81 Neuromuscular Disorders
Neuromuscular disorders include a highly variable group of diseases that affect the peripheral nervous and muscular systems on any level of the neuraxis. Pathology ranges from disorders affecting the spinal motor neuron to the muscles by way of the peripheral nerves (Figure 81-1). In this chapter, several of the more common pediatric neuromuscular disorders are reviewed and classified based on their level of involvement in the neuraxis.
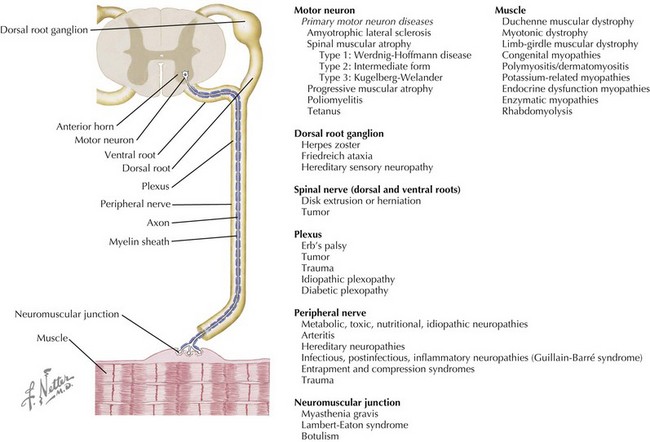
Anterior Horn Cell
Spinal Muscular Atrophy
Clinical Presentation
SMA is subclassified into four types based on age of onset and the maximal level of motor skills achieved. SMA type 1 (or Werdnig-Hoffmann disease) is the most common and severe of these disorders, accounting for approximately 60% of cases. SMA type 1 presents in the early infancy period (0–6 months) with generalized hypotonia, proximal and symmetric flaccid muscle weakness (initially lower more than upper limbs), and absent deep tendon reflexes. This frequently comes to the attention of the pediatrician with gross motor milestone delay and may present subacutely or more indolently. Most infants also have tongue fasciculations, and some have postural tremor of the fingers or joint contractures. Diaphragmatic sparing with intercostal muscle involvement results in paradoxical breathing and the classic bell-shaped torso (Figure 81-2). Importantly, these infants are alert and interactive with normal cognitive development and no sensory loss or impairment of eye movements. Systemic complications of SMA include pneumonia, scoliosis, poor weight gain, sleep difficulties, and joint contractures. These infants never achieve independent sitting. Most individuals’ expected life span is less than 2 years without invasive ventilatory and nutritional support.
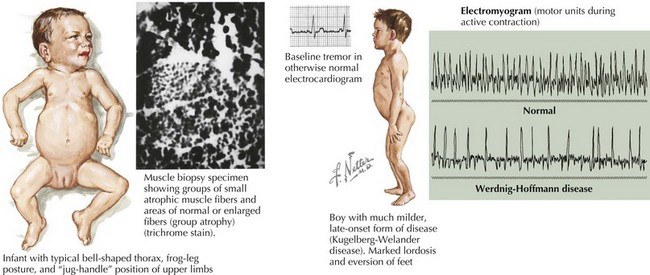
The differential diagnosis includes other disorders causing acute weakness, including poliomyelitis and infantile botulism. In addition, the general differential diagnosis for hypotonia and more chronic weakness in an infant should be considered (Table 81-1).
Table 81-1 Differential Diagnosis of the Floppy Baby, Infant, and Child
| Localization | Diagnoses (Examples) |
|---|---|
| Brain/Systemic | Chromosomal (Turner’s syndrome, trisomy 21, Prader-Willi syndrome) |
| Benign congenital hypotonia | |
| Infection (sepsis, meningitis, encephalitis, TORCH infections, tick paralysis) | |
| Metabolic (electrolyte abnormalities, hypothyroidism, hepatic encephalopathy, mitochondrial and peroxisomal disorders, amino and organic acidemias) | |
| Toxins (alcohol, narcotics, heavy metal poisoning, organophosphates, anticholinergics) | |
| Neonatal encephalopathy | |
| Trauma | |
| Spinal cord | Hypoxic-ischemic myelopathy |
| Compression | |
| Syringomyelia | |
| Anterior horn cell | Spinal muscular atrophy |
| Infection (polio, Coxsackie) | |
| Cytochrome C oxidase deficiency | |
| Peripheral nerve | Demyelinating (Guillain-Barré syndrome, hereditary motor-sensory neuropathy type I, congenital hypomyelinating neuropathy) |
| Axonal (familial dysautonomia, hereditary motor-sensory neuropathy type II, infantile neuronal degeneration) | |
| Neuromuscular junction | Infection (botulism) |
| Myasthenia gravis | |
| Muscle | Myopathies and congenital muscular dystrophies |
| Metabolic (acid maltase deficiency, hypo- or hyperthyroid myopathy, carnitine deficiency) | |
| Muscular dystrophies | |
| Inflammatory (dermatomyositis, polymyositis) | |
| Mitochondrial myopathies |
TORCH, toxoplasmosis or Toxoplasma gondii, other infections, rubella, cytomegalovirus, and herpes simplex virus.
Peripheral Nerve
Guillain-Barré Syndrome
Etiology and Pathogenesis
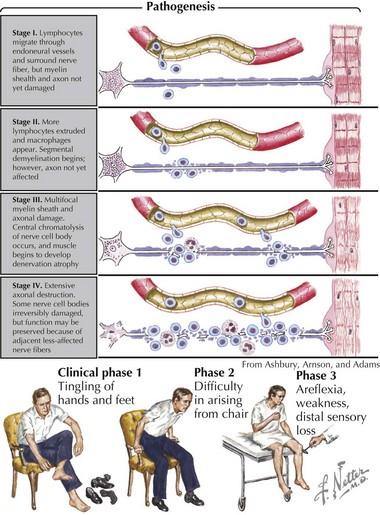
Pathologically, the two primary changes seen in GBS are acute inflammatory demyelinating polyradiculoneuropathy and acute axonal degeneration, both of which are thought to be caused by cross-reacting antibodies to the various gangliosides expressed in peripheral and cranial nerves. Campylobacter infections can result in molecular mimicry with the GM1 ganglioside, such that patients with C. jejuni infections are at 100-fold higher risk of developing GBS than the general population (Figure 81-3).
Neuromuscular Junction
Myasthenia Gravis
Etiology and Pathogenesis
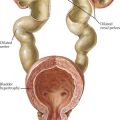
MG is caused by an antibody-mediated response attacking the nicotinic acetylcholine receptor on the postsynaptic motor endplate of the neuromuscular junction (Figure 81-4). Two specific antibodies are thought to be responsible for most MG. The first is an antibody directed against the acetylcholine receptor (anti-Ach-R Ab), which is positive in 56% of prepubertal children with JMG and 82% of children with peripubertal onset. Of the adult patients that test negative for anti-Ach-R Ab, 40% to 70% have antibodies that bind to the extracellular domain of muscle specific kinase (anti-MuSK Ab). The prevalence of anti-MuSK Ab is unclear in children.
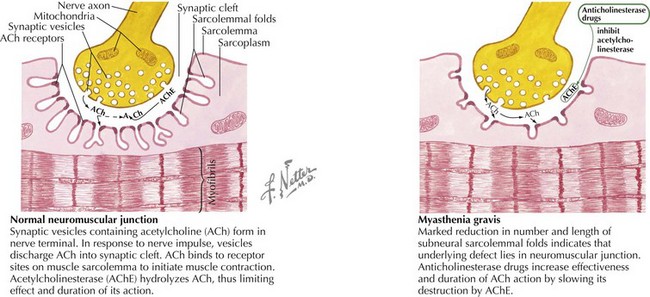
Infantile Botulism
Clinical Presentation
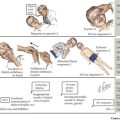
Affected infants incubate their botulinum spores for 3 to 30 days and then present with a progressive clinical picture of neuromuscular blockade, nadiring at 1 to 2 weeks. Infants initially demonstrate poor feeding and constipation followed by a subacute progression of descending bulbar and extremity hypotonia and weakness (Figure 81-5). Cranial nerve dysfunction manifests early as pupillary paralysis, ptosis, diminished extraocular movements, facial diplegia, and a weak suck and gag. Autonomic dysfunction presents with decreased salivation and tearing, widely fluctuant heart rate and blood pressure, and flushed skin. Decreased extremity movement and areflexia are later signs followed by flaccid paralysis and respiratory failure in 50% to 70%. Without treatment, the total duration of illness is approximately 1 to 2 months.
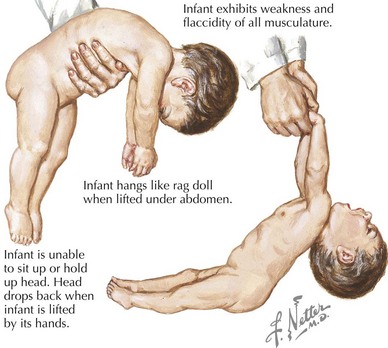
The differential diagnosis is that of the hypotonic infant (see Table 81-1). In clinical practice, SMA type 1 and metabolic disorders are the most frequent mimics of infantile botulism.
Evaluation and Management
For any case of suspected infantile botulism, the California Department of Health Services, Infant Botulism Treatment and Prevention Program should be contacted (http://www.infantbotulism.org/ or 24-hour telephone number, 510-231-7600), and the infant should be immediately treated empirically with intravenous botulism immune globulin (or “babyBIG”) while the prolonged confirmatory testing described above is pending. Prompt babyBIG therapy has been shown to decrease the mean duration of hospital stay from 5.7 to 2.6 weeks, decrease the duration of mechanical ventilation from 4.4 to 1.8 weeks, and decrease hospital costs by $89,000 (in 2004 United States dollars). Management of patients with infantile botulism is otherwise supportive. Aminoglycoside antibiotics should be avoided because they can potentiate the effects of the toxin. With appropriate treatment, the case fatality rate is under 2%, and full recovery is expected.
Muscle
Duchenne Muscular Dystrophy
Clinical Presentation
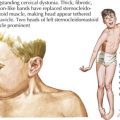
Patients with DMD classically present around the age of 3 years old with gross motor delay, excessive falling, and gait abnormalities. Calf hypertrophy and neck flexion weakness are evident by 3 to 4 years of age. Hip girdle muscles are typically affected sooner than shoulder girdle muscles, causing the classic Trendelenburg (or waddling) gait and the Gowers’ sign (Figure 81-6). As the disease progresses, weakness spreads distally. Joint contractures can develop and further worsen gait. Untreated, DMD follows a fairly predictable progressive course. Untreated, children lose the ability to walk (typically by 10 years of age and always by 13 years of age), then develop kyphoscoliosis, and finally develop cardiac and respiratory involvement and failure in the later second decade of life. The differential diagnosis includes other myopathies (see Table 81-1), dystrophies, and SMA type 3.
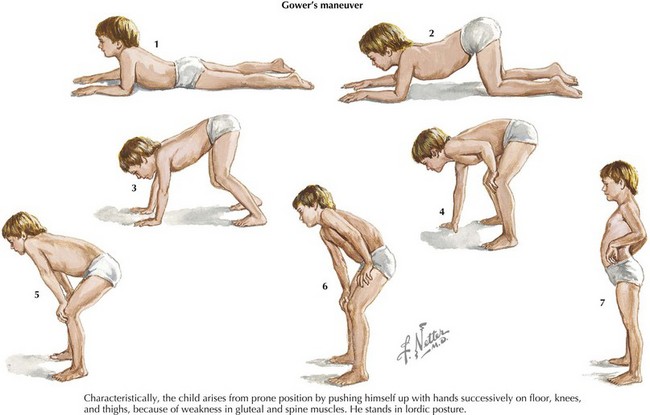
Evaluation and Management
With improved supportive care, the mean age of mortality has extended from 17 years to the mid-twenties, and some patients now survive into the fourth decade. Recent clinical care guidelines for DMD have been published (see Suggested Readings). Death is still usually from cardiomyopathy.
Arnon SS, Schechter R, Maslanka SE, et al. Human botulism immune globulin for the treatment of infant botulism. N Engl J Med. 2006;354:462-471.
Barras BT, Korf BR, Urion DK. Dystrophinopathies. Last updated March 2008. Available at http://www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=gene&part=dbmd
Bushby K, Finkel R, Birnkrant D, et al. The diagnosis and management of Duchenne muscular dystrophy–part 1. Diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 2010;9:77-93.
Bushby K, Finkel R, Birnkrant D, et al. The diagnosis and management of Duchenne muscular dystrophy–part 2. Implementation of multidisciplinary care. Lancet Neurol. 2010;9:177-189.
Chiang LM, Darras BT, Kang PB. Juvenile myasthenia gravis. Muscle Nerve. 2009;39:423-431.
Domingo RM, Haller JS, Gruenthal M. Infant botulism: two recent cases and literature review. J Child Neurol. 2008;23:1336-1345.
Hughes RA, Swan AV, Raphael JC, et al. Immunotherapy for Guillain-Barré syndrome: a systematic review. Brain. 2007;130:2245-2257.
The Infant Botulism Treatment and Prevention Program. Available at http://www.infantbotulism.org/09
Lunn MR, Wang CH. Spinal muscular atrophy. Lancet. 2008;371:2120-2133.
The Myasthenia Gravis Foundation of America. Available at http://www.myasthenia.org
Rabie M, Nevo Y. Childhood acute and chronic immune-mediated polyradiculneuropathies. Eur J Paediatr Neurol. 2009;13:209-218.
Ryan MM. Guillain-Barré syndrome in childhood. J Paediatr Child Health. 2005;41:237-241.
Wang CH, Finkel RS, Bertini ES, et al. Consensus statement for standard of care in spinal muscular atrophy. J Child Neurol. 2007;22(8):1027-1049.