[level-membership-for-basic-science-category]
Neurons and glial cells
Nerve cells
The basic structure of the neuron has been outlined in Chapter 1. It was also shown how nerve cells can be classified based upon the type of information that they transmit (afferent, efferent, interneuron) or by the number of processes that they have (unipolar, bipolar, multipolar). Neuronal excitability, impulse conduction and synaptic transmission are discussed in Chapters 6 and 7.
Cortical neurons
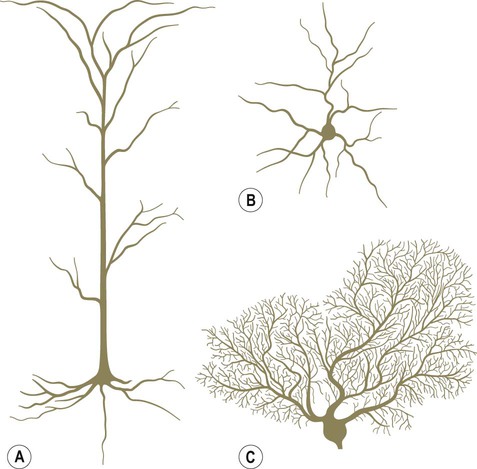
Pyramidal and granule cells (Fig. 5.1A & B)
Pyramidal cells have large, pyramid-shaped cell bodies that range from 20–120 µm in diameter. They are excitatory neurons that have numerous apical and basal dendrites and a single axon that projects out of the cortex. Pyramidal cells are particularly prominent in motor and premotor areas. Granule cells (or stellate cells) are star-shaped multipolar neurons that have short axons and make local synaptic contacts, tending to be enriched in sensory cortices. They are much smaller than pyramidal cells, with a typical diameter of less than 20 µm, and may be excitatory or inhibitory. The cerebellar cortex also contains two main types of nerve cell: granule cells (similar to those in the cerebral cortex) and Purkinje cells (large efferent neurons, equivalent to cortical pyramidal cells; see Fig. 5.1C).
Cortical lamination
More than 90% of the cerebral cortex has a characteristic six-layered structure that appeared with the evolution of the mammalian brain (Fig. 5.2). For this reason it is referred to as neocortex (Greek: neos, new). Although the same six layers can be identified in all neocortical regions at some stage of development, they are not always present in the mature brain. For instance, the motor and premotor areas of the frontal neocortex are referred to as agranular cortex since they have lost their internal granule cell layer.
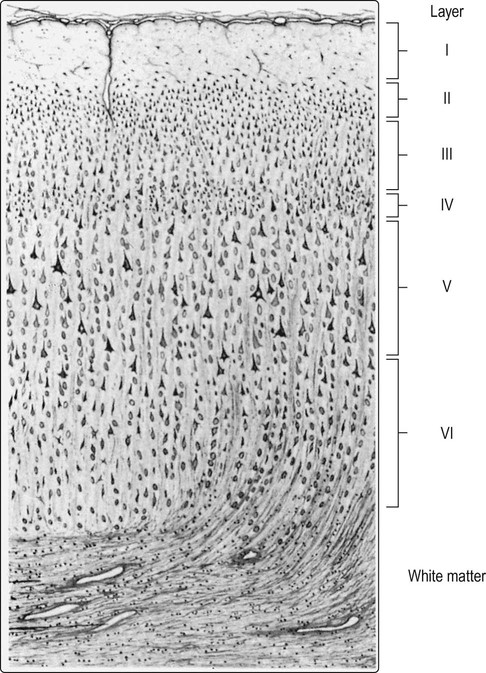
At least 90% of the cortex has six identifiable layers and is classified as neocortex. Small (granular) cells predominate in laminae that receive afferent projections, whereas large (pyramidal) cells are more numerous in layers that give rise to efferent projections. [For instance: lamina IV, the internal granule cell layer, receives projections from the thalamus; lamina V, the internal pyramidal cell layer, is the origin of projections to the brain stem and spinal cord.] From Crossman: Neuroanatomy ICT 4e (Churchill Livingstone 2010) with permission.
Different types of cortex
The cerebral cortex can be divided into more than fifty Brodmann areas based on subtle differences in the cortical structure (referred to as cytoarchitectonics) but there are three major cortical types (Fig. 5.3):
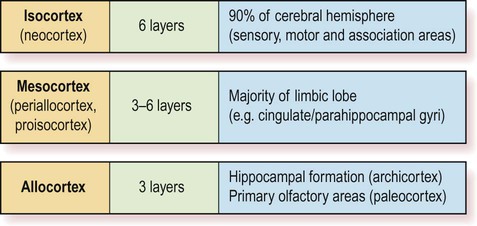
The two main types are isocortex (= neocortex) and allocortex. The mesocortex occupies a ‘transitional zone’ that lies between the six-layered isocortex and the three-layered allocortex. It has a variable number of layers and can be further subdivided into proisocortex and periallocortex, based on its similarity to the two main types.
 The isocortex has a uniform six-layered structure (Greek: isos, equal) and accounts for more than 90% of the cortical surface area. This term is synonymous with neocortex.
The isocortex has a uniform six-layered structure (Greek: isos, equal) and accounts for more than 90% of the cortical surface area. This term is synonymous with neocortex.

 The mesocortex is a transitional zone between the allocortex and isocortex and has 3–6 layers. Most of the limbic lobe (cingulate and parahippocampal gyri; see Ch. 3) is composed of mesocortex.
The mesocortex is a transitional zone between the allocortex and isocortex and has 3–6 layers. Most of the limbic lobe (cingulate and parahippocampal gyri; see Ch. 3) is composed of mesocortex.
The majority of the ‘non-neocortical’ regions belong to the limbic lobe and are primarily concerned with emotion, memory and olfaction (the sense of smell). The term paralimbic cortex is used to describe non-neocortical regions outside of the limbic lobe proper, including the posterior orbital cortex, anterior insula and temporal pole.






Features of the neuron
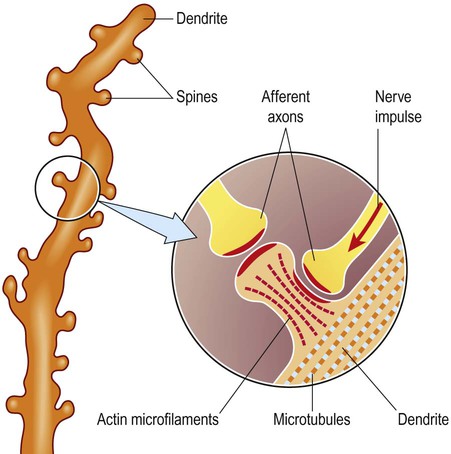
Dendritic spines
The dendrites of many neurons are studded with thousands of tiny, mushroom-shaped dendritic spines (Fig. 5.4). These include the medium spiny neurons that make up 95% of cells in the basal ganglia. In the cerebral cortex, all pyramidal cells have dendritic spines, whereas stellate cells may be spiny or smooth. Each dendritic spine is the site of an incoming excitatory synapse and a typical cortical pyramidal neuron has more than 10,000 spines. They are dynamic structures that can form, change shape or disappear altogether and are thought to be important in synaptic plasticity (Greek: plastikos, able to be moulded) and learning. Long-term memories may be mediated by the growth of new spines or the strengthening and enlargement of existing ones.
Subcellular organelles
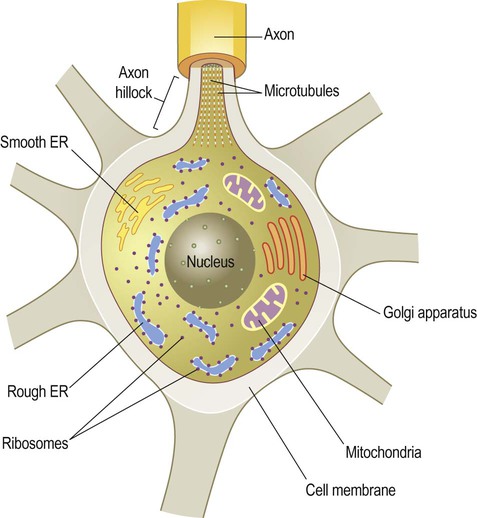
The neuronal cell body or soma (Greek: soma, body) contains the same organelles found in other cell types (Fig. 5.5) but the machinery for protein synthesis and gene transcription is particularly prominent (Fig. 5.6A). The perinuclear cytoplasm or perikaryon (Greek: peri, around; cyton, kernel) contains a well-developed network of rough endoplasmic reticulum, often arranged in clumps called Nissl bodies. The Golgi apparatus is also prominent and is the site of post-translational modification and sorting of proteins including ion channels, neurotransmitter receptors and membrane ion pumps.
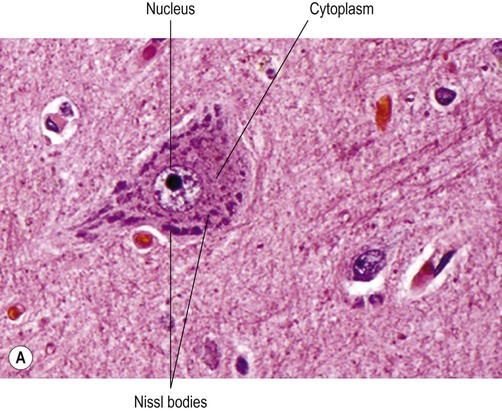
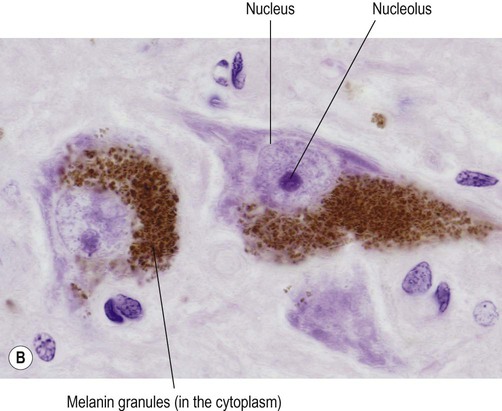
(A) Micrograph showing a pyramidal motor neuron in the cerebral cortex of the frontal lobe. It has a large nucleus with a well-defined nuclear envelope and prominent central nucleolus. The cytoplasm (stained pink) contains coarse clumps of rough endoplasmic reticulum (stained blue); these structures are referred to as Nissl bodies. [Routine haematoxylin and eosin (H&E)-stained sections.] From Prayson, Richard: Neuropathology 1e (Churchill Livingstone 2005) with permission; (B) Micrograph of two pigmented dopaminergic neurons in the substantia nigra of the midbrain (Nissl stain). The pigment is neuromelanin and is produced as a by-product of dopamine synthesis. From Standring: Gray’s Anatomy 40e (Churchill Livingstone 2008) with permission.
Lipofuscin and neuromelanin
Neurons have a high rate of membrane turnover and gradually accumulate non-digestible membrane components within lysosomal residual bodies. These contain golden-brown material called lipofuscin (pronounced: lipo-FUSKIN). This so-called ‘age pigment’ accumulates over time as a by-product of membrane turnover and is also abundant in the heart and liver. Catecholamine-synthesizing neurons contain the dark brown pigment neuromelanin (Fig. 5.6B); these include the substantia nigra of the midbrain and the locus coeruleus of the pons (see Ch. 1).
Vesicles
Neurons contain various types of membrane-bound vesicle. Neurotransmitters and neuropeptides are stored in the axon terminal (prior to release) within synaptic vesicles (see Ch. 7). Coated vesicles are derived from internalization of membrane constituents and macromolecules that have been taken up from the extracellular fluid by receptor-mediated endocytosis.
The neuronal cytoskeleton
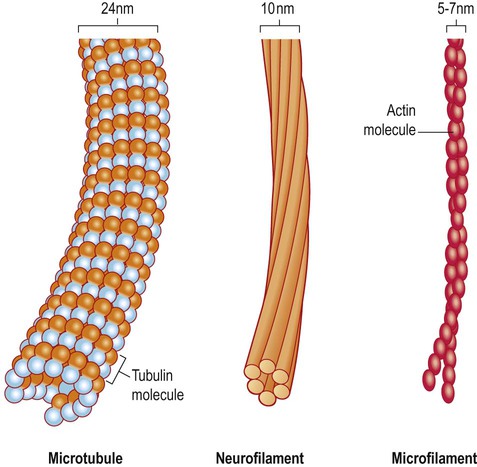
All cells have a cytoskeleton composed of an internal framework of fibrillar proteins, that gives each cell its characteristic shape. This molecular scaffold is particularly important in process-bearing cells such as neurons and glia, which have a complex structure. The cytoskeleton is also involved in the transport of materials between intracellular compartments (see below). The main components include microtubules, neurofilaments and microfilaments (Fig. 5.7).
Microtubules
Microtubules are tubular polymeric proteins that are composed of repeating subunits of alpha and beta tubulin. They are 24 nm in diameter and up to 1 mm in length. Some microtubules are stable, whereas others are dynamic and can grow or shrink by the addition or removal of tubulin subunits. Microtubules are polarized. The part that is closest to the neuronal cell body is arbitrarily termed the minus (–) end and that which is nearer to the axon terminal is designated the plus (+) end. Microtubules are stabilized by binding to a range of microtubule associated proteins (MAPs). These include the phosphoprotein tau which accumulates in various degenerative conditions including Alzheimer’s disease (Ch. 12).
Axonal transport


Fast axonal transport is mediated by a group of molecular motor proteins called kinesins (for anterograde transport) and dyneins (for retrograde transport). These proteins are ATPases that carry vesicle-bound cargo along microtubule tracks that run the length of the axon. Retrograde axonal transport is involved in some central nervous system viral infections (Clinical Box 5.1).
 Clinical Box 5.1:
Clinical Box 5.1:




Astrocytes
Astrocytes are process-bearing cells with a stellate morphology (Greek: astron, star) and are the main support cells of the central nervous system (CNS). Astrocytic processes have specialized structures called end-feet that make contact with neurons and capillaries (Fig. 5.8A).
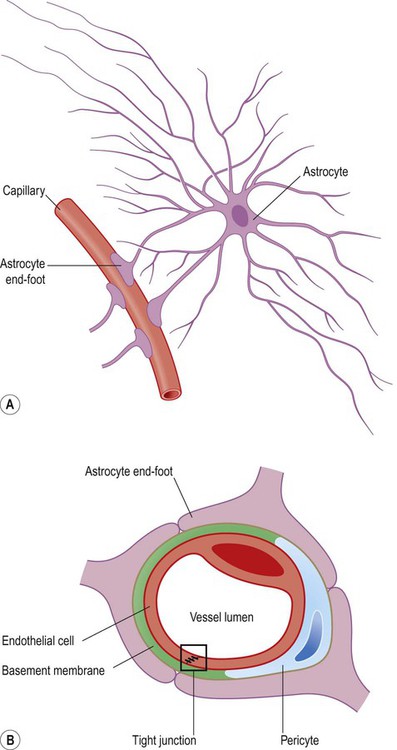
(A) Illustration of an astrocyte and its relationship to a cerebral capillary. Note the astrocytic end-feet making contact with the blood vessel; (B) Astrocytic end-feet are responsible for initiating the structural features of the blood–brain barrier, including the presence of tight junctions between capillary endothelial cells. In this figure a capillary is seen in cross-section, surrounded by three astrocytic end-feet.
Astrocyte functions
Astrocytes contribute to homeostasis, helping to maintain a constant internal environment for neurons (e.g. by removing excess potassium ions and glutamate from the extracellular fluid). They are also involved in the response to injury, releasing cytokines and growth factors and multiplying to form a glial scar (see Ch. 8). Some other key roles are discussed in more detail below.
The blood–brain barrier (Fig. 5.8B)
The existence of the blood–brain barrier requires that the brain has a number of carrier-mediated transport mechanisms for the uptake of essential nutrients. Glucose is imported by a specific glucose transporter (GLUT-1) which is present on the surface of endothelial cells. Large neutral amino acids such as phenylalanine also have a specific transporter protein, and drugs used in the treatment of Parkinson’s disease gain access to the brain via the same transporter (Ch. 13).
Energy metabolism
Following activity at an excitatory (glutamatergic) synapse, perisynaptic astrocytes take up some of the released glutamate via specific transporters. These membrane pumps import sodium ions (Na+) together with glutamate, thereby increasing the sodium ion concentration in the perisynaptic astrocytes. This in turn stimulates the sodium–potassium exchange pump (Na+/K+-ATPase) which promotes glycolysis. More lactate is thus produced, which can be made available to the metabolically active neurons. This is an example of metabolic coupling between neurons and astrocytes, which is relevant in certain neurological conditions (Clinical Box 5.2).
 Clinical Box 5.2:
Clinical Box 5.2: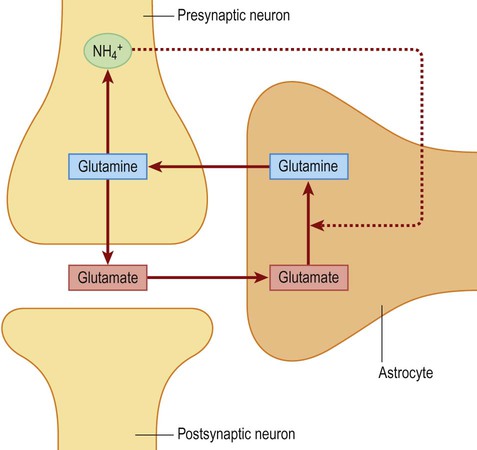




Oligodendrocytes and Schwann cells
Oligodendrocytes are small, rounded cells with relatively few cytoplasmic processes (Greek: oligo, few). They are numerous in the CNS white matter and are responsible for investing central axons with a lipid-rich myelin sheath. Each oligodendrocyte makes contact with up to a dozen neighbouring axons and provides a single segment of myelin to each (Fig. 5.10). In the peripheral nervous system, Schwann cells are responsible for myelination, but each cell makes contact with only one axon and provides a single segment of myelin. Both types of cell secrete growth factors and provide trophic support to the axons that they invest.
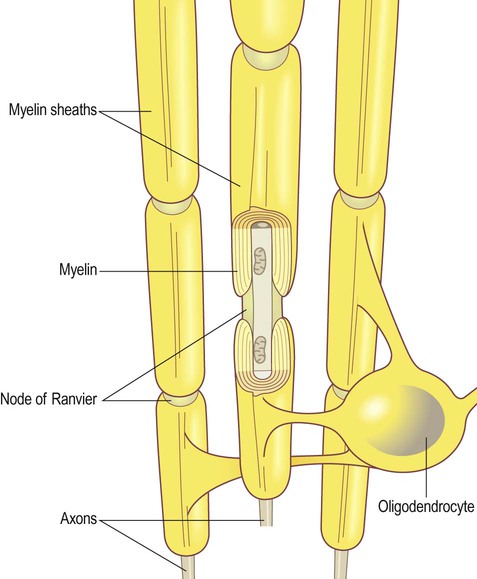
The gap between myelinated segments is the node of Ranvier.
Myelination
Myelin consists of multiple concentric layers of glial cell membrane (Figs 5.11A and 5.12). A small amount of cytoplasm is initially present between the layers but this is gradually squeezed out as the myelin is compacted and the lipid bilayers fuse. Axons are myelinated in 1 mm segments interrupted by microscopic gaps called nodes of Ranvier, at which point the axon is in direct contact with the extracellular fluid. The myelinated segments are called internodes (Latin: inter-, between). Some small-diameter axons do not have a myelin sheath, but are contained within an infolding of Schwann cell membrane, along with several other non-myelinated axons (see Fig. 5.11B).
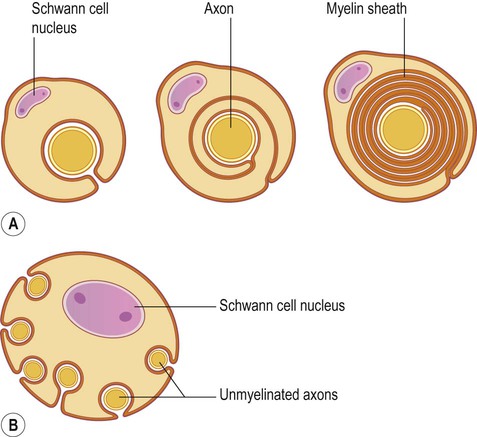
(A) In the peripheral nervous system, Schwann cells myelinate axons by investing them with up to 300 concentric layers of plasma membrane. The intervening cytoplasm between the layers is gradually ‘squeezed out’; (B) Unmyelinated fibres do not have a myelin sheath, but are associated with and enveloped by Schwann cells, which provide trophic support.
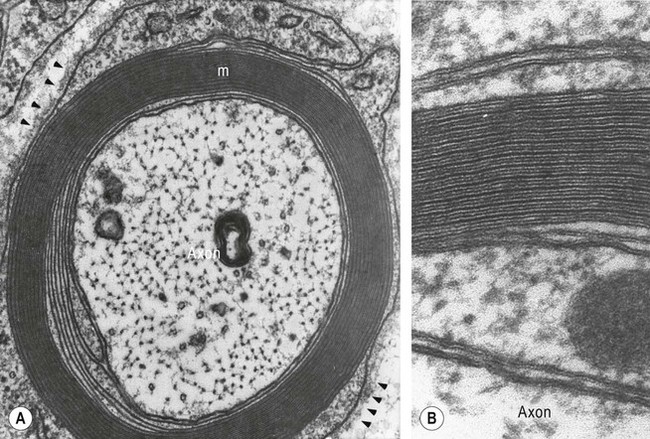
(A) An axon is shown in transverse section, together with its myelin sheath [labelled “m”] composed of multiple individual layers of cell membrane [arrowheads]; (B) Examination of the myelin sheath at higher magnification shows dark (major dense) lines between paler (intraperiod) lines. The dark lines result from fusion of the inner layers of plasma membrane after the intervening cytoplasm has been squeezed out. The pale lines represent apposition of the outer faces of the plasma membrane, which are less closely packed due to the presence of glycoproteins which project from the external surface of the plasma membrane. From SS Scherer (1997) Molecular genetics of demyelination: New wrinkles on an old membrane. Neuron 18: 13–16 with permission.
Composition of myelin
CNS myelin is rich in myelin basic protein (MBP) and proteolipid protein (PLP). The major protein component of peripheral myelin (around 80%) is myelin protein zero (P0). This is a member of the immunoglobulin superfamily and is essential for compaction of adjacent myelin lamellae which enables axons to be tightly wrapped. Another component is peripheral myelin protein 22 (PMP-22) which has been implicated in some heritable forms of peripheral neuropathy (Clinical Box 5.3).
 Clinical Box 5.3:
Clinical Box 5.3:Saltatory conduction
Myelination significantly increases axonal conduction velocity. This is because the nerve impulse ‘jumps’ from node to node (Latin: saltare, to leap) rather than spreading continuously along the axonal membrane (see Ch. 6). Focal disruption of the myelin sheath can lead to failure of impulse conduction in the denuded axon segments (termed conduction block) in part because of a paucity of voltage-gated sodium ion channels between the nodes of Ranvier. Conduction block is a feature of demyelinating conditions such as multiple sclerosis (Ch. 14).




Other glial cells





Many of these cells are related to astrocytes and have similar roles. Olfactory ensheathing cells (OECs) are of particular interest because they permit new axons to grow into the mature CNS, which only normally occurs in the olfactory bulb. These cells therefore provide some hope of encouraging axonal regrowth in the CNS after brain or spinal cord injury.
Microglia
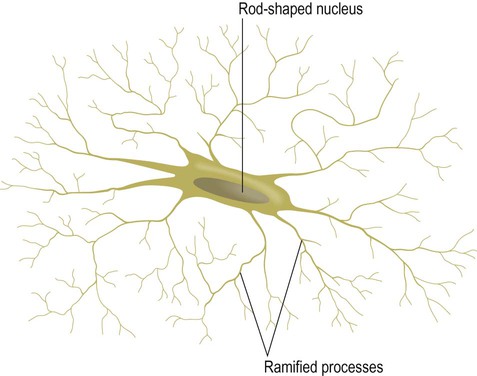
Microglia are relatively small cells (Greek: micro, small) with rod-shaped nuclei (Fig. 5.13) and are the resident phagocytes of the CNS. At rest they exist in a quiescent state and are described as ramified (entwined within the feltwork of axons and dendrites). In response to tissue damage and inflammation they transform into activated microglia which migrate to the site of injury and internalize particulate materials and microorganisms. Microglia are discussed further in Chapter 8 in the context of brain inflammation and gliosis.



Glial tumours
Intracerebral tumours can be classified as primary (arising within the brain) or secondary (metastatic tumours that have spread from another site). Secondary tumours are more common and often originate from the lung, breast or bowel. Primary CNS tumours are further classified as extrinsic or intrinsic (Fig. 5.14). Extrinsic tumours derive from the coverings of the CNS. They tend to be benign and slow-growing and include meningiomas (derived from the meninges) and peripheral nerve sheath tumours. Intrinsic tumours of the brain and spinal cord are of neuroepithelial lineage and usually show evidence of glial differentiation; for this reason they are referred to as glial tumours or gliomas.
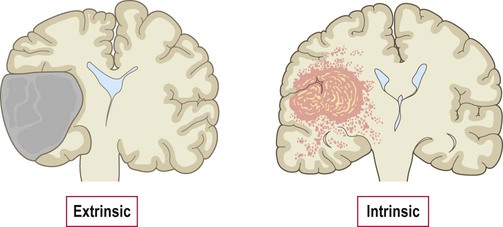
Extrinsic tumours arise from the coverings of the CNS and are usually separate from the underlying brain or spinal cord. Intrinsic tumours arise within the CNS itself and tend to be diffusely infiltrative.
Glial tumours are thought to arise from stem cells (which normally give rise to new neurons or glia) that have accumulated mutations enabling them to proliferate in an uncontrolled manner and to diffusely infiltrate brain tissue. The typical MRI appearances of a malignant glial tumour are shown in Fig. 5.15.
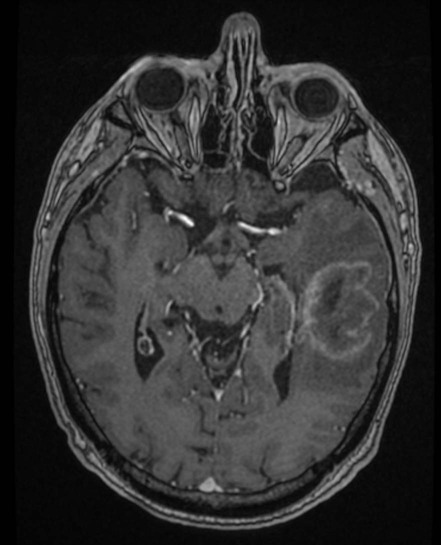
This is an axial, T1-weighted MRI scan showing an irregular, poorly circumscribed intrinsic brain lesion diffusely infiltrating the temporal lobe. The patient has been injected with the intravenous contrast agent gadolinium and the tumour shows a peripheral rim of enhancement (it is described as ‘ring enhancing’). The darker area surrounding the mass corresponds to peritumoural oedema (swelling). These appearances are typical of a glioblastoma, the most common malignant glial tumour. Courtesy of Dr Andrew MacKinnon.
General features of gliomas
Glial tumours are relatively uncommon, with a lifetime risk of less than 1 in 200, but tend to be highly malignant and carry a poor prognosis. The presenting features are discussed in Clinical Box 5.4.
 Clinical Box 5.4: Clinical features of brain tumours
Clinical Box 5.4: Clinical features of brain tumours
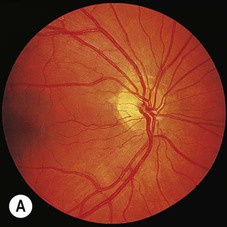
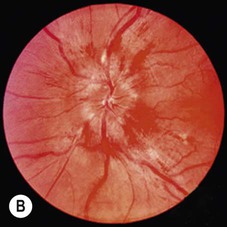
Brain tumours act as space-occupying lesions within the cranial cavity and cause raised intracranial pressure (Ch. 9). This typically presents with headaches that tend to be worse in the morning and are exacerbated by coughing, stooping or straining. There may also be nausea, vomiting or transient visual obscurations. Some patients present with seizures or focal neurological deficits. There is often papilloedema, defined as bilateral swelling of the optic discs due to raised intracranial pressure (Fig. 5.16). Drowsiness is an ominous sign that occurs in the later stages and may precede rapid neurological deterioration.
Classification and grading
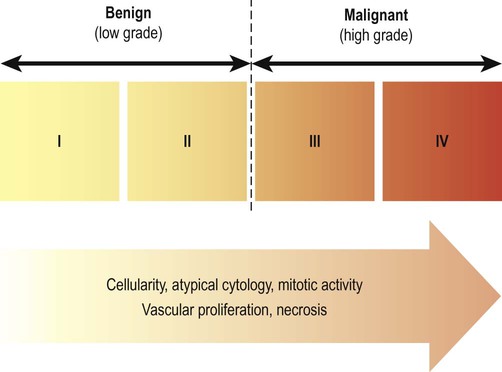
Tumours are also given a histological grade (from I to IV) that attempts to quantify their malignant potential (degree of biological aggressiveness) (Fig. 5.17). Grading is based on tumour architecture (pattern of growth), atypical cytology (cells and nuclei with abnormal structural features) and proliferation rate (speed of growth). Grade I and II tumours are termed benign (or low grade) whereas grade III and IV tumours are designated malignant (or high grade). In general, the prognosis is less favourable as the histological grade increases. The most common (and also the most aggressive) form of glial tumour is glioblastoma (grade IV) in which the median survival is less than 12 months.
Molecular genetics
Another useful molecular signature is mutation in one of the two isoforms of the Kreb’s cycle enzyme isocitrate dehydrogenase (IDH1/IDH2). This is present in 80% of low-grade astrocytic and oligodendroglial tumours and predicts a more favourable outcome. In glioblastoma, the presence of a particular molecular genetic change predicts response to chemotherapy (Clinical Box 5.5).
 Clinical Box 5.5:
Clinical Box 5.5:Management of gliomas


Complete removal of glial tumours is usually not possible (even with extensive resection) due to their highly infiltrative nature. Surgery may also be limited by involvement of eloquent (language or motor) areas. The role of radiotherapy and chemotherapy is relatively limited in primary brain tumours, but may prolong survival by a few months in high-grade gliomas.






[/level-membership-for-basic-science-category][not-level-membership-for-basic-science-category]
Neurons and glial cells
Nerve cells
The basic structure of the neuron has been outlined in Chapter 1. It was also shown how nerve cells can be classified based upon the type of information that they transmit (afferent, efferent, interneuron) or by the number of processes that they have (unipolar, bipolar, multipolar). Neuronal excitability, impulse conduction and synaptic transmission are discussed in Chapters 6 and 7.
Cortical neurons
Pyramidal and granule cells (Fig. 5.1A & B)
Pyramidal cells have large, pyramid-shaped cell bodies that range from 20–120 µm in diameter. They are excitatory neurons that have numerous apical and basal dendrites and a single axon that projects out of the cortex. Pyramidal cells are particularly prominent in motor and premotor areas. Granule cells (or stellate cells) are star-shaped multipolar neurons that have short axons and make local synaptic contacts, tending to be enriched in sensory cortices. They are much smaller than pyramidal cells, with a typical diameter of less than 20 µm, and may be excitatory or inhibitory. The cerebellar cortex also contains two main types of nerve cell: granule cells (similar to those in the cerebral cortex) and Purkinje cells (large efferent neurons, equivalent to cortical pyramidal cells; see Fig. 5.1C).
Cortical lamination
More than 90% of the cerebral cortex has a characteristic six-layered structure that appeared with the evolution of the mammalian brain (Fig. 5.2). For this reason it is referred to as neocortex (Greek: neos, new). Although the same six layers can be identified in all neocortical regions at some stage of development, they are not always present in the mature brain. For instance, the motor and premotor areas of the frontal neocortex are referred to as agranular cortex since they have lost their internal granule cell layer.
At least 90% of the cortex has six identifiable layers and is classified as neocortex. Small (granular) cells predominate in laminae that receive afferent projections, whereas large (pyramidal) cells are more numerous in layers that give rise to efferent projections. [For instance: lamina IV, the internal granule cell layer, receives projections from the thalamus; lamina V, the internal pyramidal cell layer, is the origin of projections to the brain stem and spinal cord.] From Crossman: Neuroanatomy ICT 4e (Churchill Livingstone 2010) with permission.
Different types of cortex
The cerebral cortex can be divided into more than fifty Brodmann areas based on subtle differences in the cortical structure (referred to as cytoarchitectonics) but there are three major cortical types (Fig. 5.3):
The two main types are isocortex (= neocortex) and allocortex. The mesocortex occupies a ‘transitional zone’ that lies between the six-layered isocortex and the three-layered allocortex. It has a variable number of layers and can be further subdivided into proisocortex and periallocortex, based on its similarity to the two main types.
The isocortex has a uniform six-layered structure (Greek: isos, equal) and accounts for more than 90% of the cortical surface area. This term is synonymous with neocortex.
The mesocortex is a transitional zone between the allocortex and isocortex and has 3–6 layers. Most of the limbic lobe (cingulate and parahippocampal gyri; see Ch. 3) is composed of mesocortex.
The majority of the ‘non-neocortical’ regions belong to the limbic lobe and are primarily concerned with emotion, memory and olfaction (the sense of smell). The term paralimbic cortex is used to describe non-neocortical regions outside of the limbic lobe proper, including the posterior orbital cortex, anterior insula and temporal pole.
Features of the neuron
Dendritic spines
The dendrites of many neurons are studded with thousands of tiny, mushroom-shaped dendritic spines (Fig. 5.4). These include the medium spiny neurons that make up 95% of cells in the basal ganglia. In the cerebral cortex, all pyramidal cells have dendritic spines, whereas stellate cells may be spiny or smooth. Each dendritic spine is the site of an incoming excitatory synapse and a typical cortical pyramidal neuron has more than 10,000 spines. They are dynamic structures that can form, change shape or disappear altogether and are thought to be important in synaptic plasticity (Greek: plastikos, able to be moulded) and learning. Long-term memories may be mediated by the growth of new spines or the strengthening and enlargement of existing ones.
Subcellular organelles
The neuronal cell body or soma (Greek: soma, body) contains the same organelles found in other cell types (Fig. 5.5) but the machinery for protein synthesis and gene transcription is particularly prominent (Fig. 5.6A). The perinuclear cytoplasm or perikaryon (Greek: peri, around; cyton, kernel) contains a well-developed network of rough endoplasmic reticulum, often arranged in clumps called Nissl bodies. The Golgi apparatus is also prominent and is the site of post-translational modification and sorting of proteins including ion channels, neurotransmitter receptors and membrane ion pumps.
(A) Micrograph showing a pyramidal motor neuron in the cerebral cortex of the frontal lobe. It has a large nucleus with a well-defined nuclear envelope and prominent central nucleolus. The cytoplasm (stained pink) contains coarse clumps of rough endoplasmic reticulum (stained blue); these structures are referred to as Nissl bodies. [Routine haematoxylin and eosin (H&E)-stained sections.] From Prayson, Richard: Neuropathology 1e (Churchill Livingstone 2005) with permission; (B) Micrograph of two pigmented dopaminergic neurons in the substantia nigra of the midbrain (Nissl stain). The pigment is neuromelanin and is produced as a by-product of dopamine synthesis. From Standring: Gray’s Anatomy 40e (Churchill Livingstone 2008) with permission.
Lipofuscin and neuromelanin
Neurons have a high rate of membrane turnover and gradually accumulate non-digestible membrane components within lysosomal residual bodies. These contain golden-brown material called lipofuscin (pronounced: lipo-FUSKIN). This so-called ‘age pigment’ accumulates over time as a by-product of membrane turnover and is also abundant in the heart and liver. Catecholamine-synthesizing neurons contain the dark brown pigment neuromelanin (Fig. 5.6B); these include the substantia nigra of the midbrain and the locus coeruleus of the pons (see Ch. 1).
Vesicles
Neurons contain various types of membrane-bound vesicle. Neurotransmitters and neuropeptides are stored in the axon terminal (prior to release) within synaptic vesicles (see Ch. 7). Coated vesicles are derived from internalization of membrane constituents and macromolecules that have been taken up from the extracellular fluid by receptor-mediated endocytosis.
The neuronal cytoskeleton
All cells have a cytoskeleton composed of an internal framework of fibrillar proteins, that gives each cell its characteristic shape. This molecular scaffold is particularly important in process-bearing cells such as neurons and glia, which have a complex structure. The cytoskeleton is also involved in the transport of materials between intracellular compartments (see below). The main components include microtubules, neurofilaments and microfilaments (Fig. 5.7).
Microtubules
Microtubules are tubular polymeric proteins that are composed of repeating subunits of alpha and beta tubulin. They are 24 nm in diameter and up to 1 mm in length. Some microtubules are stable, whereas others are dynamic and can grow or shrink by the addition or removal of tubulin subunits. Microtubules are polarized. The part that is closest to the neuronal cell body is arbitrarily termed the minus (–) end and that which is nearer to the axon terminal is designated the plus (+) end. Microtubules are stabilized by binding to a range of microtubule associated proteins (MAPs). These include the phosphoprotein tau which accumulates in various degenerative conditions including Alzheimer’s disease (Ch. 12).
