[level-membership-for-neurosurgery-category]
Chapter 116 Novel Targets in Deep Brain Stimulation for Movement Disorders
The surgical treatment of movement disorders has evolved considerably over the last 15 to 20 years with the increasing application of deep brain stimulation (DBS), principally for the treatment of Parkinson’s disease (PD), essential tremor, and dystonia. The sites most commonly targeted for DBS in the treatment of movement disorders have been the subthalamic nucleus (STN), the internal segment of the globus pallidus (GPi), and the motor thalamus—mainly the ventral intermediate nucleus (Vim) in Hassler’s nomenclature.1a A growing body of evidence supports DBS targeting of the GPi or STN for treatment of advanced PD with disabling motor fluctuations or dyskinesias, despite optimal medical management1,2; the Vim for disabling, medication-refractory essential tremor3; and the GPi for generalized dystonia.4 The U.S. Food and Drug Administration approved DBS of the STN or GPi for the treatment of PD, unilateral thalamic stimulation for the treatment of essential tremor, and under a Humanitarian Device Exemption, unilateral or bilateral stimulation of the GPi or STN for the treatment of dystonia. The growing literature concerning these targets increasingly outlines the limitations of each for particular diseases and for particular symptoms of a given disease, prompting the “off label” application of DBS and other methods of neuromodulation to other targets. In several respects, available treatments for movement disorders, both pharmacologic and neurosurgical, are suboptimal, including the treatment of some axial symptoms of PD, tremor syndromes that respond poorly or variably to conventional surgeries, and some dystonias with variable or poor response.
Cooper’s inadvertent laceration and ligation of the anterior choroidal artery in 1952 during a mesencephalic pedunculotomy for the treatment of a postencephalitic tremor demonstrates how an initial, serendipitous clinical finding can lead to anatomic and physiologic insights that guide the process of target selection in the treatment of movement disorders. Cooper’s hypothesis that the therapeutic effect observed after the procedure was the result of medial globus pallidus infarction led him to pursue anterior choroidal artery ligation and later chemopallidectomy and chemothalamectomy, with absolute alcohol injections into these structures.5–7 The observations that pallidotomy and thalamotomy yielded improvement in parkinsonism without weakness and with few observable deficits in motor function helped guide subsequent modeling of corticobasal ganglia function. As the responses of different movement disorders to these types of lesions were catalogued, certain observations proved difficult to reconcile with the prevailing models. These inconsistencies have been called the “paradoxes of functional neurosurgery.”8,9 The observations were made that lesions of the same target may improve different phenotypes of movement disorder (dyskinesia, dystonia, and parkinsonism all have been reported to improve with pallidal and with subthalamic DBS10,11) and that high-frequency stimulation of these structures produces clinical effects that are strikingly similar to ablative procedures. These paradoxes have posed challenges to the models of the motor system that prevailed at the time and to the various hypotheses about the mechanism or mechanisms of action of DBS. In this way, refinements of models of the motor system proceed hand in hand with testable hypotheses that guide innovation in the clinical realm. Any discussion of novel targets for the treatment of movement disorders must therefore include novel ideas about the normal and pathologic physiology of the motor system, as well as novel ideas about the mechanisms of action of therapeutic stimulation.
Current Models of Cortical–Basal Ganglia Networks
Models of the normal and pathologic physiology of the motor system, especially of corticobasal ganglia–thalamic function, have evolved significantly over the last 20 to 30 years to better account for observations such as the similarity between lesions and high-frequency stimulation of certain targets and the abnormalities observed in the parkinsonian subject and in animal models of striatal dopamine depletion. Seminal works from the 1980s and 1990s include the characterization of Albin, DeLong, and colleagues of multiple, parallel, segregated loops transmitted serially from the cortex; through the striatum, pallidum, and thalamus; and back to the cortex.12–14 Gerfen et al.15 demonstrated distinct populations of striatal medium spiny neurons that possess mainly subtype-1 dopamine receptors and project directly to the GPi or subtype-2 dopamine receptors and project indirectly to the medial pallidum via the external segment of the globus pallidus (GPe) and STN. These models conceived of pallidal outflow as a tonic, inhibitory brake on the thalamus and helped explain the observations of aberrantly high neuronal discharge frequencies in the STN and GPi in animal models of hypokinetic states such as dopamine depletion16 and in human parkinsonism. This model of hypokinesia as a result of excessive inhibition of the thalamus by the pallidum was incongruous with some observations, such as the lack of hypokinesia observed with thalamic lesions. Similarly, the therapeutic effects of pallidal, thalamic, or subthalamic lesions on hyperkinesias challenge this classical model, which, in their simplest form, posit that hyperkinetic disorders result from reduced pallidothalamic outflow. Modifications of these models have incorporated other connections in the cortical–basal ganglia network, such as extrathalamic projections of pallidal output, reciprocal GPe-STN interconnections, and direct cortical-STN projections (“hyperdirect” pathway).17 However, newer models have also significantly shifted the emphasis from abnormal frequency and patterns of neuronal discharge in basal ganglia nuclei during loss of dopaminergic innervation to an emphasis on abnormal network activity, particularly in the form of abnormal neuronal synchrony and network oscillations measured by field potentials. The crux of these newer models, sometimes referred to as “noisy signal” hypotheses,8 is that different pathologic conditions may be characterized by abnormal neuronal synchrony and dominance of particular field potential oscillations in particular networks, potentially producing tremor (5 Hz), dystonia (3-10 Hz), or akinesia (20 Hz).18,19 This shift in models owes much to investigations into the cellular and network substrates of thalamocortical oscillations performed in the 1980s, when Jahnsen and Llinas demonstrated dual behavior in thalamocortical neurons20: When simply depolarized, these cells exhibited a “classical” behavior, emitting a series of spikes and relaying their inputs on to the cortex with high fidelity. When released from hyperpolarization, these cells exhibited a “burst” mode, where afferent information was largely prevented from being relayed to the cortex. The biophysical mechanisms of the bursting were found to result from the interplay between a hyperpolarization-activated cation current (known as Ih) and a transient low-threshold calcium current (known as IT), which produces a “slow spike” lasting tens of milliseconds when released from hyperpolarization. These produce “pacemaker potentials,” which are rhythmic oscillations of thalamocortical cells in the delta frequency range (3-6 Hz).20 These cellular oscillations are important for understanding oscillating field potentials such as those measured by electroencephalogram, but the cellular oscillations alone are insufficient to generate the delta frequency oscillations measured in field potentials. The relationship between rhythmic bursting at the cellular level and oscillations measured in field potentials is complex, and a number of mechanisms may contribute to synchronization of oscillating and nonoscillating cells. Networked models of separate thalamocortical oscillators that incorporate thalamocortical connectivity and feedback pathways can account for synchronized thalamic burst firing at delta frequency and generate field potentials. Models of these pacemaker potentials interacting in a simple network among the thalamocortical cells, the corticothalamic cells, and the reticular nucleus of the thalamus have been developed to explain how these phenomena generate local field potential (LFP) oscillations in the form of delta waves and spindle waves observed during sleep.21
Similar mechanisms for cellular oscillations, rhythmic activity, and synchronization across networks of cells have been proposed for the STN and its interactions with the pallidum. STN neurons have been identified that generate rebound burst activity mediated by hyperpolarization-deinactivated calcium channels,22 and roughly half of STN neurons in rat brain slices switch from a single-spike mode to burst-firing mode when continuously hyperpolarized.23 Studies of cocultures of STN and GPe neurons demonstrate that this reciprocally interconnected network can generate correlated, rhythmic firing when isolated from the cortex and striatum.24 Models of STN-GPe reciprocal bursting, similar to those described for spindle oscillations in the thalamus, involve synchronous bursting activity in GPe neurons that hyperpolarizes STN neurons sufficiently to generate rebound burst activity, which then drives bursting activity in GPe and perpetuates the rhythm.25 The loss of dopamine, leading to overactivity in the striatal neurons of the indirect pathway would disinhibit GPe neurons, hyperpolarize STN neurons, accentuate burst firing in the STN, and generate pathologic synchrony between groups of neurons. This could result in the generation of oscillations that disturb cortical processing, or it could restrict communication among the cortex, STN, and GPe to certain bandwidths.
1. Biophysical properties of neurons can produce transitions from high-fidelity tonic states of information transfer to low-fidelity bursting and oscillating states. Such neurons can be found in the thalamus, STN, GPi, GPe, and pedunculopontine nucleus (PPN).
2. Neuromodulators (i.e., dopamine), could have direct, as well as indirect, effects on the transitions of individual neurons between these states. Thus, loss of striatal and extrastriatal dopamine could influence subthalamic neuronal-firing properties directly or indirectly (i.e., through hyperpolarization of STN neurons via increased GPe activity).
3. Populations of oscillating neurons can synchronize with one another through direct or indirect connections, and degree of synchronization could be affected by the presence or absence of neuromodulators.
4. Synchronized oscillations can contribute to the generation of specific field potentials and restrict communication among groups of cells to certain bandwidths.
5. Different pathologic conditions may be characterized by dominance of particular field potential oscillations in particular networks, potentially producing tremor (5 Hz), dystonia (3-10 Hz), or akinesia (20 Hz).19
In this type of model of subcortical pathophysiology, an intriguing hypothesis regarding the mechanism of high-frequency stimulation is to trigger tonic firing, thus preventing neurons from returning to their hypersynchronized, bursting, oscillatory firing patterns. These models can also offer possible explanations of the diverse etiologies of movement disorders: single-ion channel dysfunction that changes the biophysics of single cells; loss of neuromodulatory transmitters such as dopamine, as seen in idiopathic PD; or altered network connectivity from plastic changes at cortical or subcortical levels, as seen in post-traumatic movement disorders or models of certain dystonias.26,27 While many details of the cellular and network substrates of normal and pathologic field potential oscillations remain uncertain, some specific rhythms associated with specific symptoms are reasonably well characterized and deserve review.
Beta Oscillations and Parkinsonian Akinesia
Pathologic oscillations in movement disorders have been characterized most extensively in PD. Electrocorticographic and electroencephalographic investigations of motor tasks have long shown a decrease in signal power over motor cortices in the beta frequencies (about 20 Hz) during movement, sometimes referred to as event-related desynchronization.28 Voluntary movement is accompanied by increased power over more focal areas of the motor cortex during movement in higher-frequency bandwidths.29,30 The inability of the cortex to transition between beta oscillations and these focal higher-frequency oscillations appears to be a feature of parkinsonian akinesia, and the basal ganglia dysfunction in PD appears to be a critical part of this impediment. LFPs recorded from the STN, GPi, and motor cortex in parkinsonian patients demonstrate the presence of increased power in the beta band associated with akinesia.31 Not only is this increased power abolished with the administration of dopaminergic agonists or exogenous levodopa (l-dopa),32 but it remains suppressed in the STN immediately following high-frequency stimulation of that structure.33 Accumulating evidence of this type suggests that suppression of the “beta straightjacket” may be a key to therapeutic efficacy of medications and DBS for the treatment of Parkinsonian akinesia.
Oscillations Associated with Tremor
Tremor is one of the cardinal symptoms of PD, consists of a rhythmic (4-8 Hz) activation of mainly distal musculature, and manifests at rest and in some cases while maintaining posture. Identification of pathologic oscillations in the nervous system that correlate with the oscillatory movements of parkinsonian tremor has proved challenging. The question is complicated by the problem of defining a physiologic oscillation that is efferent in character (i.e., drives the tremor) versus one that is afferent (i.e., results from proprioceptive feedback). Physiologic models of tremor have evolved to account for the observations that dorsal rhizotomies had minimal effects on tremor (suggesting central oscillators as drivers) and that Vim DBS is often able to treat parkinsonian tremor but does not affect the other cardinal symptoms of the disease to the same degree. Tremor-frequency single-unit activity (“tremor cells”) in the ventral oral posterior nucleus, Vim, and ventral caudal nucleus thalamus has been described,34,35 as have cells with tremor-frequency bursting oscillations in the STN36 and globus pallidus.37 Pare et al. hypothesized that rhythms transmitted through inhibitory pallidal outflow to the motor thalamus were transformed into the 3- to 6-Hz tremor frequency in the thalamocortical circuit through the low-threshold calcium spikes induced by transient (T-type) calcium channels.38 Hassler et al. were able to produce contralateral resting tremor with stimulation of the internal segment of the globus pallidus at frequencies of less than 25 Hz,39 and Plaha et al. were able to elicit a 3- to 6-Hz tremor in a previously tremor-free parkinsonian patient by stimulating the caudal ZI at the beta frequency. In the latter study, low-frequency stimulation of the GPi or PPN did not produce tremor, and STN stimulation at low frequencies only produced tremor at high amplitudes.40 Because tremor in muscles of different extremities is not usually coherent, it has been concluded that independent central oscillators generate tremor separately in separate extremities.41 Simultaneous recordings of spatially separated tremor-related cells in the GPi showed that some pairs of cells oscillated in phase and some oscillated out of phase with one another,37 leading the authors to hypothesize that tremor-related activity may be propagated independently by several parallel pathways.
Surgical Targets for Tremor
Since Benabid’s demonstration of efficacy with high-frequency stimulation (more than 100 Hz) of the Vim thalamus for the treatment of parkinsonian tremor, this site has emerged as the preferred target.42 While this therapy seems to produce effects that are indistinguishable from a lesion of this target, it remains uncertain what ideal subregion of the thalamus (or possibly extrathalamic structure) provides the best tremor control, whether through stimulation or ablation. This question was posed during the ablative era of functional neurosurgery, highlighted by Laitinen’s survey of 16 neurosurgeons, who were asked to define their choice of target for treatment of a patient with PD who exhibited tremor and rigidity in equal measure. Targets differed by as much as 7 mm and included a wide span of the ventrolateral thalamus; the PSA, including the ZI; and the pallidothalamic fibers of the Forel fields.43 Some of the difficulty in defining the ideal target stems from the multiple parcellation schemes for the motor thalamus,44 though most stereotactic neurosurgeons employ Hassler’s terminology, which forms the basis for thalamic parcellation in Schaltenbrand and Wahren’s widely used atlas.45 The question of the ideal target is also complicated by questions as to how stimulation differs from ablative surgery. The ideal target for stimulation may be different from the ideal target for ablation. Among the multiple hypotheses that have been put forth regarding the mechanism or mechanisms of DBS, two intriguing ones related to tremor are the observation that adenosine release may be critical to the effect of Vim stimulation46 and data that suggest stimulation in the dorsal STN may act by directly modulating pallidothalamic or cerebellothalamic fibers in the immediate vicinity.47 Finally, while Vim DBS has been shown to be effective for parkinsonian and essential tremor,48,49 other tremor syndromes respond only inconsistently or less dramatically to Vim DBS. In addition to controversy surrounding the ideal target for a particular tremor syndrome, there is the question of the ideal target for those patients exhibiting combinations of tremor syndromes. Patients exhibiting symptoms of both PD and essential tremor may be treated with Vim DBS, but other parkinsonian symptoms would not be expected to respond. For these patients, and for patients with tremors that respond inconsistently or less dramatically to Vim DBS, it is important to describe the role of sites such as the STN, which some data suggest it can effectively treat symptoms of essential tremor,50 and the PSA.
Posterior Subthalamic Area
The PSA encompasses the ZI and the prelemniscal radiation (RaPRL). This was the target area of subthalamotomies performed frequently from the 1960s to the 1980s. Various terminologies have been used to define approximately the same target area, such as the PSA, ZI, RaPRL, subthalamic area, and posterior subthalamic white matter. In this chapter, we refer to this area as the PSA and discuss in further detail two of its most important components, the ZI and the RaPRL. The PSA is bordered anteriorly by the posterior STN, inferiorly by the dorsal substantia nigra, superiorly by the ventral thalamic nuclei, posteriomedially by the red nucleus, posteriolaterally by the ventrocaudal nucleus, posteriorly by the lateral lemniscus, and laterally by the posterior limb of the internal capsule (Fig. 116-1). As a surgical target, the PSA has been extensively reviewed by Blomstedt et al., who accounted for a total of 95 patients implanted with PSA DBS: 42 of these patients for the diagnosis of PD, 21 for multiple sclerosis, 18 for essential tremor, 7 for torticollis, 2 for dystonic tremor, 2 for post-traumatic tremor, 1 for cerebellar tremor, 1 for Holmes tremor, and 1 for spinocerebellar ataxia.51 Several reports have suggested that high-frequency stimulation of the PSA offers superior tremor control to stimulation of the Vim. Hamel et al. systematically analyzed the location of active contacts effective in suppressing intention tremor and found that those below the intercommissural plane in the subthalamic region were most effective.52
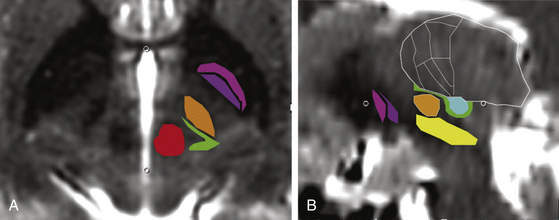
Complications of PSA DBS can include dysarthria (potentially from overly lateral placement of the electrode stimulating the corticobulbar fibers in the internal capsule or overly medial placement of the electrode stimulating the cerebellar fibers of the RaPRL); disequilibrium; lethargy; or worsening symptoms of preexistent depression.53
Analysis of lead locations following intended STN DBS electrode placement demonstrated a correlation between best clinical effect and placement of the electrode dorsal to the STN in the white matter bundles of the ZI and H1 and H2 fields of Forel, outside of the PSA.54,55
Zona Incerta
The ZI is a collection of gray matter that is continuous ventrally with the reticular nucleus of the thalamus. Functionally, it is divided into four parts: rostral, ventral, dorsal, and caudal. The rostral ZI is separated from the STN by the pallidofugal fibers crossing the internal capsule on the dorsomedial side of the STN. The ZI receives afferent input from the GPI or substantia nigra pars reticulata (SNr), the PPN, and cortical projections. Its gamma-aminobutyric acid–ergic (GABAergic) efferents target the CM/PF, the ventral anterior–ventral lateral nuclei of the thalamus, and the cortex, among others. The caudal or motor component of the ZI extends posterior to the STN and the RaPRL. Forel described it in 1877 as a “region of which nothing certain can be said,” and it remains an understudied region. Although the ZI is not considered part of the basal ganglia, the anatomy and physiology of the two regions overlap considerably. Both receive excitatory cortical input and send inhibitory projections to the thalamus, and both are involved in motor control and attention orientation, among other shared functions.56
Recent studies in rodents have demonstrated that this region plays a critical role in the gating of somatosensory input by the brain stem57 and the motor cortex.58 Like the neighboring reticular nucleus of the thalamus, cholinergic input from the brain stem results in state-dependent input from the ZI to the thalamus, which may contribute to elevated sensory thresholds during sleep. The ZI serves as an important locus for modulating sensory information from diverse sources by gating unwanted sensory input via inhibition of thalamic neurons.58 It has been proposed that the efferent GABAergic signal of ZI neurons patterned by cortical activity can play a critical role in synchronizing thalamocortical and brain stem rhythms.59 Subsequently, it has been implicated as a generator of tremor in PD and essential tremor.40
Given the similar connectivity between the ZI and the basal ganglia, it is perhaps not surprising that DBS achieves many of the same results in the ZI as it does in the basal ganglia. Indeed, high-frequency DBS may have a greater effect in the dorsal ZI than in the STN. In the largest case series published of 35 patients with refractory idiopathic PD, patients were implanted with a total of 64 high-frequency DBS electrodes. Of these, 27 DBS electrodes were implanted into the dorsal ZI, 20 into the dorsomedial STN, and 17 into the STN. This observational study suggested that high-frequency DBS of the ZI is superior to that of the STN in improving contralateral Unified Parkinson’s Disease Rating Scale motor scores. Some patients developed speech and balance disturbances following high-frequency DBS, thought to be due to the proximity of the lead to the RaPRL.60 A report by Kitagawa et al. from 2005 targeting the caudal ZI in patients with tremor-dominant PD noted 78.3% improvement in contralateral tremor and even superior improvement in rigidity.61
Prelemniscal Radiation
The ZI separates posteriorly the dorsal STN from the RaPRL located posterior to the ZI/STN. Its ascending fibers originate from the mesencephalic reticular formation and the cerebellum projecting to the thalamus. Thus, it is thought to play a role in tremor formation and muscle tone in the context of attention. Continuous stimulation of the RaPRL abolishes contralateral tremor with reduction in rigidity by inhibition of local circuits.62 With RaPRL DBS, dysarthria can be a complication by cerebellothalamic fiber stimulation and disequilibrium from the stimulation of the ascending mesencephalic reticular formation. Murata et al. targeted the RaPRL in eight patients with essential tremor of the proximal muscles using these coordinates: lateral to the red nucleus (about 10 mm from midline) and 3 to 4 mm posterior to the posterior border of the STN in the axial plane through the largest cut over the STN. Postoperative localization analysis resulted in mean stereotactic coordinates in patients with more than 71% improvement at 7.6 ± 1.2 mm posterior to the midcommissural point, 10.9 ± 0.8 mm lateral from midline, and 3.9 ± 1.7 mm inferior from the AC-PC line.63 This targeting yielded marked improvement in both proximal and distal tremor control.
Akinesia, Freezing of Gait, Postural Instability, and PPN DBS
STN and GPi stimulation have shown inconsistent benefit with respect to some axial symptoms of PD, especially those that manifest later in the course of the disease. Particularly problematic are freezing of gait, severe postural instability, and falls. In a long-term follow-up study of patients treated with STN DBS, an initial benefit in postural instability was lost after 1 year, and instability progressed significantly by 5 years.64 A survey of 55 patients who had undergone STN DBS at two centers in the Netherlands revealed that 42% described a delayed worsening of gait, despite continued improvement in global outcome scores.65 Although some measures of gait have been shown to improve with DBS, in a trial of 255 patients randomized to best medical therapy versus GPi or STN DBS, there were significantly more events for the DBS group than for the best medical therapy group for falls and gait disturbance.1
Because of the disabling nature of the symptoms of freezing and postural instability and the inconsistent effects of current therapies, novel treatments are actively pursued. Preclinical data implicating the PPN as a potential site of modulation of these symptoms propelled clinical studies with small numbers of patients. These data included findings in parkinsonian nonhuman primates that reversible inactivation of this structure with muscimol produces bilateral bradykinesia with unilateral microinjection, as well as akinesia produced with excitotoxic lesions with kainic acid.66 Akinesia was alleviated in parkinsonian nonhuman primates with the injection of the GABA antagonist bicuculline67 and with low-frequency (5-20 Hz) electrical stimulation,68,69 and PPN DBS and l-dopa treatment combined yielded significantly better results than either treatment given alone. These studies provide a rationale for the clinical application of PPN DBS in advanced PD with severe axial symptoms like gait freezing and loss of postural control. The complimentary effects to l-dopa therapy can be taken as support for PPN DBS as a potential therapy for patients not wholly responsive to l-dopa (and for possibly considering PPN DBS for other akinetic disorders like progressive supranuclear palsy and multiple system atrophy, which do not respond to l-dopa) and for the combination of PPN DBS with STN/GPi DBS to alleviate different groups of symptoms (i.e., dopaminergic and nondopaminergic).
The human PPN is located ventrocaudal from the red nucleus extending down toward the locus coeruleus, lateral to the decussation of the superior cerebellar peduncle, dorsal to the substantia nigra, and medial to the medial lemniscus (Fig. 116-2). It extends toward the laterodorsal tegmental nucleus, and it is a significant target of GPi outflow. Research across several species has revealed that the PPN is uniquely situated to play a role in the initiation and modulation of locomotion, as well as in several other functions.70 Similar to several other midbrain and brain stem motor regions, the PPN receives bilateral input—and has reciprocal connections—from the basal ganglia and motor cortex, transforms the input via local and intercommissural processing, and projects to the spinal cord. However, in addition to these descending projections, the PPN projects bilaterally to the substantia nigra pars compacta, STN, ZI, and thalamus, and it appears to influence dopaminergic activity in the striatum.70a The PPN is divided into a pars compacta (dorsolateral region with cholinergic neurons) and a pars dissipata (containing glutamatergic, GABAergic, cholinergic, dopaminergic, and noradrenergic neurons).
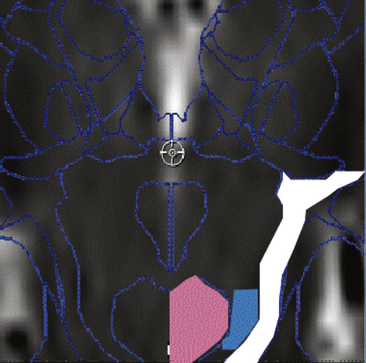
Studies in humans and other primates have demonstrated inhibitory pallidal outflow to noncholinergic cells of the PPN,72–74 and a subset of these neurons are glutamatergic and characterized by low-threshold calcium spikes mediated by T-type calcium channels.75 PPN outputs travel back along the lenticular fasciculus and ansa lenticularis and arborize in the GPi.76 Given the complex chemistry and connectivity of the PPN, it is perhaps not surprising that it is implicated in a range of functions, such as regulation of the sleep–wake cycle, reward learning, and modulation of movement.70,77
The initial human PPN DBS reports were published by two groups.78,79 Stefani et al. followed up with reporting on 6 patients in 2007 and again in 2009 with up to a 2-year follow-up on a total of 14 patients (12 with PD and 2 with progressive supranuclear palsy).80,81 Moro et al. reported their experience with 6 patients in 2009.82 Most recently, the Grenoble experience has been presented, in which 11 patients with PD and prominent freezing of gait were implanted in the region of the PPN (6 of these cases have been published83); 7 of the presented patients already had STN DBS and l-dopa therapy that had not adequately addressed their symptom. Bipolar or monopolar stimulation between 15 and 30 Hz and with 60-microsecond pulse widths was employed, and outcomes measured included a composite gait score, consisting of the Unified Parkinson’s Disease Rating Scale part 2 (items 14 and 15) and part 3 (items 29 and 30), as well as measures of falls related to freezing of gait. Of the 11 Grenoble patients, 5 patients experienced “very good” effects on these measures, 3 experienced “mild” effect, and 3 had no effect at 1-year follow-up. Through evaluation, the active contacts of responders were noted to have a more dorsal (posterior and superior) location, most consistent with placement in the nucleus cuneiformis or subcuneiformis; these, along with the PPN, are thought to serve as components of the midbrain locomotor region.84 In addition, 5 of the 11 patients experienced a modification of their level of alertness with stimulation, and this has been proposed as a potential predictor of a good outcome with respect to motor function. Thevathasan et al.85 reported outcomes and changes in reaction speed in a series of 11 patients who had undergone bilateral PPN region (one of these was described as “unilateral PPN/bilateral ZI” and two as “bilateral PPN/bilateral ZI”) electrode placements. Acute PPN stimulation in this group improved gait and balance but not akinesia scores. Chronic PPN stimulation significantly improved the frequency of falls, and improvement appeared correlated with increases in a simple reaction time task.
The methods for targeting the PPN vary among groups. Shimamoto et al. described target coordinates that were 6 to 7.5 mm lateral, 13 to 15 mm inferior, and 15 to 17 mm posterior to the midcommissural point. Adjustment based on direct measurements was made to place the lead 2 to 3 mm from the lateral edge of the brain stem at the axial level of the pontomesencephalic junction.86 Transventricular targeting has been advocated87 to maintain a trajectory parallel to the aqueduct and the floor of the fourth ventricle. This provides for the ability to span the rostral–caudal extent of the PPN with the electrode’s contacts. Pereira et al. described visualization of the decussation of the superior cerebellar peduncle on coronal and axial T2-weighted magnetic resonance imaging (MRI) scans. Diffusion tensor imaging employing a seed just lateral to the decussation delineates the connectivity of the intended site. Targeting this seed point, a trajectory along the long axis of the brain stem is chosen such that the electrode extends from a level just below the red nucleus to the level of the inferior colliculus.88
Some microelectrode recordings in parkinsonian patients have been reported, and the findings are consistent with the involvement of the PPN in the “noisy networks” described earlier for basal ganglia–thalamocortical circuits. Shimamoto et al. reported that PPN neurons had significantly more burst discharges compared with neurons from the region dorsal to PPN, as well as theta and beta peaks in the LFP power spectral density, with beta desynchronization observed during movement.86 Weinberger et al. studied LFPs recorded from microelectrodes and similarly found beta oscillations that increased in power within the PPN.89
CM/PF Complex
The CM/PF nuclei make up the caudal group of the intralaminar thalamic nuclei. Historically, the intralaminar thalamic and midline nuclei were distinguished from specific thalamic relay nuclei that receive a single source and are reciprocally interconnected with discrete sectors of the reticular nucleus of the thalamus. Unlike these specific relay nuclei, the intralaminar nuclei had been characterized as receiving converging inputs from many sources, innervating wide expanses of the reticular nucleus,90 and projecting “nonspecifically” to the cortex as part of the ascending reticulothalamocortical activating system.91 More recently, the caudal CM/PF components of the intralaminar nuclei have been shown to mediate a reciprocal thalamostriatal interaction that plays an important role in normal and pathologic movement.
The projections from the CM/PF thalamus are principally to the striatum, though sparse projections to the GPe, GPi, SNr, and STN have been described.92,93 The projections to the striatum can be subdivided into those innervating the somatosensory region of the putamen, which are derived from the centromedian thalamic nucleus, and those from the parafascicular nucleus, which innervates mainly the limbic and associative components of the striatum. The thalamostriatal projections differ from the corticostriatal projections in the sites of interaction with striatal neurons. Unlike corticostriatal neurons, which synapse principally on the heads of dendritic spines, the thalamostriatal neurons synapse on dendritic shafts, preferentially synapse with medium spiny neurons involved in the direct pathway to the GPi, and have a strong input to the cholinergic interneurons of the striatum.94 The reciprocal nature of this interaction is through the GPi/SNr, which constitutes the major input into the centromedian nucleus,95 though the CM/PF complex also receives afferents from the PPN and reticular formation.96
A body of preclinical work suggests the CM/PF as a potential target for neuromodulation for the treatment of movement disorders. Lesioning the CM in rats appears to confer protection against many cellular consequences of 6-hydroxydopamine–induced lesion of nigrostriatal dopamine neurons,97 and the CM/PF neurons themselves appear to be overactive following such 6-hydroxydopamine nigrostriatal lesioning. The CM/PF complex is a site of significant nondopaminergic degeneration in PD,98 and high-frequency stimulation of the CM/PF in 6-hydroxydopamine–treated rats alleviates the induced akinesia.97
Stereotactic surgery targeting the CM/PF has been employed for the treatment of neuropathic pain99,100 and epilepsy,101 with a few reports for the treatment of movement disorders. Studying a series of patients with chronic pain undergoing CM/PF versus somatosensory thalamic DBS, Krauss et al. found two patients with additional movement disorders that responded to CM/PF stimulation: one with resting tremor and one with stump dyskinesias.100a Caparros-Lefebvre et al. retrospectively evaluated electrode locations for two centers that achieved different results of thalamic stimulation on L-dopa–induced dyskinesias.102 Electrodes were posterior and medial in the group with better results for this symptom, and the authors suggested the effect is from greater proximity to the CM/PF complex. Mazzone et al. reported on six patients with bilateral DBS implantation of CM/PF and GPi for PD, finding superior improvement in freezing and dystonia when compared to the GPi alone, with more modest effect of CM/PF DBS on bradykinesia and rigidity.103 The same group reported on two patients with bilateral implantation of the CM/PF and STN,104 finding a greater reduction in mean and maximum accelerometer values associated with tremor during CM/PF stimulation than was achieved with STN stimulation alone.
Novel Targets for the Treatment of Dystonias
Dystonia is characterized by the simultaneous cocontraction of agonist and antagonist muscles, often producing painful, abnormal postures. Many diverse etiologies of dystonias that are characterized point to basal ganglia dysfunction, and single-cell and field potential recordings support the notion of noisy networks in these disorders. Obviously, the sources of “noise” may be different for the various etiologies. LFP recordings in dystonic patients with DBS in the pallidum reveal increased spectral power in the 4- to 10-Hz band.105 Pallidal recordings in humans and in rodent models show bursting at the single-unit level, abnormal discharge rates at rest and during movement,106 increased synchronicity among pairs of neurons, and widened receptive fields with increased responses of GPi neurons to passive movement of limbs.107 Class I evidence has been published supporting the application of GPi DBS to primary dystonias, with excellent outcomes reported particularly for primary generalized dystonias.4,108 Interest in exploring the STN as a target in part derives from empiric findings that some focal and secondary dystonias have shown less response to pallidal stimulation and from reports of pallidal stimulation–induced bradykinesia.109,110 A body of metabolic imaging and physiologic work implicates the indirect pathway in these conditions111,112 and indicates that modulation of this structure may have a therapeutic effect in cervical dystonia11,113,114 and segmental dystonia,115 with some reporting less favorable results with primary generalized dystonia.116
Spinal Cord Dorsal Column Stimulation
Promising results from a study in rodents revealed that dorsal column stimulation (DCS) has a similar effect to STN DBS in rodent models of PD.117 The authors described how electrical DCS restores locomotion in both the acute and the chronic rodent models of PD. This functional recovery was thought to be the result of disruption of pathologic beta corticostriatal oscillations from DCS, similar to what is observed from STN DBS.
Spinal cord DCS by epidural was evaluated in the 1970s for spasticity,118 with minor improvements noted in symptoms and concomitant improvement of the Hoffmann reflex. Reports of two patients with PD who were implanted with cervical epidural spinal cord stimulators failed to demonstrate a clinical benefit. Using a double-blind crossover study using frequencies ranging from 30 to 300 Hz, up to 4 V, and 240-microsecond pulse widths, neither the primary outcome measure (the motor subsection of the Unified Parkinson’s Disease Rating Scale) nor the secondary outcomes (including a hand–arm movement test, timed foot-tapping score, and test of the time to walk 10 m) were significantly improved.119,120
Cerebellar Stimulation
Cerebellar stimulation has been applied to spasticity and hyperkinetic movement disorders, with some positive preclinical and clinical results. Rats subjected to hemispheric stroke and implanted with contralateral lateral cerebellar (dentate nucleus) DBS demonstrated improved motor recovery.119 Galanda and Horvath reported on 4 human subjects with cerebral palsy and severe choreoathetoid movements who underwent suboccipital implantation of DBS electrodes into the anterior lobe of the cerebellum, with resulting improvements in spasticity, speech, swallowing, respirations, posture, ambulation, and mood with high-frequency stimulation at 185 Hz.121,122 Another study reported on 13 patients implanted with DBS targeting near the superior cerebellar peduncles.123 The most pronounced effects were observed in improvement in speech in 7 of the 13 patients; 1 patient became ambulatory. These more recent studies can be viewed as reproductions of similar results obtained with transtentorial DBS in the cerebellum in patients with cerebral palsy from the 1970s to 1980s.124,125
Cortical Stimulation
Motor cortex stimulation has been employed for the treatment of parkinsonian akinesia. Pascual-Leone et al. found that subthreshold, 5-Hz repetitive transcranial stimulation of the motor cortex shortened choice reaction times and movement times without increasing error rates in parkinsonian patients or affecting control patient performance.126 Drouot et al. performed a preclinical study of high-frequency motor cortex stimulation in a chronic 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine primate model, demonstrating reductions in akinesia and bradykinesia associated with a normalization of firing rates and reduction of synchronized oscillatory activity in the GPi and STN.127
Human clinical experience has been revealed initially in the form of case reports by Canavero and colleagues,128,129 and an Italian multicenter trial demonstrated variable and unpredictable improvement in a number of symptoms, including motor fluctuations, l-dopa–induced dyskinesias, and painful dystonia.130 This group has also reported results of motor cortex stimulation for the treatment of focal dystonias and postictal spasticity.131
Conclusions
Despite the tremendous success and increase in popularity of functional neurosurgical procedures over the last 20 years, the exploration and validation of novel targets to treat movement disorders face significant challenges. First, regulatory and financial constraints that apply to novel uses of implanted devices do not apply to ablative procedures to the same degree. Second, establishing efficacy of a given target for neuromodulation is complicated in some cases by a narrow therapeutic window for stimulation of small anatomic loci that are difficult to image. Controversies regarding optimal lead locations within these territories and difficulties addressing interindividual differences in anatomy are inevitable. Finally, establishing the relative efficacy of one therapy over another requires significant investment of resources and participation of multiple institutions to enroll sufficient patients and thus achieve statistical power of various outcomes measures. The recently published Veteran’s Administration cooperative trial comparing pallidal and subthalamic DBS in a randomized, double-blind fashion involved 13 centers, studied 299 randomized patients, and required approximately 10 years from design to publication.132 The study showed no significant difference in primary outcomes of change in motor function between the two groups, and it illustrates the challenge of obtaining class I data for novel therapeutic targets. Adding to the complexity, some investigators are pursuing strategies of targeting multiple loci in the same patient133 to address different components of the disorder. Despite the challenges, studies of these therapies and their relative efficacies will likely prove necessary for novel treatments to gain widespread acceptance and appropriate application to specific symptoms and disorders. The history of functional neurosurgery suggests that future expansion of this field depends not only on these sorts of clinical trials but also on advancement in the basic and clinical sciences through research into new devices and technologies, pursuit of serendipitous clinical findings, rediscovery and further exploration of historical targets of surgical intervention, and improved understanding of the normal and pathologic anatomy and physiology of the motor system.
Alexander G.E., Crutcher M.D., DeLong M.R. Basal ganglia–thalamocortical circuits: parallel substrates for motor, oculomotor, “prefrontal” and “limbic” functions. Prog Brain Res. 1990;85:119-146.
Bekar L., Libionka W., Tian G.F., et al. Adenosine is crucial for deep brain stimulation–mediated attenuation of tremor. Nat Med. 2008;14(1):75-80.
Blomstedt P., Sandvik U., Fytagoridis A., Tisch S. The posterior subthalamic area in the treatment of movement disorders: past, present, and future. Neurosurgery. 2009;64(6):1029-1038. discussion 1038-1042
Brown P. Bad oscillations in Parkinson’s disease. J Neural Transm Suppl. 2006;70:27-30.
Ferraye M.U., Debû B., Fraix V., et al. Effects of pedunculopontine nucleus area stimulation on gait disorders in Parkinson’s disease. Brain. 2010;133(Pt 1):205-214.
Follett K.A., Weaver F.M., Stern M., et al. Pallidal versus subthalamic deep-brain stimulation for Parkinson’s disease. N Engl J Med. 2010;362(22):2077-2091.
Fuentes R., Petersson P., Siesser W.B., et al. Spinal cord stimulation restores locomotion in animal models of Parkinson’s disease. Science. 2009;323(5921):1578-1582.
Godinho F., Thobois S., Magnin M., et al. Subthalamic nucleus stimulation in Parkinson’s disease: anatomical and electrophysiological localization of active contacts. J Neurol. 2006;253(10):1347-1355.
Jahnsen H., Llinas R. Voltage-dependent burst-to-tonic switching of thalamic cell activity: an in vitro study. Arch Ital Biol. 1984;122(1):73-82.
Jimenez F., Velasco F., Velasco M., et al. Subthalamic prelemniscal radiation stimulation for the treatment of Parkinson’s disease: electrophysiological characterization of the area. Arch Med Res. 2000;31(3):270-281.
Kitagawa M., Murata J., Uesugi H., et al. Two-year follow-up of chronic stimulation of the posterior subthalamic white matter for tremor-dominant Parkinson’s disease. Neurosurgery. 2005;56(2):281-289. discussion 281-9
Kleiner-Fisman G., Liang G.S., Moberg P.J., et al. Subthalamic nucleus deep brain stimulation for severe idiopathic dystonia: impact on severity, neuropsychological status, and quality of life. J Neurosurg. 2007;107(1):29-36.
Mazzone P., Lozano A., Stanzione P., et al. Implantation of human pedunculopontine nucleus: a safe and clinically relevant target in Parkinson’s disease. Neuroreport. 2005;16(17):1877-1881.
Mitrofanis J. Some certainty for the “zone of uncertainty”? Exploring the function of the zona incerta. Neuroscience. 2005;130(1):1-15.
Nambu A. A new dynamic model of the cortico-basal ganglia loop. Prog Brain Res. 2004;143:461-466.
Nandi D., Aziz T.Z., Giladi N., et al. Reversal of akinesia in experimental parkinsonism by GABA antagonist microinjections in the pedunculopontine nucleus. Brain. 2002;125(Pt 11):2418-2430.
Plaha P., Filipovic S., Gill S.S. Induction of parkinsonian resting tremor by stimulation of the caudal zona incerta nucleus: a clinical study. J Neurol Neurosurg Psychiatry. 2008;79(5):514-521.
Plaha P., Ben-Shlomo Y., Patel N.K., Gill S.S. Stimulation of the caudal zona incerta is superior to stimulation of the subthalamic nucleus in improving contralateral parkinsonism. Brain. 2006;129(Pt 7):1732-1747.
Plaha P., Gill S.S. Bilateral deep brain stimulation of the pedunculopontine nucleus for Parkinson’s disease. Neuroreport. 2005;16(17):1883-1887.
Salenius S., Hari R. Synchronous cortical oscillatory activity during motor action. Curr Opin Neurobiol. 2003 Dec;13(6):678-684.
Silberstein P., Kühn A.A., Kupsch A., et al. Patterning of globus pallidus local field potentials differs between Parkinson’s disease and dystonia. Brain. 2003;126(Pt 12):2597-2608.
Stefani A., Peppe A., Pierantozzi M., et al. Multi-target strategy for parkinsonian patients: the role of deep brain stimulation in the centromedian–parafascicularis complex. Brain Res Bull. 2009;78(2-3):113-118.
Vidailhet M., Vercueil L., Houeto J.L., et al. Bilateral, pallidal, deep-brain stimulation in primary generalised dystonia: a prospective 3 year follow-up study. Lancet Neurol. 2007;6(3):223-229.
Weinberger M., Mahant N., Hutchison W.D., et al. Beta oscillatory activity in the subthalamic nucleus and its relation to dopaminergic response in Parkinson’s disease. J Neurophysiol. 2006;96(6):3248-3256.
1. Weaver F.M., Follett K., Stern M., et al. Bilateral deep brain stimulation vs best medical therapy for patients with advanced Parkinson disease: a randomized controlled trial. JAMA. 2009;301(1):63-73.
1a. Hassler R. Anatomy of the thalamus. In: Schaltenbrand G., Baily P. Introduction to stereotaxis with an atlas of the humanbrain. Stuttgart: Thieme; 1959:230-290.
2. Bronstein J.M., Tagliati M., Alterman R.L., et al. Deep brain stimulation for Parkinson disease: an expert consensus and review of key issues. Arch Neurol. 2011;68(2):165.
3. Koller W.C., Lyons K.E., Wilkinson S.B., et al. Long-term safety and efficacy of unilateral deep brain stimulation of the thalamus in essential tremor. Mov Disord. 2001;16(3):464-468.
4. Vidailhet M., Vercueil L., Houeto J.L., et al. Bilateral deep-brain stimulation of the globus pallidus in primary generalized dystonia. N Engl J Med. 2005;352(5):459-467.
5. Das K., Benzil D.L., Rovit R.L., et al. Irving S. Cooper (1922-1985): a pioneer in functional neurosurgery. J Neurosurg. 1998;89(5):865-873.
6. Cooper I.S., Samra K., Bergmann L. The thalamic lesion which abolishes tremor and rigidity of parkinsonism. A radiologico-clinico-anatomic correlative study. J Neurol Sci. 1969;8(1):69-84.
7. Cooper I.S., Bergmann L.L., Caracalos A. Anatomic verification of the lesion which abolishes parkinsonian tremor and rigidity. Neurology. 1963;13:779-787.
8. Brown P., Eusebio A. Paradoxes of functional neurosurgery: clues from basal ganglia recordings. Mov Disord. 2008;23(1):12-20. quiz 158
9. Marsden C.D., Obeso J.A. The functions of the basal ganglia and the paradox of stereotaxic surgery in Parkinson’s disease. Brain. 1994;117(Pt 4):877-897.
10. Guridi J., Obeso J.A., Rodriguez-Oroz M.C., et al. L-dopa–induced dyskinesia and stereotactic surgery for Parkinson’s disease. Neurosurgery. 2008;62(2):311-323. discussion 323-5
11. Sun B., Chen S., Zhan S., et al. Subthalamic nucleus stimulation for primary dystonia and tardive dystonia. Acta Neurochir Suppl. 2007;97(Pt 2):207-214.
12. Albin R.L., Young A.B., Penney J.B. The functional anatomy of basal ganglia disorders. Trends Neurosci. 1989;12(10):366-375.
13. DeLong M.R. Primate models of movement disorders of basal ganglia origin. Trends Neurosci. 1990;13(7):281-285.
14. Alexander G.E., Crutcher M.D., DeLong M.R. Basal ganglia–thalamocortical circuits: parallel substrates for motor, oculomotor, “prefrontal” and “limbic” functions. Prog Brain Res. 1990;85:119-146.
15. Gerfen C.R., Engber T.M., Mahan L.C., et al. D1 and D2 dopamine receptor–regulated gene expression of striatonigral and striatopallidal neurons. Science. 1990;250(4986):1429-1432.
16. Bergman H., Wichmann T., Karmon B., DeLong M.R. The primate subthalamic nucleus. II. Neuronal activity in the MPTP model of parkinsonism. J Neurophysiol. 1994;72(2):507-520.
17. Nambu A. A new dynamic model of the cortico-basal ganglia loop. Prog Brain Res. 2004;143:461-466.
18. Brown P.. Bad oscillations in Parkinson’s disease. J Neural Transm Suppl, 2006;70:27-30
19. Salenius S., Hari R. Synchronous cortical oscillatory activity during motor action. Curr Opin Neurobiol. 2003;13(6):678-684.
20. Jahnsen H., Llinas R. Voltage-dependent burst-to-tonic switching of thalamic cell activity: an in vitro study. Arch Ital Biol. 1984;122(1):73-82.
21. McCormick D.A., Bal T. Sleep and arousal: thalamocortical mechanisms. Annu Rev Neurosci. 1997;20:185-215.
22. Bevan M.D., Atherton J.F., Baufreton J. Cellular principles underlying normal and pathological activity in the subthalamic nucleus. Curr Opin Neurobiol. 2006;16(6):621-628.
23. Beurrier C., Congar P., Bioulac B., Hammond C. Subthalamic nucleus neurons switch from single-spike activity to burst-firing mode. J Neurosci. 1999;19(2):599-609.
24. Bevan M.D., Magill P.J., Terman D., et al. Move to the rhythm: oscillations in the subthalamic nucleus–external globus pallidus network. Trends Neurosci. 2002;25(10):525-531.
25. Plenz D., Kital S.T. A basal ganglia pacemaker formed by the subthalamic nucleus and external globus pallidus. Nature. 1999;400(6745):677-682.
26. Byl N.N. Learning-based animal models: task-specific focal hand dystonia. ILAR J. 2007;48(4):411-431.
27. Byl N.N., Merzenich M.M., Jenkins W.M. A primate genesis model of focal dystonia and repetitive strain injury: I. Learning-induced dedifferentiation of the representation of the hand in the primary somatosensory cortex in adult monkeys. Neurology. 1996;47(2):508-520.
28. Crone N.E., Miglioretti D.L., Gordon B., et al. Functional mapping of human sensorimotor cortex with electrocorticographic spectral analysis. I. Alpha and beta event–related desynchronization. Brain. 1998;121(Pt 12):2271-2299.
29. Miller K.J., denNijs M., Shenoy P., et al. Real-time functional brain mapping using electrocorticography. Neuroimage. 2007;37(2):504-507.
30. Crone N.E., Sinai A., Korzeniewska A. High-frequency gamma oscillations and human brain mapping with electrocorticography. Prog Brain Res. 2006;159:275-295.
31. Bronte-Stewart H., Barberini C., Koop M.M., et al. The STN beta-band profile in Parkinson’s disease is stationary and shows prolonged attenuation after deep brain stimulation. Exp Neurol. 2009;215(1):20-28.
32. Weinberger M., Mahant N., Hutchison W.D., et al. Beta oscillatory activity in the subthalamic nucleus and its relation to dopaminergic response in Parkinson’s disease. J Neurophysiol. 2006;96(6):3248-3256.
33. Wingeier B., Tcheng T., Koop M.M., et al. Intra-operative STN DBS attenuates the prominent beta rhythm in the STN in Parkinson’s disease. Exp Neurol. 2006;197(1):244-251.
34. Lenz F.A., Tasker R.R., Kwan H.C., et al. Single unit analysis of the human ventral thalamic nuclear group: correlation of thalamic “tremor cells” with the 3-6 Hz component of parkinsonian tremor. J Neurosci. 1988;8(3):754-764.
35. Lenz F.A., Kwan H.C., Martin R.L., et al. Single unit analysis of the human ventral thalamic nuclear group. Tremor-related activity in functionally identified cells. Brain. 1994;117(Pt 3):531-543.
36. Rodriguez M.C., Guridi O.J., Alvarez L., et al. The subthalamic nucleus and tremor in Parkinson’s disease. Mov Disord. 1998;13(suppl 3):111-118.
37. Hurtado J.M., Gray C.M., Tamas L.B., Sigvardt K.A. Dynamics of tremor-related oscillations in the human globus pallidus: a single case study. Proc Natl Acad Sci U S A. 1999;96(4):1674-1679.
38. Pare D., Curro’Dossi R., Steriade M. Neuronal basis of the parkinsonian resting tremor: a hypothesis and its implications for treatment. Neuroscience. 1990;35(2):217-226.
39. Hassler R., Riechert T., Mundinger F., et al. Physiological observations in stereotaxic operations in extrapyramidal motor disturbances. Brain. 1960;83:337-350.
40. Plaha P., Filipovic S., Gill S.S. Induction of parkinsonian resting tremor by stimulation of the caudal zona incerta nucleus: a clinical study. J Neurol Neurosurg Psychiatry. 2008;79(5):514-521.
41. Raethjen J., Lindemann M., Schmaljohann H., et al. Multiple oscillators are causing parkinsonian and essential tremor. Mov Disord. 2000;15(1):84-94.
42. Benabid A.L., Pollak P., Louveau A., et al. Combined (thalamotomy and stimulation) stereotactic surgery of the VIM thalamic nucleus for bilateral Parkinson disease. Appl Neurophysiol. 1987;50(1-6):344-346.
43. Laitinen L.V. Brain targets in surgery for Parkinson’s disease. Results of a survey of neurosurgeons. J Neurosurg. 1985;62(3):349-351.
44. Krack P., Dostrovsky J., Ilinsky I., et al. Surgery of the motor thalamus: problems with the present nomenclatures. Mov Disord. 2002;17(suppl 3):S2-S8.
45. Schaltenbrand G., Wahren W. Atlas for Stereotaxy of the Human Brain. Stuttgardt: Thieme; 1977.
46. Bekar L., Libionka W., Tian G.F., et al. Adenosine is crucial for deep brain stimulation–mediated attenuation of tremor. Nat Med. 2008;14(1):75-80.
47. Maks C.B., Butson C.R., Walter B.L., et al. Deep brain stimulation activation volumes and their association with neurophysiological mapping and therapeutic outcomes. J Neurol Neurosurg Psychiatry. 2009;80(6):659-666.
48. Koller W.C., Lyons K.E., Wilkinson S.B., Pahwa R. Efficacy of unilateral deep brain stimulation of the VIM nucleus of the thalamus for essential head tremor. Mov Disord. 1999;14(5):847-850.
49. Koller W.C., Pahwa P.R., Lyons K.E., Wilkinson S.B. Deep brain stimulation of the Vim nucleus of the thalamus for the treatment of tremor. Neurology. 2000;55(12 suppl 6):S29-S33.
50. Stover N.P., Okun M.S., Evatt M.L., et al. Stimulation of the subthalamic nucleus in a patient with Parkinson disease and essential tremor. Arch Neurol. 2005;62(1):141-143.
51. Blomstedt P., Sandvik U., Fytagoridis A., Tisch S. The posterior subthalamic area in the treatment of movement disorders: past, present, and future. Neurosurgery. 2009;64(6):1029-1038. discussion 1038-42
52. Hamel W., Herzog J., Kopper F., et al. Deep brain stimulation in the subthalamic area is more effective than nucleus ventralis intermedius stimulation for bilateral intention tremor. Acta Neurochir (Wien). 2007;149(8):749-758. discussion 758
53. Fytagoridis A., Blomstedt P. Complications and side effects of deep brain stimulation in the posterior subthalamic area. Stereotact Funct Neurosurg. 2010;88(2):88-93.
54. Godinho F., Thobois S., Magnin M., et al. Subthalamic nucleus stimulation in Parkinson’s disease: anatomical and electrophysiological localization of active contacts. J Neurol. 2006;253(10):1347-1355.
55. Zonenshayn M., Sterio D., Kelly P.J., et al. Location of the active contact within the subthalamic nucleus (STN) in the treatment of idiopathic Parkinson’s disease. Surg Neurol. 2004;62(3):216-225. discussion 225-6
56. Mitrofanis J. Some certainty for the “zone of uncertainty”? Exploring the function of the zona incerta. Neuroscience. 2005;130(1):1-15.
57. Trageser J.C., Burke K.A., Masri R., et al. State-dependent gating of sensory inputs by zona incerta. J Neurophysiol. 2006;96(3):1456-1463.
58. Urbain N., Deschenes M. Motor cortex gates vibrissal responses in a thalamocortical projection pathway. Neuron. 2007;56(4):714-725.
59. Barthó P., Slézia A., Varga V., et al. Cortical control of zona incerta. J Neurosci. 2007;27(7):1670-1681.
60. Plaha P., Ben-Shlomo Y., Patel N.K., Gill S.S. Stimulation of the caudal zona incerta is superior to stimulation of the subthalamic nucleus in improving contralateral parkinsonism. Brain. 2006;129(Pt 7):1732-1747.
61. Kitagawa M., Murata J., Uesugi H., et al. Two-year follow-up of chronic stimulation of the posterior subthalamic white matter for tremor-dominant Parkinson’s disease. Neurosurgery. 2005;56(2):281-289. discussion 281-9
62. Jiménez F., Velasco F., Velasco M., et al. Subthalamic prelemniscal radiation stimulation for the treatment of Parkinson’s disease: electrophysiological characterization of the area. Arch Med Res. 2000;31(3):270-281.
63. Murata J., Kitagawa M., Uesugi H., et al. Electrical stimulation of the posterior subthalamic area for the treatment of intractable proximal tremor. J Neurosurg. 2003;99(4):708-715.
64. Krack P., Batir A., Van Blercom N., et al. Five-year follow-up of bilateral stimulation of the subthalamic nucleus in advanced Parkinson’s disease. N Engl J Med. 2003;349(20):1925-1934.
65. van Nuenen B.F., Esselink R.A., Munneke M., et al. Postoperative gait deterioration after bilateral subthalamic nucleus stimulation in Parkinson’s disease. Mov Disord. 2008;23(16):2404-2406.
66. Matsumura M. The pedunculopontine tegmental nucleus and experimental parkinsonism. A review. J Neurol. 2005;252(suppl 4):IV5-IV12.
67. Nandi D., Aziz T.Z., Giladi N., et al. Reversal of akinesia in experimental parkinsonism by GABA antagonist microinjections in the pedunculopontine nucleus. Brain. 2002;125(Pt 11):2418-2430.
68. Jenkinson N., Nandi D., Miall R.C., Stein J.F., Aziz T.Z. Pedunculopontine nucleus stimulation improves akinesia in a Parkinsonian monkey. Neuroreport. 2004;15(17):2621-2624.
69. Jenkinson N., Nandi D., Oram R., Stein J.F., Aziz T.Z. Pedunculopontine nucleus eletric stimulation alleviates akinesia independently of dopaminergic mechanisms. Neuroreport. 2006;17(6):639-641.
70. Mena-Segovia J., Bolam J.P., Magill P.J. Pedunculopontine nucleus and basal ganglia: distant relatives or part of the same family? Trends Neurosci. 2004;27(10):585-588.
71. Hamani C., Stone S., Laxton A., Lozano A. The pedunculopontine nucleus and movement disorders: Anatomy and the role for deep brain stimulation. Parkinsonism Relat Disord. 2007;13(suppl 3):S276-S280.
72. Noda T., Oka H. Distribution and morphology of tegmental neurons receiving nigral inhibitory inputs in the cat: an intracellular HRP study. J Comp Neurol. 1986;224(2):254-266.
73. Granata A.R., Kitai S.T. Inhibitory substantia nigra inputs to the pedunculopotine neurons. Exp Brain Res. 1991;86(3):459-466.
74. Shink E., Sidibé M., Smith Y. Efferent connections of the internal globus pallidus in the squirrel monkey: II. Topography and synaptic organization of pallidal efferents to the pedunculopontine nucleus. J Comp Neurol. 1997;382(3):348-363.
75. Takakusaki K., Kitai S.T. Ionic mechanisms involved in the spontaneous firing of tegmental pedunculopontine nucleus neurons of the rat. Neuroscience. 1997;78(3):771-794.
76. Lavoie B., Parent A. Pedunculopontine nucleus in the squirrel monkey: projections to the basal ganglia as revealed by anterograde tract-tracing methods. J Comp Neurol. 1994;344(2):210-231.
77. Winn P. How best to consider the structure and function of the pedunculopontine tegmental nucleus: evidence from animal studies. J Neurol Sci. 2006;248(1-2):234-250.
78. Mazzone P., Lozano A., Stanzione P., et al. Implantation of human pedunculopontine nucleus: a safe and clinically relevant target in Parkinson’s disease. Neuroreport. 2005;16(17):1877-1881.
79. Plaha P., Gill S.S. Bilateral deep brain stimulation of the pedunculopontine nucleus for Parkinson’s disease. Neuroreport. 2005;16(17):1883-1887.
80. Stefani A., Lozano A.M., Peppe A., et al. Bilateral deep brain stimulation of the pedunculopontine and subthalamic nuclei in severe Parkinson’s disease. Brain. 2007;130(Pt 6):1596-1607.
81. Mazzone P., Insola A., Sposato S., Scranati E. The deep brain stimulation of the pedunculopontine tegmental nucleus. Neuromodulation. 2009;12(3):191-204.
82. Moro E., Hamani C., Poon Y.Y., et al. Unilateral pedunculopontine stimulation improves falls in Parkinson’s disease. Brain. 2010 Jan;133(Pt 1):215-224.
83. Ferraye M.U., Debû B., Fraix V., et al. Effects of pedunculopontine nucleus area stimulation on gait disorders in Parkinson’s disease. Brain. 2010;133(Pt 1):205-214.
84. Jordan L.M. Initiation of locomotion in mammals. Ann N Y Acad Sci. 1998;860:83-93.
85. Thevathasan W., Silburn P.A., Brooker H., et al. The impact of low-frequency stimulation of the pedunculopontine nucleus region on reaction time in parkinsonism. J Neurol Neurosurg Psychiatry. 2010;81(10):1099-1104.
86. Shimamoto S.A., Larson P.S., Ostrem J.L., et al. Physiological identification of the human pedunculopontine nucleus. J Neurol Neurosurg Psychiatry. 2010;81(1):80-86.
87. Khan S., Javed S., Park N., et al. A magnetic resonance imaging–directed method for transventricular targeting of midline structures for deep brain stimulation using implantable guide tubes. Neurosurgery. 2010;66(6 suppl Operative):234-237. discussion 237
88. Pereira E.A., Muthusamy K.A., De Pennington N., et al. Deep brain stimulation of the pedunculopontine nucleus in Parkinson’s disease. Preliminary experience at Oxford. Br J Neurosurg. 2008;22(suppl 1):S41-S44.
89. Weinberger M., Hamani C., Hutchison W.D., et al. Pedunculopontine nucleus microelectrode recordings in movement disorder patients. Exp Brain Res. 2008;188(2):165-174.
90. Groenewegen H.J., Berendse H.W. The specificity of the “nonspecific” midline and intralaminar thalamic nuclei. Trends Neurosci. 1994;17(2):52-57.
91. Moruzzi G., Magoun H.W. Brain stem reticular formation and activation of the EEG. Electroencephalogr Clin Neurophysiol. 1949;1(4):455-473.
92. Sadikot A.F., Parent A., Smith Y., Bolam J.P. Efferent connections of the centromedian and parafascicular thalamic nuclei in the squirrel monkey: a light and electron microscopic study of the thalamostriatal projection in relation to striatal heterogeneity. J Comp Neurol. 1992;320(2):228-242.
93. Sadikot A.F., Rymar V.V. The primate centromedian–parafascicular complex: anatomical organization with a note on neuromodulation. Brain Res Bull. 2009;78(2-3):122-130.
94. Thomas T.M., Smith Y., Levey A.I., Hersch S.M. Cortical inputs to m2-immunoreactive striatal interneurons in rat and monkey. Synapse. 2000;37(4):252-261.
95. Sidibe M., Pare J.F., Smith Y. Nigral and pallidal inputs to functionally segregated thalamostriatal neurons in the centromedian/parafascicular intralaminar nuclear complex in monkey. J Comp Neurol. 2002;447(3):286-299.
96. Smith Y., Raju D., Nanda B., et al. The thalamostriatal systems: anatomical and functional organization in normal and parkinsonian states. Brain Res Bull. 2009;78(2-3):60-68.
97. Kerkerian-Le Goff L., Bacci J.J., Jouve L., et al. Impact of surgery targeting the caudal intralaminar thalamic nuclei on the pathophysiological functioning of basal ganglia in a rat model of Parkinson’s disease. Brain Res Bull. 2009;78(2-3):80-84.
98. Henderson J.M., Carpenter K., Cartwright H., Halliday G.M. Degeneration of the centre median–parafascicular complex in Parkinson’s disease. Ann Neurol. 2000;47(3):345-352.
99. Andy O.J. Parafascicular–center median nuclei stimulation for intractable pain and dyskinesia (painful dyskinesia). Appl Neurophysiol. 1980;43(3-5):133-144.
100. Krauss J.K., Simpson R.K.Jr., Ondo W.G., et al. Concepts and methods in chronic thalamic stimulation for treatment of tremor: technique and application. Neurosurgery. 2001;48(3):535-541. discussion 541-3
100a. Krauss J., Pohle T., Weigel R., Burgunder J.M. Deep brain stimulation of the centre median-parafascicular complex in patients with movement disorders. J Neurol Neurosurg Psychiatry. 2002;72(4):546-548.
101. Velasco F., Velasco M., Velasco A.L., et al. Electrical stimulation of the centromedian thalamic nucleus in control of seizures: long-term studies. Epilepsia. 1995;36(1):63-71.
102. Caparros-Lefebvre D., Pollak P., Feltin M.P., et al. Effect of thalamic stimulation of L-dopa–dyskinesias—evaluation of a new target: the centromedian and parafascicular complex. Revue Neurologique. 1999;155(8):543-550.
103. Mazzone P., Stocchi F., Galati S., et al. Bilateral implantation of centromedian–parafascicularis complex and GPi: A new combination of unconventional targets for deep brain stimulation in severe Parkinson disease. Neuromodulation. 2006;9(3):221-228.
104. Peppe A., Gasbarra A., Stefani A., et al. Deep brain stimulation of CM/PF of thalamus could be the new elective target for tremor in advanced Parkinson’s disease? Parkinsonism Relat Disord. 2008;14(6):501-504.
105. Silberstein P., Kühn A.A., Kupsch A., et al. Patterning of globus pallidus local field potentials differs between Parkinson’s disease and dystonia. Brain. 2003;126(Pt 12):2597-2608.
106. Starr P.A., Rau G.M., Davis V., et al. Spontaneous pallidal neuronal activity in human dystonia: comparison with Parkinson’s disease and normal macaque. J Neurophysiol. 2005;93(6):3165-3176.
107. Vitek J.L., Chockkan V., Zhang J.Y., et al. Neuronal activity in the basal ganglia in patients with generalized dystonia and hemiballismus. Ann Neurol. 1999;46(1):22-35.
108. Vidailhet M., Vercueil L., Houeto J.L., et al. Bilateral, pallidal, deep-brain stimulation in primary generalised dystonia: a prospective 3 year follow-up study. Lancet Neurol. 2007;6(3):223-229.
109. Berman B.D., Starr P.A., Marks W.J.Jr., Ostrem J.L. Induction of bradykinesia with pallidal deep brain stimulation in patients with cranial–cervical dystonia. Stereotact Funct Neurosurg. 2009;87(1):37-44.
110. Ostrem J.L., et al. Pallidal deep brain stimulation in patients with cranial–cervical dystonia (Meige syndrome). Mov Disord. 2007;22(13):1885-1891.
111. Black K.J., Gado M.H., Perlmutter J.S. Pet measurement of dopamine D2 receptor-mediated changes in striatopallidal function. J Neurosci. 1997;17(9):3168-3177.
112. Sani S., Ostrem J.L., Shimamoto S., et al. Single unit “pauser” characteristics of the globus pallidus pars externa distinguish primary dystonia from secondary dystonia and Parkinson’s disease. Exp Neurol. 2009;216(2):295-299.
113. Chou K.L., Hurtig H.I., Jaggi J.L., Baltuch G.H. Bilateral subthalamic nucleus deep brain stimulation in a patient with cervical dystonia and essential tremor. Mov Disord. 2005;20(3):377-380.
114. Ostrem J.L., Starr P.A. Treatment of dystonia with deep brain stimulation. Neurotherapeutics. 2008;5(2):320-330.
115. Kleiner-Fisman G., Liang G.S., Moberg P.J., et al. Subthalamic nucleus deep brain stimulation for severe idiopathic dystonia: impact on severity, neuropsychological status, and quality of life. J Neurosurg. 2007;107(1):29-36.
116. Detante O., Vercueil L., Krack P., et al. Off-period dystonia in Parkinson’s disease but not generalized dystonia is improved by high-frequency stimulation of the subthalamic nucleus. Adv Neurol. 2004;94:309-314.
117. Fuentes R., Petersson P., Siesser W.B., et al. Spinal cord stimulation restores locomotion in animal models of Parkinson’s disease. Science. 2009;323(5921):1578-1582.
118. Thoden U., Krainick J.U., Strassburg H.M., Zimmermann H. Influence of dorsal column stimulation (DCS) on spastic movement disorders. Acta Neurochir (Wien). 1977;39(3-4):233-240.
119. Machado A.G., Baker K.B., Schuster D., et al. Chronic electrical stimulation of the contralesional lateral cerebellar nucleus enhances recovery of motor function after cerebral ischemia in rats. Brain Res. 2009;1280:107-116.
120. Thevathasan W., Mazzone P., Jha A., et al. Spinal cord stimulation failed to relieve akinesia or restore locomotion in Parkinson disease. Neurology. 2010;74(16):1325-1327.
121. Galanda M., Horvath S. Effect of stereotactic high-frequency stimulation in the anterior lobe of the cerebellum in cerebral palsy: a new suboccipital approach. Stereotact Funct Neurosurg. 2003;80(1-4):102-107.
122. Galanda M., Horvath S. Stereotactic stimulation of the anterior lobe of the cerebellum in cerebral palsy from a suboccipital approach. Acta Neurochir Suppl. 2007;97(Pt 2):239-243.
123. Harat M., Radziszewski K., Rudaś M., et al. Clinical evaluation of deep cerebellar stimulation for spasticity in patients with cerebral palsy. Neurol Neurochir Pol. 2009;43(1):36-44.
124. Galanda M., Nadvornik P., Fodor S. Stereotactic approach to therapeutic stimulation of cerebellum for spasticity. Acta Neurochir Suppl (Wien). 1980;30:345-349.
125. Ratusnik D.L., Wolfe V.I., Penn R.D., Schewitz S. Effects on speech of chronic cerebellar stimulation in cerebral palsy. J Neurosurg. 1978;48(6):876-882.
126. Pascual-Leone A., Valls-Solé J., Brasil-Neto J.P., et al. Akinesia in Parkinson’s disease. II. Effects of subthreshold repetitive transcranial motor cortex stimulation. Neurology. 1994;44(5):892-898.
127. Drouot X., Oshino S., Jarraya B., et al. Functional recovery in a primate model of Parkinson’s disease following motor cortex stimulation. Neuron. 2004;44(5):769-778.
128. Canavero S., Paolotti R. Extradural motor cortex stimulation for advanced Parkinson’s disease: case report. Mov Disord. 2000;15(1):169-171.
129. Canavero S., Paolotti R., Bonicalzi V., et al. Extradural motor cortex stimulation for advanced Parkinson disease. Report of two cases. J Neurosurg. 2002;97(5):1208-1211.
130. Cioni B. Motor cortex stimulation for Parkinson’s disease. Acta Neurochir Suppl. 2007;97(Pt 2):233-238.
131. Pagni C.A., Albanese A., Bentivoglio A., et al. Results by motor cortex stimulation in treatment of focal dystonia, Parkinson’s disease and post-ictal spasticity. The experience of the Italian Study Group of the Italian Neurosurgical Society. Acta Neurochir Suppl. 2008;101:13-21.
132. Follett K.A., Weaver F.M., Stern M., et al. Pallidal versus subthalamic deep-brain stimulation for Parkinson’s disease. N Engl J Med. 2010;362(22):2077-2091.
133. Stefani A., Peppe A., Pierantozzi M., et al. Multi-target strategy for parkinsonian patients: the role of deep brain stimulation in the centromedian–parafascicularis complex. Brain Res Bull. 2009;78(2-3):113-118.
[/level-membership-for-neurosurgery-category][not-level-membership-for-neurosurgery-category]
Chapter 116 Novel Targets in Deep Brain Stimulation for Movement Disorders
The surgical treatment of movement disorders has evolved considerably over the last 15 to 20 years with the increasing application of deep brain stimulation (DBS), principally for the treatment of Parkinson’s disease (PD), essential tremor, and dystonia. The sites most commonly targeted for DBS in the treatment of movement disorders have been the subthalamic nucleus (STN), the internal segment of the globus pallidus (GPi), and the motor thalamus—mainly the ventral intermediate nucleus (Vim) in Hassler’s nomenclature.1a A growing body of evidence supports DBS targeting of the GPi or STN for treatment of advanced PD with disabling motor fluctuations or dyskinesias, despite optimal medical management1,2; the Vim for disabling, medication-refractory essential tremor3; and the GPi for generalized dystonia.4 The U.S. Food and Drug Administration approved DBS of the STN or GPi for the treatment of PD, unilateral thalamic stimulation for the treatment of essential tremor, and under a Humanitarian Device Exemption, unilateral or bilateral stimulation of the GPi or STN for the treatment of dystonia. The growing literature concerning these targets increasingly outlines the limitations of each for particular diseases and for particular symptoms of a given disease, prompting the “off label” application of DBS and other methods of neuromodulation to other targets. In several respects, available treatments for movement disorders, both pharmacologic and neurosurgical, are suboptimal, including the treatment of some axial symptoms of PD, tremor syndromes that respond poorly or variably to conventional surgeries, and some dystonias with variable or poor response.
Cooper’s inadvertent laceration and ligation of the anterior choroidal artery in 1952 during a mesencephalic pedunculotomy for the treatment of a postencephalitic tremor demonstrates how an initial, serendipitous clinical finding can lead to anatomic and physiologic insights that guide the process of target selection in the treatment of movement disorders. Cooper’s hypothesis that the therapeutic effect observed after the procedure was the result of medial globus pallidus infarction led him to pursue anterior choroidal artery ligation and later chemopallidectomy and chemothalamectomy, with absolute alcohol injections into these structures.5–7 The observations that pallidotomy and thalamotomy yielded improvement in parkinsonism without weakness and with few observable deficits in motor function helped guide subsequent modeling of corticobasal ganglia function. As the responses of different movement disorders to these types of lesions were catalogued, certain observations proved difficult to reconcile with the prevailing models. These inconsistencies have been called the “paradoxes of functional neurosurgery.”8,9 The observations were made that lesions of the same target may improve different phenotypes of movement disorder (dyskinesia, dystonia, and parkinsonism all have been reported to improve with pallidal and with subthalamic DBS10,11) and that high-frequency stimulation of these structures produces clinical effects that are strikingly similar to ablative procedures. These paradoxes have posed challenges to the models of the motor system that prevailed at the time and to the various hypotheses about the mechanism or mechanisms of action of DBS. In this way, refinements of models of the motor system proceed hand in hand with testable hypotheses that guide innovation in the clinical realm. Any discussion of novel targets for the treatment of movement disorders must therefore include novel ideas about the normal and pathologic physiology of the motor system, as well as novel ideas about the mechanisms of action of therapeutic stimulation.
Current Models of Cortical–Basal Ganglia Networks
Models of the normal and pathologic physiology of the motor system, especially of corticobasal ganglia–thalamic function, have evolved significantly over the last 20 to 30 years to better account for observations such as the similarity between lesions and high-frequency stimulation of certain targets and the abnormalities observed in the parkinsonian subject and in animal models of striatal dopamine depletion. Seminal works from the 1980s and 1990s include the characterization of Albin, DeLong, and colleagues of multiple, parallel, segregated loops transmitted serially from the cortex; through the striatum, pallidum, and thalamus; and back to the cortex.12–14 Gerfen et al.15 demonstrated distinct populations of striatal medium spiny neurons that possess mainly subtype-1 dopamine receptors and project directly to the GPi or subtype-2 dopamine receptors and project indirectly to the medial pallidum via the external segment of the globus pallidus (GPe) and STN. These models conceived of pallidal outflow as a tonic, inhibitory brake on the thalamus and helped explain the observations of aberrantly high neuronal discharge frequencies in the STN and GPi in animal models of hypokinetic states such as dopamine depletion16 and in human parkinsonism. This model of hypokinesia as a result of excessive inhibition of the thalamus by the pallidum was incongruous with some observations, such as the lack of hypokinesia observed with thalamic lesions. Similarly, the therapeutic effects of pallidal, thalamic, or subthalamic lesions on hyperkinesias challenge this classical model, which, in their simplest form, posit that hyperkinetic disorders result from reduced pallidothalamic outflow. Modifications of these models have incorporated other connections in the cortical–basal ganglia network, such as extrathalamic projections of pallidal output, reciprocal GPe-STN interconnections, and direct cortical-STN projections (“hyperdirect” pathway).17 However, newer models have also significantly shifted the emphasis from abnormal frequency and patterns of neuronal discharge in basal ganglia nuclei during loss of dopaminergic innervation to an emphasis on abnormal network activity, particularly in the form of abnormal neuronal synchrony and network oscillations measured by field potentials. The crux of these newer models, sometimes referred to as “noisy signal” hypotheses,8 is that different pathologic conditions may be characterized by abnormal neuronal synchrony and dominance of particular field potential oscillations in particular networks, potentially producing tremor (5 Hz), dystonia (3-10 Hz), or akinesia (20 Hz).18,19 This shift in models owes much to investigations into the cellular and network substrates of thalamocortical oscillations performed in the 1980s, when Jahnsen and Llinas demonstrated dual behavior in thalamocortical neurons20: When simply depolarized, these cells exhibited a “classical” behavior, emitting a series of spikes and relaying their inputs on to the cortex with high fidelity. When released from hyperpolarization, these cells exhibited a “burst” mode, where afferent information was largely prevented from being relayed to the cortex. The biophysical mechanisms of the bursting were found to result from the interplay between a hyperpolarization-activated cation current (known as Ih) and a transient low-threshold calcium current (known as IT), which produces a “slow spike” lasting tens of milliseconds when released from hyperpolarization. These produce “pacemaker potentials,” which are rhythmic oscillations of thalamocortical cells in the delta frequency range (3-6 Hz).20 These cellular oscillations are important for understanding oscillating field potentials such as those measured by electroencephalogram, but the cellular oscillations alone are insufficient to generate the delta frequency oscillations measured in field potentials. The relationship between rhythmic bursting at the cellular level and oscillations measured in field potentials is complex, and a number of mechanisms may contribute to synchronization of oscillating and nonoscillating cells. Networked models of separate thalamocortical oscillators that incorporate thalamocortical connectivity and feedback pathways can account for synchronized thalamic burst firing at delta frequency and generate field potentials. Models of these pacemaker potentials interacting in a simple network among the thalamocortical cells, the corticothalamic cells, and the reticular nucleus of the thalamus have been developed to explain how these phenomena generate local field potential (LFP) oscillations in the form of delta waves and spindle waves observed during sleep.21
Similar mechanisms for cellular oscillations, rhythmic activity, and synchronization across networks of cells have been proposed for the STN and its interactions with the pallidum. STN neurons have been identified that generate rebound burst activity mediated by hyperpolarization-deinactivated calcium channels,22 and roughly half of STN neurons in rat brain slices switch from a single-spike mode to burst-firing mode when continuously hyperpolarized.23 Studies of cocultures of STN and GPe neurons demonstrate that this reciprocally interconnected network can generate correlated, rhythmic firing when isolated from the cortex and striatum.24 Models of STN-GPe reciprocal bursting, similar to those described for spindle oscillations in the thalamus, involve synchronous bursting activity in GPe neurons that hyperpolarizes STN neurons sufficiently to generate rebound burst activity, which then drives bursting activity in GPe and perpetuates the rhythm.25 The loss of dopamine, leading to overactivity in the striatal neurons of the indirect pathway would disinhibit GPe neurons, hyperpolarize STN neurons, accentuate burst firing in the STN, and generate pathologic synchrony between groups of neurons. This could result in the generation of oscillations that disturb cortical processing, or it could restrict communication among the cortex, STN, and GPe to certain bandwidths.
1. Biophysical properties of neurons can produce transitions from high-fidelity tonic states of information transfer to low-fidelity bursting and oscillating states. Such neurons can be found in the thalamus, STN, GPi, GPe, and pedunculopontine nucleus (PPN).
2. Neuromodulators (i.e., dopamine), could have direct, as well as indirect, effects on the transitions of individual neurons between these states. Thus, loss of striatal and extrastriatal dopamine could influence subthalamic neuronal-firing properties directly or indirectly (i.e., through hyperpolarization of STN neurons via increased GPe activity).
3. Populations of oscillating neurons can synchronize with one another through direct or indirect connections, and degree of synchronization could be affected by the presence or absence of neuromodulators.
4. Synchronized oscillations can contribute to the generation of specific field potentials and restrict communication among groups of cells to certain bandwidths.
5. Different pathologic conditions may be characterized by dominance of particular field potential oscillations in particular networks, potentially producing tremor (5 Hz), dystonia (3-10 Hz), or akinesia (20 Hz).19
In this type of model of subcortical pathophysiology, an intriguing hypothesis regarding the mechanism of high-frequency stimulation is to trigger tonic firing, thus preventing neurons from returning to their hypersynchronized, bursting, oscillatory firing patterns. These models can also offer possible explanations of the diverse etiologies of movement disorders: single-ion channel dysfunction that changes the biophysics of single cells; loss of neuromodulatory transmitters such as dopamine, as seen in idiopathic PD; or altered network connectivity from plastic changes at cortical or subcortical levels, as seen in post-traumatic movement disorders or models of certain dystonias.26,27 While many details of the cellular and network substrates of normal and pathologic field potential oscillations remain uncertain, some specific rhythms associated with specific symptoms are reasonably well characterized and deserve review.
Beta Oscillations and Parkinsonian Akinesia
Pathologic oscillations in movement disorders have been characterized most extensively in PD. Electrocorticographic and electroencephalographic investigations of motor tasks have long shown a decrease in signal power over motor cortices in the beta frequencies (about 20 Hz) during movement, sometimes referred to as event-related desynchronization.28 Voluntary movement is accompanied by increased power over more focal areas of the motor cortex during movement in higher-frequency bandwidths.29,30 The inability of the cortex to transition between beta oscillations and these focal higher-frequency oscillations appears to be a feature of parkinsonian akinesia, and the basal ganglia dysfunction in PD appears to be a critical part of this impediment. LFPs recorded from the STN, GPi, and motor cortex in parkinsonian patients demonstrate the presence of increased power in the beta band associated with akinesia.31 Not only is this increased power abolished with the administration of dopaminergic agonists or exogenous levodopa (l-dopa),32 but it remains suppressed in the STN immediately following high-frequency stimulation of that structure.33 Accumulating evidence of this type suggests that suppression of the “beta straightjacket” may be a key to therapeutic efficacy of medications and DBS for the treatment of Parkinsonian akinesia.
Oscillations Associated with Tremor
Tremor is one of the cardinal symptoms of PD, consists of a rhythmic (4-8 Hz) activation of mainly distal musculature, and manifests at rest and in some cases while maintaining posture. Identification of pathologic oscillations in the nervous system that correlate with the oscillatory movements of parkinsonian tremor has proved challenging. The question is complicated by the problem of defining a physiologic oscillation that is efferent in character (i.e., drives the tremor) versus one that is afferent (i.e., results from proprioceptive feedback). Physiologic models of tremor have evolved to account for the observations that dorsal rhizotomies had minimal effects on tremor (suggesting central oscillators as drivers) and that Vim DBS is often able to treat parkinsonian tremor but does not affect the other cardinal symptoms of the disease to the same degree. Tremor-frequency single-unit activity (“tremor cells”) in the ventral oral posterior nucleus, Vim, and ventral caudal nucleus thalamus has been described,34,35 as have cells with tremor-frequency bursting oscillations in the STN36 and globus pallidus.37 Pare et al. hypothesized that rhythms transmitted through inhibitory pallidal outflow to the motor thalamus were transformed into the 3- to 6-Hz tremor frequency in the thalamocortical circuit through the low-threshold calcium spikes induced by transient (T-type) calcium channels.38 Hassler et al. were able to produce contralateral resting tremor with stimulation of the internal segment of the globus pallidus at frequencies of less than 25 Hz,39 and Plaha et al. were able to elicit a 3- to 6-Hz tremor in a previously tremor-free parkinsonian patient by stimulating the caudal ZI at the beta frequency. In the latter study, low-frequency stimulation of the GPi or PPN did not produce tremor, and STN stimulation at low frequencies only produced tremor at high amplitudes.40 Because tremor in muscles of different extremities is not usually coherent, it has been concluded that independent central oscillators generate tremor separately in separate extremities.41 Simultaneous recordings of spatially separated tremor-related cells in the GPi showed that some pairs of cells oscillated in phase and some oscillated out of phase with one another,37 leading the authors to hypothesize that tremor-related activity may be propagated independently by several parallel pathways.
Surgical Targets for Tremor
Since Benabid’s demonstration of efficacy with high-frequency stimulation (more than 100 Hz) of the Vim thalamus for the treatment of parkinsonian tremor, this site has emerged as the preferred target.42 While this therapy seems to produce effects that are indistinguishable from a lesion of this target, it remains uncertain what ideal subregion of the thalamus (or possibly extrathalamic structure) provides the best tremor control, whether through stimulation or ablation. This question was posed during the ablative era of functional neurosurgery, highlighted by Laitinen’s survey of 16 neurosurgeons, who were asked to define their choice of target for treatment of a patient with PD who exhibited tremor and rigidity in equal measure. Targets differed by as much as 7 mm and included a wide span of the ventrolateral thalamus; the PSA, including the ZI; and the pallidothalamic fibers of the Forel fields.43 Some of the difficulty in defining the ideal target stems from the multiple parcellation schemes for the motor thalamus,44 though most stereotactic neurosurgeons employ Hassler’s terminology, which forms the basis for thalamic parcellation in Schaltenbrand and Wahren’s widely used atlas.45 The question of the ideal target is also complicated by questions as to how stimulation differs from ablative surgery. The ideal target for stimulation may be different from the ideal target for ablation. Among the multiple hypotheses that have been put forth regarding the mechanism or mechanisms of DBS, two intriguing ones related to tremor are the observation that adenosine release may be critical to the effect of Vim stimulation46 and data that suggest stimulation in the dorsal STN may act by directly modulating pallidothalamic or cerebellothalamic fibers in the immediate vicinity.47 Finally, while Vim DBS has been shown to be effective for parkinsonian and essential tremor,48,49 other tremor syndromes respond only inconsistently or less dramatically to Vim DBS. In addition to controversy surrounding the ideal target for a particular tremor syndrome, there is the question of the ideal target for those patients exhibiting combinations of tremor syndromes. Patients exhibiting symptoms of both PD and essential tremor may be treated with Vim DBS, but other parkinsonian symptoms would not be expected to respond. For these patients, and for patients with tremors that respond inconsistently or less dramatically to Vim DBS, it is important to describe the role of sites such as the STN, which some data suggest it can effectively treat symptoms of essential tremor,50 and the PSA.
Posterior Subthalamic Area
The PSA encompasses the ZI and the prelemniscal radiation (RaPRL). This was the target area of subthalamotomies performed frequently from the 1960s to the 1980s. Various terminologies have been used to define approximately the same target area, such as the PSA, ZI, RaPRL, subthalamic area, and posterior subthalamic white matter. In this chapter, we refer to this area as the PSA and discuss in further detail two of its most important components, the ZI and the RaPRL. The PSA is bordered anteriorly by the posterior STN, inferiorly by the dorsal substantia nigra, superiorly by the ventral thalamic nuclei, posteriomedially by the red nucleus, posteriolaterally by the ventrocaudal nucleus, posteriorly by the lateral lemniscus, and laterally by the posterior limb of the internal capsule (Fig. 116-1). As a surgical target, the PSA has been extensively reviewed by Blomstedt et al., who accounted for a total of 95 patients implanted with PSA DBS: 42 of these patients for the diagnosis of PD, 21 for multiple sclerosis, 18 for essential tremor, 7 for torticollis, 2 for dystonic tremor, 2 for post-traumatic tremor, 1 for cerebellar tremor, 1 for Holmes tremor, and 1 for spinocerebellar ataxia.51 Several reports have suggested that high-frequency stimulation of the PSA offers superior tremor control to stimulation of the Vim. Hamel et al. systematically analyzed the location of active contacts effective in suppressing intention tremor and found that those below the intercommissural plane in the subthalamic region were most effective.52
Complications of PSA DBS can include dysarthria (potentially from overly lateral placement of the electrode stimulating the corticobulbar fibers in the internal capsule or overly medial placement of the electrode stimulating the cerebellar fibers of the RaPRL); disequilibrium; lethargy; or worsening symptoms of preexistent depression.53
Analysis of lead locations following intended STN DBS electrode placement demonstrated a correlation between best clinical effect and placement of the electrode dorsal to the STN in the white matter bundles of the ZI and H1 and H2 fields of Forel, outside of the PSA.54,55
Zona Incerta
The ZI is a collection of gray matter that is continuous ventrally with the reticular nucleus of the thalamus. Functionally, it is divided into four parts: rostral, ventral, dorsal, and caudal. The rostral ZI is separated from the STN by the pallidofugal fibers crossing the internal capsule on the dorsomedial side of the STN. The ZI receives afferent input from the GPI or substantia nigra pars reticulata (SNr), the PPN, and cortical projections. Its gamma-aminobutyric acid–ergic (GABAergic) efferents target the CM/PF, the ventral anterior–ventral lateral nuclei of the thalamus, and the cortex, among others. The caudal or motor component of the ZI extends posterior to the STN and the RaPRL. Forel described it in 1877 as a “region of which nothing certain can be said,” and it remains an understudied region. Although the ZI is not considered part of the basal ganglia, the anatomy and physiology of the two regions overlap considerably. Both receive excitatory cortical input and send inhibitory projections to the thalamus, and both are involved in motor control and attention orientation, among other shared functions.56
Recent studies in rodents have demonstrated that this region plays a critical role in the gating of somatosensory input by the brain stem57 and the motor cortex.58 Like the neighboring reticular nucleus of the thalamus, cholinergic input from the brain stem results in state-dependent input from the ZI to the thalamus, which may contribute to elevated sensory thresholds during sleep. The ZI serves as an important locus for modulating sensory information from diverse sources by gating unwanted sensory input via inhibition of thalamic neurons.58 It has been proposed that the efferent GABAergic signal of ZI neurons patterned by cortical activity can play a critical role in synchronizing thalamocortical and brain stem rhythms.59 Subsequently, it has been implicated as a generator of tremor in PD and essential tremor.40
Given the similar connectivity between the ZI and the basal ganglia, it is perhaps not surprising that DBS achieves many of the same results in the ZI as it does in the basal ganglia. Indeed, high-frequency DBS may have a greater effect in the dorsal ZI than in the STN. In the largest case series published of 35 patients with refractory idiopathic PD, patients were implanted with a total of 64 high-frequency DBS electrodes. Of these, 27 DBS electrodes were implanted into the dorsal ZI, 20 into the dorsomedial STN, and 17 into the STN. This observational study suggested that high-frequency DBS of the ZI is superior to that of the STN in improving contralateral Unified Parkinson’s Disease Rating Scale motor scores. Some patients developed speech and balance disturbances following high-frequency DBS, thought to be due to the proximity of the lead to the RaPRL.60 A report by Kitagawa et al. from 2005 targeting the caudal ZI in patients with tremor-dominant PD noted 78.3% improvement in contralateral tremor and even superior improvement in rigidity.61
Prelemniscal Radiation
The ZI separates posteriorly the dorsal STN from the RaPRL located posterior to the ZI/STN. Its ascending fibers originate from the mesencephalic reticular formation and the cerebellum projecting to the thalamus. Thus, it is thought to play a role in tremor formation and muscle tone in the context of attention. Continuous stimulation of the RaPRL abolishes contralateral tremor with reduction in rigidity by inhibition of local circuits.62 With RaPRL DBS, dysarthria can be a complication by cerebellothalamic fiber stimulation and disequilibrium from the stimulation of the ascending mesencephalic reticular formation. Murata et al. targeted the RaPRL in eight patients with essential tremor of the proximal muscles using these coordinates: lateral to the red nucleus (about 10 mm from midline) and 3 to 4 mm posterior to the posterior border of the STN in the axial plane through the largest cut over the STN. Postoperative localization analysis resulted in mean stereotactic coordinates in patients with more than 71% improvement at 7.6 ± 1.2 mm posterior to the midcommissural point, 10.9 ± 0.8 mm lateral from midline, and 3.9 ± 1.7 mm inferior from the AC-PC line.63 This targeting yielded marked improvement in both proximal and distal tremor control.
[/not-level-membership-for-neurosurgery-category]
