CHAPTER 93 NEUROLOGICAL DISORDERS ASSOCIATED WITH HUMAN IMMUNODEFICIENCY VIRUS INFECTION
Neurological disorders caused by the retrolentivirus human immunodeficiency virus (HIV), occur at all levels of the neural axis, including the central (CNS) and peripheral (PNS) nervous systems throughout the entire course of HIV infection.1 Indeed, the prevalence of neurological syndromes during HIV infection is high, affecting up to 90% of untreated patients with the acquired immunodeficiency syndrome (AIDS).2–4 Approximately 55 million people worldwide have been infected with HIV since it was first identified in the early 1980s. With the advent of highly active antiretroviral therapy (HAART) in the mid 1990s, many individuals infected with HIV are living more than 20 years after their initial infection in industrialized countries.5 Nonetheless, despite the availability of HAART, HIV-related neurological disease continues to represent substantial personal, economic, and societal burdens.
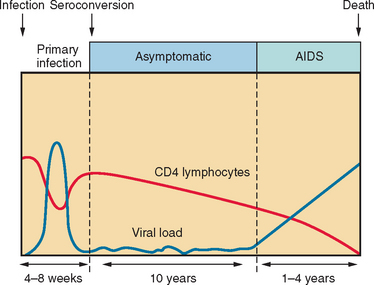
In developed countries, conversion to AIDS usually occurs approximately 10 years after initial infection (Fig. 93-1). It is defined by the Centers for Disease Control and Prevention as either a decline in CD4+ T lymphocyte levels below 200 cells/μL or an AIDS-defining illness, such as Pneumocystis carinii pneumonia, cryptococcal meningitis, or toxoplasmosis encephalitis. There are also infection-driven malignancies, such as Kaposi’s sarcoma and primary CNS lymphoma, that are commonly associated with progression to AIDS. There are two major strains of HIV: type 1 (HIV-1), which predominates globally, and type 2 (HIV-2), which is limited largely to West Africa and is less virulent. Because of HIV’s remarkable capacity for replication and subsequent mutation, significant viral molecular and antigenic diversity has occurred, which has precluded the development of an effective vaccine to date. In the industrialized world, HIV-1 B subtype (also termed B clade) predominates and is the source of most of the current understanding of HIV-related neurological disease.
NEUROLOGICAL DISORDERS OF HUMAN IMMUNODEFICIENCY VIRUS INFECTION
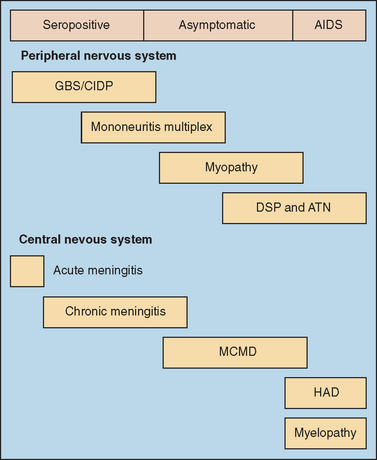
Early in the HIV epidemic, two groups of neurological disorders arising as consequences of HIV infection were recognized. The first group of neurological syndromes includes the primary HIV-induced disorders, which reflect HIV’s immediate deleterious effects on neural cells and result in damage to the brain, spinal cord, and peripheral nerves. These primary HIV-associated illnesses include HIV-associated dementia (HAD) (also termed AIDS dementia complex and HIV encephalopathy) and its antecedent syndrome, minor cognitive and motor deficit (MCMD); vacuolar myelopathy; and several types of peripheral neuropathy6 (Fig. 93-2). Indeed, other neurological disorders, including entrapment neuropathies, headache, seizures, and myopathy, appear to be more frequently encountered among patients with HIV or AIDS than noninfected persons, although these conditions are not necessarily linked to the direct effects of virus (HIV) infection. The second group is composed of the opportunistic infections of the CNS and PNS, which arise as direct consequences of HIV-induced immunosuppression. These include toxoplasmosis encephalitis, cryptococcal meningitis, cytomegalovirus encephalitis and radiculitis, primary CNS lymphoma, progressive multifocal leukoencephalopathy, and neurotuberculosis (meningitis and tuberculoma) and are dealt with in detail in other publications.7,8
HUMAN IMMUNODEFICIENCY VIRUS NEUROPATHOGENESIS AND NEUROPATHOLOGY
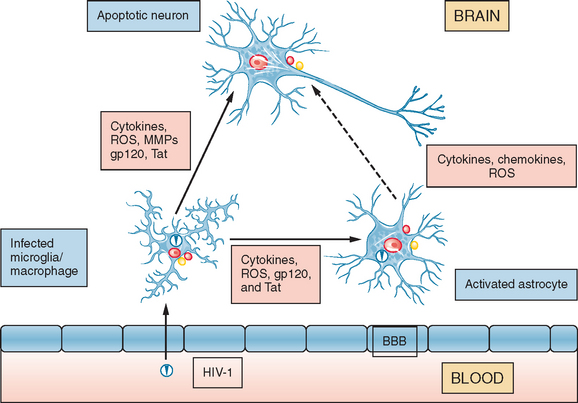
Under healthy conditions, the CNS and PNS are well protected by the blood-brain barrier or blood-nerve barrier from toxins and infectious pathogens circulating in the blood (Fig. 93-3). However, HIV is able to traverse the blood-brain barrier, and probably the blood-nerve barrier, soon after primary infection via circulating HIV-infected (and activated) leukocytes, including macrophages and lymphocytes, which adhere to endothelia within the neural compartment and subsequently enter the parenchyma. On entry, HIV establishes infection of perivascular macrophages, resident microglia, and to a lesser extent, astrocytes or Schwann cells. In contrast HIV infection of neurons is minimal or nonexistent. This process by which HIV enters the CNS or PNS is termed the “Trojan Horse” hypothesis. HIV infects macrophages and microglia through binding to the CD4+ molecule, which acts as the primary receptor in association with the chemokine receptors (CCR5 and CXCR4) as co-receptors for infection. HIV-1 exerts its neuropathogenic effects through two principal mechanisms; one is stimulation of neuroimmune cells within the CNS or PNS to produce host proinflammatory molecules, such as cytokines, chemokines, prostaglandins, redox reactants, excitotoxic amino acids or derivatives thereof, and enzymes, which damage neurons and the proximate astrocytes that support them. The alternative mechanism by which neural cells are injured is through direct (neurotoxic) interactions between HIV-encoded proteins (including glycoprotein 120, glycoprotein 41, Tat, and Vpr) and the target cell (including neurons or astrocytes). In fact, these mechanisms overlap in a complementary manner, inasmuch as perivascular macrophages and microglia are the chief sources of both the host neuropathogenic molecules and the secreted neurotoxic viral proteins; whereas some of the host molecules activate viral replication, most of the viral proteins can also activate neuropathogenic host gene responses.
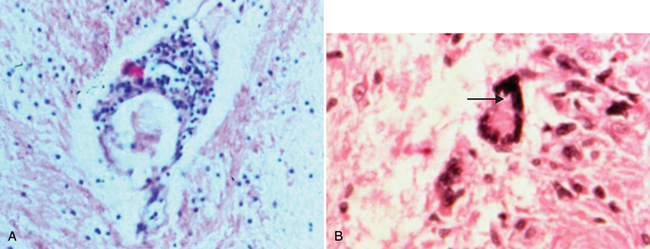
The CNS pathological hallmarks of HIV infection include multinucleated giant cells, diffuse white matter pallor, perivascular cuffs composed of monocytes and lymphocytes, microglial nodules, and the presence of HIV-1 antigens9–11 (Fig. 93-4). The diffuse white matter pallor exhibits preserved myelin protein expression but concurrent deposition of serum proteins in white matter, which implies that altered permeability of the blood-brain barrier rather than frank demyelination underlies diffuse myelin pallor.12,13 Neuronal and astrocyte injury and death are defined by dendritic “pruning,” together with cell death involving both necrotic and apoptotic mechanisms, depending on the effector molecule and the selective vulnerability of the target cell (Fig. 93-5). For example, neurons in the basal ganglia represent a highly susceptible population, in part because of the high density of microglia in this region of the CNS but also because of the intrinsic properties of this group of neurons. In the PNS, infiltrating macrophages and lymphocytes are present during HIV infection and often exhibit HIV antigens and genome. Damage to dorsal root ganglia neurons and a dying-back (wallerian) pattern of axonal injury, chiefly affecting small-diameter axons, is apparent.14,15
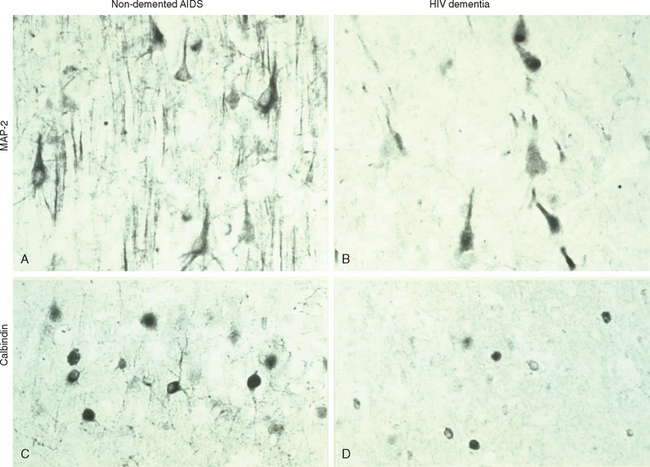
A limited correlation exists between the clinical entity HAD and the pathological entity HIV encephalitis, defined by the presence of multinucleated giant cells or the presence of viral antigens.9,16,17 There may also be a correlation between HIV antigen load and HAD,18 although other studies have suggested that macrophage and microglia presence and activation in the basal ganglia are better predictive markers for HAD.19 Although neuronal injury and death in the frontal cortex and deep gray matter occur in the brains of patients with AIDS,20–22 the degree of neuronal loss is correlated with the severity of HAD. Results of a 2001 study also imply that the astrocyte death is associated with rapidly progressive HAD.23
NEUROCOGNITIVE SYNDROMES IN HUMAN IMMUNODEFICIENCY VIRUS INFECTION
Unlike acute viral infections of the CNS, the pathogenic effects of HIV usually manifest long after initial infection, with ensuing cognitive, motor, and behavioral dysfunction among affected patients. Nevertheless, rare cases of acute encephalopathy have been reported during HIV seroconversion. HAD represents a constellation of progressive symptoms and signs associated that usually begin once an individual’s CD4+ T cell counts dips below 200 cells/μL of blood; not surprisingly, HAD is an AIDS-defining illness (Table 93-1). With the availability of HAART, HAD is now manifesting with CD4+ cell levels exceeding 200 cells/μL. Of most importance, the diagnosis of HAD heralds a worsened survival prognosis with or without HAART.24 Before the era of HAART, the annual incidence of HAD was 53%, although the overall prevalence was only 6%,24 probably a consequence of the high mortality rate after HAD onset, as survival time after diagnosis was only 5.1 months.25,26 With the advent of HAART, the incidence of HAD has fallen to less than 10%, survival time after diagnosis has leapt to 38.5 months, and longer survival times have resulted in overall higher prevalence rates. Risk factors for HAD include low CD4+ levels, high viral loads in cerebrospinal fluid or plasma, anemia, extremes of age, and intravenous drug use (Table 93-2). Also, patients with marked immunosuppression who have no experience with antiretroviral therapy may experience an exaggerated immune response, the immune reconstitution inflammatory syndrome (IRIS), after HAART introduction.27 Indeed, IRIS occasionally manifests as transient cognitive dysfunction together with signs and symptoms of acute meningoencephalitis, although preexisting neurological complications of AIDS may also be exacerbated with IRIS.28,29
TABLE 93-1 Clinical Features of HIV-Associated Dementia (Typical of a Subcortical Dementia)
HIV, human immunodeficiency virus.
TABLE 93-2 Risk Factors for HIV-Associated Dementia
HIV, human immunodeficiency virus.
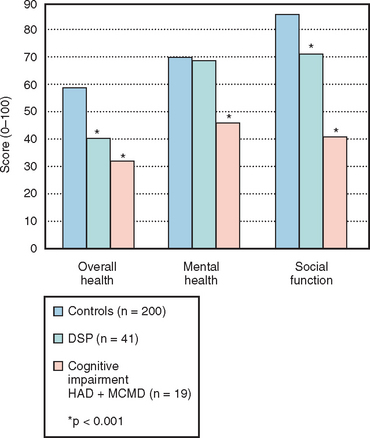
It has also been postulated that MCMD is a risk factor for progression to HAD.30 MCMD is a syndrome exhibiting many clinical aspects of HAD, although the signs and symptoms are less severe. Because MCMD has been identified in patients with higher CD4+ counts, there is some suggestion that it may be the precursor to HAD.31 MCMD may affect as many as 30% of patients with HIV or AIDS in North American clinics.32 Nonetheless, comorbid conditions, including chronic drug abuse, head injury, and other risk factors for neurocognitive impairments, may contribute to the diagnosis of MCMD. Because HIV preferentially affects the basal ganglia and deep white matter, HAD predictably manifests with many of the features of a subcortical dementia (Table 93-3). Affected patients typically display neurocognitive impairment, emotional disturbances, and progressive motor decline,33 although HAD has remarkably diverse clinical phenotypes that may include movement disorders.34–36
TABLE 93-3 Radiological, CSF, and Neuropathological Features of HIV-Associated Dementia
CSF, cerebrospinal fluid; HIV, human immunodeficiency virus; IgG, immunoglobulin G; MRI, magnetic resonance imaging; MRS, magnetic resonance spectroscopy; PCR, polymerase chain reaction.
Of importance is that HAD is difficult to detect with the Mini-Mental Status Examination unless the patient is severely demented, most likely because HAD is a subcortical dementia. If there is a suspicion that an HIV-seropositive patient is suffering from HAD, more useful screening tools are applicable, including the HIV Dementia Scale,40 the Mental Alteration Test,41 the Executive Interview,42 and the HIV Dementia Assessment.43 A widely accepted tool for clinical staging of HAD is the Memorial Sloan-Kettering Scale, which provides a qualitative measure of dementia severity, allowing the physician to track progression of the dementia over time.44
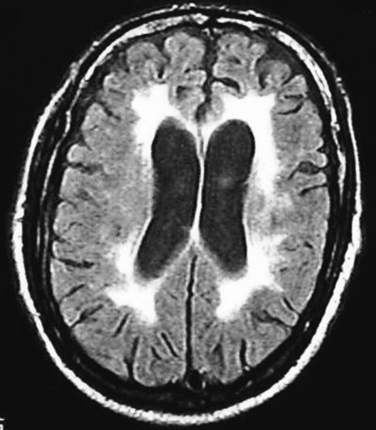
Radiological features accompanying HAD (Fig. 93-6) include cerebral and basal ganglia atrophy and diffuse periventricular white matter hyperintensities on T2-weighted MRI (Fig. 93-7).45,46 Unfortunately, it is difficult to correlate the presence of these radiological changes with the presence of HAD, as nondemented patients with HIV infection or AIDS also display these changes on neuroimaging.47 Magnetic resonance spectroscopy studies show diminished N-acetyl aspartate levels in the brain, which imply neuronal injury or loss.48 Other critical investigations include cerebrospinal fluid analyses, chiefly to exclude opportunistic processes and also to assess the levels of viral replication in the neural compartment.
The course of the dementia is variable; some individuals experience an abrupt decline in function over weeks, whereas others display a protracted course over several years that culminates in death.49 The most effective management of HAD is treatment of the underlying cause. HAART routinely consists of two nucleoside analogue reverse transcriptase inhibitors (NRTIs) and either a potent protease inhibitor (PI) or a nonnucleoside analogue reverse transcriptase inhibitor (NNRTI). Clinical trials have shown that neuropsychological testing scores improve in patients with HIV or AIDS who are treated with two NRTIs and the NNRTI nevirapine,50 as well as with two NRTIs and a protease inhibitor.51,52 Specific antiretroviral drugs, including zidovudine (AZT) (an NRTI), stavudine (an NRTI), abacavir (an NRTI), nevirapine (an NNRTI), and indinavir (a protease inhibitor), can permeate the blood-brain barrier better than others.39 The resulting high cerebrospinal fluid drug levels may act to decrease viral load in the CNS. Neuropsychological assessment is an invaluable tool in confirming the diagnosis of HAD and also in facilitating evaluation of the response to therapy among patients with HAD or MCMD. If a particular patient is having symptoms of mania or psychosis, it is best to avoid use of efavirenz (an NNRTI), because this particular drug may cause hallucinations, vivid dreams, and behavioral changes, all of which may exacerbate an individual’s existing symptoms. The addition of methylphenidate and amantidine as adjunct therapies may alleviate some symptoms of psychomotor retardation, thus increasing quality of life.53
Unfortunately, HAART has limited efficacy in reversing HAD, and thus clinicians must consider other treatments that have potential neuroprotective properties. Several agents have been investigated in the past, although few have had significant beneficial effects. Selegiline may have an antiapoptotic effect and slow the progression of HAD.31,54 Memantine has been shown to block neurotoxicity induced by the HIV viral proteins Tat and glycoprotein 120.55 A phase II multicenter trial to test the efficacy of memantine in alleviating symptoms of HAD and HIV-associated peripheral neuropathy is ongoing. Prinomastat is a matrix metalloproteinase inhibitor that has been shown to inhibit HIV Tat-associated neurotoxicity and may be a potential neuroprotective agent in HAD.56 Human growth hormone has been shown to be neuroprotective and may also be a component of HAD treatment in the future.57 CPI-1189 blocks the effect of tumor necrosis factor α but was not beneficial in clinical trials.58 Antioxidants such as OPC-14117, which is structurally similar to vitamin E, also had no effect in clinical trials.59 The L-type Ca2+ blocker nimodipine also had no effect on HAD, although it shows some promise for HIV-associated peripheral neuropathies.60
MYELOPATHY
HIV-associated vacuolar myelopathy affects 10% to 15% of untreated patients with AIDS (Table 93-4), usually manifesting as gait ataxia, leg weakness, spasticity, and incontinence.61,62 Impaired proprioception with sensory ataxia may also be present. It can occur independently or in conjunction with HAD or with opportunistic infections and malignancies. Progression is insidious over months without back pain. Physical examination reveals symmetrical spastic paraparesis, with lower extremity hyperreflexia and extensor plantar responses. Upper limb signs are less common, although hyperreflexia of the arms is occasionally present. There is no defined sensory level. Subclinical vacuolar myelopathy, indicated by hyperreflexia, spasticity, and extensor plantar reflexes, may be evident on examination in an otherwise asymptomatic patient, but other causes of myelopathy should be ruled out.
The incidence of vacuolar myelopathy has dropped with HAART to a point that it is infrequently seen in HIV clinics except in severely immunosuppressed patients.62 Although HAART appears to reduce the incidence of vacuolar myelopathy, limited reversal of the signs or symptoms is observed after therapy is implemented. The diagnosis is one of exclusion of other conditions causing chronic myelopathy, such as human T cell lymphotropic virus type I or II infection, vitamin B12 deficiency, or varicella-zoster virus–related myelopathy, all of which must be ruled out by a complete blood cell count, vitamin B12 measurement, cerebrospinal fluid analysis, and neuroimaging studies. The neuropathological correlates of vacuolar myelopathy are axonal injury and intense macrophage infiltration. The vacuolar appearance localized primarily in the lateral and dorsal columns of thoracic spinal cord may reflect intramyelinic edema and inflammation. Approximately 25% of patients with symptoms or signs suggestive of a myelopathy subsequently have pathologically confirmed vacuolar myelopathy. Symptomatic treatment for painful spasticity, clonus, and tremor, including baclofen and gabapentin, is frequently beneficial to patients with vacuolar myelopathy.
PERIPHERAL NEUROPATHIES
Peripheral neuropathy has become the major complication of HIV infection in the developed world; substantial numbers of patients with HIV or AIDS seek assistance for control of their symptoms.63 There are two major groups of neuropathy associated with HIV infection64 (Table 93-5). The first group is the HIV-associated neuropathies, which include distal sensory polyneuropathy, autonomic neuropathy, acute and chronic demyelinating neuropathies, and mononeuritides multiplex.65 The second group of neuropathies encountered among treated patients with HIV or AIDS includes the antiretroviral toxic neuropathies. These arise as a result of the use of antiretroviral agents, including didanosine, zalcitabine, and stavudine.66 Both groups of these neuropathies warrant investigation and treatment because of their remarkably disabling features and outcomes. It is also imperative to rule out other causes of painful neuropathy, including diabetes, amyloidosis, nutritional deficiency or ethanol abuse, and concomitant drugs (chemotherapeutics, metronidazole), when possible. As life expectancies among patients with HIV or AIDS increase, clinicians must be suspicious about the onset of diabetic peripheral neuropathy, especially when considering the metabolic side effects of different HAART regimens.
TABLE 93-5 Clinical Features of HIV Sensory Neuropathies, Including Distal Sensory Polyneuropathy and Antiretroviral Toxic Neuropathy
d4T, stavudine; ddC, zalcitabine; ddI, didanosine; EMG, electromyography; HIV, human immunodeficiency virus; NCS, nerve conduction study.
Sensory Neuropathies
Distal sensory polyneuropathy is the most common neuropathy, affecting 30% of patients with AIDS. It is associated with advanced HIV infection and usually manifests as neuropathic pain indicated by a subacute burning sensation, paresthesia, or dysesthesia that worsens as the day progresses, especially on the soles and dorsa of the feet. Patients often report nighttime awakening caused by foot discomfort, but symptoms in the hands can also be present, albeit less frequently. Physical examination usually reveals a stocking distribution loss of pain and temperature sensation with diminished or absent distal deep tendon reflexes, accompanied by atrophic skin changes in the feet and venous pooling.67 If proprioception is also abnormal, there is likely to be a concomitant vacuolar myelopathy. Patients often exhibit an antalgic gait and may require a cane or wheelchair in severe cases, although foot weakness is a very late component of the neuropathy. The symptoms and signs of antiretroviral toxic neuropathies are identical to those of distal sensory polyneuropathy, and the two entities are frequently indistinguishable except by a history of recent-onset neuropathy with initiation of a neurotoxic drug within several months. Nerve conduction studies with electromyography are useful for ruling out other conditions but frequently yield normal results in both distal sensory polyneuropathy and antiretroviral toxic neuropathy because both syndromes usually involve small-diameter fibers and are principally sensory neuropathies.68 Nerve biopsies are also helpful in ruling out other diagnoses; several groups have developed a quantitative evaluation of skin biopsy samples from the leg to aid in the diagnosis of sensory neuropathies.
The treatment modalities for these two latter types of neuropathy clearly require different strategies; distal sensory polyneuropathy necessitates initiation of HAART, preferably without the potentially neurotoxic antiretroviral drugs (PIs, especially indinavir),68a whereas antiretroviral toxic neuropathy entails replacing the neurotoxic antiretroviral drugs with nonneurotoxic agents. Indeed, introduction of HAART frequently improves symptoms, if not signs, of distal sensory polyneuropathy over months, and cessation of the incriminated neurotoxic antiretroviral agent also improves symptoms. Symptomatic treatment options include antiepileptic agents such as gabapentin, lamotrigine, or topiramate, although definitive studies support the use of only gabapentin.69 Despite clinical trials indicating that amitriptyline is not beneficial in the control of pain associated with distal sensory polyneuropathy, experienced clinicians continue to use it in selected patients.70 Opioids are highly effective in the control of neuropathic pain but elicit the potential for establishing or reestablishing drug dependence and may interfere with neurocognitive function in patients who are already at risk for HAD. Similarly, lidocaine gel is used by many clinicians, but it has no proven efficacy.71 A previous trial suggested that human nerve growth factor may have potential uses in the future.72
Mononeuritis Multiplex
A history of asymmetrical sensory and motor dysfunction affecting distal nerves and occurring over weeks is suggestive of mononeuritis multiplex. This condition is hypothesized to result from immune complex deposition in the vasculature surrounding the affected nerve, which leads to vasculitis.73,74 Evidence of vasculitis is occasionally found in the skin and joints. It is important to rule out hepatitis B or C infection, because cryoglobulinemia can manifest with mononeuritis multiplex in a manner similar to that of HIV during infection.75,76 A sural nerve biopsy specimen can reveal epineural and endoneural perivascular inflammatory infiltrates.77 In patients with spontaneous remission, treatment may not be necessary. Short-term prednisone has been used with some success in small groups of patients, although no large-scale clinical trials have been conducted.78 Only 1% of patients with clinically defined AIDS are affected by mononeuritis multiplex, and thus it is relatively rare among the neuropathies.
Inflammatory and Demyelinating Polyneuropathies
Patients can present with ascending neuropathy indistinguishable from Guillain-Barré syndrome at any stage of HIV infection. Analysis of the cerebrospinal fluid in HIV-associated Guillain-Barré syndrome reveals pleocytosis and raised protein levels.79,80 Treatment of HIV-associated Guillain-Barré syndrome is no different than that of sporadic Guillain-Barré syndrome, and plasmapheresis or intravenous immunoglobulin can thus be tried. The overall outcome is also no different, especially if the CD4+ count is above 200 cells/μL.81 HIV-seropositive patients can recover completely from Guillain-Barré syndrome, although some residual weakness may linger. After an episode of Guillain-Barré syndrome, patients may be at increased susceptibility to other forms of peripheral neuropathy, such as distal sensory polyneuropathy or antiretroviral toxic neuropathy. Chronic inflammatory demyelinating polyneuropathy tends to manifest in patients with CD4+ T cell counts in the range of 200 to 500 cells/μL with features typical of sporadic inflammatory demyelinating polyneuropathy, although pleocytosis and an elevated protein level are frequent findings in HIV-associated chronic disease. Electrophysiology is helpful for confirming the diagnosis, showing conduction block with slowed velocities, distal slowing, and reduced compound motor action potentials. Conventional treatments with glucocorticoids, intravenous immunoglobulin, or plasmapheresis are effective; at least 20% of patients achieve complete remission, and 80% experience at least some improvement.
Entrapment Neuropathy
HIV-seropositive patients are at increased risk for entrapment neuropathy, most commonly carpal tunnel syndrome. In addition to repetitive wrist motions, risk factors for developing carpal tunnel syndrome in the general population include metabolic syndromes such as diabetes, weight gain, and hypothyroidism. Another common entrapment neuropathy in HIV infection is meralgia paresthetica. Because treatment with protease inhibitors causes metabolic abnormalities, treatment with protease inhibitors may predispose patients to entrapment neuropathy by enhancing deposition of myxedematous material or fat in the carpal tunnel.82 Results of one case series have suggested that carpal tunnel syndrome in HIV-seropositive patients is simply related to repeated stress injury.32 A third possibility is that subclinical inflammation of the median nerve caused by HIV infection is exacerbated by friction within the carpal tunnel, causing symptoms that might not occur in a HIV-seronegative individual. Regardless of the cause, carpal tunnel syndrome can be treated with the use of splints or by surgical intervention.83
Miscellaneous Neuropathies
Two other distinct neuromuscular syndromes occur in HIV infection. Among patients with CD4+ levels in the range of 200 to 500 cells/μL, a sensorimotor neuropathy associated with diffuse infiltrative lymphocytosis syndrome (DILS) may be present. The neuropathy accompanying DILS has a subacute and frequently asymmetrical onset, together with the occurrence of parotidomegaly and sicca syndrome. The neuropathy often progresses to a symmetrical pattern with neuropathic pain. Electrophysiological study findings are usually abnormal, and nerve biopsy specimens showing CD8 lymphocyte infiltrates provide confirmation of the diagnosis. DILS responds to HAART; more than 60% of patients make an excellent recovery. Glucocorticoids may also be helpful in the treatment of DILS among patients who do not respond to HAART. A motor neuron–like condition has also been recognized among patients with HIV or AIDS.84 Affected patients are usually immunosuppressed and present with unexplained distal weakness that may progress rapidly. Electromyography discloses features indicative of symmetrical denervation and motor neuron dysfunction. This syndrome is ameliorated by HAART, with complete resolution of neurological signs in some cases.
Autonomic neuropathy is present in 12% of HIV-seropositive patients and is frequently found in conjunction with distal sensory polyneuropathy.85,86 The most common manifestations are postural hypotension, followed closely by gastroparesis and impotence. Postural hypotension can be treated with fludrocortisone, and cisapride and sildenafil can be effective in combating gastroparesis and impotence, respectively. Isolated mononeuropathies are more common in early HIV infection. Interestingly, these neuropathies tend to manifest themselves in the cranial nerves. A cranial nerve VII palsy that is indistinguishable from Bell’s palsy has been reported fre quently in patients with HIV or AIDS87 and may be recurrent. Likewise, cranial nerve V palsies can also occur during HIV infection. As with patients who do not have HIV infection, these palsies resolve over the course of several weeks, rarely leaving residual neurological deficit. A retrobulbar optic neuritis should be given special consideration in a patient with advanced HIV infection, because it may be secondary to herpes zoster infection and thus necessitate aggressive treatment.88 In addition, the possibility of underlying multiple sclerosis must be considered, because multiple sclerosis and HIV or AIDS can coexist despite the depletion of lymphocytes in HIV infection.89,90
HEADACHE
There are three general categories of headache among individuals with HIV. The first category of headache includes aseptic meningitis, which frequently manifests as part of the constellation of symptoms associated with HIV seroconversion.91 The second (and most important to rule out) is headache as a result of opportunistic infections such as cryptococcal meningitis, toxoplasmosis encephalitis, or tuberculous meningitis. The third category of HIV-associated headache is primary headache of no other known etiology, which is present in 25% of patients with HIV infection.92 There is an association between HAD and headache with an unclear mechanism,93 and primary HIV-associated headache may be merely a chronic form of aseptic meningitis that occurs long after seroconversion has taken place. Most HIV-associated primary headaches progress slowly over the course of several weeks. They are nonthrobbing and have a component of photophobia. Unfortunately, these headaches tend not to respond well to conventional pain control medications, although treatment with low-dose tricyclic medication (amitriptyline, 10 to 25 mg every hour before sleep) can be effective in some patients.94 Because the headaches frequently occur in conjunction with depression and polypharmacy, psychiatric consultation may be warranted.
SEIZURE DISORDERS
Among patients with HIV infection, new-onset seizures occur in 8% of adults and up to 20% of children. The majority of seizures are generalized tonic-clonic seizures that progress to status epilepticus in 8% to 18% of affected patients. There are many potential causes of seizures in HIV-seropositive patients, including opportunistic infections, medications, substance use or withdrawal, metabolic disturbance, and HIV infection itself. Seizures are most commonly associated with toxoplasmosis encephalitis, followed by cryptococcal meningitis and primary CNS lymphoma.95 The seizure may be the only sign that the patient is suffering from an infectious or malignant process, especially in the case of CNS lymphoma. Certain medications are associated with a decrease in seizure threshold, including selective serotonin reuptake inhibitors, tricyclic antidepressants, ganciclovir, and foscarnet. Cocaine and heroine use are also associated with an increased risk of seizures, as is alcohol withdrawal. Disturbances of electrolyte balance can also cause seizures, as in patients without HIV infection.
In up to 50% of HIV-seropositive individuals, no underlying cause of the seizure is found except HIV itself.96,97 Interestingly, 25% of patients with seizures of unknown etiology have features of HAD39 and have an increased likelihood of developing HAD within 6 months of the seizure.96 Without treatment, seizures are highly likely to recur. Because HIV-seropositive patients inherently have a higher rate of adverse drug reactions, as many as 25% of patients develop a rash with phenytoin. Because many anticonvulsants are metabolized by the cytochrome P-450 system, there is a high likelihood of drug interactions with HAART. In addition, protease inhibitors can bind to albumin and compete with the anticonvulsants, thus altering drug levels. Despite these concerns, carbamazepine is well tolerated when coupled with close monitoring of anticonvulsant levels and viral load. Valproate has been shown to induce viral replication in vitro, although this has not been demonstrated in vivo.98 The next generation of anticonvulsants, such as gabapentin, topiramate, and tiagabine, are safer choices.99 Topiramate, however, may hasten weight loss and thus should perhaps be avoided in individuals with low body mass indices.
FUTURE PERSPECTIVES
Because of the rising prevalence of HIV infection and AIDS in the developed world and the increasing incidence of HIV infection in the developing world, it is likely that new HIV-associated neurological syndromes will emerge with time. Immediate concerns include identification and characterization of the neurological illnesses associated with HIV-1 non–B subtype infections, which remain poorly defined at present. In addition, the appearance and transmission of drug-resistant HIV-1 strains is a growing problem for which there exist few data describing the accompanying neurological disorders.29 The extent to which the nervous system serves as a covert reservoir for HIV, from which it can then spread to the rest of the body, remains uncertain but is a topic of keen interest and investigation.100 As the population with HIV infection and AIDS grows and ages, neurological diseases associated with aging, including stroke and primary dementias, are becoming more prevalent. In addition, the long-term neurological effects of antiretroviral therapy remain incompletely understood. Indeed, the risk of stroke is increasing in patients with HIV or AIDS, in part because of the marked metabolic abnormalities (hyperglycemia and hyperlipidemia) associated with different antiretroviral regimens, together with advancing age and improved overall survival. Finally, the growing importance of HIV infection and co-infection with hepatitis C virus or human T cell lymphotropic virus types I and II may also have substantial effects on neurological disease manifestations and outcomes.
Estanislao L, Carter K, McArthur J, et al. A randomized controlled trial of 5% lidocaine gel for HIV-associated distal symmetric polyneuropathy. J Acquir Immune Defic Syndr. 2004;37:1584-1586.
Hahn K, Arendt G, Braun JS, et al. A placebo-controlled trial of gabapentin for painful HIV-associated sensory neuropathies. J Neurol. 2004;251:1260-1266.
Miller RF, Isaacson PG, Hall-Craggs M, et al. Cerebral CD8+ lymphocytosis in HIV-1 infected patients with immune restoration induced by HAART. Acta Neuropathol (Berl). 2004;108:17-23.
Vendrely A, Bienvenu B, Gasnault J, et al. Fulminant inflammatory leukoencephalopathy associated with HAART-induced immune restoration in AIDS-related progressive multifocal leukoencephalopathy. Acta Neuropathol (Berl). 2005;109:449-455.
World Health Organization “3 by 5” Progress Report, December 2004. Geneva: World Health Organization and Joint United Nations Programme on HIV/AIDS; 2004. Available at: http://www.who.int/3by5/progressreportfinal.pdf. (accessed April 20, 2006).
1 Janssen RS, Cornblath DR, Epstein LG, et al. Human immunodeficiency virus (HIV) infection and the nervous system: report from the American Academy of Neurology AIDS Task Force. Neurology. 1989;39:119-122.
2 Johnson RT. Viral Infections of the Nervous System, 2nd ed. Philadelphia: Lippincott-Raven, 1998.
3 Rausch DM, Davis MR. HIV in the CNS: pathogenic relationships to systemic HIV disease and other CNS diseases. J Neurovirol. 2001;7:85-96.
4 Sacktor N. The epidemiology of human immunodeficiency virus–associated neurological disease in the era of highly active antiretroviral therapy. J Neurovirol. 2002;8(Suppl 2):115-121.
5 World Health Organization “3 by 5” Progress Report, December 2004. Geneva: World Health Organization and Joint United Nations Programme on HIV/AIDS; 2004. Available at: http://www.who.int/3by5/progressreportfinal.pdf. (accessed April 20, 2006).
6 McArthur JC. Neurologic manifestations of AIDS. Medicine (Baltimore). 1987;66:407-437.
7 Mamidi A, DeSimone JA, Pomerantz RJ. Central nervous system infections in individuals with HIV-1 infection. J Neurovirol. 2002;8:158-167.
8 Roullet E. Opportunistic infections of the central nervous system during HIV-1 infection (emphasis on cytomegalovirus disease). J Neurol. 1999;246:237-243.
9 Navia BA, Cho ES, Petito CK, et al. The AIDS dementia complex: II. Neuropathology. Ann Neurol. 1986;19:525-535.
10 Sharer LR. Pathology of HIV-1 infection of the central nervous system. A review. J Neuropathol Exp Neurol. 1992;51:3-11.
11 Vinters HV, Anders KH. Neuropathology of AIDS. Boca Raton, FL: CRC Press, 1990.
12 Petito CK, Cash KS. Blood-brain barrier abnormalities in the acquired immunodeficiency syndrome: immunohistochemical localization of serum proteins in postmortem brain. Ann Neurol. 1992;32:658-666.
13 Power C, Kong PA, Crawford TO, et al. Cerebral white matter changes in acquired immunodeficiency syndrome dementia: alterations of the blood-brain barrier. Ann Neurol. 1993;34:339-350.
14 Herrmann DN, Griffin JW, Hauer P, et al. Epidermal nerve fiber density and sural nerve morphometry in peripheral neuropathies. Neurology. 1999;53:1634-1640.
15 McCarthy BG, Hsieh ST, Stocks A, et al. Cutaneous innervation in sensory neuropathies: evaluation by skin biopsy. Neurology. 1995;45:1848-1855.
16 Glass M, Faull RL, Bullock JY, et al. Loss of A1 adenosine receptors in human temporal lobe epilepsy. Brain Res. 1996;710:56-68.
17 Sharer LR, Epstein LG, Cho ES, et al. Pathologic features of AIDS encephalopathy in children: evidence for LAV/HTLV-III infection of brain. Hum Pathol. 1986;17:271-284.
18 Achim CL, Heyes MP, Wiley CA. Quantitation of human immunodeficiency virus, immune activation factors, and quinolinic acid in AIDS brains. J Clin Invest. 1993;91:2769-2775.
19 Glass JD, Fedor H, Wesselingh SL, et al. Immunocytochemical quantitation of human immunodeficiency virus in the brain: correlations with dementia. Ann Neurol. 1995;38:755-762.
20 Everall IP, Luthert PJ, Lantos PL. Neuronal loss in the frontal cortex in HIV infection. Lancet. 1991;337:1119-1121.
21 Masliah E, Ge N, Achim CL, et al. Selective neuronal vulnerability in HIV encephalitis. J Neuropathol Exp Neurol. 1992;51:585-593.
22 Masliah E, Ge N, Morey M, et al. Cortical dendritic pathology in human immunodeficiency virus encephalitis. Lab Invest. 1992;66:285-291.
23 Thompson KA, McArthur JC, Wesselingh SL. Correlation between neurological progression and astrocyte apoptosis in HIV-associated. Ann Neurol. 2001;49:745-752.
24 McArthur JC, Hoover DR, Bacellar H, et al. Dementia in AIDS patients: incidence and risk factors. Multicenter AIDS Cohort Study. Neurology. 1993;43:2245-2252.
25 Dore GJ, McDonald A, Li Y, et al. Marked improvement in survival following AIDS dementia complex in the era of highly active antiretroviral therapy. Aids. 2003;17:1539-1545.
26 Portegies P, de Gans J, Lange JM, et al. Declining incidence of AIDS dementia complex after introduction of zidovudine treatment. BMJ. 1989;299:819-821.
27 Shelburne SA3rd, Hamill RJ. The immune reconstitution inflammatory syndrome. AIDS Rev. 2003;5:67-79.
28 Miller RF, Isaacson PG, Hall-Craggs M, et al. Cerebral CD8+lymphocytosis in HIV-1 infected patients with immune restoration induced by HAART. Acta Neuropathol (Berl). 2004;108:17-23.
29 Vendrely A, Bienvenu B, Gasnault J, et al. Fulminant inflammatory leukoencephalopathy associated with HAART-induced immune restoration in AIDS-related progressive multifocal leukoencephalopathy. Acta Neuropathol (Berl). 2005;109:449-455.
30 Ellis RJ, Deutsch R, Heaton RK, et al. Neurocognitive impairment is an independent risk factor for death in HIV infection. San Diego HIV Neurobehavioral Research Center Group. Arch Neurol. 1997;54:416-424.
31 A randomized, double-blind, placebo-controlled trial of deprenyl and thioctic acid in human immunodeficiency virus–associated cognitive impairment. Dana Consortium on the Therapy of HIV Dementia and Related Cognitive Disorders. Neurology. 1998;50:645-651.
32 Asensio O, Caso JA, Rojas R. Carpal tunnel syndrome in HIV patients? AIDS. 2002;16:948-950.
33 Power C, Johnson RT. HIV-1 associated dementia: clinical features and pathogenesis. Can J Neurol Sci. 1995;22:92-100.
34 Maher J, Choudhri S, Halliday W, et al. AIDS dementia complex with generalized myoclonus. Mov Disord. 1997;12:593-597.
35 Mirsattari SM, Power C, Nath A. Parkinsonism with HIV infection. Mov Disord. 1998;13:684-689.
36 Navia BA, Jordan BD, Price RW. The AIDS dementia complex: I. Clinical features. Ann Neurol. 1986;19:517-524.
37 Alciati A, Fusi A, D’Arminio Monforte A, et al. New-onset delusions and hallucinations in patients infected with HIV. J Psychiatry Neurosci. 2001;26:229-234.
38 Koutsilieri E, Scheller C, Sopper S, et al. Psychiatric complications in human immunodeficiency virus infection. J Neurovirol 8 Suppl. 2002;2:129-133.
39 Nath A, Berger J. HIV dementia. Curr Treat Options Neurol. 2004;6:139-151.
40 Power C, Selnes OA, Grim JA, et al. HIV Dementia Scale: a rapid screening test. J Acquir Immune Defic Syndr Hum Retrovirol. 1995;8:273-278.
41 Jones BN, Teng EL, Folstein MF, et al. A new bedside test of cognition for patients with HIV infection. Ann Intern Med. 1993;119:1001-1004.
42 Berghuis JP, Uldall KK, Lalonde B. Validity of two scales in identifying HIV-associated dementia. J Acquir Immune Defic Syndr. 1999;21:134-140.
43 Grassi MP, Perin C, Borella M, et al. Assessment of cognitive function in asymptomatic HIV-positive subjects. Eur Neurol. 1999;42:225-229.
44 Price RW, Brew BJ. The AIDS dementia complex. J Infect Dis. 1988;158:1079-1083.
45 Dal Pan GJ, McArthur JH, Aylward E, et al. Patterns of cerebral atrophy in HIV-1–infected individuals: results of a quantitative MRI analysis. Neurology. 1992;42:2125-2130.
46 Simpson DM, Tagliati M. Neurologic manifestations of HIV infection. Ann Intern Med. 1994;121:769-785. [Erratum in Ann Intern Med 1995; 122:317].
47 McArthur JC, Cohen BA, Farzedegan H, et al. Cerebrospinal fluid abnormalities in homosexual men with and without neuropsychiatric findings. Ann Neurol. 1988;23:S34-S37.
48 Chang L, Ernst T, Leonido-Yee M, et al. Cerebral metabolite abnormalities correlate with clinical severity of HIV-1 cognitive motor complex. Neurology. 1999;52:100-108.
49 Power C, McArthur JC, Johnson RT, et al. Demented and nondemented patients with AIDS differ in brain-derived human immunodeficiency virus type 1 envelope sequences. J Virol. 1994;68:4643-4649.
50 Price RW, Yiannoutos T, Clifford DB, et al. Neurological outcomes in late HIV infection: adverse impact of neurological survival and protective effect of antiviral therapy. AIDS Clinical Trial Group and Neurological AIDS Research Consortium study team. AIDS. 1999;13:1677-1685.
51 Sacktor NC, Lyles RH, Skolasky RL, et al. Combination antiretroviral therapy improves psychomotor speed performance in HIV-seropositive homosexual men. Multicenter AIDS Cohort Study (MACS). Neurology. 1999;52:1640-1647.
52 von Giesen HJ, Koller H, Theisen A, et al. Therapeutic effects of nonnucleoside reverse transcriptase inhibitors on the central nervous system in HIV-1–infected patients. J Acquir Immune Defic Syndr. 2002;29:363-367.
53 Brown GR. The use of methylphenidate for cognitive decline associated with HIV disease. Int J Psychiatry Med. 1995;25:21-37.
54 Sacktor NC, Skolasky RL, Lyles RH, et al. Improvement in HIV-associated motor slowing after antiretroviral therapy including protease inhibitors. J Neurovirol. 2000;6:84-88.
55 Nath A, Haughey NJ, Jones M, et al. Synergistic neurotoxicity by human immunodeficiency virus proteins Tat and gp120: protection by memantine. Ann Neurol. 2000;47:186-194.
56 Zhang K, McQuibban GA, Silva C, et al. HIV-induced metalloproteinase processing of the chemokine stromal cell derived factor-1 causes neurodegeneration. Nat Neurosci. 2003;6:1064-1071.
57 Silva C, Zhang K, Tsutsui S, et al. Growth hormone prevents human immunodeficiency virus–induced neuronal p53 expression. Ann Neurol. 2003;54:605-614.
58 Clifford DB. Human immunodeficiency virus–associated dementia. Arch Neurol. 2000;57:321-324.
59 Safety and tolerability of the antioxidant OPC-14117 in HIV-associated cognitive impairment. The Dana Consortium on the Therapy of HIV Dementia and Related Cognitive Disorders. Neurology. 1997;49:142-146.
60 Navia BA, Dafni U, Simpson D, et al. A phase I/II trial of nimodipine for HIV-related neurologic complications. Neurology. 1998;51:221-228.
61 Dal Pan GJ, Berger JR. Spinal cord disease in human immunodeficiency virus infection. In: Berger JR, Levy RM, editors. AIDS and the Nervous System. 2nd ed. Philadelphia: Lippincott-Raven; 1997:173-187.
62 DiRocco L, Dalton T, Liang D, et al. Nonallelism for the audiogenic seizure prone (Asp1) and the aryl hydrocarbon receptor (Ahr) loci in mice. J Neurogenet. 1998;12:191-203.
63 Brinley FJJr, Pardo CA, Verma A. Human immunodeficiency virus and the peripheral nervous system workshop. Arch Neurol. 2001;58:1561-1566.
64 Fuller GN, Jacobs JM, Guiloff RJ. Nature and incidence of peripheral nerve syndromes in HIV infection. J Neurol Neurosurg Psychiatry. 1993;56:372-381.
65 Brannagan TH, McAlarney T, Latov N. Peripheral neuropathy in HIV-1 infection. In: Latov N, Wokke JH, Kelly JJ, editors. Immunological and Infectious Diseases of the Peripheral Nerves. Cambridge, UK: Cambridge University Press; 1998:285-307.
66 Dalakas MC, Cupler EJ. Neuropathies in HIV infection. Baillieres Clin Neurol. 1996;5:199-218.
67 Price RW. Neurological complications of HIV infection. Lancet. 1996;348:445-452.
68 Pardo CA, McArthur JC, Griffin JW. HIV neuropathy: insights in the pathology of HIV peripheral nerve disease. J Peripher Nerv Syst. 2001;6:21-27.
68a Pettersen JA, Jones G, et al. Sensory neuropathy in human immunodeficiency virus. Ann Neurol. 2006;59:816-824.
69 Hahn K, Arendt G, Braun JS, et al. A placebo-controlled trial of gabapentin for painful HIV-associated sensory neuropathies. J Neurol. 2004;251:1260-1266.
70 Kieburtz K, Simpson D, Yiannoutsos C, et al. A randomized trial of amitriptyline and mexiletine for painful neuropathy in HIV infection. AIDS Clinical Trial Group 242 Protocol Team. Neurology. 1998;51:1682-1688.
71 Estanislao L, Carter K, McArthur J, et al. A randomized controlled trial of 5% lidocaine gel for HIV-associated distal symmetric polyneuropathy. J Acquir Immune Defic Syndr. 2004;37:1584-1586.
72 Schifitto G, Yiannoutsos C, Simpson DM, et al. Long-term treatment with recombinant nerve growth factor for HIV-associated sensory neuropathy. Neurology. 2001;57:1313-1316.
73 Mahadevan A, Gayathri N, Taly AB, et al. Vasculitic neuropathy in HIV infection: a clinicopathological study. Neurol India. 2001;49:277-283.
74 Verma A. Epidemiology and clinical features of HIV-1 associated neuropathies. J Peripher Nerv Syst. 2001;6:8-13.
75 Cacoub P, Maisonobe T, Thibault V, et al. Systemic vasculitis in patients with hepatitis C. J Rheumatol. 2001;28:109-118.
76 Stricker RB, Sanders KA, Owen WF, et al. Mononeuritis multiplex associated with cryoglobulinemia in HIV infection. Neurology. 1992;42:2103-2105.
77 Lipkin WI, Parry G, Kiprov D, et al. Inflammatory neuropathy in homosexual men with lymphadenopathy. Neurology. 1985;35:1479-1483.
78 Bradley WG, Verma A. Painful vasculitic neuropathy in HIV-1 infection: relief of pain with prednisone therapy. Neurology. 1996;47:1446-1451.
79 Bani-Sadr F, Neuville S, Crassard I, et al. Acute Guillain-Barré syndrome during the chronic phase of HIV infection and dramatic improvement under highly active antiretroviral therapy. AIDS. 2002;16:1562.
80 Cornblath DR, McArthur JC, Kennedy PG, et al. Inflammatory demyelinating peripheral neuropathies associated with human T-cell lymphotropic virus type III infection. Ann Neurol. 1987;21:32-40.
81 Schleicher GK, Black A, Mochan A, et al. Effect of human immunodeficiency virus on intensive care unit outcome of patients with Guillain-Barré syndrome. Crit Care Med. 2003;31:1848-1850.
82 Sclar G. Carpal tunnel syndrome in HIV-1 patients: a metabolic consequence of protease inhibitor use? AIDS. 2000;14:336-338.
83 Verdugo RJ, Salinas RS, Castillo J, et al. Surgical versus nonsurgical treatment for carpal tunnel syndrome. Cochrane Database Syst Rev. (3):2003. CD001552
84 Moulignier A, Moulonguet A, Pialoux G, et al. Reversible ALS-like disorder in HIV infection. Neurology. 2001;57:995-1001.
85 Evenhouse M, Haas E, Snell E, et al. Hypotension in infection with the human immunodeficiency virus. Ann Intern Med. 1987;107:598-599.
86 Gluck T, Degenhardt E, Scholmerich J, et al. Autonomic neuropathy in patients with HIV: course, impact of disease stage, and medication. Clin Auton Res. 2000;10:17-22.
87 Schielke E, Pfister HW, Einhaupl KM. Peripheral facial nerve palsy associated with HIV infection. Lancet. 1989;1:553-554.
88 Meenken C, van den Horn GJ, de Smet MD, et al. Optic neuritis heralding varicella zoster virus retinitis in a patient with acquired immunodeficiency syndrome. Ann Neurol. 1998;43:534-536.
89 Berger JR, Sheremata WA, Resnick L, et al. Multiple sclerosis–like illness occurring with human immunodeficiency virus infection. Neurology. 1989;39:324-329.
90 Berger JR, Tornatore C, Major EO, et al. Relapsing and remitting human immunodeficiency virus-associated leukoen-cephalomyelopathy. Ann Neurol. 1992;31:34-38.
91 Cooper DA, Gold J, Maclean P, et al. Acute AIDS retrovirus infection. Definition of a clinical illness associated with seroconversion. Lancet. 1985;1:537-540.
92 Mirsattari SM, Power C, Nath A. Primary headaches in HIV-infected patients. Headache. 1999;39:3-10.
93 Singer EJ, Kim J, Fahy-Chandon B, et al. Headache in ambulatory HIV-1–infected men enrolled in a longitudinal study. Neurology. 1996;47:487-494.
94 Brew BJ, Miller J. Human immunodeficiency virus-related headache. Neurology. 1993;43:1098-1100.
95 Garg RK. HIV infection and seizures. Postgrad Med J. 1999;75:387-390.
96 Dore GJ, Law MG, Brew BJ. Prospective analysis of seizures occurring in human immunodeficiency virus type-1 infection. J Neuro-AIDS. 1996;1:59-69.
97 Holtzman DM, Kaku DA, So YT. New-onset seizures associated with human immunodeficiency virus infection: causation and clinical features in 100 cases. Am J Med. 1989;87:173-177.
98 Romanelli F, Jennings HR, Nath A, et al. Therapeutic dilemma: the use of anticonvulsants in HIV-positive individuals. Neurology. 2000;54:1404-1407.
99 Romanelli F, Ryan M. Seizures in HIV-seropositive individuals: epidemiology and treatment. CNS Drugs. 2002;16:91-98.
100 Smit TK, Brew BJ, Tourtellotte W, et al. Independent evolution of human immunodeficiency virus (HIV) drug resistance mutations in diverse areas of the brain in HIV-infected patients, with and without dementia, on antiretroviral treatment. J Virol. 2004;78:10133-10148.