CHAPTER 120 NEUROLOGY OF PULMONOLOGY AND ACIDBASE DISTURBANCE
Respiration is essential for cellular metabolism, and no other organ is more dependent on oxygen supply than is the brain. The consequences of disturbances in gas exchange are readily reflected in disturbance of neuronal function, in neuronal injury, or in death. The respiratory system is crucial for the gas exchange and plays a large part in acidbase homeostasis. Neuronal integrity and respiratory regulation are highly interdependent. A brief conceptualization of the neurological control of breathing is necessary for understanding various disordered respiratory patterns. Clinical respiratory abnormalities can be viewed in three contexts: disturbances in the neural control of respiration, in the respiratory apparatus, or in the carriage or composition of blood gases. This chapter describes abnormalities associated with neurological control of respiration and neurological consequences of respiratory dysfunction.
NEURAL CONTROL OF RESPIRATION
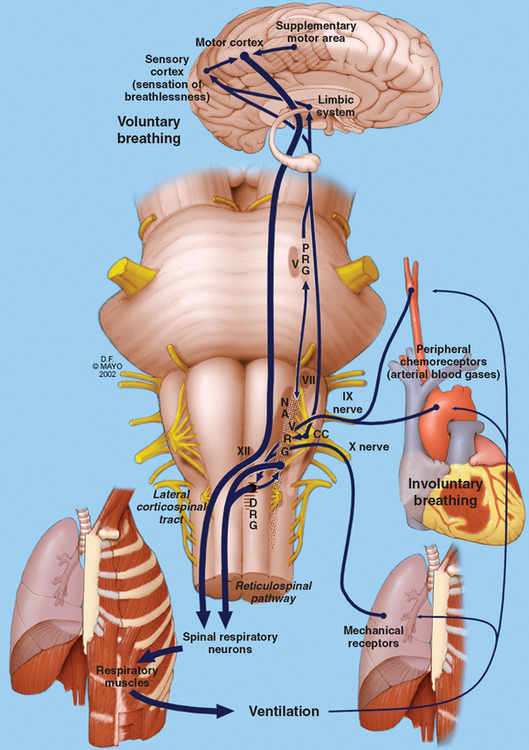
The nervous system is intricately connected to the mechanics of respiration at various levels. From the cerebral cortex via the brainstem to the level of the lower motor neurons, the nervous system regulates respiratory effort. It is aided by feedback from peripheral chemoreceptors and mechanoreceptors (Fig. 120-1).1 The anterior horn cells controlling respiratory muscles represent the lower motor neurons, which control the actual mechanics of breathing. Inspiratory muscles generate subatmospheric pressures within the thorax and induce airflow and gas exchange at the alveolar level.
Lower Motor Components of Respiration
The lower motor components are composed of neurons innervating the diaphragm, intercostal muscles, abdominal muscles, and other accessory muscles.1 During quiet breathing, only the diaphragm is vigorously active, with some contribution from the abdominal and external intercostal muscles. These muscles work in conjunction with muscles of the upper airway to maintain a patent airway and ensure an uninterrupted passage to air. The diaphragm is the most important inspiratory muscle, and it derives its nerve supply via the phrenic nerve from spinal cord segments C3-C5. Other inspiratory muscles include the external intercostal muscles, the parasternal intercostal muscles, and the scalene muscles.
Upper airway structures are crucial in maintaining the patency of airways. Dysfunction of these structures is readily evident in abnormalities of speech and swallowing, as well as in obstructive sleep apneas and other breathing disorders. The muscles of the nose, mouth, soft palate, pharynx, epiglottis, and larynx work in conjunction to ensure a free flow of air into the trachea and bronchi. Somatic innervation of these muscles is provided by the lower cranial nerves: V, VII, IX, X, and XII.
ABNORMAL RESPIRATORY PATTERNS IN NEUROLOGICAL DISEASES
Respiratory Failure
Neurological disorders can produce dyspnea or frank respiratory failure. Respiratory failure exists when arterial oxygen tension (PaO2) is less than 60 mmHg or when PaCO2 exceeds 50 mmHg when the patient is breathing air, and it is the end result of respiratory dysfunction. It can exist in acute and chronic forms. Acute respiratory failure manifests dramatically, and most patients are immediately intubated and ventilated. Nearly 300,000 cases of acute respiratory failure are encountered each year in the United States; the approximate incidence is 137 cases per 100,000 population.2 The number of cases related to neurological disorders is not known. Nevertheless, even if it is assumed that only 0.5% to 1% of cases of acute respiratory failure are related to neurological causes, they add up to 1500 to 3000 cases per year in the United States alone.
In clinical practice, the neurologist is most often consulted about patients with respiratory failure in which a clear pulmonary or medical cause is not evident. Sometimes the neurologist is consulted when there is difficulty in weaning a patient from a ventilator. This is common in the intensive care unit with critically ill patients who may display a necrotizing myopathy.3 Alternatively, respiratory failure may supervene in patients with known neurological illness such as myasthenia gravis or Guillain-Barré syndrome. Examination reveals tachypnea, brow sweating, tachycardia, weak cough, paradoxical respiration, and diminished respiratory muscle strength on pulmonary function tests. Patients with chronic respiratory failure may present with an insidious onset of sensorial alteration and coma. The commonest neurological causes of respiratory failure are neuromuscular and spinal cord disorders.4 Peripheral disorders result in respiratory muscle weakness, alveolar hypoventilation, and type II respiratory failure. Table 120-1 lists some of the common neuromuscular and spinal disorders producing respiratory weakness. Most of these disorders can manifest with acute or chronic respiratory failure. Associated findings, pulmonary function test results, neurophysiological evaluation findings, and muscle/nerve biopsy findings help identify most of these peripheral or spinal causes of respiratory failure.
TABLE 120-1 Neuromuscular Conditions Resulting in Respiratory Failure
Abnormal Respiratory Patterns
Apneustic Breathing
This pattern is characterized by brief pauses at the end of inspiration or alternating pauses at the inspiration and expiration. It is often encountered with bilateral lesions of the pontine tegmentum affecting the pontine pneumotaxic center.5 This center is involved in sending inhibitory impulses to the apneustic center and medulla that terminate inspiration and initiate passive expiration. Although readily reproducible in laboratory animals through introduction of pontine lesions, it is rarely observed in clinical practice. Isolated case reports describe reversal of apneustic breathing after stroke with buspirone.6
Hiccups
Although hiccups are by and large encountered in normal individuals in certain circumstances, they occasionally result from central nervous system lesions. The paradigm of such a lesion is a lateral medullary infarct. Hiccups are characterized by inspiratory movements in the diaphragm and external intercostal muscles, followed immediately by glottic closure. This results in ineffectual inspiration. It is more likely to supervene during inspiration than during expiration.
Disorders of Automatic Breathing
Automatic breathing may be selectively impaired in some disorders that affect the neurons of the medullary ventral respiratory group. During wakefulness, voluntary breathing may be able to sustain respiration. During slow-wave sleep, however, automatic breathing is essential for maintaining respiration. In these disorders, central apnea and hypoxemia supervene during sleep. This condition is termed Ondine’s curse, after the story of a mythical figure, Hans, who was cursed by his jilted lover, Ondine. He was doomed to lose all automatic functions and thus had to stay awake in order to ensure continued breathing. In the modern era, it was most commonly observed in bulbar poliomyelitis, in which the ventral respiratory group neurons were involved. Nowadays it is also observed after high anterior cervical lesions, which affect the reticulospinal tracts subserving automatic respiration. This is particularly common after fibrocartilaginous embolism to the C3-C4 segments of the spinal cord.7
Disorders of Voluntary Breathing
Voluntary control of breathing is mediated primarily via the corticospinal and corticobulbar tracts and is important in activities such as speech, singing, and voluntary breath holding. Disorders involving this mechanism occur mainly in bilateral pontine infarctions and lesions involving the pontine tegmentum that interrupt the descending motor pathways. The classical situation is that of patients with a “locked-in” syndrome. Such patients have a constant unvarying respiratory rhythm that cannot be modulated voluntarily. Thus, they are unable to hold their breath, breathe in deeply, or cough voluntarily. Reflex responses and responses to chemoreceptors remain intact. Partial lesions of the high cervical cord that selectively involve the corticospinal tracts at the C3-C4 segments can also produce a similar condition.7
Assorted Disorders of Breathing
Hemiplegic patients can display reduced diaphragmatic and chest wall excursions on the weak side. More often than not, they are clinically insignificant. The basal ganglia also play an important role in motor control and are involved in a wide variety of neurological disorders. Dystonia can involve muscles of the diaphragm, as well as those of the upper airway, producing subjective dyspnea.8 Chorea and dyskinesias (notably levodopa-induced dyskinesias) can interfere with breathing and produce subjective dyspnea.
Diaphragmatic flutter is a rare condition characterized by dyspnea, epigastric pulsations, abdominal pain, hyperventilation, and repetitive diaphragmatic contraction at a rate of approximately 3Hz.9 It is considered to be a form of myoclonus and, as such, responds to a number of antiepileptic drugs (phenytoin, carbamazepine, and benzodiazepines), as well as to phrenic nerve crush. It can be unilateral or bilateral and may occur after encephalitis or lesions impinging on the phrenic nerve roots. Other pulmonary disorders such as neurogenic pulmonary edema are sometimes associated with devastating neurological illnesses such as subarachnoid hemorrhage or intracerebral hemorrhage.10 These disorders, however, are outside the scope of this chapter.
NEUROLOGICAL CONSEQUENCES OF RESPIRATORY INSUFFICIENCY
Hypoxia
The paradigm of acute severe hypoxia is exemplified by altitude sickness. At high altitudes, mountain climbers experience a variety of symptoms such as headaches, delirium, hallucinations, ataxia, amnesia, gait disturbances, and seizures, which are readily reversible with oxygen therapy or descent to lower altitudes. Cerebral edema with resultant papilledema also occurs. Another commonly encountered clinical scenario is cardiac arrest. The brain is exceptionally dependent on oxygen, and patients become unconscious within 20 seconds of cardiac arrest. Neuronal adenosine triphosphate stores are depleted within seconds, and neuronal damage ensues. Neurons in selected areas such as the hippocampus, basal ganglia, cortical lamina, cerebellum, and spinal cord are particularly involved because of their lower capacity to withstand hypoxia. Nevertheless, it is believed that pure hypoxia alone is not responsible for neuronal injury unless it is accompanied by neuronal ischemia. In isolated hypoxia (PaO2 < 20 mmHg), irreversible neuronal injury is prevented by a manifold increase in cerebral blood flow. Hypoxia also induces cerebral vasodilatation directly and indirectly through the production of vasoactive factors.11 When ischemia coexists, there is a massive outpouring of excitatory neurotransmitters, such as glutamate. This activates α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptors and N-methyl-D-aspartate receptors, opens ion channels, and initiates enzymatic cascades that induce neuronal cell death. Neurological sequelae occurring after successful resuscitation from cardiac arrest vary from amnesia, motor and gait disturbances, and myoclonus to severe dementia.
Chronic hypoxia produces more insidious neurological damage. Patients with chronic obstructive pulmonary disease have an increased incidence of small-fiber peripheral neuropathy, which is linked to chronic hypoxia.12,13 Likewise, an asymptomatic autonomic neuropathy is encountered in these patients, although the etiological link is weaker with this entity.
Hyperoxia
Hyperoxia is encountered in humans exposed to hyperbaric environments. Such situations occur in patients undergoing hyperbaric therapies and in deep sea divers. These people experience short-term myopia as a result of direct hyperoxia-induced ocular lens changes. Neurological manifestations are transient and limited to headache and, in rare cases, seizures.14 Hyperoxia is also frequently encountered in patients on ventilators, in whom inadvertent hyperventilation or high fractional inspired oxygen concentrations result in elevated PaO2. The effect of hyperoxia in these clinical situations is unclear.
Hypercarbia
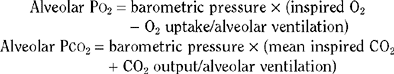
During hypoventilation, the fall in alveolar oxygen tension (PO2) can be overcome by an increase in inspired oxygen while the alveolar carbon dioxide tension continues to rise. This phenomenon is used to diagnostic advantage in the apnea test for brain death, in which supplemental high-flow oxygen is administered through a cannula placed at the level of the carina and the ventilator is disconnected. In the appropriate clinical context, the apnea test result is suggestive of brain death if a rise in PaCO2 of more than 20 mmHg does not result in respiratory efforts. Chronic hypercarbia produces milder neurological dysfunction as a result of compensatory nervous system adaptation. Nevertheless, morning headaches, asterixis or tremulousness, inattention, and cognitive dysfunction are often encountered in such patients. Cerebral edema and papilledema are often encountered in these patients. An important caveat in the treatment of these patients is that correction of hypercarbia should be attempted gradually. Rapid correction induces cerebral vasoconstriction and worsens the encephalopathy.
Air Embolism and Decompression Sickness
All of the disorders just described involve abnormalities in the partial pressures of gases dissolved in the blood. In normal circumstances, gases dissolve in blood in proportion to their partial pressure (Henry’s law). However, there are other conditions in which this gas-blood equilibrium is disturbed and bubbles of gas form. The following discussion pertains to such phenomena. A discussion on respiration would be incomplete without a reference to these disorders. Arterial air embolism occurs with trauma, arterial cannulations, and surgical procedures. Exposure to extremely high atmospheric pressures during dives can cause rupture of pulmonary alveoli and entry of air into pulmonary veins. The resultant air bubbles act as embolic fragments and produce changes in mental status and focal neurological deficits. Similarly, when deep sea divers ascend too rapidly, there is no time for arterial gases to equilibrate, and nitrogen dissolved in adipose tissue bubbles out into the blood stream, causing decompression sickness (the “bends”). Again, gas bubbles cause neurological dysfunction in myriad ways. They can act as mechanical emboli, induce coagulation cascades at the bubble-blood interface, and cause mechanical compression and distention of vascular structures and tissues. Although cutaneous, musculoskeletal, and cardiopulmonary manifestations predominate, the nervous system is another favored site of injury. Visual symptoms, spinal cord injury, labyrinthine symptoms, focal neurological deficits, and altered mental status can occur. Although air embolism and decompression sickness produce similar neurological abnormalities, the underlying pathophysiological processes are different. Nevertheless, similar treatment protocols, involving recompression with 100% oxygen in hyperbaric chambers, are used for both conditions.14
ACIDBASE BALANCE AND RESPIRATION
The respiratory system works closely with the kidneys, the liver, and an effective circulation in maintaining homeostasis and acidbase balance. Nearly 15,000 mmol of carbon dioxide is produced every day and eliminated by the respiratory system. The pH of blood is immediately altered by respiratory changes, and vice versa.15 For clinical purposes, a linear relationship can be assumed to exist between pH and acute changes in PaCO2. Hence, for every increase in PaCO2 of 20 mmHg above normal, the pH falls by 0.1, and for every decrease of PaCO2 of 10 mmHg below normal, the pH rises by 0.1.
Although metabolic acidbase disturbances are better understood with the newer concept of Stewart’s model,16 most clinicians rely on the simplified concept of anion gap and base excess. Stewart’s model postulates that acidbase balance depends on a number of factors, including PaCO2 levels, strong ion difference, and concentrations of weak acids.17,18 Metabolic acidosis from a variety of disorders results in compensatory respiratory changes. PaCO2 is “blown off’” in an attempt to normalize pH, and patients display hyperventilation. Metabolic alkalosis may result in compensatory hypoventilation and respiratory acidosis but is less commonly observed.19 The PaCO2 increases by 0.5 to 0.7 mmHg for every 1.0-mm increase in plasma HCO3 concentration up to a maximum of 55 to 60 mmHg. If PaCO2 fails to increase appropriately, a mixed acidbase disturbance such as additional respiratory alkalosis should be considered.20 Such disorders manifest less frequently in neurological practice and are hence not considered in greater detail.
CONCLUSIONS
Bolton C, Chen R, Wijdicks EFM, et al. Anatomy and physiology of the nervous system: control of respiration. In: Bolton C, Chen R, Wijdicks EFM, et al, editors. Neurology of Breathing. Philadelphia: Butterworth-Heinemann; 2004:19-35.
Corey HE. Bench-to-bedside review: fundamental principles of acidbase physiology. Crit Care. 2005;9:184-192.
Howard RS, Rudd AG, Wolfe CD, et al. Pathophysiological and clinical aspects of breathing after stroke. Postgrad Med J. 2001;77:700-702.
Laghi F, Tobin MJ. Disorders of the respiratory muscles. Am J Respir Crit Care Med. 2003;168:10-48.
Leach RM, Rees PJ, Wilmshurst P. Hyperbaric oxygen therapy. BMJ. 1998;317:1140-1143.
1 Bolton C, Chen R, Wijdicks EFM, et al. Anatomy and physiology of the nervous system control of respiration. In: Bolton C, Chen R, Wijdicks EFM, et al, editors. Neurology of Breathing. Philadelphia: Butterworth-Heinemann; 2004:19-35.
2 Behrendt CE. Acute respiratory failure in the United States: incidence and 31-day survival. Chest. 2000;118:1100-1105.
3 Maramattom B, Wijdicks EF, Sundt TM, et al. Flaccid quadriplegia due to necrotizing myopathy following lung transplantation. Transplant Proc. 2004;36:2830-2833.
4 Laghi F, Tobin MJ. Disorders of the respiratory muscles. Am J Respir Crit Care Med. 2003;168:10-48.
5 Howard RS, Rudd AG, Wolfe CD, et al. Pathophysiological and clinical aspects of breathing after stroke. Postgrad Med J. 2001;77:700-702.
6 El-Khatib MF, Kiwan RA, Jamaleddine GW. Buspirone treatment for apneustic breathing in brain stem infarct. Respir Care. 2003;48:956-958.
7 Howard RS, Thorpe J, Barker R, et al. Respiratory insufficiency due to high anterior cervical cord infarction. J Neurol Neurosurg Psychiatry. 1998;64:358-361.
8 Braun N, Abd A, Baer J, et al. Dyspnea in dystonia. A functional evaluation. Chest. 1995;107:1309-1316.
9 Cvietusa PJ, Nimmagadda SR, Wood R, et al. Diaphragmatic flutter presenting as inspiratory stridor. Chest. 1995;107:872-875.
10 Friedman JA, Pichelmann MA, Piepgras DG, et al. Pulmonary complications of aneurysmal subarachnoid hemorrhage.Neurosurgery. 2003;52:1025-1031. discussion, Neurosurgery. 2003;52:1031-1032.
11 Markus HS. Cerebral perfusion and stroke. J Neurol Neurosurg Psychiatry. 2004;75:353-361.
12 Appenzeller O, Parks RD, MacGee J. Peripheral neuropathy in chronic disease of the respiratory tract. Am J Med. 1968;44:873-880.
13 Thomas PK, King RH, Feng SF, et al. Neurological manifestations in chronic mountain sickness: the burning feet-burning hands syndrome. J Neurol Neurosurg Psychiatry. 2000;69:447-452.
14 Leach RM, Rees PJ, Wilmshurst P. Hyperbaric oxygen therapy. BMJ. 1998;317:1140-1143.
15 Williams AJ. ABC of oxygen: assessing and interpreting arterial blood gases and acidbase balance. BMJ. 1998;317:1213-1216.
16 Stewart PA. Modern quantitative acidbase chemistry. Can J Physiol Pharmacol. 1983;61:1444-1461.
17 Corey HE. Bench-to-bedside review: fundamental principles of acidbase physiology. Crit Care. 2005;9:184-192.
18 Fencl V, Jabor A, Kazda A, et al. Diagnosis of metabolic acidbase disturbances in critically ill patients. Am J Respir Crit Care Med. 2000;162:2246-2251.
19 Perrone J, Hoffman RS. Compensatory hypoventilation in severe metabolic alkalosis. Acad Emerg Med. 1996;3:981-982.
20 Galla JH. Metabolic alkalosis. J Am Soc Nephrol. 2000;11:369-375.