Nutrition Support
Malnutrition
Malnutrition can occur as a result of combined protein-calorie deficiency (marasmus), predominantly protein deficiency (kwashiorkor), and deficiencies in specific micronutrients, as well as altered metabolism arising from a disease state such as sepsis, burns, or trauma. Critically ill patients may suffer from a combination of these causes. Malnutrition is thought to be present in as many as 25% to 50% of patients on hospital admission and may affect an additional 25% to 30% of patients during their hospital stay. The National Surgical Quality Improvement Program (NSQIP) has shown preoperative serum albumin, a measure of chronic nutritional status, to be an independent predictor of mortality risk in a study of more than 400,000 surgical patients and specifically in patients undergoing colectomy, proctectomy, transurethral resection of bladder tumors, and major lung resection.1–4 Malnutrition becomes particularly important in critically ill patients, in whom the combination of bed rest and catabolic illnesses such as sepsis, multiple trauma, burns, pancreatitis, and acute respiratory distress syndrome (ARDS) hasten the malnutrition, loss of lean body mass, and organ system dysfunction.
Starvation Versus Stress Metabolism
Stress metabolism is a generalized response whereby energy and substrate are mobilized to support inflammation, immune function, and tissue repair. It occurs in response to a variety of stimuli such as sepsis, multiple trauma, burns, pancreatitis, bone marrow transplantation, and major surgery. This mobilization of energy and substrate occurs at the expense of lean body mass. It is driven by endocrine hormones such as cortisol, glucagon, and catecholamines, as well as by a multitude of inflammatory mediators. The stress response is often related to some degree of perfusion deficit (shock) and resultant microcirculatory injury. Clinically, the response is characterized by increased energy expenditure and increased oxygen consumption. The stress response is further characterized by hyperglycemia, elevated lactate, and increased urinary nitrogen excretion. The respiratory quotient is often elevated in the range of 0.80 to 0.95 as a result of the use of a mixed oxidative fuel source. Loss of lean body mass occurs more rapidly than with simple starvation because skeletal muscle protein stores become the “fuel” for the stress response. Most important, the malnutrition associated with stress metabolism is less responsive to nutrition support than is starvation metabolism. Reversal of stress-associated malnutrition is dependent not only on the provision of adequate nutrients but also on elimination of the underlying stress response (i.e., control of infection, stabilization of fractures, grafting of burns, or resolution of the inflammatory state). Starvation and stress metabolism are summarized in Table 82.1.
Table 82.1
Starvation Versus Stress Hypermetabolism
| Characteristic | Starvation | Hypermetabolism |
| Energy expenditure | Decreased | Increased |
| Respiratory quotient | Low (0.7) | High (0.85) |
| Response to feeding | +++ | + |
| Mediator activation | + | +++ |
| Primary fuels | Fat | Mixed |
| Gluconeogenesis | + | +++ |
| Proteolysis | + | +++ |
| Protein synthesis | + | ++ |
| Ureagenesis/urinary urea nitrogen | + | +++ |
| Ketone formation | ++++ | + |
Carbohydrate Metabolism in Critical Illness
Carbohydrate metabolism in critical illness is characterized clinically by hyperglycemia, often described as being due to “insulin resistance” based on increased blood glucose levels in the presence of high circulating levels of insulin. In fact, cellular glucose uptake and oxidation in the critically ill are increased,5–7 and hyperglycemia is associated with increased glucose production, decreased insulin-mediated glucose uptake, and increased non–insulin-mediated glucose uptake.5,8
Glucose production is increased primarily by increased hepatic gluconeogenesis, to a lesser extent by renal gluconeogenesis,9 and by increased glycogenolysis. Increased glucose production occurs under the influence of glucagon, catecholamines, and cortisol, with glucagon being the most important stimulant of hepatic gluconeogenesis and epinephrine being the primary stimulant of glycogenolysis.10,11 The hormonal changes observed in critical illness appear to be mediated by cytokines in both the central nervous system and peripheral tissues. Interleukin 1 (IL-1) stimulates the release of adrenocorticotropic hormone, which in turn favors the release of cortisol and glucagon, and tumor necrosis factor (TNF) stimulates increased secretion of glucagon.12 Gluconeogenic substrates include lactate and alanine, as well as glutamine, glycine, serine, and glycerol. The amino acids used for gluconeogenesis are derived largely from proteolysis in skeletal muscle.
Glucose uptake into cells is regulated by a process of facilitated transport in which a carrier protein promotes the movement of glucose across the cell membrane down its concentration gradient. Three isoforms of this glucose carrier protein (glucose transporter 1 [GLUT-1], GLUT-2, and GLUT-4) are thought to be important in glucose transport.13 Non–insulin-mediated glucose uptake is dependent on the GLUT-1 isoform, whereas insulin-mediated glucose transport is dependent on the GLUT-4 isoform. During stress, total body glucose uptake is increased, but largely via non–insulin-mediated pathways.8 Non–insulin-mediated glucose uptake is particularly important in the central nervous system and in tissues rich in macrophages and neutrophils (e.g., lung, liver, intestine, spleen, wound), which use glucose for energy and in the process generate the respiratory burst.14 Inflammatory cytokines may promote the uptake of glucose by increasing the synthesis, plasma membrane concentration, or activity of the glucose transporter.15,16 Insulin-mediated glucose transport is suppressed in the liver, heart, and skeletal muscle.17,18 Hepatic insulin resistance is characterized by elevated circulating levels of insulin-like growth factor-binding protein-1.17,18 The mechanisms of insulin resistance are incompletely understood, although proinflammatory cytokines may alter insulin receptor signaling.14,19,20
The net result of these alterations in carbohydrate metabolism in critical illness is hyperglycemia inasmuch as the liver seems to be unresponsive to high levels of circulating insulin and glucose and continues to synthesize glucose via gluconeogenesis. In other tissues, non–insulin-mediated glucose uptake is enhanced and results in increased glycolytic oxidation to pyruvate and a stoichiometric rise in lactate.5,6 Although hyperlactatemia is usually thought of as being reflective of anaerobic metabolism, it can be indicative of glycolytic flux and the level of metabolic stress when observed in conjunction with elevated pyruvate.5
Fat Metabolism in Critical Illness
In both starvation and stress metabolism, fat metabolism is characterized by increased lipolysis and decreased lipogenesis as fat stores are mobilized for energy. In the stressed state there is a marked increase in lipolytic activity in adipose tissue as a result of catecholamine-mediated stimulation of β2-receptors; cytokines may also participate in this process.21,22 Fatty acids are released from adipose tissue in quantities that exceed the amount oxidized, and approximately half of these fatty acids are re-esterified in the liver.23 In this triglyceride–fatty acid cycle, triglycerides and fatty acids are shunted back and forth between the liver and adipose tissue in an apparently futile pathway.24
Hypertriglyceridemia is common in critically ill patients, particularly in the presence of multiple organ dysfunction, and results from the combination of increased hepatic triglyceride production and decreased clearance.25,26 Hepatic steatosis has classically been attributed to overfeeding because triglycerides are constructed from fatty acids synthesized from excess carbohydrate. This theory has been called into question inasmuch as a substantial proportion of hepatic triglyceride may be synthesized from recycled fatty acids rather than from fatty acids synthesized de novo from carbohydrate.27 It is possible that fatty infiltration of the liver reflects a cytokine-mediated defect in triglyceride secretion that represents an organ-specific response to critical illness rather than an adverse effect of carbohydrate overfeeding.28,29 Regardless of the immediate source of the fatty acids used in triglyceride synthesis, experience suggests that clinically apparent fatty infiltration of the liver can be prevented by avoiding overfeeding.
Ketonemia is common in starvation, whereas ketogenesis is decreased in stress metabolism.30 Acetoacetate continues to be used as an oxidative fuel source, although the reduction of β-hydroxybutyrate to acetoacetate is impaired.31 With the development of multiple organ dysfunction, there is a progressive decrease in the acetoacetate/β-hydroxybutyrate ratio.32 This alteration in hepatic redox potential reflects a disturbance in the cellular energy charge that may be associated with hepatic mitochondrial damage by toxic oxygen radicals and other inflammatory mediators.33
Protein Metabolism in Critical Illness
Protein synthesis is increased in the stressed state relative to that seen in starvation. Protein breakdown is markedly increased in comparison to the synthetic rate, thereby resulting in net protein catabolism and a rapid decrease in lean body mass.30 There is a net efflux of amino acids from skeletal muscle as protein breakdown via the ubiquitin-proteosome pathway of protein degradation is accelerated by catecholamines, cortisol, and cytokines.34–37 Amino acid uptake by skeletal muscle is impaired despite excess amino acids circulating in the bloodstream early in the course of stress metabolism; the situation deteriorates further as plasma concentrations of specific amino acids such as leucine begin to fall.38 Decreased plasma glutamine is known to have deleterious effects on immune function and gastrointestinal barrier function.39,40 Although it has long been thought that the accelerated protein catabolism associated with critical illness is unresponsive to the provision of amino acids or other fuel sources, the fact that muscle amino acid uptake is in part compromised by decreased plasma levels of specific amino acids suggests the possibility that protein catabolism during stress may be attenuated with the provision of appropriate nutrition support.41
Amino acids mobilized from skeletal muscle are redistributed to other areas of the body to support immune function, wound healing, and tissue repair, as well as for the hepatic synthesis of acute-phase proteins, presumably in an attempt to enhance survival. It has recently been demonstrated that endotoxemia induces significant increases in protein synthesis in the liver, spleen, kidney, jejunum, diaphragm, lung, and skin at the expense of skeletal muscle catabolism.42 Amino acids such as alanine, glutamine, glycine, and serine are used as gluconeogenic substrate, and branched-chain amino acids (leucine, isoleucine, and valine) can be used as an oxidative fuel source, particularly in skeletal muscle. The net result of stress protein metabolism is a rapid decrease in lean body mass that exceeds that associated with bed rest or simple starvation, along with increased ureagenesis, azotemia, and increased urinary nitrogen excretion.
Nutrition Support in Critical Illness
Calories
In any metabolic state, energy requirements must be met to minimize the use of stored energy reserves and to decrease the loss of lean body mass. In the setting of adapted starvation, the protein-sparing effect of an adequate caloric intake is well recognized; however, in the stressed state, protein catabolism is only partially responsive to caloric intake and continues to a significant degree regardless of caloric intake.30,41 Overfeeding is of particular concern in a critically ill patient because it can result in excess carbon dioxide production and ventilator dependence,43 lipogenesis and fatty infiltration of the liver,44–46 and hyperglycemia with its attendant hyperosmolar and infectious complications.14,47–49
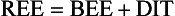
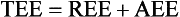
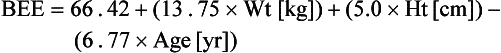
For females:
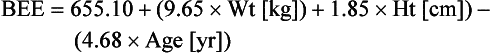
Classically, REE is then estimated by multiplying the calculated BEE by a stress factor ranging from 1.0 in a stable, mechanically ventilated patient to 2.0 in a patient with a 50% total body surface area burn. Stress factors for patients with multiple trauma or sepsis fall somewhere between the two extremes. TEE is estimated by multiplying REE by an activity factor of 1.2 to 1.3 for ambulatory patients. The caloric requirement is decreased to varying degrees (6% to 30%) in mechanically ventilated patients receiving narcotics or sedatives and is reduced by as much as 33% in chemically paralyzed patients.50 In nonventilated postoperative patients or in patients with decreased lung compliance, energy expenditure related to the work of breathing may increase. Although estimation of energy expenditure is relatively accurate in healthy people, it is much less reliable in critically ill patients because of the heterogeneity of the metabolic response and variations in sedation, work of breathing, and physical activity. Most critically ill patients should receive calories to supply 100% to 120% of calculated BEE. Estimation of energy expenditure in obese patients is particularly difficult. If indirect calorimetry is unavailable, the provision of calories should be based upon body mass index (BMI) and ideal body weight rather than on obesity adjusted body weight.51
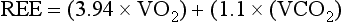
where VO2 = oxygen consumption and VCO2 = carbon dioxide production.
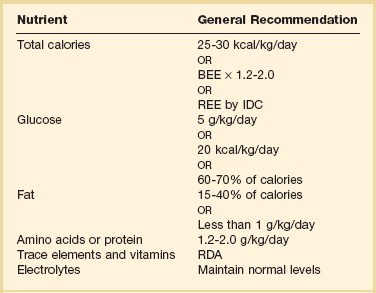
In general, critically ill patients should receive 25 to 30 kcal/kg/day, with sedated mechanically ventilated patients receiving closer to 25 kcal/kg/day. Chemically paralyzed patients generally require 20 kcal/kg/day.52 In obese patients, hypocaloric feeding may induce some degree of weight loss and improve insulin sensitivity and may decrease ventilator days and length of ICU stay.52 In patients with a BMI greater than 30, calories should not exceed 60% to 70% of target energy requirements or 11 to 14 kcal/kg actual body weight per day or 22 to 25 kcal/kg ideal body weight per day.51
Carbohydrate
Carbohydrate is usually the primary source of calories in human beings. The maximal rate of glucose oxidation is approximately 5 mg/kg/minute, or 7.2 g/kg/day.53 In stressed patients, part of this maximally tolerated glucose load will be provided by glucose synthesized endogenously from amino acids via gluconeogenesis. In a severely hypermetabolic patient, gluconeogenesis may provide as much as 4 mg/kg/minute of glucose54 and result in significant hyperglycemia when large exogenous glucose loads are administered. Insulin tends to be ineffective in controlling this hyperglycemia, in part because glucose oxidation may already be maximal, endogenous insulin levels are already high, and insulin-mediated glucose uptake in the liver and other tissues is suppressed in a septic and hypermetabolic patient.17,18 Complications of excess glucose administration include hyperglycemia and its attendant hyperosmolar/infectious complications, excess carbon dioxide production and ventilator dependence, and hepatic steatosis. In general, glucose should initially be provided at a rate of 5 g/kg/day or approximately 20 kcal/kg/day. Carbohydrate should generally constitute 60% to 70% of nonprotein calories in a hypermetabolic patient (Table 82.2).
Fat
In a hypermetabolic patient, fat should constitute 15% to 40% of daily caloric intake, both to prevent essential fatty acid deficiency and to meet caloric needs in the face of a fixed capacity to oxidize glucose. In the starved state, as little as 2% to 5% of calories can be provided as fat, primarily to prevent essential fatty acid deficiency. Substituting lipid calories for carbohydrate calories can reduce carbon dioxide production, although avoiding excess calories in general is probably most important in minimizing carbon dioxide production and ventilator dependence. Complications of excess lipid administration, particularly when given parenterally, include hyperlipemia, immunosuppression, and hypoxemia as a result of both impaired oxygen diffusion and ventilation-perfusion mismatching. In general, a hypermetabolic patient should receive 15% to 40% of calories as fat, not to exceed 1.0 to 1.5 g/kg/day (see Table 82.2).55
Protein
Protein needs in a hypermetabolic patient are increased in comparison to those in a patient with simple starvation. Protein catabolism in a stressed patient has long been thought to be unresponsive to protein or amino acid administration or glucose infusion,56 and attainment of nitrogen balance is thought to depend largely on the support of stress protein synthesis.41 However, recent evidence suggests that protein catabolism may be attenuated by the provision of appropriate nutrition support.42 Regardless of the specific mechanisms involved, lean body mass decreases during catabolic illness because of bed rest and inactivity. Amino acids are redistributed from skeletal muscle to support hepatic protein synthesis, the cellular inflammatory response, and gluconeogenesis and are used as oxidative fuel sources. The critically ill or injured patient may require 1.2 to 2.0 g/kg/day to promote nitrogen balance or at least minimize the nitrogen deficit. With the exceptions noted later, the provision of more than 2.0 g/kg/day of protein rarely promotes nitrogen balance and usually results simply in increased urinary nitrogen excretion (see Table 82.2).
To meet the protein requirements of the stressed state and at the same time avoid excess calories and the attendant complications, an injured or septic patient may require nutrition support with a nonprotein calorie-to-nitrogen ratio of 80 : 1 or 100 : 1, as compared with a patient with starvation metabolism, who may tolerate a nonprotein calorie-to-nitrogen ratio of 150 : 1 or higher. The higher protein needs of a patient with catabolic illness can be met with custom-compounded total parenteral nutrition (TPN) or with numerous commercially available enteral formulas specifically designed for such patients (Table 82.3).
In summary, most critically ill patients should receive 1.2 to 2.0 g/kg/day of protein. Obese patients with BMI 30 to 40 should receive at least 2.0 g/kg/day of protein and patients with BMI greater than 40 should receive at least 2.5 g/kg/day in conjunction with hypocaloric feeding.51 Critically ill patients with renal failure should generally receive 1.25 to 1.75 g/kg/day and those requiring continuous renal replacement therapy (CRRT) may require up to 2.5 g/kg/day.51
Electrolytes, Vitamins, and Trace Elements
Fluid and electrolytes should be provided to maintain adequate urine output and normal serum electrolyte levels. Typical daily electrolyte requirements include sodium 60 to 100 mEq/day, potassium 60 to 100 mEq/day, magnesium 10 to 20 mEq/day, calcium 10 to 15 mEq/day, chloride 80 to 120 mEq/day, and phosphorus 20 to 30 mmol/day.57 Intravenous amino acid formulations contain acetate, but additional acetate can be added in the setting of metabolic acidosis. Particular attention should be paid to the intracellular electrolytes (potassium, phosphorus, and magnesium), which are required for attainment of nitrogen balance58 and serum levels of which can fall precipitously when nutrition support is initiated.
The requirements for vitamins and trace elements in critical illness are largely unknown, and in general the recommended dietary allowance (RDA) for both should be provided. Antioxidant vitamins (including vitamins E and ascorbic acid) and trace minerals (including zinc, copper, and particularly selenium) may improve outcome in burns, in trauma, and in critically ill patients requiring mechanical ventilation59 and should be provided to all critically ill patients receiving specialized nutrition support.51 Patients with prolonged diarrhea or large burns or those undergoing dialysis have increased trace element loss and vitamin requirements, and deficiencies can develop if intake is inadequate. Initial limitation of trace elements should be considered in patients with renal failure. Routine supplementation of vitamins is necessary in patients receiving TPN. Typically, one ampule of a standard multivitamin preparation is added to TPN daily. Vitamin K, which is light sensitive, must be added separately in a light-impermeable bag or given by another route once or twice weekly. Excessive doses of vitamin C should be avoided in renal failure because of the accumulation of oxalate. Vitamin A accumulates and should not be supplemented beyond the RDA.60
Route and Timing of Administration
The gastrointestinal tract is a major interface between the host and the environment and not only regulates the ingestion and absorption of nutrients but is also responsible for defending the host against noxious microorganisms and toxins.61 Malnutrition impairs gastrointestinal barrier function,62 as does the lack of luminal nutrients independent of general nutritional status.63,64 Enteral nutrition has been shown to promote mucosal growth, improve absorptive capacity, alter digestive enzyme production, improve gut mucosal weight, generate DNA and protein synthesis, and improve the efficiency of nutrient utilization.65 Enteral nutrition stimulates intestinal contractility; the release of trophic substances such as bile salts, gastrin, and motilin; and the release of secretory IgA.65a-65d Enteral nutrition supports commensal bacteria, which in turn degrade bacterial toxins and prevent gut colonization by pathogenic organisms.65a,65e Further, enteral nutrition stimulates gut blood flow and supports gut-associated lymphoid tissue (GALT).65a-65d Enteral nutrition supports the processing of naïve CD4 helper lymphocytes by exposure to bacterial antigens in the gut. These IgA-producing lymphocytes then migrate to distant organs such as lungs, liver, and kidneys, where they form mucosal-associated lymphoid tissue (MALT) and produce secretory IgA.65a-65d TH2 CD4 helper lymphocytes, the proliferation of which are stimulated by feeding, enter the circulation and have an anti-inflammatory effect.65a,65f,65g Enteral nutrients stimulate the proliferation of TH1 and TH3 lymphocytes,65f which in turn release transforming growth factor–beta (TGF-β), resulting in a further down-regulatory or anti-inflammatory effect.65a,65f
Several randomized prospective trials in a variety of critically ill patient populations, including trauma, burns, head injury, major surgery, and acute pancreatitis, have documented the benefits of enteral compared to parenteral nutrition.66–71 Few studies show a beneficial effect on mortality rate, but most demonstrate reduced infectious morbidity, primarily pneumonia and central line infections. In trauma patients, enteral nutrition has been associated with a reduction in abdominal abscesses.67 Further, enteral nutrition has been associated with decreased length of hospital stay,69 and in head injury patients, return of cognitive function.71 Several meta-analyses comparing enteral to parenteral nutrition have shown significant reductions in infectious complications with the use of enteral nutrition.72–75
Parenteral nutrition, although able to maintain overall nutritional status, does not offer the benefits related to gut mucosal integrity and immune function mentioned earlier. Whether this is the primary reason for the increased infection risk associated with parenteral nutrition is less clear. In several of the studies mentioned in previous paragraphs, many of the patients managed with parenteral nutrition received significantly more calories and had a higher incidence of hyperglycemia73 than did their enterally fed counterparts. In a study in which parenterally fed patients had a higher incidence of sepsis, twice as many patients receiving parenteral nutrition had hyperglycemia as enterally fed patients.70 More recently, in a large prospective trial comparing tight glucose control with standard glucose control, patients with tight glucose control had significantly lower rates of morbidity, infectious complications, and mortality.49 Further analysis of these data revealed that when glucose was tightly controlled, there was no difference in infection when patients were nourished parenterally rather than enterally.77 These data suggest that control of hyperglycemia may be as important as the route of nutrition in minimizing infectious complications.
In summary, because it is less costly and associated with fewer metabolic complications and a lower incidence of infection, the enteral route continues to be the recommended route for nutrition support.51 That said, in as many as 15% of critically ill patients, enteral nutrition is either contraindicated or not tolerated, and parenteral nutrition remains a viable option, particularly when overfeeding is avoided and hyperglycemia is controlled.51
The timing for the initiation of nutrition support in critically ill patients has also been controversial. It has been suggested that enteral nutrition started early (within 24 to 72 hours of admission or the onset of the hypermetabolic insult) is associated with less gut permeability, release of inflammatory cytokines, and reduced systemic endotoxemia.69 Further, the accumulation of an energy deficit early in the course of an ICU admission has been significantly associated with the occurrence of ARDS, renal failure, pressure ulcers, and the need for surgery77a and with longer ICU length of stay and more days of mechanical ventilation.77,78 In a meta-analysis by Heyland and associates, early enteral nutrition was associated with a trend toward reduced infectious morbidity and mortality rates.69 A second meta-analysis by Marik and Zaloga demonstrated significant reductions in infectious morbidity and hospital length of stay with early enteral nutrition as compared to delayed feedings.77 In patients who are hemodynamically stable, it is recommended that enteral nutrition be started within 48 hours of admission.76
The early use of TPN to supplement enteral nutrition and thus prevent a caloric deficit has been controversial.69,71,78 In a recent prospective, randomized, multi-institutional trial 2312 patients were randomized to initiation of parenteral nutrition to supplement insufficient enteral nutrition within 48 hours of ICU admission (early initiation group) and compared to 2328 patients randomized to receive supplementary parenteral nutrition no earlier than 8 days after ICU admission (late initiation group). All patients received early enteral nutrition by protocol and had insulin infused to maintain normoglycemia. The late initiation group was slightly more likely to be discharged alive from the ICU and from the hospital without evidence of decreased functional status. Further, patients in the late initiation group had significantly fewer infections, a significant reduction in the proportion of patients requiring more than 2 days of mechanical ventilation, a significant median reduction of 3 days in the duration of renal replacement therapy, and a mean reduction in health care costs of approximately $1600. These patients were considered to be at nutritional risk but not malnourished on admission based on BMI.79 These results suggest that early parenteral nutrition, even when used to supplement enteral nutrition, may be harmful and should be avoided unless the patient is chronically malnourished.
Types of Nutritional Formulas
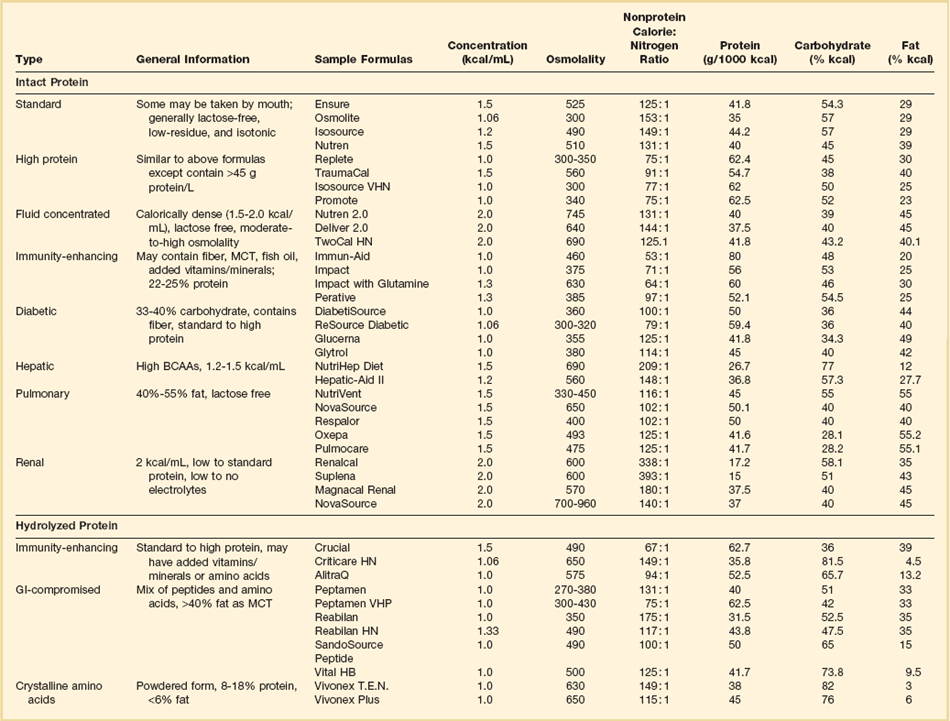
Enteral formulas are usually premixed with a fixed nonprotein calorie-to-nitrogen ratio, and the needs of a specific patient are generally met by changing the formula. Protein and carbohydrate supplements can be added at the bedside to alter premixed formulas. Enteral formulas can be classified numerous ways, including the form in which protein is provided (e.g., intact protein, hydrolyzed protein [chemically defined or peptide], or crystalline amino acids [elemental]), the quantity of protein contained, the caloric density of the formula, and the disease state for which they were designed. Examples of different formulas are shown in Table 82.3. More detailed information is available elsewhere.80
Intact protein solutions contain protein as caseinate or soy isolate–based products that are lactose-free, gluten-free, and low in residue. Such solutions contain 45% to 60% of calories as carbohydrate (oligosaccharides), 20% to 35% of calories as long-chain fats (e.g., linoleic acid), and 15% to 20% of calories as protein. Intact formulas are usually isosmotic and may contain 1 to 2 kcal/mL of solution. Some formulas contain fiber as a means to improve diarrhea or glycemic control. Hydrolyzed formulas provide protein as peptides or amino acids, are generally low in fat, and are designed for patients with gut dysfunction or malabsorption.81 Elemental solutions contain protein exclusively as amino acids and are intended for similar indications. Controlled studies comparing hydrolyzed or elemental formulas with intact formulas have not demonstrated improved tolerance or outcomes.
Organ-Specific Enteral Formulas
Pulmonary failure formulas are designed for patients with acute respiratory failure associated with chronic lung disease. They contain at least 50% of calories as fat and thus reduce CO2 production and decrease the work of breathing relative to high-carbohydrate formulas. Few data suggest a benefit with specific pulmonary formulas, and avoiding overfeeding is more important in reducing ventilatory demand.82,75
Hepatic failure formulas were developed for patients with encephalopathy. They contain high concentrations of branched-chain amino acids and reduced concentrations of aromatic amino acids. Although these solutions have been shown to correct the abnormal amino acid profile characteristic of patients with liver failure,83,84 it is less clear that they actually treat hepatic encephalopathy.84 Hepatic failure formulas should be reserved for the rare encephalopathic patient who is refractory to lactulose and luminal antibiotics.51
Immunomodulating Enteral Formulas
Arginine is a nonessential amino acid that has both beneficial and deleterious effects. It is a secretagogue for anabolic hormones, supports T-cell function, detoxifies ammonia, and supports wound healing via metabolism to polyamine and proline.85,86 An arginine deficiency may develop after trauma or major surgery that is mediated by pathologic release of arginase from granulocytes.86–88 Arginine deficiency impairs the immune response, which may result in increased infections and in impaired wound healing.86,87,89 Arginine is a precursor for the synthesis of nitric oxide, which in turn is a modulator of hepatic protein synthesis and vascular tone. This nitric oxide–induced vasodilation may be beneficial in some circumstances but in the setting of severe sepsis and hypotension may aggravate hemodynamic collapse. Further, nitric oxide is metabolized to peroxynitrite,86,90 which is a potent oxidizing and nitrating agent that may damage mitochondria, increase gut barrier permeability, and promote organ dysfunction.86,90–95
Omega-3 fatty acids are incorporated into the phospholipid fraction of cell membranes and converted to trienoic prostaglandins and pentaenoic leukotrienes, which are less “inflammatory” than their ω-6 counterparts. Omega-3 fatty acids have been shown to decrease prostaglandin E2, TNF, and IL-1 production in rat Kupffer cells96 and alter TNF and IL-1 production in human monocytes.97 Furthermore, ω-3 fats have been shown to be beneficial in animal models of chronic inflammation and in humans with psoriasis98 and rheumatoid arthritis.99
Glutamine is an important fuel for enterocytes and cells of the immune system. Considered a nonessential amino acid, glutamine may become conditionally essential when skeletal muscle stores and plasma levels become depleted during catabolic illness, thereby resulting in adverse effects on gut barrier and immune function.100–102 Glutamine has been shown to improve nitrogen balance and decrease bacterial translocation in animals.103–105 It also promotes the synthesis of glutathione, an important antioxidant. A meta-analysis of 14 randomized trials in which glutamine-supplemented nutrition was compared with standard nutrition demonstrated reduced infectious morbidity and mortality rates with glutamine supplementation, particularly in parenterally nourished surgical patients.106 Unfortunately, glutamine dipeptide, which is the optimal form for parenteral administration due to stability and the minimal added volume required for infusion, is not currently available in the United States.86
Antioxidants such as vitamins A, E, and C; selenium; and N-acetylcysteine offer the potential to reduce oxidant injury but have not been studied individually in critically ill patients in terms of clinical outcomes.107
Numerous studies comparing immunity-enhancing enteral nutrition with conventional nutrition have produced contradictory results. In a 2001 meta-analysis of 22 studies (2419 patients) in which enteral nutrition supplemented with various combinations of arginine, ω-3 fatty acids, glutamine, and nucleotides was compared with conventional enteral nutrition, the supplemented patients had decreased infectious morbidity but no difference in mortality rates when compared with patients receiving the control diet.108 The majority of studies demonstrating benefit involved elective surgery patients and not critically ill patients. In the subsequently published and largest (597 patients) randomized trial to date, immunonutrition had no benefit in terms of infectious morbidity, length of hospital stay, number of ventilator days, or mortality rate.109 In retrospect, it appears that a number of factors may have been responsible for the conflicting results mentioned earlier. First, there was considerable heterogeneity in the patient populations studied (major elective surgery, burns, trauma, sepsis, and ARDS). Second, the immunomodulating enteral formulas studied contained varying combinations of arginine, ω-3 polyunsaturated fatty acids (fish oil), glutamine, and antioxidants. These differences were addressed in a 2008 meta-analysis of 24 studies by Marik and Zaloga in which studies were analyzed by patient type (ICU, burns, and trauma) and by the combination of immunomodulating nutrients in the subject study formula (fish oil, arginine, glutamine, fish oil plus arginine, fish oil plus arginine plus glutamine).110 All enteral formulas contained antioxidants, primarily selenium, though in varying doses, and could not be studied separately. Overall, the immunomodulating diets had no effect on mortality rate but did demonstrate significant reductions in secondary infections. Mortality rates, infections, and length of stay were significantly reduced only in ICU patients with sepsis, systemic inflammatory response syndrome (SIRS), and ARDS receiving a formula supplemented with fish oil alone.111–113 Infection and length of stay were reduced in the 13 ICU studies, but this effect disappeared when the three fish oil studies were excluded.110 It appears that arginine, perhaps due to the preceding mechanisms, counteracts the beneficial effects of fish oil in trauma and ICU patients with sepsis/SIRS.
Currently, the 2009 guidelines for the Provision and Assessment of Nutrition Support Therapy in the Adult Critically Ill Patient, published jointly by the Society of Critical Care Medicine and the American Society for Parenteral and Enteral Nutrition, recommend the use of an immunomodulating enteral formula supplemented with fish oil and antioxidants for patients with acute lung injury/ARDS.75 Further, these guidelines recommend enteral formulas supplemented with arginine for patients with major surgery, burns, and trauma, but emphasize that such formulas may be harmful in severe sepsis. Finally, the guidelines describe glutamine as possibly beneficial in trauma and burns.51
Nutritional Assessment and Monitoring
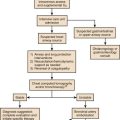
The history and physical examination remain the mainstay of nutritional assessment, although they are perhaps more useful in the ambulatory setting or in patients with chronic malnutrition.114 Pertinent historical information includes height and weight, a history of recent weight change, genetic background, recent nutritional intake, and a history of disease that might affect nutrient intake, absorption, or tolerance. The physical examination might, on occasion, demonstrate abnormal end-organ function that reflects malnutrition, but more commonly it is useful for the assessment of body mass and detection of specific nutrient deficiencies. Signs and symptoms of selected vitamin and mineral deficiencies are presented in Tables 82.4 and 82.5.
Table 82.4
| Vitamin | Function | Signs of Deficiency |
| Niacin (vitamin B5) | Component of the coenzymes NAD and NADP, which catalyze oxidation-reduction reactions and play a role in the oxidative catabolism of carbohydrates, proteins, and lipids and the biosynthesis of fatty acids | Pellagra, dermatitis, headaches, loss of memory, dementia, glossitis, diarrhea |
| Folate (vitamin B9) | Transfer of single-carbon units; DNA synthesis | Megaloblastic anemia, diarrhea, glossitis |
| Cyanocobalamin (vitamin B12) | Maintains normal folate metabolism; coenzyme in reactions involving isomerizations and reductions; participates in the metabolism of fat, carbohydrate, and protein and in myelin synthesis | Pernicious anemia, glossitis, peripheral neuropathy, spinal cord degeneration |
| Thiamine | Carbohydrate metabolism coenzyme in oxidative decarboxylation | Paresthesias, impaired memory, nystagmus, congestive heart failure, Wernicke-Korsakoff syndrome |
| Riboflavin (vitamin B2) | Electronic transport as flavin nucleotides | Mucositides, dermatitis, cheilosis, vascularization of the cornea, photophobia, decreased vision, lacrimation, impaired wound healing |
| Pyridoxine (vitamin B6) | Coenzyme in transformations of amino acids | Neuritis, dermatitis, convulsions |
| Pantothenic acid | Precursor of coenzyme A (Krebs cycle) | Headache, fatigue, malaise, insomnia, vomiting, abdominal cramps |
| Biotin | Coenzyme for carboxylation reactions | As with other B vitamins (normally synthesized and absorbed from gut) |
| Ascorbic acid (vitamin C) | Reducing agent, wound healing, integrity of blood vessels, folate metabolism | Enlargement and keratosis of hair follicles, impaired wound healing, anemia, ecchymosis, lethargy, depression, bleeding |
| Vitamin A | Normal vision, mucopolysaccharide synthesis, protease release, entry of macrophages and leukocytes into an acute wound, immune stimulation, mucosal integrity | Dermatitis, keratomalacia, xerophthalmia, night blindness |
| Vitamin D | Calcium and phosphorus homeostasis | Rickets, osteomalacia |
| Vitamin E | Antioxidant | Hemolysis |
| Vitamin K | Clotting factors II, VII, IX, X | Bleeding |
NAD, nicotinamide-adenine dinucleotide; NADP, nicotinamide-adenine dinucleotide phosphate.
Table 82.5
| Trace Element | Function | Signs of Deficiency |
| Chromium | Glucose tolerance; possible role in maintenance of normal serum lipid levels | Elevated serum lipids, insulin-resistant glucose intolerance |
| Copper | Metalloenzyme biochemical processes, component of ceruloplasmin, connective tissue metabolism, melanin formation | Anemia, neutropenia, leukopenia, depigmentation of skin |
| Iron | Constituent of hemoglobin, myoglobin, and the cytochrome enzymes | Microcytic hypochromic anemia, fatigue, faulty digestion, decreased serum iron |
| Zinc | Essential for the function of many enzymes and a component of lipid, protein, carbohydrate, and nucleic acid metabolism; cell replication and connective tissue synthesis | Dermatitis; impaired wound healing; alopecia; depressed cell-mediated immunity, taste acuity, and dark adaptation; sexual retardation; depressed visceral protein status |
| Cobalt | Biologic methylation | Pernicious anemia, methylmalonic aciduria |
| Manganese | Oxidative phosphorylation, fatty acid metabolism, protein and mucopolysaccharide synthesis | Growth retardation, bony abnormalities, central nervous system dysfunction |
Anthropometric measurements such as triceps skinfold thickness (SFT), midarm circumference (MAC), and arm muscle area, which is derived from SFT and MAC, can be used to estimate fat mass and lean body mass.115 Although serial measurements may be useful in certain patients over long periods, SFT measurements are less reliable in the elderly because of changes in fat distribution and skin compressibility associated with aging115,116 and are inaccurate in patients with peripheral edema.117 In general, these measurements are not practical for nutritional monitoring in a recumbent, critically ill patient.118
Albumin is contained in a large body pool (4 to 5 g/kg) and has a half-life of 20 days, so it is insensitive to acute changes and responds slowly to nutritional therapy. Serum albumin levels are decreased in nephrotic syndrome, enteropathies, hepatic failure, and dialysis (particularly peritoneal dialysis), as well as in the setting of acute volume expansion. Levels may be increased in the presence of dehydration, hypercortisolemia, and anabolic hormones such as insulin, growth hormone, and estrogen. Although serum albumin levels are useful in predicting surgical mortality rate and monitoring nutritional status over the long term, they are much less useful in monitoring a critically ill patient.119
Transferrin is contained in a smaller body pool and has a shorter half-life than albumin does (8 to 10 days), so it is a more sensitive indicator of nutritional status. It is subject to the same influences as mentioned for albumin and, in addition, is affected inversely by serum iron levels.120
Retinol-binding protein is a specific carrier involved in vitamin A transport and is linked with thyroxine-binding prealbumin in a constant molar ratio.121 It has a 12-hour half-life and is sensitive to synthesis and utilization rates. Levels rise in patients with renal disease122 and with excess vitamin A administration and are reduced in patients with liver disease, cystic fibrosis, hyperthyroidism, and vitamin A deficiency.
Transthyretin (thyroxine-binding prealbumin) is involved in the transport of thyroid hormone and is a carrier for retinol-binding protein. It has a small body pool and a short half-life (2 to 3 days) and as such may be a sensitive indicator of nutritional status.123,124 Levels are affected by the same variables that affect albumin and transferrin. Levels are low in patients with hyperthyroidism, cystic fibrosis, chronic illness, and acute stress. Because of its short half-life and ease of measurement, transthyretin is the visceral protein of choice for nutritional assessment and monitoring, although its use in a critically ill patient is controversial. During catabolic illness, hepatic protein synthesis is reprioritized, under the influence of cytokines, with increased synthesis of acute-phase reactant proteins and decreased synthesis of visceral proteins.125–127 Transthyretin levels fall early in the course of catabolic illness and rise with the subsequent decrease in acute-phase reactant proteins as a result of reversal of the reprioritized hepatic protein synthesis. The association of this response with specific nutrients,128–131 nitrogen balance,52,132–135 and outcomes71,134,136 has been variable. Whether transthyretin levels are reflective of appropriate nutrition support or simply a reflection of the course and severity of the inflammatory response is unclear. An initial transthyretin level lower than 50 mg/L or failure to increase by 40 mg/L per week has been associated with a poor prognosis.124
In general, because visceral protein levels are affected by a variety of non-nutritional factors, they are not recommended for the monitoring of nutritional status in critically ill patients.51
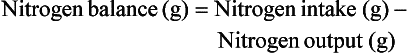
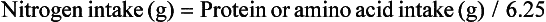

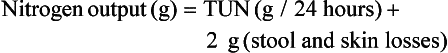
or
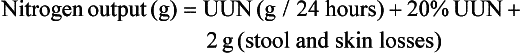
Classically, measurement of urinary nitrogen excretion involves a 24-hour urine collection, but recent evidence suggests that a carefully collected 12- or even 6-hour urine collection can be obtained and extrapolated to a 24-hour period.137 Nitrogen balance is usually calculated weekly.
Numerous techniques for assessment and monitoring of energy balance have been studied, including continuous whole-body calorimetry,138 the doubly labeled water technique,138 and nuclear magnetic resonance spectroscopy using 31P,139 but no method is ideal. Though potentially useful, these methods are either cumbersome, expensive, or impractical for use in critically ill patients.
Indirect calorimetry is used to determine the heat produced by oxidative processes by measuring oxygen consumption and carbon dioxide production, which are then used to calculate REE via the abbreviated Weir equation.140,141 Indirect calorimetry is widely used at the bedside, convenient, relatively inexpensive, and accurately estimates REE when compared with standard predictive formulas.142–148 Indirect calorimetry is performed on patients at rest and therefore does not account for energy expenditure during periods of activity. Many clinicians will increase caloric input by 20% to 25% above measured REE to account for physical activity, particularly in patients who are agitated, ambulatory, or involved in intense physical therapy.149–151
In addition to the nutrition-specific monitoring techniques mentioned, the usual laboratory values should be monitored for fluid and electrolyte composition, hepatic function, infection, and any coagulopathy that might reflect vitamin K deficiency. In a critically ill patient, serum electrolytes (Na+, K+, Cl−, and HCO3−) should be determined daily and as needed. Special attention should be paid to the intracellular electrolytes (K+, Mg2+, and PO42−), which are required for the attainment of nitrogen balance73 and can fall precipitously when nutrition support, particularly glucose, is initiated. Rapid uptake into cells can result in dangerously low serum levels acutely. Intracellular electrolyte levels should be measured before starting nutrition support, 1 or 2 days after starting support, and at least weekly thereafter. Liver function and coagulation parameters should be evaluated weekly and as needed. Glucose should be measured every 6 hours initially and then as needed; it can be measured as often as every 2 hours when continuous insulin infusions are being used (Fig. 82.1).
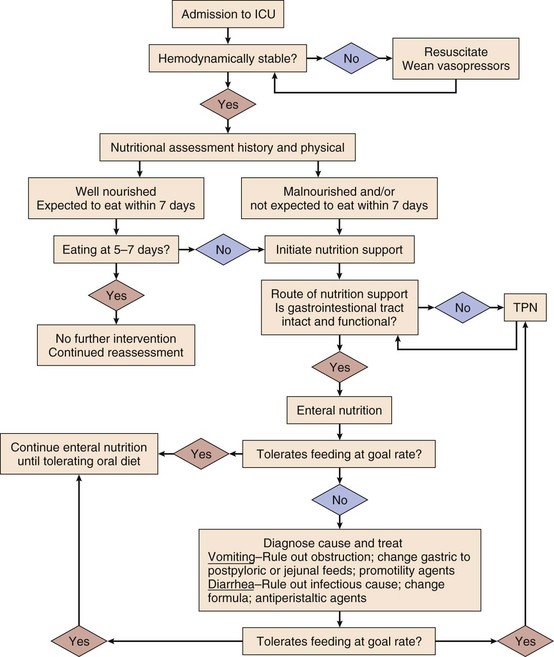
Complications of Nutrition Support
Complications of Enteral Nutrition Support
Mechanical and technical complications of enteral nutrition include feeding tube misplacement, gastrointestinal perforation, sinusitis, otitis media, ulceration of the nasal septum, and obstruction of the feeding tube. Proper placement of feeding tubes in the gastrointestinal tract should be confirmed radiographically or with pH testing before feeding is initiated. Auscultatory techniques for confirmation of proper tube placement are unreliable.152 Nasally placed feeding tubes are a potential cause of sinusitis or otitis media (or both), particularly when stiff, large-bore nasogastric tubes are used as feeding tubes. Soft, small-caliber tubes such as Miller-Frederick or Dobhoff tubes seem to cause sinusitis less frequently. In mechanically ventilated patients, feeding tubes may be placed orally to minimize the incidence of sinusitis.153 Perforation of the esophagus or other parts of the gastrointestinal tract is a disastrous complication and seems to occur most frequently in the setting of stricture, obstructing tumor, or abnormal anatomy as a result of surgery. In these situations, tube placement under fluoroscopic guidance or endoscopic tube placement should be considered. Dislodgement of the feeding tube is a frequent and frustrating problem and can be disastrous when the tube dislodged is a recent, surgically placed tube such as a gastrostomy or jejunostomy tube. When a surgically placed tube has been present for longer than 1 to 2 weeks, the stomach or intestine should be adherent to the abdominal wall and the tube can usually be replaced through the existing tract, as long as the tube is replaced within a few hours before the tract begins to close. A radiograph with contrast enhancement should be used to verify replacement of the tube in the appropriate location. Dislodgement of a surgically placed tube within a week of placement can result in peritonitis from spillage of enteral formula or gastrointestinal contents into the abdominal cavity. Care should be taken to properly secure all feeding tubes either with sutures and tape or with commercially available fixation systems, and tubes should be protected with appropriate patient restraints and caregiver attention when the patient is being moved. Feeding tube obstruction occurs most frequently when tubes are not routinely flushed or when crushed medications are delivered through the tubes. Tubes should be flushed every 6 hours and after every medication dose. Numerous techniques have been used to clear obstructed tubes, including flushing with cola, pancreatic enzymes, cranberry juice, and streptokinase, although none is universally successful. Clearing a nasoenteric tube with a stylet or wire carries the risk of perforating the gastrointestinal tract and should be avoided unless done under fluoroscopic guidance to ensure that the stylet remains within the tube lumen.
Gastrointestinal complications such as abdominal distention, nausea, vomiting, diarrhea, and constipation occur in approximately 60% of critically ill patients receiving nutrition support.154 Gastrointestinal motility is often decreased in these patients,154,155 although gastric stasis and high gastric residual volumes occur more frequently than decreased small intestinal motility does.155 Potential causes of gastroparesis include increased sympathetic tone, elevated intracranial pressure, opiates, benzodiazepines, dopamine, hyperglycemia, recent abdominal surgery, and pancreatitis. Prokinetic agents such as metoclopramide may restore gastric motility and allow gastric feeding.156 Postpyloric feeding tube placement will often allow the continuation of enteral nutrition. Advancement of the feeding tube into the proximal jejunum facilitates enteral nutrition in patients with pancreatitis.
Diarrhea occurs in as many as 70% of critically ill patients.154 Potential causes include antibiotics and other drugs, hyperosmolar formulas, infected solutions, hypoalbuminemia, chronic malnutrition and disuse mucosal atrophy, intolerance to lactose or other nutrients, pancreatic insufficiency, biliary fistula, and short-gut syndrome.
Clostridium difficile overgrowth is perhaps the most serious cause of antibiotic-associated diarrhea, and stool should be assayed for C. difficile toxin in every case of persistent diarrhea in the critically ill. C. difficile enterocolitis should be treated with appropriate antibiotics. Antibiotics may also cause diarrhea by eliminating the bacteria that ferment dietary fiber into short-chain fatty acids. Short-chain fatty acids are important for maintaining colonic mucosal integrity and enhance water and electrolyte absorption.157 Enteral formulas should be replaced daily to avoid bacterial contamination of the solution.
Aspiration pneumonia is the major infectious complication of enteral nutrition. Bolus feeding carries a higher risk of aspiration than continuous feeding does. Gastric residual volume, which has traditionally been used to assess gastric feeding tolerance, does not appear to correlate with risk for aspiration.158 Gastric feeding is routinely used in many ICUs, but because a nasogastric tube is one of the leading causes of aspiration, gastric feeding should probably be done with a soft, small-caliber tube designed for feeding. The risk of aspiration associated with gastric feeding may be lower when done via a percutaneous gastrostomy tube,158 which avoids compromising the function of the gastroesophageal junction and swallowing mechanics. Multiple studies have compared the risk of aspirating gastric versus postpyloric feedings, but the results have been contradictory.71,159,160 In a recent meta-analysis of seven randomized trials, small bowel feeding was associated with a lower risk of aspiration pneumonia than was gastric feeding.161 Nursing protocols for the bedside placement of postpyloric feeding tubes have been developed,162,163 although placement under fluoroscopic guidance is more likely to be successful and is cost-effective.164 The semirecumbent body position has been shown to significantly reduce aspiration pneumonia as compared with the supine body position.165
Complications of Parenteral Nutrition Support
The primary complications of parenteral nutrition include mechanical or technical complications and infectious complications. Mechanical and technical complications are related to central venous catheter placement and include pneumothorax, arterial injury, hemothorax, hydrothorax, cardiac arrhythmia, and cardiac perforation with tamponade. The incidence of severe complications with subclavian vein catheterization is 1% to 3%, with an overall complication rate of 5%.166 The incidence of complications with jugular venous catheterization ranges from 0.1% to 4.2%.167 Jugular venous catheterization under ultrasound guidance is increasingly being recommended.168–171 Jugular or subclavian vein thrombosis is a postprocedural complication that is manifested as neck or arm swelling and should be managed by catheter removal and anticoagulation; thrombolytics have also been recommended.172
Catheter infection and catheter-associated bloodstream infection are the major infectious complications of parenteral nutrition and occur in 2% to 8% of patients with central venous catheters. The most common infecting organisms are coagulase-negative staphylococci, Staphylococcus aureus, and Candida species. Infection is more likely when parenteral nutrition is administered through a multilumen catheter than through a single-lumen catheter,173 although the incidence of infection may be reduced when parenteral nutrition is infused through a dedicated port.174 Jugular and femoral catheters have higher rates of infection than do subclavian catheters.175 Dedicated central line care teams and regular dressing change protocols may reduce the incidence of catheter infection.174 Antibiotic-coated or antibiotic-impregnated catheters are expensive but appear to be effective in reducing catheter-related bloodstream infections.176 Guidewire exchange is associated with a decreased incidence of technical complications but an increased incidence of catheter infections.177 Although the possibility of catheter infection increases with time, routine catheter exchange is probably not indicated.176 Catheters should be replaced, ideally at a fresh site, for obvious site infection, for the evaluation and treatment of fever of unknown origin, and after any positive blood culture. If guidewire exchange is used and the removed catheter proves to be infected, the new catheter often becomes infected during the exchange and should in turn be removed and replaced at a fresh site.
Metabolic Complications of Nutrition Support
Complications related to hyperglycemia include infection, hyperosmolarity, and osmotic diuresis. The first step in the control of blood sugar is avoidance of overfeeding, as discussed previously. Insulin, whether by intermittent dosing or continuous infusion, is the mainstay for maintenance of acceptable blood glucose levels. The appropriate glucose level has been the subject of controversy. In a large, prospective, randomized trial in critically ill surgical patients, tight glucose control (80-120 mg/dL) was associated with significantly decreased mortality rate, infection, neuropathy, and renal failure.49 In medical ICU patients the same tight glucose control protocol was associated with decreased renal failure, ventilator days, and reduced ICU and hospital length of stay, but not improvements in mortality rate or infection.178 Subsequently, the NICE-SUGAR trial compared tight glucose control to glucose levels less than 180 mg/dL and demonstrated similar infection and mortality rates and fewer episodes of dangerous hypoglycemia with more liberal glucose control.179 Currently, most practitioners aim for blood glucose levels below 150 to 180 mg/dL. Symptomatic hypoglycemia has been reported in children after sudden withdrawal of TPN but is rare in adults, and weaning of TPN in adults is not usually necessary.180
Hepatobiliary complications of nutrition support include hepatic steatosis (fatty infiltration of the liver) and intrahepatic and extrahepatic cholestasis. Progression to chronic liver disease occurs in premature infants181 but is rare in adults. Hepatic steatosis may develop after 7 to 21 days of parenteral nutrition and is characterized initially by elevated transaminases. Usually asymptomatic, fatty infiltration can, in severe cases, be accompanied by hepatomegaly and right upper quadrant abdominal pain. Histologically, mild cases are characterized by periportal fat infiltration and severe cases by centrilobular infiltration. Overfeeding, hyperinsulinemia, essential fatty acid deficiency, and carnitine deficiency may predispose to fatty infiltration. Cholestasis occurs later in the course of parenteral nutrition and is characterized by elevations in bilirubin and alkaline phosphatase. Histologically, periportal or pericentral canalicular bile plugging, bile staining of surrounding hepatocytes, and lymphocytic triaditis characterize cholestasis. Cholelithiasis has been linked to long-term TPN use. Gallstone formation is almost certainly related to the lack of enteral feeding and the resultant lack of neural and hormonal stimulation of gallbladder contraction. Other potential but unproven causes of cholestasis include overfeeding, the production of toxic bile acids by intestinal bacteria, hormones, endotoxin, and taurine deficiency.31,182–185 Management of hepatobiliary complications is aimed at prevention. Overfeeding should be avoided. Cyclical TPN and enteral feeding, even if only partial, may help prevent cholestasis. A trial of oral neomycin and metronidazole may be helpful.185
Serum electrolyte levels should be monitored and electrolytes added to or eliminated from TPN as indicated, with consideration of excess losses or abnormal accumulation. Hypernatremia or hyponatremia can be managed by increasing or decreasing free water. Particular attention should be paid to the intracellular electrolytes potassium, magnesium, and phosphorus. In malnourished individuals, these electrolytes rapidly move intracellularly when nutrition support is initiated. In what is termed the refeeding syndrome, serum levels of these intracellular electrolytes fall precipitously, with hypophosphatemia resulting in hemolysis, rhabdomyolysis, and heart failure with hypokalemia and hypomagnesemia leading to cardiac arrhythmias.186,187 Serum levels of intracellular electrolytes should be corrected before and, in particular, 24 hours after the institution of nutrition support. Abnormally high levels of the intracellular electrolytes are of concern in renal failure and often need to be reduced or eliminated from TPN solutions. Hyperkalemia and hypomagnesemia may lead to weakness and cardiac arrhythmia, and hyperphosphatemia may result in hypotension, hypocalcemia, and metastatic calcification. Hyperphosphatemia is often treated with oral phosphate binders such as calcium carbonate.
Metabolic acidosis related to nutrition support is most commonly due to excess chloride administration and resultant renal bicarbonate losses, but it can also occur as a result of thiamine deficiency with resultant lactic acidosis.188,189
References
1. Khuri, SF, Daley, J, Henderson, W, et al. The Department of Veterans Affairs’ NSQIP: The first national, validated, outcome-based, risk-adjusted, and peer-controlled program for the measurement and enhancement of the quality of surgical care. National VA Surgical Quality Improvement Program. Ann Surg. 1998; 228:491–507.
2. Longo, WE, Virgo, KS, Johnson, FE, et al. Risk factors for morbidity and mortality after colectomy for colon cancer. Dis Colon Rectum. 2000; 43:83–91.
3. Hollenbeck, BK, Miller, DC, Taub, D, et al. Risk factors for adverse outcomes after transurethral resection of bladder tumors. Cancer. 2006; 106:1527–1535.
4. Harpole, DH Jr, DeCamp, MM, Jr., Daley, J, et al. Prognostic models of thirty-day mortality and morbidity after major pulmonary resection. J Thorac Cardiovasc Surg. 1999; 117:969–979.
5. Mizock, BA. Alterations in carbohydrate metabolism during stress: A review of the literature. Am J Med. 1995; 98:75–84.
6. Wolfe, RR, Jahoor, F, Herndon, DN, et al. Isotopic evaluation of the metabolism of pyruvate and related substrates in normal adult volunteers and severely burned children: Effect of dichloroacetate and glucose infusion. Surgery. 1991; 110:54–67.
7. Long, CL, Spencer, JL, Kinney, JM, et al. Carbohydrate metabolism in man: Effect of elective operations and major injury. J Appl Physiol. 1971; 31:110–116.
8. Lang, CH, Dobrescu, C. Gram-negative infection increases noninsulin-mediated glucose disposal. Endocrinology. 1991; 128:645–753.
9. Stumvoll, M, Meyer, C, Mitrakou, A, et al. Renal glucose production and utilization: New aspects in humans. Diabetologia. 1997; 40:749–757.
10. McGuinness, OP, Shau, V, Benson, EM, et al. Role of epinephrine and norepinephrine in the metabolic response to stress hormone infusion in the conscious dog. Am J Physiol. 1997; 273:E674–E681.
11. Chu, CA, Sindelar, DK, Neal, DW, et al. Comparison of the direct and indirect effects of epinephrine on hepatic glucose production. J Clin Invest. 1997; 99:1044–1056.
12. Petit, F, Bagby, GJ, Lang, CH. Tumor necrosis factor mediates zymosan-induced increase in glucose flux and insulin resistance. Am J Physiol. 268, 1995. [E219–E28].
13. Pessin, JE, Bell, GI. Mammalian facilitative glucose transporter family: Structure and molecular regulation. Annu Rev Physiol. 1992; 54:911–930.
14. McCowen, KC, Malhotra, A, Bistrian, BR. Stress-induced hyperglycemia. Crit Care Clin. 2001; 17:107–124.
15. Meszaros, K, Lang, CH, Bagby, GJ, et al. Tumor necrosis factor increases in vivo glucose utilization of macrophage-rich tissues. Biochem Biophys Res Commun. 1987; 149:1–6.
16. Bird, TA, Davies, A, Baldwin, SA, et al. Interleukin 1 stimulates hexose transport in fibroblasts by increasing the expression of glucose transporters. J Biol Chem. 1990; 265:13578–13583.
17. van den Berghe, G, Wouters, P, Weekers, F, et al. Reactivation of pituitary hormone release and metabolic improvement by infusion of growth hormone-releasing peptide and thyrotropin-releasing hormone in patients with protracted critical illness. J Clin Endocrinol Metab. 1999; 84:1311–1323.
18. van den Berghe, G, Baxter, RC, Weekers, F, et al. A paradoxical gender dissociation within the growth hormone/insulin-like growth factor I axis during protracted critical illness. J Clin Endocrinol Metab. 2000; 85:183–192.
19. Grimble, RF. Inflammatory status and insulin resistance. Curr Opin Clin Nutr Metab Care. 2002; 5:551–559.
20. Marette, A. Mediators of cytokine-induced insulin resistance in obesity and other inflammatory settings. Curr Opin Clin Nutr Metab Care. 2002; 5:377–383.
21. Herndon, DN, Nguyen, TT, Wolfe, RR, et al. Lipolysis in burned patients is stimulated by the beta 2-receptor for catecholamines. Arch Surg. 1994; 129:1301–1304.
22. Hardardottir, I, Grunfeld, C, Feingold, KR. Effects of endotoxin and cytokines on lipid metabolism. Curr Opin Lipidol. 1994; 5:207–215.
23. Wolfe, RR. Herman Award Lecture, 1996: Relation of metabolic studies to clinical nutrition—The example of burn injury. Am J Clin Nutr. 1996; 64:800–808.
24. Wolfe, R. The role of triglyceride-fatty acid cycling and glucose cycling in thermogenesis and amplification of net substrate flux in human subjects. In: Muller JM, Danforth E, Burger AG, et al, eds. Hormones and Nutrition in Obesity and Cachexia. New York: Springer Verlag, 1989.
25. Wolfe, RR, Shaw, JH, Durkot, MJ. Effect of sepsis on VLDL kinetics: Responses in basal state and during glucose infusion. Am J Physiol. 1985; 248:E732–E740.
26. Lanza-Jacoby, S, Sedkova, N, Phetteplace, H, et al. Sepsis-induced regulation of lipoprotein lipase expression in rat adipose tissue and soleus muscle. J Lipid Res. 1997; 38:701–710.
27. Aarsland, A, Chinkes, D, Wolfe, RR. Contributions of de novo synthesis of fatty acids to total VLDL-triglyceride secretion during prolonged hyperglycemia/hyperinsulinemia in normal man. J Clin Invest. 1996; 98:2008–2017.
28. Feingold, KR, Serio, MK, Adi, S, et al. Tumor necrosis factor stimulates hepatic lipid synthesis and secretion. Endocrinology. 1989; 124:2336–2342.
29. Wolfe, BM, Walker, BK, Shaul, DB, et al. Effect of total parenteral nutrition on hepatic histology. Arch Surg. 1988; 123:1084–1090.
30. Mizock, BA. Metabolic derangements in sepsis and septic shock. Crit Care Clin. 2000; 16:319–336.
31. Siegel, JH, Cerra, FB, Coleman, B, et al. Physiological and metabolic correlations in human sepsis. Invited commentary. Surgery. 1979; 86:163–193.
32. Ozawa, K, Aoyama, H, Yasuda, K, et al. Metabolic abnormalities associated with postoperative organ failure. A redox theory. Arch Surg. 1983; 118:1245–1251.
33. Fink, M. Cytopathic hypoxia in sepsis. Acta Anaesthesiol Scand Suppl. 1997; 110:87–95.
34. Lecker, SH, Jagoe, RT, Gilbert, A, et al. Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression. FASEB J. 2004; 18:39–51.
35. Tisdale, MJ. Biochemical mechanisms of cellular catabolism. Curr Opin Clin Nutr Metab Care. 2002; 5:401–405.
36. Chai, J, Wu, Y, Sheng, ZZ. Role of ubiquitin-proteasome pathway in skeletal muscle wasting in rats with endotoxemia. Crit Care Med. 2003; 31:1802–1807.
37. Costelli, P, Baccino, FM. Mechanisms of skeletal muscle depletion in wasting syndromes: Role of ATP-ubiquitin-dependent proteolysis. Curr Opin Clin Nutr Metab Care. 2003; 6:407–412.
38. Biolo, G, Fleming, RY, Maggi, SP, et al. Inverse regulation of protein turnover and amino acid transport in skeletal muscle of hypercatabolic patients. J Clin Endocrinol Metab. 2002; 87:3378–3384.
39. Wernerman, J, Skeletal muscle in the stress-induced catabolic state. Acute Catabolic State: Update in Intensive Care and Emergency Medicine. Revhaug, A, eds. Acute Catabolic State: Update in Intensive Care and Emergency Medicine; Vol. 21. Springer, Berlin, 1996.
40. Newsholme, EA, Parry-Billings, M. Properties of glutamine release from muscle and its importance for the immune system. JPEN J Parenter Enteral Nutr. 1990; 14:63S–67S.
41. Paddon-Jones, D, Sheffield-Moore, M, Creson, DL, et al. Hypercortisolemia alters muscle protein anabolism following ingestion of essential amino acids. Am J Physiol Endocrinol Metab. 2003; 284:E946–E953.
42. Orellana, RA, O’Connor, PM, Nguyen, HV, et al. Endotoxemia reduces skeletal muscle protein synthesis in neonates. Am J Physiol Endocrinol Metab. 2002; 283:E909–E916.
43. Askanazi, J, Rosenbaum, SH, Hyman, AI, et al. Respiratory changes induced by the large glucose loads of total parenteral nutrition. JAMA. 1980; 243:1444–1447.
44. Buzby, GP, Mullen, JL, Stein, TP, et al. Manipulation of TPN caloric substrate and fatty infiltration of liver. J Surg Res. 1981; 31:46–54.
45. Lowry, S, Brennan, MFB. Abnormal liver function during parenteral nutrition: Relation to infusion excess. J Surg Res. 1979; 26:300–307.
46. Sheldon, GF, Peterson, SR, Sanders, R. Hepatic dysfunction during hyperalimentation. Arch Surg. 1978; 113:504–508.
47. Furnary, AP, Zerr, KJ, Grunkemeier, GL, et al. Continuous intravenous insulin infusion reduces the incidence of deep sternal wound infection in diabetic patients after cardiac surgical procedures. Ann Thorac Surg. 1999; 67:352–360.
48. Pomposelli, JJ, Baxter, JK, 3rd., Babineau, TJ, et al. Early postoperative glucose control predicts nosocomial infection rate in diabetic patients. JPEN J Parenter Enteral Nutr. 1998; 22:77–81.
49. van den Berghe, G, Wouters, P, Weekers, F, et al. Intensive insulin therapy in the surgical intensive care unit. N Engl J Med. 2001; 345:1359–1367.
50. Barton, RG, Craft, WB, Mone, MC, et al. Chemical paralysis reduces energy expenditure in patients with burns and severe respiratory failure treated with mechanical ventilation. J Burn Care Rehabil. 1997; 18:461–468.
51. McClave, SA, Martindale, RG, Vanek, VW, et al. Board of Directors and the American College of Critical Care Medicine: Guidelines for the provision and assessment of nutrition support therapy in the adult critically ill patient. JPEN J Parenter Enteral Nutr. 2009; 33:277–316.
52. Dickerson, RN, Boschert, KJ, Kudsk, KA, et al. Hypocaloric enteral tube feeding in critically ill obese patients. Nutrition. 2002; 18:241–246.
53. Wolfe, RR, Allsop, JR, Burke, JF. Glucose metabolism in man: Responses to intravenous glucose infusion. Metabolism. 1979; 28:210–220.
54. Wolfe, RR. Carbohydrate metabolism in the critically ill patient. Implications for nutritional support. Crit Care Clin. 1987; 3:11–24.
55. Driscoll, DF, Blackburn, GL. Total parenteral nutrition 1990. A review of its current status in hospitalised patients, and the need for patient-specific feeding. Drugs. 1990; 40:346–363.
56. Elwyn, DH. Nutritional requirements of adult surgical patients. Crit Care Med. 1980; 8:9–20.
57. Monsen, ER. The 10th edition of the recommended dietary allowances: What’s new in the 1989 RDAs? J Am Diet Assoc. 1989; 89:1748–1752.
58. Rudman, D, Millikan, WJ, Richardson, TJ, et al. Elemental balances during intravenous hyperalimentation of underweight adult subjects. J Clin Invest. 1975; 55:94–104.
59. Visser, J, Labadarios, D, Blaauw, R. Micronutrient supplementation for critically ill adults: A systematic review and meta-analysis. Nutrition. 2011; 27:745–758.
60. Elia, M. Changing concepts of nutrient requirements in disease: Implications for artificial nutritional support. Lancet. 1995; 345:1279–1284.
61. Walker, W. Intestinal defenses in health and disease. In: Lifshitz F, ed. Clinical Disorders in Pediatric Growth and Nutrition. New York: Marcel Dekker; 1980:99–119.
62. Kagnoff, M. Immunology and disease of the gastrointestinal tract. In: Sleisinger MH, Fordtrain JS, eds. Gastrointestinal Disease. Philadelphia: WB Saunders; 1983:20–44.
63. Williamson, RC. Intestinal adaptation (first of two parts). Structural, functional and cytokinetic changes. N Engl J Med. 1978; 298:1393–1402.
64. Williamson, RC. Intestinal adaptation (second of two parts). Mechanisms of control. N Engl J Med. 1978; 298:1444–1450.
65. Wilmore, DW, Smith, RJ, O’Dwyer, ST, et al. The gut: A central organ after surgical stress. Surgery. 1988; 104:917–923.
65a. McClave, Stephen, A, Heyland, Daren, K. The physiologic response and associated clinical benefits from provision of early enteral nutrition. Nutrition in Clinical Practice. 2009; 3:305–315.
65b. Kudsk, KA. Importance of enteral feeding in maintaining gut integrity. Tech Gastro Endoscopy. 2001; 3:2–8.
65c. Kudsk, KA. Current aspects of mucosal immunology and its influence by nutrition. Am J Surg. 2002; 183:390–398.
65d. Dobbins, WO. Gut immunophysiology: a gastroenterologist’s view with emphasis on pathophysiology. Am J Physiol. 1982; 242:G1–G8.
65e. Alverdy, J, Zaborina, O, Wu, L. The impact of stress and nutrition on bacterial-host interactions at the intestinal epithelial surface. Curr Opin Clin Nutr Metab Care. 2005; 8:205–209.
65f. Spiekermann, GM, Walker, WA. Oral tolerance and its role in clinical disease. J Pediatr Gastroent Nutr. 2001; 32:237–255.
65g. Lebman, DA, Coffman, RL. Cytokines in the mucosal immune system. In: Ogra PL, Lamm ME, McGhee JR, eds. Handbook of mucosal immunology. San Diego, CA: Academic Press; 1994:243–249.
66. Moore, FA, Moore, EE, Jones, TN, et al. TEN versus TPN following major abdominal trauma—Reduced septic morbidity. J Trauma. 1989; 29:916–922.
67. Kudsk, KA, Croce, MA, Fabian, TC, et al. Enteral versus parenteral feeding. Effects on septic morbidity after blunt and penetrating abdominal trauma. Ann Surg. 1992; 215:503–511.
68. Windsor, AC, Kanwar, S, Li, AG, et al. Compared with parenteral nutrition, enteral feeding attenuates the acute phase response and improves disease severity in acute pancreatitis. Gut. 1998; 42:431–435.
69. Heyland, DK, Dhaliwal, R, Drover, JW, et al. Canadian clinical practice guidelines for nutrition support in mechanically ventilated, critically ill adult patients. Canadian Critical Care Clinical Practice Guidelines Committee. JPEN J Parenter Enteral Nutr. 2003; 27:355–373.
70. Kalfarentzos, F, Kehagias, J, Mead, N, et al. Enteral nutrition is superior to parenteral nutrition in severe acute pancreatitis: Results of a randomized prospective trial. Br J Surg. 1997; 84:1665–1669.
71. Taylor, SJ, Fettes, SB, Jewkes, C, Nelson, RJ. Prospective, randomized, controlled trial to determine the effect of early enhanced enteral nutrition on clinical outcome in mechanically ventilated patients suffering head injury. Crit Care Med. 1999; 27:2525–2531.
72. Moore, FA, Feliciano, DV, Andrassy, RJ, et al. Early enteral feeding, compared with parenteral, reduces postoperative septic complications. The results of a meta-analysis. Ann Surg. 1992; 216:172–183.
73. Gramlich, L, Kichian, K, Pinilla, J, et al. Does enteral nutrition compared to parenteral nutrition result in better outcomes in critically ill adult patients? A systematic review of the literature. Nutrition. 2004; 20:843–848.
74. Braunschweig, CL, Levy, P, Sheean, PM, Wang, X. Enteral compared with parenteral nutrition: A meta-analysis. Am J Clin Nutr. 2001; 74:534–542.
75. Simpson, F, Doig, GS. Parenteral vs. enteral nutrition in the critically ill patients: A meta-analysis of trials using the intention to treat principle. Intensive Care Med. 2005; 31:12–23.
76. van den Berghe, G, Wouters, PJ, Bouillon, R, et al. Outcome benefit of intensive insulin therapy in the critically ill: Insulin dose versus glycemic control. Crit Care Med. 2003; 31:359–366.
77. Marik, PE, Zaloga, GP. Early enteral nutrition in acutely ill patients: A systemic review. Crit Care Med. 2001; 29:2264–2270.
77a. Dvir, D, Cohen, J, Singer, P. Computerized energy balance and complications in critically ill patients: an observational study. Clin Nutr. 2006; 25:37–44.
78. Singer, P, Berger, MM, van den Berghe, G, et al. ESPEN guidelines on parenteral nutrition: Intensive care. Clin Nutr. 2009; 28:387–400.
79. Casaer, MP, Mesotten, D, Hermans, G, et al. Early versus late parenteral nutrition in critically ill adults. N Eng J Med. 2011; 365:506–517.
80. Product Reference Guide: Deerfield, IL, Nestle Clinical Nutrition, 2001.
81. Brinson, RR, Kolts, BE. Diarrhea associated with severe hypoalbuminemia: A comparison of a peptide-based chemically defined diet and standard enteral alimentation. Crit Care Med. 1988; 16:130–136.
82. Talpers, SS, Romberger, DJ, Bunce, SB, et al. Nutritionally associated increased carbon dioxide production. Excess total calories vs high proportion of carbohydrate calories. Chest. 1992; 102:551–555.
83. Cerra, FB, Cheung, NK, Fischer, JE, et al. Disease-specific amino acid infusion (F080) in hepatic encephalopathy: A prospective, randomized, double-blind, controlled trial. JPEN J Parenter Enteral Nutr. 1985; 9:288–295.
84. Kanematsu, T, Koyanagi, N, Matsumata, T, et al. Lack of preventive effect of branched-chain amino acid solution on postoperative hepatic encephalopathy in patients with cirrhosis: A randomized, prospective trial. Surgery. 1988; 104:482–488.
85. Bansal, V, Ochoa, JB. Arginine availability, arginase, and the immune response. Curr Opin Clin Nutr Metab Care. 2003; 6:223–228.
86. Mizock, BA. Immunonutrition and critical illness: An update. Nutrition. 2010; 26:701–707.
87. Popovic, PJ, Zeh, HJ, Ochoa, JB. Arginine and immunity. J Nutr. 2007; 137(Suppl):1681S–11686.
88. Munder, M, Schneider, H, Luckner, C, et al. Suppression of T-cell functions by human granulocyte arginase. Blood. 2006; 108:1627–1634.
89. Ochoa, JB, Makarenkova, V, Bansal, V. A rational use of immune enhancing diets: When should we use dietary arginine supplementation. Nutr Clin Pract. 2004; 19:216–225.
90. Pacher, P, Beckman, JS, Liaudet, L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007; 87:315–424.
91. Fink, MP, Delude, RL. Epithelial barrier dysfunction: A unifying theme to explain the pathogenesis of multiple organ dysfunction at a cellular level. Crit Care Clin. 2005; 21:177–196.
92. Bistrian, BR, McCowen, KC. Nutritional and metabolic support in the adult intensive care unit: Key controversies. Crit Care Med. 2006; 34:1525–1531.
93. Kirk, SJ, Barbul, A. Role of arginine in trauma, sepsis, and immunity. JPEN J Parenter Enteral Nutr. 1990; 14:226S–229S.
94. Madden, HP, Breslin, RJ, Wasserkrug, HL, et al. Stimulation of T cell immunity by arginine enhances survival in peritonitis. J Surg Res. 1988; 44:658–663.
95. Barbul, A, Lazarou, SA, Efron, DT, et al. Arginine enhances wound healing and lymphocyte immune responses in humans. Surgery. 1990; 108:331–336.
96. Billiar, TR, Bankey, PE, Svingen, BA, et al. Fatty acid intake and Kupffer cell function: Fish oil alters eicosanoid and monokine production to endotoxin stimulation. Surgery. 1988; 104:343–349.
97. Endres, S, Ghorbani, R, Kelley, VE, et al. The effect of dietary supplementation with n-3 polyunsaturated fatty acids on the synthesis of interleukin-1 and tumor necrosis factor by mononuclear cells. N Engl J Med. 1989; 320:265–271.
98. Bittiner, SB, Tucker, WF, Cartwright, I, et al. A double-blind, randomised, placebo-controlled trial of fish oil in psoriasis. Lancet. 1988; 1:378–380.
99. Kremer, JM, Jubiz, W, Michalek, A, et al. Fish-oil fatty acid supplementation in active rheumatoid arthritis. A double-blinded, controlled, crossover study. Ann Intern Med. 1987; 106:497–503.
100. McNurlan, MA, Sandgren, A, Hunter, K, et al. Protein synthesis rates of skeletal muscle, lymphocytes, and albumin with stress hormone infusion in healthy man. Metabolism. 1996; 45:1388–1394.
101. Luo, JL, Hammarqvist, F, Andersson, K, et al. Skeletal muscle glutathione after surgical trauma. Ann Surg. 1996; 223:420–427.
102. Gamrin, L, Essen, P, Forsberg, AM, et al. A descriptive study of skeletal muscle metabolism in critically ill patients: Free amino acids, energy-rich phosphates, protein, nucleic acids, fat, water, and electrolytes. Crit Care Med. 1996; 24:575–583.
103. Hammarqvist, F, Wernerman, J, Ali, R, et al. Addition of glutamine to total parenteral nutrition after elective abdominal surgery spares free glutamine in muscle, counteracts the fall in muscle protein synthesis, and improves nitrogen balance. Ann Surg. 1989; 209:455–461.
104. Wells, CL, Jechorek, RP, Erlandsen, SL, et al. The effect of dietary glutamine and dietary RNA on ileal flora, ileal histology, and bacterial translocation in mice. Nutrition. 1990; 6:70–75.
105. Zapata-Sirvent, RL, Hansbrough, JF, Ohara, MM, et al. Bacterial translocation in burned mice after administration of various diets including fiber- and glutamine-enriched enteral formulas. Crit Care Med. 1994; 22:690–696.
106. Novak, F, Heyland, DK, Avenell, A, et al. Glutamine supplementation in serious illness: A systematic review of the evidence. Crit Care Med. 2002; 30:2022–2029.
107. Bulger, EM, Maier, RV. Antioxidants in critical illness. Arch Surg. 2001; 136:1201–1207.
108. Heyland, DK, Novak, F, Drover, JW, et al. Should immunonutrition become routine in critically ill patients? A systematic review of the evidence. JAMA. 2001; 286:944–953.
109. Kieft, H, Roos, AN, van Drunen, JD, et al. Clinical outcome of immunonutrition in a heterogeneous intensive care population. Intensive Care Med. 2005; 31:524–532.
110. Marik, PE, Zaloga, GP. Immunonutrition in critically ill patients: A systematic review and analysis of the literature. Intensive Care Med. 2008; 34(11):1980–1990.
111. Gadek, JE, DeMichele, SJ, Karlstad, MD, et al. Effect of enteral feeding with eicosapentaenoic acid, gamma-linolemic acid, and antioxidants in patients with acute respiratory distress syndrome. Enteral Nutrition in ARDS Study Group. Crit Care Med. 1999; 27:1409–1420.
112. Pontes-Arruda, A, Aragao, AM, Albuquerque, JD. Effects of enteral feeding with eicosapentaenoic acid, gamma-linolenic acid, and antioxidants in mechanically ventilated patients with severe sepsis and septic shock. Crit Care Med. 2006; 34:2325–2333.
113. Singer, P, Theilla, M, Fisher, H, et al. Benefit of an enteral diet enriched with eicosapentaenoic acid and gamma-linolenic acid in ventilated patients with acute lung injury. Crit Care Med. 2006; 34:1033–1038.
114. Baker, JP, Detsky, AS, Wesson, DE, et al. Nutritional assessment: A comparison of clinical judgement and objective measurements. N Engl J Med. 1982; 306:969–972.
115. Charney, P. Nutrition assessment in the 1990s: Where are we now? Nutr Clin Pract. 1995; 10:131–139.
116. Bettany, GE, Powell-Tuck, J. Malnutrition: lncidence, diagnosis, causes, effects and indications for nutritional support. Eur J Gastroenterol Hepatol. 1995; 7:494–500.
117. Hill, GL, Windsor, JA. Nutritional assessment in clinical practice. Nutrition. 1995; 11:198–201.
118. Dark, D, Pingleton, S. Nutrition and nutritional support in clinically ill patients. J lntensive Care Med. 1993; 8:16–33.
119. Raguso, CA, Dupertuis, YM, Pichard, C. The role of visceral proteins in the nutritional assessment of intensive care unit patients. Curr Opin Clin Nutr Metab Care. 2003; 6:211–216.
120. Wessling-Resnick, M. Iron transport. Annu Rev Nutr. 2000; 20:129–151.
121. Ingenbleek, Y, Bernstein, L. The stressful condition as a nutritionally dependent adaptive dichotomy. Nutrition. 1999; 15:305–320.
122. Manelli, JC, Badetti, C, Botti, G, et al. A reference standard for plasma proteins is required for nutritional assessment of adult burn patients. Burns. 1998; 24:337–345.
123. Mears, E. Outcomes of continuous process improvement of a nutritional care program incorporating serum prealbumin measurements. Nutrition. 1996; 12:479–484.
124. Bernstain, L. Measurement of visceral protein status in assessing protein and energy malnutrition: Standard of care. Prealbumin in Nutritional Care Consensus Group. Nutrition. 1995; 11:169–171.
125. Moshage, H. Cytokines and the hepatic acute phase response. J Pathol. 1997; 181:257–266.
126. Heinrich, PC, Castell, JV, Andus, T. Interleukin-6 and the acute phase response. Biochem J. 1990; 265:621–636.
127. Pinilla, JC, Hayes, P, Laverty, W, et al. The C-reactive protein to prealbumin ratio correlates with the severity of multiple organ dysfunction. Surgery. 1998; 124:799–805.
128. Abribat, T, Nedelec, B, Jobin, N, et al. Decreased serum insulin-like growth factor-I in burn patients: Relationship with serum insulin-like growth factor binding protein-3 proteolysis and the influence of lipid composition in nutritional support. Crit Care Med. 2000; 28:2366–2372.
129. Wischmeyer, PE, Lynch, J, Liedel, J, et al. Glutamine administration reduces gram-negative bacteremia in severely burned patients: A prospective, randomized, double-blind trial versus isonitrogenous control. Crit Care Med. 2001; 29:2075–2080.
130. Houdijk, AP, Nijveldt, RJ, van Leeuwen, PA. Glutamine-enriched enteral feeding in trauma patients: Reduced infectious morbidity is not related to changes in endocrine and metabolic responses. JPEN J Parenter Enteral Nutr. 1999; 23:S52–S58.
131. Conejero, R, Bonet, A, Grau, T, et al. Effect of a glutamine-enriched enteral diet on intestinal permeability and infectious morbidity at 28 days in critically ill patients with systemic inflammatory response syndrome: A randomized, single-blind, prospective, multicenter study. Nutrition. 2002; 18:716–721.
132. Nataloni, S, Gentili, P, Marini, B, et al. Nutritional assessment in head injured patients through the study of rapid turnover visceral proteins. Clin Nutr. 1999; 18:247–251.
133. Wiren, M, Permert, J, Larsson, J. Alphaketoglutarate-supplemented enteral nutrition: Effects on postoperative nitrogen balance and muscle catabolism. Nutrition. 2002; 18:725–728.
134. Garcia-de-Lorenzo, A, Ortiz-Leyba, C, Planas, M, et al. Parenteral administration of different amounts of branch-chain amino acids in septic patients: Clinical and metabolic aspects. Crit Care Med. 1997; 25:418–424.
135. Gervasio, JM, Dickerson, RN, Swearingen, J, et al. Oxandrolone in trauma patients. Pharmacotherapy. 2000; 20:1328–1334.
136. Huang, YC, Yen, CE, Cheng, CH, et al. Nutritional status of mechanically ventilated critically ill patients: Comparison of different types of nutritional support. Clin Nutr. 2000; 19:101–107.
137. Graves, C, Saffle, J, Morris, S. Comparison of urine urea nitrogen collection times in critically ill patients. Nutr Clin Pract. 2005; 20:271–275.
138. Plank, LD, Hill, GL. Energy balance in critical illness. Proc Nutr Soc. 2003; 62:545–552.
139. Thompson, A, Damyanovich, A, Madapallimattam, A, et al. 31P-nuclear magnetic resonance studies of bioenergetic changes in skeletal muscle in malnourished human adults. Am J Clin Nutr. 1998; 67:39–43.
140. Jacobs, DO, Wong, M. Metabolic assessment. World J Surg. 2000; 24:1460–1467.
141. Weir, JB. New methods for calculating metabolic rate with special reference to protein metabolism. Nutrition. 1990; 6:213–221.
142. Long, CL, Schaffel, N, Geiger, JW, et al. Metabolic response to injury and illness: Estimation of energy and protein needs from indirect calorimetry and nitrogen balance. JPEN J Parenter Enteral Nutr. 1979; 3:452–456.
143. Liggett, SB, Renfro, AD. Energy expenditures of mechanically ventilated nonsurgical patients. Chest. 1990; 98:682–686.
144. Hwang, TL, Huang, SL, Chen, MF. The use of indirect calorimetry in critically ill patients—The relationship of measured energy expenditure to Injury Severity Score, Septic Severity Score, and APACHE II Score. J Trauma. 1993; 34:247–251.
145. Boulanger, BR, Nayman, R, McLean, RF, et al. What are the clinical determinants of early energy expenditure in critically injured adults? J Trauma. 1994; 37:969–974.
146. Raurich, JM, Ibanez, J. Metabolic rate in severe head trauma. JPEN J Parenter Enteral Nutr. 1994; 18:521–524.
147. Uehara, M, Plank, LD, Hill, GL. Components of energy expenditure in patients with severe sepsis and major trauma: A basis for clinical care. Crit Care Med. 1999; 27:1295–1302.
148. Briassoulis, G, Venkataraman, S, Thompson, AE. Energy expenditure in critically ill children. Crit Care Med. 2000; 28:1166–1172.
149. Swinamer, DL, Phang, PT, Jones, RL, et al. Twenty-four hour energy expenditure in critically ill patients. Crit Care Med. 1987; 15:637–643.
150. Weissman, C, Kemper, M, Elwyn, DH, et al. The energy expenditure of the mechanically ventilated critically ill patient. An analysis. Chest. 1986; 89:254–259.
151. Weissman, C, Kemper, M, Hyman, AI. Variation in the resting metabolic rate of mechanically ventilated critically ill patients. Anesth Analg. 1989; 68:457–461.
152. Metheny N: Verification of feeding tube placement. Current issues in enteral nutrition support: Report of the first Ross Enteral Device Conference, Sanibel Island, Florida, Oct. 9-11, 1995. Columbus, OH, Ross Products Division, Abbott Laboratories, 1996.
153. Rouby, JJ, Laurent, P, Gosnach, M, et al. Risk factors and clinical relevance of nosocomial maxillary sinusitis in the critically ill. Am J Respir Crit Care Med. 1994; 150:776–783.
154. Montejo, JC. Enteral nutrition-related gastrointestinal complications in critically ill patients: A multicenter study. The Nutritional and Metabolic Working Group of the Spanish Society of Intensive Care Medicine and Coronary Units. Crit Care Med. 1999; 27:1447–1453.
155. Dive, A, Moulart, M, Jonard, P, et al. Gastroduodenal motility in mechanically ventilated critically ill patients: A manometric study. Crit Care Med. 1994; 22:441–447.
156. Boivin, MA, Levy, H. Gastric feeding with erythromycin is equivalent to transpyloric feeding in the critically ill. Crit Care Med. 2001; 29:1916–1919.
157. Bowling, TE, Raimundo, AH, Grimble, GK, et al. Reversal by short-chain fatty acids of colonic fluid secretion induced by enteral feeding. Lancet. 1993; 342:1266–1268.
158. McClave, SA, Lukan, JK, Stefater, JA, et al. Poor validity of residual volumes as a marker for risk of aspiration in critically ill patients. Crit Care Med. 2005; 33:324–330.
159. Montejo, JC, Grau, T, Acosta, J, et al. Multicenter, prospective, randomized, single-blind study comparing the efficacy and gastrointestinal complications of early jejunal feeding with early gastric feeding in critically ill patients. Crit Care Med. 2002; 30:796–800.
160. Davies, AR, Froomes, PR, French, CJ, et al. Randomized comparison of nasojejunal and nasogastric feeding in critically ill patients. Crit Care Med. 2002; 30:586–590.
161. Heyland, DK, Drover, JW, Dhaliwal, R, et al. Optimizing the benefits and minimizing the risks of enteral nutrition in the critically ill: Role of small bowel feeding. JPEN J Parenter Enteral Nutr. 2002; 26:S51–S55.
162. Cresci, G, Martindale, R. Bedside placement of small bowel feeding tubes in hospitalized patients: A new role for the dietitian. Nutrition. 2003; 19:843–846.
163. Hernandez-Socorro, CR, Marin, J, Ruiz-Santana, S, et al. Bedside sonographic-guided versus blind nasoenteric feeding tube placement in critically ill patients. Crit Care Med. 1996; 24:1690–1694.
164. Huerta, G, Puri, VK. Nasoenteric feeding tubes in critically ill patients (fluoroscopy versus blind). Nutrition. 2000; 16:264–267.
165. Drakulovic, MB, Torres, A, Bauer, TT, et al. Supine body position as a risk factor for nosocomial pneumonia in mechanically ventilated patients: A randomised trial. Lancet. 1999; 354:1851–1858.
166. Eerola, R, Kaukinen, L, Kaukinen, S. Analysis of 13 800 subclavian vein catheterizations. Acta Anaesthesiol Scand. 1985; 29:193–197.
167. Tyden, H. Cannulation of the internal jugular vein—500 cases. Acta Anaesthesiol Scand. 1982; 26:485–488.
168. Bodenham, AR. Can you justify not using ultrasound guidance for central venous access? Crit Care. 2006; 10:175.
169. Karakitsos, D, Labropoulos, N, De Groot, E, et al. Real-time ultrasound-guided catheterisation of the internal jugular vein: A prospective comparison with the landmark technique in critical care patients. Crit Care. 2006; 10:R162.
170. Sabbaj, A, Hedges, JR. Ultrasonographic guidance for internal jugular vein cannulation: An educational imperative, a desirable practice alternative. Ann Emerg Med. 2006; 48:548–550.
171. Leung, J, Duffy, M, Finckh, A. Real-time ultrasonographically-guided internal jugular vein catheterization in the emergency department increases success rates and reduces complications: A randomized, prospective study. Ann Emerg Med. 2006; 48:540–547.
172. Haire, WD. Arm vein thrombosis. Clin Chest Med. 1995; 16:341–351.
173. Christoff, N, Watters, V, Sparks, W, et al. Use of triple-lumen subclavian catheters for administration of total parenteral nutrition. JPEN J Parenter Enteral Nutr. 1992; 18:71.
174. Corona, ML, Peters, SG, Narr, BJ, et al. Infections related to central venous catheters. Mayo Clin Proc. 1990; 65:979–986.
175. Kemp, L, Burge, J, Choban, P, et al. The effect of catheter type and site on infection rates in total parenteral nutrition patients. JPEN J Parenter Enteral Nutr. 1994; 18:71–74.
176. Mermel, LA. Prevention of intravascular catheter-related infections. Ann Intern Med. 2000; 132:391–402.
177. Cook, D, Randolph, A, Kernerman, P, et al. Central venous catheter replacement strategies: A systematic review of the literature. Crit Care Med. 1997; 25:1417–1424.
178. van den Berghe, G, Wilmer, A, Hermans, G, et al. Intensive insulintherapy in the medical ICU. N Engl J Med. 2006; 354:449–461.
179. Finfer, S, Chittock, DR, Su, SY, et al. Intensive versus conventional glucose control in critically ill patients. NICE-SUGAR Study Investigators. N Engl J Med. 2009; 360(13):1283–1297.
180. Wagman, LD, Newsome, HH, Miller, KB, et al. The effect of acute discontinuation of total parenteral nutrition. Ann Surg. 1986; 204:524–529.
181. Postuma, R, Trevenen, CL. Liver disease in infants receiving total parenteral nutrition. Pediatrics. 1979; 63:110–115.
182. Baker, AL, Rosenberg, IH. Hepatic complications of total parenteral nutrition. Am J Med. 1987; 82:489–497.
183. Fouin-Fortunet, H, Le Quernec, L, Erlinger, S, et al. Hepatic alterations during total parenteral nutrition in patients with inflammatory bowel disease: A possible consequence of lithocholate toxicity. Gastroenterology. 1982; 82:932–937.
184. von Allmen, D, Fischer, JE. Metabolic complications. In Fischer JE, ed. : Total Parenteral Nutrition, 2nd ed, Boston: Little and Brown, 1991.
185. Freund, HR, Muggia-Sullam, M, LaFrance, R, et al. A possible beneficial effect of metronidazole in reducing TPN-associated liver function derangements. J Surg Res. 1985; 38:356–363.
186. Weinsier, RL, Krumdieck, CL. Death resulting from overzealous total parenteral nutrition: The refeeding syndrome revisited. Am J Clin Nutr. 1981; 34:393–399.
187. Solomon, SM, Kirby, DF. The refeeding syndrome: A review. JPEN J Parenter Enteral Nutr. 1990; 14:90–97.
188. Nakasaki, H, Ohta, M, Soeda, J, et al. Clinical and biochemical aspects of thiamine treatment for metabolic acidosis during total parenteral nutrition. Nutrition. 1997; 13:110–117.
189. Groh-Wargo, S, Ciaccia, A, Moore, J. Neonatal metabolic acidosis: Effect of chloride from normal saline flushes. JPEN J Parenter Enteral Nutr. 1988; 12:159–161.