Non-Neoplastic Maternal Gestational Diseases
Chapter 32 emphasizes the pathologic findings. This chapter focuses on the placental findings in common clinical scenarios. Points of particular relevance in the gross examination of the placenta are highlighted. While substantial overlap exists for some entities, e.g., intrauterine growth restriction (IUGR) and uteroplacental insufficiency, the scenarios are presented separately for ease of discussion.
Presentation of the findings in a fashion intelligible to clinicians is of major importance. The terminology proposed by the Society of Pediatric Pathology has been referred to.1–3 A synopsis of the findings may be given as a ‘one-liner’ in the pathology report, and may help clinicians to categorize the histologic findings (Table 33.1).
Table 33.1
Classification System for Placental Reporting
| 1 | Normal placenta |
| 2 | Placenta with chorioamnionitis |
| 3 | Placenta with villitis |
| 4 | Placenta with materno-placental circulatory disorder |
| 5 | Placenta with fetal–placental circulatory disorder |
| 6 | Placenta with maturation disturbances |
| 7 | Placenta with findings suggestive of gene aberration |
| 8 | Placenta with placentation defects |
| 9 | Placenta with other pathology |
Courtesy of Professor Borghild Roald, Oslo, Norway.4
Early Pregnancy Loss (Spontaneous Miscarriage)
Selected tissue from all miscarriages should be examined microscopically. Tissues felt to be present macroscopically are sometimes absent histologically, and the converse is also true. Fetal parts, when present, may be examined in accordance with parental wishes. Suction termination disrupts the fetus, but measurements of a hand or foot permit comparison against tables of growth rates. Whole fetuses and large embryos may be examined according to published protocols.
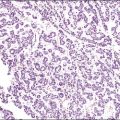
Chromosomal abnormalities in the conceptus are more frequently identified in earlier than in later fetal losses, with 65–75% of first trimester miscarriages having an abnormal karyotype.5,6 Most will be sporadic events, but a small minority result from balanced translocations in one of the parents and hence may recur. Some of the more commonly encountered karyotypic abnormalities are trisomies, especially 16 and 13, and monosomy X (45,X). Even when the karyotype is normal, there is a clear association between congenital abnormality and miscarriage. The incidence of neural tube defects and other minor abnormalities, such as cleft lip and palate or polydactyly, is higher in abortuses than in live births. Abnormalities of chromosomes 16 and 22 are prominent in early pregnancy loss (Figure 33.1A and B), and these chromosomes are preferentially involved by confined placental mosaicism.7
There have been several attempts to determine the etiology of early fetal loss by morphologic examination. Villous features thought to represent karyotypic abnormalities include vesicular change, irregular villous contour, presence of trophoblastic pseudoinclusions, trophoblastic hyperplasia, abnormal stromal cells, and, in the case of monosomy X, villous fibrosis and hypoplasia. Recognition of abnormalities at the maternal–embryonic interface (i.e., absence of interstitial trophoblast columns, absence of intravascular trophoblast plugs, and absence of physiologic changes in spiral arteries) increases the sensitivity of histology for detection of chromosomal abnormalities. Recurrent and sporadic miscarriages have similar findings, and in the majority of cases it is difficult to ascertain the cause.8 Elegant light and electron microscopic studies support the concept that many early pregnancy losses result from premature onset of the maternal–placental circulation secondary to inadequate trophoblast invasion.
Uterine abnormalities such as leiomyomas, cervical incompetence, and congenital abnormalities of müllerian duct fusion may all contribute to early pregnancy loss. Infection becomes increasingly important later in gestation as the influence of abnormal karyotype wanes. Early on, Campylobacter sp., Listeria monocytogenes, Toxoplasma gondii, rubella, cytomegalovirus (CMV), and syphilis are important. Molecular analysis suggests that coxsackie virus is one of the most common infectious agents, but placental findings are nonspecific.9 Similarly, human papillomavirus (HPV) is more commonly demonstrated in spontaneous pre-term delivery.10
The finding of a placental site trophoblastic reaction is important as it generally excludes that the pregnancy was ectopic in the fallopian tube and merely became displaced into the uterus following miscarriage. The presence of chorionic villi and decidua by themselves theoretically do not achieve this according to some reports in the literature,11 but neither of the authors has encountered an example of chorionic villi in the uterus where the origin proved to be an ectopic pregnancy. In summary, examination of the early pregnancy loss will:
Mid to Late Pregnancy Loss
If a miscarriage at 18–22 weeks is fresh, infection is a likely cause and examination should focus on the membranes. A small placenta is frequently found in trisomies and should prompt cytogenetic analysis, as karyotypic abnormalities can be expected from over 85% of placental cultures even when maceration is present.12 Second trimester miscarriages may occur following abnormal implantation, with ischemic changes up to and including infarction. Any infarction in the second trimester is likely to be clinically significant, and should be confirmed histologically. Placental infarction is associated with cerebral ischemia in the fetus, particularly in growth-restricted infants.13 The mean gestational age of pregnancy loss due to intervillositis in our practice is 25 weeks, and any increase in fibrin should prompt careful evaluation for this entity. Villitis of unknown etiology is uncommon before the third trimester.
Caution must be exercised in making the diagnosis of fetal thrombotic vasculopathy in macerated fetuses, unless there is unequivocal evidence of antemortem vascular pathology, such as clearly defined avascular villi (Figure 33.2). This is usually not possible, but, where present, enables antemortem pathology to be distinguished from the secondary effects of intrauterine death.
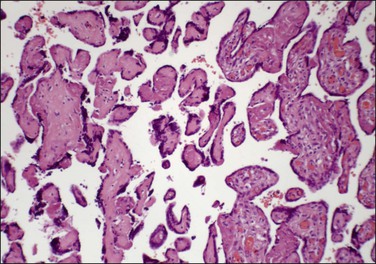
Figure 33.2 Despite changes of intrauterine death, avascular villi can be discerned (left of picture).
Abnormal cord coiling, assessed as both under- and overcoiled cords, is associated with fetal death.14,15 Cord coiling is frequently presented as an explanation for fetal death. Features of vascular compromise such as avascular villi, vascular ectasia, and mural fibrin thrombi should be sought in this context.
The role of an individual placental finding in stillbirth may be problematic. A National Institute of Child Health and Human Development workshop in 2007 evaluated maternal, fetal, and placental conditions as a cause of stillbirth.16 The proceedings also include useful overviews of the role of infectious organisms in stillbirth.
Abruption
Abruption, a condition in which the placenta detaches if not tears away from the uterine wall, can be a dramatic obstetric event. Although abruption and retroplacental hemorrhage are sometimes used interchangeably, abruption signifies a clinical event with signs and symptoms, whereas retroplacental hemorrhage is a pathologic finding, often with no clinical correlation (see Chapter 32). Occurring in 1% of pregnancies, abruption is a leading cause of vaginal bleeding in the latter half of pregnancy, and an important cause of perinatal morbidity. It accounts for 12% of perinatal deaths.17 The effect on the mother depends primarily on the severity of the abruption, whereas the effect on the fetus depends both on the severity of the abruption as well as on the fetus’s gestational age. Risk factors include prior abruption, smoking, trauma, multifetal gestation, hypertension, pre-eclampsia, thrombophilias, advanced maternal age, preterm premature rupture of the membranes, intrauterine infections, and polyhydramnios.18 Cocaine abuse may also be a cause. From the pathologist’s viewpoint, chorionic villus hemorrhage and villus edema are more frequent in cocaine users.
Questions frequently posed to the pathologist include: does an abruption exist, how extensive is it, and is it recent or old? More often than not, none of these questions can be answered solely on the pathologic examination, leading the pathologist to come away disappointed at not being able to add much of clinical use. Most are peripheral (Figure 33.3) and, if recent, will not be detected by pathologic examination, since the hemorrhage will have escaped peripherally and presented as vaginal bleeding. Retroplacental hemorrhage toward the center is far more significant (Figure 33.4), since the displaced placenta can no longer support fetal functions. In a study of over 53,000 pregnancies19 the extent of placental separation determined the chance of stillbirth. With a 75% separation, the risk of fetal death was increased 31-fold. Even a 50% separation showed profound effects. The risk for preterm delivery was also substantially increased when the separation was mild (with a separation of 25%, the relative risk was 5). The amount of bleeding is best gauged by the volume of retroplacental clot found at cesarean section, or secondarily by the amount of blood clot that accompanies the placenta to the pathology laboratory. The duration of the bleeding is best gauged by whether the parenchyma has been displaced by the blood, whether the junction of the blood and parenchyma shows discoloration from pigment breakdown, and whether the parenchyma near the abruption is infarcted.
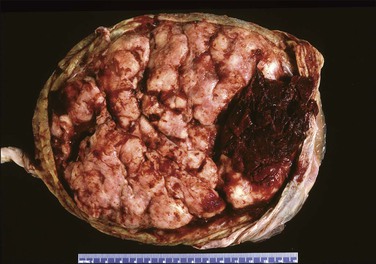
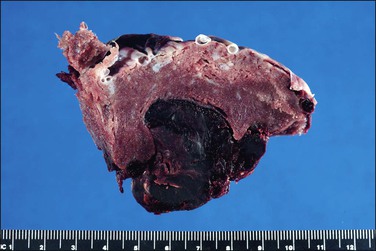
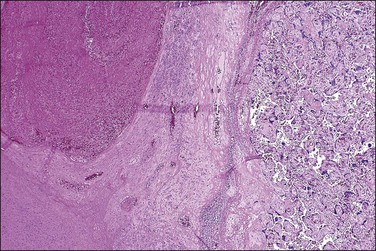
Macroscopically, the location, diameter of the involved area, depth of the crater, and thickness of the normal placenta above should be recorded, as these measurements will help determine the significance of the abruption. Microscopic sections should be taken at several areas of the junction between the blood and placental parenchyma, as the tissue reaction can help date the abruption. Neutrophils in the basal plate above the hemorrhage are the earliest finding, occurring in <4 hours.20 Nearly a day will have passed before the villi degenerate and an inflammatory response appears at the edge of the hemorrhage (Figure 33.5). Within several days there will be evidence of hemoglobin breakdown. Some placentas will show different changes at different areas. Placental abruption is generally regarded as an acute event, but it is often the end result of chronic processes that have begun earlier in pregnancy, even near the time of conception.21 In a cohort of extremely low birth weight infants (<1000 g), placental abruption was found more commonly with histologic chorioamnionitis and funisitis.22
Hypertensive Disorders
Pre-Eclampsia
Pre-eclampsia, a disorder unique to pregnancy, occurs in about 5–8% of pregnant women. Risk factors include nulliparity, women who are at the extremes of reproductive age, multiple gestation, and chronic diseases such as diabetes mellitus, kidney disease, and chronic hypertension. It usually begins insidiously after week 20 of pregnancy with excessive weight gain, marked fluid retention, increase in maternal systemic blood pressure, and the appearance of proteinuria. Pre-eclampsia is diagnosed when the blood pressure is sustained at or above 140 mmHg systolic or 90 mmHg diastolic and the urinary protein excretion exceeds 300 mg/day, but classification systems differ somewhat.23
Pre-eclampsia is classified as early onset (<34 weeks) or late onset. While late onset is by far the more common, it is not associated with placental disease and usually does not result in fetal growth restriction. The points below relate to early onset pre-eclampsia, which has been described as ‘a disease of failed interaction between two genetically different organisms.’24 Along with understanding of the role of implantation has been its description as a two-stage disease.24 Stage 1 is inadequate implantation and stage 2 is the clinical manifestations of endothelial activation in the third trimester. The sequence of events is outlined in Figure 33.6. Decidual natural killer cells, which constitute the majority of decidual white cells in the first trimester, play a key role in trophoblast invasion and successful implantation.25 Persistence of smooth muscle in uterine spiral arteries results in irregular pulsatile flow, with consequent mechanical and oxidative stress to the placenta. This results in increased shedding of trophoblast by necrosis and aponecrosis, which facilitates an inflammatory response in the mother.26 The complex interaction of abnormal placental flow and pre-eclampsia suggests that impaired invasion of deep spiral arteries might result from, rather than cause, maternal flow defects.23
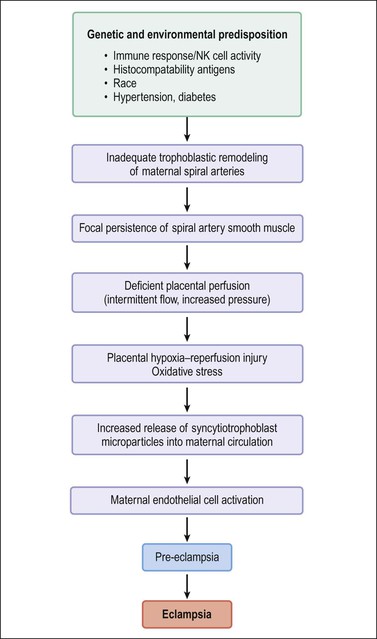
Figure 33.6 Pathogenesis of pre-eclampsia and eclampsia. (Data from Robboy SJ, Duggan M, Kurman RT. The female reproductive system. In: Rubin E, Farber J, editors. Pathology. 3rd ed. Philadelphia: Lippincott; 1999. p. 962–1028.)
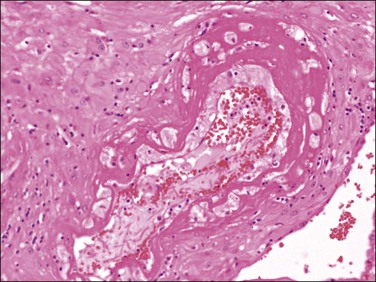
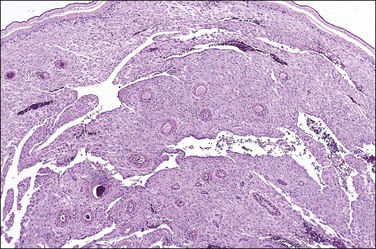
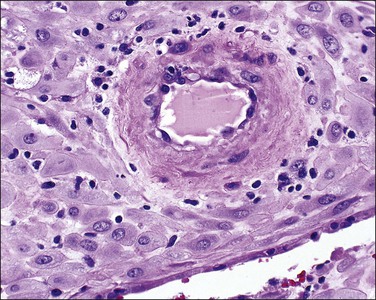
Examination of the placenta from early onset pre-eclampsia should include evaluation of acute and chronic hypoxic changes, with a comment as to the degree of severity. Expected findings include a small placenta with accelerated maturation, ischemic villous crowding, and infarction. Atherosis (Figure 33.7) will be seen in only 50% of cases, but nontransformed vessels (i.e., those whose muscularis has not been replaced by trophoblast and fibrin) (Figures 33.8 and 33.9), and hypertrophic decidual arteriopathy may be present. Laminar decidual necrosis is common. Thrombotic disease in the fetal circulation is three to four times more common when there is uteroplacental ischemic disease, and may be focal or extensive.
Intrauterine Growth Restriction
Intrauterine Growth Restriction (IUGR), also known as fetal growth restriction, is where the infant fails to achieve its biologic growth potential. This category overlaps with those who are small for gestational age. Both entities are commonly defined as fetal weight below the 10th percentile for gestational age, but approximately 70% of infants in this group are constitutionally small and have no pathology. Data on race, birth order, and parental body characteristics can help improve identification of true growth restriction.27 Agreement between customized standards and a population standard on growth restriction can be expected in approximately two-thirds of births. Birth weight below the 3rd percentile shows the strongest correlation with perinatal mortality.
IUGR is a late stage manifestation of several disease states. The etiologies can be divided into maternal causes and fetal factors. Chronic maternal hypoxia is an important cause, seen in high altitude, pulmonary hypertension, anemia, hemoglobinopathies, and cardiac failure.28 Fetal factors include fetal genetic disorders, chromosomal abnormalities, and congenital malformations. Mothers who have a history of a prior pregnancy with IUGR are also at risk for recurrence in subsequent pregnancies, including an increased risk of stillbirth.29
Pre-eclampsia is accompanied by IUGR in up to one-third of cases, but IUGR may occur in normotensive pregnancies. Differences in the pathways of spiral artery remodeling may result in the development of IUGR alone, or PET with IUGR.25
Morphologic findings in IUGR in some cases reflect the effects of stress on the placenta. Placental function is compromised by oxidative stress and complement activation, causing a decrease in the functioning trophoblast mass. This is manifest by syncytiotrophoblast apoptosis, syncytial knots, and increased perivillous fibrin.30
Early onset IUGR is less common than late onset. It is characterized by absence or reversal of end-diastolic flow in umbilical artery Doppler studies. Histologic examination shows poor peripheral villous development with nonbranching angiogenesis and increased syncytial knots.31 This cohort with persistent absence or reversal of end-diastolic flow represents a severely affected group in whom early intervention is warranted. Other abnormalities of Doppler flow include abnormalities of the systolic/diastolic ratio and of the pulsatility index. These correlate strongly with placental abnormalities, not just those reflecting maternal underperfusion, but also villous abnormalities and fetal vascular obstruction.32 Epigenetic regulation of the placenta occurs, with imprinted and nonimprinted genes influencing development.33 Overexpression of PHLDA2, a paternally imprinted gene that regulates placental growth, correlates with histologic features of maternal vascular underperfusion.34
Confined Placental Mosaicism
Confined placental mosaicism occurs when the placenta’s karyotype differs from that of the fetus. This may cause both IUGR and fetal loss. In one study of IUGR with matched controls, confined placental mosaicism was significantly higher (15.7% vs 1.4% in controls).35 While these placentas often showed decidual vasculopathy and infarction, there are no criteria for diagnosis by light microscopy. The diagnosis depends on awareness of the condition and ancillary tests such as fluorescent in situ hybridization or comparative genomic hybridization. Involvement of the cytotrophoblast as well as the mesenchymal core is required for adverse outcomes and low birth weight.7
Adverse Neurologic Outcome: Neonatal Encephalopathy and Cerebral Palsy
The area of neonatal neurologic disability is a contentious one, and the erroneous attribution of such damage to ‘birth asphyxia’ has led to obstetricians becoming uninsurable in many Western jurisdictions. The incidence of cerebral palsy has remained between 2 and 3 per 1000 live births over the last three decades, with similar figures from many centers.36 Only 4% (2 of 46) of cases of cerebral palsy in one center were related to birth asphyxia when the objective American College of Obstetricians and Gynecologists/American Academy of Pediatrics criteria were used.37
Placental examination, when combined with relevant clinical information, can add much to the understanding of a bad outcome. Any center wishing to examine such placentas must first have a mechanism in place for identifying, retrieving, and processing them, without producing an impossibly large workload. A diagnosis of cerebral palsy may often not be made until the nonprogressive nature of the impairment is clear, and, as such, placental analysis may have to proceed with a less definitive label. For term infants, neonatal encephalopathy is the best predictor of long-term neurologic disability. Over 90% of placentas from term infants who later develop cerebral palsy have a finding that may have contributed to the abnormality.38 IUGR may offer protection against the development of brain injury.39
Macroscopic description should include any findings (and relevant negatives) of the cord, membranes, fetal surface vessels, parenchyma, and basal plate. At a minimum, a section of parenchyma, cord, and membranes should be available for microscopy. Gross examination of the fixed tissue by the trained observer may identify foci of avascular villi that can be missed at a busy grossing station and may require additional sections. The key lesions to be sought, graded, and reported are those of inflammation and thrombosis.40–42 The presence of both, with lesions of different age and duration, appears to increase the risk of cerebral palsy. The coexistence of subacute and chronic pathology was significantly more likely to be identified in a cohort of medicolegal cases than in controls (24% vs 2%, respectively).42 In a series of 12 cases of neonatal stroke from a national registry, placental pathology was found in 10 (83%), with five (42%) showing mixed lesions.43 Infection plays an important role in the genesis of cerebral palsy. Clinical and histologic chorioamnionitis both increase the risk of development of cerebral palsy.44 Bacteria and other microorganisms may not be cultured, due either to antibiotic treatment or to their fastidious nature. Not uncommonly, infectious agents may exist in the placenta without histologic findings—nearly three-fourths of neonates with poor outcomes in one study had viral or bacterial disease (coxsackievirus 46%, bacteria 38%, herpes 8%, parvovirus 4%, and picornavirus 4%) when tested with in situ hybridization or reverse transcriptase polymerase chain reaction (RT-PCR), compared with none of the controls.45 In a population-based study, perinatal exposure to neurotropic viruses, in particular herpes B viruses, showed an association with cerebral palsy.45 Given the prevalence of viral nucleic acids in the control population (almost 40%), the need for a cofactor or trigger might be necessary to result in damage. Histologic findings may suggest chronic infection, with capsular deciduitis associated with periventricular leukomalacia in infants born between 23 and 34 weeks of gestation.46
Diabetes Mellitus
Both placental weight and macrosomia increase with poor control. Delayed villous maturation (villous immaturity) is significantly associated with pregestational and gestational diabetes.47 Other changes include chorangiosis. Maternal hypertensive disorders also impact on the placental morphology.48 With good control and without macrosomia, placental weight may be normal and there may be no discernible changes by routine light microscopy. With stereologic assessment, more subtle changes may be detected, even in type 1 diabetics with good glycemic control, with enhanced angiogenesis, and increased capillary length and volume.49,50
Placenta Creta
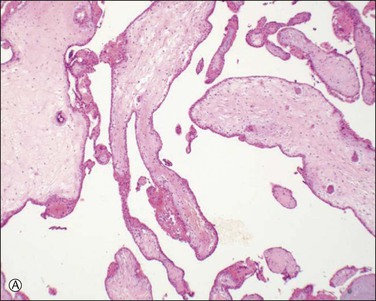
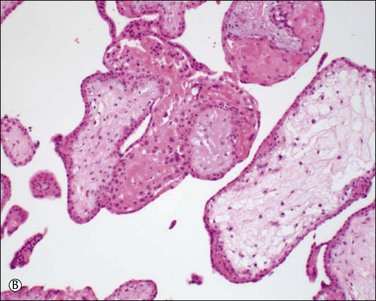
Placenta accreta, which occurs in about 0.9% of deliveries,51 substantially increases the risk of preterm delivery and small for gestational age babies. Previous cesarean section is the most common cause of a morbidly adherent placenta (Figure 33.10). The disorder is also associated with prior manual removal of the placenta, cornual implantation, leiomyomas, and prior endometrial scarring. It is frequently diagnosed clinically when there is a difficult delivery of the placenta and relates to undue adherence of the placenta to the uterine myometrium. Ultrasound with Doppler imaging of the placental vasculature and MRI of the placenta can confirm these suspicions, thus helping to guide management of the delivery. The term ‘accreta’ is used to mean an attached placenta without myometrial invasion. Increta implies a moderate degree of myometrial penetration and percreta means penetration by chorionic villi through the entire wall to the level of the serosa (Figure 33.11), sometimes with invasion of adjacent pelvic organs. In practice, all types are sometimes referred to as ‘placenta creta.’
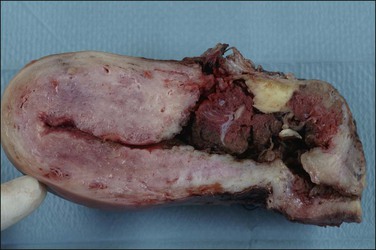
Figure 33.10 Placenta accreta: note the old cesarean section scar. (Reproduced with permission from Kelehan P, Mooney EE. Pathology of the uterus. In: Sir S. Arulkumaran et al., editors. A comprehensive textbook of postpartum hemorrhage. 2nd ed. Kirkmahoe, UK: Sapiens Publishing; 2012.)
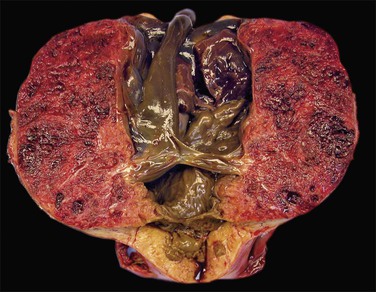
Figure 33.11 Placenta percreta. The placenta penetrates the myometrium to the level of the serosal covering.
The primary defect is deficiency of the decidua basalis with consequent apposition of chorionic villi and myometrium. The significance of myometrial cells in the basal plate in the absence of clinical evidence of placenta accreta is controversial, and the frequency of this finding will be influenced by sampling. The term ‘placenta adhesiva’ has been used for an abnormally adherent placenta that has a clear plane of separation on manual removal.52 Myometrial fibers in the basal plate were found in 78% of these placentas and multinucleate trophoblast is reduced. The term ‘occult placenta accreta’53 has been used for cases where there are myometrial fibers in the basal plate without intervening decidua. An increase in intermediate trophoblast was seen and there was an association with features of placental hypoxia.
Postpartum Hemorrhage and Subinvolution
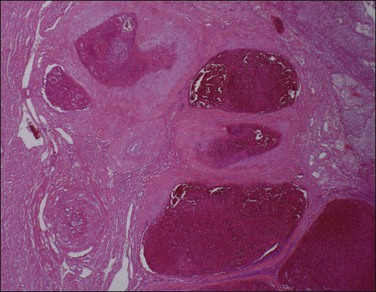
Normal arterial involution involves a decrease in the lumen size, disappearance of trophoblast, thickening of the intima, regrowth of endothelium, and regeneration of internal elastic lamina. These changes occur within 3 weeks of delivery. With subinvolution, arteries remain distended and contain red cells or fresh thrombus (Figure 33.12), and trophoblast persists in a perivascular location. In some cases, endovascular trophoblast may be present. Hemorrhage from subinvolution is maximal in the second week postpartum, although it may occur up to several months later. It is more common in older, multiparous women and may recur in subsequent deliveries. Subinvolution is not related to the method of delivery and may be regarded as a specific entity, possibly due to an abnormal immunologic relationship between trophoblast and the uterus.54 The changes may be recognized on curettage specimens, and retained products may or may not be present. The hysterectomy specimen will show a uterus that is soft and larger than expected. As normally involuted vessels may be present adjacent to subinvoluted ones, multiple blocks of placental bed should be taken to exclude this process.
Maternal Sickle Cell Trait and Disease
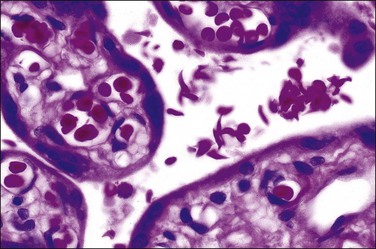
The heterozygous trait (sickle cell trait) appears not to be associated with placental pathology. In the homozygous form, maternal anemia and veno-occlusive disease result in a preplacental type of fetal hypoxia. Alterations in the endothelium of the umbilical vein, demonstrable by scanning and transmission electron microscopy, may also be due to hypoxia.55 The initial clue that the disease exists may be from the finding of sickled cells in the intervillous space of the placenta (Figure 33.13).
Twin Pregnancy
As discussed in Chapter 32, establishing chorionicity (e.g., monochorionic or dichorionic) is critical in assessing the pathology that may be present in a twin pregnancy. Twins have a higher incidence of perinatal morbidity and mortality than singleton pregnancies, and monochorionic twins are at higher risk than dichorionic. Ten percent of twins with dichorionic placentas are monozygotic, and fetal outcome is related to chorionicity rather than to zygosity. Monoamniotic monochorionic twins (MoMo) have a 20% fetal mortality rate.56 This is usually attributed to prematurity, congenital abnormalities, and rarely twin–twin transfusion syndrome. Cord entanglement is seen in 40% of MoMo twins. Monochorionic twins weigh less than twins from dichorionic gestations. Severe growth discordance between the twins (>25%) has an overall prevalence of approximately 9% in twin pregnancies57 but is more common in MoMo twins where it is associated with an increased perinatal mortality. MoMo placentas have significantly greater numbers of both superficial and deep anastomoses than do uncomplicated monochorionic diamniotic pregnancies, which may provide a vascular basis for the twin–twin transfusion syndrome that occurs on rare occasions in monoamniotic pregnancies. Monochorionic twins have an increased risk of fetal death and neurologic injury.58
Assessing growth restriction in twins may be problematic, and use of a twin nomogram with allowance for chorionicity is advised. Peripheral cord insertion reflective of a spatially limited intrauterine compartment and avascular villi indicative of occluded fetal vessels in the placenta are associated with moderate growth discordance (>15% of birth weight) or low birth weight (<10th percentile).59 Decreased placental weight, velamentous insertions, and a single umbilical artery have also been associated with severe growth discordance.60 Other findings such as thrombotic lesions differ according to chorionicity; nonocclusive mural thrombi in fetal vessels are more common in monochorionic than in dichorionic twins or in singletons, and are associated with avascular villi in the last two groups.61
A systematic review of observational studies over a 15 year period has shown that, following death of one twin, monochorionic and dichorionic twins have a death risk of 12% and 4%, respectively, with a risk of neurologic disability of 18% and 1%.62 The likelihood of survival of the co-twin decreases with increasing gestational age of the first fetal death, with mortality higher for same-sex twins.
Prolonged Pregnancy
Prolonged pregnancy is generally defined as a gestation, based on menstrual age, exceeding 42 weeks. With the estimated date of confinement, or due date, for normal pregnancies calculated as 38 weeks after conception, or 40 weeks from the first day of the last normal menstrual period, about 18% of pregnancies in the United States extend 1 week beyond the normal due date and 7% beyond 2 weeks. (Some differences between statistics in the United States and Canada seem to relate to whether menstrual or clinical dates are used.63,64) In general, the risks of post-term pregnancy include increased meconium aspiration, macrosomia, and perinatal death, with small for gestational age infants at increased risk.65 Postmature placentas may be normal. Delayed villous maturation is important to recognize in this cohort. It typically permits normal growth, but by unknown mechanisms may culminate in a normally formed macerated term stillbirth.
Maternal Infections and the Placenta
Cytomegalovirus
Cytomegalovirus (CMV) is considered one of the most common and important congenital infections in developed nations with approximately 1% of pregnant women acquiring a primary infection.66 It is also the most common agent of the toxoplasmosis, other infections, rubella, CMV infection, and herpes simplex (TORCH) group to infect the placenta and fetus.
With 40% of the women transmitting the infection to their fetuses, the risk of serious fetal injury is great. While the risk of transmission is greatest if the infection is acquired during the third trimester, the risk of serious fetal injury is greatest if acquired during the first or early part of the second trimester. The virus is transmitted in blood and body fluids, including saliva. Infection in a pregnant woman is usually asymptomatic. The virus can be transmitted following a primary infection in the mother during pregnancy or a recrudescence of a latent infection acquired at a time before pregnancy. Most (75–85%) infants suffer no adverse effects. Those that do may suffer from IUGR and serious neurologic impairment, including deafness and ocular damage. Co-infection with HIV may enhance CMV transmission and lead to more severe tissue damage.
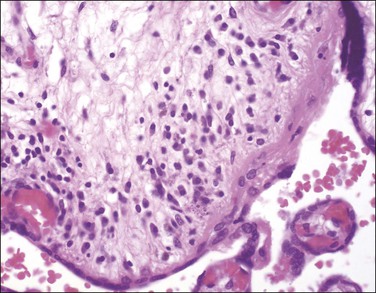
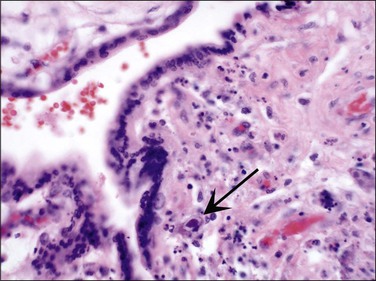
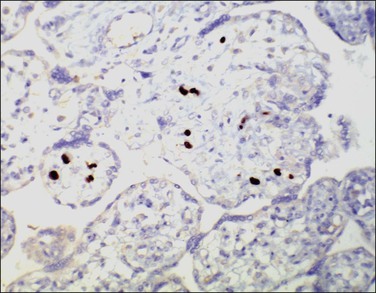
The fetus and placenta may be hydropic. The combination of a lymphoplasmacytic villitis (Figure 33.14), villous sclerosis, and hemosiderin deposition (Figure 33.15) is commonly found. The characteristic deposition of hemosiderin pigment in sclerotic villi reflects the virus’s endotheliotropic nature, although syncytiotrophoblasts are also infected.67 Intranuclear inclusions (Figure 33.16) may be seen adjacent to necrotic debris in the villous stroma, although these may be few or absent, even with immunohistochemistry (Figure 33.17).
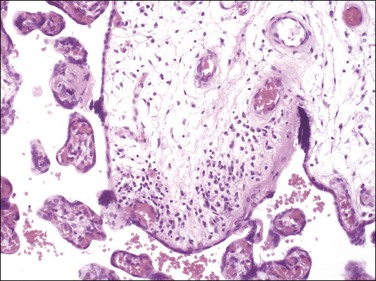
The diagnosis can be otherwise confirmed by serologic testing, viral culture, and PCR assay to detect CMV DNA in fetal and placental tissues.68 Demonstration of CMV and parvovirus B19 DNA was more common in placentas from stillbirths than controls, but does not prove causality.69
Parvovirus B19
The primary site of parvovirus B19 infections is within erythroid precursor cells and it has an affinity for the late normoblast stage (Figure 33.18). Cardiac myocytes are also infected. Hydrops is due to fetal anemia, myocarditis, and resulting cardiac failure.
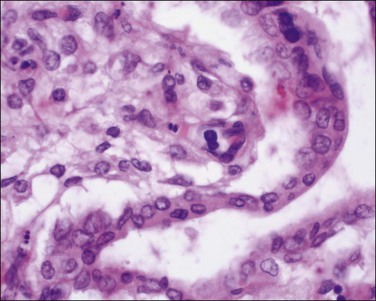
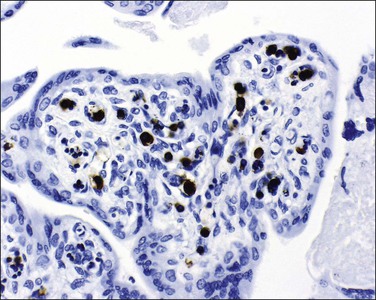
The placenta is usually bulky, pale, and edematous. Histologically, there is villous edema with villous tissue dysmaturity, villitis, and intervillitis. A fetal nucleated red cell response (erythroblastosis) is seen in capillaries. Some nucleated red blood cells show a characteristic appearance of marginated chromatin with central nuclear clearing.70 Diagnosis can also be made by immunohistochemistry (Figure 33.19).
Varicella Zoster
Primary infection with varicella zoster virus causes chickenpox. The virus remains dormant in the dorsal root ganglion and may reactivate as shingles at a later time. Chickenpox is highly contagious and is spread by droplet transmission or by direct personal contact with vesicular fluid. Primary infection in pregnancy is associated with an increased risk of complications, including maternal death and the fetal varicella syndrome. Women from tropical and subtropical areas are more likely to be seronegative for varicella zoster virus IgG and therefore are more susceptible to developing chickenpox. The risks to the fetus are greatest when the primary infection occurs prior to the 20th week of gestation or after 36 weeks. Fetal varicella syndrome does not occur at the time of the initial fetal infection, but is believed to result from subsequent herpes zoster reactivation in utero and only occurs in a minority of infected fetuses. It occurs in 1–2% of maternal varicella infections that take place prior to 20 weeks’ gestation. Placental histologic findings include chronic villitis with multinucleated giant cells, granulomatous inflammation, and fetal vessel occlusion.71 The diagnosis can be confirmed by immunohistochemistry.
Herpes Simplex
Herpes simplex virus (HSV) type 1 and 2 cause genital herpes. Direct contact with infected maternal secretions can lead to neonatal herpetic infection. The risks are greatest when a woman acquires a new infection (primary genital herpes) during late pregnancy, before the development of protective maternal antibodies. Most maternal infections are asymptomatic or go unrecognized and it may be difficult to distinguish clinically between primary and recurrent HSV infection. If primary genital herpes is present at the time of delivery and the baby is delivered vaginally, the risk of neonatal herpes is about 40%.72 Recurrent genital herpes does not pose a significant risk for the development of neonatal herpes infection. Occasionally, in utero infection of the fetus and placenta does occur, and morphologic placental features include giant cell change, usually in the decidua and extravillous trophoblast. There is often marked immaturity of the placenta with inflammatory changes in all placental areas. A heavy plasma cell infiltrate in the chorion should raise the suspicion of herpes, and there may be a chronic villitis with necrosis. Diagnosis is confirmed by any of immunohistochemistry, viral culture, or PCR.
HIV
Placentas from cases with fetal transmission of HIV-1 subtype E (the type most frequently seen in Southeast Asia) show chorioamnionitis, chronic deciduitis, and decidual necrosis.73 No specific features are seen.74 Histologic chorioamnionitis is the most common pathologic finding in placentas of HIV-1 infected women, and this may increase the risk of viral transmission to the fetus.75
Human Papillomavirus
Human Papillomavirus (HPV) is a DNA virus trophic for squamous epithelium, but may be found in the placenta. In vitro studies show that HPV can infect extravillous trophoblast and decrease extracellular matrix invasion.10 Infection with HPV was demonstrated in 6% of cases between 11 and 13 weeks’ gestation,76 and a higher rate of spontaneous abortion (60% vs 20% of controls) has been reported in HPV-positive women. HPV has been demonstrated in cord blood:77 the placentas were macroscopically normal, and no microscopic features of HPV infection have been described.
Toxoplasmosis
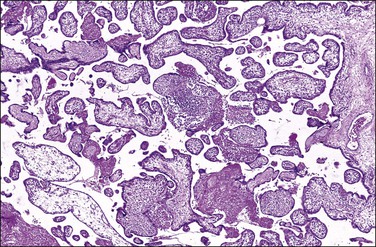
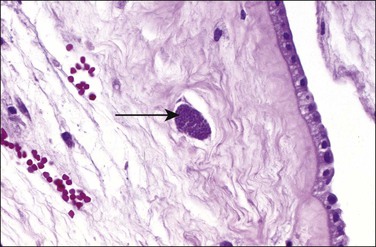
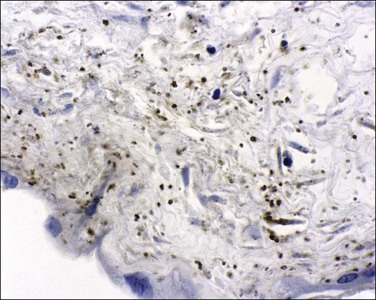
Toxoplasmosis, an infection caused by the protozoan parasite T. gondii, may be acquired by eating contaminated meat or from exposure to contaminated soil or cat litter. Immunocompetent individuals are usually asymptomatic. Acute infections in pregnant women can be transmitted to the fetus, although approximately 75% of congenitally infected newborns are asymptomatic.78 Consequences include mental retardation, blindness, and epilepsy, but, today, the classic triad of chorioretinitis, hydrocephalus, and cerebral calcifications is relatively rare. The risk of maternal–fetal transmission increases with gestational age at the time of exposure, whereas the incidence of severe disease decreases. If infection occurs just before or during the first trimester, it may cause miscarriage, intrauterine death, or severe neurologic lesions, whereas fetal infection occurring late in pregnancy may result in either congenital disease or a subclinical state. The classic finding in the placenta is villitis (Figure 33.20), but manifestations range from no sign of inflammation to necrotizing inflammation. Toxoplasma cysts may be seen in the cord and membranes (Figures 33.21 and 33.22), and fetal vessel calcification may be found.71 Toxoplasmosis is a rare cause of granulomatous villitis.79
Malaria
Pregnant women from endemic areas (especially primiparous women, who have more severe disease and significantly higher prevalence rates of malarial infection) are more prone to develop malaria than non-pregnant women. Infection in pregnancy is associated with extensive parasitic infection of the placenta, maternal anemia, a reduction in birth weight, miscarriage, preterm birth, and neonatal and maternal death.80
A mononuclear inflammatory infiltrate (intervillositis), malarial pigment, and an increase in perivillous fibrin is characteristic (Figure 33.23). In active infection, the parasitized cells attach to villi in areas of syncytiotrophoblast damage. Fibrinoid necrosis, basal membrane thickening, and increased numbers of syncytial knots are features also associated with malarial infection. Chronic infections show the most severe changes whereas placentas with acute infections exhibit a mild increase in inflammatory cells only and those with past infections show only minimal differences compared with noninfected placentas. A histologic grading system assessing the presence of pigment and inflammation in Plasmodium falciparum infection shows a correlation with birth weight.81 HIV co-infection will increase both viral and parasitic loads and decrease birth weight.
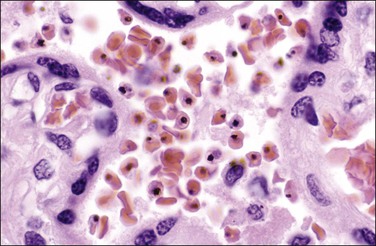
Syphilis
Grossly, the placenta may be pale, bulky, and hydropic, and necrotizing funisitis lends a ‘barber’s pole’ appearance to the cord. Classic gummas are rare today. The syphilitic ‘triad’ includes enlarged hypercellular (immature) villi, proliferative vascular changes (obliterative endarteritis), and acute or chronic villitis with plasma cells. Other features may be present, such as granulomatous, proliferative, and necrotizing villitis (Figure 33.24) with multinucleated giant cells. The decidua may have a plasma cell infiltrate. These findings are present to varying degrees in an affected placenta and are not specific for syphilis. In one cohort, necrotizing funisitis, villous enlargement, and acute villitis were significantly more common in both stillborn and liveborn infants with congenital syphilis. Placental examination improved the detection rate in both liveborn and stillborn infants.82
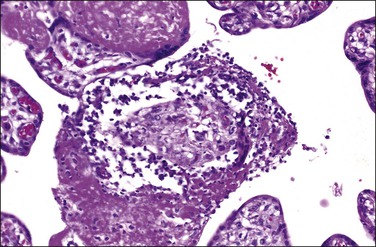
Diagnosis may be confirmed by PCR for treponemal DNA. In untreated cases, Warthin–Starry silver stain, or more recently introduced immunohistochemical methods for tissue in paraffin, may demonstrate spirochetes in placental and umbilical cord tissue. As with villitis of unknown etiology, the predominant inflammatory cells are maternal in origin and are CD8-positive T-lymphocytes.83
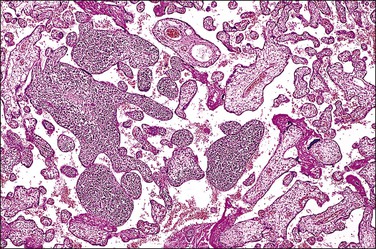
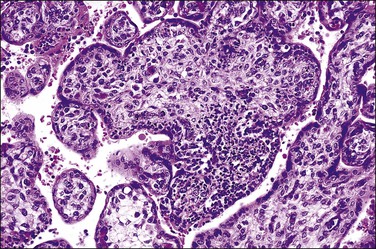
Listeriosis
This is due to infection with the bacterium L. monocytogenes. The placentas are frequently studded with microabscesses on their maternal and cut surfaces and usually have an associated chorioamnionitis. Histologically, there are multiple microabscesses with associated necrosis within the intervillous space and villi (Figures 33.25 and 33.26). Listeria may infect the placenta via the hematogenous and ascending routes. Although maternal infection produces little systemic upset, it causes devastating fetal sepsis with congenital pneumonia, gastritis, and esophagitis. Immunohistochemical84 or PCR detection of Listeria antigens may be useful in cases where culture has not been possible.
References
1. Redline, RW, Fay-Petersen, O, Heller, D, et al. Amniotic infection syndrome: nosology and reproducibility of placental reaction patterns. Pediatr Dev Pathol. 2003; 6:435–448.
2. Redline, RW, Boyd, T, Campbell, V, et al. Maternal vascular underperfusion: nosology and reproducibility of placental reaction patterns. Pediatr Dev Pathol. 2004; 7:237–249.
3. Redline, RW, Ariel, I, Baergen, RN, et al. Fetal vascular obstructive lesions: nosology and reproducibility of placental reaction patterns. Pediatr Dev Pathol. 2004; 7:443–452.
4. Turowski, G, Berge, LN, Helgadottir, LB, et al. A new, clinically oriented, unifying and simple placental classification system. Placenta. 2012; 33:1026–1035.
5. Philipp, T, Philipp, K, Reiner, A, et al. Embryoscopic and cytogenetic analysis of 233 missed abortions: factors involved in the pathogenesis of developmental defects of early failed pregnancies. Hum Reprod. 2003; 18:1724–1732.
6. Lathi, RB, Mark, SD, Westphal, LM, Milki, AA. Cytogenetic testing of anembryonic pregnancies compared to embryonic missed abortions. J Assist Reprod Genet. 2007; 24:521–524.
7. Toutain, J, Labeau-Gaüzere, C, Barnetche, T, et al. Confined placental mosaicism and pregnancy outcome: a distinction needs to be made between types 2 and 3. Prenat Diagn. 2010; 30:1155–1164.
8. Jindal, P, Regan, L, Fourkata, EO, et al. Placental pathology of recurrent spontaneous abortion: the role of histopathological examination of products of conception in routine clinical practice: a mini review. Hum Reprod. 2007; 22:313–316.
9. Nuovo, GJ, Cooper, LD, Bartholomew, D. Histologic, infectious, and molecular correlates of idiopathic spontaneous abortion and perinatal mortality. Diagn Mol Pathol. 2005; 14:152–158.
10. Gomez, LM, Ma, Y, Ho, C, et al. Placental infection with human papillomavirus is associated with spontaneous preterm delivery. Hum Reprod. 2008; 23:709–715.
11. Gruber, K, Gelven, PL, Austin, RM. Chorionic villi or trophoblastic tissue in uterine samples of four women with ectopic pregnancies. Int J Gynecol Pathol. 1997; 16:28–32.
12. Doyle, EM, McParland, P, Carroll, S, et al. The role of placental cytogenetic cultures in intrauterine and neonatal deaths. J Obstet Gynaecol. 2004; 24:878–880.
13. Burke, C, Sinclair, K, Cowin, G, et al. Intrauterine growth restriction due to uteroplacental vascular insufficiency leads to increased hypoxia-induced cerebral apoptosis in newborn piglets. Brain Res. 2006; 1098:19–25.
14. de Laat, MWM, van Alderen, ED, Franx, A, et al. The umbilical coiling index in complicated pregnancy. Eur J Obstet Gynecol Reprod Biol. 2007; 130:66–72.
15. Peng, HQ, Levitin-Smith, M, Rochelson, B, Kahn, E. Umbilical cord stricture and overcoiling are common causes of fetal demise. Pediatr Dev Pathol. 2006; 9:14–19.
16. Reddy, UM, Goldenberg, R, Silver, R, et al. Stillbirth classification—developing an international consensus for research: executive summary of a National Institute of Child Health and Human Development workshop. Obstet Gynecol. 2009; 114:901–914.
17. Ananth, CV, Wilcox, AJ. Placental abruption and perinatal mortality in the United States. Am J Epidemiol. 2001; 153:332–337.
18. Oyelese, Y, Ananth, CV. Placental abruption. Obstet Gynecol. 2006; 108:1005–1016.
19. Ananth, CV, Berkowitz, GS, Savitz, DA, Lapinski, RH. Placental abruption and adverse perinatal outcomes. JAMA. 1999; 282:1646–1651.
20. Bendon, RW. Review of autopsies of stillborn infants with retroplacental hematoma or hemorrhage. Pediatr Devel Pathol. 2011; 14:10–15.
21. Ananth, CV, Oyelese, Y, Prasad, V, et al. Evidence of placental abruption as a chronic process: associations with vaginal bleeding early in pregnancy and placental lesions. Eur J Obstet Gynecol Reprod Biol. 2006; 128:15–21.
22. Verma, RP, Kaplan, C, Southerton, K, et al. Placental histopathology in the extremely low birth weight infants. Fetal Pediatr Pathol. 2008; 27:53–61.
23. Steegers, EAP, von Dadelszen, P, Duvekot, JJ, Pijnenborg, R. Pre-eclampsia. Lancet. 2010; 376:631–644.
24. Roberts, JM, Hubel, CA. The two stage model of preeclampsia: variations on the theme. Placenta. 2009; 30(Suppl. A):S32–S37.
25. James, JL, Whitley, GS, Cartwright, JE. Pre-eclampsia: fitting together the placental, immune and cardiovascular pieces. J Pathol. 2010; 221:363–378.
26. Huppertz, B. IFPA Award in Placentology Lecture: biology of the placental syncytiotrophoblast—myths and facts. Placenta. 2010; 31(Suppl.):S75–S81.
27. Clausson, B, Gardosi, J, Francis, A, Cnattingius, S. Perinatal outcome in SGA births defined by customised versus population-based birthweight standards. BJOG. 2001; 108:830–834.
28. Hutter, D, Kingdom, J, Jaeggi, E. Causes and mechanisms of intrauterine hypoxia and its impact on the fetal cardiovascular system: a review. Int J Pediatr. 2010; 401323.
29. Surkan, PJ, Stephansson, O, Dickman, PW, Cnattingius, S. Previous preterm and small-for-gestational-age births and the subsequent risk of stillbirth. N Engl J Med. 2004; 350:777–785.
30. Scifres, CM, Nelson, DM. Intrauterine growth restriction, human placental development and trophoblast cell death. J Physiol (Lond). 2009; 587:3453–3458.
31. Kingdom, J, Huppertz, B, Seaward, G, Kaufmann, P. Development of the placental villous tree and its consequences for fetal growth. Eur J Obstet Gynecol Reprod Biol. 2000; 92:35–43.
32. Dicke, JM, Huettner, P, Yan, S, et al. Umbilical artery Doppler indices in small for gestational age fetuses: correlation with adverse outcomes and placental abnormalities. J Ultrasound Med. 2009; 28:1603–1610.
33. Nelissen, ECM, van Montfoort, APA, Dumoulin, JCM, Evers, JLH. Epigenetics and the placenta. Hum Reprod Update. 2010; 17:397–417.
34. McMinn, J, Wei, M, Schupf, N, et al. Unbalanced placental expression of imprinted genes in human intrauterine growth restriction. Placenta. 2006; 27:540–549.
35. Wilkins-Haug, L, Quade, B, Morton, CC. Confined placental mosaicism as a risk factor among newborns with fetal growth restriction. Prenat Diagn. 2006; 26:428–432.
36. Clark, SL, Hankins, GDV. Temporal and demographic trends in cerebral palsy–fact and fiction. Am J Obstet Gynecol. 2003; 188:628–633.
37. Strijbis, EMM, Oudman, I, van Essen, P, MacLennan, AH. Cerebral palsy and the application of the international criteria for acute intrapartum hypoxia. Obstet Gynecol. 2006; 107:1357–1365.
38. Redline, RW. Disorders of placental circulation and the fetal brain. Clin Perinatol. 2009; 36:549–559.
39. Wintermark, P, Boyd, T, Gregas, MC, et al. Placental pathology in asphyxiated newborns meeting the criteria for therapeutic hypothermia. Am J Obstet Gynecol. 2010; 203:579.e1–579.e9.
40. Keogh, JM, Badawi, N. The origins of cerebral palsy. Curr Opin Neurol. 2006; 19:129–134.
41. McDonald, DG, Kelehan, P, McMenamin, JB, et al. Placental fetal thrombotic vasculopathy is associated with neonatal encephalopathy. Hum Pathol. 2004; 35:875–880.
42. Redline, RW. Cerebral palsy in term infants: a clinicopathologic analysis of 158 medicolegal case reviews. Pediatr Dev Pathol. 2008; 11:456–464.
43. Elbers, J, Viero, S, MacGregor, D, et al. Placental pathology in neonatal stroke. Pediatrics. 2011; 127:e722–e729.
44. Shatrov, JG, Birch, SC, Lam, LT, et al. Chorioamnionitis and cerebral palsy: a meta-analysis. Obstet Gynecol. 2010; 116:387–392.
45. Genen, L, Nuovo, GJ, Krilov L. Davis, JM. Correlation of in situ detection of infectious agents in the placenta with neonatal outcome. J Pediatr. 2004; 144:316–320.
46. Maleki, Z, Bailis, AJ, Argani, CH, et al. Periventricular leukomalacia and placental histopathologic abnormalities. Obstet Gynecol. 2009; 114:1115–1120.
47. Higgins, M, McAuliffe, FM, Mooney, E. Clinical Associations with a Placental Diagnosis of Delayed Villous Maturation: a retrospective study. Pediatr Dev Pathol. 2011; 14:273–279.
48. Jauniaux, E, Burton, GJ. Villous histomorphometry and placental bed biopsy investigation in Type I diabetic pregnancies. Placenta. 2006; 27:468–474.
49. Mayhew, TM. Enhanced fetoplacental angiogenesis in pre-gestational diabetes mellitus: the extra growth is exclusively longitudinal and not accompanied by microvascular remodelling. Diabetologia. 2002; 45:1434–1439.
50. Higgins, M, Felle, P, Mooney, EE, et al. Stereology of the placenta in type 1 and type 2 diabetes. Placenta. 2011; 32:564–569.
51. Gielchinsky, Y, Rojansky, N, Fasouliotis, SJ, Ezra, Y. Placenta accreta—summary of 10 years: a survey of 310 cases. Placenta. 2002; 23:210–214.
52. van Beekhuizen, HJ, Joosten, I, de Groot, AN, et al. The number of multinucleated trophoblastic giant cells in the basal decidua is decreased in retained placenta. J Clin Pathol. 2009; 62:794–797.
53. Stanek, J, Drummond, Z. Occult placenta accreta: the missing link in the diagnosis of abnormal placentation. Pediatr Dev Pathol. 2007; 10:266–273.
54. Andrew, AC, Bulmer, JN, Wells, M, et al. Subinvolution of the uteroplacental arteries in the human placental bed. Histopathology. 1989; 15:395–405.
55. Decastel, M, Leborgne-Samuel, Y, Alexandre, L, et al. Morphological features of the human umbilical vein in normal, sickle cell trait, and sickle cell disease pregnancies. Hum Pathol. 1999; 30:13–20.
56. Heyborne, KD, Porreco, RP, Garite, TJ, et al. Improved perinatal survival of monoamniotic twins with intensive inpatient monitoring. Am J Obstet Gynecol. 2005; 192:96–101.
57. Tan, H, Wen, SW, Fung Kee Fung, K, et al. The distribution of intra-twin birth weight discordance and its association with total twin birth weight, gestational age, and neonatal mortality. Eur J Obstet Gynecol Reprod Biol. 2005; 121:27–33.
58. Hack, KE, Derks, JB, Elias, SG, et al. Increased perinatal mortality and morbidity in monochorionic versus dichorionic twin pregnancies: clinical implications of a large Dutch cohort study. BJOG. 2008; 115:58–67.
59. Redline, RW, Shah, D, Sakar, H, et al. Placental lesions associated with abnormal growth in twins. Pediatr Dev Pathol. 2001; 4:473–481.
60. Victoria, A, Mora, G, Arias, F. Perinatal outcome, placental pathology, and severity of discordance in monochorionic and dichorionic twins. Obstet Gynecol. 2001; 97:310–315.
61. Sato, Y, Benirschke, K. Increased prevalence of fetal thrombi in monochorionic-twin placentas. Pediatrics. 2006; 117:e113–e117.
62. Ong, SSC, Zamora, J, Khan, KS, Kilby, MD. Prognosis for the co-twin following single-twin death: a systematic review. BJOG. 2006; 113:992–998.
63. Joseph, KS, Huang, L, Liu, S, et al. Reconciling the high rates of preterm and postterm birth in the United States. Obstet Gynecol. 2007; 109:813–822.
64. Klebanoff, MA. Gestational age: not always what it seems. Obstet Gynecol. 2007; 109:798–799.
65. Nakling, J, Backe, B. Pregnancy risk increases from 41 weeks of gestation. Acta Obstet Gynecol Scand. 2006; 85:663–668.
66. Duff, P. A thoughtful algorithm for the accurate diagnosis of primary CMV infection in pregnancy. Am J Obstet Gynecol. 2007; 196:196–197.
67. Chow, SS, Craig, ME, Jacques, CF, et al. Correlates of placental infection with cytomegalovirus, parvovirus B19 or human herpes virus 7. J Med Virol. 2006; 78:747–756.
68. Satosar, A, Ramirez, NC, Bartholomew, D, et al. Histologic correlates of viral and bacterial infection of the placenta associated with severe morbidity and mortality in the newborn. Hum Pathol. 2004; 35:536–545.
69. Syridou, G, Spanakis, N, Konstantinidu, A, et al. Detection of cytomegalovirus, parvovirus B19 and herpes simplex viruses in cases of intrauterine fetal death: association with pathological findings. J Med Virol. 2008; 80:1776–1782.
70. Rogers, BB, Over, CE. Parvovirus B19 in fetal hydrops. Hum Pathol. 1999; 30:247.
71. Benirschke, K, Coen, R, Patterson, B, Key, T. Villitis of known origin: varicella and toxoplasma. Placenta. 1999; 20:395–399.
72. Brown, ZA, Selke, S, Zeh, J. The acquisition of herpes simplex virus during pregnancy. N Engl J Med. 1997; 337:509–515.
73. Bhoopat, L, Khunamornpong, S, Sirivatanapa, P, et al. Chorioamnionitis is associated with placental transmission of human immunodeficiency virus-1 subtype E in the early gestational period. Mod Pathol. 2005; 18:1357–1364.
74. Goldenberg, RL, Mudenda, V, Read, JS, et al. HPTN 024 study: histologic chorioamnionitis, antibiotics and adverse infant outcomes in a predominantly HIV-1-infected African population. Am J Obstet Gynecol. 2006; 195:1065–1074.
75. Al-Husaini, AM. Role of placenta in the vertical transmission of human immunodeficiency virus. J Perinatol. 2009; 29:331–336.
76. Weyn, C, Thomas, D, Jani, J, et al. Evidence of human papillomavirus in the placenta. J Infect Dis. 2011; 203:341–343.
77. Sarkola, ME, Grénman, SE, Rintala, MAM, et al. Human papillomavirus in the placenta and umbilical cord blood. Acta Obstet Gynecol Scand. 2008; 87:1181–1188.
78. Rorman, E, Zamir, CS, Rilkis, I, Ben-David, H. Congenital toxoplasmosis—prenatal aspects of Toxoplasma gondii infection. Reprod Toxicol. 2006; 21:458–472.
79. Yavuz, E, Aydin, F, Seyhan, A, et al. Granulomatous villitis formed by inflammatory cells with maternal origin: a rare manifestation type of placental toxoplasmosis. Placenta. 2006; 27:780–782.
80. Mens, PF, Bojtor, EC, Schallig, HDFH. Molecular interactions in the placenta during malaria infection. Eur J Obstet Gynecol Reprod Biol. 2010; 152:126–132.
81. Muehlenbachs, A, Fried, M, McGready, R, et al. A novel histological grading scheme for placental malaria applied in areas of high and low malaria transmission. J Infect Dis. 2010; 202:1608–1616.
82. Sheffield, JS, Sánchez, PJ, Wendel, GD, Jr., et al. Placental histopathology of congenital syphilis. Obstet Gynecol. 2002; 100:126–133.
83. Kapur, P, Rakheja, D, Gomez, AM, et al. Characterization of inflammation in syphilitic villitis and in villitis of unknown etiology. Pediatr Dev Pathol. 2004; 7:453–458.
84. Parkash, V, Morotti, RA, Joshi, V, et al. Immunohistochemical detection of Listeria antigens in the placenta in perinatal listeriosis. Int J Gynecol Pathol. 1998; 17:343–350.