CHAPTER 13 Neuroophthalmology
Neuroophthalmology is a broad discipline that incorporates important elements of interest to neurosurgeons, neurologists, and ophthalmologists. This chapter offers some clinical tools and examples of common neuroophthalmic disorders, with emphasis on material of importance to neurosurgeons that is not likely to be covered in other chapters of this text. Readers are referred to the authoritative, multivolume Walsh and Hoyt’s Neuro-ophthalmology1 and other excellent texts2–10 for a more detailed discussion of neuroophthalmic conditions.
From the Eye to the Visual Cortex: Afferent Aspects of Vision
History
Accompanying ophthalmic symptoms (e.g., eye pain or redness, diplopia, proptosis, ptosis) and neurological symptoms are important.11 The time that it takes to perform a thorough neuroophthalmic history is well invested because it often saves the patient (and physician) time, frustration, and health care costs.
Examination
It is key to assess vision in each eye separately. Patients are commonly unaware of or tend to minimize the importance of severe loss of vision in one eye if the opposite one is healthy. Unilateral visual loss may be “discovered” when the sound eye is momentarily covered. Frisén, in his detailed treatise, discusses the techniques for and the inherent difficulties in measuring vision.12
Visual acuity is typically measured by using a standard distance chart (Fig. 13-1) and testing one eye at a time with patients wearing their best corrective glasses or contact lenses. A pinhole frequently improves the vision tested at a distance in a patient who does not have the appropriate spectacle correction. A near-vision card is very helpful, as long as the examiner remembers that presbyopic patients (older than 45 years) may need reading glasses.
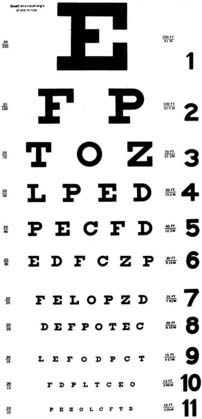
The visual field extends 60 degrees from fixation nasally to 90 degrees temporally. Even the large 20/400 “E” on the Snellen visual acuity chart occupies less than 2 degrees of this 150-degree panorama. Visual acuity alone is therefore insufficient to fully characterize visual function. Assessment of the visual field can be done with tools as simple as the examiner’s hands or can be formally plotted with kinetic or static perimetry (Fig. 13-2).13 Many patients who cannot perform well in “formal perimetry” are capable of providing remarkable diagnostic information with confrontation testing (Box 13-1).14
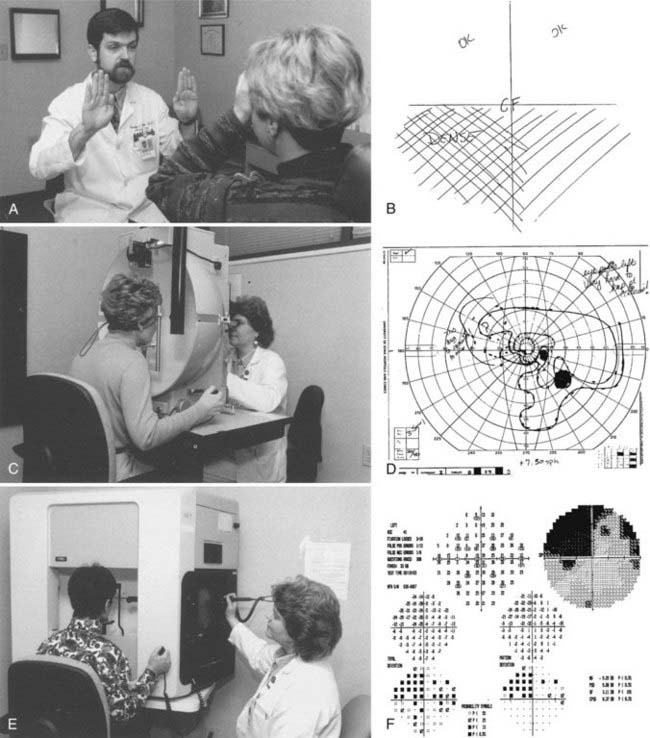
All visual field tests are subjective psychophysical instruments that require a cooperative and reasonably intelligent subject. With increasing sophistication of the various types of perimetry—confrontation, Amsler grid, tangent screen, Goldmann (kinetic), and automated (static) perimetry—the tests become more quantifiable and repeatable, but they are also more difficult for some patients to perform accurately.15,16 Quantification is essential for the management of disorders in which decision making depends on the time course of visual loss (e.g., sellar masses, optic nerve disease, idiopathic intracranial hypertension [IIH]). Furthermore, formal visual fields are necessary to document the efficacy of treatment.17
The relative afferent pupillary defect (RAPD) provides an objective comparison of the visual integrity of the two eyes. Its objectivity makes it one of the most important tools in neuro-ophthalmology (Box 13-2).18 RAPDs do not cause the pupils to be unequal in size. Unequal pupils (anisocoria) are caused by efferent disorders.
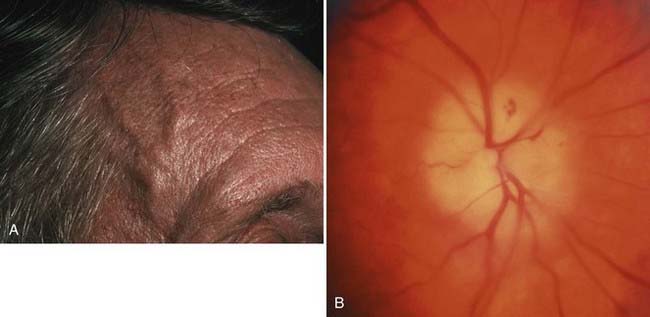
The cornea should show a crisp, clear “point of light” reflex from the penlight. Assessing the cornea is especially important when disorders affect either the trigeminal or the facial nerve, or both. Poor function of the orbicularis oculi that results in incomplete blinking can lead to corneal epithelial defects or corneal ulcers. Decreased corneal sensation can result in a similar fate. Tarsorrhaphy is a procedure that surgically apposes a portion of the upper and lower lids to protect the eye. This procedure is often required in patients with facial and trigeminal cranial nerve dysfunction, especially when both these cranial nerves are involved. It is generally too late to save vision in an eye if the clinician waits until the patient complains of decreased vision or pain (Fig. 13-3). When diseases (or procedures) affect the facial or trigeminal nerves, an ophthalmologist should be involved from the beginning to address medical or surgical management of the eye.
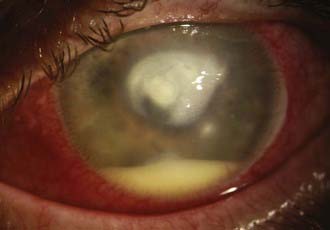
Viewing the red reflex of the eye can offer valuable information about the optical media of the eye. Cataract and corneal opacities can be seen in the red reflex, and vitreous hemorrhage (as in Terson’s syndrome) will darken or abolish the red reflex. This test requires only a few seconds with the ophthalmoscope and should become a routine step before the fundus is examined (Box 13-3).
Clinical Appearance of the Optic Nerve
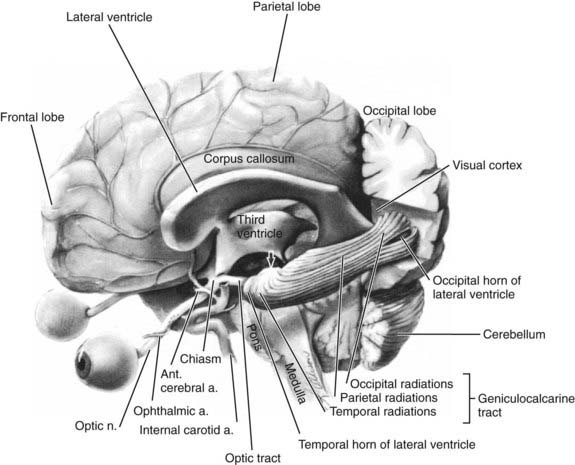
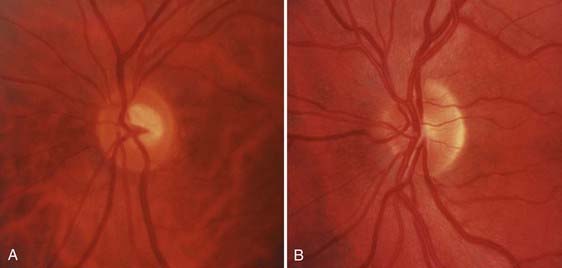
 The anatomy of the afferent visual system is reviewed in Figure 13-4. Each optic nerve consists of 1.2 million axons that have their origin in the ganglion cells of the retina, travel in the optic nerve and chiasm, and synapse in the lateral geniculate nuclei. These axons ordinarily become myelinated after passing through the plane of the sclera (i.e., lamina cribrosa), which enlarges the nerve from 1.5 mm at the optic disc to 3 to 4 mm behind the globe. The optic nerve head is the origin of the nerve proper and, when viewed through an ophthalmoscope, appears as a flat, pink ellipse from which the retinal vessels radiate (Fig. 13-E1).
The anatomy of the afferent visual system is reviewed in Figure 13-4. Each optic nerve consists of 1.2 million axons that have their origin in the ganglion cells of the retina, travel in the optic nerve and chiasm, and synapse in the lateral geniculate nuclei. These axons ordinarily become myelinated after passing through the plane of the sclera (i.e., lamina cribrosa), which enlarges the nerve from 1.5 mm at the optic disc to 3 to 4 mm behind the globe. The optic nerve head is the origin of the nerve proper and, when viewed through an ophthalmoscope, appears as a flat, pink ellipse from which the retinal vessels radiate (Fig. 13-E1).
Using the ophthalmoscope to examine the fundus (i.e., optic disc, vessels, macula, and retina) is the cornerstone for the diagnosis of afferent visual disorders. The appearance of the optic disc (i.e., edema, pallor, or cupping) is crucial in determining the cause of visual loss, as is evident in the discussion of specific disorders in the following section.19
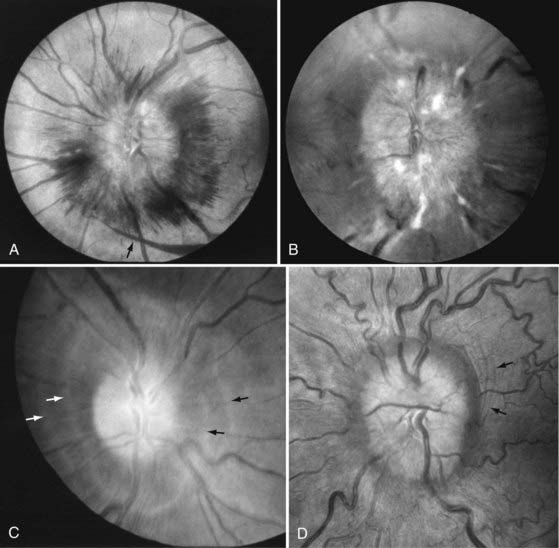
 The list of differential diagnoses for optic disc edema, or swelling, is long (Table 13-1). A variety of mechanisms such as ischemia, inflammation, metabolic disorders, and pressure differentials may produce stasis of axonal flow, which causes swelling of the axons anterior to the lamina cribrosa and elevation of the optic disc. The normally transparent peripapillary nerve fiber layer gradually opacifies and obscures the retinal vessels. The disc margins become indistinct. The optic nerve may become hyperemic as small vessels are dilated, and characteristic splinter hemorrhages may appear at the disc margin in the nerve fiber layer. In addition, deeper hemorrhages may appear in the peripapillary retinal and subretinal layers. The swelling can also compromise venous outflow by enlarging the central veins at the disc, causing tortuosity, and producing retinal hemorrhages near the optic disc (Fig. 13-E2).20 The most common causes of optic disc edema include papilledema (from elevated intracranial pressure), AION, and demyelinating optic neuritis. A common clinical challenge is distinguishing true, pathologic optic disc swelling from anomalous but otherwise normal optic discs.21
The list of differential diagnoses for optic disc edema, or swelling, is long (Table 13-1). A variety of mechanisms such as ischemia, inflammation, metabolic disorders, and pressure differentials may produce stasis of axonal flow, which causes swelling of the axons anterior to the lamina cribrosa and elevation of the optic disc. The normally transparent peripapillary nerve fiber layer gradually opacifies and obscures the retinal vessels. The disc margins become indistinct. The optic nerve may become hyperemic as small vessels are dilated, and characteristic splinter hemorrhages may appear at the disc margin in the nerve fiber layer. In addition, deeper hemorrhages may appear in the peripapillary retinal and subretinal layers. The swelling can also compromise venous outflow by enlarging the central veins at the disc, causing tortuosity, and producing retinal hemorrhages near the optic disc (Fig. 13-E2).20 The most common causes of optic disc edema include papilledema (from elevated intracranial pressure), AION, and demyelinating optic neuritis. A common clinical challenge is distinguishing true, pathologic optic disc swelling from anomalous but otherwise normal optic discs.21

 Optic atrophy is the final common pathway of optic nerve disease. Virtually all optic nerve insults eventually evolve to optic disc pallor or cupping, or both. Optic disc cupping is enlargement of the central cup as a result of loss of axons; this form of optic atrophy (without pallor of the remaining neuroretinal rim) is characteristic of glaucoma, but it can also be caused by other optic nerve disorders (Fig. 13-E3). Optic nerve pallor refers to a whitish appearance of the normally pink neuroretinal rim (Fig. 13-E4). Some forms of atrophic disc changes suggest their cause, such as the segmental pallor in AION or the temporal pallor typically present in toxic, nutritional, and hereditary optic neuropathies. However, in most cases, optic disc pallor is not diagnostic. The differential diagnosis is broad and includes all the disorders that initially cause disc edema and many entities that can lead to axonal death without first being manifested as disc swelling. Retrogeniculate insults do not cause optic disc pallor (perinatal events being an exception).
Optic atrophy is the final common pathway of optic nerve disease. Virtually all optic nerve insults eventually evolve to optic disc pallor or cupping, or both. Optic disc cupping is enlargement of the central cup as a result of loss of axons; this form of optic atrophy (without pallor of the remaining neuroretinal rim) is characteristic of glaucoma, but it can also be caused by other optic nerve disorders (Fig. 13-E3). Optic nerve pallor refers to a whitish appearance of the normally pink neuroretinal rim (Fig. 13-E4). Some forms of atrophic disc changes suggest their cause, such as the segmental pallor in AION or the temporal pallor typically present in toxic, nutritional, and hereditary optic neuropathies. However, in most cases, optic disc pallor is not diagnostic. The differential diagnosis is broad and includes all the disorders that initially cause disc edema and many entities that can lead to axonal death without first being manifested as disc swelling. Retrogeniculate insults do not cause optic disc pallor (perinatal events being an exception).
Disorders of the Afferent Visual System
Ocular Diseases
All patients who complain of decreased vision need an ophthalmologic evaluation. Even patients with known disorders that can potentially affect vision deserve a thorough ophthalmic examination if their vision changes. For example, in a patient known to have an optic nerve sheath meningioma, unrelated retinal detachment may develop. Anterior segment and retinal disorders are not always obvious on a cursory eye examination and may mimic optic nerve or intracranial disease.22 A few selected ocular disorders that may be confused with neurological vision loss are discussed in this section.
Retinal Abnormalities
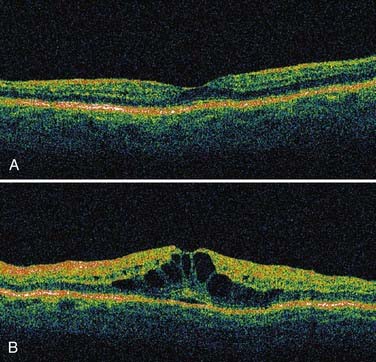
 Macular diseases cause central scotomas or metamorphopsia. Ocular coherence tomography (Fig. 13-E5) is a relatively new noninvasive tool for imaging the macula that has simplified the diagnosis of macular disease, although intravenous fluorescein angiography is still often required. Age-related macular degeneration is a common cause of painless central vision loss in patients older than 50 years; typically, macular pigment changes, whitish macular drusen, or blood is evident with the ophthalmoscope, but the findings may be subtle. Central serous retinopathy causes central scotomas in younger patients (and is often confused with optic neuritis). Epiretinal membranes tend to cause metamorphopsia rather than discrete scotomas and are not always evident on ophthalmoscopy. Macular holes can decrease visual acuity and cause tiny central visual field defects so small that the foveal threshold may be the only abnormal point with automated perimetry. Cystoid macular edema can occur in association with diabetes, uveitis, and retinitis pigmentosa and cause central vision loss. Most of the time, retinal trauma is obvious from the history and fundus examination, but permanent subtle changes from trauma can occur.
Macular diseases cause central scotomas or metamorphopsia. Ocular coherence tomography (Fig. 13-E5) is a relatively new noninvasive tool for imaging the macula that has simplified the diagnosis of macular disease, although intravenous fluorescein angiography is still often required. Age-related macular degeneration is a common cause of painless central vision loss in patients older than 50 years; typically, macular pigment changes, whitish macular drusen, or blood is evident with the ophthalmoscope, but the findings may be subtle. Central serous retinopathy causes central scotomas in younger patients (and is often confused with optic neuritis). Epiretinal membranes tend to cause metamorphopsia rather than discrete scotomas and are not always evident on ophthalmoscopy. Macular holes can decrease visual acuity and cause tiny central visual field defects so small that the foveal threshold may be the only abnormal point with automated perimetry. Cystoid macular edema can occur in association with diabetes, uveitis, and retinitis pigmentosa and cause central vision loss. Most of the time, retinal trauma is obvious from the history and fundus examination, but permanent subtle changes from trauma can occur.

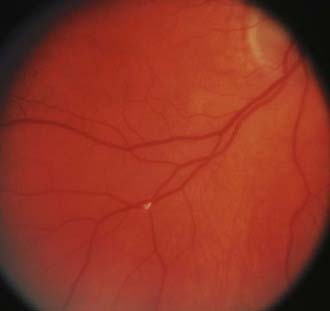
 Branch retinal artery occlusion causes acute vision loss and focal visual field defects that can be similar to those associated with optic nerve disorders.23,24 Acutely, the fundus examination reveals a whitish yellow opaque area of edematous ischemic retina; frequently, emboli can be seen in the retinal arterioles (Fig. 13-E6).25,26 However, the retinal edema typically clears in days to weeks, with only subtle changes in the caliber of the affected retinal arteriole remaining. Similarly, the retinal edema and “cherry-red spot” from central retinal artery occlusion quickly vanishes but leaves retinal arteriolar narrowing and mild diffuse optic nerve pallor (Fig. 13-E7).
Branch retinal artery occlusion causes acute vision loss and focal visual field defects that can be similar to those associated with optic nerve disorders.23,24 Acutely, the fundus examination reveals a whitish yellow opaque area of edematous ischemic retina; frequently, emboli can be seen in the retinal arterioles (Fig. 13-E6).25,26 However, the retinal edema typically clears in days to weeks, with only subtle changes in the caliber of the affected retinal arteriole remaining. Similarly, the retinal edema and “cherry-red spot” from central retinal artery occlusion quickly vanishes but leaves retinal arteriolar narrowing and mild diffuse optic nerve pallor (Fig. 13-E7).
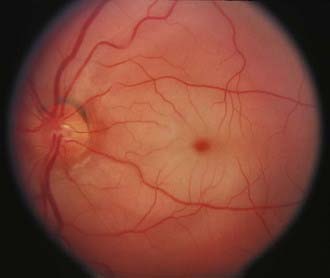
 Central retinal vein occlusion27 and branch retinal vein occlusion28 cause marked retinal hemorrhages that are impressive and difficult to miss initially (Fig. 13-E8). Sometimes, venous collateral vessels on the optic disc (opticociliary shunts), tortuous retinal veins, and slight disc pallor remain and mimic the findings of an optic nerve sheath meningioma.
Central retinal vein occlusion27 and branch retinal vein occlusion28 cause marked retinal hemorrhages that are impressive and difficult to miss initially (Fig. 13-E8). Sometimes, venous collateral vessels on the optic disc (opticociliary shunts), tortuous retinal veins, and slight disc pallor remain and mimic the findings of an optic nerve sheath meningioma.
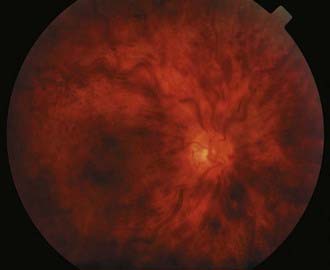
Cancer-associated retinopathy (CAR) is a paraneoplastic syndrome in which autoantibodies are directed against components of the retina; it is usually associated with small cell carcinoma or other visceral malignancies.29–31 The classic triad consists of photosensitivity, ring scotomatous visual field loss, and attenuated retinal arterioles.32 The electroretinogram is typically abnormal, and CAR antibodies may be detected in serum.33,34 A CAR-like syndrome can also occur with melanoma (melanoma-associated retinopathy) and in the setting of systemic autoimmune disorders.35
Papilledema
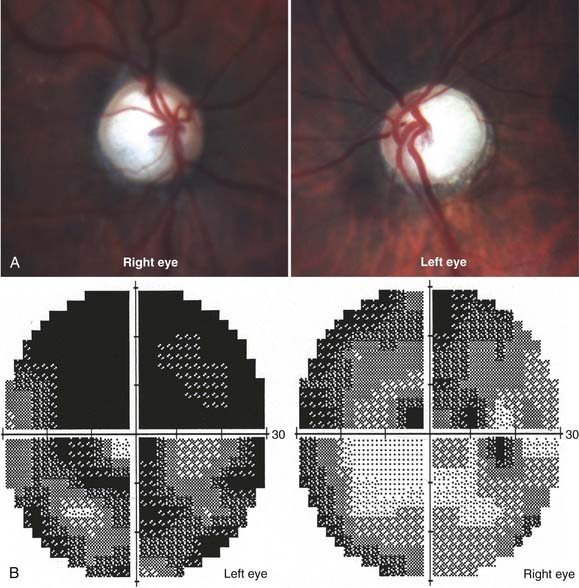
Elevated intracranial pressure from any cause can be transmitted to the optic nerve head and usually results in bilateral optic disc swelling. There are many causes of optic disc swelling, but the term papilledema refers specifically to the bilateral disc swelling associated with elevated intracranial pressure (Fig. 13-5). The optic disc may be markedly elevated, with hemorrhages of the nerve head and surrounding retina. Usually, there is no visual loss acutely, except for enlargement of the physiologic blind spot from elevation and compression of the peripapillary retina. If acute central visual loss is present, a different diagnosis such as bilateral optic neuritis or bilateral AION (i.e., giant cell arteritis) is more likely. Chronic papilledema can cause loss of the peripheral visual field, with central vision affected only very late in its course.36 The presence of bilateral optic disc edema requires immediate investigation, starting with a blood pressure measurement to evaluate the possibility of malignant hypertension and neuroimaging to rule out an intracranial mass or other abnormality. Normal findings on neuroimaging in the presence of elevated intracranial pressure suggest IIH, also called pseudotumor cerebri.37 IIH typically occurs in obese women between puberty and menopause.38 Men, as well as women who are not overweight, frequently harbor identifiable causes of elevated intracranial hypertension (such as an occult dural arteriovenous malformation, dural venous sinus occlusion, sleep apnea, and other conditions) and should not be considered to have IIH until an exhaustive clinical investigation confirms that there is no identifiable cause. IIH occurs in children, but the same careful scrutiny just noted should be applied to this group as well.39
It is important to understand that all patients with papilledema or known intracranial hypertension must be monitored by an ophthalmologist with serial optic disc evaluations (and photographs) and visual fields. The vision loss with papilledema can be insidious and unappreciated by the patient initially, with severe peripheral visual field loss even in the presence of excellent visual acuity.40
Anterior Ischemic Optic Neuropathy
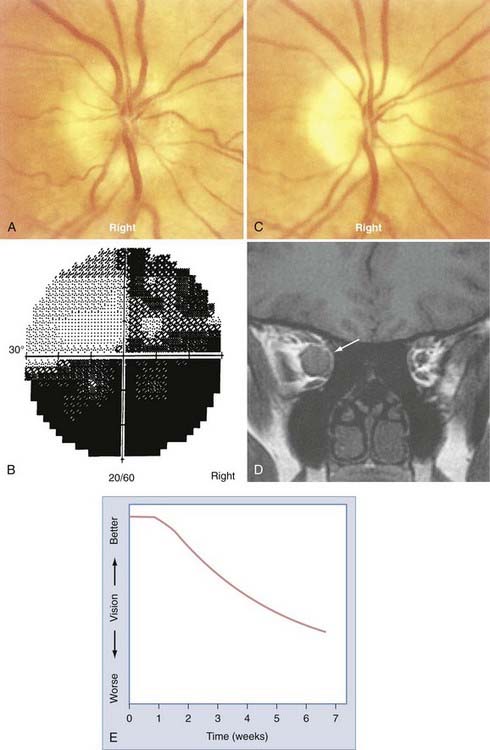
AION is characterized by sudden, painless loss of vision associated with unilateral optic disc swelling in patients 45 years or older. An infarct of the optic nerve head occurs as a result of occlusion of one or more of the posterior ciliary arteries.41 These vessels originate from the ophthalmic artery in the orbit, enter the globe around the optic nerve, and supply the peripapillary choroid and optic nerve head. AION may occur as a sequela of generalized atherosclerotic disease (i.e., nonarteritic AION)42 or be secondary to vasculitis, most commonly giant cell arteritis (i.e., arteritic AION).43
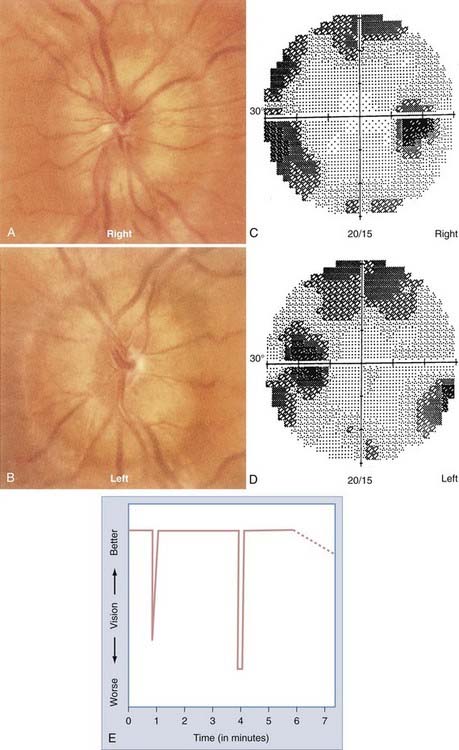
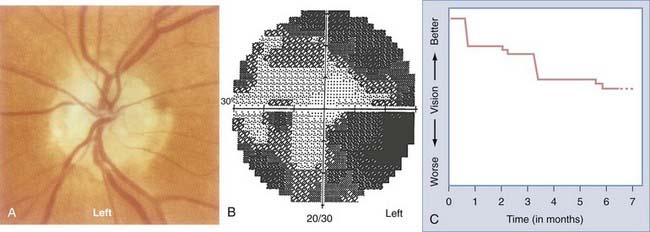
The most common form of AION is nonarteritic and it is manifested as isolated monocular vision loss. The disc edema and resulting pallor are typically confined to the upper or lower portion of the nerve, with a corresponding altitudinal visual field defect (Fig. 13-6). The visual field defect tends to be permanent, although minimal improvement may occur over time. No treatment has been shown to be effective.
The precipitating mechanisms of this multifactorial event are not fully understood.44 The architecture of the optic disc is an important factor because affected discs are almost always small and cupless. Patients with nonarteritic AION may have systemic vascular diseases such as systemic hypertension (40%) or diabetes mellitus (20%) or systemic manifestations of atherosclerotic disease such as angina pectoris, previous myocardial infarction, intermittent claudication, or a history of stroke.45,46 Hypercholesterolemia is associated with nonarteritic AION in patients 50 years or younger.47 Unilateral or bilateral nonarteritic AION can occur in predisposed patients with hypotensive episodes in the setting of surgery (including spinal cord surgery with induced hypotension) or trauma. The prognosis for vision is not good in this situation, even with prompt correction of hypotension, hypovolemia, and hypoperfusion.48
There is no proven, effective treatment of nonarteritic AION. Optic nerve sheath fenestration, once considered a potential surgical treatment in the past, was formally studied and shown to be ineffective, possibly even harmful.49,50
Thirty percent to 50% of patients have an event in the second eye, usually years later. Most clinicians advocate the daily use of aspirin because it may theoretically decrease the risk for an event in the contralateral eye.51 As with all functionally monocular patients, care should be taken to protect the unaffected eye from trauma with shatterproof polycarbonate lens eyeglasses.
 In all cases of AION, it is vital to determine whether there is any evidence of an arteritic cause (e.g., giant cell arteritis). Untreated giant cell arteritis can cause rapid, sequential, or simultaneous blindness in both eyes. For this reason, the erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP) level should be determined for all patients older than 55 years who have AION. If giant cell arteritis is suspected because of an elevated ESR or CRP, or both, or because of symptoms such as headache, scalp and temple tenderness, myalgias, arthralgias, low-grade fever, anemia, malaise, weight loss, anorexia, or jaw claudication,52 oral or intravenous steroid treatment should be instituted immediately.53 In acute cases, the patient may benefit from high-dose intravenous steroids. Biopsy of the temporal artery should follow within days of steroid therapy.54,55 In arteritic AION, the visual loss is usually profound, and the optic nerve is often diffusely swollen and pale (Fig. 13-E9).
In all cases of AION, it is vital to determine whether there is any evidence of an arteritic cause (e.g., giant cell arteritis). Untreated giant cell arteritis can cause rapid, sequential, or simultaneous blindness in both eyes. For this reason, the erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP) level should be determined for all patients older than 55 years who have AION. If giant cell arteritis is suspected because of an elevated ESR or CRP, or both, or because of symptoms such as headache, scalp and temple tenderness, myalgias, arthralgias, low-grade fever, anemia, malaise, weight loss, anorexia, or jaw claudication,52 oral or intravenous steroid treatment should be instituted immediately.53 In acute cases, the patient may benefit from high-dose intravenous steroids. Biopsy of the temporal artery should follow within days of steroid therapy.54,55 In arteritic AION, the visual loss is usually profound, and the optic nerve is often diffusely swollen and pale (Fig. 13-E9).
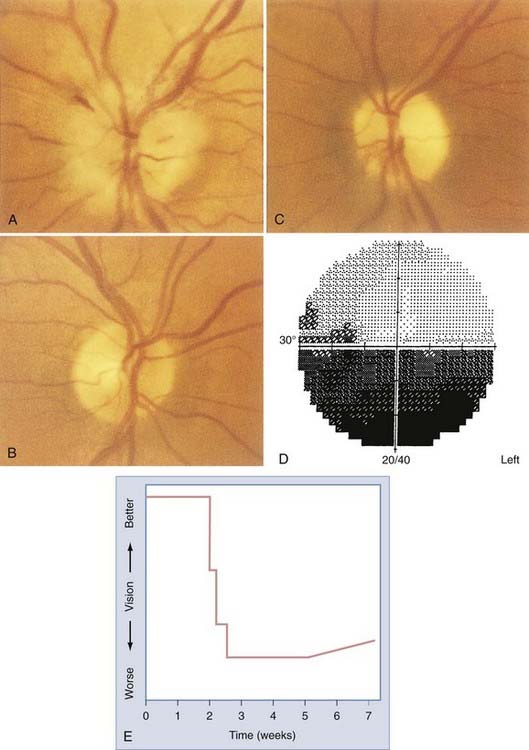
Optic Neuritis
Optic neuritis is typically manifested as unilateral visual loss (often described as a skim, scum, blur, haze, cloud, dimness, or washed-out view) in young adults (15 to 45 years old), with women outnumbering men 5 to 1. The vision worsens progressively over a period of several days and is almost always accompanied by retrobulbar pain that is made worse with eye movement (Fig. 13-7). After 1 or 2 weeks, vision begins to recover slowly. In most cases, vision gradually improves over a period of 3 to 12 months, and in more than 80% of patients, recovery is nearly complete.56 Optic neuritis is caused by demyelination of the optic nerve. Because optic neuritis is commonly associated with multiple sclerosis (MS),57–59 it is no surprise that patients may report other past or present neurological symptoms.
The optic disc initially appears normal in two thirds of adults with optic neuritis.60 An RAPD provides objective evidence of the visual loss. In addition to optic neuritis, compressive optic neuropathy, giant cell arteritis, carcinomatous meningitis, optic nerve trauma, and radiation-induced optic neuropathy can also cause acute monocular visual loss with relatively normal-appearing optic discs initially. In older patients, it may be difficult to distinguish optic neuritis with a swollen disc from AION.
The Optic Neuritis Treatment Trial (ONTT) was a multicenter national study that addressed the efficacy of steroid treatment in patients with optic neuritis. The ONTT randomized 458 patients with optic neuritis into three treatment groups: no treatment (i.e., placebo tablets), moderate-dose oral prednisone, and high-dose intravenous methylprednisolone for 3 days (followed by an 11-day taper of oral prednisone).61 The outcomes of the study and resultant treatment recommendations are as follows:
Many clinicians have modified the original high-dose intravenous regimen (250 mg every 6 hours for 3 days) to make it practical for home intravenous therapy, with 500 mg methylprednisolone administered twice each day or even 1000 mg once daily for 3 to 5 days.61
In a high percentage of women with optic neuritis and a lower percentage of men, clinically definite MS is eventually diagnosed in their lifetimes, although some studies did not find a gender predilection.62 The probability is greatly increased (50% to 80%) when white matter changes are seen on MRI.58,59
The discussion regarding long-term treatment options for patients with optic neuritis who are at risk for MS is complex. The Controlled High-Risk Subjects Avonex Multiple Sclerosis Prevention Study (CHAMPS) was one of the first to suggest that patients with isolated optic neuritis (no other neurological symptoms) and white matter lesions on MRI may benefit from initiation of interferon beta-1a. Other clinical studies have added additional information (and complexity) to treatment decisions for potential MS in patients with optic neutitis.63
Compressive and Infiltrative Optic Neuropathies
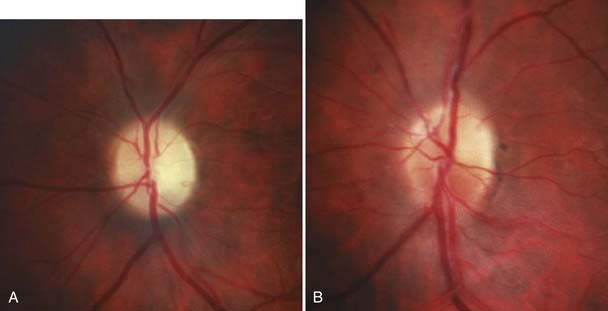
 Compression of the optic nerve in the orbit, in the optic canal, or intracranially usually causes slow, progressive loss of vision (Fig. 13-8).64–68 Common compressive optic neuropathies include optic nerve sheath meningioma and orbital Graves’ disease, although any mass lesion adjacent to the optic nerve in the orbit, in the optic canal, or intracranially can be implicated (Fig. 13-E10).
Compression of the optic nerve in the orbit, in the optic canal, or intracranially usually causes slow, progressive loss of vision (Fig. 13-8).64–68 Common compressive optic neuropathies include optic nerve sheath meningioma and orbital Graves’ disease, although any mass lesion adjacent to the optic nerve in the orbit, in the optic canal, or intracranially can be implicated (Fig. 13-E10).
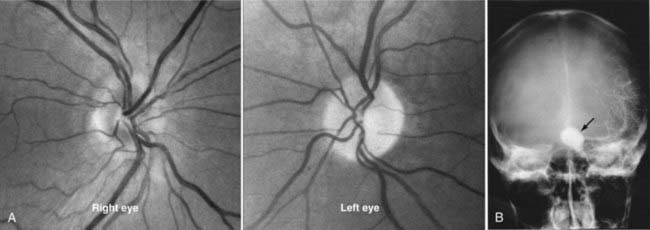
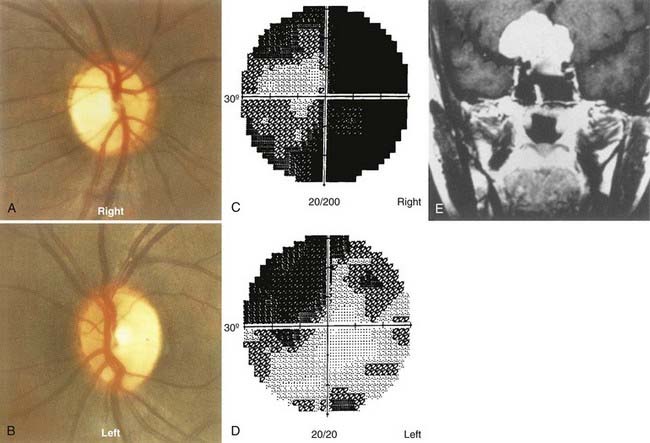
Meningiomas of the optic nerve sheath represent only 5% of all orbital tumors, but because of their anatomic proximity to the optic nerve, they are more likely than other orbital tumors to cause optic nerve compression and loss of vision. Optic nerve sheath meningiomas commonly occur in women (female-to-male ratio of 3:1) in their fourth decade. Optic atrophy and visual loss with opticociliary collateral vessels on the optic disc are characteristic of optic nerve sheath meningiomas.64,69 Meningiomas can also arise from the intracranial dura and compress the intracranial optic nerve or the chiasm. Sphenoid wing meningiomas frequently have both intraorbital and intracranial components.
Observation may be a reasonable initial treatment. The natural history of optic nerve sheath meningiomas is highly variable. They may remain static for many years, or they may progress relatively rapidly. Sequential neuroimaging plus careful monitoring of optic nerve function (sequential visual fields, visual acuity, contrast sensitivity testing, color vision testing, RAPD measurement, and other indicators of optic nerve function) in patients with an optic nerve sheath meningioma is vital because it provides a guide for management. Conformal radiation therapy is emerging as the most viable treatment option.70–73
Optic nerve gliomas usually occur in childhood (75% before 20 years of age) and are manifested as proptosis, visual loss, strabismus, or nystagmus.74 About half of optic nerve gliomas arise in the orbital portion of the optic nerve; the remainder arise intracranially. Fifty percent of patients with optic nerve gliomas have neurofibromatosis type 1 (NF1). Fifteen percent of patients with NF1 harbor an optic nerve glioma. These tumors are pilocytic astrocytomas (juvenile type) with a benign cytologic appearance. They enlarge very slowly or may appear inactive.75 Spontaneous regression of even large, symptomatic optic gliomas can occur, both in patients with and in patients without NF1.76 By contrast, malignant gliomas of the anterior visual pathway that arise in adulthood behave very differently and rapidly lead to blindness and death.77,78
Neuroimaging of childhood gliomas typically reveals a fusiform swelling of the optic nerve or chiasm, or both.79 Involvement of the chiasm may be associated with endocrine dysfunction and hypothalamic involvement. Children with optic nerve or chiasmal glioma should undergo endocrine evaluation, as well as a thorough search for systemic signs of neurofibromatosis. Most clinicians favor a conservative watch-and-wait approach given the usual static course in children,75 but surgical resection may be considered in exceptional patients with a blind eye and disfiguring proptosis. The indications for and effectiveness of radiation treatment and chemotherapy for these tumors are controversial and unresolved.
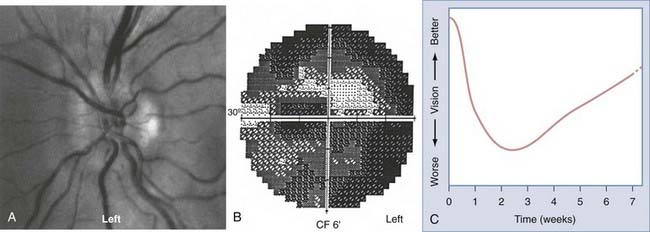
The optic nerve and disc may be directly infiltrated in lymphoproliferative80–82 or granulomatous (sarcoidosis)83–87 disorders. Leukemic infiltration with visual loss may respond to urgent radiation treatment (Fig. 13-9).
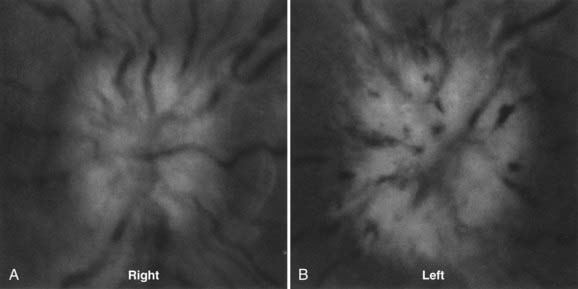
 Infectious agents can affect the optic nerve by direct infiltration, by inciting a local inflammatory response or autoimmune attack on the optic nerve (Fig. 13-E11), by local mass effect from an infectious focus, or by compromise of the vascular supply by vasospasm or vascular occlusion from the products of an inflammatory response. Many agents have been implicated, but syphilis can affect the visual pathway in so many and varied ways that it should always be included in the differential diagnosis of otherwise unexplained optic neuropathy.
Infectious agents can affect the optic nerve by direct infiltration, by inciting a local inflammatory response or autoimmune attack on the optic nerve (Fig. 13-E11), by local mass effect from an infectious focus, or by compromise of the vascular supply by vasospasm or vascular occlusion from the products of an inflammatory response. Many agents have been implicated, but syphilis can affect the visual pathway in so many and varied ways that it should always be included in the differential diagnosis of otherwise unexplained optic neuropathy.
Optic Disc Drusen and Other Anomalies
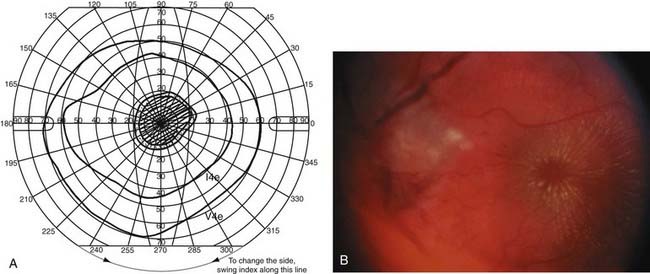
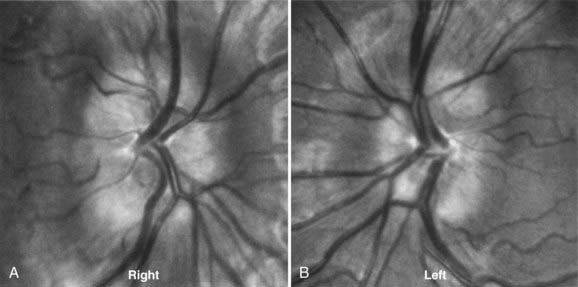
About 1% of the population may have elevated optic discs as a result of optic nerve head drusen (Fig. 13-10).88 In this condition, hyaline material is present in the substance of the optic disc and elevates the axons as they enter the optic nerve head. The rock-like, irregular yellow material is often exposed in older patients.89 This condition is bilateral in 70% of patients. Peripheral visual field defects may occur, but optic disc drusen typically do not decrease visual acuity.
In young patients, optic disc drusen may be situated deep in the substance of the optic nerve and not be visible with the ophthalmoscope. The elevated appearance of these discs with “buried” drusen can be confused with true optic disc edema. Features that help distinguish these anomalous discs from pathologic optic disc swelling include a “crowded” appearance with no central cup, irregularly scalloped disc margins, anomalous branching of the central retinal artery with many vessels crossing the disc margin, and lack of change over time (Fig. 13-11).
Hereditary Optic Neuropathies
A variety of hereditary optic neuropathies may be manifested as part of a neurological syndrome or appear as isolated findings. Kjer’s dominant optic atrophy causes relatively mild to moderate central visual loss that may not be noticed until vision-screening examinations at school. The optic discs usually show bilateral temporal pallor, but they may appear relatively normal at the onset of symptoms. The inheritance pattern may not be obvious in the family history because incomplete penetrance is common.90
Leber’s hereditary optic neuropathy causes rapid, sequential loss of central vision in young men and occasionally in women. Although the disc swelling is rarely impressive, the optic nerves often display fine peripapillary telangiectasia, which gives way to pallor after loss of vision. This disorder is inherited through at least three known mitochondrial DNA mutations and is therefore passed from (usually asymptomatic) women to their children.91
Traumatic Optic Neuropathy
Traumatic optic neuropathy may be caused by direct trauma to the optic nerve from foreign bodies or bone fragments or by indirect trauma, with visual loss resulting from force transmitted to the optic canal by blunt trauma to the brow without fracture.92,93 The optic disc may appear normal acutely, but pallor inevitably follows in 4 to 8 weeks. High-dose intravenous steroids94 and surgical decompression of the optic canal95 have been advocated for the management of acute traumatic optic neuropathy in the past, but the efficacy and safety of these treatments have been questioned.96 Retinal detachment, penetrating injuries, and other globe injury must be sought in all cases of visual loss from trauma.97
The Chiasm
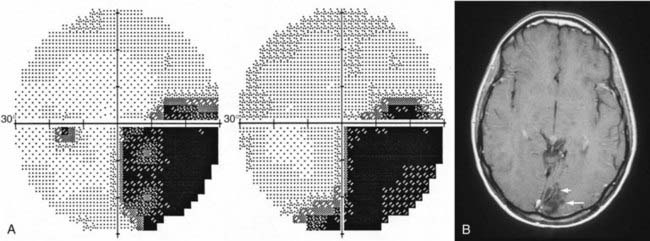
At the chiasm, axons representing each half of the visual space are routed to their respective optic tracts. Because axons from the nasal hemiretina of each eye decussate, defects in the temporal visual fields of both eyes (“bitemporal hemianopia”) result from lesions that affect the central portion of the chiasm (Fig. 13-12). However, chiasmal injury usually arises from mass lesions that do not attack with pinpoint accuracy; the visual fields may reflect damage primarily to one or both optic nerves (optic nerve–related visual field defect), the body of the chiasm (bitemporal field defects), the optic tract (incongruous homonymous field defects), or a combination of these three patterns of visual field loss.98 Mass lesions arising from the sellar and parasellar region that affect the chiasm include pituitary tumors, meningiomas, craniopharyngiomas, Rathke’s cleft cysts, and occasionally, giant aneurysms. The chiasm is located about 1 cm above the sella; thus, tumors arising in the sella (such as pituitary macroadenomas) must be relatively large to compress the chiasm and affect vision. These large pituitary macroadenomas are generally nonsecreting, given that patients with secreting adenomas usually seek medical help for their endocrine dysfunction before the tumor is large enough to compress the chiasm. The prognosis for improvement in visual fields and visual acuity is good (>80%) after transsphenoidal surgery for pituitary adenomas.99 Rarely, “herniation” of the chiasm into an empty or surgically manipulated sella may affect the body of the chiasm.100 In addition to mass lesions, conditions that commonly affect the optic nerve are also potential causes of chiasmal dysfunction: demyelination, gliomas, infiltration, vasculitis (Fig. 13-13), and trauma.101
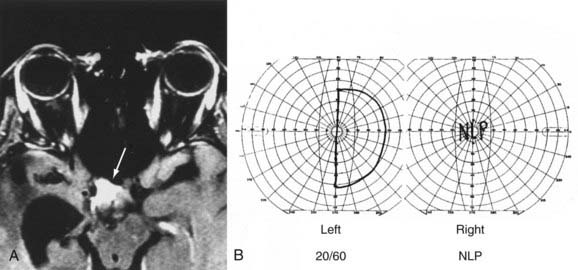
Lesions affecting the chiasm almost never cause optic disc swelling but rather result in retrograde axonal atrophy, which with time will result in visible optic disc pallor and loss of nerve fiber layers (Fig. 13-14). An RAPD is often present with chiasmal disease, depending on the density and asymmetry of the visual field loss in each of the two eyes. The RAPD will be present in the eye with the greatest visual field loss, not necessarily the eye with the worst visual acuity.
Retrochiasmal Visual Pathways
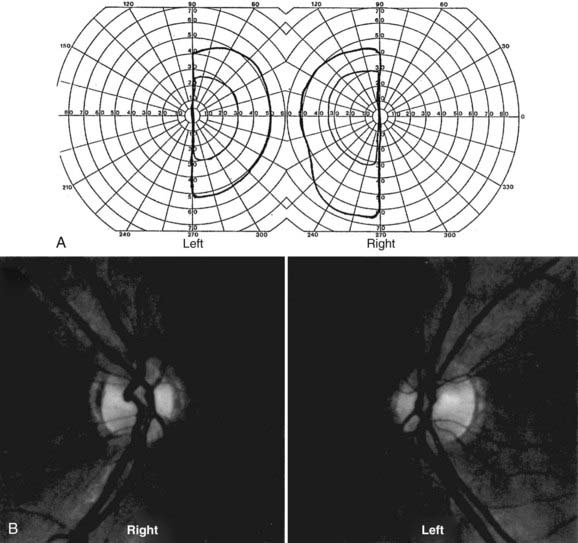
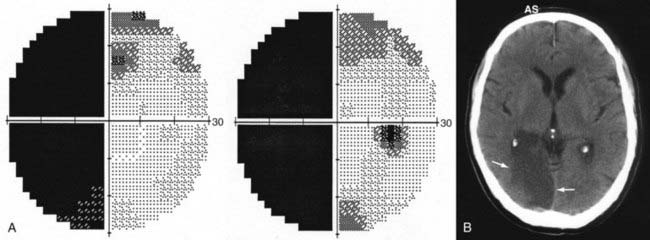
 Lesions of the visual system posterior to the chiasm affect homonymous portions of the visual fields in both eyes. Optic tract lesions are caused by the same processes that affect the chiasm. Of 21 patients with tract lesions, 17 were due to tumor or aneurysm.102 Tract lesions produce relatively incongruous homonymous visual field defects and often produce a specific pattern of optic disc pallor (bow-tie atrophy in the eye with temporal visual field loss, loss of the superior and inferior nerve fiber layer in the eye with nasal field loss). Optic tract lesions may also produce an RAPD because pupillomotor fibers travel in the optic tract before exiting at the brachium of the superior colliculus.100,103,104 Tract lesions are therefore characterized by contralateral incongruous homonymous visual field defects (Fig. 13-E12) and a small RAPD and optic disc pallor (frequently bow-tie or band atrophy105) in the eye contralateral to the lesion.103
Lesions of the visual system posterior to the chiasm affect homonymous portions of the visual fields in both eyes. Optic tract lesions are caused by the same processes that affect the chiasm. Of 21 patients with tract lesions, 17 were due to tumor or aneurysm.102 Tract lesions produce relatively incongruous homonymous visual field defects and often produce a specific pattern of optic disc pallor (bow-tie atrophy in the eye with temporal visual field loss, loss of the superior and inferior nerve fiber layer in the eye with nasal field loss). Optic tract lesions may also produce an RAPD because pupillomotor fibers travel in the optic tract before exiting at the brachium of the superior colliculus.100,103,104 Tract lesions are therefore characterized by contralateral incongruous homonymous visual field defects (Fig. 13-E12) and a small RAPD and optic disc pallor (frequently bow-tie or band atrophy105) in the eye contralateral to the lesion.103
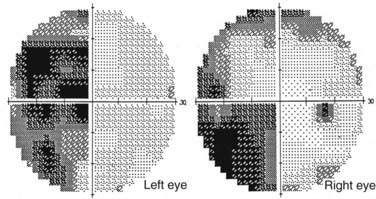
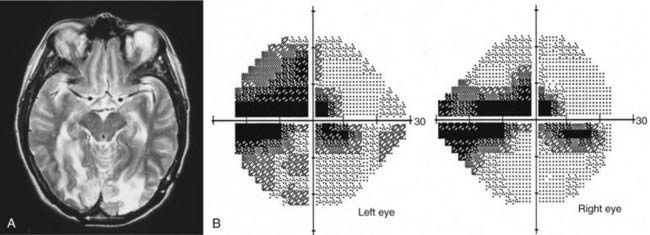
Axons in the optic tract synapse in the lateral geniculate nuclei. From there, superior retrogeniculate fibers travel directly through the parietal lobe to the occipital cortex and synapse superior to the calcarine fissure. Parietal lesions cause homonymous defects that are denser inferiorly. Inferior fibers take a more indirect route and sweep around the temporal horn of the ventricular system (i.e., Meyer’s loop) and through the temporal lobe to neurons in the inferior occipital cortex (see Fig. 13-4). Lesions in the temporal lobe produce homonymous visual field defects that are denser in the superior quadrants (Fig. 13-15). Such superior visual field defects may be created iatrogenically when pallidotomy is performed for Parkinson’s disease or temporal lobectomy is performed for seizures.106
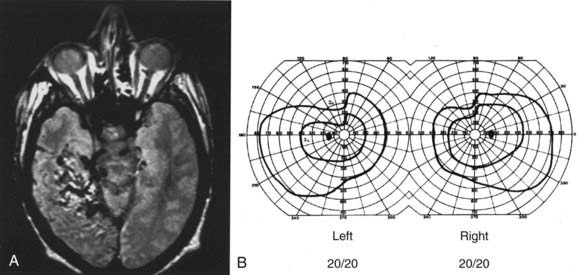


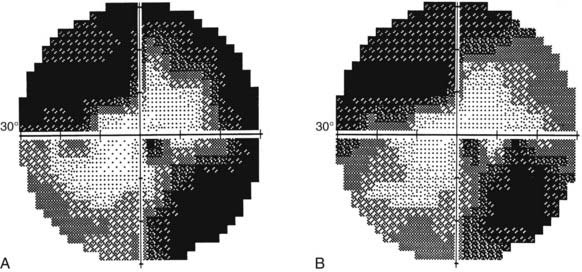
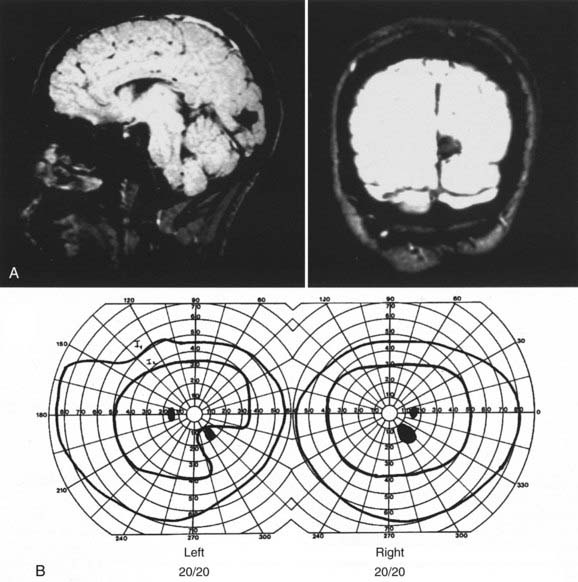
 From the chiasm posteriorly, corresponding axons move closer together as they converge on a single point in the occipital cortex; the more posterior the lesion, the more precise the homonymous defects approximate each other (i.e., greater congruity).107 Occipital lesions tend to be highly congruous (Figs. 13-16, 13-17, and Fig. 13-E13). Complete homonymous visual field defects are said to be nonlocalizing because they can be caused by lesions anywhere from the optic tract to the occipital lobe (Fig. 13-E14). A homonymous hemianopic visual field defect from a unilateral retrochiasmal lesion does not cause decreased visual acuity. However, bilateral retrochiasmal lesions (e.g., bilateral occipital infarcts) can profoundly affect vision in both eyes despite normal findings on ophthalmoscopy and normal pupillary responses (Fig. 13-E15).
From the chiasm posteriorly, corresponding axons move closer together as they converge on a single point in the occipital cortex; the more posterior the lesion, the more precise the homonymous defects approximate each other (i.e., greater congruity).107 Occipital lesions tend to be highly congruous (Figs. 13-16, 13-17, and Fig. 13-E13). Complete homonymous visual field defects are said to be nonlocalizing because they can be caused by lesions anywhere from the optic tract to the occipital lobe (Fig. 13-E14). A homonymous hemianopic visual field defect from a unilateral retrochiasmal lesion does not cause decreased visual acuity. However, bilateral retrochiasmal lesions (e.g., bilateral occipital infarcts) can profoundly affect vision in both eyes despite normal findings on ophthalmoscopy and normal pupillary responses (Fig. 13-E15).
Ocular Motility and the Pupil: The Efferent Visual System
Anatomy and Pathophysiology
Each orbit contains seven extraocular muscles (including the levator palpebrae) driven by cranial nerves III (oculomotor), IV (trochlear), and VI (abducens). Figure 13-18 reviews the course of these cranial nerves from the brainstem to the orbit.
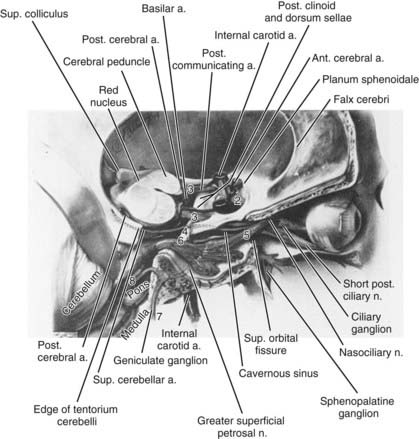
Both eyes move together to keep an object of regard imaged simultaneously on the fovea of each eye. These two retinal images are not identical because they view the object from slightly different angles. This disparity is processed by the brain to produce the perception of depth, or stereopsis. This “fusion” process can break down if there is a significant problem in the motor (efferent) visual system, with diplopia usually being produced. For example, damage to cranial nerve VI causes a weakness of the lateral rectus muscle, and as a result the eye turns too far toward the nose. An object viewed with the fovea of the normal eye is also seen by the “crossed” eye but is imaged on the nasal retina and appears to be in the patient’s temporal field. The patient has diplopia, with the object seen twice when both eyes are open: directly ahead with the fixing eye and in the temporal visual field of the crossed eye. Confusion results when there are two different superimposed foveal images or a perception of two different images seen straight ahead.108,109
History and Examination
Determining which extraocular muscle or muscles are weak is based on a logical principle: a patient’s diplopia, as well as the observed misalignment, should be greatest in the field of action of the weak muscle. Ocular alignment is observed as a patient follows the examiner’s fingertip on a prescribed course to test the individual extraocular muscles (Box 13-4).108,109
Box 13-4
How to Examine Ocular Motility
Observe the eyelid position and the pupils for asymmetry with the eyes in the primary position (third cranial nerve or Horner’s syndrome) and throughout the examination (aberrant regeneration of the third cranial nerve). Look for nystagmus and smoothness of pursuit by having the patient follow a slowly moving target. Observe horizontal versions to locate weakness or slowness of the adducting eye (intranuclear ophthalmoplegia) or abducting eye (abducents nerve deficit), or both (gaze paresis). Next, examine vertical versions. Straight up-and-down motion gives information about vertical gaze disturbances (dorsal midbrain syndrome) or orbital disturbances (Graves’ disease), but isolating the complex, vertically acting muscles requires testing in the vertical plane in right and left gaze. The superior and inferior oblique muscles become the primary vertical muscles when an eye is adducted, and the superior and inferior rectus muscles are most important vertically in abduction. For example, when the left eye looks down and right (adducted), the superior oblique muscle of that eye is being tested (the primary depressor of the abducted right eye is the right inferior rectus). Left trochlear nerve deficiencies are noted in this right and downgaze position (see Fig. 14-20). The patient’s report of which positions produce diplopia and determination of the severity of the diplopia in these positions are helpful additions to the examiner’s observations. Dissociating the two eyes with a red lens or red Maddox rod over the right eye helps patients report their observations. Ask the patient, “Are the red and white lights further apart when you look here to the right or when I move to the left?” The lens of the Maddox rod produces the image of a red line, which can be oriented to test the patient’s vision in a purely vertical or horizontal plane. Additional tests include quantitation of ocular deviations with the prism alternate cover test (which uses known prisms to neutralize the shift in eye position when a cover is moved from eye to eye) and measurement of ocular torsion with the double Maddox rod.
Disorders of the Visual Motor System
In this chapter we primarily address cranial nerve palsies. Table 13-2 shows the causes of isolated and combined cranial neuropathies in a study from the Mayo Clinic in which 4278 cases were reviewed.110 Tables 13-3 to 13-5 summarize the disorders affecting cranial nerves VI, IV, and III by anatomic location.
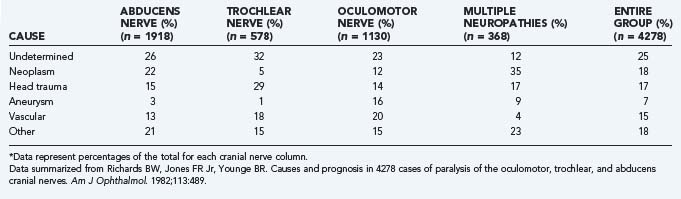
TABLE 13-3 Causes and Associated Findings in Patients with Abducens Nerve (Cranial Nerve VI) Paresis by Anatomic Location
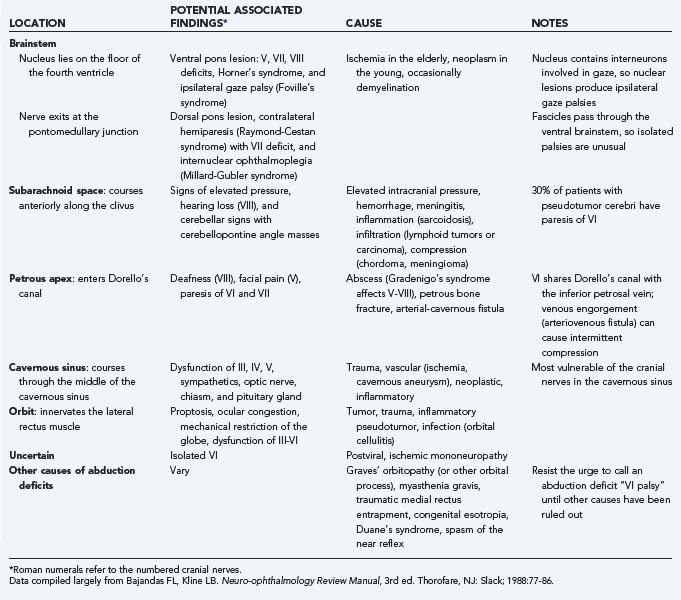
TABLE 13-4 Causes and Associated Findings in Patients with Trochlear Nerve (Cranial Nerve IV) Paresis by Anatomic Location
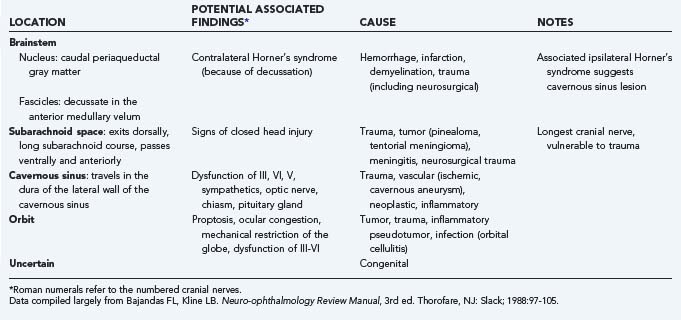
TABLE 13-5 Causes and Associated Findings in Patients with Oculomotor Nerve (Cranial Nerve III) Paresis by Anatomic Location
Abducens Nerve (Cranial Nerve VI)
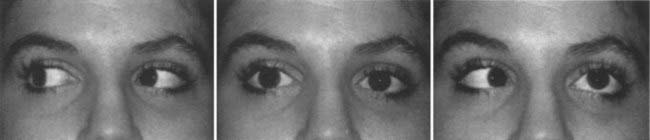
 In patients with left abducens paresis, there may be no misalignment of right gaze because the left lateral rectus muscle is relaxed in this field of gaze. However, in left gaze, the left lateral rectus muscle cannot fully pull the left eye out, and an esotropia is evident. To avoid diplopia, the patient may assume a posture with the head turned to the left. Examination of the function of cranial nerve VI is performed by having the patient look in far right and far left gaze (Fig. 13-19, Video 13-1). Trauma is a common cause of abducens palsy in all age groups (see Table 13-2). Acute, painful abducens paresis in a patient 45 years or older who has hypertension or diabetes suggests an ischemic cranial mononeuropathy. Recovery over a period of 2 to 6 months is the rule. In young patients, abducens palsies may occur as a postviral syndrome, but the clinician must remain alert for the possibility of tumor. Elevation of intracranial pressure can affect the function of one or both sixth cranial nerves (see Table 13-3).
In patients with left abducens paresis, there may be no misalignment of right gaze because the left lateral rectus muscle is relaxed in this field of gaze. However, in left gaze, the left lateral rectus muscle cannot fully pull the left eye out, and an esotropia is evident. To avoid diplopia, the patient may assume a posture with the head turned to the left. Examination of the function of cranial nerve VI is performed by having the patient look in far right and far left gaze (Fig. 13-19, Video 13-1). Trauma is a common cause of abducens palsy in all age groups (see Table 13-2). Acute, painful abducens paresis in a patient 45 years or older who has hypertension or diabetes suggests an ischemic cranial mononeuropathy. Recovery over a period of 2 to 6 months is the rule. In young patients, abducens palsies may occur as a postviral syndrome, but the clinician must remain alert for the possibility of tumor. Elevation of intracranial pressure can affect the function of one or both sixth cranial nerves (see Table 13-3).
Trochlear Nerve (Cranial Nerve IV)
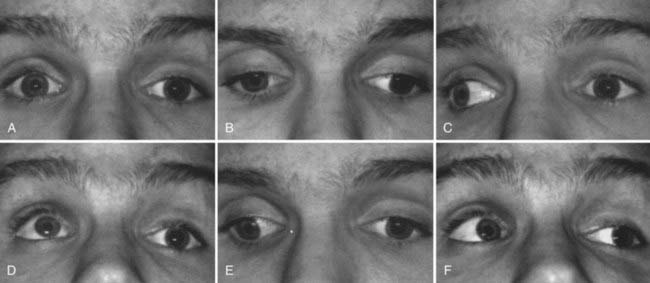
 The trochlear nerve innervates only the superior oblique muscle, but this muscle has a complex action. Because of its redirection at the trochlea and attachment to the globe, the superior oblique muscle intorts, depresses, and abducts the eye. When the globe is adducted, however, the angle of the muscle’s insertion minimizes all actions except pure depression. This fact simplifies clinical evaluation of the superior oblique muscle; the clinician looks at how well the right eye can move down in left gaze and how well the left eye moves down in right gaze (Fig. 13-20, Video 13-2). The vital role of the superior oblique muscle in ocular cyclotorsion explains why patients with trochlear palsy often describe diplopia with one image tilted.
The trochlear nerve innervates only the superior oblique muscle, but this muscle has a complex action. Because of its redirection at the trochlea and attachment to the globe, the superior oblique muscle intorts, depresses, and abducts the eye. When the globe is adducted, however, the angle of the muscle’s insertion minimizes all actions except pure depression. This fact simplifies clinical evaluation of the superior oblique muscle; the clinician looks at how well the right eye can move down in left gaze and how well the left eye moves down in right gaze (Fig. 13-20, Video 13-2). The vital role of the superior oblique muscle in ocular cyclotorsion explains why patients with trochlear palsy often describe diplopia with one image tilted.

Trauma is a common cause of trochlear palsy (also called superior oblique palsy). “Decompensation” of congenital fourth cranial nerve palsies is also common but difficult to diagnose convincingly. Neoplasm must be excluded when other causes are not evident (see Tables 13-2 and 13-4).111,112
Oculomotor Nerve (Cranial Nerve III)
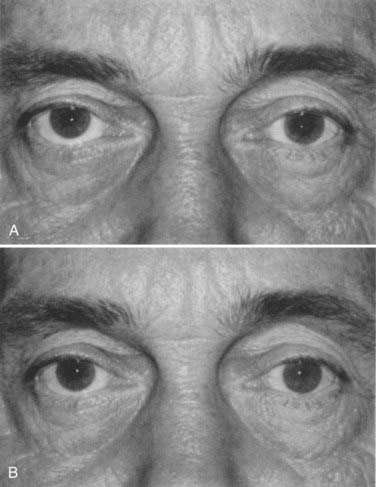
 The oculomotor nerve is complex. It innervates all the extraocular muscles (including the levator palpebrae), except for the lateral rectus and superior oblique muscles. It also carries parasympathetic efferent fibers to the pupillary sphincter and ciliary muscle through the ciliary ganglion. Complete third cranial nerve palsy produces an eye that is turned down and out (because of remaining function of the superior oblique and lateral rectus muscles) and a dilated pupil with ptosis (Video 13-3). Partial or incomplete paresis may present a more confusing picture (Fig. 13-21). The pupillary fibers run superficially in the nerve and are preferentially affected by compression, such as from a posterior communicating artery aneurysm or brainstem herniation. Ischemic cranial mononeuropathy of the vasa nervosum of the third nerve generally shows relative pupillary sparing, with the pupil being less affected than motility. Evaluation of the pupil plays a major role in clinical decision making.113,114 Oculomotor palsy occurring after relatively minor trauma may be the initial sign of a basal intracranial tumor or posterior communicating artery aneurysm.113 Other causes are outlined in Tables 13-2 and 13-5.
The oculomotor nerve is complex. It innervates all the extraocular muscles (including the levator palpebrae), except for the lateral rectus and superior oblique muscles. It also carries parasympathetic efferent fibers to the pupillary sphincter and ciliary muscle through the ciliary ganglion. Complete third cranial nerve palsy produces an eye that is turned down and out (because of remaining function of the superior oblique and lateral rectus muscles) and a dilated pupil with ptosis (Video 13-3). Partial or incomplete paresis may present a more confusing picture (Fig. 13-21). The pupillary fibers run superficially in the nerve and are preferentially affected by compression, such as from a posterior communicating artery aneurysm or brainstem herniation. Ischemic cranial mononeuropathy of the vasa nervosum of the third nerve generally shows relative pupillary sparing, with the pupil being less affected than motility. Evaluation of the pupil plays a major role in clinical decision making.113,114 Oculomotor palsy occurring after relatively minor trauma may be the initial sign of a basal intracranial tumor or posterior communicating artery aneurysm.113 Other causes are outlined in Tables 13-2 and 13-5.
Regeneration after disruption of the axons of the oculomotor nerve may result in misdirection and produce clinical signs of aberrant regeneration: pupillary constriction115 or lid elevation with attempted adduction or paradoxical motility.116 Aberrant regeneration never occurs with an ischemic (diabetic) mononeuropathy and always implies that the nerve has been injured in such a way that the myelin sheath and perineurium have been broached (i.e., aneurysm, tumor, or trauma).117 Aberrant regeneration may rarely occur without an antecedent acute third cranial nerve palsy. This is called primary aberrant regeneration and occurs as a result of slow-growing cavernous sinus masses, such as a meningioma or aneurysm.
Symptomatic Treatment of Diplopia
The Pupil
Pupillary size is determined by the net result of parasympathetic innervation to the strong pupillary sphincter (through the oculomotor nerve), sympathetic innervation of the much weaker pupillary dilator, and local iris factors that affect the “stiffness” of the iris stroma. Ptosis of the upper eyelid is an important accompanying clinical sign because the third cranial nerve and the sympathetic systems are involved in lid position. The oculomotor nerve innervates the powerful levator muscle, and the sympathetic nerves contribute a few millimeters of lid lift through Müller’s muscle.118
The Abnormally Large Pupil
When the larger pupil is abnormal, the most important concerns are oculomotor nerve compression, Adie’s pupil, or pharmacologic mydriasis. Pupillary dilation in isolation, without any other sign of oculomotor nerve dysfunction, is unlikely to be caused by an oculomotor palsy. An Adie “tonic” pupil is generally manifested as unilateral mydriasis with the pupillary response to near focus being far better than the response to light; this light-near dissociation is also seen in “blind” eyes and bilaterally in patients with neurosyphilis or the dorsal midbrain syndrome. Redilation and relaxed accommodation occur very slowly after convergence and near focus, thus the designation as a tonic pupil. This condition is the result of an insult to the ciliary ganglion, with subsequent aberrant regeneration of pupillary and accommodative fibers. Irregular reinnervation of the iris sphincter segments is seen as sector palsies or vermiform movements on slit-lamp examination. Supersensitivity of the pupillary sphincter can be identified with dilute pilocarpine testing (Fig. 13-22).119
The Abnormally Small Pupil
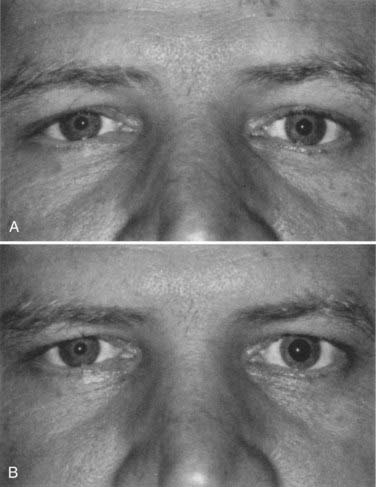
When the smaller pupil is abnormal, an oculosympathetic paresis (e.g., Horner’s syndrome) must be considered. Relative miosis and a slight ptosis (1 to 2 mm) may result from interruption of the oculosympathetic pathway anywhere along the three-neuron chain: from the hypothalamus to the ciliospinal center of Budge-Waller (C8-T1), across the lung apex to the superior cervical ganglion, or from the superior cervical ganglion by means of the carotid plexus sympathetic nerves to the pupillary dilator. Postganglionic interruption (at or distal to the superior cervical ganglion) is commonly benign or idiopathic, but preganglionic or central oculosympathetic pareses are associated with malignancy in about half the cases.118 Pharmacologic testing with topical cocaine and apraclonidine is useful in confirming an oculosympathetic paresis (Fig. 13-23).120,121 Hydroxyamphetamine, previously used to aid in localizing the level of an oculosympathetic paresis, is no longer available.
Other Disorders
Dorsal Midbrain Syndrome
 Lesions in the region of the pretectum and posterior commissure frequently produce characteristic signs. Dorsal midbrain syndrome (also known as Parinaud’s syndrome or mesencephalic syndrome) consists of vertical gaze deficiencies, convergence spasm or paresis, bilateral mid-dilated pupils with light-near dissociation (poor reaction to light and good reaction to near), and convergence-retraction nystagmus. The nystagmus is unique to this syndrome and consists of rapid convergence with retraction of the globes on attempted upgaze. This result is most evident with an optokinetic stimulus moving downward because the fast upward component induces this finding (Video 13-4). Common causes include pinealomas, aneurysms of the vein of Galen, and noncommunicating hydrocephalus with distention of the third ventricle and the anterior aqueduct of Sylvius. This syndrome may also occur with stroke as one of the “top of the basilar” syndromes.122
Lesions in the region of the pretectum and posterior commissure frequently produce characteristic signs. Dorsal midbrain syndrome (also known as Parinaud’s syndrome or mesencephalic syndrome) consists of vertical gaze deficiencies, convergence spasm or paresis, bilateral mid-dilated pupils with light-near dissociation (poor reaction to light and good reaction to near), and convergence-retraction nystagmus. The nystagmus is unique to this syndrome and consists of rapid convergence with retraction of the globes on attempted upgaze. This result is most evident with an optokinetic stimulus moving downward because the fast upward component induces this finding (Video 13-4). Common causes include pinealomas, aneurysms of the vein of Galen, and noncommunicating hydrocephalus with distention of the third ventricle and the anterior aqueduct of Sylvius. This syndrome may also occur with stroke as one of the “top of the basilar” syndromes.122
Ocular Myasthenia Gravis
Ocular myasthenia gravis can mimic a neuropathy of cranial nerves III (except pupil involvement), IV, or VI, alone or in any combination, but it is often manifested as elusive diplopia or ptosis that is not easily categorized.123,124 Ocular symptoms are generally apparent before other systemic signs and symptoms, and in some patients, the disorder is confined to the ocular muscles.124,125 Because this disorder of the neuromuscular junction causes apparent fatigue of muscular function, patients frequently report worsening symptoms (i.e., increasing ptosis or diplopia) as the day progresses and relief after a nap or rest. The orbicularis oculi is often involved and weak. This weakness may be demonstrated by having patients forcefully close their eyes against resistance. The diagnosis is sometimes difficult to make because the results of edrophonium chloride (Tensilon) testing, serum acetylcholine receptor antibody assay, repetitive electromyographic nerve stimulation, or trials of pyridostigmine bromide (Mestinon) may be unremarkable or equivocal. Single-fiber electromyography has the greatest sensitivity and specificity but is technically difficult. Correctly diagnosing this disease early in its course saves the patient a long and often expensive evaluation.
Orbital Graves’ Disease
Because the signs and symptoms of orbital Graves’ disease frequently occur in patients with normal thyroid function, normal results of thyroid function tests do not rule out the disease. Although associated with thyroid malfunction, the orbital disorder seems to run its own course, independent of success in treating the endocrine manifestations. Orbital Graves’ disease is manifested in three ways: restrictive strabismus, proptosis with external eye disease, and compressive optic neuropathy. The disturbed motility pattern can be manifested as diplopia without an evident cause. Because the inferior and medial rectus muscles are generally involved first, the eyes are usually turned in (i.e., esodeviation) and down (i.e., hypodeviation).111 Contrary to popular belief, significant muscle involvement can occur without any external manifestations. Strabismus surgery is contraindicated during the active phases of the disease, but prism correction in the spectacles is occasionally helpful.
Compressive optic neuropathy can also occur without fanfare and should always be included in the differential diagnosis of optic nerve pallor (or disc edema), despite a lack of obvious external signs. Optic nerve compression results from crowding of the orbital apex by enlarged extraocular muscles. Treatment usually consists of a trial of steroids for a defined period, followed by surgical decompression of the orbit or irradiation if the signs and symptoms persist.126
Bajandas FL, Kline LB. Neuro-Ophthalmology Review Manual, 4th ed. Thorofare, NJ: Slack; 1996.
Beck RW, Cleary PA, Anderson MM, et al. A randomized, controlled trial of corticosteroids in the treatment of acute optic neuritis. N Engl J Med. 1992;326:581-588.
, 2003 b Chan JW. Paraneoplastic retinopathies and optic neuropathies. Surv Ophthalmol. 2003;48:12-38.
Friedman DI, Jacobson DM. Idiopathic intracranial hypertension. J Neuroophthalmol. 2004;24:138-145.
Frisén L. Clinical Tests of Vision. New York: Raven Press; 1990.
Frohman LP, Grigorian R, Bielory L. Neuro-ophthalmic manifestations of sarcoidosis: clinical spectrum, evaluation, and management. J Neuroophthalmol. 2001;21:132-137.
Ghanchi FD, Dutton GN. Current concepts in giant cell (temporal) arteritis. Surv Ophthalmol. 1997;42:99-123.
Harrington DO. Visual field character in temporal and occipital lobe lesions. Localizing values of congruity and incongruity in incomplete homonymous hemianopsia. Arch Ophthalmol. 1961;66:778-792.
Kardon RH, Denison CE, Brown CK, et al. Critical evaluation of the cocaine test in the diagnosis of Horner’s syndrome. Arch Ophthalmol. 1990;108:384-387.
Leigh RJ, Zee DS. The Neuorology of Eye Movements. 4th ed. New York, NY: Oxford University Press. 2006.
Martin TJ. Horner’s syndrome, Pseudo-Horner’s syndrome, and simple anisocoria. Curr Neurol Neurosci Rep. 2007;7:397-406.
Martin TJ, Corbett JJ. Neuro-ophthalmology. In: Krachmer JH, editor. Requisites in Ophthalmology Series. St Louis: CV Mosby, 2000.
Miller NR, Newman NJ, editors. Walsh and Hoyt’s Clinical Neuro-ophthalmology, 4th ed, Vols 1-5. Baltimore: Williams & Wilkins, 1998.
Newman NJ. Hereditary optic neuropathies: from the mitochondria to the optic nerve. Am J Ophthalmol. 2005;140:517-523.
Optic Neuritis Study Group. The clinical profile of optic neuritis: experience of the optic neuritis treatment trial. Arch Ophthalmol. 1991;109:1673-1678.
Powell M. Recovery of vision following transsphenoidal surgery for pituitary adenomas. Br J Neurosurg. 1995;9:367-373.
Richards BW, Jones FRJr, Younge BR. Causes and prognosis in 4278 cases of paralysis of the oculomotor, trochlear, and abducens cranial nerves. Am J Ophthalmol. 1992;113:489-496.
Sadun AA, Rismondo V. Evaluation of the swollen disc. In: Schachat A, editor. Current Practice in Ophthalmology. Boston: Mosby–Year Book; 1992:177-186.
Thompson HS, Corbett JJ, Cox TA. How to measure the afferent pupillary defect. Surv Ophthalmol. 1981;26:39-42.
Turbin RE, Thompson CR, Kennerdell JS, et al. A long-term visual outcome comparison in patients with optic nerve sheath meningioma managed with observation, surgery, radiotherapy, or surgery and radiotherapy. Ophthalmology. 2002;109:890-899.
1 Miller NR, Newman NJ, editors. Walsh and Hoyt’s Clinical Neuro-ophthalmology, 4th ed, Vols 1-5. Baltimore: Williams & Wilkins, 1998.
2 Glaser JS. Neuro-ophthalmology, 2nd ed. Philadelphia: JB Lippincott; 1990.
3 Liu GT, Volpe NJ, Galetta SL. Neuro-Ophthalmology, Diagnosis and Management. Philadelphia: WB Saunders; 2001.
4 Burde R, Savino PJ, Trobe JD. Clinical Decisions in Neuro-ophthalmology, 2nd ed. St Louis: Mosby–Year Book; 1992.
5 Lee AG, Brazis PW. Clinical Pathways in Neuro-ophthalmology, An Evidence-Based Approach. New York: Thieme; 1998.
6 Slamovits TL, Burde R. Neuro-ophthalmology. volume eds. Podos SM, Yanoff M, editors. Textbook of Ophthalmology, Vol. 6. London: CV Mosby. 1991.
7 Rosen ES, Thompson HS, Cumming WJK, et al, editors. Neuro-ophthalmology. London: Mosby International Limited, 1998.
8 Brodsky M, Baker R, Hamid L. Pediatric Neuro-Ophthalmology. New York: Springer-Verlag; 1996.
9 Bajandas FL, Kline LB. Neuro-Ophthalmology Review Manual, 4th ed. Thorofare, NJ: Slack; 1996.
10 Martin TJ, Corbett JJ. Neuro-ophthalmology. series ed. Krachmer JH, editor. Requisites in Ophthalmology Series. St Louis: CV Mosby, 2000.
11 Lepore FE. The neuro-ophthalmologic case history: Elucidating the symptoms. In: Duane TD, editor. Duane’s Clinical Ophthalmology, vol 2. Philadelphia: Lippincott Williams & Wilkins; 1998:1-7.
12 Frisén L. Clinical Tests of Vision. New York: Raven Press; 1990. 1-105
13 Anderson DR. Testing the Field of Vision. St. Louis: CV Mosby; 1982.
14 Trobe JD, Acosta PC, Krischer JP, et al. Confrontation visual field techniques in detection of anterior visual pathway lesions. Ann Neurol. 1981;10:28-34.
15 Johnson CA, Keltner JL. Principles and techniques of examination of the visual sensory system. In: Miller NR, Newman NJ, editors. Walsh and Hoyt’s Clinical Neuro-ophthalmology. 5th ed. Baltimore: Williams & Wilkins; 1998:153-236.
16 Keltner JL, Johnson CA. Automated and manual perimetry: a six-year overview. Special emphasis on neuro-ophthalmic problems. Ophthalmology. 1983;91:68-85.
17 Mills RP. Automated perimetry in neuro-ophthalmology. Int Ophthalmol Clin. 1991;31:51-70.
18 Thompson HS, Corbett JJ, Cox TA. How to measure the afferent pupillary defect. Surv Ophthalmol. 1981;26:39-42.
19 Kline LB, editor. Optic Nerve Disorders. Ophthalmology Monographs, vol 10. San Francisco: American Academy of Ophthalmology, 1996.
20 Sadun AA, Rismondo V. Evaluation of the swollen disc. In: Schachat A, editor. Current Practice in Ophthalmology. Boston: Mosby–Year Book; 1992:177-186.
21 Miller NR, Newman NJ. Topical diagnosis of lesions in the visual sensory pathway, 5th ed. Miller NR, Newman NJ, editors. Walsh and Hoyt’s Clinical Neuro-ophthalmology, vol 1. Baltimore: Williams & Wilkins. 1998:237-386.
22 Sadun AA. Distinguishing optic nerve disease from retinal/macular disease. In: Slomowitz TL, Burde R, editors. Neuro-Ophthalmology, Vol 6. London: CV Mosby; 1991:2.1-2.9. In: Podos SM, Yanoff M, eds. Textbook of Ophthalmology vol eds
23 Young BR. The significance of retinal emboli. J Clin Neuroophthalmol. 1989;9:190-194.
24 Ros MA, Magargal LE, Uram M. Branch retinal artery obstruction: a review of 201 eyes. Ann Ophthalmol. 1989;21:103-107.
25 Hollenhorst RS. Significance of bright plaques in the retinal arterioles. JAMA. 1961;178:23-29.
26 Arruga J, Sanders MD. Ophthalmologic findings in 70 patients with evidence of retinal embolism. Ophthalmology. 1982;89:1336-1347.
27 Quinlan PM, Elman MJ, Bhatt AK, et al. The natural course of central retinal vein occlusion. Am J Ophthalmol. 1990;110:118-123.
28 Johnston RL, Brucker AJ, Steinmann W, et al. Risk factors of branch retinal vein occlusion. Ophthalmology. 1983;90:1831-1832.
29 Sawyer RA, Selhorst JB, Zimmerman LE, et al. Blindness caused by photoreceptor degeneration as a remote effect of cancer. Am J Ophthalmol. 1976;81:606-613.
30 Rizzo JF, Gittinger JW. Selective immunohistochemical staining in the paraneoplastic retinopathy syndrome. Ophthalmology. 1992;99:1286-1295.
31 Goldstein SM, Syed NA, Milam AH, et al. Cancer-associated retinopathy. Arch Ophthalmol. 1999;117:1641-1645.
32 Jacobson DM, Thirkill CE, Tipping SJ. A clinical triad to diagnose paraneoplastic retinopathy. Ann Neurol. 1990;28:162-167.
33 Thirkill CE, Roth AM, Keltner JL. Cancer-associated retinopathy. Arch Ophthalmol. 1987;105:372-375.
34 Keltner JL, Thirkill CE, Tyler NK, et al. Management and monitoring of cancer-associated retinopathy. Arch Ophthalmol. 1992;110:48-53.
35 Chan JW. Paraneoplastic retinopathies and optic neuropathies. Surv Ophthalmol. 2003;48:12-38.
36 Corbett JJ, Savino PJ, Thompson HS, et al. Visual loss in pseudotumor cerebri. Arch Neurol. 1982;39:461-474.
37 Wall M. Idiopathic intracranial hypertension. Neurol Clin. 1991;9:73-95.
38 Friedman DI, Jacobson DM. Idiopathic intracranial hypertension. J Neuroophthalmol. 2004;24:138-145.
39 Phillips PH, Repka MX, Lambert SR. Pseudotumor cerebri in children. J AAPOS. 1998;2:33-38.
40 Digre KB, Corbett JJ. Idiopathic intracranial hypertension (pseudotumor cerebri): a reappraisal. Neurologist. 2001;7:2-67.
41 Beck RW, Servais G, Hayreh SS. Anterior ischemic optic neuropathy. IX. Cup-to-disc ratio and its role in pathogenesis. Ophthalmology. 1987;94:1503-1508.
42 Salomon O, Huna-Baron R, Kurtz S, et al. Analysis of prothrombotic and vascular risk factors in patients with nonarteritic anterior ischemic optic neuropathy. Ophthalmology. 1999;106:729-742.
43 Arnold AC. Ischemic optic neuropathies. Ophthalmol Clin North Am. 2001;14:83-98.
44 Arnold AC. Pathogenesis of nonarteritic anterior ischemic optic neuropathy. J Neuroophthalmol. 2003;23:157-163.
45 Jacobson DM, Vierkant RA, Belongia EA. Non-arteritic anterior ischemic optic neuropathy. A case-control study of potential risk factors. Arch Ophthalmol. 1997;115:1403-1407.
46 Guyer DR, Miller NR, Auer CL, et al. The risk of cerebrovascular disease with anterior ischemic optic neuropathy. Arch Ophthalmol. 1985;103:1136-1142.
47 Deramo VA, Sergott RC, Augsburger JJ, et al. Ischemic optic neuropathy as the first manifestation of elevated cholesterol levels in young patients. Ophthalmology. 2003;110:1041-1046.
48 Connolly SE, Gordon KB, Horton JC. Salvage of vision after hypotension-induced ischemic optic neuropathy. Am J Ophthalmol. 1994;117:235-242.
49 The Ischemic Optic Neuropathy Decompression Trial Research Group. Optic nerve decompression surgery for non-arteritic ischemic optic neuropathy is not effective and may be harmful. JAMA. 1995;273:625-632.
50 Ischemic Optic Neuropathy Decompression Trial. Twenty-four-month update. Arch Ophthalmol. 2000;118:793-798.
51 Beck RW, Hayreh SS, Podnajsky P, et al. Aspirin therapy in nonarteritic ischemic optic neuropathy. Am J Ophthalmol. 1997;123:212-217.
52 Keltner JL. Giant-cell arteritis: signs and symptoms. Ophthalmology. 1982;89:1101-1110.
53 Ghanchi FD, Dutton GN. Current concepts in giant cell (temporal) arteritis. Surv Ophthalmol. 1997;42:99-123.
54 Weyand CM, Bartley GB. Giant cell arteritis: new concepts in pathogenesis and implications for management. Am J Ophthamol. 1997;123:392-395.
55 Boyev LR, Miller NR, Green WR. Efficacy of unilateral versus bilateral temporal artery biopsies for the diagnosis of giant cell arteritis. Am J Ophthalmol. 1999;128:211-215.
56 Beck RW, Gal RL, Bhatti MT, et al. Visual function more than 10 years after optic neuritis: experience of the optic neuritis treatment trial. for the Optic Neuritis Study Group. Am J Ophthalmol. 2004;137;:77-83. Erratum in: Am J Ophthalmol. 2004;137(4):following 793. Am J Ophthalmol. 2004;138(2):following 321
57 Beck RW, Cleary PA, Trobe JD, et al. The effect of corticosteroids for acute optic neuritis on the subsequent development of multiple sclerosis. N Engl J Med. 1993;329:1764-1769.
58 Optic Neuritis Study Group. The 5-year risk of MS after optic neuritis. Experience of the Optic Neuritis Treatment Trial. Neurology. 1997;49:1404-1413.
59 Rizzo JF, Lessell S. Risk of developing multiple sclerosis after uncomplicated optic neuritis: a long-term prospective study. Neurology. 1988;38:185-190.
60 Optic Neuritis Study Group. The clinical profile of optic neuritis: experience of the optic neuritis treatment trial. Arch Ophthalmol. 1991;109:1673-1678.
61 Beck RW, Cleary PA, Anderson MM, et al. A randomized, controlled trial of corticosteroids in the treatment of acute optic neuritis. N Engl J Med. 1992;326:581-588.
62 Ghezzi A, Martinelli V, Torri V, et al. Long-term follow-up of isolated optic neuritis: the risk of developing multiple sclerosis, its outcome, and the prognostic role of paraclinical tests. J Neurol. 1999;246:770-775.
63 Jacobs LD, Beck RW, Simon JH, et al. Intramuscular interferon beta-1a therapy initiated during a first demyelinating event in multiple sclerosis. CHAMPS Study Group. N Engl J Med. 2000;343:898-904.
64 Frisén L, Hoyt WF, Tengroth BM. Optociliary veins, disc pallor and visual loss: a triad of signs indicating sphenoorbital meningioma. Acta Ophthalmol. 1973;51:241-249.
65 Feldon SE. Tumors of the anterior visual pathways. In: Jakobiec FA, Albert DM, editors. Principles and Practice of Ophthalmology. Philadelphia: WB Saunders; 1994:2578-2592.
66 Hoyt WF, Baghdassarian SA. Optic glioma of childhood. Natural history and rationale for conservative management. Br J Ophthalmol. 1969;53:793-798.
67 Dutton JJ. Gliomas of the anterior visual pathway. Surv Ophthalmol. 1994;38:427-452.
68 Hoyt WF, Meshel LG, Lessell S, et al. Malignant optic glioma of adulthood. Brain. 1973;96:121-132.
69 Dutton JJ. Optic nerve sheath meningiomas. Surv Ophthalmol. 1994;37:167-183.
70 Andrews DW, Faroozan R, Yang BP, et al. Fractionated stereotactic radiotherapy for the treatment of optic nerve sheath meningiomas: preliminary observations of 33 optic nerves in 30 patients with historical comparison to observation with or without prior surgery. Neurosurgery. 2002;51:890-902.
71 Becker G, Jeremic B, Pitz S, et al. Stereotactic fractionated radiotherapy in patients with optic nerve sheath meningioma. Int J Radiat Oncol Biol Phys. 2002;54:1422-1429.
72 Moyer PD, Golnik KC, Breneman J. Treatment of optic nerve sheath meningioma with three-dimensional conformal radiation. Am J Ophthalmol. 2000;129:694-696.
73 Turbin RE, Thompson CR, Kennerdell JS, et al. A long-term visual outcome comparison in patients with optic nerve sheath meningioma managed with observation, surgery, radiotherapy, or surgery and radiotherapy. Ophthalmology. 2002;109:890-899.
74 Dutton JJ. Gliomas of the anterior visual pathways. Surv Ophthalmol. 1994;38:427-452.
75 Hoyt WF, Baghdassarian SA. Optic glioma of childhood. Natural history and rationale for conservative management. Br J Ophthalmol. 1969;53:793-798.
76 Parsa CF, Hoyt CS, Lesser RL, et al. Spontaneous regression of optic gliomas: thirteen cases documented by serial neuroimaging. Arch Ophthalmol. 2001;119:516-529.
77 Feldon SE. Tumors of the anterior visual pathways. In: Jakobiec FA, Albert DM, editors. Principles and Practice of Ophthalmology. Philadelphia: WB Saunders, 1994.
78 Hoyt WF, Meshel LG, Lessell S, et al. Malignant optic glioma of adulthood. Brain. 1973;96:121-132.
79 Haik BG, Louis LS, Bierly J, et al. Magnetic resonance imaging in the evaluation of optic nerve gliomas. Ophthalmology. 1987;94:709-717.
80 Kansu T, Orr LS, Savino P, et al. Optic neuropathy as initial manifestation of lymphoreticular diseases. In: Smith JL, editor. Neuro-Ophthalmology Focus. New York: Masson, 1980.
81 McFadzean R, Brosnahan D, Doyle D, et al. A diagnostic quartet in leptomeningeal infiltration of the optic nerve sheath. J Neuroophthalmol. 1994;14:175-182.
82 Strominger MB, Schatz NJ, Glaser JS. Lymphomatous optic neuropathy. Am J Ophthalmol. 1993;116:774-776.
83 Scott TF. Neurosarcoidosis: progress and clinical aspects. Neurology. 1993;43:8-12.
84 Graham EM, Ellis CJ, Sanders MD, et al. Optic neuropathy in sarcoidosis. J Neurol Neurosurg Psychiatry. 1986;49:756-763.
85 Karma A, Huhti E, Poukkula A. Course and outcome of ocular sarcoidosis. Am J Ophthalmol. 1988;106:467-472.
86 Beardsley TL, Brown SVL, Sydnor CF, et al. Eleven cases of sarcoidosis of the optic nerve. Am J Ophthalmol. 1984;97:62-77.
87 Frohman LP, Grigorian R, Bielory L. Neuro-ophthalmic manifestations of sarcoidosis: clinical spectrum, evaluation, and management. J Neuroophthalmol. 2001;21:132-137.
88 Rosenberg MA, Savino PJ, Glaser JS. A clinical analysis of pseudopapilledema. I. Population, laterality, acuity, refractive error, ophthalmoscopic characteristics, and coincident disease. Arch Ophthalmol. 1979;97:71-75.
89 Arnold AC. Optic disc drusen. Ophthalmol Clin North Am. 1991;4:505-517.
90 Newman NJ. Hereditary optic neuropathies: from the mitochondria to the optic nerve. Am J Ophthalmol. 2005;140:517-523.
91 Newman NJ. Leber’s Hereditary optic neuropathy: new genetic considerations. Arch Neurol. 1993;50:540-548.
92 Anderson RL, Panje WR, Ross CE. Optic nerve blindness following blunt forehead trauma. Ophthalmology. 1982;89:445-455.
93 Lessell S. Indirect optic nerve trauma. Arch Ophthalmol. 1989;107:382-386.
94 Spoor TC, Hartel WC, Lensink DB, et al. Treatment of traumatic optic neuropathy with corticosteroids. Am J Ophthalmol. 1990;110:665-669.
95 Joseph MP, Lesssel S, Rizzo J, et al. Extracranial optic nerve decompression for traumatic optic neuropathy. Arch Ophthalmol. 1990;108:1091-1093.
96 Levin LA, Beck RW, Joseph MP, et al. The treatment of traumatic optic neuropathy. The International Optic Nerve Trauma Study. Ophthalmology. 1999;106:1268-1277.
97 Steinsapir KD, Goldberg RA. Traumatic optic neuropathy. Surv Ophthalmol. 1994;38:487-518.
98 Glaser JS. Topical diagnosis: The optic chiasm. In: Duane TD, editor. Duane’s Clinical Ophthalmology, Vol 2. Philadelphia: Lippincott Williams & Wilkins; 1998.
99 Powell M. Recovery of vision following transsphenoidal surgery for pituitary adenomas. Br J Neurosurg. 1995;9:367-373.
100 Newman NJ. Chiasm, parachiasmal syndromes, retrochiasm, and disorders of higher visual functions. In: Slamovits TL, Burde R, editors. Neuro-ophthalmology, vol 6. London: CV Mosby; 1991:4.1-4.24. In: Podos SM, Yanoff M, series eds. Textbook of Ophthalmology
101 Savino PJ, Glaser JS, Schatz NJ. Traumatic chiasmal syndrome. Neurology. 1980;30:963-970.
102 Savino PJ, Paris M, Schatz NJ, et al. Optic tract syndrome. A review of 21 patients. Arch Ophthalmol. 1978;96:656-663.
103 Newman SA, Miller NR. Optic tract syndrome: neuro-ophthalmologic considerations. Arch Ophthalmol. 1983;101:1241-1250.
104 Bell RA, Thompson HS. Relative afferent pupillary defect in optic tract hemianopias. Am J Ophthalmol. 1978;85:538-540.
105 Unsold R, Hoyt WF. Band atrophy of the optic nerve. Arch Ophthalmol. 1980;98:1637-1638.
106 Hughes TS, Abou-Khahl B, Lavin PJM, et al. Visual field defects after temporal lobe resection. A prospective quantitative analysis. Neurology. 1999;53:167-172.
107 Harrington DO. Visual field character in temporal and occipital lobe lesions. Localizing values of congruity and incongruity in incomplete homonymous hemianopsia. Arch Ophthalmol. 1961;66:778-792.
108 Sargent JC. Nuclear and infranuclear ocular motility disorders. In: Miller NR, Newman NJ, editors. Walsh and Hoyt’s Clinical Neuro-ophthalmology. 6th ed. Philadelphia: Lippincott Williams & Wilkins; 2005:969-1040.
109 Leigh RJ, Zee DS. The Neurology of Eye Movements, 4th ed. New York: Oxford University Press; 2006. 385-474
110 Richards BW, Jones FRJr, Younge BR. Causes and prognosis in 4278 cases of paralysis of the oculomotor, trochlear, and abducens cranial nerves. Am J Ophthalmol. 1992;113:489-496.
111 Brazis PW, Lee AG. Binocular vertical diplopia. Mayo Clin Proc. 1998;73:55-66.
112 Brazis PW. Palsies of the trochlear nerve: Diagnosis and localization—recent concepts. Mayo Clin Proc. 1993;68:501-509.
113 Newman SA. Disorders of ocular motility. In: Slamovits TL, Burde R, editors. Neuro-ophthalmology, vol 6. London: CV Mosby; 1991:7.1-7.28. In: Podos SM, Yanoff M, series eds: Textbook of Ophthalmology
114 Trobe JD. Isolated third nerve palsies. Semin Neurol. 1986;6:135-141.
115 Czarnecki JSC, Thompson HS. The iris sphincter in aberrant regeneration of the third nerve. Arch Ophthalmol. 1978;96:1606-1610.
116 Sibony PA, Lessell S, Gittinger JWJr. Acquired oculomotor synkinesis. Surv Ophthalmol. 1984;28:382-390.
117 Miller NR, Newman NJ. The autonomic nervous system: Pupillary function, accommodation, and lacrimation, 5th ed. Miller NR, Newman NJ, editors. Walsh and Hoyt’s Clinical Neuro-ophthalmology, vol 1. Baltimore: Williams & Wilkins. 1998:786-853.
118 Grimson BS, Thompson HS. Horner’s syndrome: Overall view of 120 cases. In: Thompson HS, Daroff RB, Frisén L, et al, editors. Topics in Neuro-ophthalmology. Baltimore: Williams & Wilkins; 1979:151-156.
119 Thompson HS. Adie’s syndrome: some new observations. Trans Am Ophthalmol Soc. 1977;75:587-626.
120 Kardon RH, Denison CE, Brown CK, et al. Critical evaluation of the cocaine test in the diagnosis of Horner’s syndrome. Arch Ophthalmol. 1990;108:384-387.
121 Martin TJ. Horner’s syndrome, pseudo-Horner’s syndrome, and simple anisocoria. Curr Neurol Neurosci Rep. 2007;7:397-406.
122 Caplan LR. “Top of the basilar” syndrome. Neurology. 1980;30:72-79.
123 Elrod RD, Wienberg DA. Ocular myasthenia gravis. Ophthalmol Clin North Am. 2004;17:275-309.
124 Weinberg DA, Lesser RL, Vollmer TL. Ocular myasthenia: a protean disorder. Surv Ophthalmol. 1994;39:169-210.
125 Katz B. Myasthenia gravis. In: Focal Points: Clinical Modules for Ophthalmologists. San Francisco: American Academy of Ophthalmology; 1989.
126 Kazim M, Trokel S, Moore S. Treatment of acute Graves’ orbitopathy. Ophthalmology. 1991;98:1443-1448.






