Chapter 116 Occult Spinal Dysraphism and the Tethered Spinal Cord
Tethered Cord Syndrome
Pathology
The term tethered cord syndrome, as used in this chapter, signifies a pathologic fixation of the spinal cord in an abnormally low position so that the spinal cord, with activities and growth, undergoes mechanical stretching, distortion, and ischemia.1 Many conditions can cause tethering of the spinal cord, including tight filum terminale, split cord malformations, lipoma, dermal sinus, and meningomyelocele. The remainder of this section addresses the care and treatment of these conditions, excluding meningomyelocele.
Presentation
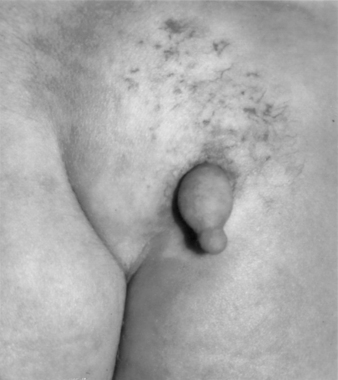
The patient with a tethered cord may be either symptomatic or asymptomatic. Both groups often, but not invariably, have a midline cutaneous dorsal abnormality such as a dimple, a hairy patch or faun’s patch, a hemangioma, a lipoma, or a skin tag (Figs. 116-1 and 116-2). If there is no external manifestation, the problem usually goes unrecognized until symptoms begin to develop.
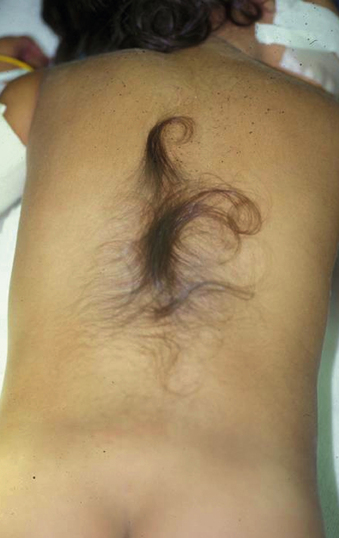
Symptoms, when present, can be grouped into three general areas: sensorimotor, sphincteric, and orthopaedic. Sensorimotor symptoms can include pain, delayed walking, sensory loss (usually in the dermatomes of the lumbosacral roots), and motor weakness of the distal leg or foot (the most common symptom, presenting in 76% of cases).2 Sphincteric symptoms usually are insidious, with frequent urinary tract infections secondary to incomplete emptying, hydronephrosis with renal involvement secondary to reflux, and fecal incontinence. In addition, these patients may develop urinary incontinence or become impotent. The orthopaedic problems are related to gait disturbance and abnormalities of the foot or scoliosis. Adult patients with tethered cords also may present with back pain that may radiate to the legs, urinary difficulties, and lower extremity weakness.3 Patients may present either as asymptomatic in childhood and symptomatic in adulthood or as having a progression of symptoms once they reach adulthood, perhaps due to repeated microtrauma to the cord.4 In adults, the disease may have an insidious onset or may have a predisposing factor such as exercise, lifting heavy loads, or even birth trauma.3,5
Diagnostic Aids
Any infant or child with a midline cutaneous lesion such as a dimple, hair patch, or hemangioma or symptoms mentioned earlier may be suspected of having a tethered cord.6 The intention of the workup is to determine the anatomy of the anomaly.
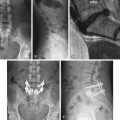
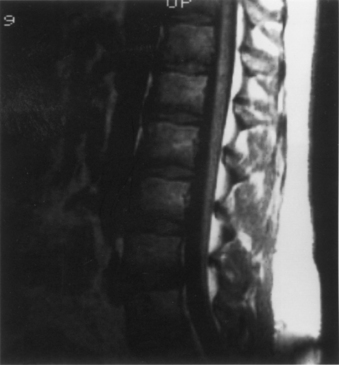
A careful neurologic examination with attention paid to evaluation of motor and sensory function as well as sphincter tone is imperative. If the history indicates possible bladder involvement, a urologic evaluation is indicated. Plain radiographs, which may be obtained as a screening procedure, may show a widened interpedicular distance and defects at one or more levels. The procedure of choice is MRI (Fig. 116-3). Often, this is the only necessary imaging study, and treatment can be planned on the basis of MRI alone. It is prudent to image the entire spinal cord at least once to rule out other associated anomalies, such as a syrinx or a type I Chiari malformation. If there are any remaining concerns or issues, a myelogram with subsequent CT may be helpful.
Treatment
Once the problem is identified, the treatment of the tethered cord is surgical. Although controversy existed in the past (over the concept of prophylactic surgery of asymptomatic patients), most surgeons now believe that the risk of waiting for deterioration to begin is not justified, because the deficit often is not reversible. Therefore, surgery is recommended, even in the asymptomatic patient,7 although a recent study8 suggests that careful follow-up and monitoring for upper motor neuron signs using urodynamic assessments, in order to time surgical intervention to coincide with the appearance of upper motor neuron signs, may be possible. In adults, patients with back pain and lower extremity pain seem to benefit more than those with sphincter problems.3
Outcome
The shorter the duration of symptoms, the better the prognosis.5 A study by Archibeck et al.9 demonstrated a 50% revision rate by 5 years after initial revision and a 57% revision rate by 2 years after the second release. In addition, 50% of patients required at least one orthopaedic procedure after tethered cord release.9 In a study by Cornette et al.8 of 12 patients operated on for tethered cord, none required a second operation. However, the series is small, although the follow-up period was reasonable (58 months). In terms of urologic outcome, improvement in symptoms as well as urologic dynamic parameters is expected in most patients, although few if any will return to normal.10 Improvements may be noted in detrusor function, EMG recordings, and pressures.11
Split Cord Malformations
Embryology and Pathology
The term split cord malformation (SCM) was introduced by Pang et al.12 in 1992 to describe diastematomyelia based on the dural tube and the nature of the septum. Two types of SCM exist: type I, diastematomyelia with septum, and type II, diastematomyelia without septum. Type II is more common.
By the end of the second week of gestation, the human embryo normally consists of a bilaminar structure: (1) an epiblast, or layer of cells next to the amnion, and (2) a hypoblast, or layer of cells next to the yolk sac. From there, the cells divide to form the primitive streak. During gastrulation, the embryo becomes trilaminar as adjacent epiblastic cells migrate medially toward the primitive streak to become mesoderm. The primitive streak begins to regress by day 16,12 and the notochordal process begins. As the notochord elongates, it canalizes, initially forming a connection through the embryo to join the amnion and yolk sac. This connection is then lost as the open notochord separates from the endoderm and again forms a blind tube.12
In the split cord malformations, an adhesion forms between the ectoderm and endoderm, leading to the formation of an “accessory neurenteric canal around which condenses an endomesenchymal tract that bisects the developing notochord and causes formation of two hemineural plates.”12 Whether a type I or type II SCM is formed depends on what happens to the endomesenchymal tract. If it develops toward bone and cartilage, the result will be two dural sacs and a type I SCM. If the tract regresses or leaves a fibrous septum, a type II SCM will develop.13
The spinal cord above and below the split is normal. The two hemicords themselves usually are the same size, but in 10% of patients, they are grossly asymmetrical. When this occurs, the spinal cord itself, above and below the bifurcation, is asymmetrical, being smaller on the side of the smaller hemicord.
The anterior spinal artery and the central canal bifurcate to accompany each hemicord,14 so that each has its own blood supply. The two hemicords give rise to the spinal nerve roots on their respective sides. Although splitting of the spinal cord at more than one site and cases of incomplete splitting of the spinal cord with a resultant partial cleft cord have been reported, most cases involve a single, complete cleft through the spinal cord and meninges. In cases in which there are two hemicords without an intervening septum, a single dural sac surrounds both. In such cases, symptoms may result from tethering of the cord by fibrous bands or a thickened filum terminale.
In cases in which the meninges themselves also are bifurcated, there almost always is an intervening septum. Its position is at the caudal end of the split; therefore, ascent of the neural elements is prohibited. The septum, or spur, usually is attached to both the dorsal elements and the dorsal aspect of the vertebral body. Because of the incidence of spina bifida, the spur may continue dorsally between unfused laminae. These spurs may present anywhere along the spine, but in 70% of cases they are between L1 and L5. They are less likely to occur in the thoracic spine and have only a 1% incidence in the cervical spine.15,16 The spur initially is cartilaginous and may mature to calcified bone with time.
Pathophysiology
The clinical symptoms most likely evolve from traction of the spinal cord against the restricting septum or bony spur.1,17 As with other forms of tethered spinal cord, ascent of the cord within the dural sac and spinal canal is prohibited. The average age of presentation is 6½ years, with neurologic symptoms first becoming evident with the onset of walking.16 With the onset of walking, however, increased traction of the distal spinal cord against the restricting septum results in new symptoms. To support this finding, Yamada et al.18 have studied the oxidative metabolism of the distal spinal cord and have found a decrease when the cord is under axial tension.
Presentation
Boxes 116-1 and 116-2 include some of the presenting symptoms and physical signs of patients with split cord malformations.19 In general, signs and symptoms fall into three categories: (1) cutaneous abnormalities, (2) pain, and (3) neurologic deficits (from spinal cord traction).
BOX 116-1 Split Cord Malformation: Common Presenting Complaints
Data from Mathern GW, Peacock WJ: Diastematomyelia. In Park TS, editor: Spinal dysraphism, Boston, 1992, Blackwell Scientific, p 91.
BOX 116-2 Split Cord Malformation: Common Physical Signs
Data from Mathern GW, Peacock WJ: Diastematomyelia. In Park TS editor: Spinal dysraphism, Boston, 1992, Blackwell Scientific, p 91.
In newborns and infants in whom neurologic deficits may not yet have developed, cutaneous lesions bring the child to the attention of the neurosurgeon. Most commonly, a patch of hair or hypertrichosis is noted in the thoracic or lumbosacral midline dorsally. This hair, usually coarse and long, is sometimes referred to as faun’s tail. The surrounding skin is associated with an intradermal angiomatous malformation, giving the skin a pinkish blue color. In addition, a dermal sinus, lipoma, abnormally protuberant spinous process, or meningocele may be associated with the spur.
If the patient with split cord malformation has successfully progressed through development with few or none of the aforementioned symptoms, the most common complaint, particularly in older children and adults, is back or leg pain.20 This pain may be due to subtle concomitant scoliosis or to the spinal bony deformity itself. The presence of unilateral symptoms is a key difference in the presentation of split cord malformation versus tethered cord syndrome.
Diagnostic Aids
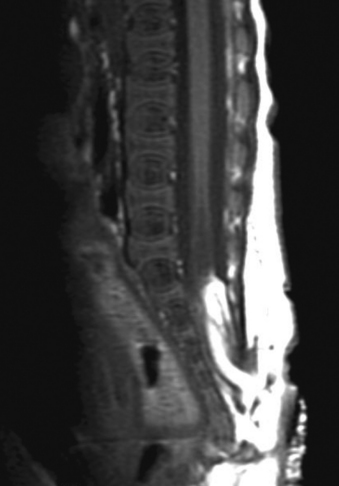
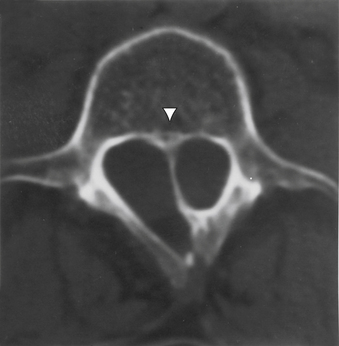
Aids that confirm the diagnosis of split cord malformation usually are radiologic. Although plain radiographs or unenhanced CT scans of the spine may reveal the bony spur, a widened interpedicular distance, spina bifida occulta, or other segmental vertebral anomalies, MRI in all three axes can be more revealing and is the preferred procedure (Figs. 116-4 to 116-7). Associated lipomas, hydromyelia, and other intraspinal and intradural defects also may be observed incidentally, allowing for a more focused treatment approach. If there are any questions or further clarification is required, myelography and postmyelographic CT best delineate the hemicords, the dural sac, and the presence and extent of the intervening bony septum (Fig. 116-8).21 Plain and CT myelography may reveal aberrant nerve roots, intradural bands, a thickened filum terminale, or a concomitant intradural lipoma. In addition, a recent study22 reported an incidence of abnormal urologic dynamic studies as high as 75% in patients with SCM, despite a lack of symptoms. Therefore, obtaining preoperative and postoperative urologic dynamic studies may be of some benefit. Again, as in the tethered cord syndrome, the entire spinal cord should be imaged.
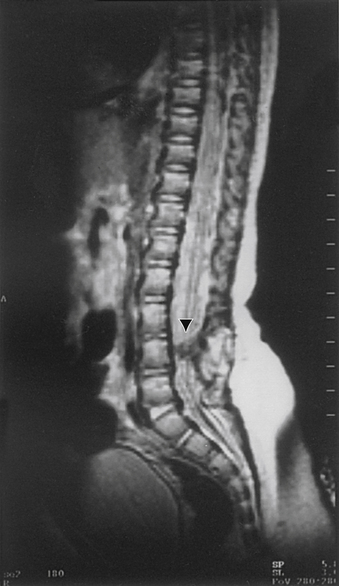
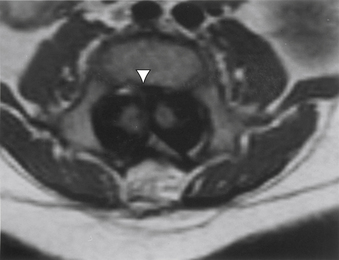
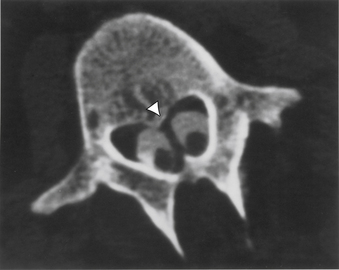
Treatment
Before making a standard midline incision to the lumbosacral fascia, the spine should be palpated. Occasionally, a protruding spinous process or bony spur may be felt, allowing for a more localized incision. In addition, a localizing plain radiograph with a skin marker is used and correlated with the preoperative MRI. If a cutaneous lesion, such as a patch of hair, is present, it may be beneficial to create an elliptical incision circumferentially around the defect. Because the underlying bony and soft tissue defect may not be clear, it is helpful to incise the fascia and perform a subperiosteal reflection of the paraspinous musculature at the levels above and below the level of the lesion, understanding that midline fusion defects may also exist here. The monopolar electrocautery should be used cautiously in retracting the muscles, because areas of expected protective bone may be missing. After the laminae above and below the lesion are exposed, their spinous processes are removed using a rongeur. A partial laminectomy is then performed at the caudal aspect of the lamina above the septum and the rostral aspect of the lamina below the septum. After careful curettage of the underside of both these laminae, the ligamentum flavum, if still intact, is elevated laterally with Penfield forceps and incised longitudinally through its outer layer. A blunt instrument is then gently inserted through the remaining ligament, and a small cottonoid patty is placed between the dura mater and the ligamentum flavum for protection. A small, angled Kerrison punch is then used to remove the ligamentum flavum until the dura mater is completely exposed laterally. With a no. 4 Penfield dissector, the septum is then felt over the dura from above and from below. A small-mouthed rongeur or angled Kerrison punch or high-speed drill is used to remove the lamina and overhanging bone of the involved level until only the spur is left. Because of the substantial epidural venous plexus associated in and around the bony spur and deep to the two hemicords, control of bleeding and cauterization of these vessels should be performed prior to and during the removal of the spur (Fig. 116-9). After decompression, with movement of the hemicords, it may be very difficult to maintain hemostasis. Any extruding segment of spur is removed using a rongeur, and a small dissector is used to probe and dissect the dural sheath away from the bony spicule down to the level of the dorsal vertebral body. A high-speed diamond-bit drill is then used to carefully thin down the spicule as far as possible.

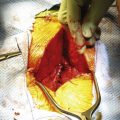
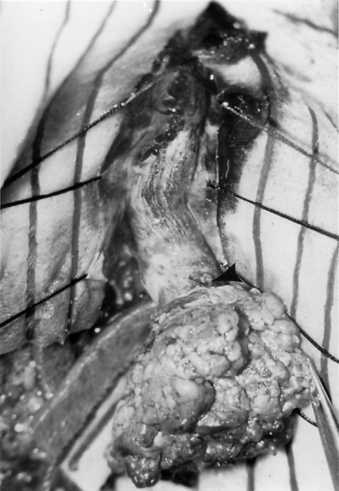
FIGURE 116-9 Intraoperative photograph showing the two hemicords (arrows) in a patient with split cord malformation.
At this point, the dura mater is opened along the midline above and below the spur and elliptically around the spur remnant. After the dural edges are tacked up, the two hemicords become evident. The ventral dural sac is then incised along the midline above the spicule and circumferentially around it. The dural “chimney” is removed, and the resultant dural edges are teased laterally so that the remainder of the spur may be completely drilled off, with the spinal cord being protected at all times. With the removal of the spur, the entire cord may migrate rostrally. Microinstruments should be used to break or cut any additional adhesions that may be tethering the spinal cord to the dura mater. A watertight closure of the dorsal dural opening is then performed, with interrupted sutures used over the elliptical incision to avoid the unraveling of a running stitch over this area of tension; again, fibrin glue is used. The ventral dura mater does not require closure. A small drain is inserted, and the soft tissues are closed in anatomic layers. The patient should then be kept flat for 3 days postoperatively to avoid CSF leak.
Outcome
In symptomatic patients, bowel and bladder dysfunction may improve up to 40% of the time; stabilization of progressive urologic symptoms also may be noted.23 Neurologic sensorimotor deficits return to normal only 5% to 10% of the time. Patients whose main complaint is pain in general improve. Also included in improvement of pain is the dysesthetic component. A higher surgical morbidity has been reported in cases in which the bony septum is present, perhaps due to removal of the bony septum.23 It is important to remember that preserving neurologic function is as important as improving it. Recent studies23,24 also report that untethering the cord may have no effect on the neuro-orthopaedic syndrome (e.g., lower limb asymmetry, foot deformities), perhaps due to irreversible changes in the ligaments.
Congenital Dermal Sinus
Pathology and Embryology
Dorsal congenital dermal sinus, a subtype of spinal dysraphism not associated with spina bifida or bony abnormalities, is defined as an epithelium-lined tract from the skin of the back, usually the lumbosacral midline (although it also may occur in the thoracic and cervical spine),13 that passes through the soft tissues toward the spine, the thecal sac, and even into the neural elements. These tracts, which usually are very thin, are thought to develop because of adhesion and failure of separation between the superficial cutaneous ectoderm and the neural tube.25 This failure of separation usually occurs at the fourth week of fetal development, after neurulation of the tail bud. At that time, the attachment between skin and spinal cord is lengthened and thinned with the ascent of the cord and fixation of the skin. Because of its small size, the surrounding bone-forming mesoderm may produce little or no spina bifida. Also, the remainder of the vertebral body at the affected levels usually is normal. Although not usually a cause of tethering of the spinal cord, the cutaneous origin of the sinus tends to be two to three vertebral levels caudal to its adhesion to the neural elements. Congenital dermal sinus is present in 1.2% of the neonatal population.
Because of the epithelial lining and potential communication with the skin, the dermal sinus may result in an expanding dermoid or epidermoid tumor in the subdural or epidural space, in the same manner that such tumors arise from iatrogenic implantation of such elements with spinal needles.26 These tumors may present as mass lesions or as a simple dermal sinus associated with a possible communication between the skin and subarachnoid space. Therefore, they are a nidus for infection, meningitis, and possibly abscess formation. Microscopically, they consist of dermal elements, such as sweat and apocrine glands and hair follicles.
Most dorsal dermal sinuses are lumbosacral; occipital sinuses are less common. Although about 60% of dermal sinuses end in dermoid or epidermoid tumors, only 30% of these tumors are associated with dermal sinuses.27,28 In addition, the dermal sinus may be involved in tethering of the spinal cord, not by itself but either by a thick band of tissue attached to the spinal cord or conus medullaris or as a result of inflammatory scarring.
Pathophysiology and Presentation
Because of the low incidence of tethering with dermal sinuses, symptoms result from infectious etiologies. Bacterial causes may include Staphylococcus aureus, Staphylococcus epidermidis, Escherichia coli, and even Proteus species.29 The tract from the bacteria-laden epidermis to the intraspinous space and even the subarachnoid space provides opportunities for intermittent, chronic, and acute infections.30,31 Although the incidence of meningitis is higher with the presence of a concomitant dermoid or epidermoid tumor, simple tracts carry this risk as well.32 In fact, dermal sinuses may be the cause of infection in up to one quarter of cases of intramedullary spinal abscesses, with approximately 70% of patients having neurologic deficits.33 Because of the dimple formed in the skin over the tract, infections are noticed and addressed, often before clinical symptoms have surfaced. The dimple, or the external ostium of the sinus, usually is in the midline and may be associated with a hemangioma, a nevus, or short tufts of hair protruding from the sinus. Parents, caretakers, or physicians may notice caseous discharge from this area or perhaps some erythema or inflammation. Because of the usual lumbosacral location, fecal and skin organisms may be the cause of meningitis. Frank neurologic symptoms or neurogenic pain rarely is present unless there is compression from an associated tumor or tethering of the spinal cord or conus, as mentioned previously.
Diagnostic Aids
MRI is the initial imaging modality of choice (Fig. 116-10). It is useful to follow the denser, low-signal sinus through the high-signal subcutaneous fat toward the dura mater. The intraspinal course of the tract, however, may not be well displayed by MRI in all cases. Dermoid and epidermoid tumors have variable signal intensity on MRI. Dermoids, with increased T1 and T2 signals compared with water (because of the cholesterol, fat, and protein content), image well. However, if chemical meningitis has been caused by leakage of the tumor, detection may be more difficult. Although epidermoid tumors contain only epithelial elements, they are equally well defined by MRI.
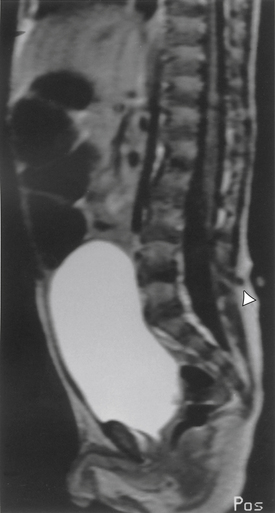
Axial CT imaging reveals the dense sinus tract through its course from the skin into the dura mater. Occasionally, the lumen of the tract may be visualized as a more hypodense line within the tract. CT scanning with intra-arachnoid myelographic contrast medium is the most useful method for imaging the course of the sinus in the subdural space as it ascends toward the conus medullaris.34 Water-soluble contrast or air should be used to avoid leaving oily droplets that could act as potential foreign bodies. A lucent mass along the tubular filling defect may represent dermoid or epidermoid tumors. The use of contrast material has been reduced dramatically since the advent of MRI, however. Even in the presence of a normal MRI, if a cutaneous lesion seems to truly represent a dermal sinus in a suspicious location, surgical exploration may be warranted.
Treatment
With gentle traction on the tract, microinstruments are used to dissect away any adhesions between the tract, the dura mater, and the nerve roots. Some adhesions, particularly postmeningitic scars, may require incision. In this way the tract is followed to its attachment, usually dorsally above the tip of the conus. The sides of the tract are completely identified by using a small blunt hook, and microscissors are used to detach it. If the stump bleeds, it is lightly coagulated with bipolar forceps. After verifying the absence of other mass lesions or areas of tethering, the subarachnoid space is copiously irrigated with warm saline, and the dura mater is closed in a watertight fashion with #4-0 silk or woven nylon suture, using a graft if necessary. Fibrin glue is then applied. A drain may be placed in the epidural space and brought through the skin for 24 hours. The soft tissues are then closed in anatomic layers. Some undermining of the subcutaneous tissue occasionally is necessary to bring together the skin edges in the area of the elliptical excision. The patient should remain flat in bed for 3 days, and antibiotics should be administered postoperatively for 3 days, or longer if a previous infection was present.
Spinal Lipomas
Pathology and Embryology
Although associated with other forms of occult spinal dysraphism, spinal lipomas are connective tissue and fat collections that are distinct, partially or completely encapsulated, and definitely attached to the spinal cord.36 It is thought that during the process of primary neurulation, improper disjunction of surface ectoderm and neuroepithelium may lead to inclusion of fat.36 Distinct from lipomyelomeningoceles, isolated lipomas technically are fibrolipomas of the filum terminale or dural fibrolipomas, as defined by Emery and London.37 In simple lipomas, the neural elements remain within the spinal canal, whereas lipomyelomeningoceles are marked by herniation of the neural elements out of the canal into the subcutaneous portion of the lipoma.38 Strictly intradural lipomas associated with an intact dura mater are lesions of subpial fat found in the cervical and thoracic spinal cord.39 In a large series reported by McLone and Naidich,40 4% of the lipomas treated surgically were intradural lipomas. More common, however, are lipomas that involve the dura mater and extend from the spinal cord to the subcutaneous tissue.39,41,42
Lipomas are associated with more severe bony changes than is the previously described dermal sinus, including scalloping of the dorsal vertebral body, widening of the interpedicular space, hemivertebrae, or even hypoplasia of the iliac wing.38 These sequelae of the mass effect associated with lipomas suggest that resultant neurologic deficits occur not only by spinal cord tethering (see Pathophysiology and Presentation) but also by direct neural compression.
Pathophysiology and Presentation
Spinal lipomas, accounting for up to 35% of skin-covered lumbosacral masses, may extend to the superficial subcutaneous tissues and present in the infant as a visible and palpable mass.43 As with other forms of the tethered cord syndrome, children with spinal lipomas may present with several complaints. At this age, before the onset of walking and the development of bladder control, a concomitant hairy patch may accompany an otherwise unnoticed lipoma. If the condition is unnoticed or disregarded, the infant without neurologic abnormalities may, with age, develop sphincter disturbance, postural and lower extremity deformities and weakness, or even verbalized discomfort.44
Classification
Lipomas of the conus medullaris may be classified into three categories: dorsal, caudal, and transitional. First described by Chapman, alone45 and with Davis,46 two distinct forms as well as a transitional form were categorized.
The dorsal variant is a lipoma that arises through a fascial defect and attaches directly to the dorsal aspect of the caudally descended conus medullaris. All nerve roots emerge from the ventral or lateral surface of the neural tissue and lie in the subarachnoid space. The lateral nerve roots are sensory, while motor roots are found more medial.
Diagnostic Aids
Lipomas constitute only 1% of primary intraspinal tumors and almost always are associated with dysraphic spines.47 Varying from intramedullary to extradural, their histologic nature and relative position to the spinal canal make them definable by both CT and MRI.48
Although spina bifida occulta, widening of the interpedicular distance, hemivertebrae, and vertebral body scalloping may all be observed on plain radiographic studies in the patient with a lipoma, these radiographs may be difficult to interpret. As with other spinal anomalies, lipomas usually are best worked up with MRI as the initial study (Fig. 116-11). CT usually is reserved for better definition of bony anatomy, if needed. Although not as detailed, myelograms may help define the extent of the mass (Figs. 116-12 and 116-13). Intradural lipomas are low density or even radiolucent on CT and have a high signal on T1-weighted MRI.49 Both techniques are useful, however, in axial section. Extradural lipomas usually are more diffuse and contiguous with the nearby epidural fat. Even when epidural lipomas are not clearly visualized on CT scan and MRI, their presence should be considered.
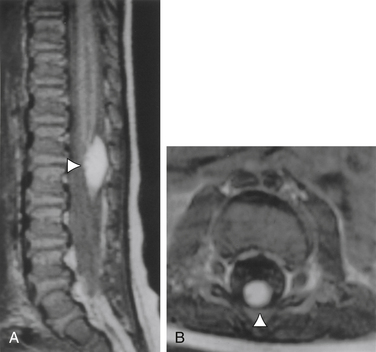
FIGURE 116-11 Sagittal (A) and axial (B) MR images of a tethered spinal cord with an intradural spinal lipoma (arrowheads).
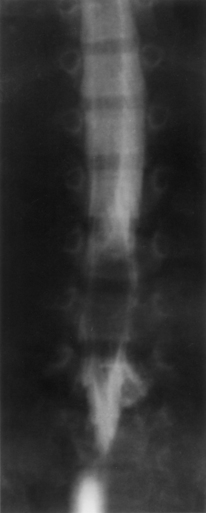
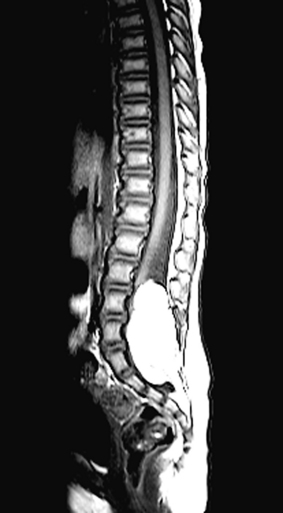
McLone and Naidich50 also support the use of ultrasonography in managing these lesions. The lack of calcium in immature bones allows for penetration and evaluation of structural detail, often well enough to verify the lesion, determine the extent of tethering, and proceed straight to surgery without the need for additional radiographic studies.
Treatment
Whether by tethering of the filum terminale and spinal cord, direct neural compression, or both, surgery for spinal lipomas is warranted in the child with neurologic deficits. Operating on the asymptomatic patient has been controversial. However, most neurosurgeons now believe that if possible, lipomas and lipomeningoceles should be operated on before neurologic sequelae occur.44,51,52 Unlike split cord malformations, in which involvement of the neural elements in the substance of the pathology is rare, such involvement is common in spinal lipomas. Therefore, even with detailed and defining preoperative studies, intraoperative electrophysiologic studies may be something that should be considered during resection of spinal lipomas. Not all surgeons consider this necessary. We, however, find it quite useful and do use intraoperative EMG monitoring, in which we insert needle electrodes in the muscles of the lower extremities as well as in the sphincter. Stimulation is performed with the same probes that are used for dorsal root rhizotomy. With this type of monitoring, one can ascertain whether there is undue traction on the conus medullaris and if it is safe to incise tissues that are near nerves or the spinal cord.
After sterilization of the skin, a midline incision should be performed over the palpable or visible subcutaneous portion of the lipoma. If there is no evidence of such a superficial lesion, needle localization with anteroposterior and lateral radiographs is useful. After incision of the fascia and lateral exposure of the laminae, microscopic enhancement, either with loupe magnification or the operating microscope, should be used. As with all occult dysraphic spines, care should be taken with both the scalpel and the electrocautery, because the unformed or bifid laminae may offer no protection for the thecal sac. Before the bony structures are reached, the extradural portions of the lipoma may require resection. If so, circumferential dissection of the lipoma is important so as to allow complete resection. The actual resection of the extradural portion of the lipoma usually is fairly straightforward. However, the location of the nerve roots relative to the lipoma-cord junction vary with the type of attachment that the lipoma has with the conus medullaris.53 If the lipoma attaches to the dorsal surface of the conus, the nerve roots are ventral to the lipoma-conus interface. If, however, the lipoma is a caudal extension of the conus medullaris, the course of the nerve roots through the lipoma can be variable, and great care has to be exerted to prevent damage to neural structures (Fig. 116-14).
At the time of the durotomy, all anesthetic muscle relaxants should be avoided. Extradural lipomas that traverse the thecal sac into the intradural compartment may require resection of some dura mater. At this point, although some surgeons advocate the use of the carbon dioxide laser to vaporize the fatty lesion, we also suggest the use of the ultrasonic aspirator. Although relief of neural compression is one principle of surgery, particularly for lipomas on the dorsal surface of the spinal cord, the primary goal is to untether the spinal cord. Therefore, the surgery should be directed at accomplishing this goal. In such cases, sectioning of the filum terminale caudal to the sacral nerve roots may be required. Electrophysiologic monitoring is particularly useful at this point, not only to determine where the filum terminale may be incised and to ensure that no neural structures are coursing within the filum, but also to take care to avoid excessive manipulation of the functional nerve roots and the spinal cord. Often it is not possible or even necessary to resect the lipoma completely (Fig. 116-15). Prudence should be the rule, and one should not take any chance of injuring the conus medullaris or the nerve roots. It should be remembered that the primary goal of surgery is to untether the spinal cord and at the same time cause no deficits.
After removal of the lipoma, untethering of the surrounding neural structures, and possible sectioning of the filum terminale, dural closure must be watertight. Although reapproximation of two of the three meningeal layers may decrease the change of retethering, the pia and arachnoid often are incompetent after removal of the lipoma. Therefore, careful closure of the dura mater, which may include a graft, is important. As with other intrathecal operations, leaving the child flat in bed for 3 days allows for tissue healing and helps avoid collection of CSF, particularly if a large “dead space” has resulted from excision of an extradural component of the lipoma. Special techniques of closure to prevent retethering have been reported, but the patients have not been followed long enough to determine whether these techniques are better than conventional methods of closure.54
Outcome
Successful resection and untethering of these lipomas is now associated with little or no morbidity and mortality.40,53,55 The best outcomes may be achieved with respect to pain, with most of the pain decreasing or disappearing within 3 months. Bladder dysfunction also may respond to resection of the lipoma in 20% to 30% of patients; of these patients, those with a spastic bladder respond best.56 In a study by La Marca et al.,51 213 patients were operated on over a 20-year period, from 1975 to 1995. In patients with filum lipomas, 28 were asymptomatic and 27 were symptomatic. None of the asymptomatic patients worsened after surgery (mean follow-up, 3.4 years), and of the symptomatic group, there were no further deteriorations noted (follow-up, 6 months to 9 years). Of the group with conus lipomas, 9 of the 71 children (12.7%) operated on prophylactically later deteriorated (mean follow-up, 6.2 years) and required a second untethering operation. Symptoms of deterioration included urinary retention, pain, gait difficulty, urinary incontinence, and spasticity. In the symptomatic group (87 patients), 36 patients (41%) further deteriorated and required further surgery. At the final follow-up, however (mean, 6.6 years), 51% remained at clinical baseline and 26% improved. In a study by Xenos et al.,57 the reoperation rate was 12% for signs of recurrent spinal cord tethering.
Emerging Technologies
Minimally Invasive Surgery
Minimally invasive or endoscopic surgical techniques have been used to treat a number of spinal pathologies in recent years. With spinal dysraphism and the tethered spinal cord presenting clinically and often repaired at a young age, minimally invasive operative techniques may be of great benefit to the patient. Recently, it has been determined that tethered spinal cords may be safely and effectively untethered using minimally invasive surgery.58 This technique provides the advantage of reduced soft tissue injury, reduced postoperative pain, reduced postoperative scarring, and lower postoperative risk of retethering.
Cavitron Ultrasonic Aspiration
Cavitron ultrasonic aspiration (CUSA; Valleylab., Inc., Boulder, CO) is one alternative to the CO2 laser. The original ultrasonic aspirator was developed in 1947 for the removal of dental plaque and was first applied to the field of eye surgery in 1967. Ultrasonic techniques became widely used in the medical and biochemical industries for sterilization, homogenization of solutions, and welding of materials. The use of the ultrasonic aspirator in neurosurgery was first reported in 1978, for the removal of intra-axial and extra-axial tumors such as meningiomas, schwannomas, and gliomas. The ultrasonic aspirator has since become a valuable tool in the neurosurgical armamentarium for the excision of intracranial and intraspinal tumors.59 As with the CO2 laser, the CUSA system provides efficient resection of target tissue with minimal injury to surrounding tissue or structures.
The ultrasonic aspirator has two tissue-disruptive effects at the tissue interface.60 The first is caused by a suction effect that couples tissue to the tip and forces impacted tissue to vibrate, accelerate, and decelerate with the tip, eventually fragmenting away from unaffected tissues. The second important effect is cavitation. In cavitation, the rapidly oscillating tip produces localized pressure waves, which cause vapor pockets around cells in tissues with high water content; the collapse of these pockets then causes the tissue cells to rupture. Tissues with weak intracellular bonds, such as tumors and lipomas, are easy to fragment, whereas tissues with strong intracellular bonds, such as nerves and vessel walls, are difficult to fragment. The speed of fragmentation depends on the amplitude setting of the system.
Akay K.M., Ershin Y., Cakir Y. Tethered cord syndrome in adults. Acta Neurochir (Wien). 2000;142:1111.
Archibeck M.J., Smith J.T., Carroll K.L., et al. Surgical release of tethered cord: survivorship analysis and orthopedic outcome. J Pediatr Orthop. 1997;17:773.
Balkan E., Kilic N., Avsar I., et al. Urodynamic findings in the tethered spinal cord: the effect of tethered cord division on lower urinary tract functions. Eur J Pediatr Surg. 2001;11:116.
Boop F.A., Russell A., Chadduck W.M. Diagnosis and management of the tethered cord syndrome. J Ark Med Soc. 1992;89:328.
Chong C., Molet J., Oliver B., et al. The tethered cord syndrome: a review of causes. Neurologia. 1994;9:12.
Garzo-Mercado R. Diastematomyelia and intramedullary epidermoid spinal tumor combined with extra-dural teratoma in an adult. J Neurosurg. 1983;58:954.
Hilal S.K., Marton D., Pollack E. Diastematomyelia in children. Radiology. 1974;112:609.
Yamada S., Zinke D.E., Sanders D.C. Pathophysiology of “tethered cord syndrome.”. J Neurosurg. 1981;54:499.
1. Guthkelch A.N. Diastematomyelia with median septum. Brain. 1974;97:729.
2. Muraszko K., Youholis A. Intramedullary spinal tumors of disordered embryogenesis. J Neurooncology. 2000;47:271.
3. Akay K.M., Ershin Y., Cakir Y. Tethered cord syndrome in adults. Acta Neurochir (Wien). 2000;142:1111.
4. Gupta S.K., Kosla V.K., Sharma B.S., et al. Tethered cord syndrome in adults. Surg Neurol. 1999;52:362.
5. Chong C., Molet J., Oliver B., et al. The tethered cord syndrome: a review of causes. Neurologia. 1994;9:12.
6. Boop F.A., Russell A., Chadduck W.M. Diagnosis and management of the tethered cord syndrome. J Ark Med Soc. 1992;89:328.
7. Pierre-Kahn A., Lacombe J., Pinchon J., et al. Intraspinal lipomas with spina bifida: prognosis and treatment in 73 cases. J Neurosurg. 1986;65:756.
8. Cornette L., Verpoorten C., Lagae L., et al. Tethered cord syndrome in occult spinal dysraphism: timing and outcome of surgical release. Neurology. 1998;50:1761.
9. Archibeck M.J., Smith J.T., Carroll K.L., et al. Surgical release of tethered cord: survivorship analysis and orthopedic outcome. J Pediatr Orthop. 1997;17:773.
10. Basar H., Aydoganli L., Yuksel M., et al. The outcome of urologic findings in operated tethered cord patients. Int Urol Nephrol. 1997;29:167.
11. Balkan E., Kilic N., Avsar I., et al. Urodynamic findings in the tethered spinal cord: the effect of tethered cord division on lower urinary tract functions. Eur J Pediatr Surg. 2001;11:116.
12. Pang D., Dias M.S., Ahab-Barmada M. Split cord malformation: part I: a unified theory of embryogenesis for double spinal cord malformations. Neurosurgery. 1992;31:451.
13. Tortori-Donati P., Rossi A., Cama A. Spinal dysraphism: a review of neuroradiological features with embryological correlations and proposal for a new classification. Neuroradiology. 2000;42:471.
14. Garzo-Mercado R. Diastematomyelia and intramedullary epidermoid spinal tumor combined with extra-dural teratoma in an adult. J Neurosurg. 1983;58:954.
15. Geremin G.K., McNeil T.W. CT demonstration of cervical diastematomyelia. J Comput Assist Tomogr. 1985;9:592.
16. Hamby W.B. Pilonidal cyst, spina bifida occulta and bifid spinal cord: report of a case and review of the literature. Arch Pathol. 1936;21:831.
17. Matson D.D., Woods R.P., Campbell J.B., et al. Diastematomyelia (congenital clefts of the spinal cord): diagnosis and surgical treatment. Pediatrics. 1950;6:98.
18. Yamada S., Zinke D.E., Sanders D.C. Pathophysiology of “tethered cord syndrome.”. J Neurosurg. 1981;54:499.
19. Mathern G.W., Peacock W.J. Diastematomyelia. In: Park T.S., editor. Spinal dysraphism. Boston: Blackwell Scientific; 1992:91.
20. Pang D., Wilberger J.F.Jr. Tethered cord syndrome in adults. J Neurosurg. 1982;57:32.
21. Hilal S.K., Marton D., Pollack E. Diastematomyelia in children. Radiology. 1974;112:609.
22. Proctor M., Bauer S.B., Scott M.R. The effect of surgery for split spinal cord malformation on neurologic and urologic function. Pediatr Neurosurg. 2000;32:13.
23. Pang D. Split cord malformation: part II: clinical syndrome. Neurosurgery. 1992;31:481.
24. Andar U.B., Harkness W.F., Hayward R.D. Split cord malformations of the lumbar region. Pediatr Neurosurg. 1997;26:17.
25. Mount L.A. Congenital dermal sinuses as a cause of meningitis, intraspinal abscess and intracranial abscess. JAMA. 1949;139:1263.
26. Van Gilder J.C., Schwartz H.G. Growth of dermoids from skin implants to the nervous system and surrounding spaces of the newborn rat. J Neurosurg. 1967;26:14.
27. Guidetti B., Gagliardi F.M. Epidermoid and dermoid cysts: clinical evaluation and late surgical results. J Neurosurg. 1977;47:12.
28. Wright R.L. Congenital dermal sinuses. Prog Neurol Surg. 1971;4:175.
29. McComb J.G. Congenital dermal sinus. In: Pang D., editor. Disorders of the pediatric spine. Raven Press; 1995:349.
30. El-Gindi S., Fairburn B. Intramedullary spinal abscess as a complication of a congenital dermal sinus: case report. J Neurosurg. 1969;30:494.
31. Walker A.R.E., Bucy P.C. Congenital dermal sinuses: source of spinal meningeal infection and subdural abscesses. Brain. 1936;57:401.
32. Matson D.D., Jerva M.J. Recurrent meningitis associated with congenital lumbo-sacral dermal sinus tract. J Neurosurg. 1966;25:288.
33. Chan C.T., Gold W.L. Intramedullary abscess of the spinal cord in the antibiotic era: clinical features, microbial etiologies, trends in pathogenesis, and outcomes. Clin Infect Dis. 1998;27:619.
34. Scott G., Harwood-Nash D.C., Hoffman H.J. Congenital thoracic dermal sinus: diagnosis by computer-assisted metrizamide myelography. J Comput Assist Tomogr. 1980;4:675.
35. Morandi S., Mercier P., Fournier H.D., Brassier G. Dermal sinus and intramedullary spinal cord abscess: report of two cases and review of the literature. Child Nerv Syst. 1999;15:202.
36. Knepper D.A., McLone D.A. Development of the spinal cord: normal and abnormal neurulation. In: Park T.S., editor. Spinal dysraphism. Boston: Blackwell Scientific; 1992:1.
37. Emery J.L., London R.G. Lipomas of the cauda equina and other fatty tumors related to neurospinal dysraphism. Dev Med Child Neurol. 1968;20:62.
38. French B.N. Abnormal development of the central nervous system. In: McLauren R.L., Schut L., Venes J.L., Epstein F.L., editors. Pediatric neurosurgery. ed 2. Philadelphia: WB Saunders; 1989:9.
39. Ammerman B.J., Henry J.M., De Girolami U., et al. Intradural lipomas of the spinal cord: clinicopathological correlation. J Neurosurg. 1976;44:331.
40. McLone D.G., Naidich T.P. Laser resection of fifty spinal lipomas. Neurosurgery. 1986;18:611.
41. Lassman L.P., James C.C.M. Lumbosacral lipomas: critical survey of 26 cases submitted to laminectomy. J Neurol Neurosurg Psychiatry. 1967;30:174.
42. Rogers H.M., Long D.M., Chou S.N., French L.A., et al. Lipomas of the spinal cord and cauda equina. J Neurosurg. 1971;34:349.
43. Villarejo F.J., Blasquez M.G., Guierrez-Diaz J.A. Intraspinal lipomas in children. Child’s Brain. 1976;2:361.
44. Bruce D.A., Schut L. Spinal lipomas in infancy and childhood. Child’s Brain. 1979;5:192.
45. Chapman P.H. Congenital intraspinal lipomas: anatomic considerations and surgical treatment. Child’s Brain. 1982;9:37.
46. Chapman P.H., Davis K.R. Surgical treatment of spinal lipomas in childhood. Pediatr Neurosurg. 1993;19:267.
47. Giuffre R. Intradural spinal lipomas. Acta Neurochir. 1966;14:69.
48. Morano J.U., Miller J.D., Connors J.J. MR imaging of spinal epidural lipoma. Am J Neuroradiol. 1989;10:102.
49. Fitz C.R. Neuroradiology of spinal dysraphism. In: Park T.S., editor. Spinal dysraphism. Boston: Blackwell Scientific; 1992:161.
50. McLone D.G., Naidich T.P. The tethered spinal cord. In: McLauren R.L., Schut L., Venes J.L., Epstein F.L., editors. Pediatric neurosurgery. ed 2. Philadelphia: WB Saunders; 1989:76.
51. La Marca F., Grant J.A., Tomita T., McLone D.G. Spinal lipomas in children: outcome of 270 procedures. Pediatr Neurosurg. 1997;26:8.
52. Reigel D.H. Tethered spinal cord. Humphreys R.P., editor. Concepts in pediatric neurosurgery. Basel: S Karger AG. 1983;vol 4:142.
53. Chapman P.H. Congenital intraspinal lipomas: anatomic considerations and surgical treatment. Child’s Brain. 1982;9:37.
54. Zide B.M. How to reduce the morbidity of wound closure following extensive and complicated laminectomy and tethered cord surgery. Pediatr Neurosurg. 1995;18:157.
55. McLone D.G., Hayashida S.F., Caldarelli M. Surgical resection of lipomyelomeningoceles in 18 asymptomatic infants. J Pediatr Neurosci. 1985;1:239.
56. Pang D. Spinal cord lipomas. In: Pang D., editor. Disorders of the pediatric spine. New York: Raven Press; 1995:175-201.
57. Xenos C., Sgouros S., Walsh R., Hockley A. Spinal lipomas in children. Pediatr Neurosurg. 2000;32:295.
58. Tredway T.L., Musleh W., Christie S.D., et al. A novel minimally invasive technique for spinal cord untethering. Neurosurgery. 2007;60(2 Suppl 1):ONS70.
59. Jagannathan J., Sanghvi N.T. High-intensity focused ultrasound surgery of the brain: Part 1—A historical perspective with modern applications. Neurosurgery. 2009;64(2):201.
60. Brock M., Ingwersen I., Roggendorf W. Ultra-sonic aspiration in neurosurgery. Neurosurg Rev. 1984;7:173.