Garrod in 1908 as a result of his studies on a condition called alkaptonuria, a rare inherited deficiency of homogentisic acid oxidase.
Inborn (usually inherited) errors of metabolism are important causes of illness presenting in infancy. Some require prompt treatment to avoid serious complications. Others defy treatment. All deserve accurate diagnosis so that parents can be counselled about the inherited risk to further pregnancies. Inborn metabolic errors are potentially chronic problems, because the primary abnormality is innate rather than due to any external cause that could be eliminated by treatment.
Inborn errors of metabolism are single-gene defects resulting in the absence or deficiency of an enzyme or the synthesis of a defective protein. Single-gene defects occur in about 1% of all births, but the diseases caused by them show geographic variations in incidence; this is exemplified by the high incidence of thalassaemias—due to defects in haemoglobin synthesis (Ch. 23)—in Mediterranean regions. These variations reflect the prevalence of specific abnormal genes in different populations.
Inborn errors of metabolism have four possible consequences:
Accumulation of an intermediate metabolite may have toxic or hormonal effects. However, in some conditions the intermediate metabolite accumulates within the cells in which it has been synthesised, causing them to enlarge and compromising their function or that of neighbouring cells; these conditions are referred to as storage disorders (e.g. Gaucher’s disease). Other inborn metabolic errors lead to the production of a protein with defective function; for example, the substitution of just a single amino acid in a large protein can have considerable adverse effects (e.g. haemoglobinopathies).
The genetic basis of the inheritance of these disorders is discussed in Chapter 3.
Inherited metabolic disorders may be classified according to the principal biochemical defect (e.g. amino acid disorder) or the consequence (e.g. storage disorder).
Disorders of carbohydrate metabolism
The commonest disorder of carbohydrate metabolism with an inherited component in its aetiology is diabetes mellitus (see p. 128). Much less common, but with an autosomal recessive pattern of inheritance and often presenting at an early age, are:
Disorders of amino acid metabolism
Several inherited disorders of amino acid metabolism involve defects of enzymes in the phenylalanine/tyrosine pathway (Fig. 7.1).
Fig. 7.1 Inborn errors of metabolism in the phenylalanine/tyrosine pathway. 1. Phenylketonuria. Lack of phenylalanine hydroxylase blocks conversion of phenylalanine to tyrosine; phenylalanine and phenylpyruvic acid appear in the urine. 2. Alkaptonuria. Lack of homogentisic acid oxidase causes accumulation of homogentisic acid. 3. Albinism. Lack of the enzyme tyrosinase prevents conversion of tyrosine via DOPA to melanin. 4. Familial hypothyroidism. Deficiency of any one of several enzymes impairs iodination of tyrosine in the formation of thyroid hormone.
Phenylketonuria
This autosomal recessive disorder affects approximately 1 in 10000 infants. It is due to a deficiency of phenylalanine hydroxylase, an enzyme responsible for the conversion of phenylalanine to tyrosine (Fig. 7.1).
In the UK and many other countries the clinical effects of phenylketonuria are now seen only very rarely, because it is detected by screening all newborn infants and treated promptly. Testing is done by analysing a drop of blood, dried on to filter paper, for phenylalanine (Guthrie test). If phenylketonuria is not tested for in this way and the affected infant’s diet contains usual amounts of phenylalanine, then the disorder manifests itself with skin and hair depigmentation, fits and mental retardation. Treatment involves a low phenylalanine diet until the child is at least 8 years old. When affected females themselves become pregnant, the special diet must be resumed to avoid the toxic metabolites damaging the developing fetus.
Alkaptonuria
This rare autosomal recessive deficiency of homogentisic acid oxidase (Fig. 7.1) is a good example of an inborn metabolic error that does not produce serious effects until adult life. The condition is sometimes recognised from the observation that the patient’s urine darkens on standing; the sweat may also be black! Homogentisic acid accumulates in connective tissues, principally cartilage, where the darkening is called ochronosis. This accumulation causes joint damage. The underlying condition cannot be cured; treatment is symptomatic only.
Homocystinuria
Like most inherited disorders of metabolism, homocystinuria is an autosomal recessive disorder. There is a deficiency of cystathionine synthase, an enzyme required for the conversion of homocystine via homocysteine to cystathionine. Homocysteine and methionine, its precursor, accumulate in the blood. Homocystine also accumulates, interfering with the cross-linking of collagen and elastic fibres. The ultimate effect resembles Marfan’s syndrome (see p. 128), but also with mental retardation and fits.
Storage disorders
Inborn metabolic defects result in storage disorders if a deficiency of an enzyme, usually lysosomal, prevents the normal conversion of a macromolecule (e.g. glycogen or gangliosides) into its smaller subunits (e.g. glucose or fatty acids). The macromolecule accumulates within the cells that normally harbour it, swelling their cytoplasm (Fig. 7.2) and causing organ enlargement and deformities. This situation is harmful to the patient because the swelling of cells often impairs their function, or that of their immediate neighbours due to pressure effects, and because of conditions resulting from deficiency of the smaller subunits (e.g. hypoglycaemia in the case of glycogen storage disorders).
Fig. 7.2 Bone marrow biopsy revealing Gaucher’s disease. Pale foamy macrophages distended with gangliosides have displaced much of the haemopoietic tissue (top left), thereby causing anaemia.
Major categories of these autosomal recessive disorders (Table 7.1) are:
Table 7.1 Examples of inborn errors of metabolism resulting in storage disorders
| Type of disease/examples | Deficiency | Consequences |
|---|---|---|
| Glycogenoses | Debranching enzyme |
McCardle’s syndromeMuscle phosphorylase von Gierke’s diseaseGlucose-6-phosphate dehydrogenase Pompe’s diseaseAcid maltase MucopolysaccharidosesLysosomal hydrolase
Hurler’s syndromeAlpha-l-iduronidase Hunter’s syndromeIduronate sulphate sulphatase SphingolipidosesLysosomal enzyme
Gaucher’s diseaseGlucocerebrosidase Niemann–Pick diseaseSphingomyelinase Tay–Sachs diseaseHexosaminidase A
Disorders of cell membrane transport
Inborn metabolic errors can lead to impairment of the specific transport of substances across cell membranes. Examples include:
Channelopathies
A channelopathy is a disease caused by the dysfunction of a specific ion channel in cell membranes. The ion channel dysfunction may result from:
Cystic fibrosis
Cystic fibrosis, a channelopathy, is the commonest serious inherited metabolic disorder in the UK; it is much commoner in Caucasians than in other races. The autosomal recessive abnormal gene is carried by approximately 1 in 20 Caucasians and the condition affects approximately 1 in 2000 births. The defective gene, in which numerous mutations have been identified, is on chromosome 7 and ultimately results in abnormal water and electrolyte transport across cell membranes.
Cystic fibrosis transmembrane conductance regulator (CFTR)
The commonest abnormality (ΔF508) in the CFTR gene is a deletion resulting in a missing phenylalanine molecule. The defective CFTR is unresponsive to cyclic AMP control, so transport of chloride ions and water across epithelial cell membranes becomes impaired (Fig. 7.3).
Fig. 7.3 Defective chloride secretion in cystic fibrosis. The normal CFTR is a transmembrane molecule with intracytoplasmic nucleotide binding folds and a phosphorylation site on the R-domain. In normal cells, interaction of the R-domain with protein kinase A results in opening of the channel and chloride secretion. In cystic fibrosis, a common defect prevents phosphorylation of the R-domain with the result that chloride secretion is impaired.
Clinicopathological features
Cystic fibrosis is characterised by mucous secretions of abnormally high viscosity. The abnormal mucus plugs exocrine ducts, causing parenchymal damage to the affected organs. The clinical manifestations are:
Porphyrias
The porphyrias, transmitted as autosomal dominant disorders, are due to defective synthesis of haem, an iron–porphyrin complex, the oxygen-carrying moiety of haemoglobin. Haem is synthesised from 5-aminolaevulinic acid. The different types of porphyrin accumulate due to inherited defects in this synthetic pathway (Fig. 7.4).
Fig. 7.4 Porphyrias. Enzyme deficiencies in the pathway of synthesis of haem from glycine and succinyl coenzyme A through 5-aminolaevulinic acid result in the accumulation of toxic intermediate metabolites. Removal of product inhibition due to deficient synthesis of haem enhances the formation of intermediate metabolites. Accumulation of 5-aminolaevulinic acid or porphobilinogen tends to be associated with neurological damage and psychiatric symptoms. Accumulation of porphyrinogens, of which there are several types (uro-, copro-, proto-), tends to be associated with photosensitivity.
Clinicopathological features
Accumulation of porphyrins can cause clinical syndromes characterised by:
The pain and psychiatric disturbances are episodic. During the acute attacks, the patient’s urine contains excess 5-aminolaevulinic acid and porphobilinogen. Consequently, the urine may gradually become dark red, brown or even purple (‘porphyria’ is derived from the Greek word ‘porphura’ meaning purple pigment) on exposure to sunlight.
Acute attacks of porphyria can be precipitated by some drugs, alcohol and hormonal changes (e.g. during the menstrual cycle). The most frequently incriminated drugs include barbiturates, sulphonamides, oral contraceptives and anticonvulsants; these should therefore be avoided.
The skin lesions are characterised by severe blistering, exacerbated by light exposure, and subsequent scarring. This photosensitivity is a distressing feature, but it has led to the beneficial use of injected porphyrins in the treatment of tumours by phototherapy with laser light.
Disorders of connective tissue metabolism
Most inherited disorders of connective tissue metabolism affect collagen or elastic tissue. Examples include:
Osteogenesis imperfecta
Osteogenesis imperfecta is a group of disorders in which there is an inborn error of type I collagen synthesis (Ch. 25). Type I collagen is most abundant in bone, so the principal manifestation is skeletal weakness resulting in deformities and a susceptibility to fractures; the other names for this condition are ‘fragilitas ossium’ and ‘brittle bone disease’. The teeth are also affected and the sclerae of the eyes are abnormally thin, causing them to appear blue. It occurs in dominantly and recessively inherited forms with varying degrees of severity.
Marfan’s syndrome
Marfan’s syndrome is a combination of unusually tall stature, long arm span, dislocation of the lenses of the eyes, aortic and mitral valve incompetence, and weakness of the aortic media predisposing to dissecting aneurysms (Ch. 13). The condition results from a defect in the FBN1 gene encoding for fibrillin, a glycoprotein essential for the formation and integrity of elastic fibres.
ACQUIRED METABOLIC DISORDERS
Many diseases result in secondary metabolic abnormalities. In others the metabolic disturbance is the primary event. For example, renal diseases almost always result in metabolic changes that reflect the kidneys’ importance in water and electrolyte homeostasis. In contrast, a disease like gout is often due to a primary metabolic disorder that may secondarily damage the kidneys. This section deals with metabolic abnormalities as both consequences and causes of disease. Acquired metabolic disorders frequently cause systemic problems affecting many organs: for example, diabetes mellitus is associated with microvascular damage in the retinas, nerves, kidneys and other organs; electrolyte imbalance compromises the function of cells in all tissues.
Two of the disorders discussed in this section—diabetes mellitus and gout—are categorised as ‘acquired’ largely because they occur most commonly in adults, but both have a significant genetic component in their aetiology.
Diabetes mellitus
Diabetes mellitus is a group of diseases characterised by impaired glucose homeostasis resulting from a relative or absolute insufficiency of insulin. Insulin insufficiency causes hyperglycaemia and glycosuria. Diabetes is covered in Chapter 17, but a brief account is relevant here.
The aetiology is multifactorial; although the disorder is acquired, there is a significant genetic predisposition. Diabetes mellitus is subclassified into primary and secondary types. Primary diabetes is much more common than diabetes secondary to other diseases.
Primary diabetes mellitus
Primary diabetes mellitus (DM) is subdivided into:
Juvenile-onset diabetes, usually appearing before the age of 20 years, is almost always of IDDM type. There is an inherited predisposition associated with human leukocyte antigens (HLA) DR3 and DR4. The initiating event may be a viral infection of the insulin-producing beta-cells of the islets of Langerhans, precipitating their immune destruction.
Diabetes mellitus developing in adults (over 25 years of age) is most likely to be NIDDM type. It is associated with an acquired resistance to insulin. There is no HLA association, but there is a familial tendency to develop the disease. The affected patients are often, but not always, obese.
Complications of diabetes mellitus
Good control of blood sugar levels reduces the risk of complications. Nevertheless, many diabetics develop complications of their disease. These are covered in more detail in the relevant chapters, but a summary is given here:
To this list should be added hypoglycaemia, which is a frequent and troublesome complication of insulin therapy in IDDM.
There are two possible biochemical explanations for the tissue damage that results from long-term diabetes mellitus:
Gout
Gout is a common disorder resulting from high blood uric acid levels. Uric acid is a breakdown product of the body’s purine (nucleic acid) metabolism (Fig. 7.5), but a small proportion comes from the diet. Most uric acid is excreted by the kidneys. In the blood, most uric acid is in the form of monosodium urate. In patients with gout, the monosodium urate concentration may be very high, forming a supersaturated solution, thus risking urate crystal deposition in tissues causing:
Fig. 7.5 Pathogenesis of gout. The metabolic pathway shows the synthesis of uric acid from nucleic acids. Primary gout can arise from an inherited (X-linked) deficiency of hypoxanthine guanine phosphoribosyl transferase (HGPRT) or excessive activity of 5-phosphoribosyl-1-pyrophosphate (PRPP). Secondary gout results either from increased tissue lysis (e.g. due to tumour chemotherapy) liberating excess nucleic acids or from inhibition of the urinary excretion of uric acid. Xanthine oxidase (XO) is inhibited by allopurinol, an effective long-term remedy for gout.
Gout occurs more commonly in men than in women, and is rare before puberty. A rare form of gout in children—Lesch–Nyhan syndrome—is due to absence of the enzyme HGPRT (hypoxanthine guanine phosphoribosyl transferase) (Fig. 7.5) and is associated with mental deficiency and a bizarre tendency to self-mutilation.
Aetiology
Like diabetes mellitus, the aetiology of gout is multifactorial. There is a genetic component, but the role of other factors justifies the inclusion of gout under the heading of acquired disorders. Aetiological factors include:
Some of these factors may, of course, be interdependent. Accordingly, gout can be subdivided into primary gout, due to some genetic abnormality of purine metabolism, or secondary gout, due to increased liberation of nucleic acids from necrotic tissue or decreased urinary excretion of uric acid.
Clinicopathological features
The clinical features of gout are due to urate crystal deposition in various tissues (Fig. 7.6). In joints, a painful acute arthritis results from phagocytosis of the crystals by neutrophil polymorphs, in turn causing release of lysosomal enzymes along with the indigestible crystals, thus accelerating and perpetuating a cyclical inflammatory reaction. The first metatarsophalangeal joint is typically affected.
Water homeostasis
Water and electrolyte homeostasis is tightly controlled by various hormones, including antidiuretic hormone (ADH), aldosterone and atrial natriuretic peptide, acting upon selective reabsorption in the renal tubules (Ch. 21). The process is influenced by the dietary intake of water and electrolytes (in food or drinking in response to thirst or social purposes) and the adjustments necessary to cope with disease or adverse environmental conditions.
Many diseases result in problems of water and electrolyte homeostasis. Disturbances can also occur in post-operative patients receiving fluids and nutrition parenterally. Fortunately, any changes are fairly easy to monitor and control by making adjustments to the fluid and electrolyte intake.
Water is constantly lost from the body—in urine, in faeces, in exhaled gas from the lungs, and from the skin. The replenishment of body water is controlled by a combination of the satisfaction of the sensation of thirst and the regulation of the renal tubular reabsorption of water mediated by ADH.
Water excess
Excessive body water may occur in patients with extensive oedema or if there is inappropriate production of ADH (e.g. as occurs with small-cell lung carcinoma) or if the body sodium concentration increases due to excessive tubular reabsorption (for example, due to an aldosterone-secreting tumour of the adrenal cortex). Water overload can be caused iatrogenically by excessive parenteral infusion of fluids in patients with impaired renal function; this should be avoided by carefully monitoring fluid input and output.
Dehydration
Dehydration results from either excessive water loss or inadequate intake or a combination of both. Inadequate water intake is a common problem in regions of the world affected by drought and famine.
Excessive water loss can be due to:
Dehydration is recognised clinically by a dry mouth, inelastic skin and, in extreme cases, sunken eyes. The blood haematocrit (proportion of the blood volume occupied by cells) will be elevated, causing an increase in whole blood viscosity. This results in a sluggish circulation and consequent impairment of the function of many organs.
The plasma sodium and urea concentrations are typically elevated, reflecting haemoconcentration and impaired renal function.
Oedema and serous effusions
[/not-level-membership-for-pathology-category]
