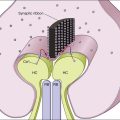
Chapter 37 Neuroprotection
History and definitions
Retinal ganglion cell glaucomatous disease
Glaucoma is a degenerative optic neuropathy characterized by loss of RGCs and their axons and warrants consideration under retinal neuroprotection strategies. Elevated IOP is a principal risk factor. However, recent evidence indicates that IOP control does not prevent glaucomatous progression in all patients and that vision loss continues in some susceptible individuals despite lowering the IOP. RGCs are terminally differentiated neurons and cannot regenerate by cell division. Optic nerve damage is irreversible. The main goal of neuroprotection is to keep these cells alive to augment the therapeutic benefit achieved by lowering the IOP. Neuroprotection in glaucoma offers a means to prevent the irreversible loss of RGC and ideally should be efficacious irrespective of the specific pathophysiology of the disease.1–3
Three decades of work have established the causal relationship between elevated IOP and the development of optic nerve damage through a series of clinical treatment studies supported by the National Eye Institute (NEI), including the Advanced Glaucoma Intervention Study trial, the Collaborative Initial Glaucoma Treatment Study Trial,4 and the Early Manifest Glaucoma Trial.5 However, glaucomatous optic neuropathy can occur despite having ocular pressures within a normal range. In some populations only half of glaucoma cases involve IOP elevated beyond the traditional demarcation of 20 mmHg pressure.6 Even in this condition of “low-tension glaucoma,” IOP remains a risk factor, as lowering the IOP further retards the loss of vision and presumptive retention of ganglion cells as monitored through nerve fiber volume in the optic nerve cup. This was shown in the Collaborative Normal Tension Glaucoma Study.7 Nevertheless, a number of individuals continue to suffer progressive glaucomatous optic nerve damage and vision impairment despite appropriate regulation of IOPs. It is these cases in particular that would benefit from an alternate strategy of neuroprotection, aimed at preventing RGC death.
Among the first studies to demonstrate pharmacologic modulation of neural damage in glaucoma was that of Bernard Becker and colleagues using oral administration of diphenylhydantoin (phenytoin).8 Diphenylhydantoin is an antiepileptic agent for controlling seizures but without the sedative effects of phenobarbital. It stabilizes the inactive state of voltage-gated sodium channels which could modulate glutamatergic transmission, and it is neuroprotective in a rat model with elevated IOP from cautery of episcleral veins.9 Diphenylhydantoin has a wide range of actions on neurons and primarily involves cell excitability, and the potential protective action on RGCs could be through ameliorating excitotoxic N-methyl-d-aspartate (NMDA) action on third-order retinal neurons. While diphenylhydantoin has numerous neurologic effects and has been tried as a central nervous system (CNS) neuroprotective agent in a number of clinical trials, it has consistently failed to meet clinical endpoints in trial.
Two neuroactive drugs that have antiexcitotoxicity action currently have Food and Drug Administration approval for CNS degenerative conditions, although clinical efficacy is limited. Riluzole extends life expectancy for amyotrophic lateral sclerosis patients by up to 3 months, and memantine is approved for advanced Alzheimer disease and provides some improvement in cognition and behavior.10 Riluzole blocks glutamatergic neurotransmission in the CNS by inhibiting release of glutamic acid, possibly through inactivation of voltage-dependent sodium channels.11 It may act at presynaptic NMDA receptors.12
Memantine is an antagonist at NMDA receptors13 and was originally developed as a dopaminergic compound derivative from amantadine, which has approval for treatment of Parkinson disease. Memantine has therapeutic benefit against excitoneurotoxicity mediated by action of NMDA receptors. A broad-spectrum blockade of NMDA channels would not be physiologically attractive. However, memantine does not compete with glutamate for the binding site. Rather, memantine binds only to open-state NMDA channels. It does not impede neuronal synaptic signal transmission in the physiological range, as it is competitive only for high levels of glutamate that lead to neuronal damage and death through excitotoxic mechanisms caused by excessive activation of NMDA receptors. The drug is well tolerated in treatment of Alzheimer dementia without apparent side-effects, suggesting that, when applied to physiologic neuronal states, it does not impede neuronal activity. Studies indicate that memantine decreases the level of free glutamate in the vitreous of a dog glaucoma disease model14 and in monkey and human glaucoma,15 although some of this work is not fully accepted. This provides a basis for considering NMDA receptor antagonists in a neuroprotection strategy for glaucoma.
Large-scale formal studies of memantine have been conducted involving patients with chronic glaucoma. In 2000–2006 Allergan ran a phase III randomized, placebo-controlled trial to evaluate the efficacy of memantine in 1179 patients aged 18–82 years old.4 Entry criteria required evidence of glaucomatous damage by examination of the optic nerve head accompanied by visual field loss. Subjects with all forms of open-angle and chronic angle closure glaucoma were entered, including having prior glaucoma filtration surgery. Two treatment arms had different doses of memantine versus an oral placebo for glaucoma patients with otherwise controlled IOP. Visual fields were monitored every 6 months for evidence of progression as the outcome measure. Unfortunately, the trial failed to meet endpoint significance versus placebo control. As the outcome has not been formally submitted for publication, information on the outcome is limited, but the memantine trials demonstrated statistically significant visual field benefit under high dose conditions compared to lower dose, although the overall trial significance failed at efficacy compared to control.4 Precise reasons for failure are not understood, but it may have had to do with study design and handling of data acquisition at the point that a subject reached the clinical endpoint. The medical glaucoma field, nonetheless, remains optimistic that the strategy of neuroprotection for RGC degenerative disease remains important for study and will ultimately yield compounds that can augment lowering of IOP in glaucoma.1,5
Neurotrophic factors in glaucoma
Recent studies suggest that neurotrophic factors may have therapeutic benefit for individuals with advanced glaucoma. Lambiase and colleagues2 demonstrated that application of nerve growth factor (NGF) in glaucomatous rats protects the RGCs from apoptotic cell death. Rats were made glaucomatous through injection of hypertonic saline into the episcleral vein, and NGF was applied topically in one of two doses (100 and 200 µg/mL). Cell biology studies indicated that molecules upstream of the apoptotic pathway, including Bax and BCL-2, were lower in treated animals, and morphometric analysis showed a significant preservation of RGC counts in the treated eyes.
These investigators then initiated a pilot human study of 3 patients with advanced glaucoma, who underwent careful electrophysiologic and psychophysical visual testing at baseline and after topical NGF eye drops applied four times daily for 3 months.2 The subjects were evaluated by multiple parameters, including visual acuity, contrast sensitivity, pattern electroretinogram (ERG), and visual-evoked potentials, along with optic disc photography. While this study was too small to provide a definitive outcome, several of these test parameters indicated improved retinal function at the level of ganglion cells, consistent with inhibiting RGC apoptosis observed in the glaucomatous rats by NGF.
The pharmaceutical industry is considering other neurotrophic factors including ciliary neurotrophic factors (CNTF) for glaucoma rescue. Van Adel and colleagues have demonstrated that continuous administration of CNTF protects RGCs in animal models.3 Neurotech has initiated preclinical studies using its encapsulated cell technology (ECT) devices in which epithelial cells transfected with the CNTF gene are sequestered and release low but therapeutic levels for months’ to years’ duration. Efficacy of other neurotrophic factors for RGC rescue has been demonstrated,16 such as brain-derived neurotrophic factor (BDNF) which can protect RGCs following optic nerve crush in cat by intravitreal injection.17 BDNF also promotes regeneration of spinal cord axons following axotomy.18
Neuroprotection through the serotonin pathway
A promising area of neuroprotection research involves the serotonin (5HT) pathway. Memantine, in addition to its direct antiexcitotoxic effects as an NMDA receptor antagonist, is a noncompetitive antagonist at the 5HT3 receptor19 which may be part of the memantine neuroprotection mechanism for Alzheimer disease.20 Recent literature indicates that serotonergic compounds, such as the selective serotonin reuptake inhibitor antidepressant agents, potentiate neurite sprouting by NGF.21 The retina has 5HT receptors, but whether this putative mechanism has relevance to retinal and/or glaucomatous disease is not currently known.
Agonists that stimulate the 5HT system are neuroprotective for RGCs.22 Unfortunately, 5HT agonists have hallucinogenic action, and broad-action agonists at 5HT receptors would not be suitable for human glaucoma treatment. Among the interesting compounds in this regard is an indazole compound AL-34662 that is a selective 5HT receptor agonist at 5-HT-2A receptors and that lowers ocular pressure but which does not cross the blood–brain barrier.23
Neurotrophic factors for retinitis pigmentosa
Roy Steinberg and Matthew LaVail and their group24 made the seminal observation that neurotrophic factors could rescue photoreceptor cell death in an animal model of retinitis pigmentosa (RP). This observation in 1990 changed the face of research in photoreceptor neurodegenerative disease. Steinberg and LaVail and their colleagues showed that a single intraocular application of basic fibroblast growth factor (bFGF) delayed the loss of rod photoreceptors in the Royal College of Surgeons (RCS) rat. The rescue of the rods was panretinal, with a gradient of rescue highest near the injection site. This observation was subsequently extended to other genetic and environmental causes of photoreceptor cell death, including the light exposure damage model,25 in which the protection was shown to extend to other cytokines and neurotrophic factors beyond bFGF alone. These observations occurred during a time of great explosion of the field of photoreceptor degeneration research, including the laboratory creation of genetic models, including the P23H rhodopsin transgenic rat that mimicked human RP.26,27 Multiple neurotrophic factors have subsequently been found to rescue the rod cells in this rodent RP model.25
Exploration of this rescue proceeded along multiple lines, including cell biology of mechanisms,28,29 and by multiple delivery approaches, including application using gene delivery with adeno-associated virus (AAV) vector.30 Experiments with control animals subjected to mechanical injury by insertion of a “dry” needle into the retina31 also showed a surprising rescue of the rod photoreceptors. It was soon recognized that mechanical injury itself led to upregulation of expression of bFGF and CNTF. The precise mechanism of rescue was studied intensely but remained difficult to elucidate, and it is currently believed that the rescue is indirect and mediated through retinal glial cells which have receptors for neurotrophic factors, whereas the adult rods do not.28 Several of the factors activate the Jak-STAT pathway.28 Both NGF29 and CNTF30 were shown in other animal models to slow down photoreceptor loss quite significantly. NGF is the original neurotrophic factor identified in the 1950s by Rita Levi-Montalcini,32,33 who subsequently received the Nobel Prize for demonstrating that NGF acts on morphological differentiation of neuronal crest-derived cells and on peripheral and CNS neuronal cells. Cayouette and colleagues34 demonstrated that CNTF rescued the retinal degeneration slow (RDS) genetic mouse model of RP when applied by intraocular gene transfer that led to prolonged and stable CNTF expression in retina. They also showed that CNTF provided functional rescue of ERG potentials deriving from photoreceptor and bipolar cells and caused significant increase of the a-wave and b-wave of the rod-driven scotopic ERG.
CNTF protein and historical selection
CNTF was first identified by Ruben Adler et al. as a factor in chick embryo extract that could support survival of chick ciliary neurons.35 The factor was subsequently purified from the chick eye.36 CNTF is a small 200-amino-acid 22-kDa protein which assembles into an α-helical tertiary structure. Adler37 and McDonald et al.38 subsequently proposed that CNTF expression is upregulated as a stress factor and released from cells, although the mechanism is not understood as the protein lacks a secretion sequence.39
Proteins interact with cells through a cellular receptor. The CNTF receptor is a trimeric construct that includes the CNTF α-receptor, the leukemia inhibitory factor receptor-β (LIFR-β), and gp130 component.40 The CNTFRα soluble factor has been detected in cerebrospinal fluid and, consequently, a cell itself needs to express only LIFR-β and gp130. Subsequent binding of CNTF to this trimeric complex activates the Jak-STAT pathway41 leading to gene transcription.42 Ultimately, it is the upregulation of gene expression by CNTF that appears to be a crucial factor in human clinical trials of rod and cone rescue in photoreceptor degenerative diseases.
The effect of CNTF on nuclear activity was seen by AAV delivery of CNTF to RDS mice with peripherin mutation leading to slow death of rod photoreceptors.43 There were several seminal observations in this study, including a further demonstration that vector-mediated gene therapy provided long-term and panretinal rescue of rod photoreceptors following a single intraocular application. Second, this particular study showed a potentially adverse consequence of CNTF, as ERG response amplitudes were diminished in the treated eyes compared to the fellow uninjected eyes. Third, the rod cell nuclei showed an increase in euchromatin and an increase in the size, resulting in increased thickness of the outer nuclear layer (ONL), which is composed exclusively of photoreceptor cell bodies containing the nuclei. They concluded that CNTF upregulated gene expression in the photoreceptor cell either directly or, more likely, indirectly. This interpretation was quickly replicated in the rabbit retina by application of CNTF using a novel delivery mode of encapsulated cell-based intraocular devices (the Neurotech ECT)44 (Fig. 37.1). These devices release a continuous low level of CNTF protein into the vitreous, and hence, to the retina over weeks and months. Following implantation of ECT devices into the eye of normal rabbit, without retinal degeneration, the nuclear morphology changes, including dispersion of the nuclear chromatin, were consistent with DNA uncoiling that occurs during gene expression. The retinal function, as monitored by ERG recordings, was not affected, although high-dose CNTF suppressed the ERG for low-intensity stimuli. Cayouette et al. found that CNTF could augment retinal ERG function,34 and Bush et al. observed44 that CNTF did not adversely affect retinal function in lower dose, whereas the studies of Bok and colleagues43 note that high-dose CNTF expression with a vigorous chick β actin promoter suppressed retinal ERG function: these observations are consistant with CNTF having a safe biological range, beyond which it may cause potential retinal toxicity. A dose-ranging study in the rod–cone dystrophy 1 (RCD1) dog model of RP indicated that the ECT delivery approach spanned a sufficiently broad range of dosing that warranted safety study in a phase I human clinical trial for RP.
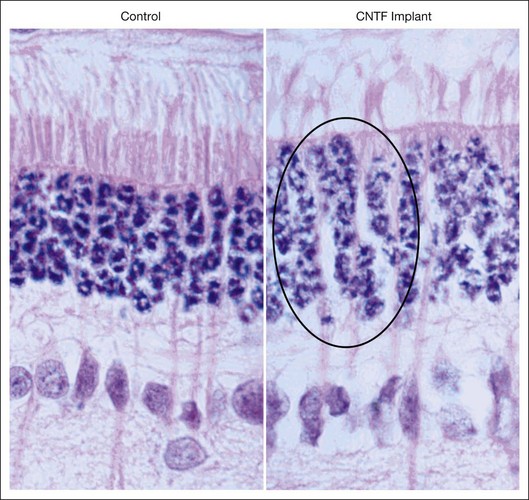
CNTF phase I trial for human photoreceptor degeneration
A human phase I trial of CNTF for RP neurodegenerative diseases was conducted and reported in 2006 using the Neurotech ECT device for intraocular production and delivery of CNTF protein.45 The device was surgically implanted in the eye of 10 study participants with advanced RP that severely limited their overall vision, including severe degradation of visual acuity (Fig. 37.2). The devices were implanted through a small pars plana incision which was closed by a single suture, causing minimal trauma to the eye, with the exception of one participant who had a transient choroidal effusion that resolved without ultimate reduction of baseline acuity. The devices were surgically explanted at the 6-month study conclusion. The initial 5 subjects received lower-dose implants delivering 0.28 ± 0.07 ng/day. The subsequent 5 participants received implants delivering 1.53 ± 0.54 ng/day, or about fivefold higher dose. The CNTF release levels were evaluated following explant. As this was a phase I safety study, the primary endpoints were safety parameters. No ocular or systemic complications ensued that met predetermined protocol adverse outcomes. The transfected cells derived from a human cultured retinal pigment epithelium (RPE) cell line. No serum antibodies to either CNTF or the transfected cells were detected in participants.
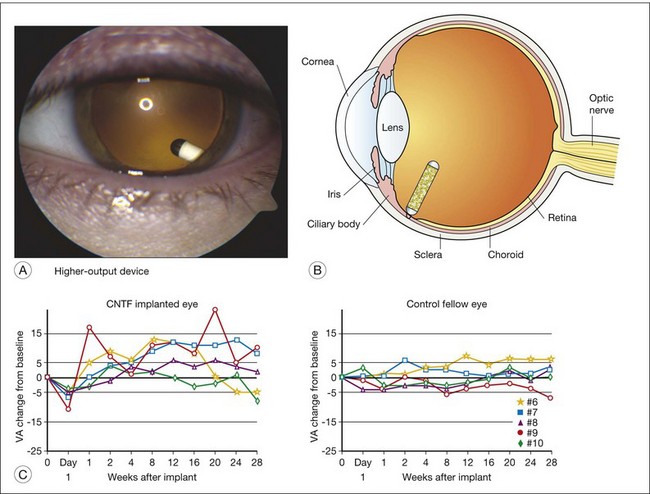
We considered the possibility of true biological rescue by CNTF and proposed a hypothesis of plausible action, as follows: visual acuity requires both close spacing of many photoreceptors and that these photoreceptors can functionally respond to light stimulation. We considered it unlikely that CNTF could “repopulate” the retina with new photoreceptors, but rather that CNTF might restore minimal function in cone photoreceptors that were present but were functionally dormant. Photoreceptors certainly can survive temporarily, even with no outer segments, in the rhodopsin knockout mouse model.46 These rod cells cannot respond to light in the absence of photopigment in the outer segments, and ultimately they die by approximately 90 days from birth. Second, both Bok et al.43 and Bush et al.44 generated evidence that CNTF elicited transcriptional activity in photoreceptor cells, consistent with CNTF stimulating higher metabolic output of photoreceptors. Finally, it is well understood that even relatively few cone photoreceptors can support some degree of visual discrimination, as deduced from retinal histology of postmortem eyes of individuals with RP and through psychophysical experiments that modeled photoreceptor loss.47 This chain of logic can be summarized as follows: first, photoreceptors become inactive before they die and go into a quiescent state and do not contribute to visual perception. Second, CNTF promotes photoreceptor transcriptional activity and metabolic function of cells, including elaboration of outer-segment material that supports photon capture and vision. Third, very few cone photoreceptors are required for a minimal level of visual acuity. Under these conditions, CNTF might partially restore visual function during a late stage of degeneration. This effect would be separate from CNTF preservation of photoreceptors in early stages of degeneration which was seen in at least 13 different animal models of degeneration, including phosphodiesterase (PDE) 6β mice, RDS peripherin mice, transgenic rats expressing P23H or S334ter mutant rhodopsin, RDY cat, RCD1 dog, rhodopsin knockout mice, and Q334ter rhodopsin transgenic mice. A study of CNTF in the S334ter rhodopsin transgenic rat model demonstrated regeneration of cone outer segments, consistent with the postulate of the CNTF phase I study group that CNTF may benefit vision by partially restoring or augmenting cone function.
Based on the successful safety study of CNTF protein released by encapsulated cell technology implants, phase II studies were developed to evaluate possible efficacy for age-related macular degeneration (AMD) involving geographic atrophy and for RP (David Birch, Retinal Foundation of the Southwest; Rafael Caruso, NEI/NIH; Paul Sieving, NEI/NIH; Weng Tao, NeuroTech; Santa Tumminia, NEI/NIH; Ronald Bush, NIDCD/NIH; and Richard Weleber, Casey Eye Instititue-OHSU). The scientific hypotheses were, first, whether one could demonstrate that CNTF slowed neurodegeneration and prolonged vision, and second, whether CNTF improves photoreceptor function in human RP. The CNTF-2 study for AMD trial had a principal outcome of visual acuity after 12 months’ treatment.48 The CNTF-3 and CNTF-4 were both for RP, with principal outcomes of visual acuity outcome at 12 months,49 or visual field sensitivity at 24 months,50 respectively. The trials involved about 60 subjects each, with asymmetric design, with 20 subjects receiving lower-dose implants and 40 subjects receiving higher-dose implants. All studies were fully enrolled.
The AMD trial (CNTF 2) reported results in 2011.51 No serious adverse events related to the CNTF implant or to surgical procedures occurred during the study. Biological activity of the implant was indicated by an increase of retinal thickness on optical coherence tomography for both the lower-dose and higher-dose CNTF groups, but not in the sham control eyes. Unfortunately, visual acuity outcome did not reach significance for the endpoint, but the higher-dose group showed a trend towards stabilization of best corrected visual acuity (BCVA) at 12 months compared to sham treatment (P = 0.078). Neither the ERG amplitudes nor Humphrey visual field sensitivity showed significant change, either for better or worse.49
New technology for endpoints for photoreceptor degenerations
In considering future trials involving disease that causes demise of rod or cone photoreceptors, the selection of clinical endpoints to demonstrate significant rescue has received intense consideration. Psychophysical measures of visual acuity, visual sensitivity, or extent of visual field appear to be insensitive measures, as they persist even after objective measures of ERG amplitudes are greatly reduced. A new potential endpoint involves adaptive optics scanning laser ophthalmoscopy, in which cone photoreceptors can be imaged in vivo by sharpening image clarity upon correction of optical aberrations inherent in the lens and cornea. Talcott et al.52 demonstrated the ability to image and count the cones in the foveal region of the human eye during CNTF treatment of 2 subjects. They found significant relative preservation of cone photoreceptors in the CNTF-treated eyes. While this approach currently is extremely labor-intensive, it may afford the best opportunity to track the natural history of photoreceptor loss by neuroprotective treatment trials in these slowly progressing diseases.
Delivery of neurotrophins
Neurotrophic factors are proteins of substantial size, and controlled delivery to target ocular tissues presents a formidable challenge. None of the neurotrophic factors are particularly amenable to crossing the blood–retinal barrier, nor can they be administered from outside the eye and achieve substantial molecular flow to intraocular tissue. While delivery of neuroprotective factors will also be considered in a separate section of this chapter, some mention is warranted here of the major approaches to delivery. Direct delivery of CNTF protein by intravitreal injection has worked well in rodent models. This approach has major limitations, however. The first is the limited half-life of therapeutic effect. A single intravitreal application in the rat eye of CNTF has a rapid clearance time. Despite this, the effect is longer-lasting than expected, following a single bolus injection, indicating that CNTF acts through a secondary mechanism, possibly by upregulating gene expression in glial cells which release some currently unknown protective factor. Persistence of treatment duration can be extended by repeated injections, as is done for anti-vascular endothelial growth factor (VEGF) compounds to treat neovascular AMD.53,54 However, even such heroic approaches are currently conducted only for 12–18 months, whereas neuroprotection for retinal photoreceptor degenerative diseases would need to continue for decades. Further, injecting a large bolus application of CNTF compound impacts retinal function and decreases the ERG amplitudes,43 whereas low-dose continuous CNTF release had the possibility of augmenting rod cell function.34
An alternative promising approach is ECT, in which low-dose protein is released continuously into the vitreous cavity following surgical implantation of the encapsulated cells transfected with the gene of choice, as used for the CNTF phase I clinical trial.45 Current indications from Neurotech are for continuous release in therapeutic quantity beyond 24 months duration. While surgical insertion of the device is a potential limitation, conversely, it is attractive to be able to remove the device by surgical explant if it is necessary to terminate the drug application.
Vector-mediated gene transfer by intravitreal or subretinal application also holds the potential for long-term chronic therapy. While adenovirus-mediated transfer has limited duration of expression of only many months, AAV vectors are capable of maintaining chronic release over the course of many years, as seen for the RPE65 trial in the dog.55 The current limitations of AAV vector-mediated transfer involve potential immunogenicity, potential for mutagenesis, and a current lack of regulation of gene expression level, including genetic control to terminate protein production. Quite likely, these limitations will be overcome in the future.
Antioxidants
Oxidative damage in light-induced and inherited photoreceptor degenerations
Absorption of light (the normal function of photoreceptor outer segments) increases oxidation of their lipids, creating morphological and functional damage as light exposure is increased.56–58 Both death and damage appear to be caused by oxidative stress, i.e., by the damaging effects of partially reduced forms of oxygen, often called reactive oxygen species. The idea that light-induced damage is caused by oxidative stress is supported by evidence that levels of endogenous antioxidants increase following light damage,58–60 and that exogenous antioxidants are protective60–67 for cones68,69 as well as rods.70–72 The stress-induced death of photoreceptors is accompanied by damage to the survivors.70,72
In several inherited photoreceptor degenerations the starting event is a mutation that leads to the death of rod photoreceptors. After the death of rods, the major consumers of oxygen in the retina, the tissue oxygen level in the outer retina is substantially increased,73 activating nicotinamide adenine dinucleotide phosphate (NADPH) oxidase and inducing accumulation of superoxide radicals.74 Superoxide radicals attack macromolecules, causing oxidative damage and production of other molecules such as nitric oxide which generates peroxynitrite, a particularly damaging free radical that amplifies the damage.75 Over time the antioxidant defense systems and repair mechanisms in cones cannot compensate for damage, leading to severe cell damage and apoptosis. According to cone density distribution, cone dysfunction and death are expected to occur first in the periphery and then spread posteriorly, a process that is common to many inherited dystrophies. The generation of free radicals is relentless and, thus, any treatment directed at preserving cones must be provided continuously for the patient’s entire life. The repair mechanisms can protect from a limited amount of damage. The aim of antioxidant treatment is to reduce the rate of oxidative damage below the rate of repair in order to prevent dysfunction and apoptosis. To determine if this can be achieved, long-term experiments are required. Alternatively, evidence of protection could be provided in a shorter time length by the rescuing effect of a candidate drug on damaged but still viable cells (Figs 37.1 and 37.2).
Preclinical evidence of antioxidant protection in photoreceptor degenerations
Inhibitors of NADPH oxidase (Nox)
Usui et al.74 investigated the hypothesis that Nox and/or xanthine oxidase serve as critical intermediaries between increased tissue oxygen and the generation of excessive reactive oxygen species that cause oxidative damage to cones. Apocynin, a blocker of Nox, but not allopurinol, a blocker of xanthine oxidase, markedly reduced the superoxide radicals in the outer retina of the retinal degeneration-1 (rd1+/+) model of RP. Compared to rd1+/+ mice treated with vehicle, those treated with apocynin, but not those treated with allopurinol, had significantly less oxidative damage in the retina. Apocynin-treated, but not allopurinol-treated, rd1+/+ mice had preservation of cone cell density, increased mRNA levels for M- and S-cone opsin, and increased mean photopic b-wave amplitude. In Q344Ter mice, a model of dominant RP in which mutant rhodopsin is expressed, apocynin treatment preserved photopic ERG b-wave amplitude compared to vehicle-treated controls. These data indicated that Nox, but not xanthine oxidase, plays a critical role in the generation of oxidative stress leading to cone cell death in RP. Inhibition of Nox provides a new treatment strategy.
Nitric oxide synthase (NOS) inhibitors
Recent studies by Komeima et al.75 have shown that treatment of rd1 mice with a mixture of NOS inhibitors significantly increased cone survival, indicating that NO-derived peroxynitrite contributes to cone cell death. Treatment with 7-nitroindazole, a specific inhibitor of neuronal NOS, also significantly reduced cone cell death, but aminoguanidine, an inhibitor of inducible NOS, did not. These data suggest that NO generated by neuronal NOS exacerbates oxidative damage to cones in RP, and that combined therapy to reduce NO and oxidative stress should be considered.
Bolstering the endogenous antioxidant defense system
In another study by the same group,76 genetically engineered rd10 mice with either inducible expression of superoxide dismutase (SOD) 2, catalase, or both in photoreceptor mitochondria were generated. Littermates with the same genetic background that did not have increased expression of SOD2 nor catalase served as controls. Coexpression of SOD2 and catalase, but not either alone, significantly reduced oxidative damage in the retinas of postnatal day (P) 50 rd10 mice, as measured by protein carbonyl content. Cone density was significantly greater in P50 rd10 mice with coexpression of SOD2 and catalase together than rd10 mice that expressed SOD2 or catalase alone, or expressed neither. Coexpression of SOD2 and catalase in rd10 mice did not slow rod cell death. These data supported the idea of bolstering the endogenous antioxidant defense system as a gene-based treatment strategy for RP, and also indicated that coexpression of multiple components may be needed for effective neuroprotection.
Following the same line of research, Lu et al.77 explored the strategy of overexpressing components of the endogenous antioxidant defense system to combat oxidative damage in RPE cells and retina. In transfected cultured RPE cells with increased expression of SOD1 or SOD2, there was increased constitutive and stress-induced oxidative damage measured by the level of carbonyl adducts on proteins. In contrast, RPE cells with increased expression of glutathione peroxidase 1 (Gpx1) or Gpx4 did not show an increase in constitutive oxidative damage. An increase in Gpx4, and to a lesser extent in Gpx1, reduced oxidative stress-induced RPE cell damage. Coexpression of Gpx4 with SOD1 or 2 partially reversed the deleterious effects of the SODs. Transgenic mice with inducible expression of Gpx4 in photoreceptors were generated, and in three models of oxidative damage-induced retinal degeneration, increased expression of Gpx4 provided strong protection of retinal structure and function. These data suggest that gene therapy approaches to augment the activity of Gpx4 in the retina and RPE should be considered in patients with RP or AMD.
Carotenoids (lutein, zeaxanthin) in combination with other antioxidants
Miranda et al.78 have shown that the use of a combination of antioxidants delayed the degeneration process in rd1 mouse retina. In an effort to understand the mechanism of action of these substances (zeaxanthin, lutein, α-lipoic acid, glutathione, and Lycium barbarum extract) the changes in the levels of several proteins and oxidative stress markers in the rd1 retina were studied. The treatment increased glutathione peroxidase activity and glutathione levels and decreased cystine concentrations in rd1 retinas. Considering all the results obtained from treated and untreated animals, a high correlation was present between glutathione concentration and glutathione peroxidase activity, and there was a negative correlation between glutathione retinal concentration and the number of TUNEL-positive cells. No difference was observed between the numbers of nNOS and NADPH diaphorase-positive cells in treated and untreated rd1 mice. Thiol contents and thiol-dependent peroxide metabolism seem to be directly related to the survival of photoreceptors in rd1 mouse retina.
Rac1
Rac1 is a component of NADPH oxidase that produces reactive oxygen species.79 Rac1 is expressed abundantly in mammalian retinal photoreceptors, where it is activated in response to light stimuli.80 Knocking down Rac1 expression, by conditional gene targeting, in mouse rod photoreceptors resulted in protection against light-induced photoreceptor death compared with wild-type (WT) littermates.81 A similar protective effect on rods was also found by using apocynin, which inhibits NADPH oxidase activity. These results implicate both neuronal Rac1 and NADPH oxidase in cell death in this model of CNS degeneration. Diminished Rac1 expression in mouse rods had no effect on retinal structure or function examined by light microscopy, electron microscopy, rhodopsin measurement, ERG activity, and visual acuity, indicating that rod outer-segment morphogenesis proceeds normally in Rac1 conditional knockout mice. The lack of structural or functional effect of Rac1 depletion on photoreceptors, but protection under conditions of stress, indicates that the Rac1 pathway warrants exploration as a target for therapy in retinal neurodegenerative diseases.
Rod-derived cone viability factor
A new signaling molecule that represents a potential therapy for these currently untreatable diseases has been identified over the past 12 years.82 This protein, called rod-derived cone viability factor (RdCVF), maintains the function and consequently the viability of cone photoreceptor cells in the retina; mice that lack this factor exhibit a progressive loss of photoreceptor cells. The gene encoding RdCVF (Nucleoredoxin-like (Nxnl1) gene) also encodes, by differential splicing, a second product that has characteristics of a thioredoxin-like enzyme and protects both photoreceptor cells and, more specifically, its interacting protein partner, the tau protein, against oxidative damage. This signaling pathway potentially links environmental insults to an endogenous neuroprotective response. The Nxnl1 gene and, most likely, the paralogous gene Nxnl2 are part of a newly discovered redox signaling pathway that involves an enzymatic product and a trophic factor, both encoded by the same gene. It has been suggested82 that this therapy, by restoring a physiological signal, may efficiently treat the effects of a broad range of RP mutations. The delivery of RdCVF into the patient’s eyes can be achieved through different routes, by injection of the protein into the eye, expression from viral vectors, or delivery of RdCVF-producing cells that are encapsulated in a semipermeable membrane to avoid attack by the immune system. RdCVF is now in translation into a possible therapeutic agent to treat a spectrum of degenerative eye diseases.
N-acetylcysteine
Very recently, Lee et al.83 determined whether orally administered N-acetylcysteine (NAC) reduced cone cell death and preserved cone function by reducing oxidative damage in two models of RP, rd1 and rd10 mice. In rd10 mice, supplementation of drinking water with NAC promoted partial maintenance of cone structure and function for at least 6 months. Topical application of NAC to the cornea also reduced superoxide radicals in the retina and promoted survival and functioning of cones. Since oral and/or topical administration of NAC is feasible for long-term treatment in humans, and NAC has a good safety profile, it is reasonable to consider clinical trials to evaluate the effects of prolonged treatment with NAC in patients with RP.
Saffron (Crocus sativus extract)
Among various antioxidants, the neuroprotective potential of the ancient spice saffron was explored.84 To test whether the saffron extract (Crocus sativus) given as a dietary supplement counteracts the effects of continuous light exposure in the albino rat retina, Sprague–Dawley rats were pre-fed either saffron or beta-carotene (1 mg extract/kg/day) before they were exposed to bright continuous light (BCL) for 24 hours. Flash ERG amplitudes, the thickness of the ONL, and the amount of apoptotic figures in the ONL were the main outcome variables. The photoreceptor layer was largely preserved in saffron-treated animals as was the ERG response. In addition, the rate of photoreceptor death induced by BCL appeared to be drastically reduced in treated animals. In beta-carotene prefeeding experiments, morphologic analysis showed preservation of the ONL similar to that obtained with saffron prefeeding, whereas the ERG response was unrecordable. Western blot analysis showed that exposure to light induced a strong upregulation of fibroblastic growth factor in control and beta-carotene-treated rats, but no change was noted in saffron-treated rats. These results showed that saffron may protect photoreceptors from retinal stress, maintaining both morphology and function and probably acting as a regulator of programmed cell death. The stigmata of C. sativus contain powerful antioxidants (crocin, crocetin) in biologically high concentrations85; their multiple C=C bonds give the stigmata their color, fragrance, taste, and antioxidant potential. To identify the genes and noncoding RNAs (ncRNAs) involved in the neuroprotective actions of saffron, Natoli et al.86 used a standardized assay of photoreceptor damage, in which albino Sprague–Dawley rats raised in dim cyclic illumination (12 hours 5 lux, 12 hours darkness) were challenged by 24 hours’ exposure to bright (1000 lux) light. Experimental groups were protected against light damage by pretreatment with dietary saffron (1 mg/kg/day for 21 days). RNA from the eye of exposed and unexposed animals was hybridized to Affymetrix rat genome ST arrays. Quantitative real-time polymerase chain reaction analysis of 14 selected genes was used to validate the microarray results. Light damage caused the regulation of 175 entities (genes and noncoding, ncRNAs) beyond criterion levels (P < 0.05 in comparison with controls, fold change >2). Saffron pretreatment reduced the expression of 53 entities. Saffron pretreatment regulated 122 entities not regulated by light damage. Saffron, given without light damage, regulated genes and ncRNAs beyond criterion levels, but in lesser numbers than during their protective action. A high proportion of the entities regulated by light damage (>90%) were known genes. By contrast, ncRNAs were prominent among the entities regulated by saffron in their neuroprotective roles (73% and 62%, respectively). Given before retinal exposure to damaging light, thus exerting its neuroprotective action, saffron regulated a large number of entities, among which ncRNAs were prominent.
Nanoceria
Cerium oxide nanoparticles, nanoceria, are inorganic antioxidants that have catalytic activities which mimic those of the neuroprotective enzymes SOD and catalase. Kong et al.87 have shown that nanoceria preserves retinal morphology and prevents loss of retinal function in a rat light damage model. The homozygous tubby mutant mouse, which exhibits inherited early progressive cochlear and retinal degeneration, was used as a model to test the ability of nanoceria to slow the progression of retinal degeneration.87 The results showed that nanoceria can protect the retina by decreasing reactive oxygen species, upregulating the expression of neuroprotection-associated genes; downregulating apoptosis signaling pathways and/or upregulating survival signaling pathways to slow photoreceptor degeneration. These data suggest that nanoceria has significant potential as global agents for the therapeutic treatment of inherited retinal degeneration and most types of ocular disease.
Clinical evidence of antioxidant protection in photoreceptor degenerations
Age-related eye disease study (AREDS)
The AREDS represents without doubt a milestone in the field of nutritional supplementation for AMD. Sponsored by the NEI and published in 2001,88 AREDS was indeed the first large-scale, randomized and controlled clinical trial to demonstrate the protective role of high doses of vitamins and zinc on the progression of AMD and visual loss. AREDS was initially conceived as a long-term multicenter, prospective study designed to assess the clinical course, prognosis, and risk factors of both AMD and cataract. However, the widespread use and marketing of antioxidants and zinc in eye-targeted formulations, not supported by consistent scientific evidence, led the researchers to incorporate a clinical trial as part of the study, to evaluate the effect of high doses of zinc and selected antioxidant vitamins on the development of advanced AMD in a cohort of older persons. A total of 3641 participants, aged 55–80, were included in the study, and randomly selected to receive daily oral tablets for one of four treatments: (1) zinc alone; (2) antioxidants alone; (3) a combination of antioxidants and zinc; and (4) placebo. The antioxidant formulation consisted of 500 mg vitamin C, 400 IU vitamin E and 15 mg beta-carotene. The specific daily amount of zinc was 80 mg as zinc oxide (formulations that included zinc also had 2 mg copper oxide added to offset potential zinc-induced copper-deficiency anemia). It should be noted that the formula was a type of active treatment, and therefore the dosages of ingredients were much higher than the current recommended daily allowance. These ingredients were thought to exert a protective effect on retinal cells by counteracting oxidative stresses. The beneficial effects of zinc supplementation were previously assessed in a small clinical trial as RPE normally has a particularly high zinc content. It was hypothesized that poor zinc intake in elderly persons might result in zinc deficiency and the loss of zinc-dependent coenzymes in the RPE, resulting in development or worsening of AMD. After a mean follow-up period of 6.3 years, AREDS researchers concluded that people with intermediate AMD (extensive intermediate drusen in one or both eyes, one or more large drusen in at least one eye, or nonsubfoveal geographic atrophy in one eye), or advanced AMD (subfoveal geographic atrophy or choroidal neovascular membrane) in one eye, treated with the formulation of “antioxidants plus zinc,” had a 25% risk reduction in progression to advanced AMD over 5 years, and the risk of losing vision of three or more lines (doubling of the visual angle) was reduced by 19%. No evidence of a direct benefit of the AREDS formulation in delaying disease progression was observed in patients with early AMD (multiple small drusen or intermediate drusen and/or pigment abnormalities in one or both eyes), and therefore AREDS supplementation was recommended only to people at high risk for developing advanced AMD.
Controlled clinical studies on antioxidants in AMD and RP employing visual function endpoints
Carotenoids alone or in combination with other antioxidants
Age-related macular degeneration
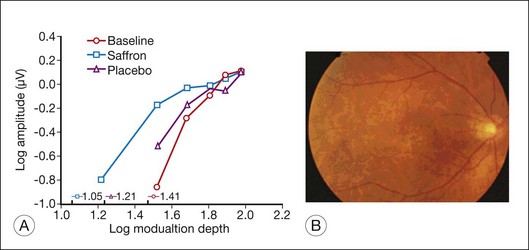
In a double-masked, placebo-controlled, randomized trial of lutein and antioxidant supplementation for intervention in atrophic AMD (the Veterans Lutein Antioxidant Supplementation Trial), Richer et al.89 determined whether nutritional supplementation with lutein or lutein together with antioxidants, vitamins, and minerals, improves visual function and symptoms in atrophic AMD. The study was a prospective, 12-month, randomized, double-masked, placebo-controlled trial. Visual function was evaluated by Snellen visual acuity, contrast sensitivity, Amsler grid, and VF-14 questionnaires. It was shown that visual function (acuity and contrast sensitivity) improved with lutein alone or lutein together with other nutrients, compared to placebo. In electrophysiological pilot studies, Falsini et al.90 and Parisi et al.91 showed that short-term supplementation of lutein or astaxantin, in combination with other antioxidants (nicotinamide, vitamin C), significantly improved macular cone-mediated function as recorded by focal or multifocal ERGs in patients with early AMD (soft drusen and/or RPE defects) and moderately reduced visual acuity. More recently,92 a randomized, double-blind, placebo-controlled study showed that 3 months of dietary saffron supplementation significantly improved the focal ERG-estimated retinal flicker sensitivity in early AMD patients (Fig. 37.3).
Retinitis pigmentosa
In a recent clinical trial of lutein supplementation in RP patients receiving vitamin A,93 it was determined whether lutein supplementation can slow visual function decline in patients with RP receiving vitamin A supplementation. The study was a randomized, controlled, double-masked trial of 225 nonsmoking patients, aged 18–60 years, evaluated over a 4-year interval. Patients received 12 mg lutein or a placebo daily. All were given 15 000 IU/day vitamin A palmitate. Randomization took into account genetic type and baseline serum lutein level. The primary outcome was the total point score for the Humphrey field analyzer (HFA) 30–2 program; prespecified secondary outcomes were the total point scores for the 60–4 program and for the 30–2 and 60–4 programs combined, 30-Hz flicker ERG amplitude, and Early Treatment Diabetic Retinopathy Study acuity. The results showed that, for the HFA 60–4 program, a decrease in mean rate of sensitivity loss was observed in the lutein plus vitamin A group (P = 0.05). Mean decline with the 60–4 program was slower among those with the highest serum lutein level or with the highest increase in macular pigment optical density at follow-up (P = 0.01 and P = 0.006, respectively). Those with the highest increase in macular pigment optical density also had the slowest decline in HFA 30–2 and 60–4 combined field sensitivity (P = 0.005). No significant toxic effects of lutein supplementation were observed. These results indicated that lutein supplementation of 12 mg/day can slow loss of midperipheral visual field on average among nonsmoking adults with RP taking vitamin A, supporting its therapeutic use as neuroprotectant.
Other antioxidants
The safety and efficacy of OT-551, a disubstituted hydroxylamine with antioxidant properties, were evaluated94 for the treatment of geographic atrophy, the advanced atrophic form of AMD. The study was a single-center, open-label phase II trial, enrolling 10 participants with bilateral geographic atrophy. Topical 0.45% OT-551 was administered in one randomly assigned eye three times daily for 2 years. Safety measures were assessed by complete ophthalmic examination, fundus photography, and review of symptoms. The primary efficacy outcome measure was the change in BCVA at 24 months. Secondary efficacy measures included changes in area of geographic atrophy, contrast sensitivity, microperimetry measurements, and total drusen area from baseline. The study drug was well tolerated and was associated with few adverse events. The mean change in BCVA at 2 years was +0.2 ± 13.3 letters in the study eyes and –11.3 ± 7.6 letters in fellow eyes (P = 0.0259). However, no statistically significant differences were found between the study and fellow eyes for all other secondary outcome measures. These results indicated that OT-551 was well tolerated by study participants and was not associated with any serious adverse effects. Efficacy measurements in this small study indicate a possible effect in maintaining visual acuity. However, the absence of significant effects on other outcome measures in this study suggests that OT-551, in the current concentration and mode of delivery, may have limited or no benefit as a treatment for geographic atrophy.
Neuroprotection with small molecules
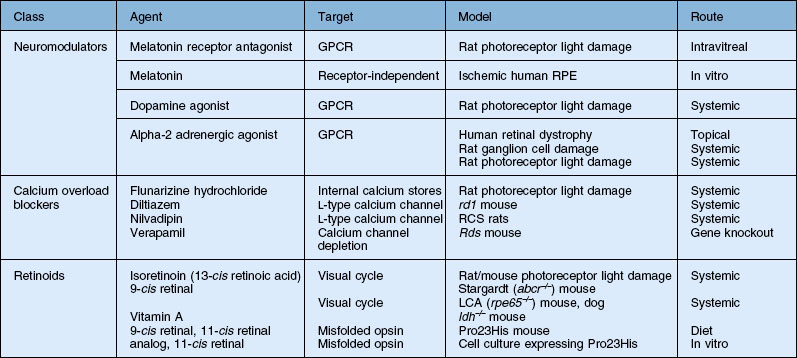
Previous sections discussed neuroprotection by proteins such as growth and neurotrophic factors (e.g., CNTF) and antibodies (e.g., anti-VEGF), gene transfer, and antioxidants. In addition, neuroprotection by glutamate receptor antagonists (e.g., memantine) in treating glaucoma was described. This section will discuss neuroprotection by small molecules, many of which target specific intracellular pathways, particularly in photoreceptor degenerations, and have been tested primarily in animal models. Small molecules, for the purpose of this section, are considered to be those with molecular weights below the limit of diffusion across the inner limiting membrane and retina, which is 40–70 kDa or 4.5 nm in radius depending on their shape.95 Thus, most can reach targets in the distal retina when injected into the vitreous. Table 37.1 includes only those discussed in this chapter, and indicates the striking variety of types of molecules and pathways explored for neuroprotection in animal models of retinal degeneration. They range in size from catecholamines, such as dopamine, and α2-adrenergic agonists, to long-chain fatty acids and small heat shock proteins. Many, as is the case with the protein growth and neurotrophic factors already discussed, are naturally occurring molecules in the retina which help maintain retinal homeostasis but may have additional function in promoting or reducing neuron survival when the retina is stressed. Others are synthetic agonists or antagonists of naturally occurring compounds.
Neuromodulators/neurotransmitters
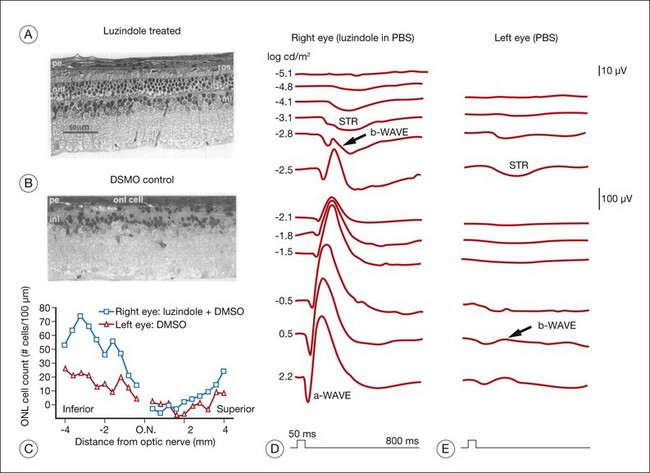
Retinal neuromodulators melatonin and dopamine negatively interact with each other in the maintenance of retinal diurnal and circadian function but also may perform various functions in the retinal response to stress. For melatonin especially, there is accumulating evidence for this dual role.96 Exogenous systemic melatonin increased photoreceptor degeneration in the light damage rat, a model of acute stress-induced photoreceptor degeneration.97–99 The rat retina is more susceptible to light damage during the dark period of the circadian cycle when retinal melatonin levels are high,100 and intravitreal injection of the melatonin receptor (MT1/MT2) antagonist luzindole prior to exposure makes the rat retina less susceptible to light damage101 (Fig. 37.4). These studies suggest that endogenous melatonin and retinal melatonin receptors play a role in photoreceptor degeneration due to acute light stress. On the other hand, loss of the melatonin MT1 receptor increased photoreceptor loss in aging mice,102 suggesting a protective role for melatonin in age-related slow degenerations. Melatonin has been shown to protect human RPE cells from ischemia-induced apoptosis by a melatonin receptor-independent mechanism,103 indicating a possible therapeutic role in prolonging visual function in AMD.104 It also performs a neuroprotective function in the CNS through its anti-inflammatory and antioxidant action.105
Dopamine, as well as being a neurotransmitter released in the light at inner retinal synapses, diffuses to the outer retina, activating receptors on photoreceptors which modulate cAMP levels through adenylate cyclase.102 Activation of these receptors may lead to reduced light sensitivity and is known to downregulate photoreceptor melatonin production, which is low in the light. Melatonin release in darkness, in turn, acts on inner retinal neuron receptors to downregulate the production and release of dopamine and may increase retinal sensitivity to light.96 Systemic administration of a dopamine agonist, bromocriptine, was found to be neuroprotective in the light damage model,97 which would be consistent with its suppressive effect on melatonin synthesis and release. Dopamine receptors are also found on Müller cells, however, and may affect photoreceptor survival through the release of neuroprotective factors from these cells.106
The alpha-2 adrenergic receptor, another catecholamine receptor, has been linked to neuroprotection in animal models. Topical administration of an alpha-2 adrenergic receptor agonist has also recently been tested for neuroprotective properties in a pilot study on humans with retinal dystrophies.107 In optic nerve reperfusion injury, alpha-2 adrenergic agonists have been shown to reduce ganglion cell degeneration,108–111 and the mechanism probably involves reducing the excitotoxic influx of calcium.112 Systemic administration of alpha-2 adrenergic agonists xylazine and clonidine can protect photoreceptors from damage due to light stress, perhaps by upregulation of bFGF and the activation of extracellular signal-regulated kinases in Müller cells.113,114
Calcium channel blockers
One of the earliest reports of neuroprotection in a photoreceptor degeneration model that offered hope for the treatment of a well-characterized form of RP using a small molecule showed that an L-type calcium channel blocker, d–cis diltiazem, could slow the course of photoreceptor cell death and loss of ERG function in the Pde6brd1 mouse.115 This followed an earlier report of protection of photoreceptors from light damage by a nonspecific Ca2+ channel blocker.116 A mutation in Pde6b, a subunit of the light-activated PDE in rod outer segments, is responsible for 5–8% of cases of RP.117 It results in a nonfunctional protein, high levels of cGMP in photoreceptors, and loss of ability to close the outer-segment light-gated cation channels. This leads to Na+ and Ca2+ overload and photoreceptor cell death by apoptosis, though the precise link between elevated cGMP and cell death is not known.118 Photoreceptor protection by d–cis diltiazem could not be replicated in other models of RP, including the Pro23His rat,119 the rcd1 dog that carries a stop codon in the Pde6b gene,120 and a different strain of rd1 mice.121 A role for calcium overload in degeneration in rd1 mice was confirmed, however, by showing slowed cell loss in rd1 mice crossed with L-type channel knockout mice.122 Subsequently, another Ca2+ channel blocker, nilvadipine, slowed photoreceptor loss in RCS rats,123 Pde6brd1 mice,124 and rds mice,125 all of which have mutations in genes causing human RP. The bulk of the evidence indicates that Ca2+ overload mediated by calcium channels “potentiates” photoreceptor degeneration in some animal models, and successful use of strategies to counter these affects for therapeutic intervention will require a further understanding of the pathways and other molecules involved.118
Retinoids
Degeneration caused by the Pde6B mutation is the result of downstream activity related to a component of the transduction cascade, i.e., chronically elevated cGMP.118 Likewise, some inherited photoreceptor degenerations result from mutations causing impairment of the first step in this cascade, the bleaching and regeneration of rhodopsin, i.e., the retinoid or visual cycle. Mutations in visual cycle enzymes result in a reduced capacity to regenerate visual pigment after bleaching. A second subset of diseases is caused by the accumulation of toxic derivatives of all-trans-retinaldehyde produced by isomerization of the visual pigment chromophore 11-cis retinaldehyde during normal photoactivation. Mutations in an ATP-binding cassette transporter called ABCR or ABCA4, which facilitates the tanslocation of all-trans-retinaldehyde from disc membrane to the cytoplasmic space, is a cause of this subset of diseases. These retinal dystrophies can be treated pharmacologically with retinoids either to supplement or to inhibit visual cycle function, respectively.126,127 An example of the former is the use of 9-cis-retinaldehyde to recover function and preserve photoreceptors in animals with a mutation in the retinoid isomerase (rpe65)128–130 and lecithin:retinol acyl transferase.131 The validity of the strategy of blocking or inhibiting the retinoid cycle to protect photoreceptors was first demonstrated in the acute light-induced model; a single intraperitoneal injection of 40 mg/kg of isoretinoin (13-cis retinoic acid) given to rats prior to light exposure resulted in significant slowing of rhodopsin regeneration and reduction in photoreceptor cell loss due to light damage.132 The effect was reversible and long-term dosing had no effect on retinal morphology or function as measured by the ERG.
In contrast to the acute effects mediated by excessive light activation of the visual cycle, inherited retinal dystrophies may take years to result in significant photoreceptor and RPE damage due to the accumulation of toxic products. For example, the slow accumulation of autofluorescent lipofuscin containing A2E derived from the condensation of all-trans-retinal and phosphatidylethanolamine in the lipid membranes of the outer-segment discs interferes with cellular metabolism and kills cells by apoptosis.126,127 Evidence for A2E accumulation is found in multiple forms of retinal and macular degeneration, including Stargardt disease, AMD, and Best vitelliform macular dystrophy, and some rod–cone dystrophies. Even mutations in some genes coding for proteins not directly involved in retinoid processing result in A2E accumulation in animal models and human diseases, implying a wider applicability of pharmacological strategies for visual cycle inhibition. Initially, the concept of modulating the visual cycle to counter the effects of A2E accumulation was done in a genetic model of Stargardt disease, the abcr–/– mouse by raising them in darkness.133 Dark-reared abcr–/– mice had levels of A2E equivalent to WT raised under cyclic light. Treatment of abcr–/– mice with isoretinoin completely blocked A2E synthesis and reduced lipofuscin deposits.134 Though isoretinoin has unacceptable side-effects at doses necessary for human therapy in retinal dystrophies, these studies validated the concept of retinal protection by pharmacologically blocking the visual cycle. Other similar molecules that directly or indirectly reduce the level of all-trans-retinaldehyde and A2E production are being explored. Though treatment would necessarily slow rod dark adaptation, patients would not notice any difference in daylight vision mediated by cones.135 Prolonging rod survival would help insure that cone vision is maintained in these rod dystrophies since the health of cones depends on the presence of rods.
Another possible application of retinoids in photoreceptor protection is as pharmacological chaperones. The term “pharmacological chaperone” refers to small molecules that bind to a protein at a specific site and help shift the folding equilibrium toward the native structure.136 These are often ligands, agonists or antagonists that bind in the ligand-binding pocket of the substrate polypeptides. Since about one-sixth of rhodopsin mutations that cause autosomal dominant RP are due to protein misfolding (see Heat shock proteins, below), retinoids are being investigated as therapy in these diseases. A series of in vitro137–139 and in vivo140 studies have demonstrated that retinoids can both stabilize the class II mutant opsin protein (e.g., Pro23His) to improve its movement through the secretory pathway and increase photoreceptor survival.
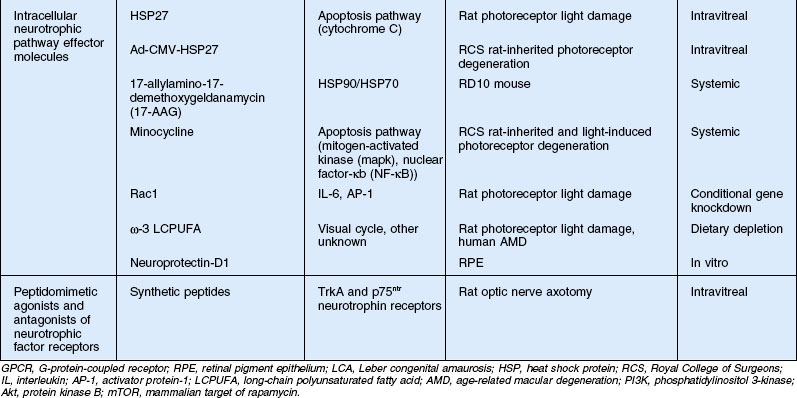
Modulation of intracellular neurotrophic pathways
Heat shock proteins
The use of neurotrophic factors, such as CNTF, in photoreceptor neuroprotection has been discussed. These proteins, released under conditions of cell stress and injury, produce their neuroprotective effect by activating intracellular pathways through cell surface receptors.141 The intracellular effector molecules of these pathways are often transcription factors which promote DNA transcription leading to the production of antioxidant enzymes, calcium regulation proteins, cell cycle proteins, and metabolic enzymes.142,143 One strategy for neuroprotection is to utilize the proteins and small molecules of these intracellular signaling pathways directly.142 Lens epithelial-derived growth factor (LEDGF), for example, is a secreted cell survival factor expressed in response to stress signals.146 Rather than binding to a cell surface receptor, it contains a Tat domain which allows it to enter cells and accumulate in the nucleus where it promotes the expression of smaller stress-related proteins, such as heat shock protein HSP27, which helps reduce caspase-dependent cell death. HSP27 binds to cytochrome c upon its release from mitochondria preventing its activation of caspase 3 and inhibiting apoptosis144 (Fig. 37.5). LEDGF has been shown to promote cell survival under stress of many cell types in culture,144 and intravitreal injection of the LEDGF protein protected photoreceptors in retinal light damage and the RCS rat model of inherited retinal degeneration.145 In the retinas treated with LEDGF, HSP25 (the rat homolog of human HSP27), and the heat shock protein αB-crystallin expression was four- to fivefold higher compared to the vehicle-injected eyes. To test whether upregulation of these small heat shock proteins could explain the protection by LEDGF, HSP25 was injected into the vitreous of RCS rats at 6 weeks of age and evaluated for retinal function by ERG and retinal morphology 4 weeks later (S Machida, PA Sieving, and RA Bush, unpublished observations). HSP25 injection resulted in significantly better ERG responses and cellular preservation at 10 weeks of age than in vehicle-injected eyes (Fig. 37.6). The retinal level of HSP25 was higher in retinas from the HSP-injected eyes (data not shown). To confirm that intracellular HSP25 expression could rescue photoreceptors comparable to LEDGF, the genes for these proteins were transferred to the RCS rat retina using adenovirus HSP27 (the human homolog) and adeno-associated LEDGF viral vectors.146 Both vectors resulted in similar degrees of rescue, which was similar to that seen with injection of the LEDGF and HSP25 protein. Protection only delayed the degeneration by about 2 weeks, however. But these results demonstrate that cell rescue can be achieved by exogenous administration of intracellular intermediates in endogenous survival-promoting pathways. Neuroprotection by small HSPs takes on added relevance with recent findings of the possible role of autoantibodies to HSP27 and HSP60 in the serum of glaucoma patients147 and the production of a pattern of ganglion cell death in rats with induced autoimmunities to heat shock proteins.148,149

Fig. 37.5 The intracellular function of heat shock protein (HSP) 25 blocking cell death.
(Reproduced with permission from Ranger AM, Malynn BA, Korsmeyer SJ. Mouse models of cell death. Nat Genet 2001;28:113–8.)

Besides mediating the stress response, HSPs function as molecular chaperones to help regulate protein folding during synthesis and to keep misfolded proteins from aggregating by directing them to the proteosomal degradation machinery.150 Many forms of autosomal dominant RP (adRP), for example, one-sixth of those caused by rhodopsin mutations, are due to mutations resulting in misfolded proteins and subsequent aggregation leading to photoreceptor cell death through a “gain of function” mechanism (e.g., Pro23His rhodopsin and Arg224Pro inosine 5′-monophosphate dehydrogenase type 1, IMPDH1, RP10, mutations).151 Thus, enhancing HSP function either by vector-mediated overexpression or by pharmacological means is being explored as a strategy for therapeutic intervention that could work in several kinds of adRP.152 One such technique shown to be successful in several neurological degeneration models is to produce an upregulation of HSP70 by pharmacologically inhibiting HSP90. This strategy has been effective in models of both Pro23His153 and Rd10154 using the partial geldanamycin derivative 17-allylamino-17demethoxygeldanamycin (17-AAG, molecular weight: 585.69). The former study was carried out in cell culture, while the latter was done using systemic administration in a mutant mouse. In addition to enhancing endogenous chaperone activity, small-molecule “pharmacological chaperones,” e.g., retinoids, are being studied for their ability to counteract both the gain of function and dominant negative phenotypes in adRP153 (see Retinoids, above).
Minocycline
Direct targeting of cell survival or death intracellular signaling pathways using small synthetic agonists or antagonists could be a valuable therapeutic approach in neuroprotection.154 A good example is the recent research on the neuroprotective possibilities of minocycline, a semisynthetic broad-spectrum tetracycline antibiotic, in several neurodegenerative disorders. Since 2004 there have been more than 20 papers describing the neuroprotective effects of minocycline in animal and cell culture models of retinal degeneration. Minocycline can easily cross cell membranes, and once in the cytoplasm, it modulates intracellular cytokine signaling pathways involving mitogen-activated kinases (MAPKs) or nuclear factor-κB (NF-κB). These pathways mediate cytokine responses to stress and injury and are linked to microglial activation in inflammation and the process of programmed cell death, or apoptosis, respectively.155 Thus, minocycline may produce neuroprotection in the CNS by direct inhibition of apoptosis or by acting as an anti-inflammatory agent through inhibition of microglial activation.156 Several animal models of inherited photoreceptor degeneration show microglia activation and migration into the ONL, suggesting that microglia contribute to photoreceptor death.157–160 Minocycline protected rods from degeneration in bright light-exposed mice and reduced the number of microglia entering the ONL.161 It also delayed both photoreceptor degeneration and microglial activation in the rds mouse model of retinal degeneration.156 In the latter study, however, depletion of microglia alone had no protective effect, suggesting the action of minocycline was microglia-independent and due to a direct effect on apoptosis pathways. Using an in vitro system of light-induced photoreceptor degeneration, Yang et al.162 found that minocycline inhibited apoptosis, partly through an NF-κB-dependent mechanism, but not through the microglial-activating MAPK pathway. Recent research has focused on a possible role for minocycline treatment of diabetic retinopathy because of its ability to inhibit mitochondrial membrane permeability transition and Müller cell osmotic swelling163 and reduce retinal edema164 in the diabetic rat.
Rho GTPases
As described earlier, activated Rac1 is a component of NADPH oxidase, which generates the superoxide anions thought to play a role in many forms of neurodegeneration.165 Specific depletion of Rac1 in photoreceptors in a conditional knockout mouse resulted in photoreceptor protection from light-induced degeneration and a reduction in membrane-bound light-activated NADPH oxidase.81 This is likely to be the mechanism of protection from light damage in the Rac1 conditional knockout. However, inhibition of Rac1 may provide neuroprotection through other mechanisms. For example, persistent activation of Rac1 leads to autocrine production of the pleiotropic cytokine interleukin-6, which is involved in immune and inflammatory responses, and activation of STAT3.166 It also stimulates activator protein-1 (AP-1), a regulator of apoptosis, through phosphorylation of c-jun.167 Acute phototoxic insult involves the induction of AP-1 and members of the interleukin-6 family of cytokines in the neural retina and phosphorylation of STAT3 in Müller glial cells.168 Small-molecule inhibitors of Rac1 that cross the blood–brain barrier have been shown to reduce the accumulation of Aβ peptide in animal models of Alzheimer disease.169 In addition, peptide inhibitors are being investigated for therapeutic effect in a variety of neurodegenerative disorders.169–172
Long-chain polyunsaturated fatty acids
Long-chain polyunsaturated fatty acids (LCPUFAs) are usually thought of as structural components of cell membranes, but they also function in a number of intracellular signaling and metabolic pathways involved in the pathogenesis of vasoproliferative and retinal neurogenerative disorders.173 Dietary omega-3 LCPUFAs are important in normal retinal function as well as neuroprotection. Docosahexaenoic acid (DHA) in particular is important for photoreceptor metabolism and transduction mechanisms and it makes up the largest fraction of the LCPUFAs in the outer-segment disc membranes.174 Lowered dietary intake of LCPUFAs reduced susceptibility of the rat retina to light-induced photoreceptor damage,175 possibly through a slowing of the regeneration rate of rhodopsin.176 Dietary effects on the levels of retinal LCPUFAs may affect a variety of retinal molecules and signaling pathways involved in neuroprotection, including dopamine, cyclic nucleotides, endocannabinoids, glutamate, and ionic transport and channel dynamics.173 Metabolic and environmental activators of LCPUFAs, phospholipase A2, cyclooxygenase, and lipoxygenase are associated with a number of retinal diseases, including diabetic retinopathy, AMD, and retinopathy of prematurity through modulation of neuronal and vascular function.173 A major dietary source of LCPUFAs is fish. Incidence of AMD was shown to decrease with increasing intake of DHA, tuna, or total fish.177 Several other studies have shown similar trends,173 but the neuroprotective mechanism is not known.
The natural recycling of membrane phospholipids produces neuroprotectin D1 (NPD1), the first known bioactive product of DHA. This molecule is produced by the action of phospholipase A2 that releases free DHA from membrane phospholipids under conditions of oxidative stress and inflammation and promotes cell survival in animal models of stroke and Alzheimer disease178,179 and protects RPE cells from oxidative stress.180,181 The latter effect is linked to activation of the phosphatidylinositol 3-kinase/Akt signaling pathway through increased phosphorylation.180 Though a direct effect on photoreceptor neuroprotection has so far not been demonstrated, NPD1 produced in RPE upon daily phagocytosis of photoreceptor membranous discs, which contain large amounts of esterified DHA, may be released and promote the survival of other retinal cells in a paracrine fashion.182
Peptide neurotrophin receptor agonists/antagonists
Neurotrophins, e.g., CNTF, and their receptors have proven to be useful as therapeutic targets in a variety of diseases, including retinal degenerations. They often fail to have the expected effect in vivo, however, in part because of their effect on multiple cell types and receptors. As members of the growth factor family of proteins, their size and complexity may also make them unstable and unable to reach the target tissues. Recently, small-molecule peptides have been designed to bind regulatory or activating subdomains of the Trk and p75NTR neurotrophin receptors to selectively activate or inhibit these receptors.183 A potential therapeutic application for these compounds in retinal degeneration has been demonstrated in ganglion cell survival after axotomy.184 While BDNF is a potent survival factor for injured ganglion cells,185 NGF does not promote survival either in vitro186 or in vivo.187 It was found, however, that peptide agonists, which selectively activate the NGF TrkA receptor on ganglion cells, provided neuroprotection following axotomy.184 Simultaneously treating with an antagonist to the proapoptotic p75NTR receptors further enhanced neuroprotection, even though these receptors are found on Müller cells but not ganglion cells. This study, carried out using intravitreal injections in the rat, highlights the potential therapeutic benefits of using small molecules that mimic selected actions of larger molecules to achieve neuroprotection and avoid unwanted effects.
1 Osborne NN. Recent clinical findings with memantine should not mean that the idea of neuroprotection in glaucoma is abandoned. Acta Ophthalmol. 2009;87:450–454.
2 Lambiase A, Aloe L, Centofanti M, et al. Experimental and clinical evidence of neuroprotection by nerve growth factor eye drops: Implications for glaucoma. Proc Natl Acad Sci U S A. 2009;106:13469–13474.
3 van Adel BA, Kostic C, Deglon N, et al. Delivery of ciliary neurotrophic factor via lentiviral-mediated transfer protects axotomized retinal ganglion cells for an extended period of time. Hum Gene Ther. 2003;14:103–115.
4 Press releases on memantine trials, fourth quarter operating results. http://agn.client.shareholder.com/releasedetail.cfm?ReleaseID=290764, Jan 30 2008. Available online at
5 Belkin M. Nerve growth factor and glaucoma. Glaucoma Today. 2009:18–19.
6 Sommer A, Tielsch JM, Katz J, et al. Relationship between intraocular pressure and primary open angle glaucoma among white and black Americans. The Baltimore Eye Survey. Arch Ophthalmol. 1991;109:1090–1095.
7 Anderson DR. Collaborative normal tension glaucoma study. Curr Opin Ophthalmol. 2003;14:86–90.
8 Becker B, Stamper RL, Asseff C, et al. Effect of diphenylhydantoin on glaucomatous field loss: a preliminary report. Trans Am Acad Ophthalmol Otolaryngol. 1972;76:412–422.
9 Hains BC, Waxman SG. Neuroprotection by sodium channel blockade with phenytoin in an experimental model of glaucoma. Invest Ophthalmol Vis Sci. 2005;46:4164–4169.
10 Kerchner GA, Tartaglia MC, Boxer A. Abhorring the vacuum: use of Alzheimer’s disease medications in frontotemporal dementia. Expert Rev Neurother. 2011;11:709–717.
11 Doble A. The pharmacology and mechanism of action of riluzole. Neurology. 1996;47:S233–S241.
12 Lamanauskas N, Nistri A. Riluzole blocks persistent Na+ and Ca2+ currents and modulates release of glutamate via presynaptic NMDA receptors on neonatal rat hypoglossal motoneurons in vitro. Eur J Neurosci. 2008;27:2501–2514.
13 Chen HS, Pellegrini JW, Aggarwal SK, et al. Open-channel block of N-methyl-d-aspartate (NMDA) responses by memantine: therapeutic advantage against NMDA receptor-mediated neurotoxicity. J Neurosci. 1992;12:4427–4436.
14 Brooks DE, Garcia GA, Dreyer EB, et al. Vitreous body glutamate concentration in dogs with glaucoma. Am J Vet Res. 1997;58:864–867.
15 Dreyer EB, Zurakowski D, Schumer RA, et al. Elevated glutamate levels in the vitreous body of humans and monkeys with glaucoma. Arch Ophthalmol. 1996;114:299–305.
16 Johnson EC, Guo Y, Cepurna WO, et al. Neurotrophin roles in retinal ganglion cell survival: lessons from rat glaucoma models. Exp Eye Res. 2009;88:808–815.
17 Chen H, Weber AJ. BDNF enhances retinal ganglion cell survival in cats with optic nerve damage. Invest Ophthalmol Vis Sci. 2001;42:966–974.
18 Song XY, Li F, Zhang FH, et al. Peripherally-derived BDNF promotes regeneration of ascending sensory neurons after spinal cord injury. PLoS ONE. 2008;3:e1707.
19 Rammes G, Rupprecht R, Ferrari U, et al. The N-methyl-d-aspartate receptor channel blockers memantine, MRZ 2/579 and other amino-alkyl-cyclohexanes antagonise 5-HT(3) receptor currents in cultured HEK-293 and N1E-115 cell systems in a non-competitive manner. Neurosci Lett. 2001;306:81–84.
20 Lipton SA. Pathologically activated therapeutics for neuroprotection. Nat Rev Neurosci. 2007;8:803–808.
21 Nishimura T, Ishima T, Iyo M, et al. Potentiation of nerve growth factor-induced neurite outgrowth by fluvoxamine: role of sigma-1 receptors, IP3 receptors and cellular signaling pathways. PLoS ONE. 2008;3:e2558.
22 May JA, Chen HH, Rusinko A, et al. A novel and selective 5-HT2 receptor agonist with ocular hypotensive activity: (S)-(+)-1-(2-aminopropyl)-8,9-dihydropyrano[3,2-e]indole. J Med Chem. 2003;46:4188–4195.
23 May JA, Dantanarayana AP, Zinke PW, et al. 1-((S)-2-aminopropyl)-1H-indazol-6-ol: a potent peripherally acting 5-HT2 receptor agonist with ocular hypotensive activity. J Med Chem. 2006;49:318–328.
24 Faktorovich EG, Steinberg RH, Yasumura D, et al. Photoreceptor degeneration in inherited retinal dystrophy delayed by basic fibroblast growth factor. Nature. 1990;347:83–86.
25 LaVail MM, Unoki K, Yasumura D, et al. Multiple growth factors, cytokines, and neurotrophins rescue photoreceptors from the damaging effects of constant light. Proc Natl Acad Sci U S A. 1992;89:11249–11253.
26 Gorbatyuk MS, Knox T, LaVail MM, et al. Restoration of visual function in P23H rhodopsin transgenic rats by gene delivery of BiP/Grp78. Proc Natl Acad Sci U S A. 2010;107:5961–5966.
27 LaVail MM, Yasumura D, Matthes MT, et al. Ribozyme rescue of photoreceptor cells in P23H transgenic rats: long-term survival and late-stage therapy. Proc Natl Acad Sci U S A. 2000;97:11488–11493.
28 Peterson WM, Wang Q, Tzekova R, et al. Ciliary neurotrophic factor and stress stimuli activate the Jak-STAT pathway in retinal neurons and glia. J Neurosci. 2000;20:4081–4090.
29 Lambiase A, Aloe L. Nerve growth factor delays retinal degeneration in C3H mice. Graefes Arch Clin Exp Ophthalmol. 1996;234(Suppl 1):S96–100.
30 Cayouette M, Gravel C. Adenovirus-mediated gene transfer of ciliary neurotrophic factor can prevent photoreceptor degeneration in the retinal degeneration (rd) mouse. Hum Gene Ther. 1997;8:423–430.
31 Cao W, Wen R, Li F, et al. Mechanical injury increases bFGF and CNTF mRNA expression in the mouse retina. Exp Eye Res. 1997;65:241–248.
32 Cohen S, Levi-Montalcini R, Hamburger V. A nerve growth-stimulating factor isolated from sarcomas 37 and 180. Proc Natl Acad Sci USA. 1954;40:1014–1018.
33 Levi-Montalcini R, Hamburger V. Selective growth stimulating effects of mouse sarcoma on the sensory and sympathetic nervous system of the chick embryo. J Exp Zool. 1951;116:321–361.
34 Cayouette M, Behn D, Sendtner M, et al. Intraocular gene transfer of ciliary neurotrophic factor prevents death and increases responsiveness of rod photoreceptors in the retinal degeneration slow mouse. J Neurosci. 1998;18:9282–9293.
35 Adler R, Landa KB, Manthorpe M, et al. Cholinergic neuronotrophic factors: intraocular distribution of trophic activity for ciliary neurons. Science. 1979;204:1434–1436.
36 Varon S, Manthorpe M, Adler R. Cholinergic neuronotrophic factors: I. Survival, neurite outgrowth and choline acetyltransferase activity in monolayer cultures from chick embryo ciliary ganglia. Brain Res. 1979;173:29–45.
37 Adler R. Ciliary neurotrophic factor as an injury factor. Curr Opin Neurobiol. 1993;3:785–789.
38 McDonald NQ, Panayotatos N, Hendrickson WA. Crystal structure of dimeric human ciliary neurotrophic factor determined by MAD phasing. EMBO J. 1995;14:2689–2699.
39 Sleeman MW, Anderson KD, Lambert PD, et al. The ciliary neurotrophic factor and its receptor, CNTFR alpha. Pharm Acta Helv. 2000;74:265–272.
40 Boulton TG, Stahl N, Yancopoulos GD. Ciliary neurotrophic factor/leukemia inhibitory factor/interleukin 6/oncostatin M family of cytokines induces tyrosine phosphorylation of a common set of proteins overlapping those induced by other cytokines and growth factors. J Biol Chem. 1994;269:11648–11655.
41 Leonard WJ, O’Shea JJ. Jaks and STATs: biological implications. Annu Rev Immunol. 1998;16:293–322.
42 Heinrich PC, Behrmann I, Müller-Newen G, et al. Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem J. 1998;334:297–314.
43 Bok D, Yasumura D, Matthes MT, et al. Effects of adeno-associated virus-vectored ciliary neurotrophic factor on retinal structure and function in mice with a P216L rds/peripherin mutation. Exp Eye Res. 2002;74:719–735.
44 Bush RA, Lei B, Tao W, et al. Encapsulated cell-based intraocular delivery of ciliary neurotrophic factor in normal rabbit: dose-dependent effects on ERG and retinal histology. Invest Ophthalmol Vis Sci. 2004;45:2420–2430.
45 Sieving PA, Caruso RC, Tao W, et al. Ciliary neurotrophic factor (CNTF) for human retinal degeneration: phase I trial of CNTF delivered by encapsulated cell intraocular implants. Proc Natl Acad Sci U S A. 2006;103:3896–3901.
46 Humphries MM, Rancourt D, Farrar GJ, et al. Retinopathy induced in mice by targeted disruption of the rhodopsin gene. Nat Genet. 1997;15:216–219.
47 Geller AM, Sieving PA. Assessment of foveal cone photoreceptors in Stargardt’s macular dystrophy using a small dot detection task. Vision Res. 1993;33:1509–1524.
48 . A study of an encapsulated cell technology (ECT) implant for patients with atrophic macular degeneration (database on the internet). Available online at http://clinicaltrials.gov/ct2/show/NCT00447954?term=CNTF+eye&rank=4
49 . A study of encapsulated cell technology (ECT) implant for patients with late stage retinitis pigmentosa (database on the internet). Available online at http://clinicaltrials.gov/ct2/show/NCT00447993?term=CNTF+eye&rank=3
50 . A study of encapsulated cell technology (ECT) implant for participants with early stage retinitis pigmentosa (database on the internet). Available online at http://clinicaltrials.gov/ct2/show/NCT00447980?term=CNTF+eye&rank=2
51 Zhang K, Hopkins JJ, Heier JS, et al. Ciliary neurotrophic factor delivered by encapsulated cell intraocular implants for treatment of geographic atrophy in age-related macular degeneration. Proc Natl Acad Sci U S A. 2011;108:6241–6245.
52 Talcott KE, Ratnam K, Sundquist SM, et al. Longitudinal study of cone photoreceptors during retinal degeneration and in response to ciliary neurotrophic factor treatment. Invest Ophthalmol Vis Sci. 2011;52:2219–2226.
53 Brown DM, Kaiser PK, Michels M, et al. Ranibizumab versus verteporfin for neovascular age-related macular degeneration. N Engl J Med. 2006;355:1432–1444.
54 Rosenfeld PJ, Brown DM, Heier JS, et al. Ranibizumab for neovascular age-related macular degeneration. N Engl J Med. 2006;355:1419–1431.
55 Acland GM, Aguirre GD, Ray J, et al. Gene therapy restores vision in a canine model of childhood blindness. Nat Genet. 2001;28:92–95.
56 Wiegand RD, Giusto NM, Rapp LM, et al. Evidence for rod outer segment lipid peroxidation following constant illumination of the rat retina. Invest Ophthalmol Vis Sci. 1983;24:1433–1435.
57 Tanito M, Yoshida Y, Kaidzu S, et al. Detection of lipid peroxidation in light-exposed mouse retina assessed by oxidative stress markers, total hydroxyoctadecadienoic acid and 8-iso-prostaglandin F2alpha. Neurosci Lett. 2006;398:63–68.
58 Organisciak DT, Darrow RM, Jiang YI, et al. Protection by dimethylthiourea against retinal light damage in rats. Invest Ophthalmol Vis Sci. 1992;33:1599–1609.
59 Penn JS, Naash MI, Anderson RE. Effect of light history on retinal antioxidants and light damage susceptibility in the rat. Exp Eye Res. 1987;44:779–788.
60 Gosbell AD, Stefanovic N, Scurr LL, et al. Retinal light damage: structural and functional effects of the antioxidant glutathione peroxidase-1. Invest Ophthalmol Vis Sci. 2006;47:2613–2622.
61 Xie Z, Wu X, Gong Y, et al. Intraperitoneal injection of Ginkgo biloba extract enhances antioxidation ability of retina and protects photoreceptors after light-induced retinal damage in rats. Curr Eye Res. 2007;32:471–479.
62 Ranchon I, Gorrand JM, Cluzel J, et al. Functional protection of photoreceptors from light-induced damage by dimethylthiourea and Ginkgo biloba extract. Invest Ophthalmol Vis Sci. 1999;40:1191–1199.
63 Tomita H, Kotake Y, Anderson RE. Mechanism of protection from light-induced retinal degeneration by the synthetic antioxidant phenyl-N-tert-butylnitrone. Invest Ophthalmol Vis Sci. 2005;46:427–434.
64 Logvinov SV, Plotnikov MB, Varakuta EY, et al. Effect of ascovertin on morphological changes in rat retina exposed to high-intensity light. Bull Exp Biol Med. 2005;140:578–581.
65 Yilmaz T, Aydemir O, Ozercan IH, et al. Effects of vitamin E, pentoxifylline and aprotinin on light-induced retinal injury. Ophthalmologica. 2007;221:159–166.
66 Stahl W, Sies H. Bioactivity and protective effects of natural carotenoids. Biochim Biophys Acta. 2005;1740:101–107.
67 Costa BL, Fawcett R, Li GY, et al. Orally administered epigallocatechin gallate attenuates light-induced photoreceptor damage. Brain Res Bull. 2008;76:412–423.
68 Komeima K, Rogers BS, Lu L, et al. Antioxidants reduce cone cell death in a model of retinitis pigmentosa. Proc Natl Acad Sci U S A. 2006;103:11300–11305.
69 Shen J, Yang X, Dong A, et al. Oxidative damage is a potential cause of cone cell death in retinitis pigmentosa. J Cell Physiol. 2005;203:457–464.
70 Jozwick C, Valter K, Stone J. Reversal of functional loss in the P23H-3 rat retina by management of ambient light. Exp Eye Res. 2006;83:1074–1080.
71 Chrysostomou V, Stone J, Stowe S, et al. The status of cones in the rhodopsin mutant P23H-3 retina: light-regulated damage and repair in parallel with rods. Invest Ophthalmol Vis Sci. 2008;49:1116–1125.
72 Valter K, Kirk DK, Stone J. Optimising the structure and function of the adult P23H-3 retina by light management in the juvenile and adult. Exp Eye Res. 2009;89:1003–1011.
73 Yu DY, Cringle SJ, Su EN, et al. Intraretinal oxygen levels before and after photoreceptor loss in the RCS rat. Invest Ophthalmol Vis Sci. 2000;41:3999–4006.
74 Usui S, Oveson BC, Lee SY, et al. NADPH oxidase plays a central role in cone cell death in retinitis pigmentosa. J Neurochem. 2009;110:1028–1037.
75 Komeima K, Usui S, Shen J, et al. Blockade of neuronal nitric oxide synthase reduces cone cell death in a model of retinitis pigmentosa. Free Radic Biol Med. 2008;45:905–912.
76 Usui S, Komeima K, Lee SY, et al. Increased expression of catalase and superoxide dismutase 2 reduces cone cell death in retinitis pigmentosa. Mol Ther. 2009;17:778–786.
77 Lu L, Oveson BC, Jo YJ, et al. Increased expression of glutathione peroxidase 4 strongly protects retina from oxidative damage. Antioxid Redox Signal. 2009;11:715–724.
78 Miranda M, Arnal E, Ahuja S, et al. Antioxidants rescue photoreceptors in rd1 mice: Relationship with thiol metabolism. Free Radic Biol Med. 2010;48:216–222.
79 Ushio-Fukai M. Localizing NADPH oxidase-derived ROS. Sci STKE. 2006;2006:re8.
80 Balasubramanian N, Slepak VZ. Light-mediated activation of Rac-1 in photoreceptor outer segments. Curr Biol. 2003;13:1306–1310.
81 Haruta M, Bush RA, Kjellstrom S, et al. Depleting Rac1 in mouse rod photoreceptors protects them from photo-oxidative stress without affecting their structure or function. Proc Natl Acad Sci U S A. 2009;106:9397–9402.
82 Leveillard T, Sahel JA. Rod-derived cone viability factor for treating blinding diseases: from clinic to redox signaling. Sci Transl Med. 2010;2:26ps16.
83 Lee SY, Usui S, Zafar AB, et al. N-Acetylcysteine promotes long-term survival of cones in a model of retinitis pigmentosa. J Cell Physiol. 2011;226:1843–1849.
84 Maccarone R, Di Marco S, Bisti S. Saffron supplement maintains morphology and function after exposure to damaging light in mammalian retina. Invest Ophthalmol Vis Sci. 2008;49:1254–1261.
85 Giaccio M. Crocetin from saffron: an active component of an ancient spice. Crit Rev Food Sci Nutr. 2004;44:155–172.
86 Natoli R, Zhu Y, Valter K, et al. Gene and noncoding RNA regulation underlying photoreceptor protection: microarray study of dietary antioxidant saffron and photobiomodulation in rat retina. Mol Vis. 2010;16:1801–1822.
87 Kong L, Cai X, Zhou X, et al. Nanoceria extend photoreceptor cell lifespan in tubby mice by modulation of apoptosis/survival signaling pathways. Neurobiol Dis. 2011;42:514–523.
88 AREDS Research Group. A randomized, placebo-controlled, clinical trial of high-dose supplementation with vitamins C and E, beta carotene, and zinc for age-related macular degeneration and vision loss: AREDS report no. 8. Arch Ophthalmol. 2001;119:1417–1436.
89 Richer S, Stiles W, Statkute L, et al. Double-masked, placebo-controlled, randomized trial of lutein and antioxidant supplementation in the intervention of atrophic age-related macular degeneration: the Veterans LAST study (Lutein Antioxidant Supplementation Trial). Optometry. 2004;75:216–230.
90 Falsini B, Piccardi M, Iarossi G, et al. Influence of short-term antioxidant supplementation on macular function in age-related maculopathy: a pilot study including electrophysiologic assessment. Ophthalmology. 2003;110:51–60. discussion 1
91 Parisi V, Tedeschi M, Gallinaro G, et al. Carotenoids and antioxidants in age-related maculopathy Italian study: multifocal electroretinogram modifications after 1 year. Ophthalmology. 2008;115:324–333. e2
92 Falsini B, Piccardi M, Minnella A, et al. Influence of saffron supplementation on retinal flicker sensitivity in early age-related macular degeneration. Invest Ophthalmol Vis Sci. 2010;51:6118–6124.
93 Berson EL, Rosner B, Sandberg MA, et al. Clinical trial of lutein in patients with retinitis pigmentosa receiving vitamin A. Arch Ophthalmol. 2010;128:403–411.
94 Wong WT, Kam W, Cunningham D, et al. Treatment of geographic atrophy by the topical administration of OT-551: results of a phase II clinical trial. Invest Ophthalmol Vis Sci. 2010;51:6131–6139.
95 El Sanharawi M, Kowalczuk L, Touchard E, et al. Protein delivery for retinal diseases: from basic considerations to clinical applications. Prog Retin Eye Res. 2010;29:443–465.
96 Wiechmann AF, Summers JA. Circadian rhythms in the eye: the physiological significance of melatonin receptors in ocular tissues. Prog Retin Eye Res. 2008;27:137–160.
97 Bubenik GA, Purtill RA. The role of melatonin and dopamine in retinal physiology. Can J Physiol Pharmacol. 1980;58:1457–1462.
98 Wiechmann AF, Chignell CF, Roberts JE. Influence of dietary melatonin on photoreceptor survival in the rat retina: an ocular toxicity study. Exp Eye Res. 2008;86:241–250.
99 Wiechmann AF, O’Steen WK. Melatonin increases photoreceptor susceptibility to light-induced damage. Invest Ophthalmol Vis Sci. 1992;33:1894–1902.
100 Grewal R, Organisciak D, Wong P. Factors underlying circadian dependent susceptibility to light induced retinal damage. Adv Exp Med Biol. 2006;572:411–416.
101 Sugawara T, Sieving PA, Iuvone PM, et al. The melatonin antagonist luzindole protects retinal photoreceptors from light damage in the rat. Invest Ophthalmol Vis Sci. 1998;39:2458–2465.
102 Baba K, Pozdeyev N, Mazzoni F, et al. Melatonin modulates visual function and cell viability in the mouse retina via the MT1 melatonin receptor. Proc Natl Acad Sci U S A. 2009;106:15043–15048.
103 Osborne NN, Nash MS, Wood JP. Melatonin counteracts ischemia-induced apoptosis in human retinal pigment epithelial cells. Invest Ophthalmol Vis Sci. 1998;39:2374–2383.
104 Yi C, Pan X, Yan H, et al. Effects of melatonin in age-related macular degeneration. Ann N Y Acad Sci. 2005;1057:384–392.
105 Esposito E, Cuzzocrea S. Antiinflammatory activity of melatonin in central nervous system. Curr Neuropharmacol. 2010;8:228–242.
106 de Melo Reis RA, Ventura AL, Schitine CS, et al. Müller glia as an active compartment modulating nervous activity in the vertebrate retina: neurotransmitters and trophic factors. Neurochem Res. 2008;33:1466–1474.
107 Merin S, Obolensky A, Farber MD, et al. A pilot study of topical treatment with an alpha-2-agonist in patients with retinal dystrophies. J Ocul Pharmacol Ther. 2008;24:80–86.
108 Ahmed FA, Hegazy K, Chaudhary P, et al. Neuroprotective effect of alpha(2) agonist (brimonidine) on adult rat retinal ganglion cells after increased intraocular pressure. Brain Res. 2001;913:133–139.
109 Chao HM, Osborne NN. Topically applied clonidine protects the rat retina from ischaemia/reperfusion by stimulating alpha(2)-adrenoceptors and not by an action on imidazoline receptors. Brain Res. 2001;904:126–136.
110 Lai RK, Chun T, Hasson D, et al. Alpha-2 adrenoceptor agonist protects retinal function after acute retinal ischemic injury in the rat. Vis Neurosci. 2002;19:175–185.
111 Yoles E, Wheeler LA, Schwartz M. Alpha-2-adrenoreceptor agonists are neuroprotective in a rat model of optic nerve degeneration. Invest Ophthalmol Vis Sci. 1999;40:65–73.
112 Dong CJ, Guo Y, Agey P, et al. Alpha-2 adrenergic modulation of NMDA receptor function as a major mechanism of RGC protection in experimental glaucoma and retinal excitotoxicity. Invest Ophthalmol Vis Sci. 2008;49:4515–4522.
113 Peng M, Li Y, Luo Z, et al. Alpha-2-adrenergic agonists selectively activate extracellular signal-regulated kinases in Müller cells in vivo. Invest Ophthalmol Vis Sci. 1998;39:1721–1726.
114 Wen R, Cheng T, Li Y, et al. Alpha-2-adrenergic agonists induce basic fibroblast growth factor expression in photoreceptors in vivo and ameliorate light damage. J Neurosci. 1996;16:5986–5992.
115 Frasson M, Sahel JA, Fabre M, et al. Retinitis pigmentosa: rod photoreceptor rescue by a calcium-channel blocker in the rd mouse. Nat Med. 1999;5:1183–1187.
116 Edward DP, Lam TT, Shahinfar S, et al. Amelioration of light-induced retinal degeneration by a calcium overload blocker. Flunarizine. Arch Ophthalmol. 1991;109:554–562.
117 McLaughlin ME, Ehrhart TL, Berson EL, et al. Mutation spectrum of the gene encoding the beta subunit of rod phosphodiesterase among patients with autosomal recessive retinitis pigmentosa. Proc Natl Acad Sci U S A. 1995;92:3249–3253.
118 Barabas P, Cutler Peck C, Krizaj D. Do calcium channel blockers rescue dying photoreceptors in the Pde6b (rd1) mouse? Adv Exp Med Biol. 2010;664:491–499.
119 Bush RA, Kononen L, Machida S, et al. The effect of calcium channel blocker diltiazem on photoreceptor degeneration in the rhodopsin Pro213His rat. Invest Ophthalmol Vis Sci. 2000;41:2697–2701.
120 Pearce-Kelling SE, Aleman TS, Nickle A, et al. Calcium channel blocker d–cis-diltiazem does not slow retinal degeneration in the PDE6B mutant rcd1 canine model of retinitis pigmentosa. Mol Vis. 2001;7:42–47.
121 Pawlyk BS, Li T, Scimeca MS, et al. Absence of photoreceptor rescue with d–cis-diltiazem in the rd mouse. Invest Ophthalmol Vis Sci. 2002;43:1912–1915.
122 Read DS, McCall MA, Gregg RG. Absence of voltage-dependent calcium channels delays photoreceptor degeneration in rd mice. Exp Eye Res. 2002;75:415–420.
123 Yamazaki H, Ohguro H, Maeda T, et al. Preservation of retinal morphology and functions in Royal College Surgeons rat by nilvadipine, a Ca(2+) antagonist. Invest Ophthalmol Vis Sci. 2002;43:919–926.
124 Takano Y, Ohguro H, Dezawa M, et al. Study of drug effects of calcium channel blockers on retinal degeneration of rd mouse. Biochem Biophys Res Commun. 2004;313:1015–1022.
125 Takeuchi K, Nakazawa M, Mizukoshi S. Systemic administration of nilvadipine delays photoreceptor degeneration of heterozygous retinal degeneration slow (rds) mouse. Exp Eye Res. 2008;86:60–69.
126 Palczewski K. Retinoids for treatment of retinal diseases. Trends Pharmacol Sci. 2010;31:284–295.
127 Travis GH, Golczak M, Moise AR, et al. Diseases caused by defects in the visual cycle: retinoids as potential therapeutic agents. Annu Rev Pharmacol Toxicol. 2007;47:469–512.
128 Gearhart PM, Gearhart C, Thompson DA, et al. Improvement of visual performance with intravitreal administration of 9-cis-retinal in Rpe65-mutant dogs. Arch Ophthalmol. 2010;128:1442–1448.
129 Van Hooser JP, Aleman TS, He YG, et al. Rapid restoration of visual pigment and function with oral retinoid in a mouse model of childhood blindness. Proc Natl Acad Sci U S A. 2000;97:8623–8628.
130 Van Hooser JP, Liang Y, Maeda T, et al. Recovery of visual functions in a mouse model of Leber congenital amaurosis. J Biol Chem. 2002;277:19173–19182.
131 Batten ML, Imanishi Y, Tu DC, et al. Pharmacological and rAAV gene therapy rescue of visual functions in a blind mouse model of Leber congenital amaurosis. PLoS Med. 2005;2:e333.
132 Sieving PA, Chaudhry P, Kondo M, et al. Inhibition of the visual cycle in vivo by 13-cis retinoic acid protects from light damage and provides a mechanism for night blindness in isotretinoin therapy. Proc Natl Acad Sci USA. 2001;98:1835–1840.
133 Mata NL, Weng J, Travis GH. Biosynthesis of a major lipofuscin fluorophore in mice and humans with ABCR-mediated retinal and macular degeneration. Proc Natl Acad Sci U S A. 2000;97:7154–7159.
134 Radu RA, Mata NL, Nusinowitz S, et al. Treatment with isotretinoin inhibits lipofuscin accumulation in a mouse model of recessive Stargardt’s macular degeneration. Proc Natl Acad Sci U S A. 2003;100:4742–4747.
135 Travis GH, Golczak M, Moise AR, et al. Diseases caused by defects in the visual cycle: retinoids as potential therapeutic agents. Annu Rev Pharmaceut Toxicol. 2007;47:469–512.
136 Mendes HF, Cheetham ME. Pharmacological manipulation of gain-of-function and dominant-negative mechanisms on rhodopsin retinitis pigmentosa. Hum Mol Genet. 2008;17:3043–3054.
137 Noorwez SM, Kuksa V, Imanishi Y, et al. Pharmacological chaperone-mediated in vivo folding and stabilization of the P23H-opsin mutant associated with autosomal dominant retinitis pigmentosa. J Biol Chem. 2003;278:14442–14450.
138 Noorwez SM, Malhotra R, McDowell JH, et al. Retinoids assist the cellular folding of the autosomal dominant retinitis pigmentosa opsin mutant P23H. J Biol Chem. 2004;279:16278–16284.
139 Saliba RS, Munro PM, Luthert PJ, et al. The cellular fate of mutant rhodopsin: quality control, degradation and aggresome formation. J Cell Sci. 2002;115:2907–2918.
140 Li T, Sandberg MA, Pawlyk BS, et al. Effect of vitamin A supplementation on rhodopsin mutants threonine-17 → methionine and proline-347 → serine in transgenic mice and in cell cultures. Proc Natl Acad Sci U S A. 1998;95:11933–11938.
141 Wahlin KJ, Campochiaro PA, Zack DJ, et al. Neurotrophic factors cause activation of intracellular signaling pathways in Müller cells and other cells of the inner retina, but not photoreceptors. Invest Ophthalmol Vis Sci. 2000;41:927–936.
142 Mattson MP. Neuroprotective strategies based on targeting of postreceptor signaling events. In: Mattson MP, ed. Neuroprotective signal transduction. Totowa, NJ: Humana Press; 1998:301–306.
143 Mattson MP. Glutamate and neurotrophic factors in neuronal plasticity and disease. Ann N Y Acad Sci. 2008;1144:97–112.
144 Shinohara T, Singh DP, Fatma N. LEDGF, a survival factor, activates stress-related genes. Prog Retinal Eye Res. 2002;21:341–358.
145 Machida S, Chaudhry P, Shinohara T, et al. Lens epithelium-derived growth factor promotes photoreceptor survival in light-damaged and RCS rats. Invest Ophthalmol Vis Sci. 2001;42:1087–1095.
146 Raz-Prag D, Zeng Y, Sieving PA, et al. Photoreceptor protection by adeno-associated virus-mediated LEDGF expression in the RCS rat model of retinal degeneration: probing the mechanism. Invest Ophthalmol Vis Sci. 2009;50:3897–3906.
147 Tezel G, Hernandez R, Wax MB. Immunostaining of heat shock proteins in the retina and optic nerve head of normal and glaucomatous eyes. Arch Ophthalmol. 2000;118:511–518.
148 Wax MB, Tezel G. Immunoregulation of retinal ganglion cell fate in glaucoma. Exp Eye Res. 2009;88:825–830.
149 Wax MB, Tezel G, Yang J, et al. Induced autoimmunity to heat shock proteins elicits glaucomatous loss of retinal ganglion cell neurons via activated T-cell-derived Fas-ligand. J Neurosci. 2008;28:12085–12096.
150 Kosmaoglou M, Schwarz N, Bett JS, et al. Molecular chaperones and photoreceptor function. Prog Retin Eye Res. 2008;27:434–449.
151 Mendes HF, van der Spuy J, Chapple JP, et al. Mechanisms of cell death in rhodopsin retinitis pigmentosa: implications for therapy. Trends Mol Med. 2005;11:177–185.
152 Tam LC, Kiang AS, Campbell M, et al. Prevention of autosomal dominant retinitis pigmentosa by systemic drug therapy targeting heat shock protein 90 (Hsp90). Hum Mol Genet. 2010;19:4421–4436.
153 Mendes HF, Cheetham ME. Pharmacological manipulation of gain-of-function and dominant-negative mechanisms in rhodopsin retinitis pigmentosa. Hum Mol Genet. 2008;17:3043–3054.
154 Martin D, Miller G, Fischer N. Neuroprotective strategies based on interleukin signaling. In: Mattson MP, ed. Neuroprotective signal transduction. Totowa, NJ: Humana Press; 1998:185–196.
155 Plane JM, Shen Y, Pleasure DE, et al. Prospects for minocycline neuroprotection. Arch Neurol. 2010;67:1442–1448.
156 Hughes EH, Schlichtenbrede FC, Murphy CC, et al. Minocycline delays photoreceptor death in the rds mouse through a microglia-independent mechanism. Exp Eye Res. 2004;78:1077–1084.
157 Hughes EH, Schlichtenbrede FC, Murphy CC, et al. Generation of activated sialoadhesin-positive microglia during retinal degeneration. Invest Ophthalmol Vis Sci. 2003;44:2229–2234.
158 Thanos S. Sick photoreceptors attract activated microglia from the ganglion cell layer: a model to study the inflammatory cascades in rats with inherited retinal dystrophy. Brain Res. 1992;588:21–28.
159 Zeiss CJ, Johnson EA. Proliferation of microglia, but not photoreceptors, in the outer nuclear layer of the rd-1 mouse. Invest Ophthalmol Vis Sci. 2004;45:971–976.
160 Zeng HY, Zhu XA, Zhang C, et al. Identification of sequential events and factors associated with microglial activation, migration, and cytotoxicity in retinal degeneration in rd mice. Invest Ophthalmol Vis Sci. 2005;46:2992–2999.
161 Zhang C, Lei B, Lam TT, et al. Neuroprotection of photoreceptors by minocycline in light-induced retinal degeneration. Invest Ophthalmol Vis Sci. 2004;45:2753–2759.
162 Yang LP, Zhu XA, Tso MO. Role of NF-kappaB and MAPKs in light-induced photoreceptor apoptosis. Invest Ophthalmol Vis Sci. 2007;48:4766–4776.
163 Krugel K, Wurm A, Pannicke T, et al. Involvement of oxidative stress and mitochondrial dysfunction in the osmotic swelling of retinal glial cells from diabetic rats. Exp Eye Res. 2011;92:87–93.
164 Zhang Y, Xu G, Ling Q, et al. Expression of aquaporin 4 and kir4.1 in diabetic rat retina: treatment with minocycline. J Int Med Res. 2011;39:464–479.
165 Raz L, Zhang QG, Zhou CF, et al. Role of Rac1 GTPase in NADPH oxidase activation and cognitive impairment following cerebral ischemia in the rat. PLoS ONE. 2010;5:e12606.
166 Faruqi TR, Gomez D, Bustelo XR, et al. Rac1 mediates STAT3 activation by autocrine IL-6. Proc Natl Acad Sci U S A. 2001;98:9014–9019.
167 Coso OA, Chiariello M, Yu JC, et al. The small GTP-binding proteins Rac1 and Cdc42 regulate the activity of the JNK/SAPK signaling pathway. Cell. 1995;81:1137–1146.
168 Wenzel A, Grimm C, Samardzija M, et al. Molecular mechanisms of light-induced photoreceptor apoptosis and neuroprotection for retinal degeneration. Prog Retin Eye Res. 2005;24:275–306.
169 Desire L, Bourdin J, Loiseau N, et al. RAC1 inhibition targets amyloid precursor protein processing by gamma-secretase and decreases Abeta production in vitro and in vivo. J Biol Chem. 2005;280:37516–37525.
170 Lu Q, Longo FM, Zhou H, et al. Signaling through Rho GTPase pathway as viable drug target. Curr Med Chem. 2009;16:1355–1365.
171 Marchioni F, Zheng Y. Targeting rho GTPases by peptidic structures. Curr Pharm Des. 2009;15:2481–2487.
172 Wang PL, Niidome T, Akaike A, et al. Rac1 inhibition negatively regulates transcriptional activity of the amyloid precursor protein gene. J Neurosci Res. 2009;87:2105–2114.
173 SanGiovanni JP, Chew EY. The role of omega-3 long-chain polyunsaturated fatty acids in health and disease of the retina. Prog Retin Eye Res. 2005;24:87–138.
174 Fliesler SJ, Anderson RE. Chemistry and metabolism of lipids in the vertebrate retina. Prog Lipid Res. 1983;22:79–131.
175 Bush RA, Remé CE, Malnoë A. Light damage in the rat retina: The effect of dietary deprivation of n-3 fatty acids on acute structural alterations. Exp Eye Res. 1991;53:741–752.
176 Bush RA, Malnoë A, Remé CE, et al. N-3 fatty acid deficiency alters rhodopsin content and function in the rat retina. Invest Ophthalmol Vis Sci. 1994;35:91–100.
177 Cho E, Hung S, Willett WC, et al. Prospective study of dietary fat and the risk of age-related macular degeneration. Am J Clin Nutr. 2001;73:209–218.
178 Bazan NG. Neuroprotectin D1-mediated anti-inflammatory and survival signaling in stroke, retinal degenerations, and Alzheimer’s disease. J Lipid Res. 2009;50(Suppl):S400–S405.
179 Stark DT, Bazan NG. Neuroprotectin D1 induces neuronal survival and downregulation of amyloidogenic processing in Alzheimer’s disease cellular models. Mol Neurobiol. 2011;43:131–138.
180 Halapin NA, Bazan NG. NPD1 induction of retinal pigment epithelial cell survival involves PI3K/Akt phosphorylation signaling. Neurochem Res. 2010;35:1944–1947.
181 Mukherjee PK, Marcheselli VL, Serhan CN, et al. Neuroprotectin D1: a docosahexaenoic acid-derived docosatriene protects human retinal pigment epithelial cells from oxidative stress. Proc Natl Acad Sci U S A. 2004;101:8491–8496.
182 Bazan NG. Cell survival matters: docosahexaenoic acid signaling, neuroprotection and photoreceptors. Trends Neurosci. 2006;29:263–271.
183 Zaccaro MC, Lee HB, Pattarawarapan M, et al. Selective small molecule peptidomimetic ligands of TrkC and TrkA receptors afford discrete or complete neurotrophic activities. Chem Biol. 2005;12:1015–1028.
184 Lebrun-Julien F, Morquette B, Douillette A, et al. Inhibition of p75(NTR) in glia potentiates TrkA-mediated survival of injured retinal ganglion cells. Mol Cell Neurosci. 2009;40:410–420.
185 Di Polo A, Aigner LJ, Dunn RJ, et al. Prolonged delivery of brain-derived neurotrophic factor by adenovirus-infected Müller cells temporarily rescues injured retinal ganglion cells. Proc Natl Acad Sci U S A. 1998;95:3978–3983.
186 Johnson JE, Barde YA, Schwab M, et al. Brain-derived neurotrophic factor supports the survival of cultured rat retinal ganglion cells. J Neurosci. 1986;6:3031–3038.
187 Cui Q, Harvey AR. At least two mechanisms are involved in the death of retinal ganglion cells following target ablation in neonatal rats. J Neurosci. 1995;15:8143–8155.