[level-membership-for-pediatrics-category]
Chapter 534 Obstruction of the Urinary Tract
Urinary tract obstruction can result from congenital (anatomic) lesions or can be caused by trauma, neoplasia, calculi, inflammatory processes, or surgical procedures, although most childhood obstructive lesions are congenital. Obstructive lesions occur at any level from the urethral meatus to the calyceal infundibula (Table 534-1). The pathophysiologic effects of obstruction depend on its level, the extent of involvement, the child’s age at onset, and whether it is acute or chronic.
Table 534-1 TYPES AND CAUSES OF URINARY TRACT OBSTRUCTION
| LOCATION | CAUSE |
|---|---|
| Infundibula | |
| Renal pelvis | |
| Ureteropelvic junction | |
| Ureter | |
| Bladder outlet and urethra |
Etiology
Ureteral obstruction occurring early in fetal life results in renal dysplasia, ranging from multicystic kidney, which is associated with ureteral or pelvic atresia (see Fig. 531-2), to various degrees of histologic renal cortical dysplasia that are seen with less severe obstruction. Chronic ureteral obstruction in late fetal life or after birth results in dilation of the ureter, renal pelvis, and calyces, with alterations of renal parenchyma ranging from minimal tubular changes to dilation of Bowman’s space, glomerular fibrosis, and interstitial fibrosis. After birth, infections often complicate obstruction and can increase renal damage.
Clinical Manifestations
Obstruction of the urinary tract generally causes hydronephrosis, which typically is asymptomatic in its early phases. An obstructed kidney secondary to a ureteropelvic junction (UPJ) or ureterovesical junction obstruction can manifest as a mass or cause upper abdominal or flank pain on the affected side. Pyelonephritis can occur because of urinary stasis. An upper urinary tract stone can occur, causing abdominal and flank pain and hematuria. With bladder outlet obstruction, the urinary stream may be weak; urinary tract infection (UTI; Chapter 532) is common. Many of these lesions are identified by antenatal ultrasonography; an abnormality involving the genitourinary tract is suspected in as many as 1/100 fetuses.
Diagnosis
Imaging Studies
Renal Ultrasonography
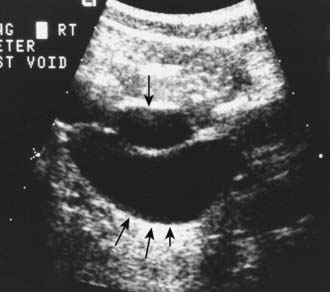
The presence of a dilated urinary tract is the most common characteristic of obstruction. Hydronephrosis is a common ultrasonographic finding (Fig. 534-1). Dilation is not diagnostic of obstruction and can persist after surgical correction of an obstructive lesion. Dilation can result from vesicoureteral reflux, or it may be a manifestation of abnormal development of the urinary tract, even when there is no obstruction. Renal length, degree of caliectasis and parenchymal thickness, and presence or absence of ureteral dilation should be assessed. Ideally, the severity of hydronephrosis should be graded from 1 to 4 using the Society for Fetal Urology grading scale (Table 534-2). The clinician should ascertain that the contralateral kidney is normal, and the bladder should be imaged to see whether the bladder wall is thickened, the lower ureter is dilated, and bladder emptying is complete. In acute or intermittent obstruction, the dilation of the collecting system may be minimal and ultrasonography may be misleading.
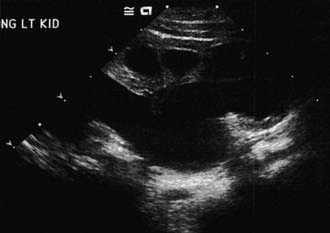
Table 534-2 SOCIETY FOR FETAL UROLOGY GRADING SYSTEM FOR HYDRONEPHROSIS
| RENAL IMAGE | ||
|---|---|---|
| GRADE OF HYDRONEPHROSIS | Central Renal Complex | Renal Parenchymal Thickness |
| 0 | Intact | Normal |
| 1 | Slight splitting | Normal |
| 2 | Evident splitting, complex confined within renal border | Normal |
| 3 | Wide splitting pelvis dilated outside renal border, calyces uniformly dilated | Normal |
| 4 | Further dilatation of pelvis and calyces (calyces may appear convex) | Thin |
After Maizels M, Mitchell B, Kass E, et al: Outcome of nonspecific hydronephrosis in the infant: a report from the registry of the Society for Fetal Urology, J Urol 152:2324–2327, 1994.
Radioisotope Studies
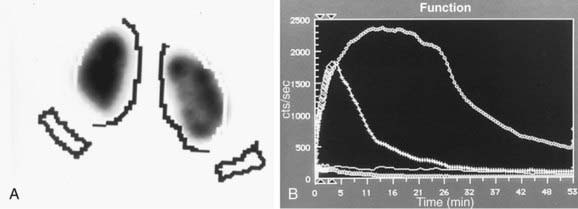
In a MAG-3 diuretic renogram, a small dose of technetium-labeled MAG-3 is injected intravenously (Figs. 534-2 and 534-3). During the first 2-3 min, renal parenchymal uptake is analyzed and compared, allowing computation of differential renal function. Subsequently, excretion is evaluated. After 20-30 min, furosemide 1 mg/kg is injected intravenously, and the rapidity and pattern of drainage from the kidneys to the bladder are analyzed. If no obstruction is present, half of the radionuclide should be cleared from the renal pelvis within 10-15 min, termed the half-time (t1/2). If there is significant upper tract obstruction, the t1/2 usually is >20 min. A t1/2 of 15-20 min is indeterminate. The images generated usually provide an accurate assessment of the site of obstruction. Numerous variables affect the outcome of the diuretic renogram. Newborn kidneys are functionally immature, and, in the first month of life, normal kidneys might not demonstrate normal drainage after diuretic administration. Dehydration prolongs parenchymal transit and can blunt the diuretic response. Giving an insufficient dose of furosemide can result in inadequate drainage. If vesicoureteral reflux is present, continuous bladder drainage is mandatory to prevent the radionuclide from refluxing from the bladder into the dilated upper tract, which would prolong the washout phase.
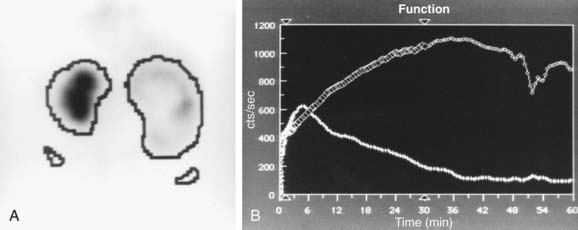
Figure 534-2 Same patient as in Figure 534-1. 6 wk old, MAG-3 diuretic renogram. The right kidney is on the right side of the image. A, Differential renal function: left kidney 70%, right kidney 30%. B, After administration of furosemide, drainage from the left kidney was normal and drainage from the right kidney was slow, consistent with right ureteropelvic junction obstruction. Pyeloplasty was performed on the right kidney.
Magnetic Resonance Urography
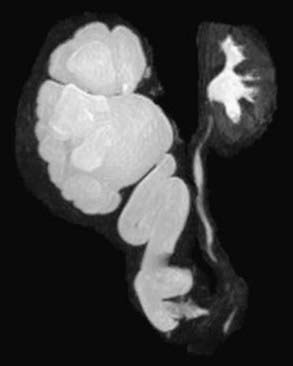
MR urography is also used to evaluate suspected upper urinary tract pathology. The child is hydrated and given intravenous furosemide. Gadolinium-DTPA is injected and routine T1-weighted and fat-suppressed fast spin-echo T2-weighted imaging is performed through the kidneys, ureters, and bladder. This study provides superb images of the pathology, and methodology permits assessment of differential renal function and drainage (Fig. 534-4). There is no radiation exposure; younger children need sedation or anesthesia. It is used primarily when renal sonography and nuclear imaging fail to delineate complex pathology.
Ancillary Studies
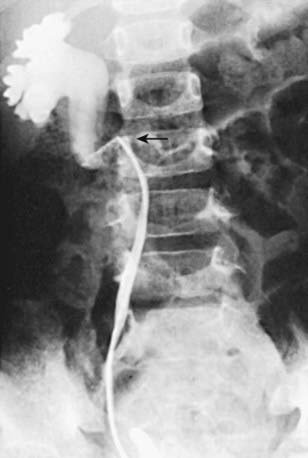
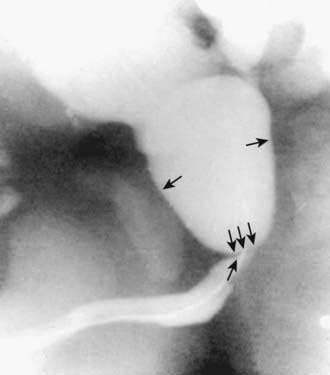
In unusual cases, an antegrade pyelogram (insertion of a percutaneous nephrostomy tube and injection of contrast agent), can be performed to assess the anatomy of the upper urinary tract. This procedure usually requires general anesthesia. In addition, an antegrade pressure-perfusion flow study (Whitaker test) may be performed, in which fluid is infused at a measured rate, usually 10 mL/min. The pressures in the renal pelvis and the bladder are monitored during this infusion, and pressure differences exceeding 20 cm H2O suggest obstruction. In other cases, cystoscopy with retrograde pyelography provides excellent images of the upper urinary tract (Fig. 534-5).
Specific Types of Urinary Tract Obstruction and Their Treatment
Ureteropelvic Junction Obstruction
Ureteropelvic junction (UPJ) obstruction is the most common obstructive lesion in childhood and usually is caused by intrinsic stenosis (see Figs. 534-1 to 534-4). An accessory artery to the lower pole of the kidney also can cause extrinsic obstruction. The typical appearance on ultrasonography is grade 3 or 4 hydronephrosis without a dilated ureter. UPJ obstruction most commonly manifests on antenatal sonography revealing fetal hydronephrosis; as a palpable renal mass in a newborn or infant; as abdominal, flank, or back pain; as a febrile UTI; or as hematuria after minimal trauma. Approximately 60% of cases occur on the left side, and the male:female ratio is 2 : 1. In 10% of cases, UPJ obstruction is bilateral. In kidneys with UPJ obstruction, renal function may be significantly impaired from pressure atrophy, but approximately half of affected kidneys have relatively normal function. The anomaly is corrected by performing a pyeloplasty, in which the stenotic segment is excised and the normal ureter and renal pelvis are reattached. Success rates are 91-98%. Pyeloplasty can be performed using laparoscopic techniques, often robotic-assisted using the da Vinci robot.
Midureteral Obstruction
Congenital ureteral stenosis or a ureteral valve in the midureter is rare. It is corrected by excision of the strictured segment and reanastomosis of the normal upper and lower ureteral segments. A retrocaval ureter is an anomaly in which the upper right ureter travels posterior to the inferior vena cava. In this anomaly, the vena cava can cause extrinsic compression and obstruction. An IVP shows the right ureter to be medially deviated at the level of the 3rd lumbar vertebra. The diagnosis may be confirmed by retrograde pyelography (see Fig. 534-5). Surgical treatment consists of transection of the upper ureter, moving it anterior to the vena cava, and reanastomosing the upper and lower segments. Repair is necessary only when obstruction is present. Retroperitoneal tumors, fibrosis caused by surgical procedures, inflammatory processes (as in chronic granulomatous disease), and radiation therapy can cause acquired midureteral obstruction.
Ectopic Ureter
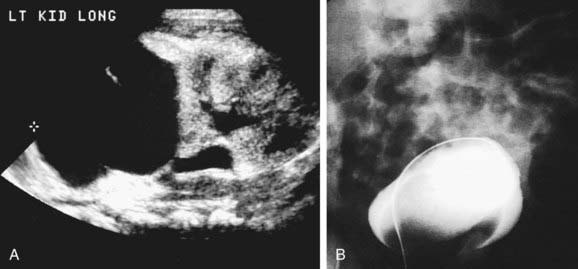
Evaluation includes a renal sonogram, VCUG, and renal scan, which demonstrates whether the affected segment has significant function. The sonogram shows the affected hydronephrotic kidney or dilated upper pole and ureter down to the bladder (Fig. 534-6). If the ectopic ureter drains into the bladder neck (female), a VCUG usually shows reflux into the ureter. Otherwise, there is no reflux into the ectopic ureter, but there may be reflux into the ipsilateral lower pole ureter or contralateral collecting system.
Ureterocele
In girls, ureteroceles nearly always are associated with ureteral duplication (Fig. 534-7), whereas in 50% of affected boys there is only 1 ureter. When associated with a duplication anomaly, the ureterocele drains the upper renal moiety, which commonly functions poorly or is dysplastic because of congenital obstruction. The lower pole ureter drains into the bladder superior and lateral to the upper pole ureter and may reflux.
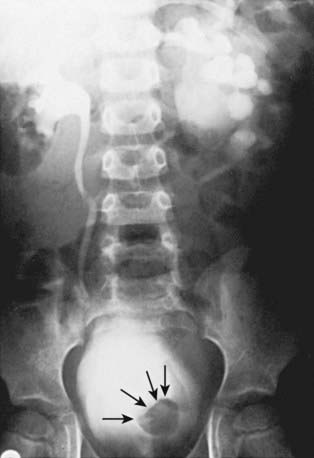
Orthotopic ureteroceles are associated with duplicated or single collecting systems, and the orifice is in the expected location in the bladder (Fig. 534-8). These anomalies usually are discovered during an investigation for prenatal hydronephrosis or a UTI. Ultrasonography is sensitive for detecting the ureterocele in the bladder and hydroureteronephrosis. IVP reveals varying degrees of ureteral and calyceal dilatation, and there is a round filling defect in the bladder. In delayed films, cystic dilatation of the ureter may be clearly visible and full of contrast material. Transurethral incision of the ureterocele effectively relieves the obstruction, but it can result in vesicoureteral reflux, necessitating ureteral reimplantation later. Some prefer open excision of the ureterocele and reimplantation as the initial form of treatment. Small, simple ureteroceles discovered incidentally without upper tract dilatation generally do not require treatment.
Megaureter
Table 534-3 presents a classification of megaureters (dilated ureter). Numerous disorders can cause ureteral dilation, and many are nonobstructive.
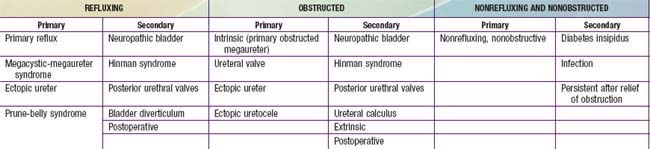
The primary obstructed nonrefluxing megaureter results from abnormal development of the distal ureter, with collagenous tissue replacing the muscle layer. Normal ureteral peristalsis is disrupted, and the proximal ureter widens. Usually there is not a true stricture. On IVP, the distal ureter is more dilated in its distal segment and tapers abruptly at or above the junction of the bladder (Fig. 534-9). The lesion may be unilateral or bilateral. Significant hydroureteronephrosis suggests obstruction. Megaureter predisposes to UTI, urinary stones, hematuria, and flank pain due to urinary stasis. In most cases, diuretic renography and sequential sonographic studies can reliably differentiate obstructed from nonobstructed megaureters. In most nonobstructed megaureters, the hydroureteronephrosis diminishes gradually (Fig. 534-10). Truly obstructed megaureters require surgical treatment, with excision of the narrowed segment, ureteral tapering, and reimplantation of the ureter. The results of surgical reconstruction usually are good, but the prognosis depends on pre-existing renal function and whether complications develop.
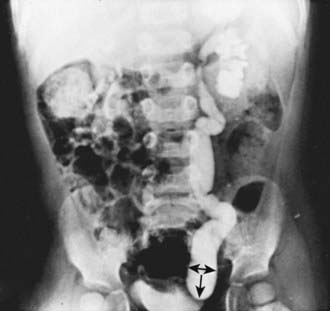
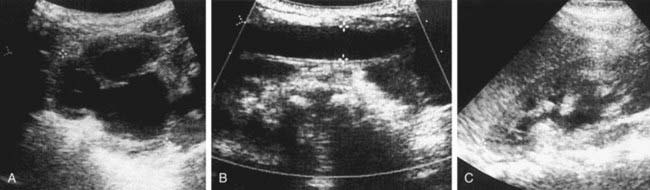
Prune-Belly Syndrome
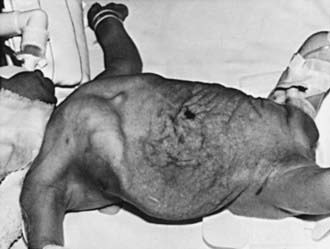
Prune-belly syndrome, also called triad syndrome or Eagle-Barrett syndrome, occurs in approximately 1 in 40,000 births; 95% of affected children are male. The characteristic association of deficient abdominal muscles, undescended testes, and urinary tract abnormalities probably results from severe urethral obstruction in fetal life (Fig. 534-11). Oligohydramnios and pulmonary hypoplasia are common complications in the perinatal period. Many affected infants are stillborn. Urinary tract abnormalities include massive dilatation of the ureters and upper tracts and a very large bladder, with a patent urachus or a urachal diverticulum. Most patients have vesicoureteral reflux. The prostatic urethra usually is dilated, and the prostate is hypoplastic. The anterior urethra may be dilated, resulting in a megalourethra. Rarely, there is urethral stenosis or atresia. The kidneys usually show various degrees of dysplasia, and the testes usually are intra-abdominal. Malrotation of the bowel often is present. Cardiac abnormalities occur in 10% of cases; >50% have abnormalities of the musculoskeletal system, including limb abnormalities and scoliosis. In girls, anomalies of the urethra, uterus, and vagina usually are present.
Posterior Urethral Valves
Affected boys with posterior urethral valves often are discovered prenatally when maternal ultrasonography reveals bilateral hydronephrosis, a distended bladder, and, if the obstruction is severe, oligohydramnios. Prenatal bladder decompression by percutaneous vesicoamniotic shunt or open fetal surgery has been reported. Experimental and clinical evidence of the possible benefits of fetal intervention is lacking, and few affected fetuses are candidates. Prenatally diagnosed posterior urethral valves, particularly when discovered in the 2nd trimester, carry a poorer prognosis than those detected after birth. In the male neonate, posterior urethral valves are suspected when there is a palpably distended bladder and the urinary stream is weak. If the obstruction is severe and goes unrecognized during the neonatal period, infants can present later in life with failure to thrive due to uremia or sepsis caused by infection in the obstructed urinary tract. With lesser degrees of obstruction, children present later in life with difficulty in achieving diurnal urinary continence or with UTI. The diagnosis is established with a VCUG (Fig. 534-12) or by perineal ultrasonography.
Urethral Strictures
In girls, true urethral strictures are rare because the female urethra is protected from trauma, particularly in childhood. In the past it was thought that a distal urethral ring commonly caused obstruction of the female urethra and UTI and that affected girls benefited from urethral dilatation. The diagnosis was suspected when a “spinning top” deformity of the urethra was found in the VCUG (see Fig. 537-3) and was confirmed by urethral calibration. There is no correlation between the radiologic appearance of the urethra in the VCUG and the urethral caliber and no significant difference in urethral caliber between girls with recurrent cystitis and normal age-matched controls. The finding usually is secondary to detrusor-sphincter dyssynergia. Consequently, urethral dilatation in girls rarely is indicated.
De Foor W, Clark C, Jackson E, et al. Risk factors for end stage renal disease in children with posterior urethral valves. J Urol. 2008;180:1705-1708.
Elder JS. Management of prenatal hydronephrosis. In: Puri P, Hollwarth M, editors. Pediatric surgery. Heidelberg, Germany: Springer-Verlag; 2009:825-838.
Elder JS, Shapiro E. Posterior urethral valves. In: Ashcraft KW, Holcomb GWIII, Murphy JP, editors. Pediatric surgery. ed 4. Philadelphia: Elsevier; 2009:781-792.
Estrada CR, Peters CA, Retik AB, et al. Vesicoureteral reflux and urinary tract infection in children with a history of prenatal hydronehprisos—should voiding cystourethrography be performed in cases of postnatally persistent grade II hydronephrosis? J Urol. 2009;181:801-806.
Grattan-Smith JD, Jones RA. Magnetic resonance urography in children. Magn Reson Imaging Clin N Am. 2008;16:515-531.
Heikkila J, Rintala R, Taskinen S. Vesicoureteral reflux in conjunction with posterior urethral valves. J Urol. 2009;182:1555-1560.
Heinlen JE, Manatt CS, Bright BC, et al. Operative versus nonoperative management of ureteropelvic junction obstruction in children. Urology. 2009;73:521-525.
Mukherjee S, Joshi A, Carroll D, et al. What is the effect of circumcision on risk of urinary tract infection in boys with posterior urethral valves? J Pediatr Surg. 2009;44:417-421.
Nguyen HT, Herndon CDA, Cooper C, et al. The Society for Fetal Urology consensus statement on the evaluation and management of antenatal hydronephrosis. J Pediatr Urol. 2010;6:212-231.
Parekh AD, Thomas JC, Trusler L, et al. Prospective evaluation of health related quality of life for pediatric patients with ureteropelvic junction obstruction. J Urol. 2008;180:2171-2175.
Pohl HG, Joyce GF, Wise M, et al. Vesicoureteral reflux and ureteroceles. J Urol. 2007;177:1659-1666.
Riccabona M, Avni FE, Blickman JG, et al. Imaging recommendations in paediatric uroradiology. Minutes of the ESPR uroradiology task force session on childhood obstructive uropathy, high-grade fetal hydronephrosis, childhood haematuria, and urolithiasis in childhood. ESPR Annual Congress, Edinburgh, UK, June 2008. Pediatr Radiol. 2009;39:891-898.
Seixas-Mikelus SA, Jenkins LC, Williot P, et al. Pediatric pyeloplasty: comparisons of literature meta-analysis of laparoscopic and open techniques with open surgery at a single institution. J Urol. 2009;182:2428-2434.
Vemulakonda VM, Cowan CA, Lendvay TS, et al. Surgical management of congenital ureteropelvic junction obstruction: a Pediatric Health Information System database study. J Urol. 2008;180:1689-1692.
Wu S, Johnson MP. Fetal lower urinary tract obstruction. Clin Perinatol. 2009;36:273-300.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]
Chapter 534 Obstruction of the Urinary Tract
Urinary tract obstruction can result from congenital (anatomic) lesions or can be caused by trauma, neoplasia, calculi, inflammatory processes, or surgical procedures, although most childhood obstructive lesions are congenital. Obstructive lesions occur at any level from the urethral meatus to the calyceal infundibula (Table 534-1). The pathophysiologic effects of obstruction depend on its level, the extent of involvement, the child’s age at onset, and whether it is acute or chronic.
Table 534-1 TYPES AND CAUSES OF URINARY TRACT OBSTRUCTION
| LOCATION | CAUSE |
|---|---|
| Infundibula | |
| Renal pelvis | |
| Ureteropelvic junction | |
| Ureter | |
| Bladder outlet and urethra |
Etiology
Ureteral obstruction occurring early in fetal life results in renal dysplasia, ranging from multicystic kidney, which is associated with ureteral or pelvic atresia (see Fig. 531-2), to various degrees of histologic renal cortical dysplasia that are seen with less severe obstruction. Chronic ureteral obstruction in late fetal life or after birth results in dilation of the ureter, renal pelvis, and calyces, with alterations of renal parenchyma ranging from minimal tubular changes to dilation of Bowman’s space, glomerular fibrosis, and interstitial fibrosis. After birth, infections often complicate obstruction and can increase renal damage.
Clinical Manifestations
Obstruction of the urinary tract generally causes hydronephrosis, which typically is asymptomatic in its early phases. An obstructed kidney secondary to a ureteropelvic junction (UPJ) or ureterovesical junction obstruction can manifest as a mass or cause upper abdominal or flank pain on the affected side. Pyelonephritis can occur because of urinary stasis. An upper urinary tract stone can occur, causing abdominal and flank pain and hematuria. With bladder outlet obstruction, the urinary stream may be weak; urinary tract infection (UTI; Chapter 532) is common. Many of these lesions are identified by antenatal ultrasonography; an abnormality involving the genitourinary tract is suspected in as many as 1/100 fetuses.
Diagnosis
Imaging Studies
Renal Ultrasonography
The presence of a dilated urinary tract is the most common characteristic of obstruction. Hydronephrosis is a common ultrasonographic finding (Fig. 534-1). Dilation is not diagnostic of obstruction and can persist after surgical correction of an obstructive lesion. Dilation can result from vesicoureteral reflux, or it may be a manifestation of abnormal development of the urinary tract, even when there is no obstruction. Renal length, degree of caliectasis and parenchymal thickness, and presence or absence of ureteral dilation should be assessed. Ideally, the severity of hydronephrosis should be graded from 1 to 4 using the Society for Fetal Urology grading scale (Table 534-2). The clinician should ascertain that the contralateral kidney is normal, and the bladder should be imaged to see whether the bladder wall is thickened, the lower ureter is dilated, and bladder emptying is complete. In acute or intermittent obstruction, the dilation of the collecting system may be minimal and ultrasonography may be misleading.
Table 534-2 SOCIETY FOR FETAL UROLOGY GRADING SYSTEM FOR HYDRONEPHROSIS
| RENAL IMAGE | ||
|---|---|---|
| GRADE OF HYDRONEPHROSIS | Central Renal Complex | Renal Parenchymal Thickness |
| 0 | Intact | Normal |
| 1 | Slight splitting | Normal |
| 2 | Evident splitting, complex confined within renal border | Normal |
| 3 | Wide splitting pelvis dilated outside renal border, calyces uniformly dilated | Normal |
| 4 | Further dilatation of pelvis and calyces (calyces may appear convex) | Thin |
After Maizels M, Mitchell B, Kass E, et al: Outcome of nonspecific hydronephrosis in the infant: a report from the registry of the Society for Fetal Urology, J Urol 152:2324–2327, 1994.
Radioisotope Studies
In a MAG-3 diuretic renogram, a small dose of technetium-labeled MAG-3 is injected intravenously (Figs. 534-2 and 534-3). During the first 2-3 min, renal parenchymal uptake is analyzed and compared, allowing computation of differential renal function. Subsequently, excretion is evaluated. After 20-30 min, furosemide 1 mg/kg is injected intravenously, and the rapidity and pattern of drainage from the kidneys to the bladder are analyzed. If no obstruction is present, half of the radionuclide should be cleared from the renal pelvis within 10-15 min, termed the half-time (t1/2). If there is significant upper tract obstruction, the t1/2 usually is >20 min. A t1/2 of 15-20 min is indeterminate. The images generated usually provide an accurate assessment of the site of obstruction. Numerous variables affect the outcome of the diuretic renogram. Newborn kidneys are functionally immature, and, in the first month of life, normal kidneys might not demonstrate normal drainage after diuretic administration. Dehydration prolongs parenchymal transit and can blunt the diuretic response. Giving an insufficient dose of furosemide can result in inadequate drainage. If vesicoureteral reflux is present, continuous bladder drainage is mandatory to prevent the radionuclide from refluxing from the bladder into the dilated upper tract, which would prolong the washout phase.
Figure 534-2 Same patient as in Figure 534-1. 6 wk old, MAG-3 diuretic renogram. The right kidney is on the right side of the image. A, Differential renal function: left kidney 70%, right kidney 30%. B, After administration of furosemide, drainage from the left kidney was normal and drainage from the right kidney was slow, consistent with right ureteropelvic junction obstruction. Pyeloplasty was performed on the right kidney.
Magnetic Resonance Urography
MR urography is also used to evaluate suspected upper urinary tract pathology. The child is hydrated and given intravenous furosemide. Gadolinium-DTPA is injected and routine T1-weighted and fat-suppressed fast spin-echo T2-weighted imaging is performed through the kidneys, ureters, and bladder. This study provides superb images of the pathology, and methodology permits assessment of differential renal function and drainage (Fig. 534-4). There is no radiation exposure; younger children need sedation or anesthesia. It is used primarily when renal sonography and nuclear imaging fail to delineate complex pathology.
Ancillary Studies
In unusual cases, an antegrade pyelogram (insertion of a percutaneous nephrostomy tube and injection of contrast agent), can be performed to assess the anatomy of the upper urinary tract. This procedure usually requires general anesthesia. In addition, an antegrade pressure-perfusion flow study (Whitaker test) may be performed, in which fluid is infused at a measured rate, usually 10 mL/min. The pressures in the renal pelvis and the bladder are monitored during this infusion, and pressure differences exceeding 20 cm H2O suggest obstruction. In other cases, cystoscopy with retrograde pyelography provides excellent images of the upper urinary tract (Fig. 534-5).
