[level-membership-for-hematology-oncology-and-palliative-medicine-category]
Chapter 42 Non–Small Cell Lung Cancer
Lung cancer killed an estimated 156,940 men and women in the United States in 2011, more than colon, breast, prostate, and pancreatic cancer combined.1 More than 80% of these cases were caused by habitual or environmental exposure to tobacco smoke.2 This clear-cut origin makes lung cancer a disease better suited to prevention than treatment. Non–tobacco-associated lung cancer (NTLC) is not a rare disease, however, with about 40,000 cases per year in the United States and an even greater percentage of total lung cancer cases in Asia. NTLC differs at a molecular level from tobacco-associated NSCLC, and these differences have clinical and therapeutic implications.
Advances in multiple disciplines include tumor biology techniques that enable both identification and selective treatment of particular molecular entities within the broad spectrum of diseases termed lung cancer, imaging and treatment delivery technologies that better identify where to put the dose and deliver it there accurately and reproducibly (or adaptively to a target size and shape that is changing over time), and a growing willingness of specialists of all modalities to work collaboratively for their patients’ benefit.3 The development of interventions to facilitate such continuity of care and communication to patients is gaining attention.4 The key to improving treatment is the integration of diagnostic and treatment modalities by a team whose members are interested in lung cancer and its treatment, well versed in the application of the appropriate modalities, and able to combine them without undue bias.5,6 The development of advocacy groups such as the Lung Cancer Alliance and their endorsement by groups such as the American Academy of Chest Physicians should hasten this becoming the standard of practice, as happened for patients with breast and prostate cancers.
Etiology and Epidemiology
More than 80% of cases of lung cancer can be attributed to carcinogens in tobacco smoke. Habitual and environmental exposures are clearly linked to lung cancer at levels ranging from descriptive epidemiology, characteristic patterns of oncogene and tumor suppressor gene mutations, and measurement of tobacco-associated carcinogens in the urine of individuals exposed to second-hand smoke.7–10 Hecht and associates11,12 showed that nonsmokers, when exposed to side-stream smoke in concentrations comparable to those found in a smoky bar, excreted metabolites of tobacco-specific carcinogens in their urine. Pipe and cigar smokers have lower lung cancer rates than cigarette smokers, but these habits are far from innocuous, particularly in former cigarette smokers, who often continue to inhale.
The risk of lung cancer is related to the amount smoked daily and the duration of smoking, leading to the term pack-years as a measure of smoking exposure. When individuals quit smoking, their risk of lung cancer declines over at least a decade but remains higher than that of those who have never smoked.13–15 Lung cancer incidence and death rates typically reflect smoking behavior from two to three decades previous. Examination of the respiratory epithelium of former smokers years after quitting shows persistence of telltale patterns of genetic and epigenetic change.16,17 Once initiated, the process of lung carcinogenesis can proceed without continued smoke exposure.
Current epidemiologic efforts are devoted to better understanding the molecular determinants of risk distal to smoke exposure, including metabolic activation of procarcinogens, degradation and excretion of carcinogens, accurate repair of damaged DNA (or apoptotic execution of cells harboring DNA damage), and other less well understood mechanisms that result in the development of lung cancer in some but not all heavy smokers.18–20 Although no single genetic abnormality has been clearly implicated in causing lung cancer, there is considerable epidemiologic evidence for an inherited component of risk, particularly for cases of lung cancer diagnosed in younger individuals.21 Polymorphisms in a variety of enzymes involved in the metabolism of potential carcinogens, their detoxification, and in DNA repair enzymes have been associated with risk of lung cancer in a number of small studies of both smokers and nonsmokers but no clear risk pattern has been replicated in large populations.
Several recent studies have reported that a polymorphism in the nicotinic receptor gene cluster in the chromosome 15q24-25 region is strongly associated both with nicotine dependence and the development of chronic obstructive pulmonary disease (COPD) and lung cancer.22 Such associations have been seen in European, African, and Asian populations. Whether this locus is directly involved in tobacco carcinogenesis or indirectly mediated through strength of nicotine dependence is not yet clear.
Smokers with significant COPD are at significantly higher risk for lung cancer than individuals with similar smoke exposure without COPD.23,24 It is not known whether this reflects a common behavioral cause (e.g., deeper smoke inhalation, longer smoke retention) in these individuals or if there are differences in metabolism of components of tobacco smoke that increase the risk for COPD and lung cancer. Chronic inflammation has also been implicated in the pathogenesis of a variety of cancers.
Human papillomavirus (HPV) infection, particularly with serotypes 16 and 18, has been associated with head and neck squamous cell carcinoma in humans, often without significant tobacco exposure.25 Such tumors are biologically distinct from the typical tobacco-associated head and neck cancers and have a better outcome when treated with chemoradiation. Several studies have investigated the association of HPV with lung cancer and have found HPV in about 25% of lung cancers studied, with considerable geographic variation (about 15% in Europe and the Americas, about 35% in Asia, and as high as 80% in parts of Japan and Taiwan26). It is not clear that the HPV virus is truly causative of lung cancer rather than associated with it, and this remains an area of active investigation. The therapeutic responsiveness and natural history of HPV-associated lung cancers has not been well characterized.
Although lung cancer used to be a predominately male disease, its incidence and death rates in women began to rise in the United States in the 1960s and the death rate from lung cancer in women has exceeded that of breast cancer since 1987.27 In the United States, the incidence of lung cancer in men increased through the 20th century until leveling off and then starting to decline in the 1980s. In women, the incidence rose steadily through the latter part of the century but may now be starting to level off in the United States and Europe.
Although most cases of lung cancer occur in individuals with a history of tobacco use, lung cancer in those who have never smoked or in minimal smokers is far from a rare disease. It represents 15% to 20% of lung cancer seen in North America and Europe and up to 40% in parts of Asia. In the United States, deaths from non–tobacco-associated lung cancer amount to about 20,000 per year, ranking in the top five causes of cancer death. There are clear data that the clinical and molecular characteristics of lung cancer arising in persons who have never smoked or in minimal smokers are quite distinct from those of tobacco-associated lung cancer and warrant separate therapeutic strategies.28,29
Molecular Biology of Lung Cancer
Multiple genetic changes have been identified in lung cancers and cell lines derived from them and several distinct pathways of oncogenic development, both for squamous cell and several distinct varieties of adenocarcinoma, have been proposed.30,31 The morphologic and clinical differences between the various types of NSCLC are reflected in decidedly different patterns of oncogene and tumor suppressor genes modifications in these disease families. Although there are no absolute differences or characteristic mutations for either type in the way that the BCR-ABL translocation is relatively pathognomonic of chronic myeloid leukemia, some molecular abnormalities frequent in one histologic type are rare in another, and vice versa.
RAS
The RAS oncogene family contains three members, HRAS, KRAS, and NRAS, which encode membrane-associated proteins involved in mediation of signals arising from binding of ligands to cell membrane receptors such as epidermal growth factor receptors (EGFRs) to nuclear transcription factors.32 Mutation in several specific sites, including codons 12, 13, and 61, lead to constitutive activation of these signaling pathways.33 In lung cancer, mutations are most commonly seen in KRAS.34 They are most frequently seen in adenocarcinoma but are also seen in other NSCLC histologies.35 They are seen almost exclusively in smokers and rarely if ever in those who have never smoked whose tumors contain EGFR mutations. Patients whose lung cancers contain mutated KRAS almost never respond to EGFR tyrosine kinase inhibition (EGFR-TKI), but it is not clear that they are resistant to cetuximab as is the situation in colorectal cancer.36 The presence of mutated RAS genes has been an adverse prognostic factor in resected and advanced lung cancer in most series in which this has been assessed.37 RAS mutations are rare in SCLC, and, when detected, they may represent an admixture of NSCLC elements or the development of an intermediate SCLC line.38
MYC
The MYC gene products are nuclear transcription factors. Structural mutations are not reported in lung cancer, but amplification of copy number and overexpression are common in SCLC and uncommon in NSCLC with the exception of large cell neuroendocrine tumors. Amplification has been more commonly reported in cell lines from patients with clinically resistant tumors after chemotherapy, but the role of MYC amplification in de novo drug resistance is uncertain.39–43
TP53
Abnormalities in TP53 function through deletion or mutation of the gene or overexpression of MDM2 leading to aberrant TP53 inactivation are among the most common genetically induced changes in human malignancies.44 Mutations are reported in a high frequency in lung cancer, about 50% of NSCLCs, and 90% of SCLCs.45,46 Different patterns of mutation (e.g., ratio of transitions to transversions) are seen in smokers and in persons who have never smoked, suggesting different mutagens. Normal TP53 plays several key roles in determining cellular response to genetic damage, particularly whether cells enter growth arrest or apoptosis. There have been conflicting reports about the prognostic implications of abnormal TP53 function in lung cancer. For a number of chemotherapeutic agents and ionizing radiation, cells lacking normal TP53 function are resistant to apoptotic cell death, although the effect on overall cell survival is controversial.47,48
RB1
The RB1 gene–encoded product (RB) is a nuclear protein that undergoes cyclic phosphorylation and dephosphorylation during the cell cycle under the control of G1 cyclins.49,50 RB regulates E2F1 transcription factor activation. Absence of normal function occurs in most SCLCs but only in about 10% of NSCLC lines examined.51–53
CDKN2A
Normally, CDKN2A (previously designated p16) regulates transcription through phosphorylation of the RB1 protein. Abnormalities of CDKN2A function are reported in a reciprocal fashion to those of RB1; they are common in NSCLC and rare in SCLC. Because either of these abnormalities can allow uncontrolled transcriptional activation through E2F, it is not surprising that they are not commonly found together.54 Although mutations in either are common in lung cancer, these mutations have not been correlated with prognosis.
Epidermal Growth Factor Receptor
EGFR mediates a number of signaling pathways critical for cell growth, survival, and response to cytotoxins, including ionizing radiation and several chemotherapeutic agents. It is overexpressed in a variety of malignancies, and overexpression has been correlated with poorer clinical outcome and resistance to radiation. Clinical attempts to target this pathway have included monoclonal antibodies targeted to the extracellular domain of the receptor (C225, ABX) and low-molecular-weight compounds that bind the adenosine triphosphate (ATP) site and inhibit the tyrosine kinase activity of the receptor. About 10% of patients with NSCLC have mutations involving the ATP-binding pocket of the EGFR, which gives rise to more prolonged signaling activity in response to ligands such as epidermal growth factor (EGF) or transforming growth factor–beta and causes them to bind these inhibitors more tightly.55–57 Such activation mutations are seen primarily in individuals with no or minimal smoking history and in Asian women and are more common in adenocarcinomas than squamous cell carcinoma. Patients whose tumors have these mutations are likely to have significant objective responses to treatment with these compounds, but it is postulated that patients with wild-type, overexpressed EGFR are more likely to show a response of growth restraint but not regression. The relative importance of mutation and overexpression of EGFR in predicting outcome of treatment with EGFR-TKI such as erlotinib is a subject of controversy and active research.58,59,60
ERBB2
Overexpression of ERBB2 (formally HER2) is reported in NSCLC, particularly adenocarcinoma, and it is correlated with decreased survival of patients with resected disease.61,62 The degree of overexpression is typically much less than seen in cancer of the breast, with few lung cancer patients showing 3+ staining. Trials of monoclonal antibodies directed against ERBB2 in unselected NSCLC patients have shown relatively little activity whether used alone or in combination with cytotoxic chemotherapy.63
In addition to overexpression of wild-type ERBB2, activating point mutations have been described in a number of patients with NSCLC.64 These patients, about 5% of all with NSCLC or 15% of those with adenocarcinoma, may be more sensitive to therapies directed against ERBB2-mediated signaling and warrant further study with these agents, as may also be appropriate for patients with unmutated but highly overexpressed (3+ by immunohistochemical staining) ERBB2.59
Recently, fusion genes that produce atypically activated transcription factors or kinases, long known in leukemias and sarcomas, have been described in a number of solid tumors.65 Fusion of EML4 and ALK has been described in a subset of patients with NSCLC.66 Patients with EML4-ALK are typically young and light smokers or persons who have never smoked with adenocarcinoma. Because these clinical features also are more commonly seen in patients with EGFR mutations but patients with EML4-ALK mutations are not sensitive to TKI targeting EGFRs such as gefitinib or erlotinib, it becomes increasingly important to select targeted therapies on the basis of molecular profiling of individual tumors rather than population statistics.67 Inhibitors of the ALK kinase have shown significant activity in vitro and in early clinical trials and are entering trials comparing them with conventional cytotoxic agents in patients with EML4-ALK mutations.68
Early Detection and Prevention
Fewer than 20% of lung cancer patients in the United States are diagnosed with localized (stage I) disease. The poor survival of patients with symptomatic lung cancer prompted efforts to diagnose the disease in asymptomatic individuals. The U.S. National Cancer Institute (NCI) sponsored several trials in the 1970s comparing active screening using annual chest radiography and sputum cytology with “routine” medical care.69,70 These studies showed increased detection of localized disease in the screened populations, with higher rates of resectability but no decrease in mortality. This led to recommendations from the American Cancer Society to abandon routine screening for lung cancer by the methods used and in the populations studied (warranted by the data) and to an unwarranted pessimism about early diagnosis in general.
Since these trials, important demographic and technologic changes have occurred. Lung cancer is increasingly a disease of women.71 Lung cancer has also increasingly become a disease of former smokers. Several large centers have reported that more than one half of their lung cancer patients are former (i.e., more than 1 year since cessation) rather than current smokers.72 This change has key implications for therapeutic interventions and in the possibility of enlisting this population, who have already demonstrated a high degree of motivation by quitting smoking, in studies of early detection and chemoprevention.
Silvestri and associates73 have reviewed the current status of screening for lung cancer in the light of this changing demography and proposed 10 essential criteria for a successful screening program: “The disease must have serious consequences and be readily detectable in the preclinical phase. The test should have a high accuracy, detect the disease before a critical point, cause little morbidity, be available and affordable, and result in little overdiagnosis. Finally, treatment for the disease must exist, and it must be effective before symptoms occur, with little risk or morbidity.”
Computed Tomography–Based Screening Studies
The availability of fast high-resolution computed tomography (CT) has prompted a new round of screening trials.74 CT is more sensitive in detecting pulmonary nodules in asymptomatic individuals than chest radiography. The percentage of detected nodules ultimately proved to be cancerous has varied considerably between different series, in part owing to the background frequency of conditions such as endemic fungal infections that may give rise to benign pulmonary nodules. Most series have shown a shift to earlier stage at diagnosis for screened patients compared with historical controls, which is potentially a marker for improved survival. However, others have argued that tumors detected through screening are likely to have a different and more indolent biology than other tumors and that only prospective trials can determine whether CT screening reduces overall or tumor-specific mortality. Several large trials of screening CT have been completed in recent years to determine whether there is a reduction in overall mortality. Results are pending, and the current National Comprehensive Cancer Network (NCCN) and American College of Chest Physicians (ACCP) recommendations do not advocate screening for any groups.75
Cytologic and Molecular Screening
Advances in molecular technology offer the promise of detecting malignant changes in exfoliated cells well before morphologic changes appear. Tockman and associates76 have shown that with monoclonal antibody staining using sera developed against SCLC and NSCLC lines, sputum cells showing atypia could predict which patients were going to develop frank malignancy with a lead time of about 2 years. This technique is being studied further in a prospective trial of patients who have undergone resection of a stage I NSCLC and are at high risk for local recurrence and development of a second primary lung cancer. Mao and co-workers77 reported similar findings using probes for mutations of KRAS and TP53 genes. Mills and colleagues78 also reported detection of KRAS mutations in fluid obtained at bronchioalveolar lavage. Others have reported the ability to detect EGFR mutations in circulating tumor cells or DNA fragments.79,80 These findings, reported initially in retrospective pilot trials, are being validated in large, prospective trials seeking to detect second malignancies in patients curatively resected for their first lung cancers. Examination of the bronchial epithelium reveals multiple areas of dysplasia and carcinoma in many of these patients at the time of their initial diagnosis, and the risk of a second invasive malignancy is about 3% per year.
Autofluorescence bronchoscopy has been used to examine the tracheobronchial tree and look for early neoplastic or preneoplastic lesions.81 This approach complements CT screening, which primarily detects peripheral lesions and has poor sensitivity for small endobronchial lesions.82
One reasonable interpretation of the results of the NCI-sponsored early-detection trials is that although they were successful in detecting disease earlier than would have been done without screening, this “early” disease was still quite advanced in terms of the natural history of the disease. Key events such as the ability of tumor cells to metastasize, induce angiogenesis, and develop resistance to chemotherapeutic agents were likely to have occurred earlier in their history. Recognition of the long promotion period in the development of lung cancer suggests that a more fruitful approach would be to detect intermediate points in this process, to screen for carcinogenesis instead of cancer.83
At present the techniques available for detection of early lung cancer are undergoing rapid development and may lead to the development of effective screening techniques, but no regimen (target population, tests, frequency) has been shown to improve overall mortality of the screened population. Until this has been shown, the recommendation of the ACCP that “individuals undergo screening only when it is administered as a component of a well designed clinical trial with appropriate human subjects’ protection” seems quite appropriate.75
Chemoprevention
Several classes of compounds have been investigated as candidate agents for lung cancer prevention.84 Epidemiologic data show a higher incidence of lung cancer in populations with low dietary intake of retinoids and carotenoids. Deficiencies of these agents can lead to squamous metaplasia in animal models. Provision of exogenous vitamin A to animals that have developed metaplasia or atypia can lead to morphologic normalization of the epithelium.
Several trials of retinoids have been conducted in patients at high risk for lung cancer. Hong and associates85,86 randomized patients with treated cancers of the head and neck to 13-cis-retinoic acid or placebo and found that although the recurrence rates of the initial lesion were unchanged, the treated group had a significantly reduced incidence of second primary tumors, particularly of the head, neck, lung, and esophagus. Pastorino and associates87 conducted a similar trial in resected lung cancer patients using retinoyl palmitate and observed fewer second primary tumors and a borderline improvement in OS. However, larger, prospective, confirmatory trials testing isotretinoin or retinyl palmitate and N-acetylcysteine88,89 have failed to confirm the initial promise of earlier trials. To further complicate matters, several trials of β-carotene as a chemopreventive agent90 have observed increased rates of cancer in populations of individuals who continue to smoke.
The epidemiologic correlation between reduced consumption of foods rich in carotenoids and an increased risk of lung cancer in smokers and the laboratory data showing a protective effect of β-carotene against lung carcinogenesis led to two large chemoprevention trials.91–94 In Finland, a trial of placebo versus β-carotene or α-tocopherol (i.e., vitamin E), or both,95 showed increased rates of and death from lung cancer in subjects receiving β-carotene. Participants in this trial were predominantly actively smoking males. In the United States, a trial of β-carotene (CARET) was closed prematurely when interim analysis also showed an increased incidence of lung cancer in the treated arm of the study.96 The increased risk of lung cancer incidence and death of lung cancer persisted even after further administration of β-carotene was discontinued.97,98 Although the popular interpretation of these trials has been negative, they offer clear proof of the principle that human lung carcinogenesis is a dynamic process amenable to pharmacologic manipulation. They also reinforce the basic principle that clever ideas must be tested in careful trials before becoming standard of practice. No agent has yet been recognized and confirmed as an effective chemopreventive agent for lung, head, or neck cancers, and an untreated control group remains appropriate and essential for future trials.
Selenium is a trace mineral whose presence in the soil varies considerably with geography. Selenium deficiency in animals can produce premalignant changes. Epidemiologic studies have suggested correlation between low dietary intake and cancer of a number of sites. With these considerations in mind, Clark and colleagues99 performed a trial of selenium supplementation versus placebo in subjects with a history of multiple skin cancers, with a reduction in subsequent skin cancers as the designed trial outcome. Such a reduction was not seen, but the number of cases of lung cancer in the treated group was one half of that in the control group. Because multiple secondary comparisons were made in the trial, this observation might reflect chance, and a confirmatory trial to compare selenium with placebo in patients with resected stage I NSCLC has been conducted in North America, with results pending.
One limitation of the chemoprevention trials is that the candidate agents were administered systemically with the attendant toxicities associated with this approach. An approach of delivering agents directly to the bronchial epithelium, where the smoke went and where carcinogenesis occurs, is appealing and is being explored with the development of agents deliverable by aerosol.100
At present no agent or combination of agents has been proven to delay or prevent the development of lung cancer of any histology in an at-risk population of current or former smokers. Although a number of the candidate agents (e.g., selenium) are of relatively low toxicity, the surprising result of increased lung cancer incidence in some retinoid trials as well as the increased risk of diabetes in patients taking selenium in a prostate cancer chemoprevention trial suggest caution rather than therapeutic adventurism in this area.101 The ACCP currently recommends that no agent be used for lung cancer chemoprevention outside of a clinical trial.102
Pathology and Pathways of Spread
The respiratory epithelium has a total surface area about the size of a tennis court. It may be divided functionally and pathologically into three zones. From the trachea through the major bronchi the normal lining is made of squamous cells with interspersed neuroendocrine cells, which appear most commonly at airway bifurcations. Terminal alveoli are lined predominantly with type I and type II pneumocytes. Intermediate bronchi and bronchioles show a transition between squamous and adenomatous lining cells and a corresponding mixture of tumor types. The exposure of this large organ to a common set of inhaled toxins sets in process a widespread set of molecular and subsequent morphologic changes. Slaughter and associates,103 who first observed this phenomenon in patients with multiple primary tumors of the head and neck, referred to it as field cancerization. Auerbach and colleagues104 described similar widespread changes in the lungs of smokers with lung cancer, and Saccomano and colleagues105 described a pattern of progressive degrees of cytologic atypia leading to frankly invasive carcinoma. The practical consequence of this is that patients who develop one neoplasm of the upper aerodigestive tract (i.e., head, neck, lung, and esophagus), if treated successfully, remain at substantial risk for developing a second or subsequent primary tumor. For patients with resected stage I NSCLC, this risk is on the order of 2% to 3% per year for at least 10 years after treatment of the first cancer.88,106,107 It can sometimes be difficult to distinguish second primary tumors from local recurrences or metastases; differences in histology location, and presence of adjacent mucosal abnormalities have been proposed as criteria. Molecular markers may allow for fingerprinting by characteristic mutations at such sites as KRAS or TP53 (which are known to have different mutational spectra in different parts of the aerodigestive tract), although it still sometimes may be difficult to distinguish between separate primary tumors and clonal variation of a metastasis and most cells in its primary tumor. Recent data have suggested, however, that multifocal lung cancers may share a common clonal origin.108,109
In addition to cell type and anatomic stage, a variety of molecular prognostic factors have been proposed to provide further prognostic or predictive information in lung cancer (Table 42-1). These include measures of cell proliferation (e.g., S-phase fraction, proliferating-cell nuclear antigen [PCNA], Ki-67, potential doubling time [Tpot]), mutated or overexpressed oncogenes and/or tumor suppressor genes (e.g., KRAS, TP53, ERBB2, BCL2), cell surface antigens (i.e., blood type antigens and their precursors), and induction of angiogenesis. Most of these have been proposed on the basis of small series of patients treated in a nonuniform fashion, and they often are based on univariate rather than multivariate analysis. Proper validation of these will require prospective study in large series of patients staged, treated, and observed in a uniform fashion.110 It is likely that patterns of expression of multiple markers as assessed by genomic or proteomic analysis will more successfully predict prognosis, as has been shown for breast cancer and intermediate-grade lymphomas. Small series have reported the success of such profiling in lung adenocarcinomas, but large validation series in uniformly treated patient groups are awaited.111 Such profiling may also be useful in determining those patients at greatest risk for specific patterns of metastatic spread, such as to the brain, for whom particular targeted adjuvant therapies such as prophylactic cranial irradiation may be more appropriate than in an unselected population.
TABLE 42-1 Proposed Prognostic and Predictive Factors for Non–Small Cell Lung Cancer
| Factor | Comment |
|---|---|
| Tumor volume | Partially incorporated into new TNM classification |
| Number of nodal sites | — |
| Clinically evident nodal involvement | Identified preoperatively as distinct from microscopic nodal involvement |
| Extranodal extension | May be prognostic for local recurrence and predictive of impact of postoperative radiation therapy on survival |
| IHC detection of nodal metastases | Nodes negative by H&E but positive by IHC |
| Histology | Predictive of response and toxicity with some agents |
| Ploidy | — |
| Proliferative rate (labeling index, Ki-67, TPOT | — |
| KRAS mutation | Predictive of resistance to EGFR-TKI |
| 3p deletion | — |
| TP53 mutation or overexpression | — |
| Angiogenesis (microvessel density) | — |
| Vascular endothelial growth factor (VEGF) overexpression | — |
| Epidermal growth factor receptor (EGFR) overexpression and/or mutation | Mutation predictive for response to EGFR-TKI |
| ERBB2 (HER/neu) oncogene expression and/or mutation | Possibly predictive for response to trastuzumab |
| EML4-ALK | Possibly predictive for response to ALK-TKI |
H&E, hematoxylin-eosin; IHC, immunohistochemical; TKI, tyrosine kinase inhibition.
Adapted from Wagner H, Bonomi P: Preoperative and postoperative therapy for non–small cell lung cancer. In Roth JA, Ruckdeschel JC, Weisenburger TH (eds): Thoracic Oncology. Philadelphia, WB Saunders, 1995, pp 147-163.
Lung cancer spreads through regional lymphatic vessels within the lung to the hilar and paratracheal nodes, which drain to the supraclavicular fossae and, to a lesser degree, the upper abdomen (Fig. 42-1). The patterns of nodal spread are partially predictable on the basis of tumor size, histology, and proximity to central airways. Involvement most often begins with intrapulmonary nodes, extends centrally to the hilum, and then extends to the mediastinum. However, noncontiguous “skip” metastases are well described. Although nodal involvement can usually be detected by preoperative CT, even for T1 primary tumors, the incidence of clinically unsuspected nodal metastases can be as high as 15% to 20% for peripheral adenocarcinomas.112,113
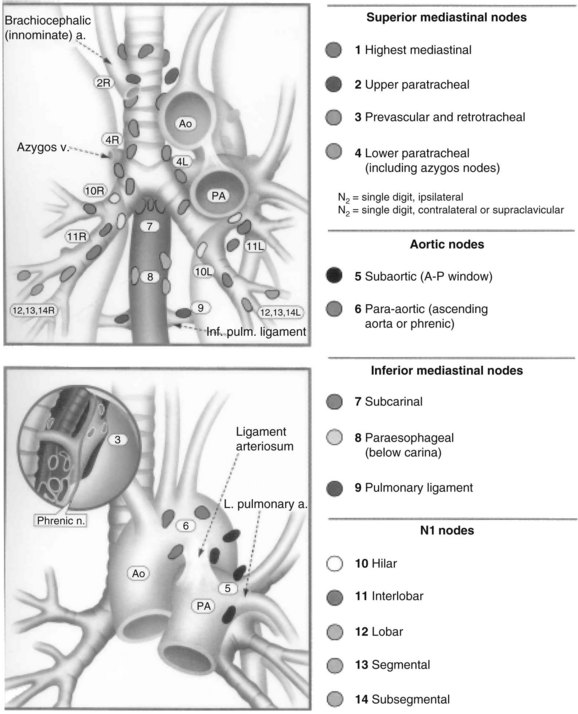
Hematogenous dissemination is common and usually is associated with nodal involvement, although about 20% of patients with resected T1N0 lesions will experience relapse in distant sites. Nodes that appear normal on routine histology, when examined by immunohistochemical staining for epithelial antigens, contain tumor cells in 15% to 25% of cases.112 Occult bone marrow metastases in resected rib at the time of thoracotomy are also found by this method, including patients without any evidence of nodal disease.114 It appears that nodal and hematogenous spread, which usually is associated, may occur independently. Routine histologic examination of nodes underestimates the true frequency of metastatic involvement.115
Although nodal drainage is primarily to the ipsilateral mediastinum, the left upper lobe drains commonly to contralateral paratracheal nodes as well as to nodes in the anterior mediastinum.116 Direct involvement of visceral and parietal pleura by peripheral tumors puts the pleural space at risk for involvement. The true frequency of this is not well known, nor is its clinical significance or appropriate therapeutic response. Several series have reported that intraoperative pleural lavage detects tumor cells in about 10% of patients and that their survival is markedly worse than that of patients with tumors of otherwise similar stage with cytologically negative lavage.117,118
Extrathoracic spread differs somewhat among the major histologic types of NSCLC. Early extrathoracic dissemination, particularly to the central nervous system (CNS), is seen more frequently with adenocarcinoma and large cell carcinoma than with squamous cell carcinoma.119 However, all histologic types are prone to early and widespread extrathoracic metastases, with brain, bone, liver, adrenal, and lung the most commonly involved sites.
Clinical Manifestations, Patient Evaluation, and Staging
Although focal neurologic symptoms most commonly result from brain metastases, CNS symptoms may also be caused by paraneoplastic cerebellar degeneration or to metabolic disturbances such as hyponatremia (most commonly seen in SCLC) or hypercalcemia (most commonly seen in squamous cell carcinoma). Nonspecific decreases in cognitive function compared with age- and gender-matched controls have also been reported in patients with lung cancer before any treatment, particularly in SCLC.120
Radiographic Procedures in Staging
Evaluation of the primary tumor is generally best accomplished by CT enhanced with intravenous administration of a contrast agent.121 The parameters of contrast enhancement (dose, infusion rate, and site of infusion [left side preferred to visualize vessels crossing in the anterior mediastinum]) can make a large difference in the quality of information obtained. It is important to examine the chest using both lung and mediastinal window settings.
Staging of the Mediastinum
Combined PET/CT is the radiographic method of choice for evaluation of the mediastinum.122,123 The false-positive and false-negative rates are about 20%, however, and this should be considered when planning therapy. In most cases, histologic confirmation of suspected nodal involvement should be done before making decisions regarding potentially curative treatment. The accessibility of mediastinal nodes to biopsy under guidance of CT or of endoesophageal or endobronchial ultrasonography makes this a simple, minimally invasive procedure for most patients, with the use of the more invasive procedures of mediastinoscopy or anterior sternotomy (i.e., Chamberlain procedure) reserved for a minority of patients.124,125 It is important to recognize that diagnostic radiologists are neither uniform nor necessarily precise in their description of the appearance and nature of mediastinal nodes. In some cases, a patient is said to have “mediastinal adenopathy” with the implication of unresectable N2 disease, although what has been shown by CT is the presence of several 1-cm nodes. The distribution of size of mediastinal nodes in normal individuals easily includes this range; and of nodes 1.5 cm in the greatest dimension, about 20% will be histologically benign. The percentage of such false-positive results is even higher among patients with obstructing endobronchial lesions or extrinsic bronchial obstruction from hilar nodes and postobstructive infection. When normal-size nodes are histologically positive, disease is usually intranodal and technically resectable. It remains unresolved whether these patients are better served by initial resection and postoperative adjuvant therapy or by detection of these N2 nodes by preoperative mediastinoscopy or mediastinotomy and treatment with neoadjuvant therapy before resection.
FDG-PET may have good sensitivity and specificity in detecting mediastinal nodal involvement and diseases in extrathoracic metastatic sites.126–132 PET has greater sensitivity and specificity than CT, but it is still imperfect. Nodes that appear involved by PET or CT should be sampled for confirmation of their status when such information will lead to alterations in clinical management, particularly when deciding whether to consider resection. If a negative PET scan is being considered to avoid performing mediastinoscopy, it is important to require that the primary tumor show good uptake (often not the case for bronchioalveolar carcinoma), that there not be a central primary tumor whose uptake could obscure small nodes, and that a dedicated PET/CT scanner be used.133 FDG-PET has also been studied for evaluating the response to preoperative chemotherapy or chemoradiation and for distinguishing between viable tumor and fibrosis in patients who have received RT or chemotherapy, or both, in assessing local disease control in patients treated nonoperatively.128,134–141
Evaluation for Extrathoracic Metastases
The use of total-body PET has been advocated as a cost-effective replacement for CT of the liver and adrenals and radionuclide bone scanning and may well be a reasonable replacement for these.122 Because the typical PET does not image the brain, it will not replace MRI for this purpose. The typical PET or fused PET/CT does not image the entire skeleton, typically omitting the calvaria and the distal extremities.
Clinically occult involvement of the CNS, especially in patients with SCLC, adenocarcinoma, and large cell carcinoma, is more common, especially in patients with involvement of mediastinal nodes. Although scanning of the brain is not indicated for patients with stage IA disease, for which the probability of involvement is about 5%, for patients with N2 disease it is in the range of 15% to 20% with high-resolution gadolinium-enhanced MRI.122 For patients being considered for aggressive chemoradiation with or without surgery, the finding of CNS metastases will lead to major changes in therapy, and scanning is warranted. Routine scanning is not done in patients with extrathoracic metastatic disease to look for CNS disease in the absence of symptoms, but the patient is instead asked about early symptoms and a scan is done at that time.
The role of CNS imaging in the patient with stage IB to II disease is controversial. Although the false-positive rate of CT and MRI is low and few patients would wrongly be excluded from surgery, the true-positive rate is also low, making this an uninformative test for most patients. However, in some series, 10% of patients with stage I to II NSCLC without neurologic symptoms had metastases demonstrated on enhanced CT.142 Such a frequency of involvement probably warrants more common CNS imaging as part of routine staging, particularly considering the cost and morbidity of a thoracotomy, which would most likely be futile in the presence of brain metastases.
Tumor-Node-Metastasis Stage Grouping
NSCLC is staged according to the principles of the tumor-node-metastasis (TNM) system. The International Staging System as outlined by Mountain in 1986143 has been the standard for reporting treatment results since that time. The broad outlines of this system are clearly valid. However, several areas emerged that indicated the need for modification of this system, and several revisions were implemented in 1997 and again in 2009.144
The TNM staging system generally is used for patients with NSCLC, and it is reasonably predictive of outcome, particularly for patients treated surgically (Tables 42-2 and 42-3). This system does not deal so well with anatomic prognostic factors important for nonsurgical treatment.
TABLE 42-2 International Staging System: TNM Classification (2009)
| Category | Definition |
|---|---|
| Tx | Primary tumor cannot be assessed, or tumor proven by the presence of malignant cells in sputum or bronchial washings but not visualized by imaging or bronchoscopy |
| T0 | No evidence of primary tumor |
| Tis | Carcinoma in situ |
| T1 | A tumor 3 cm or less in greatest dimension, surrounded by lung or visceral pleura, without radiographic or bronchoscopic evidence of invasion more proximal than the lobar bronchus (i.e., not in the main bronchus) |
| T1a | Tumor 2 cm or less in greatest dimension* |
| T1b | Tumor more than 2 cm but not more than 3 cm in greatest dimension |
| T2 | Tumor more than 3 cm but not more than 7 cm; or tumor with any of the following features†:
Associated with atelectasis or obstructive pneumonitis that extends to the hilar region but does not involve the entire lung |
* The uncommon superficial spreading tumor of any size with its invasive component limited to the bronchial wall, which may extend proximal to the main bronchus, is also classified T1a.
† T2 tumors with these features are classified T2a if 5 cm or less or if size cannot be determined and T2b if greater than 5 cm but not larger than 7 cm.
‡ Most pleural (pericardial) effusions with lung cancer are due to tumor. In a few patients, however, multiple microscopic examinations of pleural (pericardial) fluid are negative for tumor and the fluid is nonbloody and is not an exudate. When these elements and clinical judgment dictate that the effusion is not related to the tumor, the effusion should be excluded as a staging element and the patient’s disease should be classified as M0.
From Goldstraw P: Staging Manual in Thoracic Oncology. Orange Park, Fla., Editorial Rx Press, 2009.
TABLE 42-3 Stage Grouping and TNM Categories
| Stage | TNM Categories |
|---|---|
| Occult carcinoma | TXN0M0 |
| Stage 0 | TisN0M0 |
| Stage IA | T1a,bN0M0 |
| Stage IB | T2aN0M0 |
| Stage IIA | T1N1M0 |
| Stage IIB | T2bN1M0 |
| T3N0M0 | |
| Stage IIIA | T1a,bT2a,bN2M0 |
| T3N1-2M0 | |
| T4N0-1M0 | |
| Stage IIIB | T4N0-2M0 |
| Any T, N3M0 | |
| Stage IV | Any T, any N, M1 |
From Goldstraw P: Staging Manual in Thoracic Oncology. Orange Park, Fla., Editorial Rx Press, 2009.
Tumor volume is not considered an independent prognostic factor except by the proxy of unidimensional measurements. A 1-cm lesion involving the visceral pleura is T2, and a 2.9-cm lesion surrounded by lung parenchyma is T1. It might be expected that the smaller T2 lesion would be more readily controlled by RT than the larger T1 lesion, and available data are in agreement with this expectation. Data from several institutions support the hypothesis that tumor volume, independent of stage, is prognostic for local control and survival of patients with NSCLC treated with RT or chemoradiation.145,146–149 Such volumetric measurements can be performed using commercially available RT planning software, although standardization of technique is important to reduce interobserver variability.150
There are several reports that size is an important predictor of prognosis within the clinical T1N0 group, with particular importance in predicting microscopic involvement of hilar and mediastinal lymph nodes.151 This is relevant to treatment strategies that try to reduce the extent of resection or limit the radiation target volume by eliminating elective nodal irradiation (ENI) in these patients. Data suggest that only patients with lesions smaller than 1 cm in diameter have a risk of nodal involvement below 10%.
Several of the old TNM stage groups have considerable heterogeneity in outcome.154 This is true to some degree for patients with stage I disease, where the survival of patients with T1N0M0 disease (∼80%) is strikingly better than that of patients with T2N0M0 disease (∼60%). It is in the current stage group III that the heterogeneity and need for subgrouping is greatest. This was partially recognized by Mountain,143 who divided the grouping into IIIA (i.e., no invasion of the mediastinum or involvement of contralateral mediastinal or supraclavicular nodes, no pleural effusion) and IIIB (i.e., with one or more of the previous characteristics). In retrospect, this has separated a group of patients (IIIA), some of whom are treated surgically, who can undergo complete resection and have a reasonably favorable prognosis (especially T3N1M0), from those who have unresectable disease and are often treated palliatively. If patients with stage IIIA and IIIB NSCLC who are treated nonsurgically with radical RT or chemoradiation are considered, the survival differences are much less striking.152
Several other groups have proposed different revisions to the current staging system. Based on the European Lung Cancer Working Party data on patients receiving chemoradiation, Berghmans and associates153 proposed separating patients with stage T3 to T4, N3, and M0 disease from other factors that better predicted survival than the usual stage IIIA versus IIIB distinction. Andre,154 looking at patients with resected stage III NSCLC, some of whom had received induction chemotherapy, found that the number of lymph node levels and clinically detectable nodal metastases (cN2) were adverse prognostic factors.
Practical treatment decisions for patients with stage IIIA and IIIB NSCLC are often made along the following lines155:
Medical Considerations for Surgical Resection: Cardiopulmonary Evaluation
Before undergoing surgery, patients must undergo appropriate cardiopulmonary evaluation to determine their ability to tolerate the operative procedure and the expected reduction in lung function after resection.156,159 Patients with a history of cardiac artery disease should be assessed typically including a stress test along with stress radionuclide angiography. Patients in whom coronary artery calcification can be seen on CT should be similarly evaluated.
Pulmonary function evaluation should include determination of lung volumes and flow rates and of diffusing capacity and arterial blood gas analysis. A recommended preoperative workup is outlined in Table 42-4. Some patients with poor baseline pulmonary function due to extensive bullous disease and poor lung compliance may have improvement in function after combined resection of the tumor and pulmonary dead space.
| Test | Desirable Value | Comment |
|---|---|---|
| FEV1 | >1.5 L lobectomy >2 L pneumonectomy >80% predicted |
Should obtain regional ventilation and perfusion scans and calculate predicted postoperative FEV1 in borderline cases |
| PCO2 | <45 mm Hg | Recent studies suggest that this may not be an independent risk factor |
| SaO2 | >90% | Further testing indicated if <90% |
| DLCO | >80% of predicted | Further testing indicated if <80% |
 |
10 mL/kg/min | Values ≥15 mL/kg/min predict low surgical morbidity. Those >10 but <15 are borderline, and the patient’s overall clinical status needs to be considered carefully. |
DLCO, diffusing capacity of the lung for carbon dioxide; FEV1, forced expiratory volume in 1 second; PCO2, partial pressure of carbon dioxide; 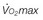 , maximum oxygen consumption.
, maximum oxygen consumption.
Data from Colice GL, Shafazand S, Griffin R, et al: Physiologic evaluation of the patients with lung cancer being considered for resectional surgery. Chest 132(Suppl):161S-177S, 2007.
Patients not meeting criteria for lobectomy should be considered for lesser resections (i.e., wedge or segmental resection) if these appear capable of obtaining negative margins. The Lung Cancer Study Group (LCSG) trial,157 which randomized patients with stage T1N0M0 NSCLC between lobectomy and more conservative resection, showed significantly better local control for lobectomy and an emergent difference in survival. The role of limited excision followed by RT to the tumor bed, either by external beam or intraoperatively placed brachytherapy mesh, is not known and is being studied by the Cancer and Leukemia Group B (CALGB) and by the American College of Surgeons Oncology Group (ACOSOG). These patients may well be better treated by stereotactic body radiation therapy (SBRT) delivering very high radiation doses to restricted volumes (see later).
Primary Therapy
Early-Stage Disease
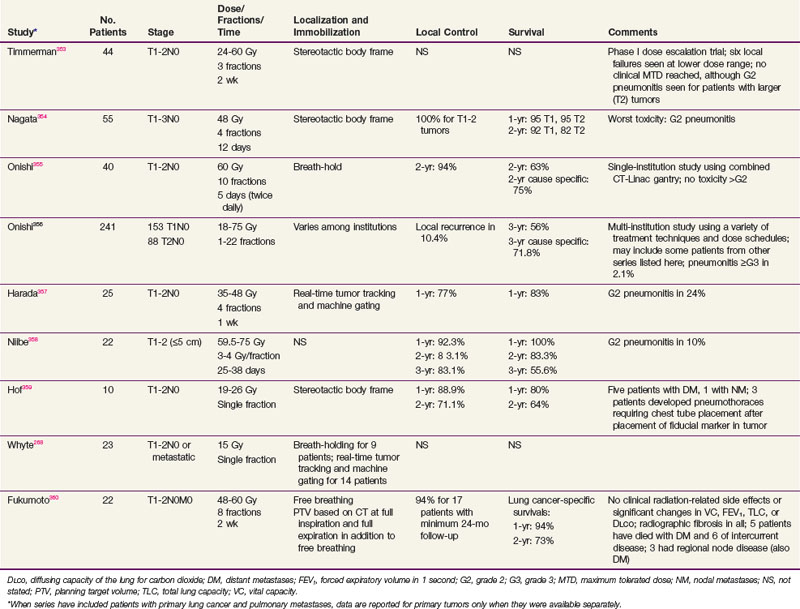
TABLE 42-5 Selected Reports of Stereotactic Radioablation of Lung Tumors: Primary Lung Cancers and Metastatic Lesions
Definitive Radiation Therapy
Single-institution reports of results of definitive RT for patients with stage I or II NSCLC who refuse surgery or are deemed inoperable for medical reasons vary in patient selection, RT technique, and outcome (Table 42-6). Some of this variation comes from different definitions of “medical unresectability,” with institutions that consider borderline disease to be unresectable in many patients, improving the apparent results of both their surgical and RT series by the Will Rogers effect (i.e., stage migration).161 However, several general conclusions may be drawn from these series:
TABLE 42-6 Radical Radiation Therapy for Patients with Medically Inoperable Non–Small Cell Lung Cancer
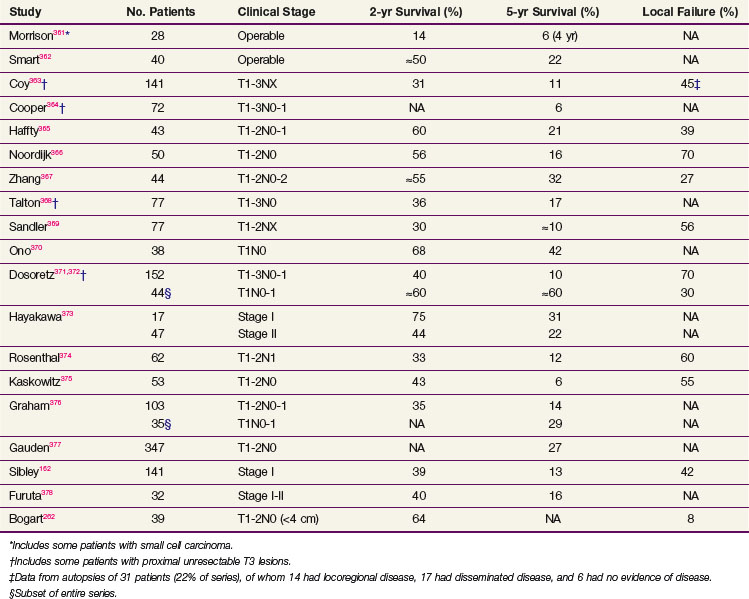
These observations suggest several strategies to improve treatment outcome. Combining RT with systemic chemotherapy in an attempt to reduce distant relapse or to improve local control is a logical approach to explore, particularly as the toxicity of current chemotherapeutic regimens has decreased. RT parameters are another fertile ground for clinical trials. Sibley162 reviewed 10 reports of definitive RT for patients with medically inoperable stage I NSCLC. About 15% of patients were long-term survivors, 25% died of intercurrent disease, 30% died of distant metastases, and 30% died of local failure. Neither time to local failure nor OS correlated with the treatment volume (i.e., primary tumor volume alone or prophylactic nodal irradiation), whereas the total tumor dose, in the range of 55 to 70 Gy, did correlate with outcome. This suggests that escalation of the biologic effective dose (by increasing the total dose or decreasing overall time with an increased fraction size or multiple fractions per day) to a small target volume, typically that of the primary tumor with margins, is a logical approach to improve the RT management of these patients. Such an approach would not preclude combination with systemic treatment, although caution should be taken because of a possible increase in pneumonitis with concurrent chemoradiation.163,164
Stereotactic Body Radiation Therapy
A number of investigators have explored the use of hypofractionated SBRT for these patients (see Table 42-5). Both frame-based and frameless systems have been used, with daily tumor localization by orthogonal images, cone-beam CT, and real-time tracking of implanted fiducial markers. Motion due to respiration presents a complicating factor not encountered in intracranial stereotactic treatment. Such motion is present in conventional large-field RT, but it is often accommodated by increasing field margins at the expense of treating increasing volumes of normal lung tissue. This is unacceptable for stereotactic large-fraction treatment. A number of approaches have been developed for minimizing respiratory motion through breath-hold and abdominal compression techniques, gating the beam-on time to a particular phase of the respiratory cycle, or tracking the tumor or radiopaque fiducial markers implanted in it and adapting the target volume to the real-time position of the tumor. These approaches are evolving, but the reported results are highly promising and suggest that, at least for T1 tumors, the results of SBRT may rival those of surgery for local control and survival (see Table 42-5). Until recently, the experience with such stereotactic approaches had been limited to single institutions, but the Radiation Therapy Oncology Group (RTOG) has conducted a phase I/II trial of fractionated SBRT for patients with medically inoperable stage I NSCLC using a regimen of three treatment fractions. Results from this and multi-institutional trials in Japan and Europe indicate local control rates of about 90% when tumors are treated to a biologically effective dose of 100BED or greater.165 A number of different fractionation schemes have been employed, ranging from a single fraction of 34 Gy to three- to five-fraction regimens over 1 to 2 weeks. At present, the optimal dose and fractionation, as well as the optimal dose homogeneity in the target volume (i.e., is a 30% hot spot at the center of the planning target volume a good thing for local control or a bad thing for complications?) remain to be clarified. Dose calculations for the multiple small fields used and tissue heterogeneities are also highly dependent on the calculation model used.166 There also have been reports of substantial toxicity, particularly with regimens using smaller numbers of fractions, when the high-dose volume includes central vascular or airway structures or the chest wall.167,168 Implementation of a program of SBRT requires careful attention to details of patient positioning, treatment planning, and dose calculation to be safe and effective, but this approach, when properly executed, appears to challenge the primacy of surgery in treating patients with early-stage lung cancer.
Another therapeutic option that has been explored for medically inoperable patients with early stage NSCLC is radiofrequency ablation (RFA). In this modality, a needle electrode is placed percutaneously under CT guidance and the tumor then heated with radiofrequency energy. Treatment is typically given in a single session. Local control rates of 50% or more seem achievable for tumors less than 3 cm, although most series are small and include a mixture of primary and metastatic tumors. Prospective trials are being conducted to better define the role of this therapy.169
Adjuvant Therapy for Early-Stage Disease
Discussing the risks and benefits of adjuvant treatment with patients is difficult. Telling a patient who is recovering from a major surgical procedure that he or she is at high risk for recurrence despite having had a complete resection, that adjuvant therapy designed to reduce this risk is of only modest benefit (typically making a difference in long-term survival for about 5% of patients), can be a daunting task. The statistical nuances of disease-free and OS curves escape many patients (Table 42-7). The format in which data are presented (i.e., whether the effect of an adjuvant treatment is expressed as a hazard ratio, a relative survival difference, or an absolute survival difference) can greatly affect their acceptance by patients.170,171
Postoperative Radiation Therapy
Several retrospective series published in the 1960s claimed that postoperative irradiation of mediastinal and hilar nodes improved survival of patients with resected NSCLC, particularly if they had nodal metastases.172–175 This issue has been investigated in several prospective trials that, although methodologically flawed, provide a consensus answer that survival is minimally if at all improved but local failure is probably reduced with such treatment.
A meta-analysis of 2128 patients treated in nine randomized trials of postoperative radiation therapy (PORT) concluded that this treatment was associated with a highly significant increase in the risk of death176 (Fig. 42-2). Overall, the risk ratio was 1.21 (p = .001). The authors concluded that PORT as used in these studies was detrimental and should not be used, at least for the time being. There are several significant differences between the treatment administered in several of the trials included in this meta-analysis and current practice patterns in the United States. One fourth of the patients had stage I disease. There has never been a strong case favoring PORT for these patients and little suggestion from their patterns of failure after surgery that such treatment would be beneficial (see Table 42-7). Details of treatment, including preoperative staging, surgical technique, and radiation dose and dose delivery, differed substantially from current practice. Several of the trials required or allowed daily fraction sizes in excess of 2.0 Gy, with the Medical Research Council (MRC) trial using 2.6 Gy/day and the Slovenian trial using 3.0 Gy/day. Larger fraction sizes would be expected to produce an increase in acute and late complications compared with slower fractionation. Seven of the nine trials also allowed the use of cobalt-60 (60Co) treatment beams, with their poorer depth-dose characteristics than higher energy accelerator beams, and only one study included CT-based treatment planning. Compared with current practice, PORT as used in these trials probably caused excess deaths from cardiac and pulmonary damage. It is notable that in this meta-analysis the increase in risk of death was most marked in those patients with stage I disease and was not significant for patients with N2 disease. This is consistent with, but does not prove, a potential benefit for properly delivered irradiation for patients with resected N2 disease.
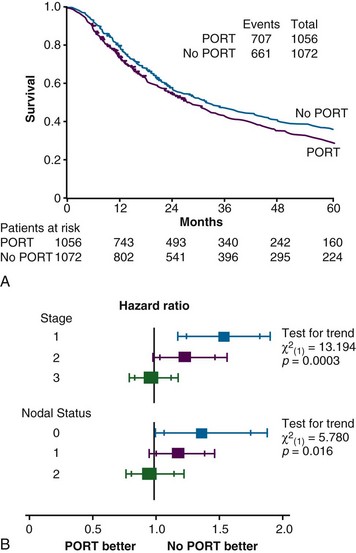
The PORT Meta-analysis Group updated their analysis with the inclusion of one additional trial.177 The overall conclusions that PORT was detrimental for patients with stage I or II NSCLC and that there was no clear evidence for benefit or detriment for patients with stage III N2 disease were not altered. However, results and updates of several other recent trials were not included in this analysis.178–180
Phlips and colleagues181 reported results of postoperative therapy given over two time periods, one with 60Co therapy and a later one with high-energy photons. For comparable patient groups, 5-year survival was 8% for those treated with 60Co but 30% for those treated on linear accelerators with CT-based treatment planning. Less cardiac and pulmonary toxicity was reported in the more recent patient group. Schraube and associates182 reported calculated complication probabilities for patients receiving postpneumonectomy RT delivered using two-dimensional (2D) or three-dimensional (3D) treatment planning. For equivalent physical doses to the mediastinal nodal target volume, the biologic mean dose to lung and heart and the associated normal tissue complication probabilities were considerably lower with 3D treatment planning. Machtay and colleagues183 compared the rates of death from intercurrent disease (DID) for patients who had received adjuvant PORT. They observed a 4-year actuarial risk of 13.5%, which was minimally but not significantly greater than the 10% expected. The total radiation dose was strongly associated with the risk of DID: 2% for doses less than 54 Gy and 17% for 54 Gy and higher doses. Wakelee and colleagues184,185 performed a retrospective analysis of DID in the INT 0115 trial, which compared PORT (50.4 Gy if no extracapsular extension, 59.4 Gy if this was documented) with or without concurrent chemotherapy with cisplatin and etoposide against that expected based on U.S. vital statistics. The actuarial rates of DID at 4 years were 12.3% for irradiation and 13.7% for irradiation plus chemotherapy (not significant) and no higher than expected given the age and smoking status of the treated patients.
Selection of patients at highest risk for mediastinal recurrence after R0 resection cannot at present be done as accurately as desired. Although the presence of metastases in resected N2 nodes clearly predicts for remaining disease in unresected nodes, the role of extracapsular extension has not been well investigated. One surgical study reported that it predicted for overall relapse in patients treated with surgery alone,185 whereas in another retrospective review186 it was found that postoperative mediastinal irradiation appeared to improve OS in patients with resected N2 disease without but not with extracapsular extension. Evidence from several institutions187–190 indicates that locoregional failure is increasingly common with a higher number of involved N2 nodes and that induction chemotherapy followed by surgery gives inadequate control of mediastinal disease.
The CALGB initiated a phase III trial of PORT in patients with resected N2 disease in an attempt to answer this question.191 Patients received four cycles of adjuvant chemotherapy with carboplatin plus paclitaxel and then were randomly assigned to observation or mediastinal irradiation. Accrual to the trial was slow, and it was closed with only 40 patients. Median failure-free survival time was 15.6 months for the nonirradiated patients and 25.9 months for those receiving mediastinal irradiation. Failure-free and OS at 1 year were 60% and 70%, respectively, on the observation arm and 56% and 72% on the irradiation arm. None of these differences reached statistical significance. The closure of this trial made it underpowered to detect a small but clinically worthwhile benefit.
Several recent nonrandomized analyses may shed some further light on the question of PORT for patients with N2 disease. Lally and associates192 analyzed data from the U.S. Surveillance, Epidemiology, and End Results (SEER) database and found that although survival for patients with resected NSCLC was not generally improved by the addition of PORT, it was significantly improved for patients with N2 disease. Douillard and colleagues193 reviewed the results of the ANITA trial in which patients with resected NSCLC, stages I through IIIA, were randomized as to whether they received adjuvant chemotherapy but received PORT or not based on institutional preference. While the numbers were small and not protected by randomization to PORT, the results were provocative. PORT was not beneficial for patients with N0 or N1 disease. But for patients with N2 disease, PORT improved both local control and survival whether or not patients received adjuvant chemotherapy, and did not significantly increase the risk of death from intercurrent disease.
A difficult clinical situation is the question of PORT for a patient without proven mediastinal lymph node involvement but whose surgery did not adequately sample or dissect the mediastinum. CT scans done postoperatively may show one or more somewhat enlarged mediastinal nodes, but this may reflect tumor or postoperative infection or inflammation. The use of PET in this setting has been explored by Roberts and colleagues, who suggested that it might be helpful in directing when to give and when to withhold further therapy.139 However positive nodes in this setting are not specific and should be considered for biopsy by endoscopic ultrasonography or endobronchial ultrasonography.
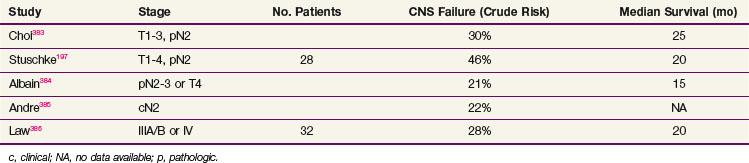
Prophylactic cranial irradiation has been less well evaluated in patients with NSCLC than in SCLC. Three reported randomized trials of prophylactic cranial irradiation in patients with NSCLC failed to show any survival benefit, but none of them included patients with early-stage, resected disease or even patients thought to be in complete remission after definitive RT (with or without chemotherapy).194–196 One retrospective series reported benefit in CNS failure rates for prophylactic cranial irradiation in patients with stage III NSCLC treated with an aggressive chemoradiation regimen. The earlier group of patients who did not receive prophylactic cranial irradiation had a CNS failure rate of 46% (13 of 28) compared with 9% (4 of 47) of a subsequent group who received prophylactic cranial irradiation.197 Others have also reported high CNS relapse rates for aggressively treated patients with stage IIIA/B NSCLC (Table 42-8). The RTOG conducted a phase III trial198 of prophylactic cranial irradiation in patients with stage IIIA/B NSCLC who had completed all other planned therapy, which included various combinations of surgery, RT, and chemotherapy, and had stable disease or better. The trial had planned to accrue 1058 patients to evaluate a potential 20% improvement in OS as its primary endpoint but was closed after accrual of 356 patients because of slow accrual. Neither overall nor disease-free survival is significantly different between the two arms, but the incidence of brain relapse at 1 year was 7.7% in the prophylactic cranial irradiation arm and 18% in the observation arm. As control of local and extrathoracic metastatic tumor improve, the potential benefit of prophylactic cranial irradiation will increase, but it cannot be considered standard at this time. As is the case for patients with SCLC, attention to details of prophylactic cranial irradiation dose and fractionation, as well as timing relative to chemotherapy, may be important in limiting neurologic toxicity.199,200
Postoperative Chemotherapy
Early trials of postoperative chemotherapy for patients with resected NSCLC failed to show significant benefit. Many of these were conducted with agents with minimal antitumor activity in this disease, and all were underpowered to detect a modest but clinically worthwhile improvement in survival on the order of 5%. A meta-analysis of all published randomized trials comparing observation with adjuvant chemotherapy showed no overall benefit, but the results differed by type of chemotherapy regimen.201 Older alkylating agent–based regimens significantly worsened survival by 5%, whereas cisplatin-based regimens gave a 5-year survival improvement of 5%, which approached statistical significance (p = .08). Although there was considerable heterogeneity of stage, surgical procedure, and planned and delivered chemotherapy, this result suggested that chemotherapy with modest activity for metastatic disease could improve survival in the adjuvant setting, as had been previously demonstrated in breast and colorectal cancers.
Several large trials of adjuvant chemotherapy have been reported in full or abstract since publication of the meta-analysis (Table 42-9). The North American Intergroup conducted a large trial designed to test the efficacy of postoperative chemotherapy in patients with completely resected N1 and N2 NSCLC.202 After extensive discussion of trial design, a two-arm comparison of PORT with or without concurrent chemotherapy (cisplatin and etoposide) was adopted. The use of PORT in both arms was designed to optimize local control, potentially increasing the ability to detect an effect of chemotherapy on systemic metastases. It was also thought that a comparison of two adjuvant regimens rather than treatment versus observation would be more acceptable to both patients and physicians. This trial (INT 0115), which enrolled 488 patients, showed no trend or significant difference in survival for the two arms, regardless of whether all 488 entered patients or 368 eligible and evaluable patients were considered. Multivariate analysis showed no benefit of the chemotherapy used in this study (i.e., four cycles of cisplatin and etoposide beginning concurrently with the start of irradiation) for any clinical subset of patients. Concurrent chemoradiation also failed to improve local mediastinal control, compared with RT alone.
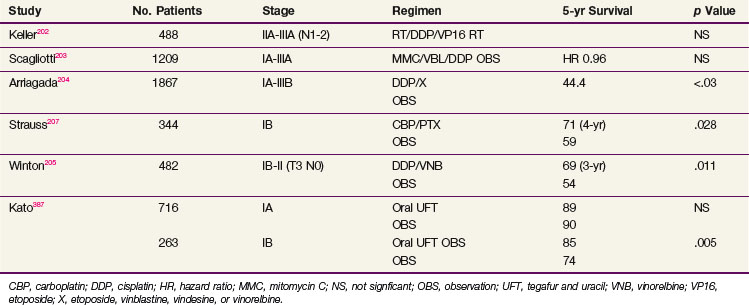
The Adjuvant Lung Project Italy (ALPI) enrolled 1209 patients with resected stage IA-IIIA NSCLC to a randomization between observation and chemotherapy with mitomycin, vindesine, and cisplatin.203 Forty-three percent of patients also received PORT (after chemotherapy if this was given). There was a survival trend favoring the chemotherapy study arm that did not reach statistical significance.
The International Adjuvant Lung Cancer Trial (IALT) is the largest trial of adjuvant chemotherapy in lung cancer that has been conducted.204 The trial was planned to study 3000 patients, but it closed after enrolling 1867 patients because of declining accrual and evidence of a significant difference between the arms of the trial on interim analysis. Patients were randomized between observation and adjuvant chemotherapy that used cisplatin and an institutional choice of second agents (i.e., etoposide, vindesine, vinblastine, or vinorelbine). RT could be given (by institutional policy) after chemotherapy. There was an overall absolute improvement in the 5-year survival rate of 4% (44.4% vs. 40.4%), which was highly significant, was present for all stage groups, and was seen whether patients also received postoperative mediastinal irradiation (not a randomized variable in this trial but done according to institutional preference). Although this trial showed a clear benefit in survival for adjuvant chemotherapy, it did not provide information on patterns of failure. To facilitate rapid accrual of a large number of patients to the trial, which was key in demonstrating the primary survival endpoint, the amount of data collected on patients was limited. Restricting the amount of information collected at randomization and during follow-up facilitated enrollment, as in previous trials.
Two other trials have demonstrated the strong, positive effects of adjuvant chemotherapy. The National Cancer Institute of Canada Cancer Treatment Group (NCIC-CTG) randomized patients with resected stage IB-IIIA NSCLC to observation or adjuvant chemotherapy with cisplatin and vinorelbine205 They found a rather large and significant improvement in OS: 69% versus 54%. Despite the availability of effective antiemetic regimens, compliance with this adjuvant chemotherapy regimen was not very good; only about 50% of patients completed all four cycles of this regimen, with variations by age, gender, extent of surgery, and nationality.206 That this was a randomized trial with a no-treatment option may have contributed to the poor compliance, with patients making the choice between known toxicity and possible benefit. With survival benefits for adjuvant chemotherapy now reasonably well established, patients may find the same regimens that were difficult to tolerate when their benefits were uncertain, much more tolerable. The CALGB randomized patients with resected stage T2N0M0 NSCLC to observation or adjuvant therapy with carboplatin plus paclitaxel. The result of this trial was initially positive, with 71% versus 59% survival at 4 years, but the difference became nonsignificant with longer follow-up.207
Hotta and associates208 published a meta-analysis of trials of adjuvant chemotherapy limited to those trials reported after the Collaborative Group meta-analysis published in 1995. Eleven trials and 5716 patients were analyzed. These investigators excluded trials in which the randomization was between PORT and chemoradiation (e.g., INT 0115). With the exception of several Japanese trials using only tegafur and uracil (UFT), the chemotherapy was cisplatin plus a variety of other agents, mostly Vinca alkaloids or etoposide. A significant survival improvement (hazard ratio = 0.872; p = .001) was seen overall, and in subset analysis this held for the cisplatin-containing chemotherapy and for UFT. The latter results are not well understood in view of the rather low response rates (6.3%) seen with UFT in the setting of metastatic disease.209 Hamada and associates210 also analyzed six published Japanese trials of UFT adjuvant chemotherapy with a highly significant improvement in 5-year OS (absolute magnitude of 5%; p = .01).
Potentially Resectable Disease: Stages IIIA and IIIB
Preoperative Treatment for Patients with Resectable Non–Small Cell Lung Cancer
Several trials conducted in the 1960s211–213 showed that for patients with initially resectable lung cancers, preoperative RT did not improve survival, and for those with initially unresectable disease, it improved resectability with no effect on survival. Surgical complications, particularly bronchopleural fistulas, were increased. The approach was largely discarded until recently, when the flurry of trials of preoperative chemotherapy or chemoradiation prompted renewed interest in preoperative RT. Unlike the trials of the 1960s, these trials have used strict criteria for resectability, surgical staging of the mediastinal nodes, and modern attention to RT planning.
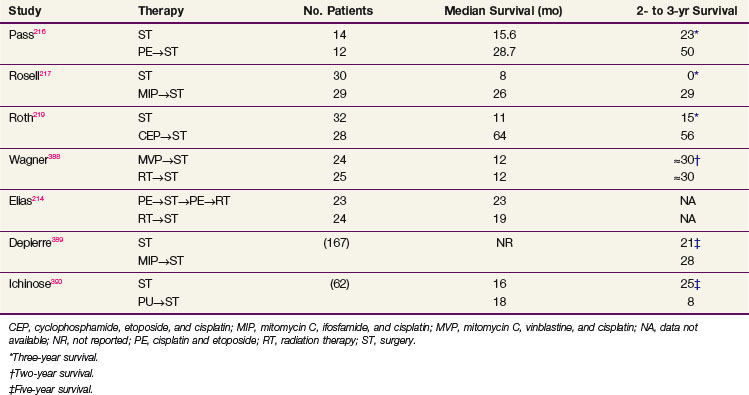
Two small, randomized, phase II trials have been reported from the CALGB and the LCSG.214,215 Both studies randomized patients with unresectable stage IIIA/B NSCLC between preoperative RT followed by surgery or preoperative chemotherapy followed by surgery. Postoperative treatment was not specified in the LCSG trial but included RT to varying doses depending on tumor status at time of resection in the CALGB trial. Table 42-10 shows the results of these two trials, as well as those of several other randomized trials that compared immediate resection to induction chemotherapy followed by surgery for patients with stage IIIA NSCLC.216–219 Results of the trials reported by Roth and Rosell were significantly positive, and the trials were closed early because of survival differences seen on interim analysis, but the LCSG, Japan Clinical Oncology Group (JCOG), and CALGB trials failed to show any difference in survival or resectability between preoperative RT, a strategy that might improve local but not systemic control, and preoperative chemotherapy. The trial by Depierre included patients with stages IB through IIIB; and although it demonstrated a survival benefit for patients with stage IB and IIA/B disease, it did not show a significant difference for patients with stage IIIA disease. Because all of these studies were small there is no clear consensus on the optimal management of patients with operable stage IIIA NSCLC. In the United States, many centers advocate the use of preoperative treatment with chemotherapy or chemoradiation, but whether this is superior to resection and postoperative therapy remains unclear, although a number of theoretical considerations ranging from better patient tolerance to earlier treatment of occult systemic disease favor the preoperative approach.
TABLE 42-10 Randomized Phase II and Phase III Trials of Neoadjuvant Therapy in Patients with Stage IIIA/IIIB Non–Small Cell Lung Cancer
A Spanish trial that compared surgery alone to either preoperative or postoperative chemotherapy (the NATCH trial) failed to show a progression-free survival benefit for either timing of chemotherapy, and a meta-analysis of trials of preoperative or postoperative chemotherapy for patients with resectable NSCLC did not find any difference between these strategies in OS.220 An indirect comparison meta-analysis of trials of either preoperative or postoperative chemotherapy compared with surgery alone failed to show differences in the hazard ratios for either disease-free or OS between the preoperative and postoperative approaches.221
The role of surgery in patients with stage IIIA and IIIB disease remains controversial. A small RTOG/Eastern Cooperative Oncology Group (ECOG) trial of induction chemotherapy followed by RT or surgery found no difference but was inadequately powered.222 The North American Intergroup Trial INT 0139 that randomized patients with pathologic N2 disease to definitive chemoradiation (cisplatin/etoposide concurrent with 61 Gy) or concurrent preoperative chemoradiation to 45 Gy showed better disease-free but not OS with the inclusion of surgery, but an unplanned subset analysis of patients who underwent lobectomy compared with patients who were thought to be candidates for lobectomy had they been randomized to surgery did show a survival benefit.223 Advocates of surgery argue that better exclusion of patients at poor risk for surgery would have shown an OS benefit, whereas partisans of nonsurgical treatment will point out that there have been significant improvements in radiographic staging (e.g., PET/CT) and RT planning and delivery since this trial was conducted (1994-2001).
A German Lung Cancer Group trial of preoperative CT or CT plus RT showed no progression-free or OS benefit to the irradiation-containing arm of this study.224 However, PORT was given to the patients in the study arm not receiving preoperative treatment. The overall resectability rate in the trial was low, only about 50%, in a population that was composed of about two thirds of patients with stage IIIB disease. By including RT in both arms of the trial, the issue becomes one of timing rather than amount of treatment, and the similarity in overall outcome is perhaps not surprising.
Betticher and colleagues225 reported 90 patients with stage IIIA N2 NSCLC given induction chemotherapy with cisplatin and docetaxel, of whom 75 underwent resection, with 59 R0 resections (i.e., microscopically negative margins). The overall median survival was 27.6 months and 33 months for resected patients. Among patients with resected disease, 38 had experienced relapse (10 local, 10 local plus distant, and 18 distant only). An RTOG neoadjuvant trial took the Betticher regimen and added RT in one arm to determine whether preoperative RT in this setting improves local control, but this trial closed prematurely due to low accrual.
Unresectable Stage IIIA/IIIB Disease
There are few good prospective data on the dependence of local control and survival of patients with NSCLC on radiation dose. In the 1980s it was believed by many authorities that local control could be achieved in about one half of patients treated to doses to 60 Gy given in 30 fractions over 6 weeks. However, the data on which that assumption was based (RTOG 73-01) indicated failure of progression rather than long-term control.226 Patients were randomized to 40, 50, or 60 Gy given as a continuous course with 2 Gy/day and to a fourth arm of 40 Gy given by split course (i.e., 20 Gy in five fractions, a 2-week break, repeated dosage). The 50- and 60-Gy arms showed modest improvement in local control and survival compared with the 40-Gy split or continuous arms, but this advantage was lost after 2 years. The lack of durable benefit for the higher doses may be because local control was poor with even the highest doses, to the lack of CT treatment planning, and the use of posterior spinal cord blocks that shielded mediastinal lymph nodes. Patterns of failure in this trial were reported in terms of crude risk of clinically detected local failure: 33% with 60 Gy and 44% to 49% with 40 Gy. It is incorrect to assume that local control is achieved in 67% of patients with 60 Gy. Le Chevalier and colleagues227–229 reported actuarial local control, assessed radiographically and bronchoscopically for patients treated with definitive RT to a dose of 65 Gy in 26 fractions in 6.5 weeks) alone or preceded and followed by chemotherapy with cyclophosphamide, cisplatin, lomustine, and vindesine, of only 17% to 20% at 1 year and falling to about 10% at 4 years. In addition to its necessity for cure, local control is also important for quality of life, because even patients dying of systemic disease are benefited if they can avoid the complications of local disease progression (e.g., airway obstruction with dyspnea or postobstructive infection, hemoptysis, compression of mediastinal structures such as the superior vena cava and phrenic or recurrent laryngeal nerves, or pain from chest wall invasion.
Combining Radiation Therapy and Chemotherapy
One risk of multimodality therapy of lung cancer is the potential for extending the time from initial presentation to start of treatment. Several reports have shown inconsistent effects of time to treatment on average and long-term survival of lung cancer patients. This may in part stem from starting patients with a disease with a poor prognosis on simple palliative treatment quickly, with more delay for both thorough staging and planning of more complex treatment for patients treated with curative intent. In the study by Wang and co-workers,230 no benefit in average survival was seen but there was a significant association between short time to treatment and survival for patients who lived more than 5 years, who comprised about 10% of the entire population.
Chemotherapy and RT can be combined in several ways. One can try to achieve additive cytotoxicity by treating the known local disease with RT and the local and systemic disease with chemotherapy. Such an approach is epitomized by the use of induction chemotherapy before local treatment with RT or surgery. This strategy may allow greater sparing of normal tissue by virtue of smaller RT target volumes or lesser resections performed of the postchemotherapy tumor volume. Another approach is to administer RT and chemotherapy concurrently in the hope of shortening the overall time of treatment and avoid delay of starting either modality. Both approaches were superior to RT alone in prospective trials conducted in the 1980s. Concurrent therapy has more acute toxicity (myelosuppression and esophagitis), but its superiority to sequential therapy has been demonstrated. At least six randomized trials comparing the sequential and concurrent use of chemotherapy and RT for patients with stage IIIA/B NSCLC have been reported (Table 42-11). Although all trials addressed the same general question of sequencing, they differed in many details. In some, different chemotherapy regimens were used in the two arms. The number of cycles of induction chemotherapy used before RT ranged from two to four. It is not clear in all trials whether the prechemotherapy or postchemotherapy tumor volume was taken as the gross tumor volume or whether, if the prechemotherapy volume was to be used, the treatment planning was completed before the start of chemotherapy.
The overall tendency confirmed in a meta-analysis of individual patient data, although not reaching statistical significance in most of these small trials, is for better results with concurrent rather than sequential combination of irradiation and chemotherapy.231 An alternate interpretation is that the overall treatment time is key and that this is shorter in concurrent regimens. With protraction of the overall treatment time, accelerated repopulation of the tumor, which may have begun during chemotherapy, may outpace the ability of daily fractionated radiation to kill remaining tumor clonogens. Data from the U.K. Continuous Hyperfractionated Accelerated Radiation Therapy (CHART) trial and ECOG 2597 are consistent with this hypothesis. The median survival for the ECOG sequential chemotherapy followed by accelerated irradiation arm was 20.3 months compared with 14 months for patients receiving conventional irradiation after chemotherapy.232
The CALGB has reported a trial comparing induction chemotherapy with carboplatin and paclitaxel followed by concurrent chemoradiation with initial concurrent therapy. Unlike the Locally Advanced Multimodality Protocol (LAMP) trial, this was planned as a phase III comparison. This large cooperative group trial failed to confirm the earlier phase II results, with somewhat disappointing median survival times of 11.4 months for the initial concurrent regimen and 14 months for induction followed by concurrent therapy (p = .154).233
The SWOG conducted a phase II trial of consolidation with docetaxel rather than additional cycles of cisplatin plus etoposide after concurrent irradiation with cisplatin plus etoposide. Median survival was 26 months, and the 3-year survival rate was 37%. In the prior trial with cisplatin plus etoposide, consolidation values were 15 months and 17%.234 This is a striking difference, but it does represent two studies done at different times, with potentially different staging, patient allocation, and treatment planning. The Hoosier Oncology Group (HOG) attempted to confirm the SWOG results in a phase II trial: this showed no survival benefit for consolidation therapy with docetaxel but a significant increase in toxicity and hospitalization.
Integration of Molecular Targeted Agents with Radiation Therapy
Identification of radioresistant cells in human tumors has led to several approaches to sensitize them while not altering the radiation response of normal tissues. Tumor cells may be resistant for reasons of local environment (e.g., hypoxia) or because of genetic changes (e.g., mutations in TP53 or KRAS). Past strategies have looked largely to reverse environmental hypoxia without great success, but some newer agents show promise. Several approaches that exploit specific tumor genetic changes are being explored.235,236
Epidermal Growth Factor Receptor Inhibitors
Exposure of cells to ionizing radiation induces phosphorylation of EGFR and activation of its kinase activity in much the same fashion as binding of ligands such as EGF or transforming growth factor-α. This leads to activation of several signaling cascades with resultant increased cellular proliferation, decreased apoptosis, and effects on angiogenesis and DNA repair.237,238–240 Inhibition of these processes through anti-EGFR monoclonal antibodies, expression of a dominant-negative truncated version of EGFR, or low-molecular-weight compounds that bind to the ATP site of EGFR and block its kinase activity result in significant radiosensitization of a number of cell lines in vitro and in vivo.241,242 A completed phase III trial comparing RT with or without the anti-EGFR antibody C225 in patients with squamous cell carcinoma of the head and neck, which commonly overexpresses EGFR, has demonstrated clinically significant improvements in local control and survival.243 Similar trials in NSCLC using monoclonal antibodies and the low-molecular-weight tyrosine kinase inhibitors (TKIs) erlotinib and gefitinib are being conducted. Although several groups have shown that objective regressions of NSCLC in response to treatment with these TKIs occur almost exclusively in patients with specific point mutations in the ATP-binding region, which are present in only a minority of patients, it is not clear that such mutations are required for response to monoclonal antibodies that target the extracellular domain of the receptor or for radiosensitization by TKIs. The RTOG conducted a phase II trial investigating the combination of cetuximab with concurrent weekly carboplatin and paclitaxel with irradiation to 63 Gy that showed acceptable toxicity and an encouraging median survival of about 22 months.
The incremental activity of bevacizumab in combination with chemotherapy in patients with metastatic NSCLC and theoretical considerations and demonstration in colorectal cancer or increased tumor perfusion after bevacizumab treatment prompted interest in its combination with chemoradiation for patients with stage III NSCLC and limited SCLC. Several small trials have been conducted using concurrent treatment, with disturbing reports of formation of tracheobronchial fistulas, prompting early trial closure.244 Such combinations, and by extrapolation other strategies combining RT or chemoradiation with antivascular agents, should be approached with caution and only in the context of a clinical trial.
Radiation Dose and Fractionation
In North America, dose and fractionation for curative treatment of patients with unresectable NSCLC has generally been 1.8 to 2.0 Gy per fraction, with one fraction per day, 5 days per week, to a total dose of 60 to 65 Gy. Such a low total dose has been recognized as unlikely to achieve a high rate of local control, but simple dose escalation was limited by acute and late toxicity to normal tissues. Several series have challenged this assumption by using conformal treatment planning and quantitative evaluation of doses to various normal tissues. Retrospective data from the University of Michigan found a relation between radiation dose and survival for patients with stage III NSCLC, which was seen both for patients receiving irradiation as a single modality as well as those having concurrent or sequential chemoradiation.245 Some of these trials have also limited the target volume by eliminating ENI.147,246 Others have treated more traditional elective volumes, at least to doses of 50 Gy or so for possible microscopic disease.247 These trials have clearly demonstrated the feasibility of dose escalation substantially above what had been previously believed limiting, to at least 74 to 78 Gy. Retrospective review suggests improvement in clinical outcome, particularly in patients with bulky disease, with such higher doses.248 Whether such doses will truly result in better local control and survival when used as a component of combined-modality therapy remains to be evaluated in prospective trials. The RTOG is currently randomizing patients with stage IIIA/B NSCLC to doses of 60 Gy or 74 Gy in combination with concurrent and consolidation chemotherapy (± cetuximab in a second randomization) to address the dose question. In both trial arms only gross disease is targeted; there is no ENI.
The use of altered fractionation schedules is an attempt to exploit differences between tumor and normal tissues in the fraction-size dependence of their radiation sensitivity.249 Several variations on standard fractionation have been proposed and evaluated:
Hyperfractionation
The RTOG conducted a series of trials exploring the use of altered fractionation in NSCLC.250,251 The first group-wide trial (RTOG 81-08) evaluated acute toxicity of several radiation dose levels using fractions of 1.2 Gy given 4 to 6 hours apart. Dose to the initial large volume encompassing known and electively treated tissues was always 50.4 Gy, whereas the dose to known disease was increased from 50.4 to 60, 69.6, and 74.4 Gy. This was a dose-seeking and toxicity study, but comparison of the group of patients treated to 69.6 Gy with contemporaneous controls from other trials (RTOG 78-11 and 79-17) suggested a late survival benefit (8.3 ± 4.0% vs. 5.6 ± 1.5% survival at 5 years). After this, in RTOG 83-11 the total dose was increased in two sequential randomizations from 60 to 79.2 Gy.252 Late toxicities were similar in all trial arms, and OS did not differ among arms. However, when a favorable subset of patients, identified retrospectively by the CALGB criteria of good performance status and minimal weight loss, was analyzed, the 69.6-Gy arm appeared superior to other arms and to patients treated to 60 Gy in 6 weeks on other RTOG trials (13 months on median survival time [MST], with 1- and 2-year survival rates of 56% and 29%, compared with 9 months on RTOG 83-21, with rates of 35% and 10%, respectively). The higher-dose arms did not show this benefit, nor were they associated with greater toxicity, which might have offset a better therapeutic effect.
An Intergroup RTOG/ECOG trial made two comparisons: conventional RT alone or preceded by induction chemotherapy (as in CALGB 8433) and, as a third arm, the 69.6-Gy, twice-daily regimen, which had been the most favorable dose level in prior RTOG trials. It confirmed the CALGB demonstration of benefit for induction chemotherapy, with a statistically significant difference between this and standard RT alone.253,254 The twice-daily arm was numerically superior to daily fractionation, but the differences did not reach statistical significance.
The North Central Cancer Treatment Group conducted a trial of similar design. Instead of using a continuous twice-daily regimen, they used a split-course regimen of 1.5 Gy given twice daily for 20 fractions over 2 weeks, a 2-week break, and another 1.5 Gy given in 20 fractions.255 The total dose was 60 Gy in 20 fractions, but the overall time was no shorter than with conventional fractionation. This was compared with standard fractionation of 60 Gy in 30 fractions over 6 weeks. A third arm combined the twice-daily split-course regimen with concomitant cisplatin plus etoposide chemotherapy. Accrual to this trial was slow, and it was closed short of the planned sample size. The results trend toward favoring the twice-daily arm (with or without chemotherapy) for freedom from progression and survival. For patients with nonsquamous histology, survival was significantly improved (p = .02). In the North Central Cancer Treatment Group (NCCTG) trial, freedom from local progression was better for the twice-daily regimen with or without concomitant chemotherapy, although this did not quite reach statistical significance (p = .06).
Fu and associates256 reported a trial in which 109 patients with stage I-IIIB NSCLC were randomized between a standard daily regimen of 63.9 Gy and a twice-daily regimen of 69.6 Gy. Although survival in the two arms did not differ for the entire group, in a subgroup of patients with stage I to IIIA disease (number not specified), there were significant benefits for local control and survival for the twice-daily arm (2-year survival, 32% vs. 6%; local control, 28% vs. 13%).
In RTOG 83-01,257 11% of patients had significant treatment interruptions. When compared with those completing treatment in the planned time, survival at 2 and 5 years was significantly less for those with treatment interruption (24% vs. 13% at 2 years; 10% vs. 3% at 5 years). However, the length of treatment was not determined randomly and may reflect other prognostic factors. Survival differences, like the benefit for hyperfractionation, were seen only for the subset of patients with good performance status and minimal weight loss.
Accelerated Fractionation: Chart, Hart, and Chartwel Regimens
Saunders and colleagues258,259 compared an accelerated regimen called continuous hyperfractionated accelerated radiation therapy (CHART) with a conventional regimen of 60 Gy in 30 fractions in a mixed group of patients who had locally advanced (stage IIIA/IIIB) or medically unresectable (stage I or II) disease. The CHART regimen consisted of 54 Gy given in 36 fractions over 12 consecutive days (weekends included). The interfraction interval was 6 hours. Results showed a survival advantage (30% vs. 20% at 2 years; p <.001) for the CHART regimen, with local control also significantly improved. Modifications of this regimen that increased total dose, shortened interfraction interval to 4 hours, and omitted weekend treatments (HART in the United States260 and CHARTWEL in the United Kingdom261) have reported similar encouraging results in phase II trials.
ECOG conducted a trial of HART compared with standard daily fractionation in patients with stage IIIA and IIIB NSCLC.232 Patients received two cycles of induction chemotherapy with carboplatin and paclitaxel and were then randomized to conventional RT or HART. The study was designed to look for an improvement in median survival from 14 to 21 months, but it was closed early because of slow patient entry. The results, a median survival of 21 months on the HART arm and 13 months on the standard arm, are provocative, but the difference did not reach statistical significance.
Physical Considerations in Fractionation Trials
The previously discussed trials evaluating radiation dose and fractionation were done during the 1980s and 1990s, when CT and 3D planning were not uniformly employed. There was considerable uncertainty and error about the extent of disease and the adequacy of its coverage, particularly with the use of lateral and oblique beams. If a different proportion of the total dose is administered through anterior-posterior/posterior-anterior and oblique beam arrangements in the two fractionation regimens and the rate of tumor miss was greater for oblique than for anterior-posterior/posterior-anterior beams, it is reasonable to expect a difference in local control based on technique that is independent of any difference due to fractionation. Even in trials incorporating fairly straightforward patient eligibility criteria and irradiation technique requirements, errors in staging and field size or placement are commonly in the range of 15%. Because more complex techniques were typically used in the hyperfractionated regimens that gave a higher total dose, the net effect might have been to mask a possible biologic benefit of the higher-dose regimen. The use of large fields and ENI also increased acute toxicity. The role of accelerated fractionation to smaller target volumes in patients with CT/PET staging (PET was not performed in the majority of patients in CALGB 39904) is worthy of further study and has given promising results in some early trials.262
[/level-membership-for-hematology-oncology-and-palliative-medicine-category][not-level-membership-for-hematology-oncology-and-palliative-medicine-category]
Chapter 42 Non–Small Cell Lung Cancer
Lung cancer killed an estimated 156,940 men and women in the United States in 2011, more than colon, breast, prostate, and pancreatic cancer combined.1 More than 80% of these cases were caused by habitual or environmental exposure to tobacco smoke.2 This clear-cut origin makes lung cancer a disease better suited to prevention than treatment. Non–tobacco-associated lung cancer (NTLC) is not a rare disease, however, with about 40,000 cases per year in the United States and an even greater percentage of total lung cancer cases in Asia. NTLC differs at a molecular level from tobacco-associated NSCLC, and these differences have clinical and therapeutic implications.
Advances in multiple disciplines include tumor biology techniques that enable both identification and selective treatment of particular molecular entities within the broad spectrum of diseases termed lung cancer, imaging and treatment delivery technologies that better identify where to put the dose and deliver it there accurately and reproducibly (or adaptively to a target size and shape that is changing over time), and a growing willingness of specialists of all modalities to work collaboratively for their patients’ benefit.3 The development of interventions to facilitate such continuity of care and communication to patients is gaining attention.4 The key to improving treatment is the integration of diagnostic and treatment modalities by a team whose members are interested in lung cancer and its treatment, well versed in the application of the appropriate modalities, and able to combine them without undue bias.5,6 The development of advocacy groups such as the Lung Cancer Alliance and their endorsement by groups such as the American Academy of Chest Physicians should hasten this becoming the standard of practice, as happened for patients with breast and prostate cancers.
Etiology and Epidemiology
More than 80% of cases of lung cancer can be attributed to carcinogens in tobacco smoke. Habitual and environmental exposures are clearly linked to lung cancer at levels ranging from descriptive epidemiology, characteristic patterns of oncogene and tumor suppressor gene mutations, and measurement of tobacco-associated carcinogens in the urine of individuals exposed to second-hand smoke.7–10 Hecht and associates11,12 showed that nonsmokers, when exposed to side-stream smoke in concentrations comparable to those found in a smoky bar, excreted metabolites of tobacco-specific carcinogens in their urine. Pipe and cigar smokers have lower lung cancer rates than cigarette smokers, but these habits are far from innocuous, particularly in former cigarette smokers, who often continue to inhale.
The risk of lung cancer is related to the amount smoked daily and the duration of smoking, leading to the term pack-years as a measure of smoking exposure. When individuals quit smoking, their risk of lung cancer declines over at least a decade but remains higher than that of those who have never smoked.13–15 Lung cancer incidence and death rates typically reflect smoking behavior from two to three decades previous. Examination of the respiratory epithelium of former smokers years after quitting shows persistence of telltale patterns of genetic and epigenetic change.16,17 Once initiated, the process of lung carcinogenesis can proceed without continued smoke exposure.
Current epidemiologic efforts are devoted to better understanding the molecular determinants of risk distal to smoke exposure, including metabolic activation of procarcinogens, degradation and excretion of carcinogens, accurate repair of damaged DNA (or apoptotic execution of cells harboring DNA damage), and other less well understood mechanisms that result in the development of lung cancer in some but not all heavy smokers.18–20 Although no single genetic abnormality has been clearly implicated in causing lung cancer, there is considerable epidemiologic evidence for an inherited component of risk, particularly for cases of lung cancer diagnosed in younger individuals.21 Polymorphisms in a variety of enzymes involved in the metabolism of potential carcinogens, their detoxification, and in DNA repair enzymes have been associated with risk of lung cancer in a number of small studies of both smokers and nonsmokers but no clear risk pattern has been replicated in large populations.
Several recent studies have reported that a polymorphism in the nicotinic receptor gene cluster in the chromosome 15q24-25 region is strongly associated both with nicotine dependence and the development of chronic obstructive pulmonary disease (COPD) and lung cancer.22 Such associations have been seen in European, African, and Asian populations. Whether this locus is directly involved in tobacco carcinogenesis or indirectly mediated through strength of nicotine dependence is not yet clear.
Smokers with significant COPD are at significantly higher risk for lung cancer than individuals with similar smoke exposure without COPD.23,24 It is not known whether this reflects a common behavioral cause (e.g., deeper smoke inhalation, longer smoke retention) in these individuals or if there are differences in metabolism of components of tobacco smoke that increase the risk for COPD and lung cancer. Chronic inflammation has also been implicated in the pathogenesis of a variety of cancers.
Human papillomavirus (HPV) infection, particularly with serotypes 16 and 18, has been associated with head and neck squamous cell carcinoma in humans, often without significant tobacco exposure.25 Such tumors are biologically distinct from the typical tobacco-associated head and neck cancers and have a better outcome when treated with chemoradiation. Several studies have investigated the association of HPV with lung cancer and have found HPV in about 25% of lung cancers studied, with considerable geographic variation (about 15% in Europe and the Americas, about 35% in Asia, and as high as 80% in parts of Japan and Taiwan26). It is not clear that the HPV virus is truly causative of lung cancer rather than associated with it, and this remains an area of active investigation. The therapeutic responsiveness and natural history of HPV-associated lung cancers has not been well characterized.
Although lung cancer used to be a predominately male disease, its incidence and death rates in women began to rise in the United States in the 1960s and the death rate from lung cancer in women has exceeded that of breast cancer since 1987.27 In the United States, the incidence of lung cancer in men increased through the 20th century until leveling off and then starting to decline in the 1980s. In women, the incidence rose steadily through the latter part of the century but may now be starting to level off in the United States and Europe.
Although most cases of lung cancer occur in individuals with a history of tobacco use, lung cancer in those who have never smoked or in minimal smokers is far from a rare disease. It represents 15% to 20% of lung cancer seen in North America and Europe and up to 40% in parts of Asia. In the United States, deaths from non–tobacco-associated lung cancer amount to about 20,000 per year, ranking in the top five causes of cancer death. There are clear data that the clinical and molecular characteristics of lung cancer arising in persons who have never smoked or in minimal smokers are quite distinct from those of tobacco-associated lung cancer and warrant separate therapeutic strategies.28,29
Molecular Biology of Lung Cancer
Multiple genetic changes have been identified in lung cancers and cell lines derived from them and several distinct pathways of oncogenic development, both for squamous cell and several distinct varieties of adenocarcinoma, have been proposed.30,31 The morphologic and clinical differences between the various types of NSCLC are reflected in decidedly different patterns of oncogene and tumor suppressor genes modifications in these disease families. Although there are no absolute differences or characteristic mutations for either type in the way that the BCR-ABL translocation is relatively pathognomonic of chronic myeloid leukemia, some molecular abnormalities frequent in one histologic type are rare in another, and vice versa.
RAS
The RAS oncogene family contains three members, HRAS, KRAS, and NRAS, which encode membrane-associated proteins involved in mediation of signals arising from binding of ligands to cell membrane receptors such as epidermal growth factor receptors (EGFRs) to nuclear transcription factors.32 Mutation in several specific sites, including codons 12, 13, and 61, lead to constitutive activation of these signaling pathways.33 In lung cancer, mutations are most commonly seen in KRAS.34 They are most frequently seen in adenocarcinoma but are also seen in other NSCLC histologies.35 They are seen almost exclusively in smokers and rarely if ever in those who have never smoked whose tumors contain EGFR mutations. Patients whose lung cancers contain mutated KRAS almost never respond to EGFR tyrosine kinase inhibition (EGFR-TKI), but it is not clear that they are resistant to cetuximab as is the situation in colorectal cancer.36 The presence of mutated RAS genes has been an adverse prognostic factor in resected and advanced lung cancer in most series in which this has been assessed.37 RAS mutations are rare in SCLC, and, when detected, they may represent an admixture of NSCLC elements or the development of an intermediate SCLC line.38
MYC
The MYC gene products are nuclear transcription factors. Structural mutations are not reported in lung cancer, but amplification of copy number and overexpression are common in SCLC and uncommon in NSCLC with the exception of large cell neuroendocrine tumors. Amplification has been more commonly reported in cell lines from patients with clinically resistant tumors after chemotherapy, but the role of MYC amplification in de novo drug resistance is uncertain.39–43
TP53
Abnormalities in TP53 function through deletion or mutation of the gene or overexpression of MDM2 leading to aberrant TP53 inactivation are among the most common genetically induced changes in human malignancies.44 Mutations are reported in a high frequency in lung cancer, about 50% of NSCLCs, and 90% of SCLCs.45,46 Different patterns of mutation (e.g., ratio of transitions to transversions) are seen in smokers and in persons who have never smoked, suggesting different mutagens. Normal TP53 plays several key roles in determining cellular response to genetic damage, particularly whether cells enter growth arrest or apoptosis. There have been conflicting reports about the prognostic implications of abnormal TP53 function in lung cancer. For a number of chemotherapeutic agents and ionizing radiation, cells lacking normal TP53 function are resistant to apoptotic cell death, although the effect on overall cell survival is controversial.47,48
RB1
The RB1 gene–encoded product (RB) is a nuclear protein that undergoes cyclic phosphorylation and dephosphorylation during the cell cycle under the control of G1 cyclins.49,50 RB regulates E2F1 transcription factor activation. Absence of normal function occurs in most SCLCs but only in about 10% of NSCLC lines examined.51–53
CDKN2A
Normally, CDKN2A (previously designated p16) regulates transcription through phosphorylation of the RB1 protein. Abnormalities of CDKN2A function are reported in a reciprocal fashion to those of RB1; they are common in NSCLC and rare in SCLC. Because either of these abnormalities can allow uncontrolled transcriptional activation through E2F, it is not surprising that they are not commonly found together.54 Although mutations in either are common in lung cancer, these mutations have not been correlated with prognosis.
Epidermal Growth Factor Receptor
EGFR mediates a number of signaling pathways critical for cell growth, survival, and response to cytotoxins, including ionizing radiation and several chemotherapeutic agents. It is overexpressed in a variety of malignancies, and overexpression has been correlated with poorer clinical outcome and resistance to radiation. Clinical attempts to target this pathway have included monoclonal antibodies targeted to the extracellular domain of the receptor (C225, ABX) and low-molecular-weight compounds that bind the adenosine triphosphate (ATP) site and inhibit the tyrosine kinase activity of the receptor. About 10% of patients with NSCLC have mutations involving the ATP-binding pocket of the EGFR, which gives rise to more prolonged signaling activity in response to ligands such as epidermal growth factor (EGF) or transforming growth factor–beta and causes them to bind these inhibitors more tightly.55–57 Such activation mutations are seen primarily in individuals with no or minimal smoking history and in Asian women and are more common in adenocarcinomas than squamous cell carcinoma. Patients whose tumors have these mutations are likely to have significant objective responses to treatment with these compounds, but it is postulated that patients with wild-type, overexpressed EGFR are more likely to show a response of growth restraint but not regression. The relative importance of mutation and overexpression of EGFR in predicting outcome of treatment with EGFR-TKI such as erlotinib is a subject of controversy and active research.58,59,60
ERBB2
Overexpression of ERBB2 (formally HER2) is reported in NSCLC, particularly adenocarcinoma, and it is correlated with decreased survival of patients with resected disease.61,62 The degree of overexpression is typically much less than seen in cancer of the breast, with few lung cancer patients showing 3+ staining. Trials of monoclonal antibodies directed against ERBB2 in unselected NSCLC patients have shown relatively little activity whether used alone or in combination with cytotoxic chemotherapy.63
In addition to overexpression of wild-type ERBB2, activating point mutations have been described in a number of patients with NSCLC.64 These patients, about 5% of all with NSCLC or 15% of those with adenocarcinoma, may be more sensitive to therapies directed against ERBB2-mediated signaling and warrant further study with these agents, as may also be appropriate for patients with unmutated but highly overexpressed (3+ by immunohistochemical staining) ERBB2.59
Recently, fusion genes that produce atypically activated transcription factors or kinases, long known in leukemias and sarcomas, have been described in a number of solid tumors.65 Fusion of EML4 and ALK has been described in a subset of patients with NSCLC.66 Patients with EML4-ALK are typically young and light smokers or persons who have never smoked with adenocarcinoma. Because these clinical features also are more commonly seen in patients with EGFR mutations but patients with EML4-ALK mutations are not sensitive to TKI targeting EGFRs such as gefitinib or erlotinib, it becomes increasingly important to select targeted therapies on the basis of molecular profiling of individual tumors rather than population statistics.67 Inhibitors of the ALK kinase have shown significant activity in vitro and in early clinical trials and are entering trials comparing them with conventional cytotoxic agents in patients with EML4-ALK mutations.68
Early Detection and Prevention
Fewer than 20% of lung cancer patients in the United States are diagnosed with localized (stage I) disease. The poor survival of patients with symptomatic lung cancer prompted efforts to diagnose the disease in asymptomatic individuals. The U.S. National Cancer Institute (NCI) sponsored several trials in the 1970s comparing active screening using annual chest radiography and sputum cytology with “routine” medical care.69,70 These studies showed increased detection of localized disease in the screened populations, with higher rates of resectability but no decrease in mortality. This led to recommendations from the American Cancer Society to abandon routine screening for lung cancer by the methods used and in the populations studied (warranted by the data) and to an unwarranted pessimism about early diagnosis in general.
Since these trials, important demographic and technologic changes have occurred. Lung cancer is increasingly a disease of women.71 Lung cancer has also increasingly become a disease of former smokers. Several large centers have reported that more than one half of their lung cancer patients are former (i.e., more than 1 year since cessation) rather than current smokers.72 This change has key implications for therapeutic interventions and in the possibility of enlisting this population, who have already demonstrated a high degree of motivation by quitting smoking, in studies of early detection and chemoprevention.
Silvestri and associates73 have reviewed the current status of screening for lung cancer in the light of this changing demography and proposed 10 essential criteria for a successful screening program: “The disease must have serious consequences and be readily detectable in the preclinical phase. The test should have a high accuracy, detect the disease before a critical point, cause little morbidity, be available and affordable, and result in little overdiagnosis. Finally, treatment for the disease must exist, and it must be effective before symptoms occur, with little risk or morbidity.”
Computed Tomography–Based Screening Studies
The availability of fast high-resolution computed tomography (CT) has prompted a new round of screening trials.74 CT is more sensitive in detecting pulmonary nodules in asymptomatic individuals than chest radiography. The percentage of detected nodules ultimately proved to be cancerous has varied considerably between different series, in part owing to the background frequency of conditions such as endemic fungal infections that may give rise to benign pulmonary nodules. Most series have shown a shift to earlier stage at diagnosis for screened patients compared with historical controls, which is potentially a marker for improved survival. However, others have argued that tumors detected through screening are likely to have a different and more indolent biology than other tumors and that only prospective trials can determine whether CT screening reduces overall or tumor-specific mortality. Several large trials of screening CT have been completed in recent years to determine whether there is a reduction in overall mortality. Results are pending, and the current National Comprehensive Cancer Network (NCCN) and American College of Chest Physicians (ACCP) recommendations do not advocate screening for any groups.75
Cytologic and Molecular Screening
Advances in molecular technology offer the promise of detecting malignant changes in exfoliated cells well before morphologic changes appear. Tockman and associates76 have shown that with monoclonal antibody staining using sera developed against SCLC and NSCLC lines, sputum cells showing atypia could predict which patients were going to develop frank malignancy with a lead time of about 2 years. This technique is being studied further in a prospective trial of patients who have undergone resection of a stage I NSCLC and are at high risk for local recurrence and development of a second primary lung cancer. Mao and co-workers77 reported similar findings using probes for mutations of KRAS and TP53 genes. Mills and colleagues78 also reported detection of KRAS mutations in fluid obtained at bronchioalveolar lavage. Others have reported the ability to detect EGFR mutations in circulating tumor cells or DNA fragments.79,80 These findings, reported initially in retrospective pilot trials, are being validated in large, prospective trials seeking to detect second malignancies in patients curatively resected for their first lung cancers. Examination of the bronchial epithelium reveals multiple areas of dysplasia and carcinoma in many of these patients at the time of their initial diagnosis, and the risk of a second invasive malignancy is about 3% per year.
Autofluorescence bronchoscopy has been used to examine the tracheobronchial tree and look for early neoplastic or preneoplastic lesions.81 This approach complements CT screening, which primarily detects peripheral lesions and has poor sensitivity for small endobronchial lesions.82
One reasonable interpretation of the results of the NCI-sponsored early-detection trials is that although they were successful in detecting disease earlier than would have been done without screening, this “early” disease was still quite advanced in terms of the natural history of the disease. Key events such as the ability of tumor cells to metastasize, induce angiogenesis, and develop resistance to chemotherapeutic agents were likely to have occurred earlier in their history. Recognition of the long promotion period in the development of lung cancer suggests that a more fruitful approach would be to detect intermediate points in this process, to screen for carcinogenesis instead of cancer.83
At present the techniques available for detection of early lung cancer are undergoing rapid development and may lead to the development of effective screening techniques, but no regimen (target population, tests, frequency) has been shown to improve overall mortality of the screened population. Until this has been shown, the recommendation of the ACCP that “individuals undergo screening only when it is administered as a component of a well designed clinical trial with appropriate human subjects’ protection” seems quite appropriate.75
Chemoprevention
Several classes of compounds have been investigated as candidate agents for lung cancer prevention.84 Epidemiologic data show a higher incidence of lung cancer in populations with low dietary intake of retinoids and carotenoids. Deficiencies of these agents can lead to squamous metaplasia in animal models. Provision of exogenous vitamin A to animals that have developed metaplasia or atypia can lead to morphologic normalization of the epithelium.
Several trials of retinoids have been conducted in patients at high risk for lung cancer. Hong and associates85,86 randomized patients with treated cancers of the head and neck to 13-cis-retinoic acid or placebo and found that although the recurrence rates of the initial lesion were unchanged, the treated group had a significantly reduced incidence of second primary tumors, particularly of the head, neck, lung, and esophagus. Pastorino and associates87 conducted a similar trial in resected lung cancer patients using retinoyl palmitate and observed fewer second primary tumors and a borderline improvement in OS. However, larger, prospective, confirmatory trials testing isotretinoin or retinyl palmitate and N-acetylcysteine88,89 have failed to confirm the initial promise of earlier trials. To further complicate matters, several trials of β-carotene as a chemopreventive agent90 have observed increased rates of cancer in populations of individuals who continue to smoke.
The epidemiologic correlation between reduced consumption of foods rich in carotenoids and an increased risk of lung cancer in smokers and the laboratory data showing a protective effect of β-carotene against lung carcinogenesis led to two large chemoprevention trials.91–94 In Finland, a trial of placebo versus β-carotene or α-tocopherol (i.e., vitamin E), or both,95 showed increased rates of and death from lung cancer in subjects receiving β-carotene. Participants in this trial were predominantly actively smoking males. In the United States, a trial of β-carotene (CARET) was closed prematurely when interim analysis also showed an increased incidence of lung cancer in the treated arm of the study.96 The increased risk of lung cancer incidence and death of lung cancer persisted even after further administration of β-carotene was discontinued.97,98 Although the popular interpretation of these trials has been negative, they offer clear proof of the principle that human lung carcinogenesis is a dynamic process amenable to pharmacologic manipulation. They also reinforce the basic principle that clever ideas must be tested in careful trials before becoming standard of practice. No agent has yet been recognized and confirmed as an effective chemopreventive agent for lung, head, or neck cancers, and an untreated control group remains appropriate and essential for future trials.
Selenium is a trace mineral whose presence in the soil varies considerably with geography. Selenium deficiency in animals can produce premalignant changes. Epidemiologic studies have suggested correlation between low dietary intake and cancer of a number of sites. With these considerations in mind, Clark and colleagues99 performed a trial of selenium supplementation versus placebo in subjects with a history of multiple skin cancers, with a reduction in subsequent skin cancers as the designed trial outcome. Such a reduction was not seen, but the number of cases of lung cancer in the treated group was one half of that in the control group. Because multiple secondary comparisons were made in the trial, this observation might reflect chance, and a confirmatory trial to compare selenium with placebo in patients with resected stage I NSCLC has been conducted in North America, with results pending.
One limitation of the chemoprevention trials is that the candidate agents were administered systemically with the attendant toxicities associated with this approach. An approach of delivering agents directly to the bronchial epithelium, where the smoke went and where carcinogenesis occurs, is appealing and is being explored with the development of agents deliverable by aerosol.100
At present no agent or combination of agents has been proven to delay or prevent the development of lung cancer of any histology in an at-risk population of current or former smokers. Although a number of the candidate agents (e.g., selenium) are of relatively low toxicity, the surprising result of increased lung cancer incidence in some retinoid trials as well as the increased risk of diabetes in patients taking selenium in a prostate cancer chemoprevention trial suggest caution rather than therapeutic adventurism in this area.101 The ACCP currently recommends that no agent be used for lung cancer chemoprevention outside of a clinical trial.102
Pathology and Pathways of Spread
The respiratory epithelium has a total surface area about the size of a tennis court. It may be divided functionally and pathologically into three zones. From the trachea through the major bronchi the normal lining is made of squamous cells with interspersed neuroendocrine cells, which appear most commonly at airway bifurcations. Terminal alveoli are lined predominantly with type I and type II pneumocytes. Intermediate bronchi and bronchioles show a transition between squamous and adenomatous lining cells and a corresponding mixture of tumor types. The exposure of this large organ to a common set of inhaled toxins sets in process a widespread set of molecular and subsequent morphologic changes. Slaughter and associates,103 who first observed this phenomenon in patients with multiple primary tumors of the head and neck, referred to it as field cancerization. Auerbach and colleagues104 described similar widespread changes in the lungs of smokers with lung cancer, and Saccomano and colleagues105 described a pattern of progressive degrees of cytologic atypia leading to frankly invasive carcinoma. The practical consequence of this is that patients who develop one neoplasm of the upper aerodigestive tract (i.e., head, neck, lung, and esophagus), if treated successfully, remain at substantial risk for developing a second or subsequent primary tumor. For patients with resected stage I NSCLC, this risk is on the order of 2% to 3% per year for at least 10 years after treatment of the first cancer.88,106,107 It can sometimes be difficult to distinguish second primary tumors from local recurrences or metastases; differences in histology location, and presence of adjacent mucosal abnormalities have been proposed as criteria. Molecular markers may allow for fingerprinting by characteristic mutations at such sites as KRAS or TP53 (which are known to have different mutational spectra in different parts of the aerodigestive tract), although it still sometimes may be difficult to distinguish between separate primary tumors and clonal variation of a metastasis and most cells in its primary tumor. Recent data have suggested, however, that multifocal lung cancers may share a common clonal origin.108,109
In addition to cell type and anatomic stage, a variety of molecular prognostic factors have been proposed to provide further prognostic or predictive information in lung cancer (Table 42-1). These include measures of cell proliferation (e.g., S-phase fraction, proliferating-cell nuclear antigen [PCNA], Ki-67, potential doubling time [Tpot]), mutated or overexpressed oncogenes and/or tumor suppressor genes (e.g., KRAS, TP53, ERBB2, BCL2), cell surface antigens (i.e., blood type antigens and their precursors), and induction of angiogenesis. Most of these have been proposed on the basis of small series of patients treated in a nonuniform fashion, and they often are based on univariate rather than multivariate analysis. Proper validation of these will require prospective study in large series of patients staged, treated, and observed in a uniform fashion.110 It is likely that patterns of expression of multiple markers as assessed by genomic or proteomic analysis will more successfully predict prognosis, as has been shown for breast cancer and intermediate-grade lymphomas. Small series have reported the success of such profiling in lung adenocarcinomas, but large validation series in uniformly treated patient groups are awaited.111 Such profiling may also be useful in determining those patients at greatest risk for specific patterns of metastatic spread, such as to the brain, for whom particular targeted adjuvant therapies such as prophylactic cranial irradiation may be more appropriate than in an unselected population.
TABLE 42-1 Proposed Prognostic and Predictive Factors for Non–Small Cell Lung Cancer
| Factor | Comment |
|---|---|
| Tumor volume | Partially incorporated into new TNM classification |
| Number of nodal sites | — |
| Clinically evident nodal involvement | Identified preoperatively as distinct from microscopic nodal involvement |
| Extranodal extension | May be prognostic for local recurrence and predictive of impact of postoperative radiation therapy on survival |
| IHC detection of nodal metastases | Nodes negative by H&E but positive by IHC |
| Histology | Predictive of response and toxicity with some agents |
| Ploidy | — |
| Proliferative rate (labeling index, Ki-67, TPOT | — |
| KRAS mutation | Predictive of resistance to EGFR-TKI |
| 3p deletion | — |
| TP53 mutation or overexpression | — |
| Angiogenesis (microvessel density) | — |
| Vascular endothelial growth factor (VEGF) overexpression | — |
| Epidermal growth factor receptor (EGFR) overexpression and/or mutation | Mutation predictive for response to EGFR-TKI |
| ERBB2 (HER/neu) oncogene expression and/or mutation | Possibly predictive for response to trastuzumab |
| EML4-ALK | Possibly predictive for response to ALK-TKI |
H&E, hematoxylin-eosin; IHC, immunohistochemical; TKI, tyrosine kinase inhibition.
Adapted from Wagner H, Bonomi P: Preoperative and postoperative therapy for non–small cell lung cancer. In Roth JA, Ruckdeschel JC, Weisenburger TH (eds): Thoracic Oncology. Philadelphia, WB Saunders, 1995, pp 147-163.
Lung cancer spreads through regional lymphatic vessels within the lung to the hilar and paratracheal nodes, which drain to the supraclavicular fossae and, to a lesser degree, the upper abdomen (Fig. 42-1). The patterns of nodal spread are partially predictable on the basis of tumor size, histology, and proximity to central airways. Involvement most often begins with intrapulmonary nodes, extends centrally to the hilum, and then extends to the mediastinum. However, noncontiguous “skip” metastases are well described. Although nodal involvement can usually be detected by preoperative CT, even for T1 primary tumors, the incidence of clinically unsuspected nodal metastases can be as high as 15% to 20% for peripheral adenocarcinomas.112,113
Hematogenous dissemination is common and usually is associated with nodal involvement, although about 20% of patients with resected T1N0 lesions will experience relapse in distant sites. Nodes that appear normal on routine histology, when examined by immunohistochemical staining for epithelial antigens, contain tumor cells in 15% to 25% of cases.112
[/not-level-membership-for-hematology-oncology-and-palliative-medicine-category]
