66 Congenital Anomalies of the Kidney and Urologic System
This chapter reviews several common congenital and inherited anomalies of the kidney and urologic system. Congenital abnormalities are a frequent cause of renal failure in children, accounting for more than 30% of end-stage renal disease (ESRD). Congenital anomalies can be subdivided into three categories based on the stage of the primary abnormality in renal embryologic development (Figure 66-1). The first category refers to a failure to form a functional nephron, leading to renal parenchyma malformations, such as renal agenesis or cystic dysplasia. The second group of defects relates to a failure of the developing kidney to migrate to its appropriate destination. This may lead to renal ectopy (pelvic or thoracic kidneys) or fusion abnormalities, such as a horseshoe kidney. The third category describes defects of the urinary collecting system, such as double ureters or posterior urethral valves (PUVs). Here, changes of the renal parenchyma (hydronephrosis, dysplasia) are often secondary to obstructive uropathy or urinary reflux disease. Inherited conditions such as the polycystic kidney diseases (PKD) are the result of specific genetic mutations that may present with detectable renal abnormalities at birth or may not develop until later in life. The following sections detail common and exemplary conditions representative of these subcategories.
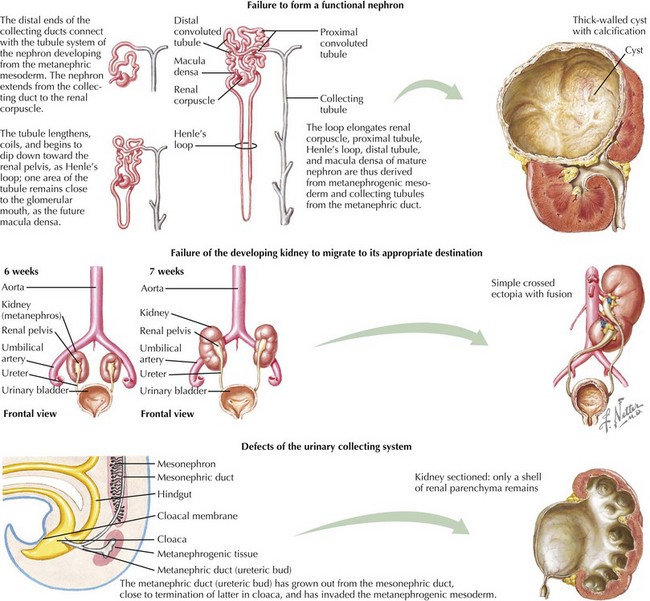
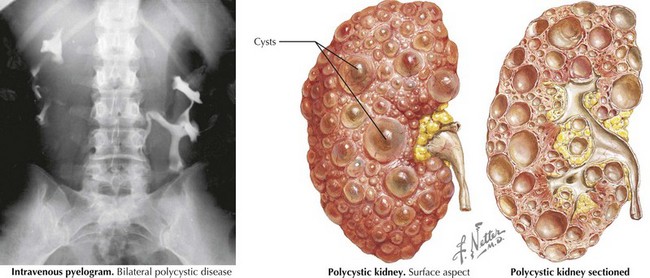
Polycystic Kidney Disease
PKDs are a group of genetically inherited conditions in which cyst formation and renal parenchymal replacement can occur at anytime from fetal life to adulthood. There are two major forms, autosomal dominant PKD (ADPKD) and autosomal recessive PKD (ARPKD) (Table 66-1). In addition, a number of rare pleiotropic disorders exist, which are loosely associated because of similar clinical and pathophysiologic features, including renal cystic and hepatobiliary disease (Table 66-2). All of these disorders share ciliary dysfunction as a common principle in their pathogenesis. PKD proteins have been localized to the cilia or basal body, and loss or abnormalities of cilia in the kidney are associated with cyst development (Figure 66-2).
Table 66-1 Autosomal Recessive Polycystic Kidney Disease versus Autosomal Dominant Polycystic Kidney Disease
| Autosomal Recessive Polycystic Kidney Disease | Autosomal Dominant Polycystic Kidney Disease | |
|---|---|---|
| Gene | Chromosome 6p21.2-p12 | Chromosome 16p13.3, chromosome 4q13-q23 |
| Protein | Fibrocystin | Polycystin 1, polycystin 2 |
| Age of presentation | Commonly prenatally, childhood, adolescence | Highly variable |
| Renal cysts | Radial pattern | Anywhere in kidney, varying size |
| Extrarenal manifestations | Biliary obstruction, hepatic fibrosis with portal hypertension, liver failure | Liver cysts, pancreas cysts, vascular cysts (“Berry aneurysms”) |
| Things in common | Renal cysts do not communicate. Disease may present at any age, including prenatally. Gene defect leads to malfunction of the ciliary body. The kidneys are usually enlarged. Usually bilateral disease (exception: early diagnosis ADPKD in childhood with positive family history) | |
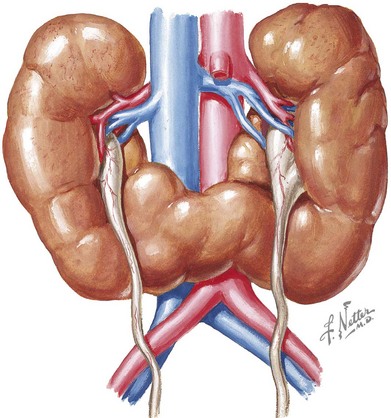
Horseshoe Kidney
Horseshoe kidney is the most prevalent fusion abnormality of the developing kidney. The fusion occurs commonly at the lower poles, and two separate excretory urinary systems are maintained (Figure 66-3) The incidence is reported between one in 400 and one in 1600. The isthmus may be located at or lateral to the midline (symmetric vs. asymmetric horseshoe kidney). It may contain actual parenchyma or a fibrous band. The kidneys arrest during their embryologic ascension toward the dorsolumbar position, usually between the fourth to ninth weeks of gestation. This is caused by the inferior mesenteric artery holding the isthmus and preventing further rostral migration. As a consequence, blood supply of the fused kidney is variable and may come from the iliac arteries or aorta or at times, the hypogastric and middle sacral arteries. Horseshoe kidneys may occur in isolation or as part of a syndrome, such as Turner syndrome, trisomy 18, or less commonly trisomy 13 and 21. It is occasionally associated with other genitourinary findings, such as bicornuate or septate uterus in girls and hypospadias and undescended testis in boys. Most patients with horseshoe kidneys are asymptomatic and are diagnosed incidentally by ultrasonography. However, some patients may present with pain, hematuria, obstruction, or UTIs. Hydronephrosis may be seen in up to 80% of children with horseshoe kidneys. The etiologies include vesicoureteral reflux (VUR), obstruction of the collecting system by external ureteric compression (from blood vessels, renal calculi), or ureteropelvic junction (UPJ) obstruction caused by a relatively high insertion of ureters. Twenty percent of patients have urolithiasis.
Urologic Malformations
Double Ureter
Double ureters are part of a duplicated collecting system complex, defined as two pyelocaliceal systems within one renal unit. Both ureters may either drain separately or jointly over a single orifice into the bladder. Double ureters can be unilateral or bilateral and can be associated with a variety of congenital genitourinary tract abnormalities. Most patients are asymptomatic, with double ureters being detected incidentally on imaging studies. Double ureters occur when two ureteral buds arise from the Wolffian duct. In cases of complete duplication, the lower renal unit typically drains into the normal ureteric insertion and the ureter of the upper renal unit drains ectopically in the bladder, urethra, or elsewhere. The ectopic ureter usually enters the bladder inferiorly and medially to the normal ureter. In girls, if the ectopic ureter inserts into the vagina, urethra, or uterus, urinary dribbling or incontinence may be the presenting sign. Most patients are asymptomatic. Symptoms usually occur in ureteric duplication if there is reflux into the ureter of the lower renal unit or if the ectopic upper renal unit ureter becomes obstructed (Figure 66-4). The typical complications observed with complete double ureter systems are summarized in Table 66-3. Recurrent UTIs can occur in obstructed or refluxing ureters. It is noteworthy that up to 80% of ureteroceles are associated with double ureters. In duplex kidneys, ureteroceles are usually an outgrowth of the ectopic ureter. Double ureters are common and noted to occur in 0.2% of live births. There is a 12% chance of a double ureter occurring in first-degree relatives of persons with this condition. Bilateral duplication is noted in 15% to 40% of individuals with complete duplication. The diagnostic evaluation includes renal ultrasound and VCUG. The goal of imaging is to identify VUR, ureteroceles, and obstructive uropathy. If these are found, surgical correction may be indicated.
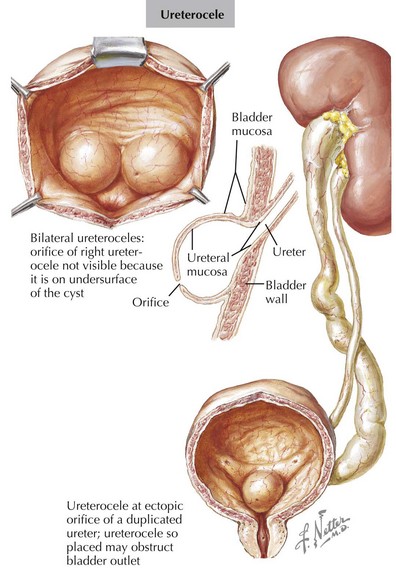
| Renal upper pole | Urinary obstruction from ectopic insertion of the ureter leading to upper pole hydronephrosis Ureterocele |
| Renal lower pole | Urinary reflux from maldevelopment of the ureter’s valve mechanism |
Posterior Urethral Valves
PUVs are the most common cause of lower urinary tract obstruction in male neonates (Figure 66-5). The reported incidence ranges from one in 8000 to one in 25,000 live births. The etiology of PUV is still disputed. Autopsy studies on stillborn fetuses with PUVs have found a congenital obstructing posterior urethral membrane (COPUM). It is possible that the valves classically seen on ultrasound and VCUG occur as a result of the perforation of the COPUM by an advancing Foley catheter.
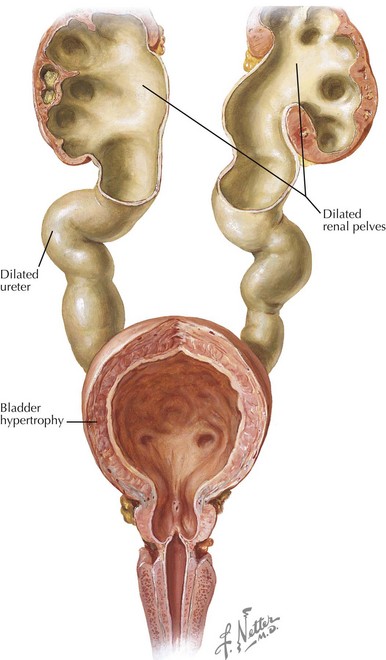
The posterior urethra is formed by the cloacae and the urogenital sinus, and its lining contains transitional epithelium. The theory is that abnormal integration of the Wolffian ducts into the posterior urethra might lead to formation of the COPUM. PUV has a wide spectrum of clinical presentation largely depending on the initial degree of urethral obstruction. The associated morbidity is not limited to renal abnormalities and may be related to impaired lung development in utero (Potter sequence), bladder wall thickening and dilatation, hydroureters, urinomas, and hydronephrosis. The obstructive uropathy may result in renal dysplasia, renal insufficiency, and end-stage renal disease. Up to 15% of patients who undergo renal transplantation in childhood have PUV as the underlying condition. The widespread use of prenatal ultrasound has resulted in most cases being diagnosed in the fetal or neonatal period. Some patients may be missed and present later in life. In those cases, a voiding history is of particular importance because patients with PUV usually have a diminished or abnormal urinary stream. Treatment consists of fulguration of the PUV by a urologist. Children commonly develop postobstructive diuresis and should be monitored carefully for electrolyte and volume instabilities. The prognosis depends on the degree of preserved renal function after the obstruction is relieved (see Figure 66-5).
Harris PC. 2008 Homer W. Smith Award: insights into the pathogenesis of polycystic kidney disease from gene discovery. J Am Soc Nephrol. 2009;20(6):1188-1198.
North American Pediatric Renal Trials and Collaborative Studies. NAPRTCS 2008 Annual Report. Available at https://web.emmes.com/study/ped/index.htm
Neville H, Ritchey ML, Shamberger RC, et al. The occurrence of Wilms tumor in horseshoe kidneys: a report from the National Wilms Tumor Study Group (NWTSG). J Pediatr Surg. 2002;37(8):1134-1137.
Hildebrandt F, Attanasio M, Otto E. Nephronophthisis: disease mechanisms of a ciliopathy. J Am Soc Nephrol. 2009;20(1):23-35.
Krishnan A, de Souza A, Konijeti R, Baskin LS. The anatomy and embryology of posterior urethral valves. J Urol. 2006;175(4):1214-1220.