[level-membership-for-neurology-category]
CHAPTER 113 NEUROLOGY OF CARDIOLOGY
The neurology of cardiology may be divided into three categories: (1) the cardiac complications of neurological disease; (2) the neurological complications of cardiac disease (e.g., cardiac-source embolic stroke, neurological complications of cardiac surgery); and (3) neurocardiac syndromes (mitochondrial diseases; Friedreich’s ataxia; muscular dystrophies affecting the heart). The latter two categories are covered in other chapters of this book. This chapter deals with the cardiac manifestations of neuropsychiatric illnesses.
In 1942, Walter Cannon reported several incidents of “voodoo” death,1 by which he meant death from fright. These events had several features in common. They were all induced by an absolute belief that an external force could cause demise and that the victim had no power to alter this course. Cannon postulated that death was caused by intense action of the sympathetic adrenal system. Evidence has accumulated to support the concept that voodoo death is a real phenomenon and is not limited to ancient peoples. Rather, it may be a basic biological principle that provides an important clue to understanding the phenomenon of sudden death in modern society, as well as opening a window into the world of neurovisceral disease. George Engel collected 160 accounts from the lay press of sudden death that was attributed to disruptive life events2 and concluded that such events could be divided into eight categories (Fig. 113-1), the common feature of which is life-threatening stress without escape or control.

Richter reported a series of experiments aimed at elucidating the mechanism of “voodoo” death.3 He found that domesticated rats could swim for about an hour in 93°F water, but if the animals’ whiskers were trimmed, they would invariably drown within a few minutes. When similar experiments were performed with wild rats, restraint contributed significantly to the tendency for demise, whereas, in the case of the calm, domesticated animals in which restraint and confinement were apparently not significant stressors, shaving the whiskers rendered these animals as fearful as wild rats, with a corresponding tendency for sudden death. Adrenalectomy did not protect the animals.
In humans, one of the easily accessible windows into autonomic activity is the electrocardiogram (ECG). Byer and associates reported six patients with neurological disease in whom ECGs showed large upright T waves and long Q-T intervals.4 On the basis of experimental results of cooling or warming the endocardial surface of the dog’s left ventricle, they concluded that these electrocardiographic changes resulted from subendocardial ischemia. Levine reported a patient with a subarachnoid hemorrhage who had electrocardiographic changes reminiscent of coronary disease.5 Burch and colleagues reported 17 patients with various types of stroke who had long Q-T intervals, large and usually inverted T waves, and frequent U waves.6 Cropp and Manning reported the abnormalities on ECGs in 29 patients with subarachnoid hemorrhage; in five of these patients, autopsy study verified the absence of coronary artery disease and myocardial infarction, which suggested that the abnormalities on ECGs were neurogenic.7
Selye described electrolyte-steroid cardiopathy with necrosis and argued that this lesion was distinct from the coagulation necrosis that occurred as a result of ischemic disease.8 Certain steroids and other hormones created a predisposition for the development of electrolyte-steroid cardiopathy with necrosis, but other factors were necessary for its development, including stress. Raab and associates found that cardiac lesions may be produced in rats by pretreatment with fluorocortisol, calciferol, or thyroxine, followed by restraint or cold stress, and that pharmacological blockade of sympathetic activity was cardioprotective.9 Intracoronary infusions of adrenaline reproduce the characteristic electrocardiographic pattern of neurocardiac disease, which is reminiscent of subendocardial ischemia, although no ischemic lesion could be found in the hearts of dogs sacrificed after several months of infusions.10
Melville and coworkers produced changes on ECG and myocardial necrosis by stimulating the hypothalamus of cats; autopsy studies revealed evidence of open coronary arteries.11 The cardiac lesion was characterized by intense cytoplasmic eosinophilia with loss of cross-striations and some hemorrhage, a condition now most commonly known as contraction band necrosis. Oppenheimer and Cechetto mapped the chronotropic organizational structure in the rat insular cortex, demonstrating that sympathetic innervation arises from a more rostral part of the posterior insula than does parasympathetic innervation.12
Despite the fact that myocardial damage had been produced in animals, it was not until Koskelo and associates reported on three patients with electrocardiographic changes caused by subarachnoid hemorrhage13 that contraction band necrosis was demonstrated in humans with neurological disease. Connor reported focal myocytolysis in 8% of 231 autopsy studies; the incidence was highest in patients who had died of intracranial hemorrhages.14 Connor pointed out that prior pathological reports probably overlooked the lesion because of the fact that it was multifocal, each individual focus being quite small, necessitating extensive tissue sampling. It is clear now that even Connor underestimated the prevalence of the lesion and that serial sections are required to rigorously rule out its presence. Greenshoot and Reichenbach reported nine patients with subarachnoid hemorrhage, all of whom had cardiac lesions, and demonstrated that the cardiac pathology could be reproduced in cats given mesencephalic reticular formation stimulation.15 Adrenalectomy did not protect the hearts. This finding supported the contention that the electrocardiographic changes and cardiac lesions result from direct intracardiac release of catecholamines. Hawkins and Clower injected blood intracranially into mice, thereby producing the characteristic myocardial lesions, which could be ameliorated but not prevented by pretreatment with adrenalectomy.16 This finding indicated that humorally delivered catecholamines had some role in the genesis of neurocardiac necrosis.
Jacob and colleagues produced subarachnoid hemorrhage experimentally in dogs and carefully studied the sequential hemodynamic and ultrastructural changes that occurred.17 The hemodynamic changes occurred in four stages, directly corresponding to the effects seen with intravenous noradrenaline injections: hypertension, tachycardia, rise in left ventricular pressure, and increased coronary blood flow. Ultrastructurally, a series of three stereotyped events, which could be imitated exactly with noradrenaline injections, occurred: migration of calcium-containing granules to the periphery of mitochondria, disappearance of these granules, and myofilament disintegration at the I bands. Partially successful efforts to modify the developments of neurocardiac lesions were made by McNair and coworkers, using reserpine pretreatment in mice subjected to simulated intracranial hemorrhage,18 and by Hunt and Gore, who pre-treated a group of rats with propranolol and then attempted to produce cardiac lesions with intracranial blood injections.19
The phenomena of the various types of myocardial cell death were clarified by Baroldi,20 who described three patterns: coagulation necrosis, the fundamental lesion of infarction; colliquative myocytolysis, the fundamental lesion of low output syndromes; and coagulative myocytolysis (now known as contraction band necrosis), the fundamental lesion of catecholamine-induced necrosis.
Contraction band necrosis may be seen in reperfused areas around regions of coagulation necrosis, in sudden unexpected and accidental death, and in hearts exposed to high levels of catecholamines, as in people with pheochromocytoma. This is probably the major lesion described by Selye as electrolyte-steroid cardiopathy with necrosis8 and is clearly the lesion seen in animals and people suffering acute neurological or psychiatric catastrophes (Fig. 113-2).
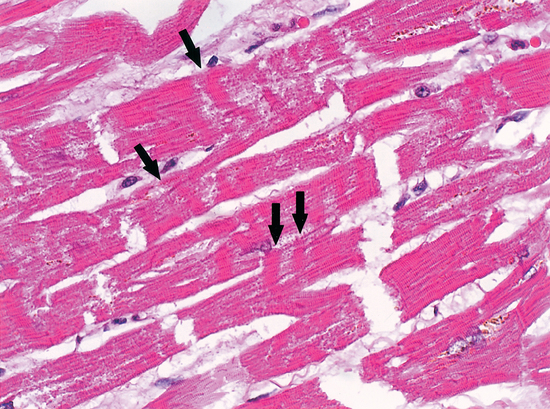
It is likely that the subcellular mechanisms underlying the development of contraction band necrosis involve calcium entry. Zimmerman and Hulsmann reported that the perfusion of rat hearts with calcium-free media for short periods creates a situation such that upon readmission of calcium, there is a massive contracture, followed by necrosis and enzyme release.21 This phenomenon, known as the calcium paradox, can be imitated almost exactly with reoxygenation after hypoxemia and reperfusion after ischemia. The latter, called the oxygen paradox, has been linked to the calcium paradox by pathological calcium entry.22 This major ionic shift is probably the cause of the dramatic changes seen on ECG in the context of neurological catastrophe, a fact that could explain the phenomenon of sudden unexpected death in many contexts: for example, sudden death in middle-aged men; sudden infant death syndrome; sudden unexpected nocturnal death syndrome; frightened to death (“voodoo” death); sudden death in epilepsy; sudden death during natural catastrophe; sudden death associated with drug abuse; sudden death in wild and domestic animals; sudden death during asthma attacks; sudden death during the alcohol withdrawal syndrome; sudden death during grief after a major loss; sudden death during panic attacks; sudden death from mental stress; and sudden death during war. The connection between the nervous system and the cardiopulmonary system provides the unifying link that allows a coherent explanation for most, if not all, of the forms of sudden unexpected death. Powerful evidence from multiple disparate disciplines allows for a neurological explanation of sudden unexpected death.23
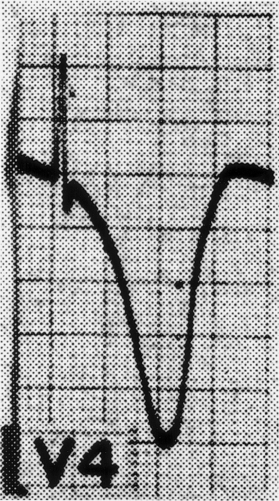
A wide variety of changes in the ECG is seen in the context of neurological disease. Two major categories of change are arrhythmias and repolarization changes. It is likely that the increased tendency for life-threatening arrhythmias found in patients with acute neurological disease arises from the repolarization change, which increases the vulnerable period during which an extrasystole would be likely to result in ventricular tachycardia and/or ventricular fibrillation. Thus, the essential and potentially most lethal electrocardiographic features, which are known to change in the context of neurological disease, are changes in the ST segment and T wave, reflecting abnormalities in repolarization. Most often, the changes are observed best in the anterolateral or inferolateral leads (Fig. 113-3). The electrocardiographic abnormalities usually improve, often dramatically, with brain death.
Josué first showed in 1907 that adrenaline infusions could cause cardiac hypertrophy.24 This observation has been reproduced on many occasions, which documents the fact that systemically administered catecholamines are associated not only with electrocardiographic changes reminiscent of widespread ischemia but also with a characteristic pathological picture in the cardiac muscle (contraction band necrosis) that is distinct from myocardial infarction. An identical situation may be found in humans with chronically elevated catecholamines, as with pheochromocytoma.
An identical cardiac lesion can be produced in various models of stress. A few autopsy studies on patients who experienced sudden death have shown myofibrillar degeneration. Cebelin and Hirsch reported a careful retrospective analysis of the hearts of 15 victims of physical assault who died as a direct result of the assault but without sustaining internal injuries.25 In 11 of the 15 individuals, contraction band necrosis was present. Wittstein and colleagues reported a stunned myocardium in people who suffered a severely stressful event.26 This is the human stress cardiomyopathy. As the contraction band necrosis is predominantly subendocardial, it may involve the cardiac conducting system, thus predisposing to cardiac arrhythmias. This lesion, combined with the propensity of catecholamines to produce arrhythmias even in a normal heart, may well raise the risk of a serious arrhythmia. This may be the major immediate mechanism of sudden death in many neurological circumstances, such as subarachnoid hemorrhage, stroke, epilepsy, head trauma, psychological stress, and increased intracranial pressure. Stress-induced myocardial lesions may be prevented by sympathetic blockade with many different classes of antiadrenergic agents (e.g., ganglionic blockers, catecholamine-depleting agents such as reserpine, and β-blockers). This suggests that catecholamines, either released directly into the heart by sympathetic nerve terminals or reaching the heart through the bloodstream after release from the adrenal medulla, may be excitotoxic to myocardial cells.
Nervous system stimulation also produces contraction band necrosis. Stimulation of the lateral hypothalamus produces hypertension and electrocardiographic changes reminiscent of those seen in patients with central nervous system disorders. Furthermore, this effect on the blood pressure and ECG can be completely prevented by C2 spinal section and stellate ganglionectomy, but not by vagotomy, which suggests that the mechanism of the electrocardiographic changes is sympathetic rather than parasympathetic or humoral. Prolonged bilateral hypothalamic stimulation produces contraction band necrosis indistinguishable from that produced by catecholamines and stress. Stimulation of the left insula (as occurs in some seizures) or ablation of the right (as occurs with some strokes) may produce neurogenic electrocardiographic changes and contraction band necrosis. Other methods of producing cardiac lesions of this type include stimulation of the limbic cortex, the mesencephalic reticular formation, the stellate ganglion, and regions known to elicit cardiac reflexes such as the aortic arch. High levels of circulating catecholamines exaggerate the electrocardiographic findings and myocardial lesions, but high circulating catecholamine levels are not required for the production of pathological changes.
Contraction band necrosis also occurs with cardiac reperfusion, as is commonly seen in patients who die after a period of time on a left ventricular assist pump for cardiac surgery. Similar lesions are seen in hearts that were reperfused by angioplasty or fibrinolytic therapy. The mechanism whereby reperfusion of ischemic cardiac muscle produces contraction band necrosis involves entry of calcium after a period of relative deprivation.27 Sudden calcium influx by one of several possible mechanisms (e.g., a period of calcium deficiency with loss of intracellular calcium; a period of anoxia followed by reoxygenation of the electron transport system; a period of ischemia followed by reperfusion, or opening of the receptor-operated calcium channels by excessive amounts of locally released noradrenaline) may be the final common pathway whereby the irreversible contractures occur, leading to contraction band necrosis. Thus, reperfusion-induced myocardial cell death may be a form of apoptosis (programmed cell death) analogous to that seen in the central nervous system wherein excitotoxicity with glutamate results in a similar, if not identical, series of events.28
The precise cellular mechanism for the electrocardiographic change and the histological lesion may well reflect the effects of large volumes of norepinephrine released into the myocardium from sympathetic nerve terminals.29 The fact that the cardiac necrosis is greatest near the nerve terminals in the endocardium and is progressively less severe closer to the epicardium provides further evidence that catecholamine toxicity produces the lesion. This locally released norepinephrine is known to stimulate synthesis of adenosine 3′,5′-cyclic phosphate, which in turn results in the opening of the calcium channel with influx of calcium and efflux of potassium. The actin and myosin filaments interact under the influence of calcium but do not relax unless the calcium channel closes. Continuously high levels of norepinephrine in the region may result in failure of the calcium channel to close, which leads to cell death and, finally, to leakage of enzymes (troponin, creatine kinase) out of the myocardial cell. Free radicals released as a result of reperfusion after ischemia or by the metabolism of catecholamines to the known toxic metabolite adrenochrome may contribute to cell membrane destruction, leading to leakage of cardiac enzymes into the blood.30,31 Thus, the cardiac toxicity of locally released norepinephrine represents a continuum ranging from a brief reversible burst of abnormalities on ECG to an irreversible failure of the muscle cell with enzyme leak and permanent repolarization abnormalities.
Histological changes would also represent a continuum ranging from complete reversibility in a normal heart through mild changes seen best with electron microscopy to severe myocardial cell necrosis with mononuclear cell infiltration and even hemorrhages. The level of cardiac enzymes released and the electrocardiographic changes would be correlated approximately with the severity and extent of the pathological process. In the most severe circumstance, neurogenic myocardial dysfunction may be sufficiently severe to substantially reduce cardiac output, producing acute heart failure with or without chest pain. The pattern of cardiac dysfunction corresponds to the density of catecholamines, producing the characteristic finding of left ventricular apical ballooning, which on echocardiogram or ventriculogram resembles the Japanese octopus trapping pot, the takotsubo, leading to the term takotsubo-like cardiomyopathy (broken heart syndrome).32,33
Armour JA, Ardell JL. Neurocardiology. Oxford, UK: Oxford University Press, 1994.
Brillman J. Neurocardiology in Neurologic Clinics, vol 11, no. 2. Philadelphia: WB Saunders, 1993.
Johnson RH, Lambie DG, Spalding JMK. Neurocardiology. Philadelphia: WB Saunders, 1984.
Kulbertus HE, Franck G. Neurocardiology. Mt. Kisco, NY: Futura, 1988.
Meerson FZ. Adaptation, Stress and Prophylaxis. Berlin: Springer-Verlag, 1984.
Samuels MA. “Voodoo” death revisited: the modern lessons of neurocardiology. Neurologist. 1997;3:293-304.
1 Cannon WB. “Voodoo” death. Am Anthropol. 1942;44:169-181.
2 Engel G. Sudden and rapid death during psychological stress. Ann Intern Med. 1971;74:771-782.
3 Richter CP. On the phenomenon of sudden death in animals and man. Psychosom Med. 1957;19:191-198.
4 Byer E, Ashman R, Toth LA. Electrocardiogram with large upright T wave and long Q-T intervals. Am Heart J. 1947;33:796-801.
5 Levine HD. Non-specificity of the electrocardiogram associated with coronary heart disease. Am J Med. 1953;15:344-350.
6 Burch GE, Myers R, Adildskov JA. A new electrocardiographic pattern observed in cerebrovascular accidents. Circulation. 1954;9:719-726.
7 Cropp CF, Manning GW. Electrocardiographic change simulating myocardial ischaemia and infarction associated with spontaneous intracranial haemorrhage. Circulation. 1960;22:24-27.
8 Selye H. The Chemical Prevention of Cardiac Necrosis. New York: Ronald Press, 1958.
9 Raab W, Stark E, MacMillan WH, et al. Sympathogenic origin and antiadrenergic prevention of stress-induced myocardial lesions. Am J Cardiol 1961;. 1958;8:203-211.
10 Barger AC, Herd JA, Liebowitz MR. Chronic catheterization of coronary artery induction of ECG pattern of myocardial ischaemia by intracoronary epinephrine. Proc Soc Exp Biol Med. 1961;107:474-477.
11 Melville KI, Blum B, Shister HE, et al. Cardiac ischemic changes and arrhythmias induced by hypothalamic stimulation. Am J Cardiol. 1963;12:781-791.
12 Oppenheimer SM, Cechetto DF. Cardiac chronotropic organization of the rat insular cortex. Brain Res. 1990;533:66-72.
13 Koskelo P, Punsar SO, Sipila W. Subendocardial haemorrhage and ECG changes in intracranial bleeding. BMJ. 1964;1:1479-1483.
14 Connor RCR. Myocardial damage secondary to brain lesions. Am Heart J. 1969;78:145-148.
15 Greenshoot JH, Reichenbach DD. Cardiac injury and subarachnoid haemorrhage. J Neurosurg. 1969;30:521-531.
16 Hawkins WE, Clower BR. Myocardial damage after head trauma and simulated intracranial haemorrhage in mice: the role of the autonomic nervous system. Cardiovasc Res. 1971;5:524-529.
17 Jacob WA, Van Bogaert A, DeGroot-Lasseel MHA. Myocardial ultrastructural and haemodynamic reactions during experimental subarachnoid haemorrhage. J Mol Cell Cardiol. 1972;4:287-298.
18 McNair JL, Clower BR, Sanford RA. The effect of reserpine pretreatment on myocardial damage associated with stimulated intracranial haemorrhage in mice. Eur J Pharmacol. 1970;9:1-6.
19 Hunt D, Gore I. Myocardial lesions following experimental intracranial hemorrhage: prevention with propranolol. Am Heart J. 1972;83:232-236.
20 Baroldi F. Different morphological types of myocardial cell death in man. Fleckstein A, Rona G, editors. Recent Advances in Studies in Cardiac Structure and Metabolism: Pathophysiology and Morphology of Myocardial Cell Alteration. vol 6. Baltimore: University Park Press; 1975:385-397.
21 Zimmerman ANA, Hulsmann WC. Paradoxical influence of calcium ions on the permeability of the cell membranes of the isolated rat heart. Nature. 1966;211:616-647.
22 Hearse DJ, Humphrey SM, Bullock GR. The oxygen paradox and the calcium paradox: two facets of the same problem? J Mol Cell Cardiol. 1978;10:641-668.
23 Samuels MA. Neurally induced cardiac damage. Neurol Clin. 1993;11:273-292.
24 Josué O. Hypertrophie cardiaque causee par l’adrenaline et la toxine typhique. C R Soc Biol (Paris). 1907;63:285-287.
25 Cebelin M, Hirsch CS. Human stress cardiomyopathy. Hum Pathol. 1980;11:123-132.
26 Wittstein IS, Thiemann DR, Lima JA, et al. Neurohumoral features of myocardial stunning due to sudden emotional stress. N Engl J Med. 2005;352:539-548.
27 Braunwald E, Kloner RA. Myocardial reperfusion: a double-edged sword? J Clin Invest. 1985;76:1713-1719.
28 Gottlieb R, Burleson KO, Kloner RA, et al. Reperfusion injury induces apoptosis in rabbit cardiomyocytes. J Clin Invest. 1994;94:1621-1628.
29 Eliot RS, Todd GL, Pieper GM, et al. Pathophysiology of catecholamine-mediated myocardial damage. J S Carolina Med Assoc. 1979;75:513-518.
30 Singal PK, Kapur N, Dhillon KS, et al. Role of free radicals in catecholamine-induced cardiomyopathy. Can J Physiol Pharmacol. 1982;60:1390-1397.
31 Meerson FZ. Pathogenesis and prophylaxis of cardiac lesions in stress. Adv Myocardiol. 1983;4:3-21.
32 Kawai S, Suzuki H, Yamaguchi H, et al. Ampulla cardiomypathy (“Takotsubo” cardiomyopathy)—reversible left ventricular dysfunction. Jpn Circ J. 2000;64:156-159.
33 Gianni M, Dentali F, Grandi AM, et al. Apical ballooning syndrome or takotsubo cardiomyopathy: a systematic review. Eur Heart J. 2006;27:1523-1529.
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]
CHAPTER 113 NEUROLOGY OF CARDIOLOGY
The neurology of cardiology may be divided into three categories: (1) the cardiac complications of neurological disease; (2) the neurological complications of cardiac disease (e.g., cardiac-source embolic stroke, neurological complications of cardiac surgery); and (3) neurocardiac syndromes (mitochondrial diseases; Friedreich’s ataxia; muscular dystrophies affecting the heart). The latter two categories are covered in other chapters of this book. This chapter deals with the cardiac manifestations of neuropsychiatric illnesses.
In 1942, Walter Cannon reported several incidents of “voodoo” death,1 by which he meant death from fright. These events had several features in common. They were all induced by an absolute belief that an external force could cause demise and that the victim had no power to alter this course. Cannon postulated that death was caused by intense action of the sympathetic adrenal system. Evidence has accumulated to support the concept that voodoo death is a real phenomenon and is not limited to ancient peoples. Rather, it may be a basic biological principle that provides an important clue to understanding the phenomenon of sudden death in modern society, as well as opening a window into the world of neurovisceral disease. George Engel collected 160 accounts from the lay press of sudden death that was attributed to disruptive life events2 and concluded that such events could be divided into eight categories (Fig. 113-1), the common feature of which is life-threatening stress without escape or control.
Richter reported a series of experiments aimed at elucidating the mechanism of “voodoo” death.3 He found that domesticated rats could swim for about an hour in 93°F water, but if the animals’ whiskers were trimmed, they would invariably drown within a few minutes. When similar experiments were performed with wild rats, restraint contributed significantly to the tendency for demise, whereas, in the case of the calm, domesticated animals in which restraint and confinement were apparently not significant stressors, shaving the whiskers rendered these animals as fearful as wild rats, with a corresponding tendency for sudden death. Adrenalectomy did not protect the animals.
In humans, one of the easily accessible windows into autonomic activity is the electrocardiogram (ECG). Byer and associates reported six patients with neurological disease in whom ECGs showed large upright T waves and long Q-T intervals.4 On the basis of experimental results of cooling or warming the endocardial surface of the dog’s left ventricle, they concluded that these electrocardiographic changes resulted from subendocardial ischemia. Levine reported a patient with a subarachnoid hemorrhage who had electrocardiographic changes reminiscent of coronary disease.5 Burch and colleagues reported 17 patients with various types of stroke who had long Q-T intervals, large and usually inverted T waves, and frequent U waves.6 Cropp and Manning reported the abnormalities on ECGs in 29 patients with subarachnoid hemorrhage; in five of these patients, autopsy study verified the absence of coronary artery disease and myocardial infarction, which suggested that the abnormalities on ECGs were neurogenic.7
Selye described electrolyte-steroid cardiopathy with necrosis and argued that this lesion was distinct from the coagulation necrosis that occurred as a result of ischemic disease.8 Certain steroids and other hormones created a predisposition for the development of electrolyte-steroid cardiopathy with necrosis, but other factors were necessary for its development, including stress. Raab and associates found that cardiac lesions may be produced in rats by pretreatment with fluorocortisol, calciferol, or thyroxine, followed by restraint or cold stress, and that pharmacological blockade of sympathetic activity was cardioprotective.9 Intracoronary infusions of adrenaline reproduce the characteristic electrocardiographic pattern of neurocardiac disease, which is reminiscent of subendocardial ischemia, although no ischemic lesion could be found in the hearts of dogs sacrificed after several months of infusions.10
Melville and coworkers produced changes on ECG and myocardial necrosis by stimulating the hypothalamus of cats; autopsy studies revealed evidence of open coronary arteries.11 The cardiac lesion was characterized by intense cytoplasmic eosinophilia with loss of cross-striations and some hemorrhage, a condition now most commonly known as contraction band necrosis. Oppenheimer and Cechetto mapped the chronotropic organizational structure in the rat insular cortex, demonstrating that sympathetic innervation arises from a more rostral part of the posterior insula than does parasympathetic innervation.12
Despite the fact that myocardial damage had been produced in animals, it was not until Koskelo and associates reported on three patients with electrocardiographic changes caused by subarachnoid hemorrhage13 that contraction band necrosis was demonstrated in humans with neurological disease. Connor reported focal myocytolysis in 8% of 231 autopsy studies; the incidence was highest in patients who had died of intracranial hemorrhages.14 Connor pointed out that prior pathological reports probably overlooked the lesion because of the fact that it was multifocal, each individual focus being quite small, necessitating extensive tissue sampling. It is clear now that even Connor underestimated the prevalence of the lesion and that serial sections are required to rigorously rule out its presence. Greenshoot and Reichenbach reported nine patients with subarachnoid hemorrhage, all of whom had cardiac lesions, and demonstrated that the cardiac pathology could be reproduced in cats given mesencephalic reticular formation stimulation.15 Adrenalectomy did not protect the hearts. This finding supported the contention that the electrocardiographic changes and cardiac lesions result from direct intracardiac release of catecholamines. Hawkins and Clower injected blood intracranially into mice, thereby producing the characteristic myocardial lesions, which could be ameliorated but not prevented by pretreatment with adrenalectomy.16 This finding indicated that humorally delivered catecholamines had some role in the genesis of neurocardiac necrosis.
Jacob and colleagues produced subarachnoid hemorrhage experimentally in dogs and carefully studied the sequential hemodynamic and ultrastructural changes that occurred.17 The hemodynamic changes occurred in four stages, directly corresponding to the effects seen with intravenous noradrenaline injections: hypertension, tachycardia, rise in left ventricular pressure, and increased coronary blood flow. Ultrastructurally, a series of three stereotyped events, which could be imitated exactly with noradrenaline injections, occurred: migration of calcium-containing granules to the periphery of mitochondria, disappearance of these granules, and myofilament disintegration at the I bands. Partially successful efforts to modify the developments of neurocardiac lesions were made by McNair and coworkers, using reserpine pretreatment in mice subjected to simulated intracranial hemorrhage,18 and by Hunt and Gore, who pre-treated a group of rats with propranolol and then attempted to produce cardiac lesions with intracranial blood injections.19
The phenomena of the various types of myocardial cell death were clarified by Baroldi,20 who described three patterns: coagulation necrosis, the fundamental lesion of infarction; colliquative myocytolysis, the fundamental lesion of low output syndromes; and coagulative myocytolysis (now known as contraction band necrosis), the fundamental lesion of catecholamine-induced necrosis.
Contraction band necrosis may be seen in reperfused areas around regions of coagulation necrosis, in sudden unexpected and accidental death, and in hearts exposed to high levels of catecholamines, as in people with pheochromocytoma. This is probably the major lesion described by Selye as electrolyte-steroid cardiopathy with necrosis8 and is clearly the lesion seen in animals and people suffering acute neurological or psychiatric catastrophes (Fig. 113-2).
[/not-level-membership-for-neurology-category]
