CHAPTER 116 NEUROLOGY OF COMMON ELECTROLYTE DISORDERS
HYPEROSMOLALITY AND HYPERTONICITY
Normal serum, and therefore body fluid, osmolality is in the range of 275 to 295 mOsm/kg; clinically significant effects are generally seen at levels greater than 325 mOsm/kg. Osmolality may be measured directly by the freezing point depression or calculated as serum osmolarity in milliosmoles per liter with the following formula, which accounts for the millimolar quantities of major serum solutes (where BUN is blood urea nitrogen):
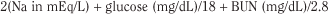
Azotemia is caused by renal failure or inadequate renal perfusion (prerenal azotemia).
The treatment of hyperosmolality requires calculation of apparent water losses:
HYPONATREMIA
Some patients with SIADH may become more hyponatremic with saline infusion as the water is retained and the salt excreted. This response can be predicted if the urinary [Na + K] level exceeds the serum [Na] level. In such a case, furosemide may be a useful adjunct for diluting the urine.