CHAPTER 74 Neuropathology of Movement Disorders
Movement disorders, can be divided into four major groups according to clinical phenomenology (Table 74-1); only the first two are discussed in this chapter. Most rigid-kinetic and hyperkinetic forms have their origin in dysfunction of the dorsal basal ganglia (BG), which work in tandem with the cortex via complex information circuits of the brain, although virtually the entire nervous system is engaged in motor control. Recent progress has provided insight into the anatomy, functional organization, and pathophysiologic significance of BG in specific types of movement disorders, as well as the role of different neuron subpopulations in mediating different aspects of motor control.1–9
TABLE 74-1 Clinical Classification of Movement Disorders
Functional Anatomy of Basal Ganglia
The interconnections of the nuclei of the BG are shown schematically in Figure 74-1. The three main transmitter systems involved in the integration of BG function are glutamate, γ-aminobutyric acid (GABA), and dopamine (DA).10 Normal movement is controlled by cortico-BG-thalamocortical circuits, in which the striatum receives glutamatergic input from the cerebral cortex. It sends GABAergic output to the substantia nigra reticulata (SNr) and globus pallidus (pars) interna (GPi), which release projections to specific thalamic nuclei and, to a lesser extent, to the deep layers of the superior colliculus and mesencephalic reticular formation. The respective thalamic nuclei have an excitatory glutamatergic input to specific regions of the cerebral cortex involved in motor function. In this major circuit, the GABAergic output of the substantia nigra compacta (SNc) and GPi diminishes the glutamatergic projections from the thalamus back to the cortex. Projections of the globus pallidus (pars) externa (GPe), the dopaminergic SNc, and the subthalamic nucleus (STN) remain primarily within the realm of the BG, and these nuclei modulate the main flow of information through the BG. The functional specialization of the striatum is closely related to its chemical heterogeneity along the dorsoventral and mediolateral axes.11 The topography of cortico-BG projections has led to a model of their function based on parallel and segregated pathways operating through discrete functional channels that are represented in specific regions of each BG structure, whereas others have suggested a more complicated pattern of BG connections that indicate potential complex interactions between these channels.
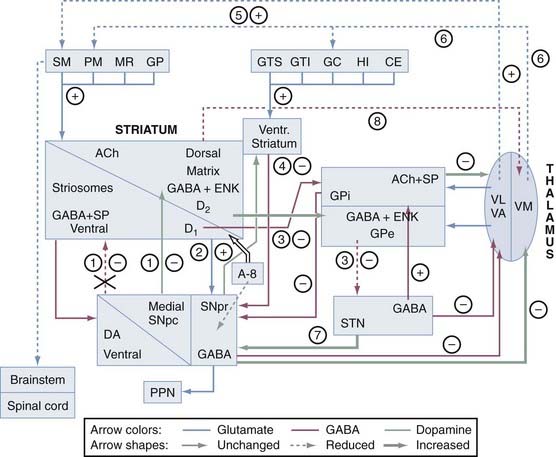
Corticobasal Ganglia–Thalamocortical Circuits
The BG are tightly linked to the frontal cortex and are thought to be involved not only in motor control but also in learning processes, behavior motivation, and planning.9,12 A bidirectional pattern of cortico-BG communication is differentially patterned across bands and during changes in movement.13 These circuits involve, in a sequential manner, specific parts of the prefrontal cortex, striatum, pallidonigral complex, medial and ventral thalamus, and the frontal or prefrontal cortical area. Five such BG-thalamocortical circuits have tentatively been defined: the motor and oculomotor circuits and the dorsolateral prefrontal, lateral orbitofrontal, and anterior cingulate or limbic circuits involving different parts of the striatum, the pallidonigral complex, and the medial and ventral thalamus.
The indirect pathway includes projections from the glutamatergic cortex to the striatal MSNs (containing enkephalin [ENK] and GABA and expressing the DA D2 receptor) along with sequential striatal projections to the GPe, GABAergic GPe projections to the STN, and glutamatergic STN projections to the GPi and SNr. Activation of striatal neurons in this pathway leads to inhibition of the tonically active neurons in the GPe, thereby inducing decreased inhibition (disinhibition) of the STNs and their thalamic and mesencephalic targets and causing suppression of motor and behavioral output. It is the “stop” pathway. Experimental evidence indicates that the STN is a critical component of complex networks controlling not only motor function but also emotion, cognition, and corticothalamic excitability.1,5
Balance between these two pathways at the level of the pallidum and substantia nigra (SN) appears to be crucial for normal functioning of the BG-thalamocortical circuits, and in pathologic situations (in particular in movement disorders), this equilibrium is disrupted. The circuits subserving abnormal movements in primates and humans with specific lesions may be different from those governing normal movements in intact subjects. This core model has helped explain some of the pathophysiologic mechanisms for the major movement disorders, in which there is either increased inhibition of the thalamocortical pathway, which results in hypokinetic disorders, or decreased inhibition of thalamacortical output, which results in hyperkinetic disorders. DA has opposite effects on the two pathways. The direct pathway has D1 receptors, and when DA binds to them, this pathway is activated. The indirect pathway has D2 receptors, and when DA binds to them, this pathway is inhibited. Therefore, the overall effect of DA is to decrease GPi activity, thereby promoting movement. In contrast, DA depletion, as in Parkinson’s disease (PD), leads to higher neuron activity in the output structures and consequently to inhibition of their thalamic and midbrain targets, with reduced activity in “direct” cortical-putamen-GPi projections. This model provides a reasonable explanation for the origin of the akinetic features in PD and the response to drugs and surgery.14 However, the concept of direct and indirect pathways is likely to be far too simplistic and will probably be modified as more complex organization emerges. Recent studies have led to refinement of the model and the development of a novel hypothesis for better understanding how DA regulates the BG and contributes to BG pathophysiology in PD. Although the striatum remains the main functional target of DA, it is now appreciated that there is dopaminergic involvement of the globus pallidus (GP), STN, and SN. The differential distribution of D1 and D2 receptors on neurons in the direct and indirect striatopallidal pathway has been re-emphasized, and cholinergic interneurons are recognized as an intermediary mediator of DA-mediated communication between the two pathways.15 Recently, two “hyperdirect” pathways were reported. One is a direct excitatory connection from the cortex to the STN, which has an excitatory connection to the GPi. Activity in this pathway will increase GPi activity and reduce thalamic and cortical activity.16 A new dopaminergic-thalamic system has also been uncovered that sets the stage for direct DA action on thalamocortical activity.17 Its degeneration in the monkey model of PD provides further evidence for a critical extrastriatal site whereby DA depletion could induce pathologic changes in neuronal activity and behavior.15 Another ultra-short DA pathway from SNc to SNr regulates the intensity and pattern of these BG outputs by dendritically released DA that excites SNr GABA neurons via D1-5 receptor activation enhancing active TRPC3 channels.17a Parallel processing and integrative networks probably work together rather than in conflict to allow coordinated behavior and motor control to be maintained, as well as to be modified and changed according to the appropriate external and internal stimuli, which are key deficits in BG disorders.8 A model for altered neural network dynamics related to movement disorders in PD has recently been presented.18
Classification of Movement Disorders
Most movement disorders related to BG dysfunction are neurodegenerative diseases that are morphologically characterized by neuronal degeneration and loss accompanied by astrocytosis in various, often disparate parts of the central nervous system (CNS). According to recent genetic and molecular-biologic data, movement disorders can be classified into several groups (Table 74-2).16 They may or may not be associated with cytoskeletal abnormalities, which represent important histologic signposts pointing to the diagnosis (Table 74-3). For some of these disorders, consensus criteria for their clinical and neuropathologic diagnoses have been established.19–25
TABLE 74-2 Morphologic and Biochemical Classification of Degenerative Diseases with Movement Disorders
| α-SYNUCLEINOPATHIES |
| Invariable Forms (Consistent α-Synuclein Deposition) |
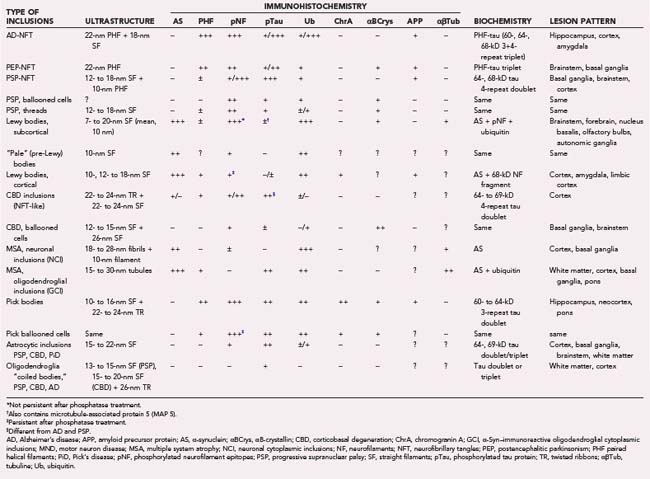
TABLE 74-3 Ultrastructure, Immunohistochemistry, Biochemistry, and Distribution of Neuroglial Inclusions
Movement disorders are classified into synucleinopathies, a heterogeneous group of neurodegenerative disorders caused by misfolded α-synuclein (α-Syn) protein that forms amyloid-like filamentous inclusions in many brain areas. They include Lewy body (LB) disorders—sporadic and rare familial forms of PD (brainstem type of LB disease [LBD]), dementia with Lewy bodies (DLB), and pure autonomic failure (PAF)—multisystem atrophy (MSA), and Hallervorden-Spatz disease, renamed neurodegeneration with brain iron accumulation type I (NBIA-I) or pathothenate kinase–associated neurodegeneration (PKAN). Other major groups are tauopathies, all of which feature neurofibrillary pathology (progressive supranuclear palsy [PSP], corticobasal degeneration [CBD], and so on); polyglutamine (CAG) disorders, such as Huntington’s disease (HD) and related disorders; the recently described TDP-43 proteinopathies, such as frontotemporal lobe dementia with ubiquitin (Ub) inclusions (FTLD-U); and other neurodegenerative movement disorders without hitherto detected genetic or specific disease markers (Table 74-4). Movement disorders have additional importance in differentiating Creutzfeld-Jakob disease from Alzheimer’s dementia (AD) and DLB.26
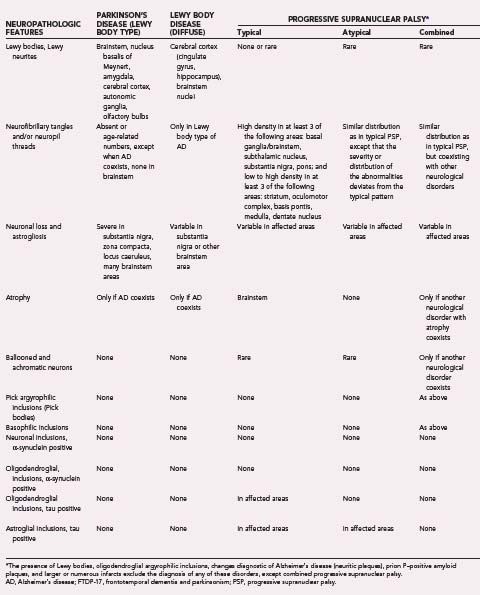
Synucleinopathies
α-Syn is a partially unfolded, 140–amino acid presynaptic protein with potential for self-oligomerization and fibrillary aggregation under pathologic conditions. Its gene, located on chromosome 4, is mutated in rare familial forms of PD.27 For its molecular basis, functions, aggregation modes, interaction with DA metabolites, and relevant animal models, see other sources.28–31 α-Syn assembles into special oligomers and is potentially prone to misfold,32 which may lead to neuronal death,33 but other modes of toxicity have also been proposed.34 The lysosomal protease cathepsin D influences α-Syn processing, aggregation, and toxicity in vivo.35 α-Syn was demonstrated to be a major component of LBs, Lewy-related dystrophic neurites (LNs), and astroglia in PD and DLB36,37 and neuronal and glial inclusions in MSA.38 Given the fundamental nature of the α-Syn–containing lesions, these and other disorders are referred to as synucleinopathies.39 The reliability of assessment of α-Syn pathology and its dysfunction in LBD has been reviewed.29,31,40–44 α-Syn phosphorylated at serine 129 has a central role in both familial and nonfamilial forms of PD in dysregulating the DA synthesis pathway.45 Recently, elevated levels of soluble α-Syn oligomers have been detected in postmortem brains of patients with DLB.46
Akinetic-Rigid Movement Disorders—Parkinsonism
This group includes various forms of parkinsonism, tauopathies, and other hereditary degenerative disorders causing atypical parkinsonian syndromes.47 Parkinsonism describes the presence of extrayramidal movement disturbances manifested by a combination of rigidity and bradykinesia with or without resting tremor and postural instability. It has many causes (Table 74-5), with frequent clinical misclassification even if strict diagnostic criteria are used.
| COMMON CAUSES OF PARKINSONISM |
| Idiopathic Parkinson’s disease |
| Drug-induced parkinsonism |
| Multiple system atrophy |
| Dementia with Lewy bodies |
| Progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome) |
| UNCOMMON NEURODEGENERATIVE CAUSES OF PARKINSONISM |
| Vascular pseudoparkinsonism |
| Corticobasal degeneration |
| Alzheimer’s disease, Pick’s disease |
| Frontotemporal lobe degeneration (FTDP-17) |
| Wilson’s disease |
| Neuroacanthocytosis |
| Huntington’s disease |
| Multisystem degeneration |
| Dentatorubral-pallidoluysian atrophy |
| Lubag (X-linked dystonia-parkinsonism) |
| Dopa-responsive dystonia |
| Pallidal degenerations |
| Neuronal inclusion body and neurofilament inclusion body disease |
| SECONDARY CAUSES OF PARKINSONISM |
| Space-occupying lesions |
| Hydrocephalus (normal pressure) |
| Drugs (especially dopamine receptor blocker) |
| Toxin-induced disease (manganese, carbon monoxide, carbon disulfide, MPTP, rotenon |
| Boxer’s encephalopathy (dementia pugilistica) |
| Infections and postinfectious diseases—HIV encephalopathy, Creutzfeldt-Jakob disease, neurosyphilis, Japanese B encephalitis |
| Anoxic brain injury |
| Metabolic disorders (e.g., Wilson’s disease) |
| Basal ganglia calcification (Fahr’s syndrome, hypoparathyroidism) |
HIV, human immunodeficiency virus; MPTP, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine.
Lewy Body–Associated Disorders
The prominent cytoskeletal lesions in this group are α-Syn–positive LBs, cytoplasmic inclusions occurring in many regions of the CNS and autonomic nervous system.48,49 They are the morphologic hallmarks of PD and DLB but are also found in a variety of neurodegenerative disorders, for example, in 7% to 71% of sporadic and familial forms of AD50,51 and in 2% to 61% of aged individuals with or without dementia.52–55 The variation in the estimated prevalence of LB pathology in the older population depends mainly on case selection and the methods used for detecting LBs.53 α-Syn pathology has been encountered in autonomic nuclei, plexuses, and nerves49 and more recently in cutaneous nerves56 as an essential or coincidental feature.
Lewy Bodies
There are two types of LBs: the classic brainstem and the cortical type. Classic LBs are spherical cytoplasmic intraneuronal inclusions 8 to 30 µm in diameter with a hyaline eosinophilic core, concentric lamellar bands, and a narrow pale-stained halo. Although most LBs are single, some neurons contain multiple or polymorphic inclusions. In some brain regions, such as the dorsal motor nucleus of the vagus (DMX), similar inclusions within neuronal processes are intraneuritic LBs. They can be detected in routine histopathologic preparations and should be distinguished from LNs, which are not visible on routine histology. Ultrastructurally, classic LBs are non–membrane-bound, granulofilamentous structures composed of radially arranged, 7- to 20-nm intermediate filaments associated with electron-dense granule material and vesicular structures, with the core showing densely packed filaments and dense granular material and the periphery having radially arranged 10-nm filaments. Cortical LBs—eosinophilic, rounded, angular, or reniform structures without a halo—are more difficult to detect by routine histology. Ultrastructurally, they are poorly organized, granulofibrillary structures with a felt-like arrangement composed of 7- to 27-nm–wide filaments, mostly devoid of a central core.57 They are found in small nonpyramidal neurons in the lower cortical layers, with densest accumulation in the insular cortex, amygdala, and parahippocampal and cingulate gyri. Similar lesions—rounded areas of granular, pale-staining eosinophilic material displacing neuromelanin (NM) in brainstem neurons—are referred to as “pale bodies” and have been considered precursors of LBs.58
Both classic and cortical LBs share immunocytochemical and biochemical characteristics, the major components being α-Syn, Ub, and phosphorylated neurofilaments associated with many other substances, including parkin and synphilin-1 isoforms.22,25,59,60 LBs, the morphologic hallmark of PD and DLB, have a distinct central parkin- and Ub-positive domain with α-Syn in the periphery,61 but it is incorporated into LBs and dystrophic neurites before ubiquitination.62,63 Colocalization of α-Syn and parkin within LBs suggests that parkin plays a role in ubiquitination and posttranslational modification of α-Syn.64 The latter results in changes in protein size, structure, or charge28 (e.g., phosphorylation and nitration, both enhancing fibrillation and formation of LBs65,66). A functional Ub–3-ligase complex consisting of early-onset familial PD associated with parkin, PINK-1, and DJ-1 mutations promotes the degradation of unfolded or misfolded proteins and may be a pathogenetic mechanism for PD.67,68 Full-length leucine-rich repeat kinase 2 (LRRK2) is not a major component of the LBs and tau inclusions in AD. Proteomic analysis of cortical LBs has revealed 296 proteins related to multiple or unknown functions.69 In brainstem LBD, 90 proteins were identified that differ from those in Pick bodies, thus suggesting a complex formation process.70 Classic LBs show an initial intraneuronal appearance of dust-like particles related to NM or lipofuscin that are cross-linked to α-Syn, with homogeneous deposition of α-Syn and Ub in the center. Septin 4 (SEPT4), a polymerizing guanosine triphosphate–binding protein that serves as a scaffold for diverse molecules, has been found to colocalize with α-Syn in LBs. Because it serves as a substrate for parkin, it may play a central role in the etiopathogenesis of PD.71 In the MPTP model of parkinsonism, no Lewy pathology was seen in old monkeys.72
Cortical LBs show diffuse α-Syn and Ub labeling, whereas subcortical LBs have a distinct, central Ub-positive domain with α-Syn occurring primarily in the periphery and ubiquitination being the later event. The developmental stages of cortical LBs include granular accumulation of α-Syn in the neuronal cytoplasm initially, stepwise accumulation of dense filaments, spreading to dendrites, later deformation of LBs, and final degradation by astroglial processes.73 Extraneuronal LBs are related to death and disappearance of the involved neurons.
LBs are associated with coarse, dystrophic neurites—LNs—and also contain α-Syn and Ub as inclusions in axonal processes, which according to recent three-dimentional studies may evolve into LBs.74 They occur in many regions of the CNS and peripheral and autonomic nervous systems; absence of tyrosine hydrolase (TH) immunoreactivity suggests that many of these neuritic processes are not derived from dopaminergic neurons.
Although not all LBs contain Ub, 7% to 10% have only α-Syn, which is more widespread than Ub staining, thus making specific antibodies against α-Syn the best markers for diseases with LBs and other synucleinopathies and for differentiating LBs and LNs from negative neurofibrillary tangles (NFTs), Pick bodies, and other protein inclusions.75 Despite the presence of high Aβ affinity binding sites on α-Syn filaments, no discernible interaction of [3H]–Pittsburgh Compound B (PIB) was detected on amygdala sections from patients with PD that contained frequent α-Syn–immunoreactive LBs and LNs, thus indicating that LB pathology is unlikely to contribute significantly to the retention of PIB in positron emission tomography (PET) studies.76
Intranuclear inclusions, referred to as Marinesco bodies, are found at higher frequency in elderly individuals in the pigmented neurons of the SN and locus caeruleus (LC) that contain LBs than in those without such inclusions, and their frequency appears to have an inverse relationship with striatal concentrations of DA transporter (DAT) and TH.77
Lewy Bodies and Neuronal Cell Death
The biologic significance of these insoluble proteinaceous cytoplasmic inclusions, the mechanism of LB formation, and their impact on neurodegeneration await further elucidation. LBs, which are the sequelae of frustraneous proteolytic degradation of abnormal cytoskeletal elements, may represent—similar to other inclusions such as NFTs in AD or Pick bodies—end products or reactions to unknown neuronal degenerative processes.78 Inhibition of complex I (reduced nicotinamide adenine dinucleotide ubiquitinone oxidoreductase) may be a central cause of sporadic PD, and derangement of complex I causes aggregation of α-Syn, which contributes to the impairment in protein handling and detoxification,79 whereas mitochondrial accumulated α-Syn may interact with complex I and interfere with its functions.80 Complex I deficiency in PD brain is not confined to the SN but has also been demonstrated in the frontal cortex.81
Involvement of the Ub-proteasome system (UPS) and the autophagy-liposome pathway (ALP) suggests that LBs are structural manifestations of a cytoprotective process. Inhibition of proteasomal function or generation of misfolded proteins exceeding the degradative capacity of the UPS causes the formation of aggresomes, which are cytotoxic inclusions formed in the centrosome, or a cytoprotective response to sequester and degrade excess levels of potentially cytotoxic proteins.61,82,83 Aggresomal proteins such as β-tubulin and others have been demonstrated in LBs.84,85 There is no correlation between the density of LB formation and cell loss,86 and the comparatively low number of neurons containing LBs in any brain region would not be expected to result from altered synaptic function. Oligomerization of α-Syn at the initial stage of LB development is well documented,87 and accumulation of α-Syn oligomeres coincides with behavioral and pathologic changes, thus indicating that these oligomeres may initiate protein aggregation, disrupt cellular function, and eventually lead to neuronal death.88 Accumulation of small α-Syn aggregates at presynaptic terminals has been linked to synaptic pathology in LBD,89 a finding suggesting that PD is caused by presynaptic accumulation of α-Syn aggregates and resultant synaptic degeneration and that loss of dopaminergic neurons is rather an epiphenomenon after the loss of synapses. LRRK2 expression is found widely in the human brain and may be associated with early-age α-Syn pathology in the brainstem in PD.90 Fragmentation of the Golgi apparatus, seen in 5% of PD nigral neurons with LBs and 3% of those without LBs and in 19% of neurons containing pale bodies, suggests that the cytotoxicity of α-Syn aggregates is reduced by the process of LB formation,91 whereas SN neurons showing DNA fragmentation have no somal LBs. LBs bear similarities to some intermediate filament inclusions, such as Mallory bodies, Rosenthal fibers, and others, which have been proposed as a structural manifestation of a cytoprotective response designed to confine and eliminate damaged cellular elements.91,92a Nevertheless, significant intracellular protein aggregation, such as LB formation, is a pathologic process reflecting changes in the cellular environment that may contribute to dysfunction of the involved cells. Recent studies have shown the development of LBs in grafted neurons in individuals with PD, thus suggesting host-to-graft disease propagation.93,94 In the SN, the proportion of LB-bearing neurons appears to be stable, with 3.6% of neurons involved on average. This suggests that destruction of LBs may be equal to their production, and with the hypothesis that neuronal death is related to LBs, their life span was estimated to be around 6.2 months (15.9 months for any type of α-Syn inclusion).95 Thus, neuronal loss of 71%, necessary for the manifestation of motor symptoms, would be reached after about 20 years, which is in line with standard progression of the disease.95
Deposition of tau can be demonstrated in a proportion of LBs and is greatest in neurons vulnerable to both LB and NFT formation, such as in the LC, nucleus basalis of Meynert (NBM), and amygdala.96,97 Tau phosphorylation at Ser396 has been observed in synaptic-enriched fractions of the frontal cortex in patients with PD and DLB and in advanced stages of AD with and without amygdala LBs.98 Aβ inhibits the proteasome and enhances amyloid and tau accumulation.99 This suggests that α-Syn and tau may be related to several pathologic processes (bystander effect), which may explain the frequent overlap between synucleinopathies and tauopathies.100–102 Interactions between Aβ, α-Syn, and tau may be a molecular mechanism in the overlapping pathology of AD and PD/DLB.103,104
Sporadic Parkinson’s Disease
PD or the brainstem type of LBD, the most common neurodegenerative disorder in patients with advanced age, is manifested clinically by bradykinesia, rigidity, tremor at rest, postural imbalance, and various nonmotor features.105 Subtle cognitive dysfunction and depression are often present early in the disease,105 whereas dementia is common in later stages.106 PD is characterized by progressive degeneration of the dopaminergic nigrostriatal system and other cortical and subcortical networks associated with widespread α-Syn pathology, and the resultant striatal DA deficiency and multiple other biochemical deficits produce a heterogeneous clinical phenotype.107 Accepted clinical criteria for the diagnosis of possible and probable PD108–110 have high sensitivity but a specificity of just 75% for identifying and differentiating PD from other LBDs.111 For the diagnosis of definite PD, histopathologic confirmation is required. Although LBs are not specific to PD and occur in a variety of conditions as secondary pathology, a positive diagnosis of PD can usually be made by inspecting two unilateral sections from the midportion of the SN and finding LBs. If no LBs are found, two further sections should be examined. If LBs are not seen in either the SN or LC, the diagnosis of PD of the LB type can be excluded. In case of cell loss in the SN and LC in the absence of LBs or other α-Syn–positive inclusions, an alternative cause of parkinsonism should be pursued.22,25 Several clinicopathologic studies have shown that LBD accounts for 73% to 83% of cases of parkinsonism, including 42% to 63% of cases of PD, whereas other degenerative disorders masquerading as PD, such as DLB, MSA, or PSP, account for 9% to 33%.59 Awareness of the high rate of misdiagnosis and refinements in the clinical diagnostic criteria for PD seem to have improved the accuracy of diagnosis.109 Although data on the lesion pattern of α-Syn pathology and the multisystem degeneration in PD have provided insight into its course and the pathophysiology of its clinical subtypes, the cause and pathogenesis of PD remain unclear.112,113
Neuropathology of Parkinson’s Disease
The brain is usually grossly unremarkable or may show mild cortical atrophy, enlargement of the ventricles, and pallor of the SN and LC. Histopathologic examination reveals widespread α-Syn–immunoreactive deposits in neurons (LBs) and dystrophic neurites throughout the CNS. Recent studies have demonstrated α-Syn–positive deposits in presynaptic terminals of the cerebral cortex.89 Although PD is generally considered a disease of the CNS, LBs may also be found in sympathetic and parasympathetic neurons in PD patients, including the heart83,114 and the enteric nervous system.49 These findings have been related to gut dysmobility and cardiac disorders in many PD patients. For the distribution of LBs in PD, see other sources.25,59
Glial pathology is present in PD, with argyrophilic, α-Syn–positive, tau-negative inclusions in both oligodendroglia and astrocytes, including Bergmann glia.115–117 Ultrastructurally, they are composed of approximately 23- to 40-nm filamentous structures.118
There is variable neuronal loss in the midbrain and other subcortical nuclei, in particular the SNc, LC, and NBM: severe depletion of melanized neurons (45% to 66%) and dopaminergic neurons immunoreactive for TH (60% to 85%) in the A9 group of the SNc, particularly in the ventrolateral tier (area A, 91% to 97%) projecting to the striatum, followed by the medioventral, dorsal, and lateral areas. The susceptibility of dopaminergic neurons, among others, depends on their distribution within compartments of the SN defined by calbindin (CAB) immunoreactivity. The CAB-rich matrix is separated from the CAB-poor zones of nigrosomes, which show greater cell loss in the caudal and mediolateral region (98%) than in the adjacent matrix. From there it spreads to other nigrosomes and finally to the matrix along a caudorostral, lateromedial, and ventrodorsal progression.119 This temporospatial disorder corresponds to a somatotopic pattern of dopaminergic terminal loss that is more severe in the dorsal and caudal putamen than in the caudate nucleus (CN). The degree of A9 SNc cell loss and the resulting reduction of TH and DAT immunoreactivity in the putamen followed by the CN and nucleus accumbens show close correlation with the duration and severity of motor dysfunction.120,121 DAT immunoreactivity in the striatum is inversely correlated with the total α-Syn burden in the SN, but not with the LB count; nigral TH immunoreactivity does not correlate with α-Syn immunopositivity.122 These data support the concept of synaptic dysfunction and impairment of axonal transport by pathologic α-Syn aggregation.
A close relationship between decreased TH-negative neurons, LBs, and neuronal loss has been shown for the SN.123 The reduced intensity of DAT mRNA in the remaining SNc neurons is associated with decreased α-Syn mRNA expression in the SN and cortex with loss of the vesicular monoamine tranporter VMAT2 (a dopaminergic neuronal marker) in the striatum, orbitofrontal cortex, and amygdala, but not in the SN in the early stages of PD,124 whereas α-Syn inclusions and neuritic changes in the neostriatum increase with progression of PD.125 Akinesia and rigidity are linked to neuronal loss, but the percentage of LB-bearing and α-Syn–positive neurons in the SN is not correlated with disease duration and is apparently stable over time, with 39% of the neurons being involved on average. Such stability suggests that during the course of disease the destruction of LBs is equal to their production and that they are destroyed with the afflicted neurons.
The A10 group of dopaminergic neurons—ventral tegmental area, nucleus parabrachialis, and nucleus parabrachialis pigmentosus—projecting to the striatal matrix,126 thalamus,127 and cortical and limbic areas (mesocorticolimbic system) show less severe involvement (40% to 50% cell loss), whereas the periretrorubral A8 region, which contains only a few dopaminergic but CAB-rich neurons, and the central periventricular gray matter show little or no degeneration.128 Others have reported greater cell loss in the LC (area A6) than in the SN in both PD and AD patients.129 These changes differ from the age-related lesions in the dorsal tier of the SNc, which is involved only in the late stages of PD.130,131 Morphometric studies have shown a 35% to 41% reduction in pigmented SN cells, with severe loss of DAT-immunoreactive neurons in older persons132 and an increase in the volume of these cells.133 Some studies have estimated the loss to be 4.3% per decade,133 whereas others have reported almost 10% per decade.134 Recent morphometric stereologic studies of the human SN have revealed a significant loss of pigmented (−28.3%) and TH+ (−36.2%) neurons in older controls versus younger individuals, with hypertrophy of cells in older controls being intepreted as a compensatory mechanism to allow normal motor function despite neuronal loss. Patients with PD had a massive loss of SN neurons with significant atrophy of the remaining cells (20% of controls), but most of the patients examined were in the end stage of the disease.135
Degeneration of the nigrostriatal system causes dopaminergic denervation in the striatum progressing from the ventrorostal to the posterior putamen and CN. These changes are preceded by a preclinical phase ranging from 4.6 years for the anterior putamen to 6.6 years for the posterior putamen,136 with an annual decline of striatal DA intake of 8% to 10% and of DAT between 5.7% and 6.4% or 10% to 13%. Higher striatonigral dopaminergic neuron loss is suggested in early-onset than in late-onset PD.137 There is marked loss of DA (−89%) in the CN and more severe loss in the putamen (−98.4%), whereas DA loss in the GPi (−89%) and GPe (−51%) is not related to the pattern of putaminal DA loss.138 Reduction of striatal DA by 57% to 80%139 and DAT loss of 56% cause motor symptoms. Therefore, about 50% of dopaminergic striatal innervation appears to be sufficient for normal motor function.140 Striate DA release was reduced by 60% in PD patients, whereas frontal DA release was within the normal range, thus indicating that it remained preserved even in severe stages of disease.141 Sprouting of DA terminals and decreased DAT, which prevent the appearance of parkinsonian symptoms until about 60% loss of SN neurons takes place, also contribute to altered DA release and increased DA turnover and predispose to the occurrence of motor complications and dyskinesias as the disease progresses.142
SN cell degeneration is preceded by loss of neurofilament proteins; neuronal TH immunoreactivity; TH, DAT, and neurofilament mRNA; TH and DAT proteins; and cytochrome c oxidase—findings indicative of functional neuronal damage.143 Neuronal loss is accompanied by extracellular release of NM with uptake into macrophages, rare neuronophagia or phagocytosis of neurons by macrophages, astroglial reaction, and an increase in major histocompatibility complex class II–positive microglia, which may release proinflammatory cytokines and other substances that mediate immune reactions.144,145 Microglial reaction, together with the 35% to 80% pigmented neuronal loss reported in normal aging human SN, indicates the presence of a pathologic process that may be additive with specific age-related changes.146 Activated microglia may also be a source of trophic factors that upregulate neurotrophins in response to signals received from failing nigral neurons and may protect against reactive oxygen species and glutamate.147 Demonstration of microglial activation and corresponding dopaminergic terminal loss in the affected nigrostriatal pathology in early PD (and DLB) by PET and in the rat SN suggests that neuroinflammatory reaction contributes to the progressive degenerative process.145,148–150
Development of Lewy Body–Related Pathology
A hypothetic staging of brain pathology related to sporadic PD with ascending progression has been proposed.151–153 LB pathology may begin in the lower brainstem and involve the DMIX/DMX, intermediate reticular zone, and anterior olfactory nucleus, with the NBM and midbrain regions being preserved (stage 1), and then extend to the caudal raphe nuclei, gigantocellular reticular nucleus, and ceruleus-subceruleus complex (stage 2). These initial stages are considered asymptomatic or presymptomatic and may explain the early nonmotor (autonomic and olfactory) symptoms that precede the somatomotor dysfunctions.154,155 In stage 3, the LC, the central nucleus of the amygdala, the nuclei of the basal forebrain, and the posterolateral and posteromedial SNc are the focus of cytoskeletal changes and neuronal depletion, whereas the allocortex and isocortex are preserved. In stage 4, the anteromedial temporal limbic and neocortex and amygdala are additionally affected. Stages 3 and 4 have been correlated with clinically symptomatic stages, whereas in the terminal stages 5 and 6, the pathologic process reaches the neocortex, with the high-order sensory association cortex and prefrontal areas being affected first and later progressing to the primary sensory and motor areas or involving the entire neocortex (Fig. 74-2).
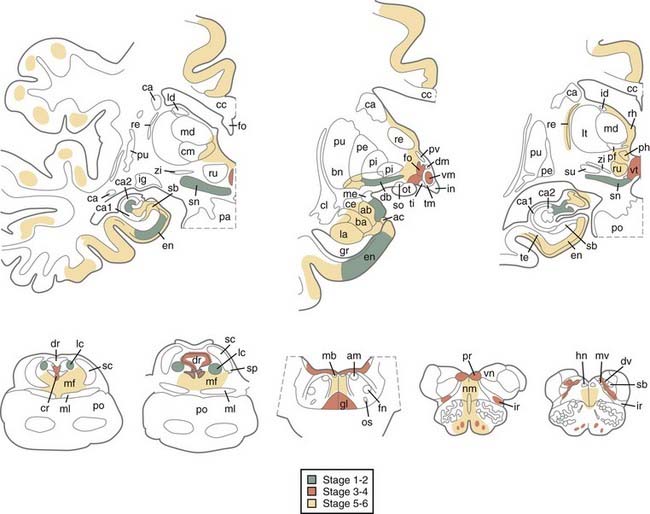
Recent studies have only partly confirmed this staging by showing that all brains of individuals with clinical PD reveal α-Syn–positive inclusions and neuronal loss in the medullary and pontine nuclei and SN and additional lesions in the NBM (90% to 98.5%), olfactory bulb (70%), limbic cortex (50% to 60%), cingulate area (32% to 46%), frontal cortex (29% to 31%), and amygdala (25%), which corresponds to LB stages 4 to 6.156 Although one study revealed significant interrater and intrarater reliability and supported the suitability of the staging procedure for application in routine neuropathology and brain banking,157 more recent studies have shown that some early PD symptoms may occur in rare patients with LB stage 2 (e.g., autonomic and bladder dysfunction, sleeping disorders, constipation, orthostatic hypotension, and depression) and more often in stage 3, in which most patients clinically manifested stiffness, asymmetric rigidity, and mild hypomimia but no tremor.153 In one study, only 6.3% of PD brains diverged from the hypothetic staging scheme of α-Syn pathology,61 whereas others revealed that between 17% and 47% of all cases of autopsy-proven PD did not follow the predicted spread of α-Syn inclusions and that in 7% to 8.3% of cases, the DMX was not involved despite definite α-Syn inclusions in the higher brainstem or even cortical regions.53,156,158,159 In contrast, in large autopsy samples, 49% to 55% of individuals with widespread α-Syn pathology were neurologically intact and lacked clinical symptoms or were not classifiable.53,160
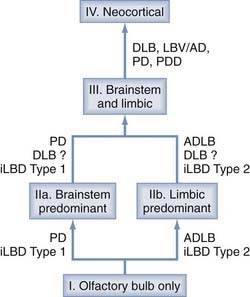
Therefore, the predictive validity of these concepts was suggested to be doubtful because there was no relationship between Braak stage and the clinical severity of PD,161 and their relationships to coincidental other pathologies are unclear.61,62 A new unifying system for LB disorders was proposed recently that correlates with nigrostriatal degeneration, cognitive impairment, and motor dysfunction.163 Although the previous classifications left 42% to 50% of elderly individuals unclassified, all were classifiable into one of the following stages: I, olfactory bulb only; IIa, brainstem predominant; IIb, limbic predominant; III, brainstem and limbic; and IV, neocortical (Fig. 74-3). Progression of individuals through these stages was accompanied by stepwise deterioration in terms of striatal TH concentration, SN pigmented cell loss, Mini-Mental Status Examination (MMSE) score, and Unified Parkinson’s Disease Rating Scale (UPDRS) part 3. There were significant correlations between these measures and LB-type α-Syn pathology. If validated in a greater proportion of patients, the proposed staging system would improve on its predecessors by allowing classification of a greater proportion of patients.
Early brainstem cell loss in PD is mainly confined to A9 neurons in the SN and is associated with more widespread formation of LBs that rapidly infiltrate the brain, particularly in patients with short survival, whereas those with disease onset at a younger age and longer survival usually have a typical clinical course consistent with the Braak PD staging scheme, which is not consistent with the unitary concept of the pathogenesis of LB pathology.164
Incidental LB disease (iLBD) is the term used when LBs are found in the nervous system in individuals without clinically documented parkinsonism. The distribution of LBs is similar to that in PD, with one or multiple brain areas involved, and some also have sparing of LBs in the limbic or temporal cortex (average Braak PD stage of 2.7), whereas in definite PD cases, more numerous LBs are found in all regions and the Braak PD stage is significantly higher (4.4). Decreased TH immunoreactivity was shown in the striatum and epicardial nerve fibers in comparison to normal controls, but not to the same extent as in PD.165,166 These findings suggest that iLBD is probably a precursor to or a preclinical form of PD and that the lack of symptoms is due to subthreshold pathology.
Single clinicopathologic case reports suggested that random eye movement (REM) sleep behavior disorder (RBD) may represent iLBD or an early clinical manifestation of PD167 or may precede or coincide with Parkinson’s disease dementia (PDD).168
Neuronal Vulnerability
The neurodegenerative lesions in PD show a selective vulnerability of SN neurons rich in NM and caspase-3, which have high expression of DAT mRNA, unrelated to their intrinsic capacity for DA synthesis.143 The majority of midbrain neurons severely affected in PD are melanized cells located in the densely packed ventral tier of the SNc; they contain CAB and glycolytic enzymes but are poor in DAT and arborize profusely in the extrastriatal components of the BG and sparsely in the striatum. Dopaminergic neurogenesis, intracellular and extracellular substances, and interactions among these factors have been discussed as essential causes of selective death of dopaminergic neurons in PD.169 The susceptibility of nigral dopaminergic neurons may further lie within the transcription profile of these cell populations.170 Neurons in the STN and GABAergic cells in the SNr, rich in calcium-binding proteins (calcineurin and parvalbumin) are either not affected or involved only in the terminal stages of PD. A close relationship among SN cell loss, α-Syn accumulation, and decreased TH immunoreactivity was seen, whereas the majority of pigmented SN but not LC neurons bearing α-Syn aggregates lacked TH reactivity, which leads to a decrease in cytotoxic α-Syn oligomeres. The decreased TH immunoreactivity in pigmented neurons can be considered a cytoprotective mechanism in PD,123 but it can also be preserved in neurons with early α-Syn accumulation.122
In the midbrain A9 area, the region of greatest vulnerability in PD, intracellular NM lipid changes, increased concentrations of α-Syn, and interactions with increased iron make dopaminergic nigral neurons susceptible to oxidative stress.171–175 Dysfunction of the BG circuitry in PD may affect the iron content not only in the SN but also in other BG as well.176 In the later stages of degeneration, SN neurons show a significant reduction in intracellular pigment, whereas those of normal morphologic appearance exhibit increased pigment density associated with an increased concentration of α-Syn with respect to its lipid component and loss of cholesterol. No such changes were observed in other NM-containing neurons in the A2, A6, and A10 areas in early PD, which emphasizes the selectivity of the early NM changes in A9 neurons.20
Symptom-Related Specific Lesion Patterns in Parkinson’s Disease
The major clinical subtypes of PD show specific morphologic patterns of pathophysiologic importance.
In the rigid-akinetic type, which occurs in about 50% of all patients, the ventrolateral SNc projecting to the dorsal putamen degenerates more severely than the medial parts projecting to the CN and anterior putamen. There is ventromedial gradient loss of TH- and DAT-immunoreactive fibers and endings from the dorsal to the ventral putamen, with prominent involvement of the met-ENK and SP-rich acetylcholinesterase-poor striosomes of the putamen projecting to the severely involved ventrolateral SNc. DA loss in the GPe and GPi does not match the more severe DA loss in the putamen.138 Preservation of the CAB-positive somatostatin-rich matrix, which shows increased somatostatin mRNA expression and projects to the GABAergic neurons of the SNr and motor thalamus, suggests that the endings richest in DAT are most sensitive to degeneration.177 Dopaminergic denervation of the striatum causes severe loss of dendrites on type I MSNs, the principal targets of dopaminergic input from the SN,178 and loss of convergent nigrostriatal DA and corticostriatal glutamate axon integrity, and the abundant α-Syn pathology in the neostriatum,125,179 dystrophic neurites in the CN, progressive loss of TH- and DAT-immunoreactive nigrostriatal fibers, and reduced met-ENK immunostaining suggest transsynaptic degeneration as a substrate for the severe motor deficits and decreased efficacy of DA-mimetic treatment in the late stages.180,181 Reduced dopaminergic input to the putamen causes increased activity of the GABAergic indirect striatal efferent loop via the SNr and GPi to the ventrolateral thalamus projecting to the cortex (Fig. 74-4; also see Fig. 74-1). Excessive excitatory glutamatergic drive from the STN and Gpi/SNr leads to an akinetic-rigid syndrome through reduced cortical activation. The increased GABAergic activity is reduced by levodopa treatment and disappears in the course of the disease, and changes in N-methyl-D-aspartate (NMDA) receptors and glutamatergic synapses may be generated, events favoring drug resistence and motor complications.182,183 Free endogenous DA may induce a relative hyperstimulation of dopaminergic receptors, which may account for the develoment of motor fluctuations and dyskinesias after levodopa treatment (see Fig. 74-4); it has also been related to increased pro-ENK mRNA levels in the striatum.184,185 Dyskinesia is critically related to the levodopa dosage via loss of synaptic depotentiation.186
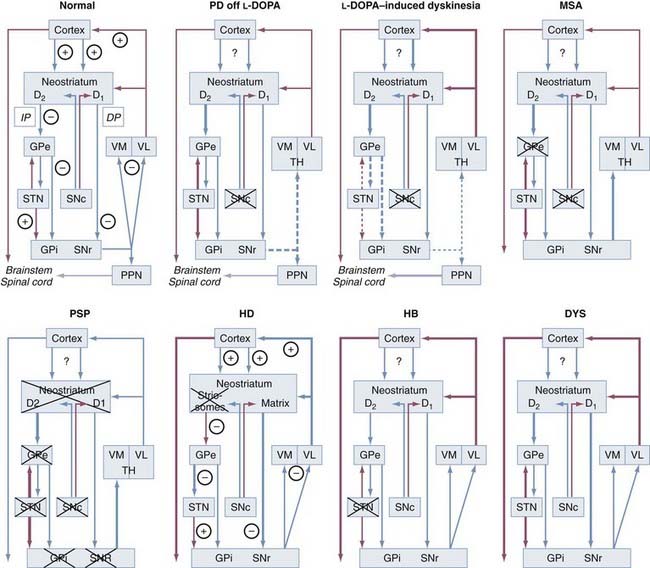
The tremor-dominant type of PD occurs in about 25% of patients and shows less severe total cell loss and less severe depletion of the lateral SNc, but damage to the retrorubral A8 field, which is usually preserved in rigid-akinetic PD. It projects to the matrix of the dorsolateral striatum and ventromedial thalamus and influences striatal efflux via the SNc and thalamus to the prefrontal cortex (see Fig. 74-1).187 In contrast to rigid-akinetic PD, DA levels in the ventral GPi were normal in PD with prominent tremor, a finding suggesting functional disequilibrium between GABAergic and dopaminergic influences in favor of DA in the caudoventral parts of the GPi, which may contribute to the resting tremor.138 Functional neuroimaging of patients with resting tremor suggests increased activity of the ventral intermediate thalamus and dysfunction of cerebellar connections,188 and recent morphometric magnetic resonance imaging (MRI) studies have for the first time demonstrated volume reduction in the cerebellum of PD patients with rest tremor, thus documenting involvement of the cerebellar-thalamic-cortical circuit in the pathogenesis of PD rest tremor,189 which has considerable implications for stereotactic treatment of tremor (deep stimulation of the ventral intermediate thalamus).190
Involvement of Extranigral Systems
PD is a multisystem disorder with involvement of many extranigral systems.22,25,59,151 The extranigral lesions in PD correlate well with early premotor symptoms, later nonmotor fluctuations, and advanced non–DA-responsive nonmotor features.191 The olfactory bulb has a special vulnerability for LB–α-Syn pathology, and early involvement has been reported in patients with PD, DLB, iLBD, and AD with LBs.151,163,192–197 Recent studies have found differences in the severity of LB pathology across the olfactory cortex: more severe involvement of the temporal than the frontal division of the piriform cortex, the olfactory tubercle, and the anterior entorhinal cortex.198 Most lesions are region specific and do not affect all neurons containing a specific transmitter or harboring LBs, which may explain the complex pattern of deficits of the disorder seen in PD. Both LBDs and PSP are associated with marked neuronal loss (40% to 55%) in the glutamatergic center median (CM) and parafascicular (PF) thalamic nuclei, which together with involvement of dopaminergic SN and cholinergic pedunculopontine neurons, may contribute to the movement and cognitive dysfunctions in both disorders.199,200 Both diseases further include the mesocortical dopaminergic system, fibers originating from the medial SNc, the ventral tegmental area, and the retrorubral area, and lesions in these areas are related to cognitive and behavioral impairment; the noradrenergic system, with loss of 40% to 50% of LC neurons, is more severely affected in PD patients with depression and dementia, whereas the dorsal vagal nucleus shows little but severe SP-positive neuronal loss and early involvement by α-Syn pathology. The adrenergic nuclei A1 and A2 in the medulla remain intact, whereas noradrenaline-synthesizing cells in the C11 area are depleted. The serotoninergic system suffers loss of TH-immunoreactive neurons in the central raphe nucleus and a 50% reduction in PH-8 serotonin-synthesizing neurons in the caudal midbrain and pons, which causes a reduction in serotonin transporters in both the striatum and midbrain, regions that are not affected in the early stages of PD.201,202 In the cholinergic system, the magnocellular part of the NBM shows a 30% to 40% reduction that does not correlate with age or disease duration; the reduction is less severe in patients with PD without dementia and in control individuals than in patients with PDD or AD. It is associated with a decrease in cortical and hippocampal cholinergic innervation, but its character as primary or secondary (retrograde) degeneration is under discussion.59,203 The nucleus tegmentalis pedunculopontinus (PPNc), a cholinergic loop nucleus in the caudal mesencephalic tegmentum, suffers 36% to 57% cell loss, which is strongly correlated with SN cell depletion but not with the duration of illness and LB counts, as well as unaltered parameters in the thalamus and STN, thus suggesting retrograde rather than primary degeneration of the nucleus. Overactivity of the PPNc in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced parkinsonism indicates dysfunction of the tegmentonigrosubthalamocortical loops, which contributes to disorders of gait, locomotion, posture, and cognition.204 The Westphal-Edinger nucleus, a cholinergic subdivision of the oculomotor complex that regulates pupillary constriction, suffers 55% neuronal loss in PD, which together with damage to the periaqueductal gray matter and nucleus interstitialis of Cajal, explains the neuro-ophthalmic and REM sleep dysfunctions. Pathologic lesions of the amygdala affect mainly the accessory cortical and lateral nuclei,205 which are involved in endocrine and autonomic dysfunctions. Other systems involved in PD are the reticular brainstem nuclei controlling the somatomotor and autonomic systems,151,206 the posterolateal hypothalamus, the CM-PF thalamus,207 the intralaminar thalamic nuclei,208 and the intermediolateral nuclei and Clark’s column in the spinal cord. The GABAergic system suffers a reduction in the activity of glutamate decarboxylase (GAD) in the BG, a decrease in GAD mRNA in the GPe, and a decline in GABA receptors because of degeneration of dopaminergic neurons, which increases GABAergic activity in the early stages of PD but disappears in the course of the disease and with the introduction of levodopa therapy. The peptidergic system shows a reduction in ENK, somatostatin, and neuropeptide receptors in the BG, an increase in somatostatin and mRNA expression, and a reduction of SP in the thalamus and NMDA receptors in the CN, whereas increased phosphorylation of NMDA receptors in striatal neurons is related to levodopa-induced complications.182 There is early involvement of the autonomic plexuses and nerves by α-Syn pathology,49 but the cutaneous nerves are affected in rather late stages of the disease.56
Etiology and Pathogenesis of Parkinson’s Disease
The etiology of PD has long been thought to involve both genetic and environmental factors, but until recently there has been no direct evidence to support either one as a causative factor.209,210 A molecular interaction between environmental risk factors and genetic factors has been implicated in the etiology of sporadic PD.211 The pathogenesis of the neurodegeneration in PD and other LBDs has been related to a cascade of multiple noxious factors, including misfolded aggregated α-Syn, the formation of free radicals, lipid peroxidation, oxidative and proteolytic stress, mitochondrial dysfunction and nuclear RNA deficits, protein-iron and NM-iron interactions, excitotoxicity, iron and transcriptional dysregulation, disorders in calcium homeostasis, neuroinflammation, impaired bioenergetics, inhibition or loss of neuroprotective mechanisms, perturbation of protein degradation systems such as the UPS and ALP, excitotoxicity from increased glutamatergic input, and interaction between these and other factors.84,112,209,210,212–216 The demonstration that α-Syn is degraded by both proteasome and autophagy pathways indicates a possible linkage between the UPS and ALP, and the fact that mutated α-Syn inhibits ALP functioning by binding to the receptor on the lysosomal membrane for the autophagy pathway further supports the assumption that the ALP may be related to the development of PD.83 Recent microarray analyses of dopaminergic SN neurons in PD patients revealed downregulation of members of the PARK gene family and dysregulation of multiple genes associated with programmed cell death (PCD), providing a “molecular fingerprint identity” of late-stage PD.217 All possible pathogenetic factors need to be carefully analyzed and are consistent with the multiple-hit hypothesis of PD.218,219
Pathology of Dementia in Parkinson’s Disease
PDD, which has an incidence rate of 95 to 112.5 per 1000 patient-years, a point prevalence close to 30%, and a cumulative prevalence of 48% to 83% after 15 to 20 years of follow-up, respectively, is suggested to have a lifetime incidence rate that is increased four to six times over that of age-matched controls.106,220 CNS lesions contributing to the cognitive impairment in PD are dysfunction of the subcorticocortical networks as a result of neuronal loss in the brainstem and limbic areas, cholinergic deficits in the cortex and thalamus associated with neuronal loss in the NBM and decreased striatal dopaminergic function,25,59,221 decreased nicotinic acetylcholine receptors (nAChRs),222 and degeneration of the medial SN and nuclei of other ascending pathways causing dysfunction of the striatosubfrontal and mesocorticolimbic loops. The cognitive deficits in early PD are associated with impaired nigrostriatal dopaminergic function, which results in abnormal processing in the cortico-BG circuit with reduced prefrontal and parietal metabolism, whereas mesocortical DA transmission initially appears to be preserved.223 Frequent lesions are cortical and hippocampal LBs and AD pathology with loss of synapses and neurons, presynaptic α-Syn aggregates,89 or variable combinations of these changes. They may have a common origin with mutual triggering because of synergistic reactions between α-Syn, amyloid-β, and tau protein, with frequent morphologic overlap or co-occurrence of lesions.101,103,104,224–226 However, epidemiologic, neuroimaging, and neuropathologic data support PDD as being distinct from AD.227
Although a few cortical LBs are found in virtually all cases of sporadic PD, the impact of cortical LBs and AD pathology on cognitive impairment is a matter of discussion. Some studies have demonstrated that the number of LBs in the frontal cortex or the number of LB densities in the limbic cortex is a better predictor of dementia in PD than AD pathology is.228–231 Cognitive impairment is often correlated with the density of LNs and neuritic degeneration in the hippocampus and periamygdaloid cortex, which causes disruption of the limbic loop and “disconnection” from key areas, as described in AD,232 and is a major basis for the dementia and visual hallucinations.233,234 The density of both limbic LBs and neuritic plaques correlated well with the severity of the dementia,235 although hippocampal atrophy and cell loss are not necessarily involved in the memory impairment in PD.236 The increasing cognitive decline with increasing pathologic LB stages from 3 to 6 secondary to progression of α-Syn pathology237 was not confirmed by others.224,238,239 PD patients without dementia may have AD pathology largely restricted to the limbic system (neuritic Braak stages <4), whereas patients with PDD often have severe AD lesions, with or without neocortical atrophy. However, quantitative stereologic studies found no global loss of neocortical neurons but could not exclude local neuronal loss in specific subpopulations in small but essential subregions in PD.240 In PDD, increased atrophy of the hippocampal head and amygdala is observed.241
In a large autopsy series of elderly patients with clinical parkinsonism (37.6% with dementia), only 3.2% of patients with dementia had LB Braak stages 3 to 5, whereas 7% of PDD patients had LB stages 4 or 5 with additional severe AD pathology (neuritic Braak stages 5 and 6). More than half of them showed a strong relationship between the severity of α-Syn and tau pathology. Other degenerative disorders with superimposed AD or vascular pathology accounted for 7% and 17%, respectively, and more than 31% had DLB with or without AD. Mild cerebrovascular lesions (lacunar state, few microinfarcts) were almost never associated with PDD. PDD patients had significantly more severe AD lesions than did patients without dementia, but LB Braak scores were only moderately increased in PDD.224 In the Sydney Multicenter Study of PD, 47% of 17 PDD brains had diffuse LBs as the only cause of dementia, whereas the others had mixed pathology.220 11C-PIB PET studies showed cortical Aβ deposits comparable to AD in some PDD patients, and fluorescence microscopy in postmortem sections revealed binding of PIB to LBs and NM in the SN of both PD and PDD brainstem, which was not seen in controls.242 The association among cognitive impairment, moderate LB scores, and AD lesions suggests an influence of AD-related pathology on the progression of neurodegeneration and on cognitive decline in PD.159
Genetic Forms of Parkinson’s Disease
Although familial parkinsonism with clear mendelian inheritance is rare (5% to 10%), the importance of genetic factors is increasingly being recognized.243 Molecular analysis of familial PD has identified point mutations and abnormalities in gene copy number in multiple genes, including SNCA (on chromosome 4q21), RKH, UCHL-1, DJ-1, the more common PTEN-induced kinase 1 (PINK1), LRRK2, and MAPT, many of them coding for proteins found in LBs or implicated in mitochondrial function, or both.22,244,245 To date, 15 genetic loci, PARK1 to PARK15 (“Park loci”), have been linked to familial forms of parkinsonism.246,247 Mutations in other genes have been linked to parkinsonism in small numbers of families or in individual cases but have not (yet) been assigned a PARK locus number.248 Several genes in which mutations have been linked to familial PD have been implicated as possible risk factors for sporadic PD.249,250 Genetic models contributed to understanding of the pathomechanisms of PD.251–253 In pathologically proven PD, glucocerebrosidase (GBA) gene mutations have been suggested to be the most common genetic factor for this disease.254
Different mutations in a single gene exhibit considerable clinical and neuropathologic variables both within and between kindreds. Neuropathologic studies of brains with α-Syn/SNCA mutations showed cell loss in pigmented brainstem nuclei with widespread LBs, many individuals with cerebral cortex A53T mutations (e.g., in the Contursi family) had conspicuous α-Syn neuritic pathology, tau-positive neuritic and perikaryal inclusions, and some had both tau and α-Syn pathology.96,255,256 Pathologically confirmed LBD with progressive parkinsonism and dementia caused by SNCA duplication results in hyperaccumulation of phosphorylated α-Syn in the brain.257 A recent neuropathologic study of a patient with familial PD secondary to A30P mutant α-Syn showed findings identical to those of idiopathic PD (IPD).258 Individuals with SNCA gene triplication have unusual neuronal loss and gliosis in the hippocampus, with pleomorphic LBs, α-Syn–positive glial inclusions, and widespread severe neuritic pathology.259 Autosomal recessive juvenile parkinsonism related to mutations in the PRKN gene on chromosome 6q25.2-27 shows severe cell loss in the nigrostriatal tract and LC with remarkable absence of LBs or α-Syn pathology but occasional cortical tau pathology.260 In compound heterozygous cases, LB pathology or NFTs have been identified.259,261 A Japanese family with autosomal dominant parkinsonism (PARK7) showed nigral neuronal lesions without LBs,262 reminiscent of some cases of familial juvenile PD caused by mutations of the PARK2 gene.260 One autopsy case with a UCH-L1 (PARK5) gene mutation had α-Syn pathology similar to IPD.263 Three patients with heterogeneous mutation of PARK6 have shown LB pathology, although this may be coincidental. No histopathologic studies of brains of homocygotic DJ1 (PARK7) mutation and of ATP13A2 (PARK9) carriers have been reported. Other autosomal dominant forms of PDD, with or without dystonia, may pathologically resemble PD with neuronal loss in the SN and striatum, with or without subcortical or cortical LBs, amyloid plaques, and NFTs.264–266 LRRK2 mutations (PARK8), a major cause of late-onset parkinsonism, and patients with multiple mDNA deletions have pathology comparable to sporadic PD247,250,263,267,268 but display variable neuropathology, including α-Syn and tau inclusions,252,269,270 thus suggesting an upstream role of LRRK2 in protein aggregation.90 LRRK2 is considered a key player in the pathogenesis of PD.271 The pathology in Japanese patients demonstrated nigral degeneration without LBs.272 Others have shown disparate pathologies such as nonspecific neuronal loss and gliosis in the SN to LBs and LNs or tau-positive pathology similar to PSP.273–276 Thus, PARK9 seems to be a form of MSA. Recent studies have raised the possibility of a role of TDP-43—which has been reported in FTLD-U, amyotrophic lateral sclerosis (ALS), some AD cases,277,278 and in familial LB disorders279—that is potentially analogous to the association of tau pathology in LB disorders. The combination of autosomal dominant parkinsonism, hypoventilation, depression, and severe weight loss (Perry’s syndrome280) is an early-onset (40 to 56 years of age), rapidly progressing disease morphologically characterized by massive neuronal loss in the SN without LBs and involvement of putative respiratory neurons in the ventral medulla.281 Recent studies have found TDP-43–positive, highly pleomorphic neuronal inclusions, dystrophic neurites, and axonal spheroids in a predominantly pallidonigral distribution. The pathologic forms of TDP-43 were neurochemically similar to those found in FTLD-U, and there were no mutations in progranulin (GRN) or TDP-43 (TARDBP) genes. These data indicate that Perry’s syndrome is a unique TDP-43 proteinopathy selective to the extrapyramidal system, with sparing of the neocortex and motor neurons.282 Mutations of the DCTN1 gene coding for the large subunit of dynactin, which is involved in microtubule-associated intracellular transport, have been found to be associated with Perry’s syndrome.283 The phenotypic variablity observed in familial PD reflects genetic interactions arising from differences in genetic backgrounds.
Dementia with Lewy bodies
DLB describes a progressive syndrome in the elderly with the core neuropsychiatric features of fluctuating levels of consciousness, visual hallucinations, and cognitive impairment associated with parkinsonism. The pathologic features include a variable burden of α-synucleinopathy with widespread cortical LBs and various degrees of AD-type pathology. Based on current clinical diagnostic criteria23,24,284,285 that have low sensitivity, early discriminatory diagnosis of DLB has been discussed,286 but there are still no generally accepted biomarkers to distinguish DLB from other dementias. In population-based clinical studies of people older than 65 years, its prevalence was reported to be 0.3%, which suggests that it could account for up to 10% of all dementia cases, consistent with DLB rates of 10% to 15% from hospital-based autopsy series. In a community-based study of individuals older than 65 years, 5% met consensus criteria for DLB (3.3% probable, 1.3% possible) and represented 22% of all dementia cases,287 which is consistent with estimates of LB prevalence in a dementia register confirmed by autopsy.288 In recent autopsy series, DLB was the second most frequent cause of dementia in the elderly after AD and accounted for 7% to 30%, with a mean incidence of 15%.289 Other population-based autopsy studies have found that LBs were evenly distributed between individuals with dementia and those without or showed no relationship between α-Syn–positive lesions and clinical findings.53 No classic epidemiologic studies for DLB have been reported.
No single gene determinant of DLB has been described, although a few families with autosomal dominant inheritance have been reported290–292 and patients with autopsy-confirmed DLB have an increased frequency of a familiy history of dementia.293 There is evidence that DLB and familial PD are related to an E46K mutation of α-Syn with autopsy findings of diffuse DLB without AD-type pathology,294 which has recently also been associated with GBA mutations.295 Mutations in the α-Syn gene may predispose to familial DLB.296 In a Belgian DLB family, a novel locus at 2q35-q36 has been identified,297 and in a family with pathologically confirmed early-onset DLB with extensive tauopathy, mutations in known genes were absent.298 Twin pairs may be discordant for neuropathologically confirmed DLB.299 The fact that many members of kindreds with mutations in the SNCA gene have some features of DLB, as well as the frequent occurrence of LBs in familial and sporadic AD, may suggest an overlap in the genetic factors of these disorders, but its pathogenesis is unknown.
Neuropathology of Dementia with Lewy Bodies
The macroscopic appearance of the brain in patients with DLB is usually similar to that in PD, including some degrees of diffuse cerebral atrophy and variable pallor of the SN and LC. The histologic hallmark is α-synucleinopathy manifested as LBs of the classic and cortical type and neuritic degeneration with or without AD-type pathology according to three main patterns: (1) widespread LBs associated with (sometimes numerous) cortical diffuse Aβ plaques and low Braak NFT stages, (2) widespread LBs with sufficient neuritic plaques and NFTs for an independent diagnosis of AD, and (3) “pure” LB disease involving widespread cortical areas without significant AD-type pathology.300 According to the revised consensus pathologic guidelines, LB density is assessed semiquantitatively, based on α-Syn immunohistochemistry, in five cortical regions (Tables 74-6 and 74-7). This protocol has been simplified by excluding the frontal region as the common occurence of occasional LBs in this region in patients with PD in the absence of dementia.235 According to the severity and anatomic distribution of LBs, patients are allocated to the brainstem-predominant (PD), limbic (or transitional), and neocortical type with widespread cortical LBs.23,301 These guidelines did not provide definite diagnostic criteria as is sometimes mistakenly assumed, and they were not included in the protocol of the Consortium to Establish a Registry for Alzheimer’s Disease (CERAD), which is used for the semiquantitative evaluation of neuritic plaques and NFTs.302 For revised neuropathologic criteria of DLB see Fujishiro and colleagues.303

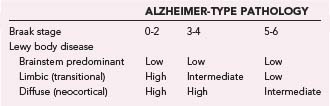
Cortical involvement by α-Syn pathology varies in DLB. In some cases, LBs are relatively restricted to the limbic structures (“transitional” LBD), whereas in others they are widespread in the cortical areas (“diffuse” or “neocortical” LBD); most numerous in the limbic structures, deep layers of the temporal and frontal lobe, and anterior cingulate cortex; less frequent in the parietal and occipital cortex; and absent in the primary sensory or motor cortex. The anatomic distribution of LBs in DLB does not follow the hierarchical spread of NFTs,303 although in some cases, clusters of LBs appear to be more closely related spatially to clusters of senile plaques than to NFTs. They affect various groups of neurons, including pyramidal cells and GABAergic interneurons, whereas cortical neurons expressing calcium-binding proteins are spared.304 The upper cerebral cortex and amygdala frequently show spongiform changes with loss of neurons and apical dendrites.
AD-type pathology of variable intensity and extent is frequent (see Table 74-7). Thirty-two percent to 89% (depending on the criteria used to define AD) of neuropathologically defined DLB cases have concomitant Alzheimer changes; in the Hisayama study, for example, about 60% of autopsy-confirmed DLB cases had severe AD pathology.305 Approximately 80% have numerous diffuse plaques and few or no neuritic plaques, around 60% have NFTs in the entorhinal cortex in moderate to severe intensity and rare neocortical NFTs, and about 30% have advanced AD-type changes with Braak NFT stages 5 to 6; a subgroup with minimal diffuse Aβ deposition and no neuritic AD lesions or lesions restricted to the hippocampus is referred to as “pure” DLB. They may show a preponderance of diffuse plaques with different proportions of Aβ subtypes (DLB with less Aβ-40 and AD with more frequent Aβ-40 than Aβ-42 deposits).306,307 In a personal series of 103 autopsy-proven DLB cases, 62% were classified as “pure” DLB without considerable neuritic AD lesions (68% transitional, 22% diffuse cortical forms), and 38% were associated with severe AD-type pathology (Braak stages 5 and 6). Reclassification of 51 Japanese autopsy cases gave similar results: 66.7% fulfilled the definition of DLB with Braak AD stages 1 to 4, whereas 33.3% had AD criteria (Braak stages 4 to 6; CERAD C) that did not meet these LB stages.308 Although the Aβ load and plaque density are consistently higher in patients with AD and DLB than in controls, the Aβ pathology of DLB patients with frequent single large clusters of diffuse plaques differs from that in “pure” AD.309 Neuritic plaques are frequently present at a burden equivalent to that in definite AD according to the CERAD protocol,310 and many patients have diffuse Aβ plaques with few neuritic elements228 or only minimal cerebral Aβ deposition. LBD and the LB variant of AD (LBV/AD) were found to differ from “pure” AD in that neuritic plaques generally do not contain paired helical filaments (PHFs) unless they are accompanied by neocortical NFTs.311 Patients with significant neuritic AD pathology sufficient for the diagnosis of definite AD with LBs can be divided into those with the clinical features of DLB, in whom LBV/AD should be diagnosed,312,313 and those with more prominent AD pathology and minor α-Syn pathology limited to the amygdala, which is considered a distinct form of α-synucleinopathy.314 Most DLB brains have an excess of AD-typical phosphorylated tau protein in the hippocampus and are considered to have higher Braak stages of AD pathology than seen in PD patients without dementia and age-matched controls but lower LB stages than those with “pure” AD.315,316 Biochemical evalution of tau, Aβ, and α-Syn overlap in sporadic DLB cases showed that all brains were associated with important deposits of all three proteins that were similar in quality to those in AD, thereby confirming less severe NFT pathology in DLB versus AD. Tau pathology was less severe in DLB (+AD) than in “pure” AD.317 Recent studies have shown co-occurrence of abnormal deposition of α-Syn, tau, and TDP-43 in AD, specific subtypes of FTLD-U, and DLB, which suggests common pathogenic pathways, probably triggered by genetic factors.318,319 Clinical diagnostic accuracy was higher for DLB cases with low Braak AD stages but only 15% to 39% in those with severe AD pathology.239,320 These data indicate that in DLB, AD pathology has more influence than the cortical LB distribution on both phenotype and diagnostic accuracy.320,321 Cortical PIB binding in DLB is associated with AD-like characteristics.322
| HEREDITARY |
| Huntington’s disease |
| Neuroacanthocytosis |
| Huntington’s disease–like syndromes |
| Benign hereditary chorea |
| Dentatorubral-pallidoluysian atrophy |
| Paroxysmal choreoathetosis |
| Wilson’s disease |
| Lesch-Nyhan syndrome |
| Hallervorden-Spatz disease (pantothenate kinase–associated neurodegeneration) |
| Ataxia-telangiectasia |
| Lesch-Nyhan syndrome |
| Hereditary ferritinopathy |
| Spinocerebellar ataxia (especially SCA-17) |
| SPORADIC |
| Sydenham’s chorea |
| Chorea gravidarum |
| Autoimmune disease (systemic lupus erythematosus) |
| Antiphospholipid syndrome |
| Behçet’s syndrome |
| Metabolic derangements |
| Drug induced (dopamine receptor blocking drugs, stimulants, levodopa, anticonvulsants) |
| Focal lesions (vascular) |
| Infectious (human immunodeficiency virus, abscesses in the basal ganglia, Creutzfeldt-Jakob disease) |
| Paroxysmal chorea (paroxysmal nonkinesigenic/kinesigenic dyskinesia) |
“Pure” DLB cases usually show no significant differences in neocortical synapse density and synaptophysin reactivity versus controls, whereas severe synapse protein loss comparable to AD is seen in LBV/AD.310,311,323 Despite comparable neocortical LB counts and choline acetyltransferase (ChAT) losses, the DLB patients had significantly less dementia than did the LBV/AD patients.324 There are differences in the expression of α-Syn and 20S proteasome isoforms in DLB, AD, and aged controls.325,326 DLB cortex does not show overexpression of α-Syn, but there may be a primary defect in clearance of the protein,327 and insoluble α-Syn did not correlate with the number of LBs but did correlate strongly with the expression of several heat shock proteins.328
Hippocampal pathology in DLB is usually less prominent than in AD, and neuronal loss in the perforant pathway is milder and more variable than that in AD.329 Diffuse neuritic lesions in the CA2/3 region of the hippocampus were initially regarded as a means to discriminate diffuse DLB from PD,330 but recent studies have shown more frequent involvement of the CA2/3 subareas by α-Syn deposits in DLB than in PD/PDD (79% versus 36%).25,159 These data suggest a specific involvement of hippocampal projections in DLB.331
The cerebellum in DLB and PD shows Aβ-positive inclusions in the white matter, with most being located in Purkinje cell axons and not observed in MSA.332 Loss of cholinergic pedunculopontine tegmental neurons occurs in DLB but is less severe than in MSA, probably because it does not represent the primary mechanism of RBDs in these conditions.333 In DLB, LBs, Lewy neurites, and dystrophic axons were observed in the ventrolateral medulla (VLM), which controls the sympathetic output maintaining arterial pressure, but the number of catecholaminergic and serotoninergic neurons was not significantly depleted, thus suggesting that the orthostatic hypotension in DLB is due to involvement of sympathetic ganglia neurons rather than VLM neurons.334
Glial lesions in DLB include α-Syn–positive, tau-negative, thorn-shaped astrocytes or coiled bodies,118,335 but the glial cytoplasmic inclusions (GCIs) of MSA are not seen. The role of microglia in the evolution of DLB is unresolved,336 but neuroinflammatory reactions have been implicated in the neuronal damage, including LB formation.337
Dementia with Lewy Bodies versus Parkinson’s Disease/Parkinson’s Disease Dementia
The question whether DLB and PD/PDD are different disorders or represent distinct phenotypes in a continuum within the spectrum of LBDs and their relationships to AD have been a matter of controversy.25,285,338–340 Their neuropathologies show both similarities and slight differences. Morphology, molecular isoforms, and immunohistochemistry of cortical and subcortical LBs and the ascending spreading pattern of α-Syn pathology do not significantly differ between both phenotypes, with the late stages 5 and 6 of LB pathology suggesting a transition between PD and DLB, although DLB has a higher density of cortical LBs and AD lesions than PDD does.341 The SN and other subcortical nuclei in DLB show variable neuronal loss that is often indistinguishable from sporadic PD, except for the occasionally more severe loss in the ventrolateral or dorsolateral tier as opposed to the predominant cell loss in the medioventral parts of the SNc in PD/PDD. A major morphologic difference is the significantly more frequent and severe load of diffuse amyloid plaques in the striatum in DLB, dissociated from cortical and limbic AD-type lesions, than that seen in PD patients without dementia, who are virtually free of Aβ pathology.341–344 Tau pathology in the striatum is also more frequent in DLB. No correlations have been found between LB density in any brain area of DLB patients with cognitive changes or parkinsonism, between LB density and Braak AD stages or the frequency of neuritic plaques,224 or between LBs in the cortex and SN, thus suggesting that DLB should not be considered a severe form of PD. Although LB densities, in general, cannot separate DLB from PDD, there is more frequent involvement by α-Syn deposits in the limbic system in DLB, in particular the CA2/3 subareas of the hippocampus.159 The severity and duration of dementia appear to be related to both increased parahippocampal LB density and neuritic plaque grade. A screening algorithm suggesting that LB density thresholds in the parahippocampus may distinguish PD with dementia from PD without dementia independent of other pathologies205 awaits further confirmation. However, individuals can show significant cognitive disturbances with minimal cortical LBs and, conversely, widespread cortical LB pathology without cognitive decline.237,238
Differences in the proportion of Aβ-40 deposits and LB pathology in AD and DLB, the lack of relationship of LB formation to the number of AD lesions, variation in the distribution of tau pathology and cholinergic biochemistry, and genetic differences in apolipoprotein ε4 and ε2 frequency345,346 argue for separation of DLB and AD. However, both PD and cortical LBD manifested the same seven α-Syn isoforms but with changes in expression.347 Conversely, more severe LB pathology is found in DLB with severe AD than in diffuse forms, and the various biochemical and morphologic overlapping between PD, DLB, and AD, including colocalization of tau and α-Syn epitopes in LBs, suggests that the process of LB formation is triggered, at least in part, by AD pathology.25,348,349 This collision of two processes may occur in the same brain region or even within single cells in the human brain, for example, in LRRK2 mutations,273 in animal models of PD,350 and in rare familial forms of DLB,298 with association of phospho-tau and α-Syn in both NFTs and LBs351 and in vitro promotion of tau aggregation by α-Syn and vice versa.352 Others have suggested that amyloid rather than tau enhances α-Syn pathology in the human brain and tg mice.225,353 These interactions highlight the interface between these and other misfolded proteins,354 which may represent molecular mechanisms in the overlapping pathology of AD and PD/DLB,103,104 and together with recent biochemical data on tau and Aβ317 challenge the view of DLB as a distinct entity. The global cortical amyloid burden is high in DLB but low in PDD, thus suggesting that Aβ may contribute to the cognitive impairment in DLB.355
The frequency of associated cerebrovascular pathology in DLB is lower than in both PD and AD but higher than in age-matched controls, which suggests less susceptibility to stroke in the DLB population. Conversely, as in PD and AD, the cognitive impairment appears to be independent of coexisting vascular pathology and is related mainly to AD or cortical LB pathology, or a combination of both.224 In conclusion, the pathology underlying the cognitive impairment in PDD and DLB is heterogeneous, but there are some differences in the topography and severity of lesions between both phenotypes that need further elucidation.
Biochemistry of Dementia with Lewy Bodies
In addition to nigrostriatal degeneration with disruption of dopaminergic input to the striatum and low DAT in DLB in comparison to AD, the cholinergic system is abnormal356 as a result of NBM pathology.357 Although neuronal loss in the cholinergic basal forebrain is commonly found in these disorders, early and more widely spread cholinergic losses (reduced neocortical ChAT levels and ChAT-immunoreactive neurons as a result of NBM pathology) differentiate DLB from AD.358 Muscarinic M1 and M2 receptors in DLB are affected differently from those in AD,359 thus indicating differences in the underlying extent of pathology in the cholinergic projection from the basal forebrain. Deficits in neuronal nAChRs in the thalamus, CN, and putamen related to the loss of dopaminergic neurons360 may contribute to the neuropsychiatric features in DLB.361 Involvement of different cholinergic nuclei in the amygdala in DLB and AD may be due to involvement of both the basal forebrain and brainstem nuclei,362 although the cholinergic projective neurons are intact in DLB and provide a rationale for cholinergic therapy.363 Reduction of nAChR binding in the putamen did not correlate with α-Syn expression in both PD and DLB364 or with plaques and NFTs in AD and DLB, so it is not a reliable marker of cognitive loss in these disorders365; however, other researchers have shown a different pattern of nAChR loss in AD and DLB.366 The striatum in DLB exhibits a variable reduction (more than in AD, less than in PD) in TH immunoreactivity and loss of DA markers, findings reflecting the degeneration of DA neurons in SN.367
Multiple System Atrophy
This adult-onset, progressive neurodegenerative disorder of unknown etiology is clinically characterized by autonomic failure and motor impairment with variable combinations of poorly levodopa-responsive parkinsonism, cerebellar ataxia, and corticospinal tract dysfunction. Diagnostic consensus criteria recommend classification into MSA-P (predominant parkinsonism, 80% in the western world) and MSA-C (predominant cerebellar features associated with olivopontocerebellar atrophy [OPCA], around 20%),21 and red flag categories had a specificity of 98.3% and sensitivity of 84.2%.368 These criteria were simplified recently.369 Autonomic dysfunction (urogenital dysfunction, orthostatic hypotension) is common in both variants and reflects degeneration of the central and peripheral autonomic pathways.370 MSA is less common than PD, with a prevalence of 1.9 to 4.9 per 100,000 and an incidence of 3 per 100,000 individuals.371 Familial clustering is uncommon, but familial forms with autosomal recessive inheritance may occur,372 and a single patient with MSA-C had an abnormal expansion in one of the two spinocerebellar ataxia 3 (SCA3) alleles.373,374 No mutation in the α-Syn gene or effect of genetic variation in other genes related to PD and no clearly established environmental risk factors have been identified,373 but in the rostral pons of MSA patients, significant changes in the expression of 254 genes (180 downregulated, 74 upregulated) were found, some similar to those in the SN of patients with PD and others related to oligodendrocyte function. SNCA variants have been shown to be associated with increased risks for MSA.376
The brain grossly reveals atrophy and green-gray discoloration of the putamen in MSA-P and atrophy of the cerebellum, middle cerebellar peduncles, and pons in MSA-C. The pigmented brainstem nuclei are pale, and cerebral cortical atrophy may be present. The histologic hallmark is cytoplasmic α-Syn–immunoreactive oligodendroglial inclusions (GCIs) within oligodendroglial cells, demonstration of which is required for the diagnosis of definite MSA.22,377 Less frequent are neuronal cytoplasmic inclusions (NCIs), neuronal intranuclear inclusions, astroglial cytoplasmic inclusions of similar composition, rare Ub-positive nuclear or cytoplasmic inclusions (or both) within neurons resembling those in motor neuron diesease, and Ub-positive neuritic dystrophy reminiscent of neuropil threads but lacking tau immunoreactivity. These changes are associated with neuronal cell loss, atrophy, reactive gliosis, iron deposition, and demyelination; they occur in an antomically selective manner and affect the pons, medulla, putamen, cerebellum, SNc, and preganglionic autonomic structures.22,370,378,379 Degeneration of the striatonigral system is most severe in the dorsolateral caudal putamen and lateral SN, thus suggesting transsynaptic degeneration of striatonigral fibers. Additional damage to the GP and STN leads to dysfunction of these inhibitory nuclei projecting to the motor thalamus, a mechanism similar to that in PSP (see Fig. 74-4). Microglial proliferation and the nonheme iron (Fe3+) content in the SNc and GP are more prominent than in LBD and controls, similar to PSP.380 Neuronal loss in the hypothalamus, catecholaminergic and noradrenergic groups in the VLM arcuate nucleus, and the intermediate zone of the anterior spinal horn and vagal autonomic nuclei contributes to parkinsonism and the autonomic and endocrine dysfunction in MSA,377,381 whereas mesopontine cholinergic neuron involvement (PPTNc, laterodorsal tegmental nuclei) may contribute to REM behavior disorders (RBDs).333 In the cerebellum, the Purkinje cells are more severely affected in the vermis, with atrophy of the olivary nucleus, cerebellopontine fibers, and basis pontis. The degree and pattern of striatonigral and olivopontocerebellar degeneration correlate with the density and distribution of GCIs, the duration of illness, and the clinical subtype of MSA,382,383 but there is no clear correlation between α-Syn glial burden and neuronal disease. Based on semiquantitative assessment of neuronal loss, astrocytosis, and GCIs in various brain areas, four degrees of severity were distinguished for both striatonigral degeneration and OPCA.383 Both grading systems were shown to reflect the initial symptoms, disease progression, and clinical key features, but a low correlation between involvement of the two major systems and the natural history of the disorder was observed. Postmortem MRI changes in the putamen (type 1, mild atrophy and isointensity; type 2, atrophy and diffuse hyperintensity with a hyperintense putaminal rim [HPR]; type 3, putaminal atrophy and isointensity or hypointensity with HPR) reflect various degrees of histologic changes.384 Fluorodeoxyglucose (FDG)-PET studies showed a stable metabolic brain network characterized by decreased metabolism in the putamen and cerebellum, significantly different from healthy controls.385 These changes range from “minimal-change MSA” with degeneration restricted to the SN386,387 to fully developed lesions. Widespread occurrence in the initial stages with a short disease duration underlines the multisystem character of “glial degeneration” of the disease.388 Involvement of other neuronal populations may be linked to frontal lobe dysfunction and cortical motor involvement,389,390 but AD-type lesions were less frequent in MSA than in age-matched control brains.25 LBs were seen in 10.7% to 22.7% of autopsy-proven MSA cases,25,373 whereas AD-type lesions were rare.391 Patients with MSA-P show more severe and more widespread cognitive dysfunctions than do those with MSA-C, which may be associated with prefrontal involvement.392 Loss of mesopontine cholinergic neurons in MSA is more severe than in DLB but may not be associated with RBD.333
The GCIs are argyrophilic, triangular, sickle- or half moon–shaped, oval or conical cytoplasmic aggregates composed of fibrillary α-Syn, Ub, and a large number of multifunctional proteins, including 14-3-3 protein and LRRK2347,393,394; they form a meshwork of loosely packed filaments or tubules 15 to 30 nm in diameter with a periodicity of 70- to 90-nm and straight filaments, both consisting of polymerized α-Syn, granular material, and variable types of filaments.36,375 Tau protein appears not to be a principal component.395 The soluble α-Syn in GCIs differs from the insoluble form in LBs, probably because of different processing of α-Syn.396 An early change is the accumulation of p25α (tubulin polymerization–promoting protein [TPPP]), with its localization and pattern being virtually identical to that of α-Syn.397–399 TPPP is a potent stimulator of α-Syn agregation400 and may decrease myelin basic protein, thereby favoring both the deposition and fibrillation of α-Syn and altering myelin metabolism.397,400 Myelin lesions are well documented in MSA and are most frequent in the external capsule, striatopallidal fibers, cerebellar white matter, middle cerebellar peduncle, and transverse pontine tracts, but they can be identified in otherwise apparently normal areas.401 GCIs and microglial burden are greatest in mild to moderate white matter lesions and decrease with progression of myelin damage, but they showed no correlation with the severity of gray matter damage in the putamen and SN, which is in line with previous findings of decreased GCIs in severely affected areas.402 Oligodendroglial changes are more widespread than α-Syn–positive GCIs, thus suggesting that primary oligodendroglial pathology is the main engine that drives the disease process, with secondary degeneration of the oligodendroglia-myelin-axon-neuron complex.403 MSA can be considered a primary oligodendrogliopathy and is a unique entity associated with synucleinopathy, early myelin dysfunction, and axonal damage leading to secondary neurodegeneration.404 Incidental MSA showing widespread GCIs without clinical neurological disease is rare,386,405,406 as is the coexistence of MSA and PSP.407
Tauopathies
Tau abnormalities are increasingly common in the neurodegenerative disorders known as tauopathies, with filamentous neuronal and glial tau inclusions associated with the degeneration of affected brain areas being morphologic hallmarks.408,409 Human tau proteins are encoded by a single gene consisting of 16 exons on chromosome 17q21. The adult human brain has six tau isoforms that differ by the presence of either three (3R) or four (4R) carboxy-terminal tandem repeat sequences of 31 to 32 amino acids that are encoded by exons 9 to 12. The triplets of 3R- and 4R-tau isoforms differ as a result of alternative splicing to generate isoforms with or without 29– or 58–amino acid inserts.410 The splice site mutations result in increased inclusion of exon 10, which causes a relative release of tau isoforms containing 4R domains over those containing 3R domains. This could be a central mechanism in several tauopathies.411 Western blot binding has distinguished patterns of soluble and insoluble tau different from those of other disorders, and these patterns have formed the basis for biochemical classification of the major tauopathies.221 In contrast to AD, postencephalitic parkinsonism (PEP), and Guamanian ALS-Parkinson’s disease complex (ALS-PDC) (which have 3R and 4R triplets of 68, 64, and 60 kD), PSP and CBD contain predominantly 4R-tau doublets with two 68- and 64-kD insoluble tau bands at exon 10; they are the most common sporadic tauopathies and are often manifested as atypical parkinsonism. The morphology of the neuronal and glial inclusions is distinctive (see Table 74-2), but there is frequent overlap between different disorders; DJ-1 protein is present in neuronoglial inclusions in tau diseases and is associated with both 3R- and 4R-tau isoforms.412 It is suggested that the isoform composition of sporadic tauopathies may have a spectrum of findings in individual cases and that the cellular isoform composition may differ in various brain regions.413
Progressive Supranuclear Palsy
PSP, or Steele-Richardson-Olszewski syndrome, a predominantly sporadic progressive movement disorder, is the most common atypical parkinsonian disease,371,414 with incidence rates increasing with age from 0.3 to 14 per 100,000 per year and a prevalence between 1.4 and 6 per 100,000.415 The mean age at disease onset is around 60 years, and mean survival is 5 years. PSP is clinically manifested by progressive postural instability and falls, supranuclear vertical gaze palsy, and frontal cognitive disturbances,416,417 but the presence of atypical cases with a variety of clinical syndromes is indicative of the heterogeneity of PSP.418,419 Two clinical phenotyes have been termed Richardson’s syndrome (RS) for typical expression of the disease with a rapid course and PSP-parkinsonism (PSP-P), which often mimics PD.420,421 Given these variants, it is not surprising that overall diagnostic accuracy is just 70% to 75%.422 The clinical syndrome of PSP may arise through several pathologic processes: RS, PSP-P, FTDP-17, FTLD-U, FTLD-MND (FTLD with motor neuron disease), CBD, progressive subcortical gliosis, and MSA.22 Research pathologic criteria for PSP have been proposed.20,421,423
Typical cases show atrophy of the STN, midbrain, and pontine tegmentum, loss of pigment from the SN, and atrophy of the superior cerebral peduncle424; in addition, there may be mild cortical atrophy. Histologically, PSP is characterized by MSA, globose tangles (different from the flame-shaped cortical NFTs), neuronal threads, and tau deposits in glia in specific BG, diencephalon, and brainstem regions, including the SNc, SNr, LC, STN, pallidum, striatum, periaqueductal gray matter, red nucleus, raphe nuclei, oculomotor complex, trochlear nuclei, pontine tegmentum and basis, dentate and inferior olivary nuclei, and spinal gray matter.22 They differ ultrastructurally and biochemically from those in AD or PEP in that they overexpress 4R-tau with a polymorphous tandem repeat allele located in the intron of the tau gene and are composed of 12- to 15-nm straight tubules/filaments containing 4R-tau with a sequence encoded on exon 10.425 Swollen achromatic neurons in the cortex and BG contain tau aggregates with straight filaments, which are also present in “tufted” or thorn-shaped astrocytes (stellate with fine radiation processes and straight, irregular 22-nm filaments, in contrast to the “astrocytic plaques” of CBD) and in oligodendroglia as “coiled bodies” (straight 14-nm filaments with a relatively smooth surface) throughout the neuraxis, in particular, the white matter. Only few tangle-bearing neurons but many tau-positive oligodendrocytes are seen in the brainstem tegmentum and pontine nuclei, but not in the SNc, and they exhibit DNA fragmentation and may express caspase-3.426 The astrocytic tau pathology and microglial activation in PSP correlate with NFT density and neuronal loss.427,428 Cortical pathology predominates in the precentral gyrus, entorhinal cortex, hippocampus, dentate granule cells, and extranigral A10 midbrain cell groups429; the distribution of NFTs is similar to that in PEP and Guamanian ALS/PDC. Severe damage to the GPi, GPe, SNr, and STN causes dysfunction of striatal efflux to the motor thalamus, thereby accounting for akinesia-rigidity and its resistance to dopaminergic treatment (see Fig. 74-4). There are significant morphologic and biochemical differences between the two clinical phenotyes: PSP-P has a significantly lower tau pathology score with more restricted involvement of the SN, STN, and Gpi and a mean 4R-tau/3R-tau ratio of 2.8, whereas RS has more severe and more widely distributed tau pathology, the score of which correlates negatively with disease duration,430,431 and a mean 4R-tau/3R-tau ratio of 1.6. The cortical tau pathology in PSP differs from that in AD, with the highest density seen in the prefrontal and limbic areas and the major location being in the deeper cortical layers, as compared with a bimodal distribution in AD. In patients with PSP and cortical symptoms, tau pathology is more excessive in the cortex because of loss of synapses.432 Hippocampal and amygdala pathology is usually minimal, but 20% of patients have ballooned neurons and argyrophilic grains (AGs) in the limbic region.433 LBs and cerebrovascular lesions are rare,425,430,434 although vascular PSP has been described as a multi-infarct disorder.435 The LBs in PSP are suggested to represent an independent disease process.436 The major genetic risk factor for sporadic PSP, which represents around 85% of all cases, is a common variant in the gene encoding tau protein, with a prevalence of A0/A0 genotypes and the presence of the H1/H1 genotype being a genetic predisposition marker.437 Recent studies have suggested that this may result in altered expression for specific tau protein isoforms.425 In PSP-P patients, no mutations of microtubule-associated protein (MAP) tau in exons 1 and 10 were found.438 Most, but not all cases of familial PSP are considered to be part of the spectrum of disorders of FTDP-17, which is associated with MAPT mutations, whereas small kindreds of PSP are linked to chromosome 1.439
Neurochemically, nigrostriatal dysfunction in PSP is associated with an 80% to 90% reduction in DA and a 40% to 50% reduction in homovanillic acid (HVA) in the CN and putamen, whereas the mesocortical and mesolimbic dopaminergic systems remain intact in comparison to PD. FDG-PET studies showed a specific metabolic brain network characterized by metabolic decreases in the brainstem and medial frontal cortex that is significantly different from healthy controls.385 Cell loss from the cholinergic NBM, striatum, and thalamus is less severe than in AD, but the cholinergic systems are severely affected, with a 40% to 80% reduction in ChAT activity, which may play a role in the motor and cognitive dysfunction in PSP, and the 60% loss of neurons in the PPNc may correlate with disequilibration in PSP.440,441 Mental decline is often ascribed to subcortical pathology related to dysfunction of the striatofrontal or prefrontal circuits as a result of degeneration of the BG and brainstem tegmental nuclei affecting the hippocampal and prefrontal structures,442 but there is no difference in subcortical tau pathology between PSP patients with and without cognitive impairment.432 Loss of postsynaptic DA D2 receptors from the BG accounts for the failure of response to levodopa treatment.443
Corticobasal Degeneration
CBD, previously described as corticodentatonigral degeneration with neuronal achromatism,444 is a rare, sporadic, late-onset progressive disorder of unknown etiology that is clinically manifested as non–levodopa-responsive rigidity with focal asymmetric cortical signs (apraxia and aphasia; “alien hand syndrome”) and frontal lobe dementia.445 The clinical syndrome is not specific for CBD, and clinical features of pathologically proven CBD, PSP, Pick’s disease (PiD), and FTLD overlap.446 Neuropathologic evaluation reveals depigmentation of the SN and asymmetric atrophy of the posterior frontal, parietal, and perirolandic cortex along with neuronal loss, superficial laminar spongiosis, and gliosis, with the temporal and occipital lobes being unaffected. The histologic hallmarks of CBD are prominent neuronal and glial cytoplasmic tau inclusions (ballooned/achromatic neurons) in the cortex, BG, brainstem, and cerebellum and extensive accumulation of tau-positive thread-like processes throughout the brain, which are more widespread than in PSP. The ballooned neurons are similar to those seen in PiD and contain phosphorylated neurofilament protein and αB-crystallin. The aggregates of CBD are composed of predominantly 4R-tau isoforms with exclusively exon 10 isoforms,447 identical to PSP and certain forms of FTDP-17, and they do not stain with antibodies to 3R isoforms and Ub.19,448,449 Ultrastructurally, they consist of 10- to 15-nm filaments, with fewer 25- to 30-nm filaments, granular material, and lipofuscin, resembling those seen in PSP.450 The twisted ribbons in CBD are different from the PHFs in AD. In the white matter, “astroglial plaques” and numerous inclusions involve both astrocytes and oligodendroglia (“coiled bodies”). They do not stain for α-Syn or Ub and thus differ from the GCIs in MSA. Astrocytic plaques, typical for and of significant diagnostic value in CBD,19 resemble the neuritic plaques in AD, but instead of clustering around amyloid cores, the tau-positive processes surround unstained neuropil. They are frequent in the prefrontal and orbital regions and can be found throughout the striatum, but are uncommon in the brainstem.451 AGs, which also have a predominance of 4R-tau452,453 and show no differences in frequency of the extended haplotype between PSP and CBD, occur in both disorders more frequently than in controls without dementia and patients with AD.433 Although the tau isoforms in CBD may differ from those in PSP, they share as a common risk factor the extended H1 tau haplotype, which is overrepresented in both disorders, thus reinforcing molecular commonality of the two conditions.416,454 Cases described in the literature as familial CBD are now regarded as FTDP-17 associated with tau gene mutation.455 Minimal research pathologic criteria for CBD are cortical and striatal tau-positive neuronal and glial incusions, particularly astrocytic plaques and thread-like lesions in both the gray and white matter, as well as neuronal loss from the SN,19 but these criteria alone do not allow distinction from familial tauopathies (FTDP-17, PiD), so additional genetic and molecular information is necessary. An increased apparent diffusion coefficient of the putamen (by diffusion-weighed brain imaging) provides good discrimination between PD and atypical parkinsonism (e.g., Richards’ PSP syndrome and CBD and involvement of the superior cerebellar peduncle in PSP).456
Postencephalitic Parkinsonism
This progressive neurodegenerative disorder, a sequela of encephalitis lethargica and other viral encephalitides, is clinically featured by rigid parkinsonism, oculomotor lesions (ocular palsy and oculogyric crises), and cognitive impairment.457 Sporadic cases have been reported.458,459 In addition to depigmentation of the SN, neuropathologic evaluation reveals marked neuronal loss and astrocytosis in the brainstem—particularly in the SN (diffuse and more marked than in PD)—and widespread occurrence of tau-positive globose NFTs, neuropil threads, and glial inclusions in the brainstem, BG, NBM, and amygdaloid complex but less severe occurrence in the striatopallidum, thalamus, hypothalamus, and cerebellum. NFTs and neutrophil threads, composed of 22-nm twisted tubules with occasional straight filaments showing 3R- plus 4R-tau and Ub immunoreactivity, are identical to those in AD. Tau-immunoreactive astroglia are seen in affected areas, whereas tufted astrocytes, oligodendroglial inclusions, astrocytic plaques, and ballooned neurons (all typical of CBD) or Pick bodies are absent. Perivascular aggregates of lymphocytes and plasma cells can be found in the midbrain for many years after the initial encephalitic illness, although they are sparse in long-surviving patients. Microglial activation may be striking. Cortical pathology is common, with NFTs mainly in the hippocampus and less often in layers II and III of other cortical areas, different from that in AD.460 Neither LBs nor α-Syn pathology was detected in PEP.461 The distribution of lesions shows similarities and overlapping with PSP, and it is extremely difficult to distinguish the two disorders by histopathology alone,457,458 although there are distinctive clinical differences and subtle deviations in the distribution of lesions, with rare involvement of the red nucleus, cranial nerves IV and XII, inferior olivae, and striatopallidum, different cortical involvement, and less tau pathology in the white matter in PEP. Lesions in the cholinergic subcortical supranuclear centers of gaze movement in some cases of PEP cause gaze palsy and lid apraxia similar to that in PSP.459 There is total absence of α-Syn–positive deposits in any brain areas of patients with PEP, thus classifying it as a “pure” tauopathy.457,461,461a Despite epidemiologic evidence of a viral infection, the etiology and pathogenesis are unknown, and recent molecular-biologic studies have failed to identify influenza virus in archival material from PEP brain.462,463
Pick’s Disease
This progressive dementia with personality deterioration and signs of frontal disinhibition exhibits rare extrapyramidal symptoms. Most cases are sporadic, but familial cases, usually with autosomal dominant inheritance as a result of MAPT mutations, have been reported.464 Gross inspection shows frontotemporal atrophy, often with a “knife blade” appearance of the cortical gyri, dilated ventricles, and degeneration of the striatum and SN. Histologic examination shows loss of neurons, astrocytosis, and extensive spongiosis of the outer cortical layers; swollen neurons (“Pick cells”), indistinguishable from the swollen achromatic (ballooned) neurons in other conditions; and characteristic argyrophilic intraneuronal cytoplasmic inclusions (Pick bodies), abundant in the granule neurons of the dentate fascia and pyramidal neurons of the hippocampus. Their major component is 3R-tau,411,465 but some patients have a mixture of 3R-tau and 4R-tau.453 Ultrastructurally, they have loosely arranged, 10- to 16-nm straight filaments and 22- to 24-nm twisted filaments with a periodicity either longer or similar to the PHFs of AD.466 Some patients have extensive loss of pigmented neurons in the SN,467 whereas in others the SN is preserved.
Frontotemporal Dementia with Parkinsonism Linked to Chromosome 17-Tau
This group, linked to chromosome 17 associated with mutations of the MAPT gene for tau protein, referred to as FTDP-17, includes a variety of cases characterized by disturbances in behavior and personality with parkinsonian features.221,468 The gross features are focal temporal atrophy with rust-colored appearance of the GP as a result of increased iron pigment and depigmented SN.469 The dominant histologic feature is diffuse tau immunoreactivity of the pretangles in neurons, with some patients also having NFTs resembling those in AD, globose tangles and astrocytic lesions similar to those in PSP or CBD, and tau-positive glial inclusions also resembling those in PSP, CBD, and argyrophilic grain disease. Ultrastructurally, the filaments found in different mutations vary in structure and appearance, with PHFs, 15- to 27-nm-wide twisted ribbons, and 12- to 15-nm or 15- to 20-nm straight tubules. Neuronal loss in the cortical and subcortical gray matter is associated with astrogliosis. The hippocampal lesions resemble those of hippocampal sclerosis.221 Despite significant pathologic heterogeneity between different mutations, some broad correlations have been suggested, including tau pathology resembling AD, PSP, CBD, or PiD. There are similarities and differences between cases of FTDP linked to mutations in MAPT and progranulin (PGRN).468 A recent report of a tau S305S mutation in a family with autopsy-proven FTDP-17 provides further evidence of the clinical and pathologic variability in patients with mutations in the tau gene.470 There is no relationship to TDP-43, the signature protein of FTLD-U.471
Guamanian and Other Forms of Western Pacific Parkinsonism
A high incidence of ALS and PDC was recognized in three regions of the western Pacific, the Mariana islands of Guam and Rota, the Muro district on the Kii peninsula in Japan, and western New Guinea. The incidence of ALS/PDC in Guam has declined since the 1960s.472 Guamanian PDC and ALS-PDC of the Chamorro population may appear clinically similar to FTLD-U and ALS. Neuropathologic evaluation shows cerebral and BG atrophy, depigmentation of the SN and LC, and widespread loss of neurons and gliosis in the hippocampus, amygdala, NBM, brainstem tegmentum, and dentate nucleus, accompanied by abundant NFTs, granulovacuolar degeneration, and Hirano bodies in the hippocampus.473–475 The severe atrophy of the frontal and temporal cortices and base of the brainstem differs from the brainstem lesions in PSP and AD. The loss of large neurons in neostriatum and nucleus accumbens in Guamanian PDC is more severe than in PSP and may be linked to marked degeneration of the limbic areas. The NFTs in the cortex show a predilection for layers II and III, similar to that in PSP.460 Ultrastructural and biochemical analysis of NFTs in both the Japanese and Guamanian forms of PDC shows similarities to AD, including all six tau isoforms.474 Glial pathology is prominent in the PDC of Guam and includes granular astrocytes, coiled inclusions in the oligodendroglia, and tau-positive fine granules in the frontal white matter that are composed of 4R-tau isoforms.476 α-Syn–positive aggregates in the amygdala in the PDC of Guam often colocalize with neurons harboring NFTs, and spherical α-Syn–positive structures occur in the molecular layer of the cerebellar cortex.476,477 The cortex in PDC is distinguished from that in AD and PSP by accumulation of α-Syn, thus suggesting that PDC should be considered a synucleinopathy, as well as a tauopathy.478 Guamanian PDC was associated with cortical TPD-43–positive dystrophic neurites and neuronoglial inclusions in the gray and white matter. Biochemical analysis showed the presence of FTLD-U–like insoluble TPD-43, and the spinal cord exhibited tau-positive tangles and TDP-43–positive inclusions. These results indicate that Guamanian PDC and ALS are associated with pathologic TDP-43, the major disease protein in FTLD-U.479 The western Pacific clusters of neurodegenerative disease may reveal factors similar to the cause of AD and other tauopathies, but genome-wide analysis has failed to identify a single gene locus for Guamanian PDC, thus supporting the hypothesis of a mixed genetic/environmental or pure environmental etiology,480 but the cause remains enigmatic. The cycad hypothesis suggests that dietary consumption of cycad toxins or sterol glucosides is causative but has not been substantiated.481
Secondary Parkinsonism
About 10% of all patients with parkinsonism have secondary forms caused by certain drugs and toxins, metabolic disorders, viral infections, multiple infarcts, brain tumors, trauma, or hydrocephalus (see Table 74-5).
Vascular Parkinsonism (Pseudoparkinsonism)
The term vascular parkisonism (arteriosclerotic pseudoparkinsonism)482 implies a rare akinetic-rigid syndrome resulting from cerebrovascular disease, but with a variety of clinical and pathologic features distinct from those of sporadic PD. It accounts for 3% to 6% of all parkinsonian syndromes and is difficult to diagnose with clinical certainty. Symptoms include bilateral symmetrical rigidity, bradykinesia predominantly involving the lower limbs (“lower body” parkinsonism), postural instability, shuffling gait, falls, dementia, and corticospinal disorders, but resting tremor is unusual.483 Neuropathologic evaluation shows multiple ischemic lesions secondary to small-vessel disease in the striatum, pallidum, white matter, and less often the SN that involve the corticostriatopallidal (nigral), thalamocortical, and other loops in the absence of coexisting pathologic lesions linked to neurodegenerative disease.484–486 The postmortem demonstration of LBs in 13% of patients with multi-infarct encephalopathy, an incidence that is twice as common as in age-matched controls, suggests subclinical PD, whereas vascular lesions in the BG and white matter are observed in 44% to 58% of individuals without dementia and in up to 94% of individuals with dementia.224 The vascular damage should be evaluated as a possible additional, but independent, pathogenic factor.487
Drug- and Toxin-Related Parkinsonism
Drug-induced parkinsonism (DIP), which can be clinically confused with rigid-akinetic IPD, is most often associated with neuroleptic drugs, calcium channel blocking agents, and other substances causing DA depletion, blockage of postsynaptic D1 and D2 receptors, or transient loss of striatonigral TH immunoreactivity.488,489 DIP affects 15% to 60% of patients treated with typical neuroleptics, depending on their type, dose, and the underlying susceptibility of the patients.490–492 It is a common form of parkinsonism that is under-recognized, especially in the elderly.493 Many of them show age-related SNc cell loss and iLBD and are predisposed to adverse drug effects as a result of relative DA deficiency.
The pathology of parkinsonism resulting from carbon monoxide and carbon disulfide intoxication or postnarcotic encephalopathy consists of anoxic lesions or necrosis of the pallidum and SN.494 Methanol intoxication causes bilateral putaminal necrosis and variable necrosis of the subcortical white matter.495 Chronic lead intoxication causes SN damage, and manganese encephalopathy is characterized by widespread neuronal loss and gliosis in the pallidum, particularly the GPi, and in the striatum with little or no SN damage and absence of LBs, which contrasts with the typical findings in PD.496,497 Severe parkinsonism was reported after poisoning with potassium cyanide and was due to neuronal loss and gliosis in the GP, putamen, and SNr, but the SNc was spared.498 Individuals in whom severe levodopa-responsive parkinsonism developed after exposure to MPTP—a synthetic heroin drug that leads to mitochondrial damage and neuronal death—show diffuse neuronal loss and gliosis in the SN along with extracellular NM and activated microglia but without typical LBs.499 Eosinophilic inclusion bodies resembling LBs have been seen in the SN and LC of MPTP-treated aged monkeys, but their ultrastructure differed from that of typical human LBs.500 Other toxins that may cause parkinsonism include paraquat, rotenone, and other herbicides and pesticides.501–504
Other Lesions Causing Parkinsonism
Parkinsonism has been observed in a wide variety of disorders involving the brainstem or SN, or both, that affect the dopaminergic projections; it can occur after conditions such as head trauma with direct destruction of the SN by bullet injury, after direct traumatic impact, or after herniation-contusion of the upper brainstem or secondary damage to the midbrain caused by vascular compression.505 Pugilistic encephalopathy, or boxer’s dementia, which is often accompanied by parkinsonian symptoms, is characterized by diffuse cortical atrophy; degeneration of the corpus callosum and cerebellum; severe cell loss in the SN, LC, and striatum with widespread NFTs in the superficial cortical layers that are often absent in the hippocampus; and widely distributed Aβ deposits. In contrast to AD, however, only sparse or absent neuritic plaques are present,506,507 although the NFTs show the same tau isoform profile and phosphorylation state as in AD.508 Parkinsonism has also been obsrved in rare cases of tuberculoma, tumors of the brainstem, solid tumors causing brainstem compression, calcification of the BG (Fahr’s disease), viral encephalitis, subacute sclerosing panencephalitis, multiple sclerosis, and normal-pressure hydrocephalus.509
Hyperkinetic Movement Disorders
Conditions characterized by excessive movement are grouped together as hyperkinetic disorders, in contrast to the poverty of movements seen in akinetic-rigid movement disorders. The clinical range includes chorea, myoclonus, ballism, dystonia, and tics (see Table 74-1).
Chorea
Chorea is typified by nonrhythmic, rapid, involuntary movements. It may be divided into two main groups: hereditary and sporadic (Table 74-8).
Huntington’s Disease
This autosomal dominant disorder, clinically manifested as chorea, involuntary movements, dystonia, emotional disturbances, and psychiatric symptoms progressing to dementia and cachexia,510 is caused by an unstable expansion of CAG (trinucleotide) repeats in the coding region of the gene IT15 (for “interesting transcript,” referred to as HD-IT15 CAG repeats) on chromosome 4p16.3. It encodes the 350-kD protein huntingtin, which has important functions in healthy brain.511,512 The disease occurs when the critical threshold of 37 polyQ is exceeded.513 The age at onset is inversely related to CAG repeat length. Middle-aged and late-onset patients are seen with a hyperkinetic disorder, whereas juvenile or early-onset ones have bradykinesia and rigidity.
Macroscopic changes in the brain vary with the duration and stage of the disease. Early stages show no gross changes, whereas the late stages are characterized by severe cerebral atrophy, gyral shrinkage, and bilateral atrophy in the neostriatum with enlargement of the frontal horns of the lateral ventricles and atrophy of other brain areas, thus suggesting that HD is a polytypic disorder. Histologically, there is loss of neurons with astrocytosis and microgliosis in the striatum, and increased oligodendrocytic density may precede the onset of symptoms by years.514 The striatal degeneration involves stereotypic neuronal loss progressing in a caudal to rostral direction, dorsomedially to ventrolaterally in the CN, and dorsally to ventrally in the putamen with sequential involvement of the striatum, GPe, and GPi. The severity of anatomic lesions correlates with clinical severity and has been classified into five grades.515,516 Grade 0 (<1% of all HD brains) is assigned to individuals with clinical signs and normal-appearing brains but dysfunction of vulnerable striosomal spiny neurons and gliosis in the neostriatum preceding neuronal loss. Grade 1 (4% of all HD brains) shows atrophy of the CN tail with neuronal loss and gliosis; the bodies of the CN and putamen appear grossly normal but may show focal variations. Grade 2 (16% of HD brains) is associated with atrophy of the head of the CN (still slightly convex and bulging into the ventricle) and mild to moderate gross atrophy of the putamen. Grade 3 (52% of all HD brains) exhibits severe atrophy of the head of the CN and putamen. The microscopic changes in grades 2 and 3 are more severe than those in grade 1. In grade 4, severe atrophy of the total neostriatum (95% loss of CN volume) and pallidum occurs along with involvement of the striosomes and matrix in a dorsoventral progression517 and a concave contour of the head of the CN. In at least 50% of grade 4 HD brains, the nucleus accumbens remains relatively preserved.
In grades 1 and 2, nonstriatal structures of the brain show no or mild atrophy, whereas in grades 3 and 4, the GP, neocortex, thalamus, STN, white matter, and cerebellum show atrophy with neuronal loss. The GPe is more involved than the GPi. Neuronal loss with or without astrocytosis is seen in the center median of the thalamus,199 in the SNr without involvement of the SNc, and in the STN with little gliosis. The cerebellar and hippocampal atrophy, often reported in patients with juvenile onset, is probably due to hypoxia resulting from seizures. Cortical degeneration is variable, depending on the stage of the disease, and requires morphometric evaluation.516 Cell type–specific vulnerability correlates with triplet repeat mutation gains. Neostriatal pathology starts with the loss of ENK- and GABA-containing medium and spiny neurons projecting to the GPe, with relative sparing of the large cholinergic interneurons and medium-sized aspiny neurons and interneurons containing NO synthase, nicotinamide adenine dinucleotide phosphate (NADPH) diaphorase, and various neuropeptides.516,517 This is consistent with current models of BG function in which hyperkinesia results from interruption of the indirect pathway involving the striatal GPe, STN, and GPi as a result of increased glutamatergic stimulation of the cortex secondary to the reduced inhibitory effects of the GPi and SNr (see Fig. 74-4).518 In the later stages of HD, the decreased motor activity (bradykinesia) and rigidity are the result of damage extending to the nucleus accumbens, GP (showing loss of SP- and calcineurin-immunoreactive fibers and neurons), amygdala, ventrolateral thalamus, lateral hypothalamus, STN, and SNc (with 40% to 50% cell loss), disappearance of corticostriatal neurons from cortical layer V, and additional loss of striatal GPi efferents (direct pathway).519 In juvenile cases, corresponding to early rigid Westphal variants, the striatal GPe and GPi efferents degenerate, thus suggesting that degeneration of the direct pathway is responsible for the rigidity (see Fig. 74-4). The differences between rigid and choreiform HD are not related to presynaptic SN damage but to involvement of striatal GABA- and SP-containing neurons projecting to the GPi that inhibit increased dopaminergic activity of the neostriatum. The coexistence of hyperkinetic and hypokinetic movement disorders in HD may be explained by involvement of the direct and indirect pathways in the BG-thalamocortical circuit, but the models of striatal connectivity and pathology are insufficient to explain the nonmotor features often seen in early HD.520 The cognitive changes in HD are related to diffuse cortical atrophy with cell loss in the deep layers and loss of corticostriatal neurons in frontal layer V and in the entorhinal cortex and subiculum, which causes disorders of the striatofrontal and limbic circuitry. There is an increasing prevalence of nonneuritic tau pathology in the limbic areas and Aβ deposits in the neocortex in young patients with early stages of HD but less rapid progress in advanced age, thus explaining a rare coexistence of HD and AD.521
Neuronal intranuclear inclusions composed of aggregates of abnormal huntingtin protein have been demonstrated by immunohistochemistry studies with antibodies against huntingtin protein and Ub in both humans and tg mouse models.522,523 They are present in the cerebral cortex, hippocampus, and to a lesser extent, the neostriatum, amygdala, and dentate and red nuclei. Dystrophic neurites with similar immunohistochemical properties in the cortex, medial temporal lobe, and striatum are more common than nuclear inclusions. The frequency of cortical intranuclear inclusions correlates with the extent of CAG expansion and is inversely related to the age at onset and death, whereas no such relationships were detected for the striatum, which reflects the advanced neuronal loss accrued by the time of death.524 In earlier stages of HD, accumulation of N-terminal huntingtin protein occurs in the cytoplasm together with dystrophic neurites inducing degeneration of the corticostriatal pathway.525 The role of mutant huntingtin protein in neuronal degeneration and the pathogenic mechanisms of HD are not yet fully understood, but it is presumed that huntingtin protein is cleaved by caspases, with toxic gain of function leading to cytoskeletal defects, synaptic dysfunction, and reduced brain-derived neurotrophic factor and neurotrophic receptor signaling.526 Evidence suggests that conformational changes in the expanded polyglutamine induce a cascade of cellular mechanisms, including excitotoxicity; altered proteasome degradation, cell signaling, and regulation of transcription; increased free radical and oxidative damage products; early mitochondrial calcium defects; impaired energy metabolism; transport dysfunctions; and apoptosis.22,516,527,528 Recent studies showed that a protein called Rhes, which is specific to the stratum, mediates the neurotoxic effect of mutant-huntingtin.529
Other Hereditary Choreas
Neuroacanthosis
This rare, genetically heterogeneous condition shows an association of erythrocytic acanthocytosis with chorea, dystonia, and the later development of akinetic-rigid parkinsonism, the severity of which is not correlated with the degree of acanthosis. The most prevalent form is autosomal recessive with mutation of the CHAC gene on chromosome 9q21,530 but there are various genetic and clinical subtypes. Neuropathologic data are sparse and not well correlated with genetics. Neuroacanthosis is characterized by gross atrophy of the neostriatum with loss of small and medium-sized neurons, severe pallidal involvement, and less consistent lesions in the thalamus, SN, and spinal anterior horns. The absence of changes in the cerebral cortex, STN, and cerebellum may help in differetiating this condition from HD.22 Peripheral nerves may show chronic axonal neuropathy.531
Huntington’s Disease–Like Syndromes
Approximately 1% of patients with a clinicopathologic picture of HD have no mutation of the huntingtin gene, and three separate mutations or linkages have been identified. Autosomal dominant HD-like type 1 (HDL-1), caused by an expanded octapeptide repeat in the prion gene at chromosome 20pter-p12, with valine at codon 129, shows atrophy of the BG, variable cortical atrophy, and prion-specific changes, including typical prion plaques.532
Autosomal dominant HDL-2 occurs in patients of African ancestry and exhibits features similar to HD: frontal inhibition with parkinsonism but absence of choreoathetosis. It is due to a CAG/CTG repeat expansion in the juntophilin-3 gene on chromosome 16q243. Degeneration of the dorsal and lateral CN and putamen and the neocortices is associated with Ub- and TATA box binding protein–immunoreactive neuronal intranuclear inclusions. Its neuropathologic similarity to HD suggests partial pathogenetic overlap of both disorders.533
Benign Hereditary Chorea
Benign hereditary chorea is clinically and genetically heterogeneous. A number of kindreds with autosomal dominant inheritance, related to mutations in the TTF1 (thyroid transcription factor 1) gene on chromosome 14q,534 have an onset of chorea in childhood with little progression and without mental deterioration. Functional imaging studies reveal changes in the BG similar to HD, but the neuropathology of benign hereditary chorea is not well documented.
Sporadic Chorea
Nongenetic forms include those of autoimmune pathogenesis, such as Sydenham’s chorea, which occurs after streptococcal infection and is frequently linked to classic rheumatic fever or systemic lupus erythematosus (antiphospholipid syndrome). Affected patients have unilateral involuntary movements and other symptoms, but the neuropathology is uncertain.535 Other forms include polycythemia vera or vascular chorea caused by small strokes in the striatum, toxoplasmosis in patients with acquired immunodeficiency syndrome,536 herpes simplex encephalitis,537 and paraneoplastic chorea.538
Hereditary Striatal Necrosis
This rare, autosomal dominant or recessive progressive disorder with gait disturbance, dystonia, rigidity, ataxia, and optic atrophy morphologically shows bilateral necrosis of the neostriatum and GP with vascular proliferation, as well as similar lesions variably involving the thalamus, SN, and brainstem tegmentum, but rarely the cerebral cortex. Pathologic evaulation shows overlap with mitochondrial encephalopathies such as Leigh’s disease and Leber’s optic neuropathy.539 In childhood, however, bilateral striatal lesions may occur in a variety of disorders.22
Dentatorubral-Pallidoluysian Atrophy
This rare autosomal dominant disorder, largely confined to populations with Japanese ancestry, is caused by an abnormally expanded CAG triplet in the gene for atrophin-1 secondary to a CTG-B37 mutation on chromosome 12p13.31.540 The pathogenic range of CAG repeats is 54 to 79, with normal alleles having fewer than 26 repeats. The diverse clinical picture includes chorea, athetosis, ataxia, myoclonus, epilepsy, and dementia, with considerable intrafamilial variations. Early-onset patients with larger CAG repeat expansions tend to have prominent myoclonic epilepsy. Adult-onset cases may be mistaken for HD and other types of SCA. Neuropathologic examination reveals MSA with neuronal loss and gliosis involving the cerebellar dentate nucleus, GPe, STN, and red nucleus, with disruption of the dentatorubral, pallidofugal, spinocerebellar, and motor systems. Milder changes involve the striatum, thalamus, SN, inferior olivae, midbrain tegmentum, spinocerebellar tracts, and posterior spinal columns, often associated with neuroaxonal dystrophy, but the cerebral cortex is largely spared.22 Patients with the Haw River syndrome have smaller expansions of the atrophin-1 gene with calcification of the pallidum, demyelination of cerebral white matter, and atrophy of the dentate nucleus and pallidum.541 The intranuclear and cytoplasmic inclusions in the neurons and oligodendroglia of various brain sites are immunoreactive for Ub and atrophin-1 and have expanded polyglutamine tracts.542,543 Their density correlates with CGA repeat length, thus suggesting that these protein aggregates may be a common feature in the pathogenesis of glutamine repeat neurodegenerative disorders.544
Machado-Joseph Disease
Machado-Joseph disease/SCA3 is a fatal, autosomal dominant progressive ataxia in Europe and Japan that results from CAG expansion in the MJC1 gene and toxic overexpression of ataxin-3, which maps to chromosome 14q.32.1.545 It was subdivided into different clinical types. Type 1, the least common, has an onset from 5 to 20 years of age and features spasticity, rigidity, and bradykinesia but little ataxia. Type 2, the most frequent, is characterized by progressive ataxia and spasticity. Type 3 (late onset) is typified by ataxia, distal amyotrophy, and areflexia. Type 4 shows prominent parkinsonism. There is involvement of the cerebellar afferent and efferent pathways, extrapyramidal structures, and lower motor neurons, with cell loss in the SN, STN, dentate and red nuclei, Clark’s column, raphe nuclei, cranial nerve motor nuclei, and anterior horns; the striatum and cerebral cortex are less affected. The degenerative changes are often accompanied by intranuclear neuronal inclusions immunoreactive with ataxin-3 and Ub.22 Its pathogenesis is related to aggregation of ataxin-3, which causes selective neuronal loss.
Progressive Pallidal Degeneration
Pallidal degeneration is characterized by isolated bilateral atrophy of the GP with gliosis and degeneration of the efferent pallidal fibers and pallidoluysian atrophy by bilateral atrophy and gliosis of the GPe and STN. Pallidonigral and pallidonigroluysian degeneration, which may be associated with levodopa-resistent akinesia, is typified by degeneration of the GP, STN, and SN and occasional involvement of the ventromedial thalamus and iron deposition in the affected nuclei.546 In single cases with progressive choreiform or dystonic disorders or nonprogressive cerebral palsy, autopsy has revealed symmetrical degeneration of the GPe or status marmoratus of the BG associated with polyglucosan bodies in neuronal perikarya, axons, and dendrites (Bielschowsky bodies).22 The combination of recessive early-onset parkinsonism and pyramidal tract signs is known as pallidopyramidal syndrome and has recently been discussed critically.547
Hallervorden-Spatz Disease
NBIA-I or PKAN defines a group of genetic autosomal-recessive degenerative disorders with progressive dystonia, akinetic rigidity, optic atrophy, retinitis pigmentosa, seizures, and dementia.548,549 The majority of classic cases are caused by mutations in the pantothenate kinase 2 (PANK2) gene linked to chromosome 20p12.3-13 (Online Mendelian Inheritance in Man [OMIM] 234200),550 whereas others bear mutations in the PLA2G6 gene on chromosome 22.551 Classic PKAN with infantile onset consists of dystonia, early pyramidal signs, cognitive decline, and retinal changes; the atypical juvenile form features neurobehavioral involvement and slower progress; and adult forms are typically extrapyramidal disorders with dementia. Different subtypes can be reliably distinguished by T2* and T2 fast spin-echo brain MRI, which provides an accurate clinical and subsequent molecular diagnosis.552 Neuropathologic evaluation reveals a rust-brown discoloration of the GP and SN, loss of neurons, gliosis, and iron pigment deposits; histologic markers are axonal spheroids (dystrophic 20- to 120-µm swellings) immunoreactive for neurofilament and Ub in many areas of the CNS and peripheral nervous system. Ultrastructurally, the terminal axons and presynaptic endings are filled with granulovesicular and tubulomembranous material with a paracrystalline appearance and a central cleft. Detection in biopsy material as the method of choice for in vivo diagnosis of the disorder553 was replaced by genetic studies to detect mutations in the PANK2 gene,554 the relationships of which to neurodegeneration and iron deposition in the brain are obscure. Although the pathogenic cascade involving PANK2 deficiency, iron deposition, and accumulation of insoluble α-Syn remains to be delineated, dysregulation of neuronal lipid metabolism appears to play a key role in PKAN. PANK2 is one of four human genes that encode PANK activity, which catalyzes the rate-controlling first step in the pathway of coenzyme A synthesis—and thereby fatty acid metabolism—by phosphorylation of pantothenate (vitamin B5) to phosphopantothenate. In addition to PD and DLB, PKAN is likely to be the third human synucleinopathy that is associated with dysregulation of fatty acid metabolism. Widespread α-Syn–immunoreactive inclusions and extensive tau pathology with NFTs often coexisting with LBs have been observed in juvenile- and adult-onset Hallervorden-Spatz disease and have been related to disturbances caused by axonal damage.555,556 Atypical adult forms of cortical α-Syn and tau pathology are considered a distinctive clinicopathologic entity.557
The disease is thought to be part of a widespread spectrum of neuroaxonal dystrophies that include infantile, late infantile, juvenile, adult, and senile forms, all of which share the presence of axonal spheroids; however, there are neuroimaging and ultrastructural differences between the infantile and other forms.549,552 Mutations in the PLA2G6 gene cause both infantile neuroaxonal dystrophy and, more rarely, an atypical neuroaxonal dystrophy that overlaps clinically with other forms of NBIA.558
Neuroferritinopathy
This hereditary multisystem disorder is caused by mutations in the gene encoding ferritin light chain (FTL) in various pedigrees. Its clinical features include choreoathetosis, parkinsonism, focal dystonia, cerebellar signs, and cognitive impairment.559 The distribution of its hallmark cytoplasmic lesion, the iron/ferritin body, which is probably formed in oligodendrocytes, depends on the underlying genetic subtype. It involves the BG and is associated with cystic degeneration, particularly of the GP. A tg mouse model expressing a pathogenic FTL mutation shows pathology similar to the disease in humans, but its pathogenesis is not fully understood.22 It may be diagnosed by muscle or nerve biopsy.560
Neuronal Intranuclear Inclusion Disease and Basophilic Inclusion Body Disease
Both neuronal intranuclear inclusion disease (NIID) and basophilic inclusion body disease (BIBD) are rare, progressive, fatal sporadic and rarely familial neurodegenerative conditions whose heterogeneous clinical phenotype includes motor neuron, extrapyramidal, and cerebellar signs, as well as choreoathetosis, tremor, levodopa-responsive dystonia, rigidity, dysarthria, behavioral changes, cognitive impairment, and autonomic dysfunctions. Patients with infantile or juvenile onset have cardiomyopathy, and adult-onset cases exhibit dementia with parkinsonism and autonomic dysfunction.22,561–563 The neuropathology of both NIID and BIBD is quite similar: the brain shows various degrees of generalized atrophy that is accentuated in the frontotemporal and frontoparietal regions. The striking morphologic feature in both disorders is the presence of neuronal and glial intracytoplasmic and intranuclear inclusions in many areas of the central, peripheral, and visceral nervous systems and in parenchymal cells of the adrenal medulla.22 The intraneuronal eosinophilic inclusions in NIID are immunopositive for Ub, neurofilament, α-internexin, and ataxin-3 but negative for tau, amyloid precursor protein, Aβ, and TPD-43, whereas the basophilic inclusions in BIBD are positive for α-internexin and p62 protein but negative for tau, α-Syn, polyglutamine, and TPD-43. These disorders are associated with multisystemic degeneration with neuronal loss and gliosis in the neocortices, hippocampus, basal ganglia, SN, cerebellum, and spinal cord, often accompanied by pyramidal tract degeneration.22,563 NIID or neuronal intermediate filament inclusion disease (NIFID) was recently classified as a new entity of frontotemporal lobar degeneration.564 Familial cases are well documented, but no causative gene has been identified; polyglutamine extension is unlikely in the pathogenesis of NIID. The differential diagnosis includes various types of FTLD, atypical parkinsonism, and trinucleotide repeat disorders. The etiology of both disorders is unclear, but an unknown protein besides α-internexin and neurofilament may play a pivotal pathogenic role, at least in some NIID cases.
Wilson’s Disease (Hepatolenticular Degeneration)
Wilson’s disease (WD), an autosomal recessive disorder characterized by liver disease (cirrhosis) and cerebral degeneration along with abnormal hepatic copper metabolism and excretion, is caused by mutation of the ATP7B gene on chromsome 13q14.3, which encodes a copper-transporting P-type adenosine triphosphatase (ATPase).565 The protean clinical manifestations of WD vary with pediatric or adult onset. A constant, diagnostically important sign in the eye is the Kayser-Fleischer ring, which is caused by deposition of copper in the limbus of the cornea. Its clinical features include tremor, dystonia, parkinsonism, abnormal behavior, and schizophrenia-like symptoms.566 Neuropathologic studies show copper deposition, especially in the basal ganglia, with cavitation, neuronal loss, gliosis, and occasionally subcortical myelin degeneration and neuronal loss in the deep frontal cortex.22,567 Many mutations have been described in the WD gene, which is expressed primarily in the liver.568 The most common mutation (H1069Q) replaces a histidine residue in a cytoplasmic loop adjacent to the adenosine triphosphate–binding domain, which is essential for normal function of both the WD gene product and the related gene mutated in Menkes’ disease.569
Menkes’ Disease
The X-linked recessive disorder known as Menkes’ disease (MD) is due to mutations of the ATP7A gene, which encodes a copper-transporting P-type ATPase and shows more than 75% homology with the WD gene.570 ATP7A has a dual function: incorporation of copper into copper-dependent enzymes and maintenance of intracellular copper levels by removing excess copper from the cytosol.571 The reduced activity of these enzymes results in systemic copper deficiency, which accounts for most of the features of MD: abnormal hair, mental retardation, hypotonia, and cerebellar degeneration.572 MRI shows brain atrophy and abnormal myelination. Neuropathologic evaluation reveals diffuse cerebral atrophy, multilocal neuronal degeneration, and abnormal Purkinje cell dendritic arborization with axonal swellings.22 Clinical differences from WD probably arise from the tissue-specific functions of the gene products, with the MD gene being expressed predominantly in the placenta, gastrointestinal tract, and blood-brain barrier; mutation of ATP7A results in failure of copper transport through the placenta with resultant deficiency of brain-specific cuproenzymes.
Myoclonus and Startle Syndromes
Myoclonus, defined as brief, electric shock–like jerks caused by rapid, involuntary contraction of a single muscle or muscle group, is a nonspecific sign of CNS disease that occurs in a wide range of degenerative disorders, including advanced AD, Creutzfeldt-Jakob disease, dentorubral-pallidoluysian degeneration (DRPLD), LBD, CBD,573 and other CNS conditions, including those induced by hypoxia, trauma, and drugs. Distinction should be made between cortical and subcortical forms. Cortical forms are related to hyperexcitability of the somatosensory and primary motor cortex and are due to point mutations in the gene encoding GLRA1, which causes abnormalities in channel gating. Subcortical forms are associated with lesions in the brainstem, such as palatal myoclonus secondary to lesions in the central tegmental tract and dentate nucleus. Opsoclonus (ocular myclonus) is seen in children with neuroblastoma or in adults with CNS infections, but the pathogenic mechanisms are unknown.574
Ballism and Hemiballism
Ballism is a severe form of chorea characterized by involuntary, violent flinging movements of the limbs. When unilateral, it is called hemiballism. Most cases are caused by damage to the STN or its outflow tracts as a result of vascular disease (infarct or hemorrhage). Rarely, other focal lesions are caused by metastases, demyelination, infections, or tumors. Bilateral ballism is rare.575 Experimental lesions in monkeys showed that more than 20% of the STN must be destroyed to produce hemiballism. Lesions of the STN reduce the normal inhibitory output from the GPi and SNr and result in reduced activity of the inhibitory GABAergic STN-pallidal pathway to the thalamic nuclei. The increased glutamatergic drive in the cortex produces ballism through enhanced synchronization of neuronal activity (see Fig. 74-4).
Dystonia
Dystonia, a syndrome of sustained muscle contractions that frequently causes abnormal posture or twisting and repetitive movements, comprises a heterogeneous group of phenotypes that may be due to different hereditary degenerative, metabolic, or genetic diseases.576,577 It is classified by age of onset, severity, distribution of abnormal movements, and cause, and one must distinguish between primary, generalized, focal or segmental, and mixed dystonias, dystonia-plus syndromes, and hereditary degenerative and paroxysmal dystonias.578,579
Primary dystonias include predominantly generalized and focal ones. Seventeen different types have been differentiated according to genetic aspects.16,22,580,581,581a Predominantly generalized dystonias include autosomal dominant early-onset or primary (idiopathic) torsion dystonia (PTD/DYT1), autosomal recessive dystonia (DYT1), and dominant (DYT4) forms. Most PTD cases are caused by mutations in the DYT1 gene on chromosome 9q32.q34, which encodes the protein torsin A,582 familial-onset dystonia is due to deletion of the torsin A gene in exon 5 or mutation of the gene encoding ε-sarcoglycan, and a minority of patients have no abnormalities in the torsin A gene. Other adult-onset familial forms of torticollis have been mapped to DYT7 on chromosome 18p, to DYT6 on chromosome 8p21.q22, or to other loci. Neuroimaging studies show increased metabolism of the presupplementary motor and parietal association cortices in DYT1– and DYT6-associated dystonia and increased metabolism in the striatum, anterior cingulate gyrus, and cerebellum of carriers.583 Evidence suggests that dystonia results from functional BG disturbances, particularly those involving striatal control of the GPi and SNr, which causes altered thalamic control of cortical motor planning in the supplementary motor area and abnormal regulation of the brainstem and spinal inhibitory intraneuronal mechanism,584 as shown by thalamofrontal disinhibition and abnormal central sensorimotor processing (see Fig. 74-4).585 Recent neuroimaging studies showing reduction of axons in the pontine brainstem and white matter of the sensorimotor region suggested disturbances in the cerebellothalamocortical pathways,586 and genotype-phenotype interaction in PTDs were revealed by different changes in brain structures.589a Earlier neuropathologic studies reported no abnormalities in dystonia 1 and 5, nor cell loss or gliosis in the striatum SN, LC, and PPNc; a patchy pattern of neuronal loss and gliosis in the CN and putamen; and infrequent NFTs and LBs in the brainstem nuclei, all of which are obviously independent of the genotype.587 Analysis of patients with DYT1 frontotemporal dementia revealed neuronal inclusions and immunoreaction for torsin A, Ub, and lamin A/C in brainstem nuclei, findings not present in late-onset focal forms, which show normal brain structures in most patients.588,589
Focal or segmental dystonias include autosomal dominant focal dystonia (DYT7) and adult-onset PTD; 25% of patients have a family history, including blepharospasm, writer’s cramp, and other focal dystonias. Patients with blepharospasm associated with oromandibular dystonia (Meige’s syndrome) may be inclined to the development of parkinsonism, and LB pathology has been reported.590 An unusual familial phenotype showed an association of focal dystonia and cerebellar atrophy.591 For mixed dystonia (adult-onset primary type [DYT6] and PTD with craniocervical or upper limb onset [DYT13]), no neuropathologic data are available.22 Spasmodic dystonia (SD) is a primary task-specific focal dystonia characterized by involuntary spasms in the laryngeal muscles during speech, frequently related to brainstem intraneuronal excitability and probably caused by modulation of descending input from higher brain centers such as the BG.592 No neuropathologic data are available. Recent studies have found small clusters of microglia in the reticular formation surrounding the solitary tract nucleus, spinal trigeminal nucleus, nucleus ambiguus, inferior oliva, and pyramids, along with mild neuronal degeneration and depigmentation in the SN and LC, but no abnormal protein deposits or signs of demyelination or axonal degeneration were found. These subtle abnormalities of the brainstem in patients with SD may represent changes secondary to abnormalities in the supramedullary regions.593
Dystonia-plus syndromes include dopa-responsive dystonia (DRD) (DYT5), inherited myoclonus-dystonia-parkinsonism (DYT12), and rapid-onset dystonia-parkinsonism, which may be focal or generalized. Autosomal dominant DRD (Segawa’s syndrome) is associated with mutations in GCH1 on chromosome 14q, which encodes guanosine triphosphate cyclohydrolase I, the rate-limiting enzyme in the biosynthesis of tetrahydrobiopterin, a cofactor of TH that regulates DA synthesis. Neuropathologic evaluation reveals hypopigmentation of the SN and LC without neuronal loss and absence of LBs.587 A new locus, DYT16, shows phenotypic similarities to DYT12.
Myoclonus-dystonia is a rare autosomal dominant disorder characterized by a combination of myoclonic jerks and dystonia, with onset in the first or second decade of life. A major culprit gene, the ε-sarcoglycan gene (SGCE), is located on chromosome 7q21 (DYT11, OMIM 604140), but mutations or deletions of SGCE are detected in less than 40% of patients with a typical course, thus suggesting that the disorder is genetically heterogeneous; its neuropathology and pathogenesis are unclear.581
Complicated recessive dystonia-parkinsonism syndromes with overlapping features include conditions that have been classified under the PARK and DYT genes and the NBIA disorder.594
Heredodegenerative dyskinesias include X-linked dystonia-parkinsonism (DYT3 with the gene locus at X13.1), also known as “Lubag’s disease“; it is prevalent in the Philippines and is characterized by segmental or generalized focal dystonia and the later development of parkinsonism as a result of reduced expression of the D2 receptor gene (DRD2) in neurons.595 CN atrophy and a patchy pattern of neuronal loss and gliosis, with preferential loss of striosome projections to the GP and SN, suggest that this dystonia may result from an imbalance in activity between the striosomal and matrix pathways.596
Paroxysmal dyskinesias are classified as kinesigenic or nonkinesigenic (DYT8) with episodes of dystonia, chorea, and athetosis lasting minutes to hours; these disorders are associated with mutation of the myofibrillogenesis regulator 1 gene (MR1 on chromosome 2q33-35; no pathologic abnormalities are seen in the brain). Hypogenic and exercise-induced forms (DYT9 and DYT10 mapping to 16p12-q2; no reported mutations) have also been described, but there are few neuropathologic data.22
Secondary dystonias can be caused by a variety of neurological disorders, including PD, parkinsonism syndromes, HD, lysosomal storage and mitochondrial disorders, organic aminoacidurias, and others.580 They are rarely associated with pontomesencephalic lesions.597 Primate models have revealed that in brainstem areas, various affected BG nuclei of the thalamus causing disruption of sensory receptive fields in the supplementary motor area (SMAp) may lead to dystonia.598
Tic Disorders
Tics are involuntary, brief, stereotyped movements or vocalizations that can be suppressed at the expense of mounting inner tension. They are classified as motor tics (brief movements), vocal tics (uttering brief sounds), and sensory tics (brief sensations).599 Tics may be simple or complex and may appear semipurposeful (e.g., obscene gestures). They may be associated with several neurodegenerative disorders or may be complications of drug therapy or CNS infection. Transient tics are the most common and mild form. Chronic tic disorders are severe and include Tourette’s syndrome, which is clinically defined by the presence of motor and vocal tics. Those not meeting the criteria are known as “secondary tic disorders” and can be due to infections, drugs, toxins, stroke, or trauma. Genetic studies have shown links to chromosomes 2, 8, and 11. Imaging studies indicate a reduction in CN volume and increased density of type 2 vesicular monoamine transporter (VMAT2).600 No specific anatomic CNS lesions have been observed, except reduced dynorphin-like staining in the GPe and SN, decreased parvalbumin-positive GABAergic interneurons in the striatum, and increased parvalbumin-positive projection neurons in the GPi.601
Tremor Syndromes
Tremor is a common disorder characterized by rhythmic involuntary oscillatory movements of the body. For currently proposed phenomenology and syndrome classification of tremors and their pathophysiology, see other sources.16,190,602 Essential tremor (ET), one of the most common neurological disorders, may occur at any age and is occasionally associated with rest and intention tremor. In addition to environmental agents, genetic factors may contribute to its onset. Three gene loci (ETMI1 and ETMI2 on 3q.13 and 2p24.1 and a locus on 6p23) have been identified in families with this disorder.603 Functional MRI found overactivity in the cerebellum, red nucleus, and GP without activation of the inferior olivary nucleus.604 Until recently, no consistent morphologic abnormalities have been identified, but postmortem studies have demonstrated a reduction in Purkinje cell number and the occurrence of torpedo bodies (early axonal swellings indicating neuronal degeneration) in the cerebellar cortex without LBs in the majority of patients with ET; a small proportion have brainstem LBs. Others have observed cerebellar gliosis and LC depletion, thus suggesting that ET seems to be a heterogeneous degenerative disorder.605–608 Determination of the clinical differences between patients with and without LBs and elucidation of the pathophysiology of ET require further study.






