Chapter 21 Neurological disorders – epilepsy, Parkinson’s disease and multiple sclerosis
• This chapter focuses on several common neurological disorders, each of which has a wide range of therapeutic strategies available. These disorders are: epilepsy, Parkinson’s disease and multiple sclerosis. The treatments of other common neurological disorders are covered in other sections: namely: headaches (Pain section: Ch. 18), stroke (Ch. 24) and dementia (Ch. 20).
• It also touches on the pharmacological principles of other neurological disorders, including: movement disorders other than Parkinson’s disease; spasticity (that is a physical sign characteristic of certain diseases such as stroke or multiple sclerosis); peripheral neuropathy; motor neurone disease; tetanus.
Epilepsy
Definitions
A seizure is a clinical symptom or sign caused by abnormal electrical discharges within the cerebral cortex.1 For example, a tonic–clonic seizure refers to a pattern by which a patient loses consciousness, becomes generally stiff (tonic), and subsequently jerks all limbs (clonus); whereas a complex partial seizure refers to a constellation of impaired consciousness, déjà vu sensations, epigastric rising sensation, olfactory hallucinations and motor automatisms, e.g. lip smacking.2 By contrast, epilepsy refers to the clinical syndrome of recurrent seizures, and implies a pathological state that predisposes to further future seizures. Hence having one, or even a single cluster of seizures (i.e. over a few days) does not in itself qualify as epilepsy, since these seizures may have been due to a febrile illness or drug intoxication that themselves later resolve. By contrast, having at least two seizures, separated by at least a few weeks, is usually sufficient to signify epilepsy. Only one-third of people having seizures develop chronic epilepsy.
Pathology and seizure types
Epilepsy affects 0.5–1% of the general population, while the lifetime risk of having a seizure is 3–5 %. There are both multiple causes and multiple seizure types.3 Approximately half of adult epilepsy is believed to be due to genetic or early developmental causes, although the exact nature of these – e.g. sodium channel mutations or cerebral palsy – are determined in only a small minority. The other half of adult epilepsy is due to acquired causes, such as alcohol, stroke, traumatic head injury or brain tumours.
The cause of epilepsy determines the seizure type. Genetic causes (i.e. ‘primary’) predispose to generalised seizures4 characterised by tonic–clonic or absence seizures (lapses of consciousness lasting seconds), myoclonus (random limb jerk at other times), photosensitivity (seizures triggered by flashing lights), EEG showing a 3-Hz spike-and-wave pattern, and a normal MRI brain. Conversely, where focal brain injury has occurred, e.g. brain tumour or stroke, and the brain scan is abnormal, focal epileptic discharges occur within the brain leading to a partial seizure – i.e. when only a narrow set of brain functions are disturbed, e.g. causing single limb jerking (implying motor cortex involvement). Importantly, partial seizures can propagate very quickly to become a ‘secondary generalised seizure’. Another common cause of adult-onset partial epilepsy is maldevelopment of the medial temporal lobes (‘mesial temporal sclerosis’) believed to be due to injury, e.g. hypoxia or infection, during fetal or early childhood life, and sometimes apparent as atrophic hippocampi and amygdala on high-resolution MRI.
Principles of management
• Identification of underlying cause and treatment of this where possible, e.g. cerebral neoplasm or arteriovenous malformation.
• Educate the patient about the disease, duration of treatment and need for compliance.
• Counselling the patient about avoiding harm from seizures, e.g. driving regulations, swimming or bathing alone and climbing, as well as other dangerous pursuits, to be avoided.
• Avoid precipitating factors, e.g. alcohol, sleep deprivation, stroboscopic light.
• Anticipate natural variation, e.g. fits may occur particularly or exclusively around menstruation in women (catamenial5 epilepsy).
• For most cases with recurrent seizures, an antiepileptic drug is prescribed with subsequent monitoring and adjustment of dosage or drug type (see below).
• Consider surgical therapies in patients with refractory seizures, e.g. vagal nerve stimulation, temporal lobectomy. For childhood refractory epilepsy, a ketogenic diet – i.e. high fat:carbohydrate ratio – is useful, as ketone bodies are antiepileptogenic.
• Acute treatment of generalised convulsive seizures consists of ensuring the patient lies on the floor away from danger, and is postictally manoeuvred into the recovery position. If a seizure continues for more than a few minutes, rectal or buccal diazepam or intranasal midazolam can be given. If convulsive seizures last for more than 5 min, patients should be transferred to hospital for consideration of intravenous benzodiazepine and phenytoin.
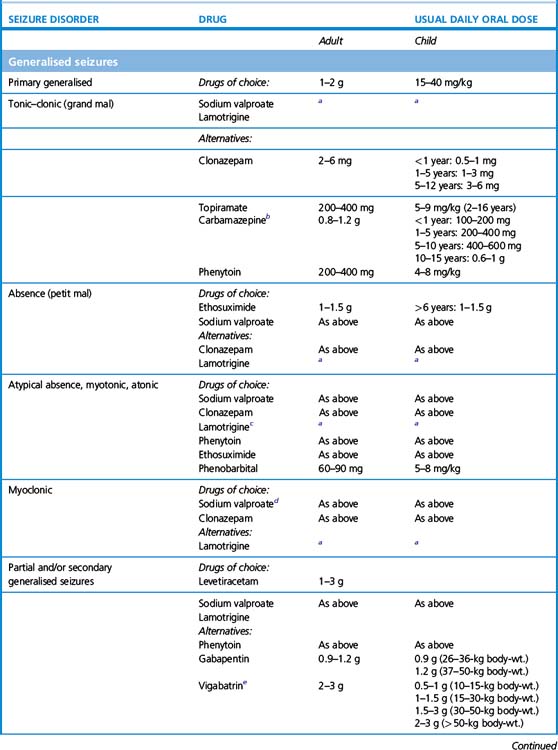
Practical guide to antiepilepsy drugs
1. When to initiate. Following a single seizure the chance of a further seizure is approximately 25% over the following 3 years. Furthermore, only 33% of single-seizure patients develop chronic epilepsy. Hence the majority of first seizures are provoked by a reversible, and often recognisable, factor, e.g. infection, drug toxicity, surgery. For these reasons, following a single seizure6 anticonvulsants are not generally prescribed, whereas after two or more distinct seizure episodes (i.e. with more than a few weeks apart between episodes), they generally are prescribed. Immediate treatment of single or infrequent seizures does not affect long-term remission but introduces the potential for adverse effects. Patients need to be made aware that anticonvulsant therapy reduces harm caused by generalised seizures, and may also reduce the risk of sudden death in epilepsy (SUDEP), that usually occurs during sleep.
2. Monotherapy. Although the choice of anticonvulsants is large (approximately 20), first–line therapy is generally restricted to one of only a few drugs that have a good track record and are relatively safe and well-tolerated. Initial therapy is confined to a single drug (i.e. monotherapy) that is usually effective in stopping seizures or at least significantly decreasing their frequency. The majority of epilepsy patients (70%) can remain on monotherapy for adequate control, although sometimes the choice of monotherapy may need to be switched to allow for tolerance or optimisation of seizure control. As the number of single anticonvulsants tried increases, the incremental likelihood that any new one will offer a significant reduction in seizures decreases: from 50% response to a first drug, to an additional 30% to a second drug, to an extra 10% to a third drug, and less than 5% for any subsequent drug tried.
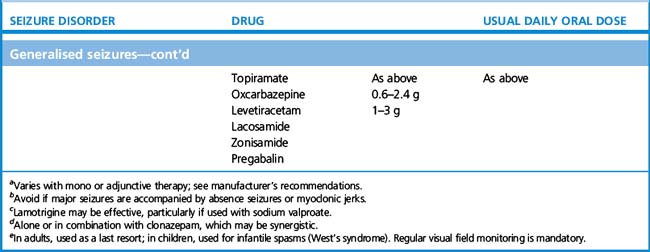
3. What drug to initiate. For older types of anticonvulsants, knowing the seizure type – i.e. whether partial or primary generalised – mattered, because in certain cases the spectrum of seizure efficacy is limited, and, moreover, certain seizure types can be worsened by ill-chosen drugs. For example, carbamazepine is an effective first-line therapy for partial seizures but may worsen primary generalised, absence or myoclonic seizures; similarly phenytoin can worsen absence and myoclonic seizures. Ethosuximide, by contrast, is only effective in primary generalised, and not partial, seizures.
More modern anticonvulsants, by contrast, are in general effective over a much broader range of seizure types allowing for more confidence of use even when seizure type is uncertain. Thus sodium valproate, lamotrigine and levetiracetam are active against both primary and secondary generalised epilepsy, and being relatively well tolerated, account for most first-line prescriptions. In one head-to-head study comparing popular first-line therapies for generalised and partial seizures, lamotrigine was generally tolerated better than other drugs, while valproate was the most efficacious; carbamazepine and topiramate were more likely to cause unwanted effects.7
4. Women of reproductive age and children. These categories of patients prompt selection of particular drugs and avoidance of others (see below for more detail).
5. Polytherapy. If a trial of three or so successive anticonvulsants (i.e. taken as monotherapy at adequate dosage for at least several months) does not control a patient’s epilepsy, it may be worthwhile trying dual therapy. Polytherapy offers the theoretical advantage of controlling neuronal hyperexcitability by more than one mechanism, that can be synergistic. In reality, increasing polytherapy often adheres to the law of diminishing returns, viz. the proportion of uncontrolled patients who show a positive response decreases at each addition of drug number And at the same time, adverse effects become more likely.
6. Abrupt withdrawal. Effective therapy must never be stopped suddenly, as this is a well-recognised trigger for status epilepticus, which may be fatal. But if rapid withdrawal is required by the occurrence of toxicity, e.g. due to a severe rash or significant liver dysfunction, a new drug ought to be started simultaneously. The speed by which the dose of a new drug can be raised varies according to drug type and urgency.
7. Circumstantial seizures. In cases where fits are liable to occur at a particular time, e.g. the menstrual period, adjust the dose to achieve maximal drug effect at this time or confine drug treatment to this time. For example, in catamenial epilepsy, clobazam can be useful given only at period time.
Failure to respond
• Non-compliance, diarrhoea and vomiting, patients instructed to be ‘nil by mouth’ (revealed by measuring blood concentrations of drug).
• Inadequate dosing, including the possibility of drug interaction, e.g. another drug reducing the effective dose of the anticonvulsant by hepatic enzyme induction.
• Pregnancy also causes hepatic induction, and reduces the effective dose of lamotrigine.
• Increase in the severity of an underlying disease, e.g. enlargement of a brain tumour, or new disease.
• Drug resistance, e.g. genetic polymorphisms in hepatic cytochromes (such as CYP 2 C9) that metabolise drugs, sodium channel subunit SCN1A, or the P glycoprotein drug transporter (ABCB1 gene) that expels drugs from neurones.
Drug withdrawal
If patients have remained seizure-free for more than a few years, it is reasonable to consider withdrawal of antiepilepsy drug therapy.8,9 The prognosis of a seizure disorder is determined by:
• Type of seizure disorder – benign rolandic epilepsy, solely petit mal or grand mal seizures confer a high chance of full remission, whereas juvenile myoclonic epilepsy, temporal or frontal lobe epilepsies often require lifelong treatment.
• Time to remission – early remission carries a better outlook.
• Number of drugs required to induce remission – rapid remission on a single drug is a favourable indicator for successful withdrawal.
• MRI brain scan findings – presence of an underlying lesion predicts difficult control.
• EEG findings – epileptogenic activity is a predictor of poor outcome for drug withdrawal.
• Associated neurological deficit or learning difficulty – control is often difficult.
• Length of time of seizure freedom on treatment – the longer the period, the better the outlook.
Pregnancy and epilepsy
Pregnancy worsens epilepsy in about a third of patients, but also improves epilepsy in another third. One of the main concerns in this patient group is that all anticonvulsants increase the chance of teratogenicity slightly, with valproate, phenytoin and phenobarbital carrying most risk. The toxicological hazard must be weighed against the risk of seizures which themselves can be harmful to mother and unborn baby, and are likely to worsen if anticonvulsants are discontinued. For instance, the risk of major congenital anomalies in the fetus is 1% for healthy mothers, 2% in untreated epileptic mothers (in observational studies, so generally not severe epileptics), and 2–3% in mothers on epilepsy monotherapy. Valproate, by contrast, has been associated with a malformation rate of approximately 10%,10 while 20–30% of children are subsequently found to have mild learning disabilities or require ‘special needs’ education. The UK maintains a national drug monitoring register of all pregnant women taking antiepileptic drugs.
• neural tube defects are related to deficiencies in folic acid stores before pregnancy, so that antiepileptic drugs that affect stores, e.g. valproate, can be avoided, and folic acid 5 mg per day given for several months in advance, and
• adjustments in dose and type of drug can be avoided in the early stages of pregnancy as there is a higher risk of toxicity and seizure breakthrough during this critical phase of fetal development. In general, patients having seizures with blackouts should be on an effective dose of an anticonvulsant, because of the risks of anoxia, lactic acidosis and trauma.
Status epilepticus
Always investigate and treat the cause of a generalised seizure. Give aciclovir i.v. if viral encephalitis is suspected or, if status is triggered by removing an antiepileptic drug, it must be re-instituted. Magnesium sulphate is the treatment of choice for seizures related to eclampsia (see also p. 125).11
| Status | Treatment |
|---|---|
| Early | Lorazepam 4 mg i.v., repeat once after 10 min if necessary, or clonazepam 1 mg i.v. over 30 s, repeat if necessary, or diazepam 10–20 mg over 2–4 min, repeat once after 30 min if necessary |
| Established | Phenytoin 15–18 mg/kg i.v. at a rate of 50 mg/min, and/or phenobarbital 10–20 mg/kg i.v. at a rate of 100 mg/min |
| Refractory | Thiopental or propofol or midazolam with full intensive care support |
Pharmacology of individual drugs
Modes of action
Enhancement of gamma-aminobutyric acid (GABA) transmission
Examples: benzodiazepines, phenobarbital, valproate, vigabatrin, tiagabine.12 By enhancing GABA, the principal inhibitory transmitter of the brain, neuronal membrane permeability to chloride ions is increased, which secondarily reduces cell excitability. Benzodiazepines and barbiturates activate the GABA receptor via specific benzodiazepine and barbiturate binding sites.
Sodium channel blockers
Phenytoin
GABA-potentiators
Sodium valproate
The main concerns, particularly to women, are weight gain, impaired glucose tolerance, teratogenicity (see p. 119), polycystic ovary syndrome and loss of hair, which grows back curly.13 Nausea and dyspepsia may be a more general problem, ameliorated by using an enteric-coated formulation. Some patients exhibit a rise in liver enzymes, which is usually transient and without sinister import, but patients should be closely monitored until the biochemical tests return to normal as, rarely, liver failure occurs (risk maximal at 2–12 weeks); this is often indicated by anorexia, malaise and a recurrence of seizures. Other reactions include pancreatitis, coagulation disorder due to inhibition of platelet aggregation or thrombocytopenia, and hyperammonaemia that can present with acute confusion.
Parkinson’s disease and parkinsonism
Definitions
Parkinson’s disease 14 refers to a specific neurodegenerative disease characterised pathologically by intracellular accumulation of Lewy bodies and subsequent neuronal loss, predominantly within the substantia nigra pars compacta (SNpc)15 (see Fig. 20.3, p. 323). This midbrain nucleus normally provides dopaminergic input to the neostriatum (caudate and putamen), a critical aspect to motor control; one cardinal feature of Parkinson’s disease is that motor symptoms are reversible by administration of dopaminergic drugs. By contrast, parkinsonism refers to the clinical symptoms and signs of Parkinson’s disease (tremor, rigidity, bradykinesia and postural imbalance) that may arise either from Parkinson’s disease or one of its mimics; the latter include extensive small-vessel cerebral ischemia due to hypertension, neuroleptic drugs or other neurodegenerative disease such as multiple-systems atrophy (MSA) or Huntington’s disease. Note that parkinsonism due to conditions other than Parkinson’s disease generally responds poorly to dopaminergic therapies. Another distinguishing feature is that symptoms and signs of Parkinson’s disease, but not other causes, are often asymmetric.
Pathophysiology of Parkinson’s disease
Parkinson’s disease16 is the second commonest neurodegenerative disease after Alzheimer’s disease. Both diseases show an exponentially increasing risk with age, with the risk of Parkinson’s disease rising from approximately 0.2% under the age of 60 to 1% over the age of 60 and 4% of people over 85 years old. At the time that a clinical diagnosis first becomes apparent, radioactive dopamine uptake scans (sensitive to dopaminergic neurones) reveal that approximately 70% of the patient’s nigrostriatal dopaminergic neurones have already been lost. The implication of this finding is that treatments which might actually halt neuronal death (‘disease-modifying drugs’ as opposed to ‘symptomatic treatments’) should ideally be used in pre-symptomatic cases, e.g. as identified by dopamine scanning of the elderly, or relatives of affected individuals. In only about 15% of Parkinson’s cases is there a clear family history, and not more than 10% of cases are caused by a recognised gene mutation.17 Furthermore, no current treatment strategies have been shown to prevent disease progression, with the possible exception of rasagiline (see below). Rather, existing drug therapies primarily serve to enhance dopaminergic neurotransmission, whose deficiency underlies the main symptoms of Parkinson’s disease.
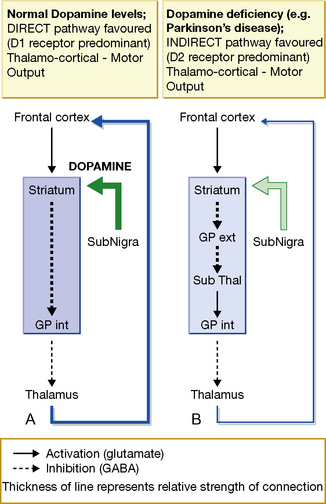
The pathophysiology of Parkinson’s disease, and its critical dependency upon dopamine, arises from an imbalance between two striatal–thalamic–cerebral cortical circuits (see Fig. 21.1). To understand this, it is useful to remember that under normal circumstances the globus pallidus INternus (GPi) INhibits thalamic communication with the frontal cortex, thereby braking voluntary movements. Since dopaminergic input to the striatum itself suppresses GPi (via a ‘direct’ pathway), loss of dopaminergic input allows the GPi to be overactive, thereby favouring inhibition of motor cortex, and so causing slowness of movement (bradykinesia) or freezing. Loss of dopamine also encourages an indirect pathway between striatum, subthalamic nucleus and GPi, which has the net effect of increasing GPi braking of movement. Therefore replacing dopamine can increase the relative contribution of the ‘direct’ relative to ‘indirect’ pathway. Furthermore, inducing neurosurgical stereotactic against the globus pallidus or subthalamus can offer a reduction in bradykinesia.18
Objectives of therapy
Enhancement of dopaminergic neurotransmission
• Administering the dopamine precursor levodopa.
• Decreasing endogenous clearance and breakdown of dopamine by inhibiting monoamine oxidase (selegiline, rasagiline) or catechol-O-methyltransferase (entacapone, tolcapone).
• Stimulating dopamine receptors with agonists (ropinirole, pramipexole, cabergoline, rotigotine, apomorphine).
• Increasing pre-synaptic dopamine release (amantadine – this mechanism being only one of many by which this drug may be effective).
• Avoiding dopamine antagonists (especially traditional neuroleptics such as haloperidol, and antiemetics such as prochlorperazine).19
Pre-empting and treating levodopa-induced motor complications
Although levodopa is the most effective symptomatic treatment at all stages of the disease, its main drawback is that it usually results in treatment-induced fluctuations in motor control (‘on–off’ phenomena) and dyskinesias (i.e. excessive purposeless movements appearing as restlessness or rocking) by about 5–10 years from treatment initiation. One of the main factors associated with this complication is the total previous exposure to levodopa. Hence, in patients under approximately 70 years old, it is preferable to begin therapy with drugs other than levodopa, typically dopamine agonists or an MAO inhibitor. As the disease progresses, and symptoms become more severe in spite of non-levodopa therapies,20 levodopa is introduced, albeit in small doses, and increasing only very gradually so that only the smallest dose providing reasonable symptom relief is used. In patients over 70 years old, long-term dyskinesias are not so relevant, and moreover alternatives to levodopa, namely dopamine agonists, anticholinergics and amantadine, are more prone to cause confusion in this age group.
One of the theories for why motor fluctuations occur is related to the pulsatile nature by which levodopa is traditionally administered, which results in brain concentrations of levodopa (and dopamine) that rise and fall several times over a typical day. This is in contrast to the ‘physiological mode’ of dopamine release that is to a large extent tonic. Pulsatile, rather than smooth, administration of levodopa or dopamine agonists has been found in animal models to result in a gradual shortening of treatment response and dyskinesias. Consequently, methods which ‘smooth’ circulating levodopa concentrations may protect against eventual development of motor complications. Furthermore, a more sustained and steady supply of levodopa compared to conventional levodopa preparations can be achieved by using catechol-O-methyltransferase (COMT) inhibitors (entacapone or tolcapone) taken at the same time as levodopa, which has the effect of inhibiting dopamine catabolism, and so increasing dopamine availability. Using this strategy from the outset of Parkinson’s disease is controversial, with one study21 showing that levodopa-COMT inhibitors started as first-line therapy result in the sooner development of dyskinesias relative to standard levodopa therapy, possibly due to the fact that patients are exposed to higher effective concentrations of levodopa from early on.
Drugs for Parkinson’s disease
Dopaminergic drugs
Levodopa and dopa decarboxylase inhibitors
Levodopa (‘dopa’ stands for dihydroxyphenylalanine) is the natural amino acid precursor of dopamine.22,23 It is readily absorbed from the upper small intestine24 by active amino acid transport, and traverses the blood–brain barrier by a similar active transport mechanism. Within the brain levodopa is decarboxylated (by dopa decarboxylase) to dopamine.
A major disadvantage is that levodopa is also extensively decarboxylated to dopamine in peripheral tissues, such that only 1–5% of an oral dose of levodopa reaches the brain. This means that large quantities of administered levodopa would be required for a meaningful antiparkinsonian effect. Such high doses cause a high rate of adverse effects caused by peripheral actions of levodopa and dopamine, notably nausea, cardiac arrhythmia and postural hypotension. Furthermore, high-dose levodopa inhibits gastric emptying and results in erratic delivery to the absorption site and fluctuations in plasma concentration. This problem has been largely circumvented by the development of peripheral decarboxylase inhibitors, which do not enter the CNS, and so selectively prevent peripheral conversion of levodopa to dopamine. Thus by combining levodopa with a peripheral decarboxylase inhibitor, unwanted effects due to peripheral dopamine production are minimised,25 while the proportion of ingested levodopa available for export into the CNS is maximised. Decarboxylase inhibitors are given in combination with levodopa in one of several formulations:
• Co-careldopa (carbidopa + levodopa in respective proportions 12.5/50, 25/100 and 50 mg/200 mg26) (Sinemet).
• Co-beneldopa (benserazide + levodopa in proportions 12.5/50, 25/100, 50 mg/200 mg) (Madopar).
• Co-careldopa with entacapone (carbidopa + levodopa + entacapone in same proportions as co-careldopa plus entacapone 200 mg with each tablet) (Stalevo).
Levodopa-induced dyskinesias are dose-related, so that in general the lowest dose of levodopa is used that achieves a reasonable degree of symptom relief. Further strategies that can be used against motor fluctuations/dyskinesias include: avoiding taking levdopa with meals;27 using small doses more often; slow-release levodopa preparations taken before sleep,28 and use of adjunctive medications such as dopamine agonists;29 amantadine,30 and COMT inhibitors (taken together with levodopa). In advanced cases, a nasoduodenal feeding tube can be placed that allows for continuous enteric infusion of levodopa gel (Duodopa), or patients can be offered deep brain stimulation. All these methods, in enhancing the effectiveness of dopamine or providing alternative antiparkinsonian methods, enable a reduction in levodopa dose, and so provide strategies in cases where dyskinesias are prominent.
In the short term, the main unwanted effect is nausea, which can be minimised by increasing dosage gradually, and offering patients a safe antiemetic such as domperidone (a peripherally confined dopamine antagonist) or cyclizine (an antihistamine), taken half an hour before levodopa. This is usually only required for the first few weeks. Postural hypotension may occur,31 although this can develop as a feature of advanced Parkinson’s disease due to degeneration of noradrenergic nuclei within the brainstem and cord. Agitation and confusion, including visual hallucinations, may occur but it may be difficult to decide whether these are due to drug or to disease. Mental changes are particularly likely in the elderly, especially when there is pre-existing dementia. If acute confusion occurs, other Parkinson’s drugs that cause confusion – antimuscarinics, amantadine or dopamine agonists should be progressively withdrawn before levodopa. Alternatively, the anticholinesterase rivastigmine, or atypical neuroleptics such as quetiapine or clozapine, may be of benefit.
Dopamine agonists
• Reproducing the right balance of D1 and D2 stimulation (dopamine itself is slightly D1 selective, in test systems, but its net effect in vivo is determined also by the relative amounts and locations of receptors – which differ in parkinsonian patients from normal).
• Avoiding the undesired effects of peripheral, mainly gastric, D2 receptors.
(ropinirole, pramipexole, rotigotine). These drugs are currently preferred as first-line therapy in newly diagnosed patients under the age of 70 years old. Beyond this age group, the long-term complications of levodopa are not felt to be so relevant (and are milder in older-onset Parkinson’s patients) allowing for levodopa use from the outset. Furthermore, psychosis32 or confusion (unwanted effects of all dopamine agonists) are more likely in elderly patients taking these drugs.
Apart from psychosis in elderly patients, the other notable unwanted effects of the non-ergot dopamine agonists are: postural hypotension, ankle oedema, daytime somnolence (including sudden sleep attacks in a minority of patients33), and impulse-control disorders in about 15%, including punding,34 hypersexuality, gambling, eating binges and compulsive shopping. Some of these behaviours may be more likely in Parkinson’s disease patients, independent of a treatment effect, due to a dysregulated dopamine-prefrontal reward system. Where obsessionality develops on dopamine agonist therapy, this usually occurs in patients already taking a high daily levodopa dose, and indeed can lead to addiction and patient-initiated escalation of drug doses. The approach in these cases is gradually to withdraw the dopamine agonist.
Adverse effects of apomorphine follow from the fact that it is both a dopamine agonist and a morphine derivative. An antiemetic, e.g. domperidone,35 should accompany initial dosing as nausea is almost a universal accompaniment, at least initially. Overdose causes respiratory depression, while naloxone antagonises its action. Apomorphine can also cause confusion, psychosis and dysphoria, induce penile erection (without causing sexual excitement36), Raynaud’s phenomenon and yawning. Autoimmune-mediated haemolytic anaemia is a rare complication in patients taking concurrent levodopa, and their blood counts should be monitored.
Inhibition of dopamine metabolism: MAO-inhibitors and COMT inhibitors
Monoamine oxidase (MAO) enzymes have an important function in modulating the intraneuronal content of neurotransmitters. The enzymes exist in two principal forms, A and B, defined by specific substrates, some of which cannot be metabolised by the other form (Table 21.3). The therapeutic importance of recognising these two forms arises because they are to some extent present in different tissues, and the enzyme at these different locations can be selectively inhibited by the individual inhibitors: moclobemide for MAO-A (used for depression, see p. 311) and selegiline or rasagiline for MAO-B (used in Parkinson’s disease; Table 21.3).
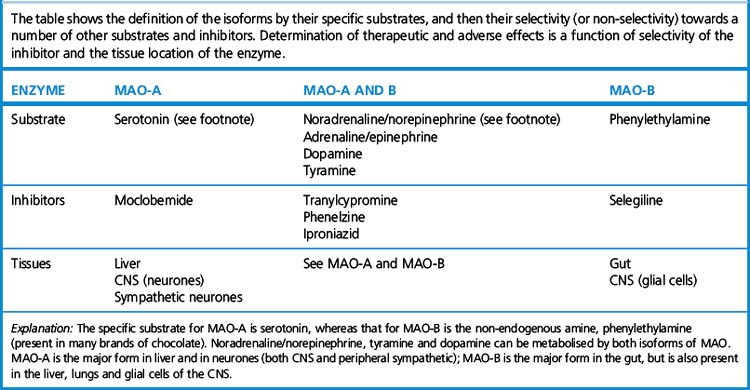
is a selective, irreversible inhibitor of MAO type B. The problem with non-selective MAO inhibitors is that they prevent degradation of dietary amines, especially tyramine, which may then act systemically as sympathomimetics (causing the so-called hypertensive ‘cheese reaction’37). Selegiline does not cause the cheese reaction, because MAO-A in the liver and sympathetic nerve endings is unaffected, allowing tyramine to be metabolised. In the CNS, selegiline reduces intraneuronal degradation of dopamine, but has no effect on synaptic cleft concentrations of other neuromodulatory amines, such as serotonin and noradrenaline/norepinephrine. The effects of these amines can be enhanced by MAO-A inhibitors used as antidepressants. Rasagiline38 is another MAO type B inhibitor, but has an advantage over selegiline in not producing amfetamine metabolites, which are believed to be part of the reason for confusion in susceptible patients. Furthermore, two trials39 of rasagiline have suggested that patients who take this early in their disease course show an enduring benefit (of up to 6 years), relative both to patients on placebo, and to those who are treated with rasagiline only after a delay of 9 months. While potentially evidence for a ‘disease-modifying’, rather than merely a symptomatic treatment effect, longer-term follow-up of these patients, and further independent trials will be required to confirm this.
Drug-induced parkinsonism
The classical antipsychotic drugs (see p. 328) block dopamine receptors, and their antipsychotic activity relates closely to this action, which notably involves the D2 receptor, the principal target in Parkinson’s disease. It comes as no surprise, therefore, that these drugs, as well as certain calcium antagonists, e.g. flunarizine (used for migraine) and valproate, can induce a state with clinical features very similar to those of idiopathic Parkinson’s disease. An important distinguishing measure between drug-induced parkinsonism and Parkinson’s disease is that the former shows a normal dopamine-transporter binding using a DAT scan, since the nigrostriatal terminals (on which the transporter is located) are intact; in Parkinson’s disease there is typically a (asymmetric) reduction in transporter binding. The piperazine phenothiazines, e.g. trifluoperazine, and the butyrophenones, e.g. haloperidol, are most commonly involved, whereas neuroleptics with high antimuscarinic blocking activity, e.g. thioridazine, are less likely to cause this.
Treatment of drug-induced parkinsonism firstly involves consideration of whether the offending drug can be withdrawn, or replaced, e.g. by an atypical antipsychotic such as quetiapine, olanzapine or clozapine, that provokes fewer extrapyramidal effects (see p. 325). After withdrawal of the offending drug, most cases resolve completely within 7 weeks. When drug-induced parkinsonism is troublesome, an antimuscarinic drug, e.g. trihexyphenidyl, is beneficial, while levodopa and dopamine agonists are not (and risk provoking psychosis).
Other movement disorders
Drug-induced dystonic reactions are seen:
• As an acute reaction, often of the torsion type, and occur following administration of dopamine receptor-blocking antipsychotics, e.g. haloperidol, and antiemetics, e.g. metoclopramide. An antimuscarinic drug, e.g. biperiden or benzatropine, given i.m. or i.v. and repeated as necessary, provides relief.
• In some patients who are receiving levodopa for Parkinson’s disease.
• In younger patients on long-term antipsychotic treatment, who develop tardive dyskinesia (see p. 325).
Multiple sclerosis
Symptomatic therapies
therapy is often used for acute, disabling attacks of MS. The drugs decrease the length of an attack but do not reduce the number of recurrent attacks or the final disability. Methylprednisolone (0.5–1 g) is given i.v. or by mouth over a 3–5-day period. Intravenous treatment appears to benefit relapses of optic neuritis more than do oral steroids but usually requires hospital admission.40
Miscellaneous neurological disorders
Motor neurone disease
The cause of the progressive destruction of upper and lower motor neurones is unknown. The only drug available, riluzole, acts by inhibiting accumulation of the neurotransmitter, glutamate. Riluzole prolongs survival time from 13 to 16 months, with no effect on motor function.41 It may cause neutropenia. Its use in the UK is limited to neurologists.
Tetanus
• Immediate neutralisation of any toxin that has not yet become attached irreversibly to the CNS. Human tetanus immunoglobulin 150 units/kg is given intramuscularly at multiple sites to neutralise unbound toxin.
• Wound debridement and destruction of Clostridium tetani with metronidazole.
• Control of convulsions while maintaining respiratory function. Midazolam or diazepam is given for spasms and rigidity, and tracheal intubation and mechanical ventilation for prolonged spasms with respiratory dysfunction (in severe cases, a neuromuscular blocking drug, e.g. intermittent doses of pancuronium, may be required).
• Control of cardiovascular function (tetanus toxin often causes disturbances in autonomic control, with sympathetic overactivity). First-line treatment is by sedation with a benzodiazepine and opioid; infusion of the short-acting β-blocker esmolol, or the α2-adrenergic agonist clonidine, helps to control episodes of hypertension.
• Severe cases generally require admission to an intensive care unit for fluid and electrolyte management; enteral nutrition (weight loss is universal in tetanus); and monitoring for infection (usually aspiration pneumonia), thromboembolism, and pressure sores.
Baker G.A., Lane S., Benn E.K., et al. Adverse antiepileptic drug effects in new-onset seizures: a case-control study. Neurology. 2011;76:273–279.
Drivers Medical Group, At a glance guide to the current medical standards of fitness to drive. DVLA, Swansea. Available online at: http://www.dft.gov.uk/dvla/medical/ataglance.aspx (accessed February 2011).
Fahn S., Oakes D., Shoulson I., Parkinson Study Group. Levodopa and the progression of Parkinson’s disease. N. Engl. J. Med.. 2004;351:2498–2508.
Marson A.G., Al-Kharusi A.M., Alwaidh M., et alSANAD Study Group. The SANAD study of effectiveness of valproate, lamotrigine, or topiramate for generalised and unclassifiable epilepsy: an unblinded randomised controlled trial. Lancet. 2007;369:1016–1026.
Marson A.G., Al-Kharusi A.M., Alwaidh M., et alThe SANAD Study Group. The SANAD study of effectiveness of carbamazepine, gabapentin, lamotrigine, xcarbazepine, or topiramate for treatment of partial epilepsy: an unblinded randomised controlled trial. Lancet. 2007;369:1000–1015.
Meador K.J., Baker G.A., Browning N., et alThe NEAD Study Group. Effects of breastfeeding in children of women taking antiepileptic drugs. Neurology. 2010;75:1954–1960.
Rascol O., Fitzer-Attas C.J., Hauser R., et al. A double-blind, delayed-start trial of rasagiline in Parkinson’s disease (the ADAGIO study): prespecified and post-hoc analyses of the need for additional therapies, changes in UPDRS scores, and non-motor outcomes. Lancet Neurol.. 2011;10:415–423.
Rudzinski L.A., Meador K.J. Epilepsy: five new things. Neurology. 2011;76(Suppl. 2):S20–S25.
Siddiqui A., Kerb R., Weale M.E., et al. Association of multidrug resistance in epilepsy with a polymorphism in the drug-transporter gene ABCB1. N. Engl. J. Med.. 2003;348:1442–1448.
1 Epilepsy has been recognised since early times. A Babylonian medical text dated about 650 BC gives the following description: ‘while he is sitting down, his left eye moves to the right, a lip puckers, saliva flows from his mouth, and his hand, leg and trunk on the left side jerk like a slaughtered sheep …’. Because of its unusual manifestations epilepsy was known as the ‘sacred disease’. Wilson J V K, Reynolds E H 1990 Medical History 34:192.
2 For a first-hand description of what it is like to experience a seizure, the reader is referred to many passages in the works of a lifelong epilepsy sufferer, Fyodor Dostoevsky, e.g. in The Idiot (1869): ‘all at once everything seemed to open up before him: an extraordinary inner light flooded his soul. That lasted half a second … he clearly remembered the beginning, the first sound of a dreadful scream which burst from his chest of its own accord and which no effort of his could have suppressed.’
3 Some people with epilepsy make pilgrimages to Terni (Italy) to seek intercession from Saint Valentine to relieve their condition. There was more than one Saint Valentine and it is unclear whether he was also the patron saint of lovers.
4 So-called ‘primary’ or ‘idiopathic’ generalised epilepsies that reflect the fact that the specific cause is usually undetermined, although presumed to be developmental (e.g. in utero) or genetic.
6 Or single cluster of seizures, i.e. if they all occurred on one day or over a few consecutive days, without any recurrence.
8 Medical Research Council 1991 Antiepileptic Drug Withdrawal Study Group. Lancet 337:1175–1180.
9 Medical Research Council 1993 Antiepileptic Drug Withdrawal Study Group. British Medical Journal 306:1374–1378.
10 As well as spina bifida, cleft palates, cardiac and urogenital anomalies in the fetus, valproate during early pregnancy or pre-conception is associated with a particular dysmorphic appearance of the newborn (‘fetal valpraote syndrome’) characterised by wide, flat nasal bridge, long philtrum, thin lip, widely spaced eyes (hypertelorism) and epicanthic folds.
11 Eclampsia Trial Collaborative Group 1995 Which anticonvulsant for women with eclampsia? Evidence from the Collaborative Eclampsia Trial. Lancet 345:1455–1463.
12 The last two of these examples have names that help to recall their mechanisms: vigabatrin (as well as valproate) being a GABA TRansamine INhibitor, and TiaGABINe being an INhibitor of GABA Transporter.
13 ‘We thought the change might be welcomed by the patients, but one girl preferred her hair to be long and straight, and one boy was mortified by his curls and insisted on a short hair cut’ (Jeavons P M, Clark J E, Harding G F 1977 Valproate and curly hair. Lancet i:359).
14 Sometimes referred to as idiopathic Parkinson’s disease (IPD) – a name that is likely to move out of favour as its pathophysiology becomes increasingly elucidated.
15 Substantia nigra is (Latin) black substance. A coronal section at this point in the brain shows the distinctive black areas, visible with the naked eye in the normal brain, but absent from the brains of patients with Parkinson’s disease.
16 James Parkinson (1755–1824), physician; he described paralysis agitans in 1817.
17 The commonest mutations associated with Parkinson’s disease (including sporadic disease – i.e. not apparently inherited) are leucine-rich repeat kinase (LRRK) and glucocerebrosidase (GBA), the latter of which in homozygous form causes the severe childhood disorder Gaucher’s disease.
18 Deep brain stimulation, a more popular neurosurgical technique, causes functional inactivity (i.e. ‘virtual lesion’) of either of these same target brain regions.
19 Atypical neuroleptics such as quetiapine or olanzapine, and antiemetics that only minimally cross the blood–brain barrier, such as domperidone, can be used relatively safely.
20 The CALM-PD study, for example, randomised 300 early PD patients to levodopa or pramipexole, where either group could subsequently have the other drug added in if clinically required. At 6 years the pramipexole group had less wearing off and dyskinesias than the levodopa group, but more oedema and sleepiness. Overall quality of life was no different between groups.
22 Levodopa is derived from the fava bean (Vicia faba); compare this with the prototypical drug for Alzheimer’s disease, physostigmine, which is derived from the calabar bean. The discoverer of natural levodopa, Marcus Guggenheim, explored its physiological effects by ingesting 2.5 g of the compound. He subsequently became violently ill with vomiting – a fact not too surprising given that the recommended starting dose now is one-tenth this amount.
23 The reason why the levo form of dopa is used is because racemic (d/l) mixtures of dopa were found to cause agranulocytosis in up to 25% of patients.
24 Explaining why nasoduodenal tube insertion for continuous levodopa instillation (Duodopa) can be effective.
25 For example, nausea frequency is reduced from 80% with levodopa alone to less than 15% with a decarboxylase inhibitor, for the same dopamine brain concentration achieved.
26 The dosage commonly referred to is the sum of both drugs: hence: 62.5 mg, 125 mg and 250 mg.
27 Amino acids compete for levodopa uptake and so absorption may vary depending on meal size and type.
28 Slow-release preparations are not preferable during the day as they are generally insufficient in achieving peak levels required for normal motor activity.
29 Dopamine agonists enable a reduction in the total levodopa dose, and in the cases of long-acting formulations, e.g. rotigotine, provide a relatively continuous and smooth level of dopaminergic stimulation.
30 Which has an independent antidyskinesia effect.
31 Note that dopamine causes hypotension, whereas further metabolism of levodopa to adrenaline/epinephrine and noradrenaline/norepinephrine causes hypertension – that is not seen with levodopa therapy.
32 This unwanted effect can be predicted from the fact that dopamine antagonists are used as antipsychotics.
33 Patients who drive, or engage in other potentially dangerous pursuits, need to be warned about this (and to stop driving etc. if they were to experience this).
34 Punding refers to behaviours such as sorting or hoarding objects to an extreme degree; the phrase was first coined to describe similar behaviour in chronic amfetamine abusers.
35 Domperidone is preferred as it does not cross the blood–brain barrier, unlike metoclopramide or prochlorperazine.
36 It also enhances the penile response to visual erotic stimulation, allegedly.
37 Tyramine is an indirectly acting amine which displaces noradrenaline/norepinephrine from nerve terminals.
38 This drug is unique in PD for having a very easy dosing schedule: 1 mg per day with no titration upwards necessary.
39 TEMPO and ADAGIO trials with approximately 400 and 1100 mild PD patients enrolled, respectively.
40 Some MS centres have arrangements in place for patients to receive intravenous corticosteroids in their own home, e.g. by employing community nurses who can monitor the hourly infusion. This is often preferred by patients, and reduces overall medical costs.
41 Lacomblez L, Bensimon G, Leigh P N et al 1996 A controlled trial of riluzole in ALS. Lancet 347:1425–1431.