Chapter 5 Normal Mechanisms of Vascular Hemostasis
Endothelial Function and Platelet Activation
Platelets are anucleate cells produced by megakaryocytes in the bone marrow. Once they have traversed from the bone marrow to the general circulation, their lifespan is approximately 10 days. They function mainly to limit hemorrhage after trauma resulting in vascular injury. Normally in the vasculature, platelets are in a resting state and only become activated after exposure to a stimulus leads to a shape change and release reaction that causes the platelet to export many of its biologically important proteins. Some of the agonists that can initiate this response include thromboxane A2, adenosine diphosphate (ADP), thrombin, and serotonin. In areas of vascular injury, platelets are attracted to the impaired site by collagen through binding with von Willebrand factor (vWF) via the glycoprotein (GP) Ib/V/IX complex. This initial binding results in platelet activation, with a subsequent feedback mechanism in which ADP, thrombin, and thromboxane A2 further activate the platelets and recruit additional platelets to the area. The complex firmly binds the platelet to the area of injury so there is no disruption by the high shear forces of turbulent blood flow that occur with vessel disruption. This amplification of the response is essential to form a hemostatic plug and represents the first stage in the hemostatic process. When vWF is not present, hemostatic abnormalities result, with deficiencies leading to von Willebrand’s disease, which can be associated with severe bleeding. Hemostasis issues also arise when the platelet receptor complex GPIb/V/IX is mutated, resulting in inability of vWF to bind, a disorder termed Bernard-Soulier’s syndrome.1,2
Additional platelet aggregation occurs through activation of G protein–coupled receptors (GPCRs), with the final pathway relying on the GP IIb/IIIa complex, the main receptor for platelet aggregation and adhesion.3,4 Fibrinogen tethers GP IIb/IIIa complexes on different platelets, stabilizing the clot. The integral role of this receptor is manifest in Glanzmann thrombasthenia, a disorder in which fibrinogen binding is impaired, leading to spontaneously occurring mucocutaneous bleeding episodes.5
Vascular endothelium is essential to this hemostatic process; this is the cellular site where regulation and initiation of coagulation begins. Endothelial cells (ECs) modulate vascular tone, generate mediators of inflammation, and provide a resistant surface that allows for platelets to experience laminar flow with minimal shear. Endothelial cells regulate hemostasis by releasing a number of inhibitors of platelets and inflammation. Vascular endothelium is essential for regulating uncontrolled platelet activity through mechanisms of inhibition including the arachidonic acid–prostacyclin pathway, L-arginine–nitric oxide pathway, and endothelial ectoadenosine diphosphatase (ecto-ADPase) pathway6 (Table 5-1).
| Prohemostatic | Antihemostatic |
|---|---|
| Circulating | |
| α2-Antiplasmin | Antithrombin III |
| Thrombin | Protein C |
| Thrombin-activatable fibrinolysis inhibitor (TAFI) | Protein S |
| Tissue factor pathway inhibitor (TFPI) | |
| Endothelium-Derived | |
| Plasminogen activator inhibitor-1 (PAI-1) | Ectoadenosine diphosphatase (Ecto-ADPase)/CD39 |
| Tissue factor (TF) | Heparan sulfate (HS) |
| von Willebrand factor (vWF) | Nitric oxide (NO) |
| Thrombomodulin | |
| Tissue plasminogen activator (tPA) | |
| Urokinase plasminogen activator (uPA) | |
Nitric oxide (NO) is produced constitutively by (ECs) via an endothelial isoform of nitric oxide synthase (eNOS) in a process dependent on conversion of L-arginine to L-citrulline. Vascular tone is regulated by NO as it controls smooth muscle cell (SMC) contraction. It also inhibits platelets directly, blocking platelet aggregation through stimulation of guanylyl cyclase and cyclic guanosine monophosphate (cGMP) and inhibition of platelet phosphoinositol3-kinase (PI-3 kinase). Nitric oxide functions by decreasing the intracellular Ca2 + level through cGMP, which inhibits the conformational change in GP IIb/IIIa suppressing fibrinogen’s ability to bind to the receptor, thereby attenuating platelet aggregation.7
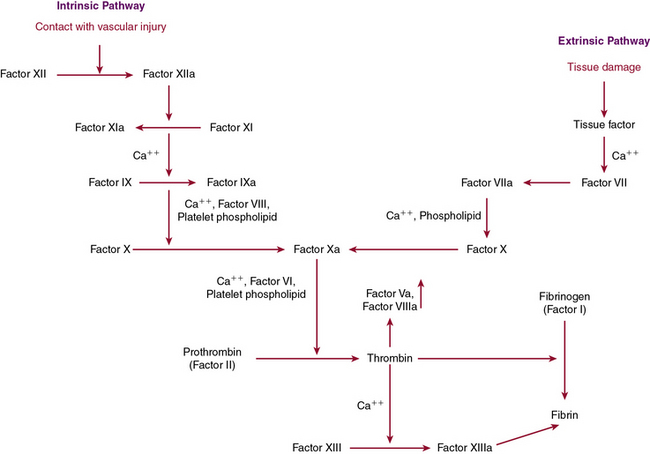
The last pathway important in modulating vascular endothelium’s interaction with platelets is the endothelial ecto-ADPase pathway, which impairs ADP-mediated platelet activation. By hydrolyzing ADP, this enzyme inhibits the critical state of platelet recruitment to a growing aggregate, thereby limiting thrombus formation. Once the platelet aggregate has been stabilized by fibrin with red cells to the vessel wall, the next stage of hemostasis involves activation of the highly regulated coagulation cascade (Fig. 5-1).
Coagulation Cascade Leading to Fibrin Formation
Disruption in the endothelium not only recruits platelets for plug formation, it also stimulates activation of the coagulation cascade, which is essential for secondary clot formation through fibrin generation. The coagulation cascade is a dynamic integrated process in which each step is dependent on another step for activation of proenzymes or zymogens to their active forms through proteolytic cleavage. This process is dependent upon calcium and the phospholipid bilayer allowing inactive clotting factors to be converted to active enzymes through serine protease activity. These coagulation proteins function in a step-by-step fashion to activate downstream members of the cascade, leading to production of the penultimate clotting factor, thrombin. Thrombin is versatile, playing a role in many of the essential stages of hemostasis. Not only is it important for platelet activation, it is also necessary for the cross-linking of fibrin. Recently there have been attempts to limit thrombus formation by directly inhibiting thrombin activity through anticoagulants such as ximelagatran and the oral medication, dabigatran, which is now available for clinical use.8
The clotting cascade is divided into two main pathways, the intrinsic and extrinsic pathways. The extrinsic pathway begins with establishment of a complex between tissue factor, found on the cell surface or on microparticles, and factor VIIa. This complex leads to activation of factor X to Xa, which can then further the response by looping back and converting factor VII to VIIa in a feedback mechanism. When factor Xa is present, it binds to factor Va on the membrane surface and again generates prothrombinase, which converts prothrombin to thrombin and then generates fibrin as detailed earlier. The activity of factor Xa is accelerated by the presence of factor Va through calcium and formation of a noncovalent association γ-carboxyglutamate residues of factor Xa and the phospholipid surface of activated platelets.9 The extrinsic pathway is measured by prothrombin time (PT), which is determined by adding an extrinsic substance such as tissue factor or thromboplastin.10
The extrinsic pathway, which is dependent on tissue factor, appears to be the main pathway responsible for hemostasis, with the intrinsic pathway playing a supporting role. Tissue factor is a membrane-bound GP that is constitutively expressed by SMCs and fibroblasts but selectively expressed by ECs when there is vessel wall injury. The “encrypted” activated form of factor VIIa is made functional through a conformational change that occurs at cysteines 186 and 209, leading to disulfide bond formation upon vessel wall injury. Protein disulfide isomerase, glutathione, and NO all may have a role in these allosteric changes; however, recent studies have questioned the importance of “de-encryption” in this process.11–14 Tissue factor functions through activation of factors X and IX after interactions with factor VII as a complex. Factor VII, although at low levels in an active state (factor VIIa) in the circulation, only becomes biologically important after it is bound to tissue factor in complex with factors X and IX. This complex formation is essential for activation of thrombin.9
The role of tissue factor has recently been expanded. It circulates in the blood in association with microvesicles that are derived from cellular membranes produced from lipid rafts on monocytes and macrophages.15 These tissue factor–bearing microvesicles can directly initiate the coagulation cascade on activated platelets in a process that may be important for understanding the hypercoagulable state.16,17
The extrinsic pathway, described earlier, joins up with the intrinsic pathway through factor X to form the common pathway. The intrinsic pathway is initiated by contact and results in activation of factor IXa, which then goes on to activate factor X as described. It is generally accepted that the intrinsic pathway is of less importance in coagulation than the tissue factor–mediated extrinsic pathway, although it plays an essential role in inflammation and fibrinolysis. The intrinsic pathway is based on exposure of blood to a negatively charged surface, and is classically initiated by activation of factor XIIa by kallikrein, which is facilitated by kininogen. Kallikrein is generated from prekallikrein through proteolytic cleavage by activated factor XII in a reaction dependent on the presence of high-molecular-weight kininogen (HMWK). When kallikrein has been generated, it also functions to cleave HMWK to bradykinin, which functions as an inflammatory mediator to potentiate vasodilation and vascular permeability, thereby expanding the role of factor XIIa to inflammation, regulation of vascular tone, and fibrinolysis.18 Activated factor XII catalyzes conversion of factor XI to the active enzyme form, factor XIa. When calcium is present, factor XIa next functions to convert IX to IXa, which then binds to VIIIa on membrane surfaces, converting X to its active form, factor Xa. Factor Xa then binds to Va on the membrane surface to generate prothrombinase, which converts prothrombin to thrombin. As thrombin is formed, two small prothrombin fragments, termed molecules F1 and F2, are released and can be used as markers of serum thrombin formation.19 The intrinsic pathway is monitored through the activated partial thromboplastin time (APTT), which relies on foreign substances such as glass or silicates to activate factor XII to initiate the pathway. Deficiencies in the earliest states of the intrinsic pathway, when prekallikrein, HMWK, and factor XII are involved, are not associated with bleeding tendencies and therefore do not lead to a bleeding diathesis, even though there is an elevation in partial thromboplastin time. Mutations in factor XII have been reported in a group of patients with hereditary angioedema, although there does not appear to be a bleeding diathesis with this disorder. Some initial studies have suggested that factor XII polymorphisms may be associated with an increased propensity for thrombosis, but this has not been validated.20,21
When factor Xa generates thrombin, the intrinsic and extrinsic pathways have merged into the common pathway. Thrombin is essential for fibrinogen to generate fibrin, which is released through proteolytic cleavage.22 The fibrin molecules that are generated have polymerization sites exposed, making it easier for fibrin to cross-link noncovalently. This cross-linking enables platelets to be entrapped in a meshwork of fibrin strands to form the secondary clot through the action of factor XIII, activated by thrombin.23 In the process of cross-linking, there is also an inherent mechanism of autoregulation, with the binding sites necessary to initiate fibrinolysis being blocked so the clot does not self-destruct.
Antithrombin is a serine protease inhibitor that binds specifically to factors IXa, Xa, and thrombin, thereby inactivating them. Antithrombin has two main binding sites that maintain its functionality: the reactive center at Arg 393/Ser 394 and the heparin binding site at the amino-terminal end of the molecule. Binding of both endogenous and exogenous heparins at this site causes a conformational change in antithrombin that enables it to inactivate its targets at an accelerated rate. The glycosaminoglycan heparan sulfate (HS), present on the surface of ECs, mediates antithrombin’s ability to increase its activity and functions as the physiological equivalent to heparin.24 Deficiency of antithrombin is associated with a genetic propensity to form venous thrombosis, discussed in Chapter 10.25
Activated protein C (APC) and protein S are also important mechanisms for preventing excessive clotting. During the clotting process, thrombin binds to thrombomodulin, which is also present on the EC surface. It then undergoes a conformational change leading to activation of protein C.26 Activated protein C complexes with protein S and proteolytically cleaves factors Va and VIIIa, resulting in their inactivation and a decrease in generation of factors Xa and thrombin. Cleavage of factor Va occurs at Arg 506, Arg 306, and Arg 679 by APC in a sequential manner such that the cleavage at Arg 506 exposes cleavage sites at the other sites through a conformational change. Mutation of the arginine located at position 506 to glutamine leads to factor V Leiden, which is associated with a hypercoagulable state.
Fibrinolysis
The importance of fibrinolysis lies in its removing blood clots and maintaining hemostasis without excessive clotting. The mechanism of serine protease activity is preserved in the fibrinolytic system and accounts for the mechanism of action of many of its components (Fig. 5-2). The main factor responsible for fibrinolysis is plasmin. The process begins when plasminogen in its inactive form is converted to the active enzyme, plasmin, which functions to covert fibrin to soluble fibrin degradation products. Two molecules that mimic this function include tissue-type plasminogen activator (tPA) and urokinase-type plasminogen activator (uPA). The motif responsible for its action is the kringle domain, which resides in the amino-terminal end. Kringles are 80 amino acids in length and have a unique folded sheet structure that results from disulfide linkages, which yields a homotypic binding site specific for plasminogen, fibrinogen, and fibrin. There is homology between the kringles contained in all three of these molecules.
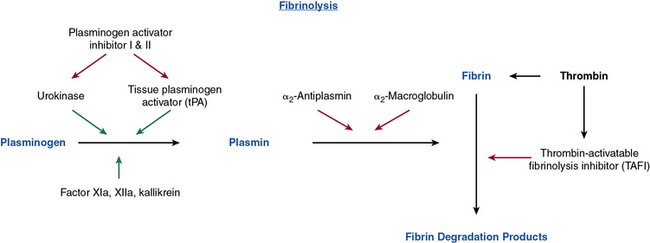
Figure 5-2 Pathway of fibrinolysis.
Inhibition is signified by red arrows and stimulation is signified by green arrows.
These kringle domains are essential for providing a mechanism for binding many components of the developing thrombus, including fibrinogen and fibrin. The kringle domains shared by tPA and plasminogen allow fibrinogen and fibrin to bind and therefore be incorporated into the developing clot. Plasminogen is converted to plasmin through the proteolytic cleavage achieved by tPA and uPA at the Arg 560 and Val 561 sites.27 The plasmin generated can then bind to a number of proteins involved in the process of fibrinolysis. Relevant properties include its high affinity for fibrin, ability to cleave Glu-plasminogen to Lys-plasminogen, ability to activate factor XII, and ability to inactivate factors V and VIII in the coagulation cascade. Plasmin cleaves the fibrin molecule into differentially sized degradation or split products (FDP), the smallest of which is D-dimer, which is used as a marker of venous thromboembolism and disseminated intravascular coagulopathy (DIC).
The plasminogen pathway is complex and tightly regulated.28 The main proteins involved in its modulation are plasminogen activator inhibitors (PAI)-1 and PAI-2. The activators and inhibitors of plasminogen regulate fibrinolysis upon release from ECs. These activators of the fibrinolytic process are under the control of PAIs, which complex with tPA and uPA to inactivate them and therefore block plasmin generation.
Evidence for why tPA is more important than uPA for normal hemostasis is how ECs up-regulate production of this protein when injured. It is stimulated by a variety of substances, including thrombin, serotonin, bradykinin, cytokines, and epinephrine. This binding affords tPA some protection from degradation and enables it to survive for longer than its expected half-life of only 4 minutes. Its role in hemostasis is of such significance that recombinant tPA (alteplase) and its derivatives that incorporate the kringle domains (e.g., reteplase, tenecteplase) are used as thrombolytic agents in patients with acute thrombotic events, including myocardial infarction.29
The other essential plasminogen activator in this process is uPA, which exists in a high-molecular-weight and a low-molecular-weight form, both of which have the ability to activate plasminogen through cleavage at Arg 560/Val 561. Urokinase is present in high concentration in urine. Whereas tPA is mainly important for intravascular fibrinolysis, urokinase has more of a role in the extravascular compartment. Unlike tPA, however, uPA does not bind to fibrin and therefore is not involved in activation of plasminogen incorporated into clots through fibrin binding.30 As its name implies, uPA is derived from urokinase, which consists of a single-chain precursor molecule termed scuPA that is hydrolyzed by plasmin or kallikrein to the two-chain active uPA, which is biologically active.31 In plasma, scuPA does not activate plasminogen, but in the presence of fibrin, it is actually scuPA that induces clot lysis. Interestingly, the role of urokinase has been expanded to include support of invasion and metastasis in malignancy32,33; uPA has been shown to play a role in extracellular matrix degradation, allowing for migration and invasion of metastatic cells. There is now a growing interest in developing targeted therapy that blocks this pathway as a means of controlling metastasis.
Streptokinase does not participate in normal hemostasis but is used as a therapeutic agent for acute thrombosis. It is isolated from β-hemolytic streptococci, and since it is not an enzyme, must complex with plasminogen to form an active molecule which then has the ability to cleave plasminogen to plasmin.34 Its use as a therapeutic agent, however, is limited; as a foreign substance, it is often recognized by the immune system, and antistreptokinase antibodies are generated.
There are multiple endogenous proteins that can rapidly inhibit the fibrinolytic response. These include PAI-1, α2-antiplasmin, α2-antitrypsin, and C1 inhibitor. Most of these inhibitors act through serine protease inhibition (serpin) and therefore affect many aspects of coagulation. The most important of these inhibitors is PAI-1, which is expressed by ECs or platelets after exposure to thrombin; inflammatory mediators such as tumor necrosis factor alpha (TNF-α); and growth factors, lipids, insulin, angiotensin II (ANGII), and endotoxin.35 Recently the role of PAI-1 as an inhibitor of tissue factor has been postulated to regulate hemostasis in inflammatory conditions such as sepsis or acute lung injury.36 It has been shown that platelets release PAI-1 as a mechanism of preventing premature clot dissolution. Patients who are deficient in PAI-1 have a bleeding diathesis when confronted with trauma or surgery.
Another important mechanism for regulation of fibrinolysis is thrombin-activatable fibrinolysis inhibitor (TAFI), which is not a member of the serpin family. It is known for its ability to cleave the carboxy-terminal lysine in fibrin, impairing plasminogen binding.37 Activation of TAFI is dependent upon the thrombin-thrombomodulin complex, which can expedite the inhibitory process in a similar manner to thrombin.38 This process has recently been shown to be inhibited by platelet factor 4, which is secreted by activated platelets.39 If the feedback mechanisms of thrombin generation through factors V, VIII, and XI is impaired—leading to diminution of the thrombin-thrombomodulin complex and therefore decreased activation of TAFI—clinical consequences can occur. It has been suggested that in chronic liver disease where coagulation factors are decreased, low amounts of TAFI may account for the low-grade fibrinolysis typically observed.40 The opposite can also occur, as is seen in patients with the G20210A prothrombin gene mutation in which thrombin generation is increased leading to increased activation of TAFI and an increased thrombotic propensity through a inhibition of fibrinolysis.41
Recently it has been shown that there is yet another important mechanism by which to regulate the fibrinolytic process via matrix metalloproteinases (MMPs). Matrix metalloproteinases (including MMP-3, -7, -9, and -12) are found in ECs and have the ability to cleave uPA and plasminogen. The importance of MMPs in down-regulating cellular fibrinolysis remains to be elucidated, but it is clear they function by reducing availability of plasminogen. MMP-3 and -7 also have the ability to degrade fibrinogen and cross-linked fibrin; MMP-11 can degrade fibrinogen but not fibrin. Matrix metalloproteinases also can modulate the activity of many inhibitors of fibrinolysis, including α2-antiplasmin and PAI-1.42,43
1 Kunishima S., Kamiya T., Saito H. Genetic abnormalities of Bernard-Soulier syndrome. Int J Hematol. 2002;76(4):319–327.
2 Sadler J.E. Von Willebrand disease type 1: a diagnosis in search of a disease. Blood. 2003;101(6):2089–2093.
3 Offermanns S. Activation of platelet function through G protein-coupled receptors. Circ Res. 2006;99(12):1293–1304.
4 Kulkarni S., Dopheide S.M., Yap C.L., et al. A revised model of platelet aggregation. J Clin Invest. 2000;105(6):783–791.
5 Bellucci S., Caen J. Molecular basis of Glanzmann’s thrombasthenia and current strategies in treatment. Blood Rev. 2002;16(3):193–202.
6 Davi G., Patrono C. Platelet activation and atherothrombosis. N Engl J Med. 2007;357(24):2482–2494.
7 Moncada S. Adventures in vascular biology: a tale of two mediators. Philos Trans R Soc Lond B Biol Sci. 2006;361(1469):735–759.
8 Mehta R.S. Novel oral anticoagulants. Part II: direct thrombin inhibitors. Expert Rev Hematol. 2010;3(3):351–361.
9 Mann K.G., Butenas S., Brummel K. The dynamics of thrombin formation. Arterioscler Thromb Vasc Biol. 2003;23(1):17–25.
10 Bajaj S.P., Joist J.H. New insights into how blood clots: implications for the use of APTT and PT as coagulation screening tests and in monitoring of anticoagulant therapy. Semin Thromb Hemost. 1999;25(4):407–418.
11 Mandal S.K., Pendurthi U.R., Rao L.V. Cellular localization and trafficking of tissue factor. Blood. 2006;107(12):4746–4753.
12 Chen V.M., Ahamed J., Versteeg H.H., et al. Evidence for activation of tissue factor by an allosteric disulfide bond. Biochemistry. 2006;45(39):12020–12028.
13 Kothari H., Nayak R.C., Rao L.V., et al. Cystine 186-cystine 209 disulfide bond is not essential for the procoagulant activity of tissue factor or for its de-encryption. Blood. 2010;115(21):4273–4283.
14 Bach R.R., Monroe D. What is wrong with the allosteric disulfide bond hypothesis? Arterioscler Thromb Vasc Biol. 2009;29(12):1997–1998.
15 Bogdanov V.Y., Balasubramanian V., Hathcock J., et al. Alternatively spliced human tissue factor: a circulating, soluble, thrombogenic protein. Nat Med. 2003;9(4):458–462.
16 Del Conde I., Shrimpton C.N., Thiagarajan P., et al. Tissue-factor-bearing microvesicles arise from lipid rafts and fuse with activated platelets to initiate coagulation. Blood. 2005;106(5):1604–1611.
17 Panes O., Matus V., Saez C.G., et al. Human platelets synthesize and express functional tissue factor. Blood. 2007;109(12):5242–5250.
18 Skidgel R.A., Alhenc-Gelas F., Campbell W.B. Prologue: kinins and related systems. New life for old discoveries. Am J Physiol Heart Circ Physiol. 2003;284(6):H1886–H1891.
19 Horan J.T., Francis C.W. Fibrin degradation products, fibrin monomer and soluble fibrin in disseminated intravascular coagulation. Semin Thromb Hemost. 2001;27(6):657–666.
20 Cochery-Nouvellon E., Mercier E., Lissalde-Lavigne G., et al. Homozygosity for the C46T polymorphism of the F12 gene is a risk factor for venous thrombosis during the first pregnancy. J Thromb Haemost. 2007;5(4):700–707.
21 Reuner K.H., Jenetzky E., Aleu A., et al. Factor XII C46T gene polymorphism and the risk of cerebral venous thrombosis. Neurology. 2008;70(2):129–132.
22 Mosesson M.W., Siebenlist K.R., Meh D.A. The structure and biological features of fibrinogen and fibrin. Ann N Y Acad Sci. 2001;936:11–30.
23 Ariens R.A., Lai T.S., Weisel J.W., et al. Role of factor XIII in fibrin clot formation and effects of genetic polymorphisms. Blood. 2002;100(3):743–754.
24 Weitz J.I. Heparan sulfate: antithrombotic or not? J Clin Invest. 2003;111(7):952–954.
25 Patnaik M.M., Moll S. Inherited antithrombin deficiency: a review. Haemophilia. 2008;14(6):1229–1239.
26 Esmon C.T. The protein C pathway. Chest. 2003;124(3 Suppl):26S–32S.
27 Miles L.A., Castellino F.J., Gong Y. Critical role for conversion of glu-plasminogen to Lys-plasminogen for optimal stimulation of plasminogen activation on cell surfaces. Trends Cardiovasc Med. 2003;13(1):21–30.
28 Kolev K., Machovich R. Molecular and cellular modulation of fibrinolysis. Thromb Haemost. 2003;89(4):610–621.
29 Kunadian V, Gibson CM: Thrombolytics and myocardial infarction, Cardiovasc Ther doi: 10.1111/j.1755-5922.20a0.00239.x. [Epub ahead of print].
30 Rijken D.C., Sakharov D.V. Basic principles in thrombolysis: regulatory role of plasminogen. Thromb Res. 2001;103(Suppl 1):S41–S49.
31 Colman R.W. Role of the light chain of high molecular weight kininogen in adhesion, cell-associated proteolysis and angiogenesis. Biol Chem. 2001;382(1):65–70.
32 Mekkawy A.H., Morris D.L., Pourgholami M.H. Urokinase plasminogen activator system as a potential target for cancer therapy. Future Oncol. 2009;5(9):1487–1499.
33 Hildenbrand R., Allgayer H., Marx A., et al. Modulators of the urokinase-type plasminogen activation system for cancer. Expert Opin Investig Drugs. 2010;19(5):641–652.
34 Bell W.R. Present-day thrombolytic therapy: therapeutic agents–pharmacokinetics and pharmacodynamics. Rev Cardiovasc Med. 2002;3(Suppl 2):S34–S44.
35 Kohler H.P., Grant P.J. Plasminogen-activator inhibitor type 1 and coronary artery disease. N Engl J Med. 2000;342(24):1792–1801.
36 Sen P., Komissarov A.A., Florova G., et al. Plasminogen activator inhibitor-1 inhibits factor VIIa bound to tissue factor. J Thromb Haemost. 2010;9:531–539.
37 Zhao L., Buckman B., Seto M., et al. Mutations in the substrate binding site of thrombin-activatable fibrinolysis inhibitor (TAFI) alter its substrate specificity. J Biol Chem. 2003;278(34):32359–32366.
38 Mosnier L.O., Meijers J.C., Bouma B.N. Regulation of fibrinolysis in plasma by TAFI and protein C is dependent on the concentration of thrombomodulin. Thromb Haemost. 2001;85(1):5–11.
39 Mosnier L.O. Platelet factor 4 inhibits thrombomodulin-dependent activation of thrombin-activatable fibrinolysis inhibitor (TAFI) by thrombin. J Biol Chem. 2010;286:502–510.
40 Van Thiel D.H., George M., Fareed J. Low levels of thrombin activatable fibrinolysis inhibitor (TAFI) in patients with chronic liver disease. Thromb Haemost. 2001;85(4):667–670.
41 Colucci M., Binetti B.M., Tripodi A., et al. Hyperprothrombinemia associated with prothrombin G20210A mutation inhibits plasma fibrinolysis through a TAFI-mediated mechanism. Blood. 2004;103(6):2157–2161.
42 Lijnen H.R., Van Hoef B., Collen D. Inactivation of the serpin alpha(2)-antiplasmin by stromelysin-1. Biochim Biophys Acta. 2001;1547(2):206–213.
43 Lijnen H.R., Arza B., Van Hoef B., et al. Inactivation of plasminogen activator inhibitor-1 by specific proteolysis with stromelysin-1 (MMP-3). J Biol Chem. 2000;275(48):37645–37650.