[level-membership-for-pediatrics-category]
Chapter 13 Neurology
Long Cases
Cerebral palsy
Children with cerebral palsy (CP) often are used for both long- and short-case examinations. The following is a very brief listing of some of the major issues raised in the CP long case, plus a short-case approach.
Background information
Classification
CP is classified according to the clinical type of neuromotor dysfunction:
Causes
1. Prenatal problems; for example, cerebral malformations, intrauterine TORCH infection (toxoplasma, other [e.g. syphilis], rubella, CMV, herpes [simplex or varicella] or HIV infection), toxins (e.g. drugs), placental insufficiency, fetal coagulation and autoimmune disorders, cerebrovascular accidents, trauma or chromosomal anomalies.
2. Perinatal problems; for example, neonatal encephalopathy, hypoglycaemia, toxins (e.g. kernicterus from hyperbilirubinaemia), viral (e.g. HSV) encephalitis, bacterial (e.g. early onset Group B streptococcus) meningitis, prematurity (babies born before 28 weeks gestation have 50 times the risk of CP compared with babies born at term; mechanisms can include intraventricular haemorrhage and periventricular leucomalacia).
3. Postnatal problems; for example, head trauma (accidental or non-accidental), hypoxic insult (from near-drowning, choking or poisoning), cerebrovascular accident, toxic encephalopathy (e.g. lead), meningitis or hypoglycaemia.
Diagnostic assessment
EEGs are only worthwhile if the patient has had fits, to determine classification of any epilepsy syndrome; they are not useful in determining the aetiology of the child’s CP.
Metabolic conditions. Treatable conditions should not be missed. Example: glutaric acidaemia type 1; mutation in the gene encoding glutaryl-CoA dehydrogenase [GCDH]; alters catabolism of amino acids lysine, hydroxylysine and tryptophan; can impersonate dyskinetic CP; treatable—low-protein (low lysine and tryptophan) diet (avoiding milk, cheese, dairy, meat, poultry, fish, dried beans and legumes, nuts, peanut butter), special low-protein formula, supplements: riboflavin and L-carnitine.
Other metabolic conditions are not treatable but have a different prognosis to CP. First example: Lesch–Nyhan syndrome (X-linked deficiency of hypoxanthine guanine phosphoribosyl-transferase, HGPRT); patients eventually self-mutilate, and diagnosis is made. Second example: Sjogren–Larsson syndrome (mutation in gene coding for fatty aldehyde dehydrogenase); patients eventually develop ichthyosis, and diagnosis is made.
Muscular dystrophies (e.g. Becker muscular dystrophy; eventually proximal myopathy and Gowers sign, and calf hypertrophy).
Mitochondrial disorders (e.g. Leigh syndrome; eventually developmental regression becomes apparent).
Malformation syndromes. Example 1: Miller–Dieker lissencephaly syndrome (MDLS), chromosome 17p13.3; subtle dysmorphic findings (e.g. wrinkled skin over glabella, downward-slanting palpebral fissures) are eventually noticed. Example 2: Rett syndrome; eventually acquired microcephaly, hand-wringing and hyperventilation become apparent.
Level I: Ambulatory in all settings
Level II: Walks without aides, but has limitations in community settings
Most children with CP are born with the condition, but abnormal features may not be noted for months; most are diagnosed by 3 years. Diagnosis of CP before 12 months is fraught with uncertainty, due to the plasticity of the newborn brain; infants can be seen who have apparently clear-cut signs of CP that then resolve by the age of 1 or 2 years. The ‘ideal’ diagnostic age for CP may be 2 years.
History
Current symptoms/functioning
1. Intellectual abilities (or present developmental status), current placement regarding education, domestic situation, employment.
2. Behaviour (e.g. hyperactivity), affect (e.g. depression).
3. Vision (e.g. cortical visual impairment, strabismus, myopia, hemianopia).
4. Speech, hearing and communication problems (e.g. expressive or receptive dysphasia, dysarthria, athetoid movements, use of aids such as communication boards and computers).
5. Activities of daily living (e.g. bathing, cleaning teeth, combing hair, dressing, writing and other hand usage, toileting, menses).
6. Feeding and nutrition (e.g. sucking and swallowing ability, tube feeds, gastrostomy, gastro-oesophageal reflux, aspiration, failure to thrive).
7. Seizures (e.g. type, duration, frequency, usual treatment including side effects, compliance and drug levels, last seizure).
8. Mobility (e.g. walking ability [‘community’, ‘household’ or ‘non-functional’ ambulator], gait pattern, wheelchair mobility), skeletal problems (e.g. kyphoscoliosis, lumbar lordosis, spondylolisthesis, hip subluxation and dislocation, pseudoacetabulum formation, contractures, unequal leg length), abnormal posturing.
9. Other problems: urinary incontinence, constipation, management of menses, chest infections, pressure sores.
Birth history
1. Maternal past history of miscarriages or infertility.
2. Pregnancy: hyperemesis, hypertensive disease of pregnancy, teratogenic medications, placental problems, clinical evidence of, or exposure to, infection (e.g. TORCH), quality of fetal movements, gestational age.
3. Delivery: presentation (e.g. breech, face), instrumental delivery, Apgar score, resuscitation required, birth weight, need for oxygen, nursery care.
4. Neonatal period: respiratory distress, feeding difficulties, seizures, hyperbilirubinaemia (phototherapy or exchange transfusion), intraventricular haemorrhage (IVH), periventricular leukomalacia (PVL), hydrocephalus, retinopathy of prematurity (ROP).
Important signs in examination of the child with CP
See the short case on hemiplegia in this chapter for a suggested examination procedure.
General observations
1. Dysmorphic features (e.g. chromosomal anomalies).
2. Parameters: head circumference (often obvious microcephaly), weight (often failing to thrive), height (usually decreased), progressive percentile charts.
3. Posture (e.g. fisting, increased extensor tone, asymmetric tonic neck reflex [ATNR], hemiplegic, quadriplegic).
4. Movement: (a) involuntary (e.g. choreoathetoid movements, dystonic spasms, seizures); (b) voluntary (e.g. immature gait pattern with wide base, up on toes, arms out for balance; hemiplegic, diplegic gaits; note posturing of arms when walking).
5. Asymmetry (e.g. hemiatrophy: look at the size of the thumbnails and the great toenails for subtle clues to asymmetry).
6. Behaviour (e.g. lack of interaction with environment, crying).
7. Eye signs (e.g. squint, nystagmus).
8. Bulbar signs (e.g. dysarthria, drooling).
9. Interventions (e.g. nasogastric tube, gastrostomy tube, scars of orthopaedic procedures).
Demonstration of signs of CP
1. If possible, perform a standard gait examination.
2. If the child cannot walk, but can crawl, look for abnormal crawling: (a) those with spastic diplegia or quadriplegia—buttock crawling and ‘bunny-hopping’ (jumping while on knees); (b) those with hemiplegia—asymmetrical crawl.
3. Gross motor ‘180° manoeuvre’, incorporating primitive reflexes:
4. Inspect carefully for tendon release scars.
5. Palpate muscle bulk in each muscle group.
6. Tone: as above, plus assessment of upper and lower limbs, and evaluation of contractures (e.g. tight hip adductors, and tendo achilles).
7. Power: voluntary movement, functional power (grasp of toys, cloth cover test).
8. Reflexes: the head should be held in the midline (e.g. by an examiner) so that an ATNR does not give a false impression of unilateral hypertonia; note whether there is any crossed adductor reflex, spread of reflexes, clonus or upgoing plantar responses.
Complications of CP
1. Measure the head (for microcephaly, or macrocephaly due to hydrocephalus).
2. Check the vision, visual fields and extraocular movements (for myopia, squint).
3. Check the hearing (for sensorineural deafness).
4. Check the ears (for chronic serous otitis media).
5. Ask to check the gag reflex (bulbar dysfunction).
6. Look at the teeth (for dental caries).
7. Look at the back (for kyphoscoliosis).
8. Inspect and auscultate the chest (for chest infection).
9. Palpate the abdomen (for constipation).
10. Examine the hips (for dislocation).
11. Screen nutritional status (demonstrate fat and protein stores).
12. Perform a functional assessment for activities of daily living (e.g. offer cup, spoon, fork, knife, comb, toothbrush; ask the child to put on a piece of clothing).
Investigations
1. Brain imaging (MRI) may show abnormalities in some 90% of patients with CP. It may show the basis of CP (e.g. gross malformations, hydrocephalus, intracranial calcification from congenital infection). It may suggest the timing of the aetiology (e.g. cortical dysplasias develop around 12–20 weeks’ gestation; periventricular leukomalacia around 28–34 weeks’ gestation; cortical and subcortical gliosis and atrophy in parasagittal watershed areas in term babies with intrapartum hypoxia) or show unexpected degenerative disorders (e.g. one of the leukodystrophies).
2. TORCH screen (including HIV), in infants, for intrauterine infection.
3. Urinary metabolic screen (various inborn errors of metabolism).
4. Chromosomes (various anomalies).
5. Lumbar puncture in dyskinetic CP, to diagnose the treatable genetic condition, glucose transporter 1 deficiency syndrome, especially if there are associated refractory seizures (see below).
Management
Physiotherapy, occupational therapy, splints/orthoses
Serial casting is useful for reversing ankle/foot equinus in younger children. Over 2–6 weeks, the calf muscle is stretched gradually, with the foot and ankle held in position by a below-the-knee plaster.
Management of spasticity
In 2010, the American Academy of Neurology released an evidence-based review of pharmacological treatments for childhood spasticity due to CP, where a multidisciplinary panel reviewed relevant literature from 1966 to 2008. In the case of localised/segmental spasticity that warrants treatment, the AAN recommendation was that botulinum toxin A (BTX-A) is effective and generally safe in reducing spasticity in the upper and lower extremities, but there is conflicting evidence regarding functional improvement, and severe generalised weakness can occur; there is insufficient data to support or refute BTX-A use to improve motor function. There is insufficient data to support or refute other medications used for the same indication (and these are not discussed further): phenol, alcohol and BTX type B. In the case of generalised spasticity that warrants treatment, the AAN recommendation was that diazepam could be considered for short-term treatment, and also tizanidine may be considered; but, again, there is insufficient data to support or refute other medications used for the same indication: dantrolene, oral baclofen or continuous intrathecal baclofen. The latter is discussed because patients who have received this treatment may present in the examination, so a candidate should have some knowledge of the side effects.
Intrathecal baclofen (IT-BLF)
In view of the lack of data to support this treatment, it may well become of historical interest only. There are many and varied side effects, several related to the mode of delivery: an implantable drug-delivery pump system for continuous infusion. Significant complications include central side effects such as apnoea, respiratory depression, bradycardia, hypotension and sedation, mechanical complications including pump or side-port failure, catheter kinks, extrusions or dislodgement, cerebrospinal fluid fistula, local infection and meningitis; also, rapidly progressive scoliosis has been reported.
Orthopaedic procedures
Surgical procedures for spastic hip displacement
• Preventative (soft-tissue surgery): adductor longus release, gracilis release, adductor brevis release, iliopsoas lengthening, obturator neurectomy (anterior branch only).
• Reconstructive (redirectional osteotomies): femoral varus derotation osteotomy, pelvic osteotomy, combined femoral and pelvic osteotomy with or without open reduction.
• Salvage: excision of proximal femur, valgus osteotomy, interpositional arthroplasty, replacement arthroplasty, arthrodesis.
These procedures lead to significant improvements in function, including improved walking speed.
Other orthopaedic procedures
Upper limb surgery may be offered to correct a flexion–pronation deformity of the wrist, to achieve a better functional position; however, usually only cosmetic improvement is achieved, with little gain functionally, largely because of associated cortical sensory loss. Some children will require surgery for scoliosis (e.g. placing of Luque rods).
Excessive salivation (sialorrhoea)
Anticholinergic drugs (e.g. glycopyrrolate) have been used for excessive drooling with some success, but can have unpleasant side effects, such as urinary retention, constipation, blurred vision, headache, drowsiness, dizziness and behaviour changes.
Cognition/learning and communication
The risk of associated cognitive and learning difficulties varies with the type of CP. Those with quadriplegic CP have the highest risk of cognitive impairment and those with hemiplegic CP have the lowest risk. In children with movement impairment hindering their verbal communication, augmentative and alternative communication systems are available. Aided formal communication systems include communication boards and books, where pictures and symbols are used to communicate specific messages, and electronic devices of varying complexity, some allowing the person to communicate with recorded speech; unaided formal communication systems include key word signing (Makaton vocabulary), and informal communication systems include facial expression, eye contact, vocalisations, body language and gestures, to convey feelings, wants and needs.
Pain
This is a common complaint, which can be very challenging in the non-verbal child with severe CP. The most common sites of pain seem to be gastrointestinal (constipation, GORD), and orthopaedic/musculoskeletal (scoliosis, hip dislocation, patella alta); other possibilities include: neuromuscular—muscle spasms; head and neck—migraine, raised intracranial pressure, glaucoma, corneal abrasions, tooth abscess, temporomandibular joint pain; urological—urolithiasis, bladder spasm; skin—boils, decubitus ulcers. A thorough history (e.g. temporal association with feeding, or nappy changes) and thorough general examination (e.g. inspecting the mouth with a torch and spatula to find tooth abscesses) may find the cause, but often empirical interventions are tried before the patient is eventually free of discomfort.
Prognosis
A few ‘rules of thumb’ are worth knowing when parents ask about prognosis; most parents ask whether their child will walk. While unlikely to be an active discussion issue with a patient in a long case, examiners may ask hypothetical questions such as how to approach answering the parents’ concerns when the concept of permanent neurological damage has been mentioned. Note the following: children with hemiplegic or diplegic CP usually walk, those with quadriplegic CP rarely walk; those with the dyskinetic subtype are more difficult to predict. Most children who can sit independently by 2 years will walk, but most children who cannot sit by 4 years are unlikely to walk. In terms of life span, only the most profound degrees of CP are associated with a decreased life expectancy. If a child cannot lift the head to prone, and requires tube feeding, the median survival is around 17 years.
Dystrophinopathies: Duchenne muscular dystrophy (DMD)
Background information: genetics of DMD
DMD is an X-linked recessive disorder, due to mutations (often deletions) in the dystrophin gene on the X chromosome (Xp21.2). It has a frequency of 1 in 3600–6000 male births. The DMD gene is the largest known human gene. It is around 2000 kilobases in size and codes for dystrophin, a large, rod-like 427-kD protein containing 3685 amino acids and located at the inner face of the muscle cell membrane. Dystrophin binds with a group of ‘dystrophin-associated proteins’ (DAPs) (e.g. sarcoglycans, dystroglycans, merosin) that span the muscle membrane, linking the muscle cytoskeleton and the extracellular matrix (see Figure 13.1). Absence of dystrophin disrupts the link, making the muscle membrane susceptible to damage from shearing stresses. Hence, dystrophin-deficient muscle is very susceptible to muscle injury, and degeneration of muscle fibres is a feature of dystrophic muscle. Dystrophin is undetectable in the muscle of DMD patients. Virtually all males with DMD have identifiable mutations. Over 4700 mutations in the DMD gene have been identified. Disease-causing alleles can include deletion of the complete gene, deletion or duplication of exons, small deletions, insertions and single base changes. Around two thirds of patients with DMD have intragenic out-of-frame (gross rearrangements) deletions, and around 10% have duplications of one or more exons of the gene; the remaining 25% have point mutations or other small rearrangements, including intronic deletions, insertions of repetitive sequences and splice site mutations. Generally out-of-frame mutations cause lack of dystrophin (and DMD), whereas in-frame mutations cause abnormal but partially functional dystrophin, resulting in BMD. The tissue distribution of dystrophin correlates with clinical features. It is found in skeletal, cardiac and smooth muscle, and results in skeletal muscle weakness and cardiomyopathy. In DMD-associated DCM, dystrophin expression is abnormal in myocardium, but may be normal or mildly abnormal in skeletal muscle. Dystrophin is found within the central nervous system, resulting in a static encephalopathy and cognitive deficits. Various forms of dystrophin are expressed in neurons and glia in the brain, especially the cortex, hippocampus, cerebellum and retina. Molecular genetic testing of DMD can confirm diagnosis of a dystrophinopathy without need for muscle biopsy in most patients ith DMD or BMD. There is a high incidence of new mutations, and two thirds of new patients have no positive family history.
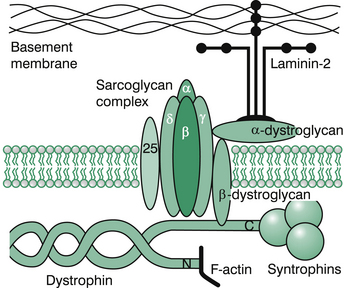
Figure 13.1 Subsarcolemmal cytoskeleton.
Redrawn from; South, Isaacs, Roberton 2007. Practical Paediatrics 6th edition, p. 609, Figure 17.3.4.
Molecular tests for DMD
1. Multiplex PCR, Southern blotting and FISH are utilised to detect deletions, which account for two thirds of mutations in DMD.
2. Southern blotting and quantitative PCR analysis are utilised to detect duplications, which account for 6–10% mutations in males with DMD.
3. Multiple ligation probe amplification (MLPA) is utilised to analyse deletion/duplication of the gene in probands and carrier females.
4. Mutation scanning or sequence analysis is utilised to detect small deletions or insertions, single base changes, and splicing mutations that make up around a third of mutations in DMD.
5. More recent methods, including single-condition amplification internal primer sequencing (SCAIP) and denaturing gradient gel electrophoresis (DGGE)-based whole-gene mutation scanning, are utilised to detect the remaining third of mutations not yet elucidated by the above tests.
6. Muscle biopsy-based approaches utilising protein- and RNA-based analyses in combination with direct cDNA sequencing increase the mutation detection frequency to almost 100%.
7. Prenatal diagnosis is available by chorionic villus biopsy or amniocentesis by direct testing for abnormalities in the dystrophin gene.
Recent advances
In the last few years, the natural history of this disease has been changed by interventions (e.g. steroids, cardiac, respiratory, orthopaedic, rehabilitative), quality of life has improved and affected patients may now reach their fourth decade (a significant advance on the previous life expectancy of 19 years). Research into DMD has continued to accelerate, with a number of different treatment strategies being proposed. The role of steroids has become clearer. Glucocorticoids remain the only medication currently available that can slow down the decrease in muscle strength and function in DMD; this then reduces scoliosis and stabilises respiratory function. Low-dose steroids are very useful when boys are still walking, to improve motor function. The time of commencement of steroids remains controversial; currently, recommendations are to wait until identifying a plateau in the child’s motor development, where there is no longer progress in motor skills. Once that plateau is identified, steroids are commenced; they are not recommended for children still gaining motor skills, especially under 2 years. Typically, an affected male will progress with motor skills until 4–6 years old. Presently (2010), corticosteroid therapy is the treatment of choice for affected patients between 5 and 15 years of age. Some studies, however, suggest that the ideal window for treatment could be under 5 years. These patients need close monitoring, adjusting dose and timing to avoid unwanted side effects, especially weight gain.
Primitive stem cells in bone marrow have been shown to migrate into muscle and become new muscle cells. Stem cell therapy is under investigation but still experimental. Gentamicin has been found to permit cells to ignore an abnormal stop codon in the dystrophin gene and to proceed and synthesise the protein in the 15% of DMD patients who have premature stop codons as their underlying mutation. PTC124 is a new agent that may permit ribosomal read through of nonsense mutations. Morpholino antisense oligonucleotides permit exon skipping.
History
Current problems
1. Functional abilities with activities of daily living (e.g. dressing, writing, toileting); aids required (e.g. splints, supportive prostheses, computer-assisted communication/learning).
2. Mobility (e.g. long leg braces, wheelchair use, school and home access).
3. Home modifications required (e.g. ramps, bathroom fittings, specialised beds and mattresses).
4. Transport needs (e.g. van with hoist).
5. Scoliosis (e.g. progression, any planned surgery).
6. Joint contractures (AFOs, night splints).
7. Respiratory problems (e.g. symptoms of respiratory failure at later stage, sleep disordered breathing, non-invasive ventilation).
8. Cardiac symptoms (e.g. arrhythmias, symptoms of cardiac failure).
9. Gastrointestinal problems (e.g. incontinence, vomiting).
10. Difficulties with micturition.
11. School (e.g. any access problems, educational problems, any help needed or provided with toileting; attitudes of class teacher, headmaster, fellow students).
Past history
1. Initial diagnosis: when, where, presenting symptoms (e.g. late walking, tendency to fall, method of rising after fall, clumsiness, muscle cramps, learning delay, cognitive impairment, global or gross motor developmental delay).
2. The time period between onset of symptoms and diagnosis.
3. What investigations were undertaken to make the diagnosis.
4. Stages of deterioration (e.g. age at which the child lost the ability to climb stairs, stand from the floor or walk independently).
5. Number of hospitalisations.
6. Development of complications (e.g. scoliosis, cardiac failure) and their management.
Social history
1. Impact on child (e.g. difficulties at school, poor job prospects, limitations on lifestyle, body image, self-esteem, peer reactions).
2. Impact on family (e.g. parental coping, difficulties between mother and father, genetic implications for further children; financial considerations, such as cost of wheelchairs, home modifications, transport, hospitalisations, private health insurance; physical burden of helping children in and out of wheelchairs, cars, bed, bath).
4. Social supports (e.g. social worker, DMD Family Support Group provided by the Muscular Dystrophy Association, visits to school by occupational therapist and community liaison nurse from hospital muscle clinic to meet class and teachers).
Family history
Other known family members with DMD, other males with developmental delay or late walking.
Examination
Gait
Boys with DMD can walk well on their toes, but are unable to walk on their heels, and if they try they end up inverting the feet. On squatting, these children are slow to return to standing, need to extend the knee before the hip, and lean on their thighs to assist extension at the hip.
Of particular importance is the elicitation of Gower’s sign. The boy is asked to lie supine on the floor and then to get up. This first leads to rolling over to be prone (he cannot sit up because of weak neck and spine flexion), then on to the knees, then on ‘all fours’—that is, hands and feet—then ‘climbing up’ the legs to stand. Gower’s sign is often used as a functional timed test: timing how long it takes to arise from the floor is useful to monitor deterioration over time. See Figure 13.2.
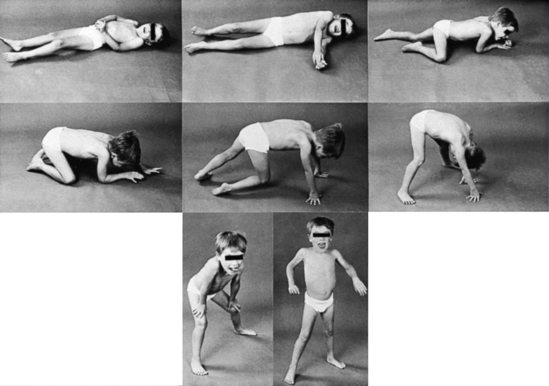
Muscle power
After comprehensive assessment of muscle power, check for tone and reflexes (knee jerks often lost) and contractures, especially at the ankles, knees and hips.
Management
The main management areas are described below.
Psychosocial
1. Education of parents and patient. Clarification of misinformation from other sources requires multiple informative sessions with the various disciplines involved in the comprehensive management of these patients. Regular feedback is needed to ensure that no misunderstandings occur. Other family members (e.g. siblings, grandparents, aunts, uncles and cousins) are often very much involved emotionally and tend to be neglected.
2. Expectation of a grief reaction to the diagnosis and its implications. Preparatory explanation to parents of probable feelings such as guilt, anger and depression. Explanation regarding any misapprehensions or strange beliefs about the nature of the illness.
3. Ensuring sufficient social supports, both from professionals (such as social workers) and non-professionals (such as other affected patients, families of other patients and groups such as the Muscular Dystrophy Association [MDA], Montrose Access [Qld] or Northcott [NSW]). The MDA provides parents’ groups and a useful handbook that deals with the home situation, recreational activities, education and vocational possibilities. There are excellent sites on the internet, including http://mdausa.org>and <http://www.treat-nmd.eu/patients/DMD/family guide/
4. Informing about financial assistance measures, such as any government benefits (which may assist directly with the child’s care, transport costs, accommodation costs and provision of aids). The illness places a very large financial burden for home modification (such as ramps, bathroom modification, lifting machines) on the family.
5. Discussion of treatment difficulties (such as non-compliance), seeking other opinions, and understanding and acceptance of alternative therapies sought by parents.
6. A major point is helping the family to determine how various people (especially the affected son) are to be informed. The paediatrician may not be the person who will help most, but he or she does have responsibility to ensure that it is done adequately. The consequences of failure in this aspect of management may include: parents who will not discuss the disease with their son, siblings, teachers or each other; one parent (usually the father) who withdraws from reality; and help being refused (because the parents believe that the child will be cured by divine intervention).
Therapies, orthoses and orthopaedic surgery
Physiotherapy
1. Physiotherapy to encourage walking and avoid joint deformity is very important.
Knee–ankle–foot orthoses (KAFOs) can be offered to patients at the end of independent walking and can prolong ambulation for 2 years, although these are not used much in Australia, where AFOs are used more often. Serial casting to reduce ankle contracture may be needed before fitting of the KAFOs can occur.
2. Stretches for contractures (e.g. flexion contractures at knees, hips, elbows, wrists, fingers); to minimise contractures, daily passive range-of-motion exercises of all joints of upper and lower limbs.
3. Exercises for strength (against gravity or resistance). Once the muscles are very weakened, these have little effect.
4. Chest physiotherapy for respiratory problems; teaching effective coughing. The use of inspiratory and expiratory exercises against resistance has increased over the last decade (‘motivational breathing exercises’/‘incentive spirometry’ are used to open the alveoli). Other devices include the intrapulmonary percussionator, the In-Exsufflator (i.e. cough assist devices).
Occupational therapy
1. A very important function is prescribing specifications for equipment (e.g. wheelchairs).
2. Home visits and visits to the school (meetings with teaching staff, and question and answer sessions with the affected boy’s class). These visits are very valuable.
3. Advice regarding aids to activities of daily living (e.g. getting in and out of wheelchairs, bed, bath or cars).
4. Aiding posture: avoidance of certain positions (e.g. postural scoliosis, flexion), orthoses (e.g. arm supports).
5. Assessment and advice on installation of ramps for adequate mobility at home. Assess the need for lifting machines (dependent on the strength of the caregiver at home), modifications to home (e.g. bathroom access).
Equipment
1. Wheelchairs: generally a manual type is used from around 9 years of age, and a powered type from 11 years. An appropriate vehicle is required for transport (e.g. between home and school). New-generation wheelchairs have been developed, including one that has gyroscopic electronic sensors, and the ability to switch between four wheels and two to go through sand, up and down stairs and over kerbs, as well as a lift to tackle high counters and allow eye-level interaction with standing adults.
2. Inclined standing board, to stretch tendo achilles.
4. Standing desks, or lying on inclined prone board.
Scoliosis management
1. Scoliosis commonly develops around 13–15 years, usually in the thoracolumbar region, becoming most apparent after loss of walking, when in a wheelchair, and rapidly progresses during the pubertal growth spurt. This adversely affects respiratory function, appearance, body image, posture, feeding, sitting and comfort. The advent of corticosteroid treatment, leading to prolonged walking and increased truncal muscle strength, has also lead to a reduced incidence and severity in scoliosis.
2. An important area is fitness for surgery. Adequate presurgical assessment of respiratory function is essential (if respiratory function parameters are over 50% of predicted values for the child’s size, surgery is possible). Postoperatively, early mobilisation should be undertaken to prevent deterioration in function while confined to bed. The occupational therapist helps to accommodate activities of daily living to increase sitting height (increased shoulder and elbow height) and rigid spine.
3. In any surgical procedure, neuromuscular depolarising agents such as succinylcholine must be avoided.
Common medical problems
Monitoring steroid therapy and bone health
1. Assess benefits: timed muscle function tests, pulmonary function tests; note the age when independent walking is lost.
2. Assess risks: look for steroid side effects (Cushingoid appearance, decreased linear growth, acne, behavioural change).
3. There is an increased risk of vertebral and long bone fractures with prolonged steroid use. However, immobilisation of fractures should be avoided in DMD. This is an additional burden on bones adversely affected by lack of mobility; these boys already have decreased bone density. Corticosteroids increase the already present risk of vertebral compression fractures, which can be asymptomatic. It is prudent to recommend sunshine and a balanced diet, with adequate vitamin D and calcium. Current recommendations are for annual bloods (bone minerals, PTH), urine for hypercalciura, and daily supplementation of all boys with 1000 IU vitamin D and 600 mg calcium.
4. Patients should have a MedicAlert bracelet to identify their diagnosis of DMD in case they need emergency anaesthesia, and it should also note whether they are receiving steroids.
Restrictive lung disease (RLD)
1. RLD is secondary to weakness of the diaphragm, chest wall and abdominal musculature. In teenage years, this is associated with worsening respiratory reserve and sleep hypoventilation, episodes of REM-sleep-related hypoxaemia and obstructive sleep apnoea.
2. Patients tend to have recurrent chest infections, which should be treated aggressively, as they can tip these boys into overt respiratory failure. They may also require inhaled treatment (nebuliser or pressurised metered dose inhaler with spacer) with bronchodilators, mucolytics or antibiotics. They must not be exposed to tobacco smoke. They should be fully immunised, including receiving yearly influenza vaccine.
3. They may develop cor pulmonale secondary to hypoxia, due to ventilatory difficulties and ventilation–perfusion inequalities associated with scoliosis.
4. Monitor with regular (6-monthly) pulmonary function tests (PFTs), including forced vital capacity (FVC) and maximal inspiratory and expiratory pressure, the former reflecting diaphragmatic strength, the latter reflecting the strength of the chest wall and abdominal muscles, correlating with the ability to cough and clear secretions. FVC predicts the development of hypercapnia and survival. When the FVC falls below 50%, this is the time to consider non-invasive ventilation (NIV; see below). Other useful aspects of respiratory monitoring used to attempt to predict respiratory failure are: peak cough flows (PCFs), which measure the capacity for mucociliary clearance; maximum expiratory mouth pressure (MEP), which can measure effective cough capacity; and maximum inspiratory mouth pressure (MIP) testing, which can indicate the need to consider starting NIV. All these tests can be carried out after the age of 5 years. Sleep studies (overnight polysomnography) should be done when the patient is first in a wheelchair, or when he first exhibits symptoms of sleep disordered breathing (SDB) or hypoventilation.
5. Symptomatic respiratory failure can supervene, with hypoventilation, hypercapnoea (e.g. restless at night, somnolent by day, morning headaches, anorexia at breakfast, nausea, general malaise, fatigue, poor concentration at school, and cyanosis possibly occurring during meals or on transfer from the wheelchair). Untreated, hypercapnoea can prove fatal within a year. Symptoms can resolve with nocturnal ventilatory support, such as bimodal positive airway pressure (BiPAP), a mode of positive pressure ventilation.
6. In the last few years, domiciliary non-invasive ventilation (NIV) has been proven effective in relieving symptoms of OSA and hypercapnoea, improving quality of life and prolonging survival. A compact portable ventilator is used, with a snugly fitting face or nose mask; the requirement for a good face or lip seal on the mask or nasal/oral orthotic interface cannot be overemphasised. NIV corrects hypoventilation and OSA, and with cough assist devices, has extended average survival to the mid-twenties, and occasionally to the fourth decade. There is now a widely held view that withholding NIV from hypercapnoeic patients with DMD is unethical. The benefits of NIV at night, may lead, within a couple of years, to a decision about daytime support and then 24-hour ventilation.
7. Decisions about whether or not to go into long-term 24-hour ventilation have to be made in consultation with the respiratory physician, the parents and the teenage boy, with the decision based on patient and family preference. Some units point out that waiting for daytime hypercapnoea/ventilator failure, until suggesting daytime NIV, is unsafe, as it exposes the patient to the risk of a rapid deterioration should he be exposed to a minor viral respiratory tract infection. Education and early discussion of options are the keys.
8. Volume ventilators may be used when vital capacity is below 40% of normal. These use a soft plastic nasal or face mask (for connection to the airway), with Velcro head and chin straps to hold them in position. The ventilator can fit on a ventilator tray on the bottom of a powered wheelchair.
9. Eventually, a tracheostomy may be appropriate, as masks may cause skin irritation due to constant skin pressure. A Passy–Muir valve can allow air through the vocal cords and improve voicing. The respiratory support area remains a major current issue in management of DMD.
10. NIV use in DMD is usually considered if the patient has symptoms (e.g. fatigue, morning headache) and any of the following: PaCO2 of 45 mmHg or more; nocturnal oxygen saturation of below 88% for 5 minutes consecutively; maximal inspiratory pressure below 60 cmH2O, or FVC below 50% predicted. Long-term NIV has been shown to reduce respiratory tract infections and hospital admissions in children with neuromuscular disorders
11. The main determinant of operative risk in DMD is respiratory function.
12. The usual cause of death: the median age of death was 22 years in 2005; the mean age of survival has increased from 19 years, 10 years ago, to 27 years currently (2010), mainly due to NIV and corticosteroids.
Cardiac disease
Current research is assessing whether ACE inhibitors and beta blockers should be given prophylactically to patients with DMD to prevent cardiac deterioration; the optimal time to commence therapy is still not agreed upon, although it is agreed that early therapy is clearly superior to late therapy. In patients with mild BMD, or in patients with DMD-related DCM, cardiac transplantation can be offered in severe cases. Without aggressive treatment, DCM can be rapidly progressive after onset in the teenage years, and cause death from cardiac failure within 2 years. It is important to treat any coexisting nocturnal hypoventilation, which aggravates cardiac dysfunction. Around 10% of carriers develop DMD-related cardiac disease, which can occur in the absence of muscle weakness.
Schooling, career prospects, lifestyle
1. Most children with DMD are integrated into a normal school, or may attend a special unit in a regular school. Special schools are less often required.
2. Dystrophin is expressed in the brain (role not known). Boys with DMD have a decrease in IQ of 1 standard deviation (around 15 IQ points) compared with unaffected siblings. Thus the mean IQ is around 85, and a higher percentage have intellectual impairment (around 20–30% have an IQ below 70). Encephalopathy is static, unlike muscle disease. A problem is underestimating the abilities of DMD patients (and disabled people generally).
3. The outlook for work is poor.
4. Some children, or young adults, can have what they feel is a fairly reasonable lifestyle, such as those pursuing courses of tertiary education, working in quadriplegic workshops or working with computers.
5. Sex education is a very valid area, which should not be ignored. Often, these patients can be quite ignorant of sexual matters, and education, such as group sessions discussing sexuality in disabled people, can be very useful.
6. Special accommodation needs should be addressed, such as the need for wide doors, special fittings and the availability of lifting hoists, as these may represent an enormous financial burden to the family.
Carrier females
Carrier females should be assessed for cardiomyopathy at least every 5 years from the age of 16 years. Mothers should be advised to seek cardiac review with ECG and echocardiogram.
Genetic counselling
1. Recent advances allow improved detection of carriers and prenatal diagnosis in families with a child known to have DMD (see earlier, ‘Background information on genetics of DMD’).
2. A female is defined an obligate carrier if she has an affected son and at least one other affected male relative in a pedigree that corresponds to X-linked-recessive inheritance.
3. Mothers of isolated DMD patients are possible carriers, as are non-obligate carrier females related to affected males. Analysis of pedigrees and DNA analysis may clarify carrier status.
4. One third of DMD patients represent new mutations. Effective genetic counselling will increase this proportion.
Seizures and epileptic syndromes
Background information
Currently recognised electroclinical syndromes by age of onset are listed below, using ILAE grouping, reorganised to simplify recall, causes including (if known) the chromosome location, gene, mechanism and salient clinical features. The ones that are asterisked are worth knowing in detail. These are thumbnail summaries, attempting only to give an overview, with some new mnemonics for those obsessed with memorising lists.
Neonatal period
Ohtahara syndrome (early infantile epileptic encephalopathy with suppression burst): Causes: cerebral malformations; for example, Aicardi syndrome, linear sebaceous naevus syndrome, porencephaly, hemimegaloencephaly, focal cortical dysplasia and cerebral dysgenesis. It is an age-dependent reaction to an insult, and can be seen as the start of a continuum, which can change to West syndrome and thence to Lennox–Gastaut syndrome. Clinically: tonic spasms (forward flexion), last 1–10 seconds, often in clusters up to 100 times per day, EEG shows burst suppression; 50% mortality rate within weeks or months; survivors have severe cognitive/neurological deficits; no treatment effective.
BFNS (benign familial neonatal seizures): Chromosomes/gene: 20q13.3/KCNQ2, and 8q24/KCNQ3. These genes code for potassium channel subunits, which combine to form the ‘M’ channel. Onset day 3, brief, various features (apnoea, deviation of head/eyes, tonic/clonic, autonomic changes), remit in days to months, normal intelligence long term.
EME (early myoclonic encephalopathy): Multifactorial. Commonest causes: inborn errors of metabolism (IEMs); for example, non-ketotic hyperglycinaemia, Menkes disease, Zellweger syndrome and methylmalonic acidaemia. Onset usually before day 10, clinical triad: myoclonus (randomly shifting, fragmentary), then focal fits, then tonic (infantile) spasms; devastating psychomotor developmental regression; all have bilateral pyramidal signs.
Infancy
(Mnemonic to Dr West, who described the condition in his son: MB, MB, MD, West)
Migrating partial seizures of infancy: Cause unknown. Clinical: onset average 3 months, multifocal seizures, frequent (almost continuous) seizures, associated psychomotor/developmental regression, quadriplegia, death usually by 12 months.
Benign infantile seizures (Watanabe–Vigevano syndrome): Non-familial and familial forms. Clinically almost identical: onset average 5 months, focal, brief (under 3 minutes), diurnal seizures, in clusters of 5–10 daily (first fit being longer) for 1–3 days, recur in 1–3 months, familial cases have longer fits; altered consciousness, motor arrest, unilateral clonic seizures, automatisms. Familial form, chromosomes: 2q24,16p12–q12, 19q12–13.1.
Myoclonic epilepsy in infancy (MEI): An idiopathic generalised epilepsy. Onset between 6 months and 3 years; myoclonic jerks may be spontaneous or reflex (to photic, auditory, somatosensory stimuli); usually head nodding, upper limbs flinging outward; duration brief (1–2 seconds); consciousness often intact; responds well to AEDs (valproate).
Benign familial neonatal–infantile seizures: Chromosome/gene: 2q23–24/SCN2A. This is a sodium channelopathy, the gene being a sodium channel subunit gene; onset between day 2 and 7 months; focal seizures; can resolve by 12 months.
Myoclonic encephalopathy in non-progressive disorders: Causes: chromosomal disorders (especially Angelman and 4p syndromes), cerebral malformations, ischaemic encephalopathy. Onset average 12 months; pre-existing encephalopathy; repetitive long periods of myoclonic-absence status epilepticus; frequent startle episodes; severe cognitive deficits. AEDs of choice: benzodiazepines, valproate, ethosuximide, ACTH.
Dravet syndrome (severe myoclonic epilepsy in infancy, SMEI): Chromosome/gene: 2q24/SCN1A, 5q34/GABRG2; the former a sodium channel gene, the latter a gamma-aminobutyric acid receptor subunit gamma-2 gene; 70% patients have mutation, 95% de novo. Onset average 6 months; can present in status (hemiclonic, general or febrile); can have hemimotor status (but involve other side next time) plus complex partial (pallor, automatisms, absence), in clusters, often head turn and flexed upper limbs, mainly myoclonic by 4 years. Precipitants: hyperthermia, water [bathing], light, CBZ; frequent fits until 12 months, multiple seizure types between 1 and 4, mainly myoclonic by 4; most have normal development initially, but after 12 months, developmental/neurocognitive regression. Ataxia and pyramidal signs evolve. IQ outcome poor, fits may continue. AEDs used (suggested sequence): first try valproate, then topiramate, then clobazam, then levetiracetam, then stiripentol [chemically unrelated to other AEDs, approved in Europe for Dravet syndrome alone]. Avoid lamotrigine, as can aggravate seizures. Currently (2010) the only epilepsy with a gene test commercially available; test child and parents. Expensive, but stops the need to do other tests (for mitochondrial disorders etc) and affects the parents’ decision to have further children.
West syndrome∗: Epileptic encephalopathy with multiple causes (around 80% symptomatic: cerebral ischaemia [pre-, peri- or postnatal], chromosomal anomalies, cerebral malformations, tuberous sclerosis [TS], infections [congenital or acquired, e.g. TORCH, pertussis, meningitis]; 20% idiopathic). Onset average 5 months, comprises epileptic (infantile) spasms (which are age-dependent reaction to an insult), EEG changes of hypsarrhythmia, and in many, developmental regression. Spasms occur in clusters of up to 30 per day, with each cluster having 20–150 spasms, these being short (2 seconds or less), usually fairly violent, bilateral tonic flexor ‘jack-knife’ or ‘salaam spasms’, on arousal or awake states, and Moro-like extensor spasms. The prognosis is related to the cause. A trial of pyridoxine is worthwhile to exclude pyridoxine dependent seizures if there is a previous history of neonatal or focal seizures. AEDs of choice are ACTH, steroids or vigabatrin. If TS is the cause, then vigabatrin is first line; if non-TS, then ACTH or prednisolone are first line.
Childhood
Febrile seizures plus (FS+)∗ (can start in infancy): also called GEFS+ (generalised epilepsy with FS+): Chromosomes/genes: 2q24/SCN1A, 2q23–24/SCN2A, 19q/SCN1B, 5q34/GABRG2, 1p36.3/GABRD [Gamma AminoButyricAcid receptor delta gene, codes for subunit of a ligand-gated chloride channel for GABRDelta]. The term describes a family, not a patient. Can be simple febrile seizures, febrile seizures older than 6 years, absences, TLE, myoclonic astatic epilepsy, and SMEI. Phenotype guides treatment and prognosis, not finding mutation.
Early-onset benign childhood occipital epilepsy (Panayiotopoulos type): Onset peak 4–5 years; autonomic symptoms (especially emetic: nausea, retching, vomiting—others include pallor and mydriasis); consciousness and speech preserved at onset; behaviour change (ictal: restlessness, terror); syncope-like unresponsiveness and loss of tone; unilateral eye deviation; autonomic status can occur, may end with hemiconvulsions, or generalised convulsions. Interictal EEG multifocal, high-amplitude, sharp-slow-wave complexes. Prognosis benign:, 25% have one seizure only, 50% have 2–5 only. AEDs rarely needed.
Epilepsy with myoclonic atonic (previously astatic) seizures (EM-AS, also called Doose syndrome): Cause: likely genetically determined in multifactorial polygenic pattern. Onset peak 2–4 years, in 2/3, febrile and afebrile GTCSs occur initially, months before myoclonic–atonic; all patients have symmetrical myoclonic jerks followed immediately by loss of tone (post-myoclonic inhibition); can also have pure atonic or absence, and non-convulsive (myoclonic–atonic) status. Prognosis varies: 50% eventually seizure free, normal development; other 50% usually symptomatic or part of other syndromes, continue to have seizures and neurocognitive deterioration, with ataxia, dysarthria and language problems. AEDs of choice: valproate, lamotrigine, topiramate, levetiracetam. Note contraindicated AEDs: carbamazepine, vigabatrin, phenytoin.
Late-onset childhood occipital epilepsy (Gastaut type): Phenotype of ‘benign childhood seizure susceptibility syndrome’; often family history of epilepsy or migraine. Onset average 8 years; pure occipital seizures, with visual hallucinations (usually small multicoloured circular patterns, compared to ‘fuzzy’ confetti, or sequins), blindness or a combination; short (few seconds to 3 minutes), frequent often diurnal; non-visual features may include deviation of eyes with ipsilateral turning of head, eyelid blinking, eyelid closure; consciousness preserved; interactal EEG shows occipital paroxysms. AED of choice: carbamazepine. Secondary GTCS can occur if not treated.
Benign epilepsy with centrotemporal spikes (BECTS)∗: Also called benign rolandic epilepsy (after region involved; lower part of central gyrus of Rolando). Commonest syndrome; chromosome 11p13; peak age 8–9 years; nocturnal orofaciobrachial focal seizures, short (1–3 minutes), involving the vocal tract, with oropharyngolaryngeal guttural sounds (‘chuggers’ and ‘gluggers’), and hemifacial sensorimotor symptoms that spread to the tongue, mouth and face, causing hypersalivation and speech arrest; consciousness preserved; secondarily GTCSs in 50%. Occur during non-REM sleep, around sleep onset or just before waking, especially 5–7 a.m., in boys more than in girls (3:2). EEG: interictal CTS. Neurodevelopmental disorder, can lead to learning disabilities, behaviour problems. May not need AEDs if seizures infrequent. If frequent or secondary GTCS, AEDs used: carbamazepine, levetiracetam.
Absence, myoclonic—epilepsy with myoclonic absences (MAE): Idiopathic. Onset peak 7 years; seizures comprise rhythmic myoclonic jerks (shoulders, arms, legs), with tonic contraction, usually unilateral; impairment of consciousness, short (8–60 seconds), frequent (several times a day). Around 70% have other seizure types, GTCS or atonic, which may portend a poor prognosis. EEG generalised or multifocal spike and slow wave; around 50% have decreased cognition prior to absences, although 50% of the normal (prior) patients develop cognitive and behavioural impairment as well. Often hard to treat; half the patients have seizures as adults, some develop other forms of epilepsy (e.g. LGS, JME). AEDs tried: valproate plus ethosuximide or lamotrigine, clonazepam, acetazolamide.
Lennox–Gastaut syndrome (LGS)∗: Causes: not genetic; similar to those for West syndrome, but more often cerebral malformations, and less often Aicardi and lissencephaly; can be third part of an encephalopathic continuum (mnemonic OWL); Ohtahara to West to LGS. Onset peak 3–5 years. LGS is an epileptic encephalopathy, with polymorphic intractable seizures (tonic [symmetrical, brief (2–10 seconds)], atypical absences [clouding of consciousness, tone changes, myoclonic jerks], atonic [sudden brief (1–2 seconds) loss of postural tone], EEG slow spike wave discharges); impaired cognition and behaviour. Prognosis very poor: 80–90% have seizures into adult life. AEDs used: valproate (beware hepatic failure, acute haemorrhagic pancreatitis), clonazepam, lamotrigine, levetiracetam, vigabatrin, topiramate.
Absence—childhood absence epilepsy (CAE)∗: Susceptibility conferred by several genes; termed ECA (epilepsy childhood absence). Chromosome/gene: ECA1, 8q24; ECA2, 5q31.1/GABRG2; ECA4, 5q34/GABRA1; ECA5, 15q11–q12/GABRB3; ECA6, 16p13/CACNA1H [calcium channel, voltage dependent, T-type, alpha-1H subunit]. Onset peak 6–7 years, more in girls; very frequent (dozens per day) absences; brief (under 20 seconds); abrupt loss of consciousness; may be automatisms. EEG ictal 3 Hz generalised high-amplitude spike and spike/slow wave. Prognosis excellent: under 10% go on to have GTCSs in adolescence. AEDs of choice: valproate, ethosuximide, lamotrigine. Note contraindicated AEDs: most others; that is, carbamazepine, oxcarbazepine, phenytoin, phenobarbitone—ones with ‘gab/a’ in their name, such as pregabalin, vigabatrin, gabapentin, and tiagabine. Can withdraw AEDs over 6 months after 2–3 years seizure-free.
Nocturnal—autosomal-dominant nocturnal frontal lobe epilepsy (ADNFLE): Chromosome/gene: type 1, 20q13.3/CHRNA4 [cholinergic receptor, neuronal nicotinic, alpha polypeptide 4; gene encoding the alpha 4 nicotinic acetylcholine receptor (nAChR) subunit]; type 2, 15q24; type 3, 1q21/CHRNB2; type 4, 8p21/CHRNA2. Onset average 11 years; frequent (almost nightly) clusters of short (20–50 seconds) hyperkinetic motor seizures with dystonic posturing; may be aura; may be thrown out of bed; consciousness preserved; may be precipitated by movement, sound; often misdiagnosed as obstructive sleep apnoea (OSA), night terrors, nightmares, parasomnia. AEDs of choice: carbamazepine, levetiracetam, clobazam, lamotrigine, topiramate.
CSWS—epileptic encephalopathy with continuous spike-and-wave during sleep (CSWS): Cause unknown, although 50% have pre-existing problem (e.g. cerebral malformations); problem is CSWS causing neurocognitive regression rather than the seizures. Onset of seizures peak 4–5 years, EEG finding of CSWS onset peak 8 years; seizures may be nocturnal unilateral motor seizures, diurnal absences or atonic; regression of IQ, language, behaviour and psychological state; deficits depend on spike localisation. Frontal lobe targeting leads to disinhibition, aggressiveness, inattention, cognitive decline, psychosis (frontal lobe dementia); temporal lobe involvement causes expressive aphasia. Motor problems: ataxia, hemiparesis, dyspraxia. Treatment: spike suppression; similar to approach in LKS treatment (see below). EEG and clinical remission occurs in second decade, including improvement in psychological well being, but not back to normal.
Epileptic encephalopathy, including Landau–Kleffner syndrome (LKS)∗: Also called acquired epileptic aphasia. Cause unknown. Onset peak 5–7 years; normal prior development or isolated language delay; regression in receptive and expressive language abilities, auditory agnosia (cannot identify sounds in environment, e.g. dog barking), global aphasia; seizures tend to be mild, infrequent, nocturnal, resolving by 15 years. EEG: focal/bilateral slow spike wave foci over temporal regions; can be continuous, unilateral or bilateral synchronous; NREM sleep accentuates EEG abnormalities. AEDs used: valproate with lamotrigine, ethosuximide, clonazepam, clobazam, levetiracetam; if unsuccessful, then ACTH or prednisolone; if intractable deterioration, surgical treatment with multiple subplial intracortical transections can succeed. Prognosis: earlier onset imparts worse prognosis.
Adolescence–adult
Juvenile absence epilepsy (JAE): Susceptibility conferred by several genes; termed EJA (epilepsy juvenile absence). Chromosome/gene: EJA1, 6p12–p11/EFHC1 [EF hand domain (C-terminal)-containing 1; gene product is called myoclonin 1]; EJA2, 3q26/CLCN2 [voltage-gated chloride channel 2]. Onset average 9–13 years; absences similar to CAE, severe, frequent, short (4–30 seconds); GTCS and myoclonic jerks commence up to 10 years after absences start. Ictal EEG: 3–4 Hz generalised polyspike wave discharge; lifelong, but good control in 80% with AEDs valproate and lamotrigine; control of absences generally means control of GTCSs.
Juvenile myoclonic epilepsy (JME)∗: Also called Janz syndrome. Susceptibility conferred by several genes; termed EJM (epilepsy juvenile myoclonus). Chromosome/gene: EJM1, 6p12–p11/EFHC1; EJM2, 15q14/CHRNA7 [alpha 7 subunit of nAChR]; EJM3, 6p21/BR2 [bromodomain-containing protein 2, a nuclear transcriptional regulator]; EJM4, 5q12–q14; EJM5, 5q34–q35/GABRA1 [GABA receptor alpha 1]; EJM6, 2q22–q23/CACNB4 [calcium channel, voltage dependent beta-4 subunit]; EJM7, 1p36.3/GABRD [GABA receptor delta]; EJM8, 3q26/CLCN2 [voltage-gated chloride channel 2]. Onset around 5–16 years for absences, and 14–15 years for myoclonic jerks (typically within an hour of waking; often drop their breakfast cereal bowl), with GTCSs appearing some months after jerks; precipitants include fatigue, sleep deprivation. EEG: 3–6 Hz polyspike wave discharge; probably lifelong treatment needed. It is lifestyle dependent: if take medication but awake all night, will fit; patients with this can drown in the surf. AEDs of choice: valproate, levetiracetam. Note contraindicated AEDs: all the ‘gab/a’ ones (vigabatrin, gabapentin, pregabalin, tiagabine), plus phenytoin, carbamazepine, oxcarbazepine.
Temporal lobe—other familial temporal lobe epilepsies: These include: familial mesial temporal lobe epilepsies (chromosomes: 1q, 4q, 18q); familial occipito-temporal lobe epilepsy (9q); familial partial epilepsy with variable foci (22q12).
Autosomal dominant partial epilepsy with auditory features (ADPEAF): Also called autosomal dominant lateral temporal lobe epilepsy (ADLTLE). Chromosome/gene: 10q24/LGI1 [leucine-rich gene, glioma inactivated]/epitempin. The first non-ion channel familial epilepsy described; onset peak teenage years; mainly simple auditory hallucinations (humming, ringing); also may be visual, olfactory, vertiginous; infrequent nocturnal GTCSs. AEDs: carbamazepine, levetiracetam. Prognosis excellent.
Progressive myoclonus epilepsies (PME): This is a group of rare, genetic disorders (mainly autosomal recessive); details beyond this section. Examples: Unverricht disease (commonest PME); mitochondrial encephalopathy with red ragged fibres (MERRF); all causes have devastating neurocognitive morbidity.
Epilepsy with generalised tonic–clonic seizures alone (EGTCSA)∗: Also called idiopathic generalised epilepsy. Chromosome/gene: 3q26/CLCN2 [voltage gated chloride channel]. Onset peak age 16–17 years; probably lifelong, with 80% relapse if off AEDs. AEDs used: valproate, levetiracetam, lamotrigine, topiramate, phenobarbitone.
Distinctive constellations
Mesial temporal lobe epilepsy with hippocampal sclerosis (MTLE with HS)∗: Cause unknown, although many have an early childhood history of febrile convulsions, head injury, meningoencephalitis or hypoxic insults. This is the commonest epileptic syndrome. Simple focal seizures commence with an ascending epigastric aura, a ‘welling up’ from the stomach; impaired ability to speak, but retained ability to comprehend conversations, last 1–2 minutes; can progress to complex with automatisms, restlessness or motor arrest, staring, head deviation (when early, to side of lesion; when later, can be to contralateral side and precede (infrequent) secondary GTCS), dystonic posturing, autonomic features, lasting 2–3 minutes; complex partial status epilepticus involves ongoing disturbance of consciousness, also called dyscognitive or psychomotor status. High-resolution MRI shows hippocampal sclerosis. Interictal EEG may show focal spike/sharp and slow complexes, in a third, thus it may well be normal in two thirds. Prognosis variable. AEDs used: carbamazepine, levetiracetam. Neurosurgical treatment available if AEDs fail, with anterior temporal lobe resection with hippocampectomy having a success rate of 60% patients seizure free; however, 10% get no benefit and 10% get worse (e.g. neurosurgical complications).
SCN1A-related seizure disorders
Commonly associated phenotypes: febrile seizures (FS), generalised seizures with febrile seizures plus (GEFS+), Dravet syndrome (SMEI), and three that are not in the ILAE list above—severe myoclonic epilepsy, borderline (SMEB), intractable childhood epilepsy with generalised tonic–clonic seizures (ICE–GTC), and infantile partial seizures with variable foci (also called severe infantile multifocal epilepsy).
• Voltage-gated ion channels: sodium (GEFS+, severe myoclonic epilepsy of childhood, benign neonatal–infantile convulsions), potassium (neonatal seizures), chloride (generalised epilepsies), calcium (absence epilepsy).
• Ligand-gated ion channels: GABA receptors (GEFS+, absence epilepsy, juvenile myoclonic epilepsy), ACh receptors (autosomal dominant [AD] nocturnal frontal lobe epilepsy).
• Non-ion channel genes: temporal lobe epilepsy with auditory features (LGI1).
• Idiopathic epilepsies with simple inheritance (usually quite benign) are now well recognised but are relatively rare: AD syndromes of early infancy (benign familial neonatal, neonatal–infantile and infantile; three different ages of onset; three different genes), AD nocturnal frontal lobe epilepsy, AD partial epilepsy with auditory features and familial temporal lobe epilepsies. In contrast, most common epilepsies have quite complex inheritance.
History
1. Any prodromal symptoms (e.g. irritability, pallor). The setting in which the events occur (e.g. from sleep, during exercise, when ill, when sleep-deprived). Any precipitating factors (seizure triggers): tiredness, lack of sleep, fever, infectious illness, hot water, having a hot bath, change of dosage or type of anticonvulsant, intake of other substances (in adolescents), falls or blows to the head, movements, sensory stimuli such as flashing lights, television, computer games, patterns (e.g. stripes), elimination of central vision or fixation, sounds, music, startling by sudden noises or touch, reading, calculating, decision-making, playing chess. Reflex seizures are consistently elicited by a particular stimulus. The term ‘catamenial’ refers to seizures that increase in relation to the menstrual cycle, whether perimenstrual, periovulatory or luteal (oestrogens generally are proconvulsant, and progesterones anticonvulsant). Adolescent girls may be asked about this.
2. Any aura (e.g. a specific psychic or sensory symptom, as distinct from a prodromal symptom). This includes running to the parent for comfort. This is applicable to children with partial seizures.
4. Initial localising signs (e.g. twitching of one hand).
5. Description of all the manifestations (motor and autonomic) of the seizure (e.g. eyes ‘rolling back’, altered awareness, cyanosis, jerking movements of limbs, urinary and/or faecal incontinence).
6. The duration of the seizures—the range (e.g. between 5 and 20 minutes) and the ‘usual’ time (e.g. 5 minutes). Any episodes of status epilepticus (duration >30 minutes).
7. The frequency of the seizures—range (e.g. none for 6 weeks to 6 in a day) and the ‘usual’ (e.g. once every 3 weeks).
8. Time of occurrence of seizures (e.g. on waking, or on going to sleep).
9. The date and time of the last seizure.
10. Postictal events (e.g. sleeping, confusion, headache, vomiting, Todd’s paralysis).
11. Presence of neurological dysfunction (other than seizure).
Finally, remember to take a full social history, including any benefits the child is receiving (e.g. Children’s Disability Allowance), social supports (e.g. Epilepsy Association) and the impact of the disease on the child, schooling, parents and siblings. An enquiry into social isolation or bullying at school can be revealing. If the patient is an adolescent, this involves a whole range of new issues, including compliance, effects on career prospects, driver’s licence, and menstrual and reproductive issues. Mental health issues often come to the fore with the high rates of teenage depression and anxiety seen.
Investigations
Imaging: structural
Brain-imaging studies by either magnetic resonance imaging (MRI) or computerised tomographic (CT) scanning may be indicated in the following circumstances. The present gold standard is MRI. Thin cuts can identify atrophy of the hippocampus, developmental anomalies (e.g. cortical dysplasia), and low-grade tumours. Fluid attenuation inversion recovery (FLAIR) sequences can detect very subtle abnormalities of the cortex:
1. Focal seizures, except for BECTS (rolandic seizures), which is diagnosed on clinical and EEG features that are characteristic enough to make MRI/CT scanning unnecessary. However, a scan should be done if atypical features are present.
2. Focal EEG findings (again, except for those of BECTS).
Imaging: functional
1. Positron emission tomography (PET) scanning, which assesses cerebral metabolism (mnemonic: PET checks METabolism), using fluorodeoxyglucose. PET shows a decreased metabolic rate of the epileptogenic area during the interictal period. It is especially useful in identifying temporal lobe epilepsy (TLE).
2. Single photon emission computed tomography (SPECT) scanning, which evaluates cerebral perfusion, using hexamethyl-propyleneomine axime (HMPOA) or iodoamphetamine or technetium-labelled Ceretec. There is interictal hypoperfusion, but ictal intense hyperperfusion at onset, with depression of perfusion after seizure. (Mnemonic for SPECT: Seizure [area] Perfusion Evaluated by Ceretec with Technetium.) SPECT is ideally performed during a seizure to demonstrate regional hyperperfusion.
3. Functional MRI: remains investigational, but regional localisation of primary language, motor and visual cortical areas is possible with this technique.
Common management issues
Increasing frequency of seizures and intractable epilepsy
Question the medication
The dose may be wrong, the drug may be wrong or adverse drug interactions may be occurring. The dosage may be too low, usually related either to non-compliance or to ‘outgrowing’ the dosage given (weight increase). The drug may not be the most appropriate choice for the types of seizure the child has (see below), or a drug appropriately chosen for one seizure type may worsen, or even ‘unmask’, another type of seizure (e.g. carbamazepine can worsen absence seizures). Insufficient numbers of medications may have been trialled.
Advice to parents (and schoolteachers)
Everyday childhood/adolescent activities: safety considerations
1. No swimming alone. Swimming pools, and dams should be fenced off.
2. No bathing alone for younger children. No unattended buckets of water.
3. Showers rather than baths for older children (with the bathroom door open and someone nearby). Retractable taps to avoid head injury. Never lock the bathroom door from inside.
4. No diving or surfing if JME, but tandem parachute diving is acceptable.
5. Strategies to prevent burns: putting guards in front of fires and heaters, use of microwaves rather than stoves or conventional ovens, stove barriers, anti-spill mugs, insulated plastic (rather than metal) kettles that switch off automatically. Avoidance of: hand-held hair dryers, electric irons, accessible bar heaters, open fires, hotplates, barbeques, cigarettes, cigars, pipes [avoiding the latter three also to prevent fires], thermostats to control the temperature of the hot water supply. Always run cold water before hot.
6. No climbing ladders or similar pursuits with a risk of falls.
7. No skateboard or bicycle riding on busy roads.
8. Use of helmets when riding on less busy roads.
9. Use of helmets for drop attacks (as in atonic seizures).
11. Adolescent females taking AEDs, who are sexually active, should take folate 5 mg per day, as AEDs increase risk of neural tube defects in their offspring (valproate 2%, carbamazepine 1%).
Avoid overprotection
Allow participation in normal sports and school activities. There is no contraindication to body-contact sports. Some neurologists prefer to encourage participation in non-contact sports such as tennis and athletics.
Anticonvulsant medications
Which drugs can have serum levels measured?
The candidate should know which drugs can have serum levels measured, but also recognise the limitations of such levels. A drug can be at the recommended range, but the patient still has fits and/or side effects; this is not an adequate ‘therapeutic’ drug level, as it is not providing adequate therapy. A level is only ‘therapeutic’ if there are no fits and no side effects. Of the ‘older’ AEDs, carbamazepine (CBZ), phenytoin (PHT) and phenobarbitone (PB) all have recommended ‘optimal’ ranges of serum drug levels, which may be useful in monitoring therapy. Valproate (VPA) is an exception: levels can be measured, but they do not correlate with efficacy or toxicity and so are unhelpful (except in suspected non-compliance, where a level of zero may confirm this suspicion). The same applies to the benzodiazepines (BDZs). Of the newer drugs, serum levels are important only for lamotrigine, topiramate, and zonisamide.
Which drug is preferable in which type of seizure?
Table 13.1 is an incomplete list of epilepsy syndromes and useful drugs in each.
Table 13.1 Selected epilepsy syndromes and examples of drugs used
| Epilepsy syndromes | Drugs |
|---|---|
| West syndrome/infantile spasms | First line: if not caused by tuberous sclerosis: ACTH, prednisolone |
| First line: if caused by tuberous sclerosis: VGB | |
| Second/third line: VGB, VPA, CLZ, TPM | |
| Dravet syndrome (SMEI) | VPA, TPM, clobazam, LEV, stiripentol [** avoid CBZ**] |
| Doose syndrome (MAE) | VPA, LTG, TPM, LEV, clobazam, CLZ, ESM, ketogenic diet, steroids (non- convulsive status) [**avoid CBZ and VGB**] |
| Lennox–Gastaut syndrome | VPA, LTG, TPM, CLZ, clobazam, LEV, PHT (childhood epileptic encephalopathy) |
| Childhood absence epilepsy (CAE) | ESM, VPA |
| BECTS | CBZ, VPA, LEV |
| Landau–Kleffner syndrome (LKS) | VPA and LTG, ESM, CLZ, clobazam, LEV, ACTH, prednisolone |
| Juvenile myoclonic epilepsy (JME) | VPA |
| Symptomatic partial epilepsy | CBZ, VPA, LTG, clobazam |
ACTH = adrenocorticotropic hormone; BDZs = benzodiazepines; CBZ = carbamazepine; CLZ = clonazepam; ESM = ethosuximide; LEV = levetiracetam; LTG = lamotrigine; PHT = phenytoin; VGB = vigabatrin; VPA = valproate.
What side effects are likely?
It is beyond the scope of this book to list all the side effects of the commonly used anticonvulsants. Behavioural and cognitive effects are among the most common side effects reported in children. Candidates should be familiar with the acute toxicities (e.g. drowsiness, nystagmus and ataxia with carbamazepine and phenytoin), acute idiosyncratic reactions (e.g. skin manifestations such as Stevens–Johnson syndrome), chronic toxicities (e.g. various effects on the haematological system, bones, connective tissue, cosmetic effects and teratogenic effects) and drug interactions, both between anticonvulsants (e.g. CBZ lowers PHT levels, VPA increases PB levels) and with other drugs (e.g. erythromycin increases CBZ levels).
Are any of the newer AEDs likely to be of use here?
Vigabatrin (VBT)
An irreversible inhibitor of GABA transaminase, VBT increases the availability of GABA. Around 50% of patients have a 50% reduction in intractable partial seizures. It remains the drug of choice for West syndrome/infantile spasms, when caused by tuberous sclerosis, despite great concern involving the risk of some loss of peripheral visual fields, which can be irreversible despite cessation of the drug when detected. Side effects: sedation and fatigue, weight gain, agitation and hyperkinesis.
Surgical treatment
Management of the prolonged seizure
In ambulance/at hospital
• Initial minute: intravenous (IV) diazepam or midazolam; if no access, intramuscular (IM) midazolam or rectal (PR) diazepam.
• 5 minutes: intravenous phenytoin infusion over 20 minutes, with ECG monitoring; if no access, repeat IM midazolam or PR diazepam.
• 10 minutes (still fitting): repeat intravenous diazepam or midazolam; if no access, PR paraldehyde diluted 50:50 in normal saline.
• 25 minutes (still fitting, phenytoin infusion complete): phenobarbitone can be commenced, while contacting the anaesthetist/intensivist/emergency physician to prepare for intubation (rapid sequence induction including thiopentone).
Management of the first non-febrile seizure
The chances of further seizures occurring after a single seizure, at 5 years of age, are as follows:
Psychosocial issues
Parents
The parents of an epileptic child often have concerns in the following areas:
2. Being uncomfortable with the label/stigma of ‘epilepsy’.
3. False beliefs that the cause relates to themselves (e.g. ‘stress’ during pregnancy).
4. Equating an EEG with a treatment modality such as electroconvulsive therapy.
5. Difficulty accepting the need for medication: ambivalence (forgetting to give medications); the belief that the need for drugs equates with, or causes, ‘retardation’; and suddenly stopping the medication when the supply is used up.
Spina bifida
In September 2009, mandatory folic acid fortification was introduced into Australia, requiring Australian millers to add folic acid to wheat flour for bread-making purposes. Current recommendations include folic acid supplementation 1 month before conception and through at least the first 8–12 weeks of pregnancy. All women contemplating pregnancy should ingest a daily multivitamin containing 0.5 mg folic acid. If there is a family history of neural tube defect, one needs to advise that 5 mg is taken. It has been suggested that NTDs are not due to folate deficiency per se, but due to enzymatic abnormalities involving metabolic processes that depend on folate and its metabolites, tetrahydrofolate and 5-methyltetrahydrofolate, abnormalities that could be overcome with folate supplementation.
1. How does this child function in day-to-day life (i.e. a brief outline of the child’s practical problems)? This will make clear how the disability affects the child’s life.
History
Current history
How does this child function?
1. Mobility: type, aids, therapy.
3. Education: What type of schooling does the child have (e.g. regular school, special class, special school)? What particular problems are there with learning?
4. Developmental problems: related to age (e.g. awareness of disability, making friends, self-esteem, adolescent problems of identity, independence, sexuality, employment). How independent is this child in self-help skills such as bathing, dressing and feeding?
Specific medical problems
1. Hydrocephalus: Does this child have a shunt? Are there any problems with the shunt?
2. Urinary system: Infections, operations, any concerns on routine tests (reflux, kidney damage), any medications (antibiotics, anticholinergics).
3. Orthopaedic problems: Feet and hips (e.g. deformity, pressure areas, problems with splints, calipers, crutches or wheelchairs); back (e.g. scoliosis, kyphosis).
4. Other medical complications: Ask about (a) eyes (squint, amblyopia); (b) skin care: pressure ulcers; burns; latex allergy; (c) growth and development—precocious puberty; short stature; obesity; (d) problems with executive functioning, impaired ability to organise; (e) coordination problems; (f) sleep issues—any history suggestive of sleep apnoea.
5. Issues of adolescence: Problems with motivation; transition to adult care.
Social history
What assistance does the family receive?
1. Community supports: community nurse, community therapy, respite care, social worker, Spina Bifida Association, Crippled Children’s Society.
2. Financial supports: Child Disability Allowance, Isolated Patients Transport and Accommodation Assistance Scheme, Provision of Aids for Disabled People (in Victoria this is now called the Victorian Aids and Equipment Program). After the age of 16, catheters are accessed via the Commonwealth CASS scheme.
Management
Major disabilities
Paralysis
Associated problems include immobility, dependence versus independence, joint contractures, anaesthetic skin risks and pressure necrosis of soft tissues. Barriers to mobility include the level of the lesion, associated spasticity, poor balance, the Arnold–Chiari II malformation and orthopaedic deformities (including scoliosis, oblique pelvis, hip flexion), as well as behavioural problems and some degree of cognitive impairment.
Management involves a team approach:
1. Physiotherapy (e.g. ambulation training, wheelchair mobility).
2. Occupational therapy (e.g. functional training, activities of daily living).
3. Clinical motion analysis: Paediatric Gait Analysis Laboratory (see CP section).
4. Positioning orthoses (e.g. ankle–knee orthoses, hip-positioning brace).
5. Orthopaedic surgical procedures (e.g. tendon releases or transfers, surgery for dislocated hips). Orthopaedic surgery can be performed on many areas during the same operation.
6. Mobility aids (e.g. crutches, reciprocating gait orthoses (usually younger patients with higher lesions [lower thoracic]), hip–knee–ankle–foot orthoses (HKAFOs [for lesions at lower thoracic level, or L1–L2, adds to ability to be ambulatory]), walkerette, parapodium, wheelchairs).
7. Education regarding risks to skin integrity (e.g. tight clothes, temperature of bath water, sunburn, overtight tying of booties in babies or even hospital identification labels on ankles, ill-fitting orthoses, standing on hot sand at the beach). Aggressive management of pressure necrosis.
Bladder and renal function
Urinary tract infections (UTIs) need a full course of antibiotics for the acute infection. However, a positive urine culture in the absence of significant symptoms probably reflects urinary tract colonisation rather than infection per se. It is prudent to only treat for a UTI with antibiotics if the child is unwell; otherwise, antibiotic resistance would increase. Only give prophylactic antibiotics in the neonatal period when there is hydronephrosis or vesico-ureteric reflux (VUR). Urine specimens (MSUs) should be obtained to check for infection, when parents suspect (urine odour, child unwell). Surgery may be required for several urinary problems: augmentation cystoplasty if the bladder is small and hypertonic, bladder neck reconstruction or artificial external urinary sphincter. Surgery for VUR may be needed if recurrent urinary tract infections occur despite treatment, and if there is renal scarring or persistent hydronephrosis. Urolithiasis can occur secondary to immobilisation.
CKD-mineral bone disorder (renal osteodystrophy) in this population is of interest: children with spina bifida are at increased risk of fracturing their paraplegic lower limbs, even with normal renal function, so that with renal impairment, bone mineralisation is compromised further by metabolic acidosis, secondary hyperparathyroidism and impaired hydroxylation of vitamin D. This can be treated with dietary restriction of phosphate, administration of phosphate binders, and 1-OH-vitamin D. The correction of metabolic acidosis is particularly important, as spina bifida patients may have urinary conduits, bladder augmentation or urinary reservoirs, each of which can cause hyperchloraemic acidosis requiring alkali therapy. For more details on CKD, dialysis and RTx, see Chapter 12 (Nephrology).
Hydrocephalus
This is seen in 90% of all children with spina bifida, and is treated with a ventricular shunt. The caudal hindbrain anomaly of the Arnold–Chiari II malformation is present in almost all patients. Associated problems include an increased chance of intellectual impairment, and shunt complications such as infection, obstruction (underdrainage), low-pressure syndrome (overdrainage) or seizures. An average of two shunt changes are needed in the first 10 years. Management includes appropriate schooling for intellectual impairment, and education regarding complications of shunts and their presentation.
The Arnold–Chiari II malformation, syringomyelia and scoliosis
Symptoms can include: swallowing difficulties (due to lower cranial nerve or brainstem dysfunction), choking on foods (especially liquids), nasal regurgitation or gastro-oesophageal reflux when drinking or vomiting, repeated aspiration pneumonia episodes, dysarthria, obstructive sleep apnoea, cyanosis, stridor (inspiratory), hoarse or high-pitched cry, weakness or spasticity of upper limbs, neck pain, headache, scoliosis, dizziness, clumsiness or poor coordination (the last three being cerebellar symptoms).
The tethered cord
This is present in virtually all children with spina bifida (seen on MRI dorsally in the area of the previous defect), because of the nature of the closure of the MMC, but only causes problems requiring surgical intervention in around one third, who manifest symptoms or signs of tethering generally during growth spurts, as in adolescence. These include gait changes (especially crouched gait), back (at site of defect) or lower limb pain, worsening of motor function (decreased muscle strength or increased tone), change in sensory level, change in bladder or bowel habit, altered gait, or progressive orthopaedic deformities of the spine or lower limbs, particularly foot deformities (e.g. unilateral equinus deformity). Imaging includes MRI to confirm tethering and define the regional anatomy, and to identify any associated lesions (syringomyelia, lipomas) that could produce similar symptomatology or signs. Surgical untethering involves reopening the original repair wound, dissecting the scarred part of the cord from the dura and checking for any other areas of tethering (e.g. thickened filum terminale). Tethered cord can recur.
Other significant disabilities (the six Ss)
Skin
Anaesthetic areas of skin can also lead to missing significant pathology that would normally present with pain (e.g. significant infections or orthopaedic injuries that are not felt, and only noticed by swelling or redness). Poorly fitting calipers, crutches or wheelchairs can cause substantial skin breakdown before it is noticed. All of these areas become more of a problem with increasing independence and self-care. In treating pressure areas, avoid film dressings, as these, in combination with the excessive sweating often seen in these children, worsen skin breakdown. The majority of commercially available wound dressings are adequate for pressure ulcers. Management also requires comprises pressure relief.
Other problems
Sex education and pregnancy
Most males with lesions below L3 have penile sensation, and can sustain an erection and achieve ejaculation, although the latter may be abnormal and many males will be infertile. Females, however, have normal fertility. Almost all females with lesions below L3 have vulval sensation and most can experience orgasm. Contraception must be discussed with any adolescent female patient. Group discussions regarding sexuality in disabled people may be very beneficial. Group outings may improve socialisation skills, and encourage appropriate friendships.
Prognosis
Around 80% of children with spina bifida have IQs in the normal range. Among those with normal intelligence, 60% have some form of learning disability, particularly problems with mathematics, sequencing of information, problem-solving and visual perception. Problems with attention and impulsivity, of ADHD proportions, are often seen. Approximately 70% of surviving children walk well enough to function in the community. Social continence (free of urinary incontinence in social situations) is achieved in 85% of school-aged children. Of adult patients, 80% are independent in the community, 30% attend or finish tertiary education, but only one third are gainfully employed.
Short Cases
Developmental assessment
Begin by introducing yourself to the parent and patient. Inspect for the following:
1. Growth parameters: e.g. failure to thrive, associated with syndromal or chromosomal anomalies. Undernutrition or chronic illnesses can be associated with developmental delay, as may be small or large head size.
2. Evidence of any dysmorphic features (various syndromal diagnoses).
3. Appearance of the ‘ex-premature’ infant (beware the ‘ex-premmie’ whose age is not corrected for prematurity).
4. Obvious neurological abnormalities (including ‘floppy infant’ posturing, hemiplegic posturing and involuntary movements).
Testing hearing, with the infant on the mother’s lap, requires initial distraction with a non-noisemaking (i.e. purely visual) stimulus, directly in front of the child. This is then hidden, at which time the noise-maker (e.g. bell) is brought towards the ear from behind (out of range of visual fields) by an assistant (e.g. the chief examiner). On a signal given by yourself, the assistant makes a sound (e.g. rings the bell) at a certain distance from the ear (this varies for different ages), testing each ear in turn and noting whether the child’s facial expression, or activity (in babies), changes, and if the head turns towards the stimulus, localising the sound (in older children). If the conditions are not optimal for testing hearing (e.g. fractious toddler), say so. If there is an equivocal result, it is reasonable to suggest a formal audiological assessment.
1. Sucking and rooting (birth to 4 months, when awake, and to 6 months when sleeping).
2. Palmar grasp (birth to 3 months).
3. Placing, stepping (both from birth to 6 weeks).
4. Landau reflex, a two-stage reflex. With the child supported prone (with your hand under the abdomen), the child should (normally) extend head, trunk and hips. This is the first, and more important, stage. Next, flex the head and neck; normally the response is flexion of trunk and hips, but this is less constant than the first stage (first stage from 4 months, plus second stage from 9 months; gone by 2 years).
5. ATNR. With the child supine, the head is rotated to one side. A ‘fencing’ posture develops, with extension of the ipsilateral upper and lower limb (i.e. the side towards which the head is turned) and flexion of the opposite side (2–6 months). Persistence beyond 6 months is indicative of upper motor neurone problems, especially CP. Maintaining the ATNR posture throughout the time that the head is held turned, such that the child cannot ‘break’ from that position, is similarly significant.
6. Neck-righting reflex. Rotation of the trunk to conform with the position of the head when the head is rotated to one side (6 months to 2 years).
7. Moro reflex (birth to 4 months). As with most primitive reflexes, persistence beyond the usual time of disappearance is pathological. Make a point of focusing not only on the limb movements but also the facial response, for asymmetry (e.g. in hemiplegic CP).
8. Parachute reflex. With the infant held in the prone position, move him or her rapidly, face downwards, towards the floor. The normal reaction is to extend both upper limbs as if to break the fall (appears between 6 and 12 months, usually at 9 months, and persists; its absence beyond 12 months is abnormal). Asymmetry occurs with hemiplegia.
Eye examination
Background information: some important eye conditions
Lids
Ptosis (short for blepharoptosis)
The muscle involved in elevation of the lid is the levator palpebrae superioris, supplied by the third cranial nerve. Congenital causes of ptosis are the most common. Acquired causes include myopathies and third cranial nerve palsy (and Horner’s syndrome causes a partial ptosis). Mechanical problems such as orbital cellulitis or haemangioma may also cause ptosis. Bilateral ptosis should prompt examination for a neuromuscular cause.
Retina
Retinopathy of prematurity (ROP)
Stage 1: flat white demarcation line between avascular and vascularised retina.
Stage 2: ridge due to arteriovenous shunting (demarcation line raised into vitreous).
Stage 3: extraretinal fibrovascular proliferation (new vessels elevated into vitreous).
Stage 4A: partial retinal detachment, macula attached (visual prognosis still hopeful).
Stage 4B: partial retinal detachment, macula detached (visual prognosis poor).
‘Plus’ is added to each stage if there are dilated and tortuous vessels in the posterior retina. This indicates a worse prognosis and more progressive disease.
Squint (strabismus)
Certain terms should be known by the candidate:
• Pseudostrabismus: false appearance of squint (often due to epicanthal folds).
• Orthophoria: perfect condition of ocular balance.
• Heterophoria: latent tendency to squint (e.g. when tired).
• Heterotropia: permanent tendency to squint.
• Non-paralytic squint: not due to a problem with any extraocular muscles or the cranial nerves supplying them.
• Paralytic squint: due to a problem with extraocular muscles or cranial nerves.
Squint can be divided into non-paralytic and paralytic types.
Paralytic
1. Third nerve palsy (frequently congenital; divergent squint, plus downward deviation of eye and ptosis).
2. Fourth nerve palsy (congenital or acquired from head trauma; accompanying head tilt towards opposite shoulder to eliminate a vertical deviation).
3. Sixth nerve palsy (frequently acquired from head trauma; convergent squint).
Procedure
Irrespective of the child’s age, eye examination is always started by inspection for external abnormalities. Commence by looking from in front, from the side and from above to detect any proptosis. Next, focus successively on each of the anatomical structures of the eye to detect any abnormalities (i.e. look at the eyebrows, eyelids, cornea, iris, sclera and conjunctivae). This systematic approach should prevent important signs being overlooked. If the child wears glasses, examine these also.
Finally, for completeness, offer to test the corneal reflex (do not just go ahead and do it, as children tend to find this very distressing), palpate for evidence of raised intraocular pressure (glaucoma) and then auscultate over each closed eyelid with the bell of the stethoscope (with the child holding his or her breath while you auscultate, if possible) for bruits.
1. Finding: papilloedema. The next steps could include a neurological examination for signs of raised intracranial pressure, measuring the blood pressure to detect hypertension and checking for asterixis from hypercapnia.
2. Finding: retinopathy consistent with diabetes. The next steps may include an endocrine assessment and urinalysis.
3. Finding: nystagmus. The next steps would include a full neurological examination; for example, for cerebellar signs, if there is horizontal nystagmus.
Stages of visual development (in relation to clinically applicable testing)
• Neonates turn their head towards a diffuse light source.
• By 6 weeks of age, babies follow a face or large, coloured (especially red) object (which should be silent, so that turning to sound is not misinterpreted). Also by this age the ‘blink to menace’ response is present.
• By 3 months, the eyes converge for finger-play.
• By 4 months, the infant follows objects through 180°, turning the head.
• By 5 months, infants reach for a toy within their visual field and are able to regard a small raisin on a table.
• By 6 months, they move the eyes together in all directions. A squint at this stage is abnormal. This is the earliest age at which the Stycar graded balls test can be used to test the vision.
• By 9 months, they are able to pick up a raisin (raking grasp), look for fallen toys and play peek-a-boo.
• By 12 months, babies are able to pick up a raisin with neat pincer grasp. At this stage, it is possible to test with rolling balls as small as 3 mm at a distance of 3 m. Also, the Stycar mounted balls test for ability to fixate and peripheral vision can be used. In the examination setting, a ball of red wool can be used for peripheral vision testing, bringing it from behind the child and watching the head turn towards the ball when it enters the child’s visual field.
Motor cranial nerves
Next, examine the fifth nerve. You may ask the child the following, demonstrating each move as you proceed. ‘Open your mouth’: the jaw will deviate towards the weak side with a unilateral fifth nerve lesion, pushed by the normal pterygoid. ‘Now keep it open; don’t let me close it’: this tests the pterygoids. ‘Clench your teeth tight’: feel the muscle bulk on each side. Then with your hand against the lateral aspect of the chin, ‘Move your chin towards my hand’, on each side. This tests each pterygoid in turn. Finally check the jaw jerk: ‘Open your mouth a little; I’m just going to tap on your chin’. The jerk will be increased in pseudobulbar palsy.
At the completion of the examination, summarise your findings and give a differential diagnosis. The remainder of the examination should be directed towards confirming any suspected diagnosis, which may entail examination of the gait, lower limbs, upper limbs or neuromuscular assessment (see Figure 13.3).
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]
Chapter 13 Neurology
Long Cases
Cerebral palsy
Children with cerebral palsy (CP) often are used for both long- and short-case examinations. The following is a very brief listing of some of the major issues raised in the CP long case, plus a short-case approach.
Background information
Classification
CP is classified according to the clinical type of neuromotor dysfunction:
Causes
1. Prenatal problems; for example, cerebral malformations, intrauterine TORCH infection (toxoplasma, other [e.g. syphilis], rubella, CMV, herpes [simplex or varicella] or HIV infection), toxins (e.g. drugs), placental insufficiency, fetal coagulation and autoimmune disorders, cerebrovascular accidents, trauma or chromosomal anomalies.
2. Perinatal problems; for example, neonatal encephalopathy, hypoglycaemia, toxins (e.g. kernicterus from hyperbilirubinaemia), viral (e.g. HSV) encephalitis, bacterial (e.g. early onset Group B streptococcus) meningitis, prematurity (babies born before 28 weeks gestation have 50 times the risk of CP compared with babies born at term; mechanisms can include intraventricular haemorrhage and periventricular leucomalacia).
3. Postnatal problems; for example, head trauma (accidental or non-accidental), hypoxic insult (from near-drowning, choking or poisoning), cerebrovascular accident, toxic encephalopathy (e.g. lead), meningitis or hypoglycaemia.
Diagnostic assessment
EEGs are only worthwhile if the patient has had fits, to determine classification of any epilepsy syndrome; they are not useful in determining the aetiology of the child’s CP.
Metabolic conditions. Treatable conditions should not be missed. Example: glutaric acidaemia type 1; mutation in the gene encoding glutaryl-CoA dehydrogenase [GCDH]; alters catabolism of amino acids lysine, hydroxylysine and tryptophan; can impersonate dyskinetic CP; treatable—low-protein (low lysine and tryptophan) diet (avoiding milk, cheese, dairy, meat, poultry, fish, dried beans and legumes, nuts, peanut butter), special low-protein formula, supplements: riboflavin and L-carnitine.
Other metabolic conditions are not treatable but have a different prognosis to CP. First example: Lesch–Nyhan syndrome (X-linked deficiency of hypoxanthine guanine phosphoribosyl-transferase, HGPRT); patients eventually self-mutilate, and diagnosis is made. Second example: Sjogren–Larsson syndrome (mutation in gene coding for fatty aldehyde dehydrogenase); patients eventually develop ichthyosis, and diagnosis is made.
Muscular dystrophies (e.g. Becker muscular dystrophy; eventually proximal myopathy and Gowers sign, and calf hypertrophy).
Mitochondrial disorders (e.g. Leigh syndrome; eventually developmental regression becomes apparent).
Malformation syndromes. Example 1: Miller–Dieker lissencephaly syndrome (MDLS), chromosome 17p13.3; subtle dysmorphic findings (e.g. wrinkled skin over glabella, downward-slanting palpebral fissures) are eventually noticed. Example 2: Rett syndrome; eventually acquired microcephaly, hand-wringing and hyperventilation become apparent.
Level I: Ambulatory in all settings
Level II: Walks without aides, but has limitations in community settings
Most children with CP are born with the condition, but abnormal features may not be noted for months; most are diagnosed by 3 years. Diagnosis of CP before 12 months is fraught with uncertainty, due to the plasticity of the newborn brain; infants can be seen who have apparently clear-cut signs of CP that then resolve by the age of 1 or 2 years. The ‘ideal’ diagnostic age for CP may be 2 years.
History
Current symptoms/functioning
1. Intellectual abilities (or present developmental status), current placement regarding education, domestic situation, employment.
2. Behaviour (e.g. hyperactivity), affect (e.g. depression).
3. Vision (e.g. cortical visual impairment, strabismus, myopia, hemianopia).
4. Speech, hearing and communication problems (e.g. expressive or receptive dysphasia, dysarthria, athetoid movements, use of aids such as communication boards and computers).
5. Activities of daily living (e.g. bathing, cleaning teeth, combing hair, dressing, writing and other hand usage, toileting, menses).
6. Feeding and nutrition (e.g. sucking and swallowing ability, tube feeds, gastrostomy, gastro-oesophageal reflux, aspiration, failure to thrive).
7. Seizures (e.g. type, duration, frequency, usual treatment including side effects, compliance and drug levels, last seizure).
8. Mobility (e.g. walking ability [‘community’, ‘household’ or ‘non-functional’ ambulator], gait pattern, wheelchair mobility), skeletal problems (e.g. kyphoscoliosis, lumbar lordosis, spondylolisthesis, hip subluxation and dislocation, pseudoacetabulum formation, contractures, unequal leg length), abnormal posturing.
9. Other problems: urinary incontinence, constipation, management of menses, chest infections, pressure sores.
Birth history
1. Maternal past history of miscarriages or infertility.
2. Pregnancy: hyperemesis, hypertensive disease of pregnancy, teratogenic medications, placental problems, clinical evidence of, or exposure to, infection (e.g. TORCH), quality of fetal movements, gestational age.
3. Delivery: presentation (e.g. breech, face), instrumental delivery, Apgar score, resuscitation required, birth weight, need for oxygen, nursery care.
4. Neonatal period: respiratory distress, feeding difficulties, seizures, hyperbilirubinaemia (phototherapy or exchange transfusion), intraventricular haemorrhage (IVH), periventricular leukomalacia (PVL), hydrocephalus, retinopathy of prematurity (ROP).
Important signs in examination of the child with CP
See the short case on hemiplegia in this chapter for a suggested examination procedure.
General observations
1. Dysmorphic features (e.g. chromosomal anomalies).
2. Parameters: head circumference (often obvious microcephaly), weight (often failing to thrive), height (usually decreased), progressive percentile charts.
3. Posture (e.g. fisting, increased extensor tone, asymmetric tonic neck reflex [ATNR], hemiplegic, quadriplegic).
4. Movement: (a) involuntary (e.g. choreoathetoid movements, dystonic spasms, seizures); (b) voluntary (e.g. immature gait pattern with wide base, up on toes, arms out for balance; hemiplegic, diplegic gaits; note posturing of arms when walking).
5. Asymmetry (e.g. hemiatrophy: look at the size of the thumbnails and the great toenails for subtle clues to asymmetry).
6. Behaviour (e.g. lack of interaction with environment, crying).
7. Eye signs (e.g. squint, nystagmus).
8. Bulbar signs (e.g. dysarthria, drooling).
9. Interventions (e.g. nasogastric tube, gastrostomy tube, scars of orthopaedic procedures).
Demonstration of signs of CP
1. If possible, perform a standard gait examination.
2. If the child cannot walk, but can crawl, look for abnormal crawling: (a) those with spastic diplegia or quadriplegia—buttock crawling and ‘bunny-hopping’ (jumping while on knees); (b) those with hemiplegia—asymmetrical crawl.
3. Gross motor ‘180° manoeuvre’, incorporating primitive reflexes:
4. Inspect carefully for tendon release scars.
5. Palpate muscle bulk in each muscle group.
6. Tone: as above, plus assessment of upper and lower limbs, and evaluation of contractures (e.g. tight hip adductors, and tendo achilles).
7. Power: voluntary movement, functional power (grasp of toys, cloth cover test).
8. Reflexes: the head should be held in the midline (e.g. by an examiner) so that an ATNR does not give a false impression of unilateral hypertonia; note whether there is any crossed adductor reflex, spread of reflexes, clonus or upgoing plantar responses.
Complications of CP
1. Measure the head (for microcephaly, or macrocephaly due to hydrocephalus).
2. Check the vision, visual fields and extraocular movements (for myopia, squint).
3. Check the hearing (for sensorineural deafness).
4. Check the ears (for chronic serous otitis media).
5. Ask to check the gag reflex (bulbar dysfunction).
6. Look at the teeth (for dental caries).
7. Look at the back (for kyphoscoliosis).
8. Inspect and auscultate the chest (for chest infection).
9. Palpate the abdomen (for constipation).
10. Examine the hips (for dislocation).
11. Screen nutritional status (demonstrate fat and protein stores).
12. Perform a functional assessment for activities of daily living (e.g. offer cup, spoon, fork, knife, comb, toothbrush; ask the child to put on a piece of clothing).
Investigations
1. Brain imaging (MRI) may show abnormalities in some 90% of patients with CP. It may show the basis of CP (e.g. gross malformations, hydrocephalus, intracranial calcification from congenital infection). It may suggest the timing of the aetiology (e.g. cortical dysplasias develop around 12–20 weeks’ gestation; periventricular leukomalacia around 28–34 weeks’ gestation; cortical and subcortical gliosis and atrophy in parasagittal watershed areas in term babies with intrapartum hypoxia) or show unexpected degenerative disorders (e.g. one of the leukodystrophies).
2. TORCH screen (including HIV), in infants, for intrauterine infection.
3. Urinary metabolic screen (various inborn errors of metabolism).
4. Chromosomes (various anomalies).
5. Lumbar puncture in dyskinetic CP, to diagnose the treatable genetic condition, glucose transporter 1 deficiency syndrome, especially if there are associated refractory seizures (see below).
Management
Physiotherapy, occupational therapy, splints/orthoses
Serial casting is useful for reversing ankle/foot equinus in younger children. Over 2–6 weeks, the calf muscle is stretched gradually, with the foot and ankle held in position by a below-the-knee plaster.
Management of spasticity
In 2010, the American Academy of Neurology released an evidence-based review of pharmacological treatments for childhood spasticity due to CP, where a multidisciplinary panel reviewed relevant literature from 1966 to 2008. In the case of localised/segmental spasticity that warrants treatment, the AAN recommendation was that botulinum toxin A (BTX-A) is effective and generally safe in reducing spasticity in the upper and lower extremities, but there is conflicting evidence regarding functional improvement, and severe generalised weakness can occur; there is insufficient data to support or refute BTX-A use to improve motor function. There is insufficient data to support or refute other medications used for the same indication (and these are not discussed further): phenol, alcohol and BTX type B. In the case of generalised spasticity that warrants treatment, the AAN recommendation was that diazepam could be considered for short-term treatment, and also tizanidine may be considered; but, again, there is insufficient data to support or refute other medications used for the same indication: dantrolene, oral baclofen or continuous intrathecal baclofen. The latter is discussed because patients who have received this treatment may present in the examination, so a candidate should have some knowledge of the side effects.
Intrathecal baclofen (IT-BLF)
In view of the lack of data to support this treatment, it may well become of historical interest only. There are many and varied side effects, several related to the mode of delivery: an implantable drug-delivery pump system for continuous infusion. Significant complications include central side effects such as apnoea, respiratory depression, bradycardia, hypotension and sedation, mechanical complications including pump or side-port failure, catheter kinks, extrusions or dislodgement, cerebrospinal fluid fistula, local infection and meningitis; also, rapidly progressive scoliosis has been reported.
Orthopaedic procedures
Surgical procedures for spastic hip displacement
• Preventative (soft-tissue surgery): adductor longus release, gracilis release, adductor brevis release, iliopsoas lengthening, obturator neurectomy (anterior branch only).
• Reconstructive (redirectional osteotomies): femoral varus derotation osteotomy, pelvic osteotomy, combined femoral and pelvic osteotomy with or without open reduction.
• Salvage: excision of proximal femur, valgus osteotomy, interpositional arthroplasty, replacement arthroplasty, arthrodesis.
These procedures lead to significant improvements in function, including improved walking speed.
Other orthopaedic procedures
Upper limb surgery may be offered to correct a flexion–pronation deformity of the wrist, to achieve a better functional position; however, usually only cosmetic improvement is achieved, with little gain functionally, largely because of associated cortical sensory loss. Some children will require surgery for scoliosis (e.g. placing of Luque rods).
Excessive salivation (sialorrhoea)
Anticholinergic drugs (e.g. glycopyrrolate) have been used for excessive drooling with some success, but can have unpleasant side effects, such as urinary retention, constipation, blurred vision, headache, drowsiness, dizziness and behaviour changes.
Cognition/learning and communication
The risk of associated cognitive and learning difficulties varies with the type of CP. Those with quadriplegic CP have the highest risk of cognitive impairment and those with hemiplegic CP have the lowest risk. In children with movement impairment hindering their verbal communication, augmentative and alternative communication systems are available. Aided formal communication systems include communication boards and books, where pictures and symbols are used to communicate specific messages, and electronic devices of varying complexity, some allowing the person to communicate with recorded speech; unaided formal communication systems include key word signing (Makaton vocabulary), and informal communication systems include facial expression, eye contact, vocalisations, body language and gestures, to convey feelings, wants and needs.
Pain
This is a common complaint, which can be very challenging in the non-verbal child with severe CP. The most common sites of pain seem to be gastrointestinal (constipation, GORD), and orthopaedic/musculoskeletal (scoliosis, hip dislocation, patella alta); other possibilities include: neuromuscular—muscle spasms; head and neck—migraine, raised intracranial pressure, glaucoma, corneal abrasions, tooth abscess, temporomandibular joint pain; urological—urolithiasis, bladder spasm; skin—boils, decubitus ulcers. A thorough history (e.g. temporal association with feeding, or nappy changes) and thorough general examination (e.g. inspecting the mouth with a torch and spatula to find tooth abscesses) may find the cause, but often empirical interventions are tried before the patient is eventually free of discomfort.
Prognosis
A few ‘rules of thumb’ are worth knowing when parents ask about prognosis; most parents ask whether their child will walk. While unlikely to be an active discussion issue with a patient in a long case, examiners may ask hypothetical questions such as how to approach answering the parents’ concerns when the concept of permanent neurological damage has been mentioned. Note the following: children with hemiplegic or diplegic CP usually walk, those with quadriplegic CP rarely walk; those with the dyskinetic subtype are more difficult to predict. Most children who can sit independently by 2 years will walk, but most children who cannot sit by 4 years are unlikely to walk. In terms of life span, only the most profound degrees of CP are associated with a decreased life expectancy. If a child cannot lift the head to prone, and requires tube feeding, the median survival is around 17 years.
Dystrophinopathies: Duchenne muscular dystrophy (DMD)
Background information: genetics of DMD
DMD is an X-linked recessive disorder, due to mutations (often deletions) in the dystrophin gene on the X chromosome (Xp21.2). It has a frequency of 1 in 3600–6000 male births. The DMD gene is the largest known human gene. It is around 2000 kilobases in size and codes for dystrophin, a large, rod-like 427-kD protein containing 3685 amino acids and located at the inner face of the muscle cell membrane. Dystrophin binds with a group of ‘dystrophin-associated proteins’ (DAPs) (e.g. sarcoglycans, dystroglycans, merosin) that span the muscle membrane, linking the muscle cytoskeleton and the extracellular matrix (see Figure 13.1). Absence of dystrophin disrupts the link, making the muscle membrane susceptible to damage from shearing stresses. Hence, dystrophin-deficient muscle is very susceptible to muscle injury, and degeneration of muscle fibres is a feature of dystrophic muscle. Dystrophin is undetectable in the muscle of DMD patients. Virtually all males with DMD have identifiable mutations. Over 4700 mutations in the DMD gene have been identified. Disease-causing alleles can include deletion of the complete gene, deletion or duplication of exons, small deletions, insertions and single base changes. Around two thirds of patients with DMD have intragenic out-of-frame (gross rearrangements) deletions, and around 10% have duplications of one or more exons of the gene; the remaining 25% have point mutations or other small rearrangements, including intronic deletions, insertions of repetitive sequences and splice site mutations. Generally out-of-frame mutations cause lack of dystrophin (and DMD), whereas in-frame mutations cause abnormal but partially functional dystrophin, resulting in BMD. The tissue distribution of dystrophin correlates with clinical features. It is found in skeletal, cardiac and smooth muscle, and results in skeletal muscle weakness and cardiomyopathy. In DMD-associated DCM, dystrophin expression is abnormal in myocardium, but may be normal or mildly abnormal in skeletal muscle. Dystrophin is found within the central nervous system, resulting in a static encephalopathy and cognitive deficits. Various forms of dystrophin are expressed in neurons and glia in the brain, especially the cortex, hippocampus, cerebellum and retina. Molecular genetic testing of DMD can confirm diagnosis of a dystrophinopathy without need for muscle biopsy in most patients ith DMD or BMD. There is a high incidence of new mutations, and two thirds of new patients have no positive family history.
Figure 13.1 Subsarcolemmal cytoskeleton.
Redrawn from; South, Isaacs, Roberton 2007. Practical Paediatrics 6th edition, p. 609, Figure 17.3.4.
Molecular tests for DMD
1. Multiplex PCR, Southern blotting and FISH are utilised to detect deletions, which account for two thirds of mutations in DMD.
2. Southern blotting and quantitative PCR analysis are utilised to detect duplications, which account for 6–10% mutations in males with DMD.
3. Multiple ligation probe amplification (MLPA) is utilised to analyse deletion/duplication of the gene in probands and carrier females.
4. Mutation scanning or sequence analysis is utilised to detect small deletions or insertions, single base changes, and splicing mutations that make up around a third of mutations in DMD.
5. More recent methods, including single-condition amplification internal primer sequencing (SCAIP) and denaturing gradient gel electrophoresis (DGGE)-based whole-gene mutation scanning, are utilised to detect the remaining third of mutations not yet elucidated by the above tests.
6. Muscle biopsy-based approaches utilising protein- and RNA-based analyses in combination with direct cDNA sequencing increase the mutation detection frequency to almost 100%.
7. Prenatal diagnosis is available by chorionic villus biopsy or amniocentesis by direct testing for abnormalities in the dystrophin gene.
Recent advances
In the last few years, the natural history of this disease has been changed by interventions (e.g. steroids, cardiac, respiratory, orthopaedic, rehabilitative), quality of life has improved and affected patients may now reach their fourth decade (a significant advance on the previous life expectancy of 19 years). Research into DMD has continued to accelerate, with a number of different treatment strategies being proposed. The role of steroids has become clearer. Glucocorticoids remain the only medication currently available that can slow down the decrease in muscle strength and function in DMD; this then reduces scoliosis and stabilises respiratory function. Low-dose steroids are very useful when boys are still walking, to improve motor function. The time of commencement of steroids remains controversial; currently, recommendations are to wait until identifying a plateau in the child’s motor development, where there is no longer progress in motor skills. Once that plateau is identified, steroids are commenced; they are not recommended for children still gaining motor skills, especially under 2 years. Typically, an affected male will progress with motor skills until 4–6 years old. Presently (2010), corticosteroid therapy is the treatment of choice for affected patients between 5 and 15 years of age. Some studies, however, suggest that the ideal window for treatment could be under 5 years. These patients need close monitoring, adjusting dose and timing to avoid unwanted side effects, especially weight gain.
Primitive stem cells in bone marrow have been shown to migrate into muscle and become new muscle cells. Stem cell therapy is under investigation but still experimental. Gentamicin has been found to permit cells to ignore an abnormal stop codon in the dystrophin gene and to proceed and synthesise the protein in the 15% of DMD patients who have premature stop codons as their underlying mutation. PTC124 is a new agent that may permit ribosomal read through of nonsense mutations. Morpholino antisense oligonucleotides permit exon skipping.
History
Current problems
1. Functional abilities with activities of daily living (e.g. dressing, writing, toileting); aids required (e.g. splints, supportive prostheses, computer-assisted communication/learning).
2. Mobility (e.g. long leg braces, wheelchair use, school and home access).
3. Home modifications required (e.g. ramps, bathroom fittings, specialised beds and mattresses).
4. Transport needs (e.g. van with hoist).
5. Scoliosis (e.g. progression, any planned surgery).
6. Joint contractures (AFOs, night splints).
7. Respiratory problems (e.g. symptoms of respiratory failure at later stage, sleep disordered breathing, non-invasive ventilation).
8. Cardiac symptoms (e.g. arrhythmias, symptoms of cardiac failure).
9. Gastrointestinal problems (e.g. incontinence, vomiting).
10. Difficulties with micturition.
11. School (e.g. any access problems, educational problems, any help needed or provided with toileting; attitudes of class teacher, headmaster, fellow students).
Past history
1. Initial diagnosis: when, where, presenting symptoms (e.g. late walking, tendency to fall, method of rising after fall, clumsiness, muscle cramps, learning delay, cognitive impairment, global or gross motor developmental delay).
2. The time period between onset of symptoms and diagnosis.
3. What investigations were undertaken to make the diagnosis.
4. Stages of deterioration (e.g. age at which the child lost the ability to climb stairs, stand from the floor or walk independently).
5. Number of hospitalisations.
6. Development of complications (e.g. scoliosis, cardiac failure) and their management.
Social history
1. Impact on child (e.g. difficulties at school, poor job prospects, limitations on lifestyle, body image, self-esteem, peer reactions).
2. Impact on family (e.g. parental coping, difficulties between mother and father, genetic implications for further children; financial considerations, such as cost of wheelchairs, home modifications, transport, hospitalisations, private health insurance; physical burden of helping children in and out of wheelchairs, cars, bed, bath).
4. Social supports (e.g. social worker, DMD Family Support Group provided by the Muscular Dystrophy Association, visits to school by occupational therapist and community liaison nurse from hospital muscle clinic to meet class and teachers).
Family history
Other known family members with DMD, other males with developmental delay or late walking.
Examination
Gait
Boys with DMD can walk well on their toes, but are unable to walk on their heels, and if they try they end up inverting the feet. On squatting, these children are slow to return to standing, need to extend the knee before the hip, and lean on their thighs to assist extension at the hip.
Of particular importance is the elicitation of Gower’s sign. The boy is asked to lie supine on the floor and then to get up. This first leads to rolling over to be prone (he cannot sit up because of weak neck and spine flexion), then on to the knees, then on ‘all fours’—that is, hands and feet—then ‘climbing up’ the legs to stand. Gower’s sign is often used as a functional timed test: timing how long it takes to arise from the floor is useful to monitor deterioration over time. See Figure 13.2.
Muscle power
After comprehensive assessment of muscle power, check for tone and reflexes (knee jerks often lost) and contractures, especially at the ankles, knees and hips.
Management
The main management areas are described below.
Psychosocial
1. Education of parents and patient. Clarification of misinformation from other sources requires multiple informative sessions with the various disciplines involved in the comprehensive management of these patients. Regular feedback is needed to ensure that no misunderstandings occur. Other family members (e.g. siblings, grandparents, aunts, uncles and cousins) are often very much involved emotionally and tend to be neglected.
2. Expectation of a grief reaction to the diagnosis and its implications. Preparatory explanation to parents of probable feelings such as guilt, anger and depression. Explanation regarding any misapprehensions or strange beliefs about the nature of the illness.
3. Ensuring sufficient social supports, both from professionals (such as social workers) and non-professionals (such as other affected patients, families of other patients and groups such as the Muscular Dystrophy Association [MDA], Montrose Access [Qld] or Northcott [NSW]). The MDA provides parents’ groups and a useful handbook that deals with the home situation, recreational activities, education and vocational possibilities. There are excellent sites on the internet, including http://mdausa.org>and <http://www.treat-nmd.eu/patients/DMD/family guide/
4. Informing about financial assistance measures, such as any government benefits (which may assist directly with the child’s care, transport costs, accommodation costs and provision of aids). The illness places a very large financial burden for home modification (such as ramps, bathroom modification, lifting machines) on the family.
5. Discussion of treatment difficulties (such as non-compliance), seeking other opinions, and understanding and acceptance of alternative therapies sought by parents.
6. A major point is helping the family to determine how various people (especially the affected son) are to be informed. The paediatrician may not be the person who will help most, but he or she does have responsibility to ensure that it is done adequately. The consequences of failure in this aspect of management may include: parents who will not discuss the disease with their son, siblings, teachers or each other; one parent (usually the father) who withdraws from reality; and help being refused (because the parents believe that the child will be cured by divine intervention).
