[level-membership-for-critical-care-medicine-category]
Objectives
• Describe the etiology and pathophysiology of selected neurological disorders.
• Identify the clinical manifestations of selected neurological disorders.
• Explain the treatment of selected neurological disorders.
• Discuss the nursing priorities for managing a patient with selected neurological disorders.
• Discuss the concept of cerebral autoregulation.
• Describe the therapies commonly used to treat intracranial hypertension.

Be sure to check out the bonus material, including free self-assessment exercises, on the Evolve web site at
http://evolve.elsevier.com/Urden/priorities/.
To accurately anticipate and plan nursing interventions, the critical care nurse must have an understanding of the disease pathology, determine the areas of focused assessment, and be well acquainted with the medical management of the neurological patient. Fortunately, despite a wide array of neurological disorders, only a few routinely require the critical care environment.
Coma
Normal consciousness requires awareness and arousal. Awareness is the combination of cognition (mental and intellectual) and affect (mood) that can be construed based on the patient’s interaction with the environment.1 Alterations of consciousness may be the result of deficits in awareness, arousal, or both.2 There are four discrete disorders of consciousness: coma, vegetative state, minimally conscious state, and locked-in syndrome. Coma is characterized by the absence of both wakefulness and awareness, whereas a vegetative state is characterized by the presence of wakefulness with the absence of awareness. In the minimally conscious state, wakefulness is presence and awareness is severely diminished but not absent. Locked-in syndrome is characterized by the presence of wakefulness and awareness, but with quadriplegia and the inability to communicate verbally; thus the patient appears to be unconscious.3 Box 18-1 lists the disorders of consciousness in descending order of wakefulness.
Coma is the deepest state of unconsciousness; arousal and awareness are lacking.1–3 The patient cannot be aroused and does not demonstrate any purposeful response to the surrounding environment.4 Coma is a symptom rather than a disease, and it occurs as a result of some underlying process.1,2 The incidence of coma is difficult to ascertain because a wide variety of conditions can induce coma.1,2 This state of unconsciousness is unfortunately very common in critical care, and it is the focus of the following discussion.
Etiology
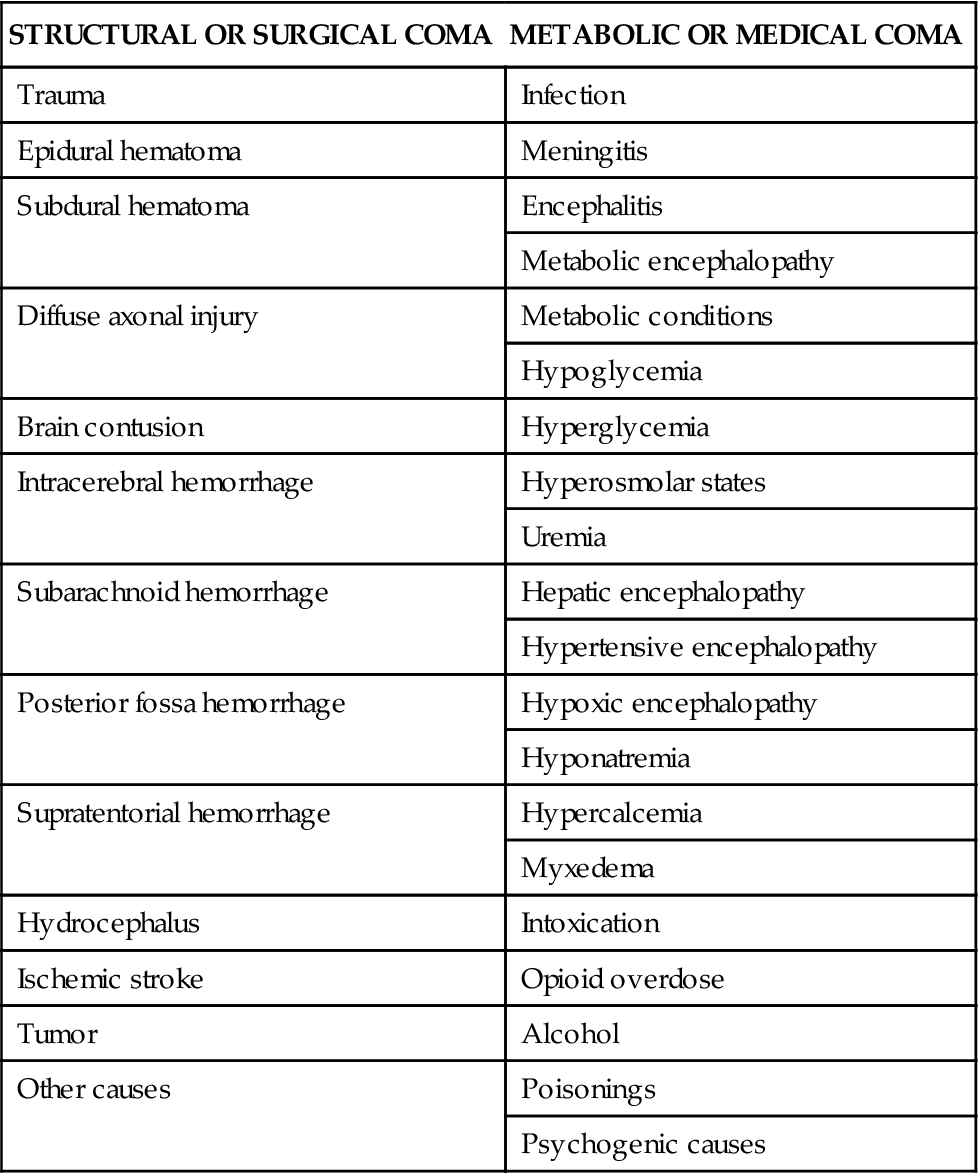
The causes of coma can be divided into two general categories: structural or surgical and metabolic or medical. Structural causes of coma include ischemic stroke, intracerebral hemorrhage (ICH), trauma, and brain tumors.5 Metabolic causes of coma include drug overdose, infectious diseases, endocrine disorders, and poisonings.5 Coma demands immediate attention, resulting in a high percentage of admissions to all hospital services.6 Table 18-1 provides a list of the possible causes of coma.
TABLE 18-1
| STRUCTURAL OR SURGICAL COMA | METABOLIC OR MEDICAL COMA |
| Trauma | Infection |
| Epidural hematoma | Meningitis |
| Subdural hematoma | Encephalitis |
| Metabolic encephalopathy | |
| Diffuse axonal injury | Metabolic conditions |
| Hypoglycemia | |
| Brain contusion | Hyperglycemia |
| Intracerebral hemorrhage | Hyperosmolar states |
| Uremia | |
| Subarachnoid hemorrhage | Hepatic encephalopathy |
| Hypertensive encephalopathy | |
| Posterior fossa hemorrhage | Hypoxic encephalopathy |
| Hyponatremia | |
| Supratentorial hemorrhage | Hypercalcemia |
| Myxedema | |
| Hydrocephalus | Intoxication |
| Ischemic stroke | Opioid overdose |
| Tumor | Alcohol |
| Other causes | Poisonings |
| Psychogenic causes |
Pathophysiology
Consciousness involves arousal, or wakefulness, and awareness. Neither of these functions is present in the patient in coma. Ascending fibers of the reticular activating system (ARAS) in the pons, hypothalamus, and thalamus maintain arousal as an autonomic function. Neurons in the cerebral cortex are responsible for awareness. Diffuse dysfunction of both cerebral hemispheres and diffuse or focal dysfunction of the reticular activating system can produce coma.1,6,7 Structural causes usually produce compression or dysfunction in the area of the ARAS, whereas most medical causes lead to general dysfunction of both cerebral hemispheres.8 Trauma, hemorrhage, and tumor can damage the ARAS, leading to coma. Destruction of large regions of bilateral cerebral hemispheres can be the result of seizures or viral agents. Toxic drugs, toxins, or metabolic abnormalities can suppress cerebral function.5–7
Assessment and Diagnosis
The clinical diagnosis of the coma state is readily established by assessment of the level of consciousness. However, determining the full nature and cause of coma requires a thorough history and physical examination. A medical history is essential, because events immediately preceding the change in level of consciousness can often provide valuable clues to the origin of the coma. When limited information is available and the coma is profound, the response of the patient to emergency treatment may provide clues to the underlying diagnosis; for example, the patient who becomes responsive with the administration of naloxone can be presumed to have ingested some type of opiate.6
Detailed serial neurological examinations are essential for all patients in coma. Assessment of pupillary size and reaction to light (normal, sluggish, or fixed), extraocular eye movements (normal, asymmetrical, or absent), motor response to pain (normal, decorticate, decerebrate, or flaccid), and breathing pattern yields important clues for determining whether the cause of the coma is structural or metabolic.1,6
The areas of the brainstem that control consciousness and pupillary responses are anatomically adjacent. The sympathetic and parasympathetic nervous systems control pupillary dilation and constriction, respectively. The anatomic directions of these pathways are known, and changes in pupillary responses can help identify where a lesion may be located. For example, if damage occurs in the midbrain region, pupils are slightly enlarged and unresponsive to light. Lesions that compress the third nerve result in a fixed and dilated pupil on the same side as the neurological insult. Pupillary responses are usually preserved when the cause of the coma is metabolic in origin. Pupillary light responses are often the key to differentiating between structural and metabolic causes of coma.1,6,7,9
Areas of the brainstem adjacent to those responsible for consciousness also control the oculomotor eye movement. The ability to maintain conjugate gaze requires preservation of the internuclear connections of cranial nerves III, VI, and VIII by means of the medial longitudinal fasciculus (MLF).9 As with pupillary responses, structural lesions that impinge on these pathways cause oculomotor dysfunction such as a disconjugate gaze. Deficits in extraocular eye movements usually accompany a structural cause.1,5,9
Focal or asymmetric motor deficits usually indicate structural lesions.1,5 Abnormal motor movements may also help pinpoint the location of a lesion. Decorticate posturing (abnormal flexion) can be seen with damage to the diencephalon. Decerebrate posturing (abnormal extension) can be seen with damage to the midbrain and pons. Flaccid posturing is an ominous sign and can be seen with damage to the medulla.9
Abnormal breathing patterns may also assist in differentiating structural from metabolic causes of coma. Cheyne-Stokes respirations are seen in patients with cerebral hemispheric dysfunction or metabolic suppression. Central neurogenic hyperventilation, or Kussmaul breathing, occurs with metabolic acidosis or damage to the midbrain and upper pons. Apneustic breathing may occur with damage to the pons, hypoglycemia, and anoxia. Ataxic breathing occurs with damage to the medulla. Agonal breathing occurs with failure of the respiratory centers in the medulla.6,9
In addition to physical assessment, laboratory studies and diagnostic procedures are done. Structural causes of coma are usually readily apparent with computed tomography (CT) or magnetic resonance imaging (MRI).4,8 Evoked potentials are also useful in facilitating a differential diagnosis between the disorders of consciousness and in evaluating a patient’s prognosis.3 Generally a patient in a coma with an absence of brainstem auditory evoked responses (BAERs) is considered to have a poor prognosis of recovery.3 Laboratory studies are also used to identify metabolic or endocrine abnormalities.7 Occasionally, the cause of coma is never clearly determined.
Medical Management
The goal of medical management of the patient in a coma is identification and treatment of the underlying cause of the condition. Initial medical management includes emergency measures to support vital functions and prevent further neurological deterioration. Protection of the airway and ventilatory assistance are often needed. Administration of thiamine (at least 100 mg), glucose, and an opioid antagonist is suggested when the cause of coma is not immediately known.1,6 Thiamine is administered before glucose, because the coma produced by thiamine deficiency, Wernicke’s encephalopathy, can be precipitated by a glucose load.1
The patient who remains in a coma after emergency treatment requires supportive measures to maintain physiological body functions and prevent complications. Intubation for continued airway protection and nutritional support are essential. Fluid and electrolyte management is often complex because of alterations in the neurohormonal system. Anticonvulsant therapy may be necessary to prevent further ischemic damage to the brain.1,5,6
The health care team and the patient’s family make decisions jointly regarding the level of medical management to be provided. Family members require informational support in terms of probable cause of the coma and prognosis for recovery of consciousness and function. Prognosis depends on the cause of the coma and the length of time unconsciousness persists. Only 15% of patients in nontraumatic coma make a satisfactory recovery.7 Metabolic coma usually has a better prognosis than coma caused by a structural lesion, and traumatic coma usually has a better outcome than nontraumatic coma.5,7
Nursing Management
Nursing management of the patient in a coma incorporates a variety of nursing diagnoses (Nursing Diagnosis Priorities box on Coma) and is directed by the specific cause of the coma, although some common interventions are used. The patient in a coma totally depends on the health care team. Nursing priorities are directed toward (1) monitoring for changes in neurological status and clues to the origin of the coma, (2) supporting all body functions, (3) maintaining surveillance for complications, (4) providing comfort and emotional support, and (5) initiating rehabilitation measures. Measures to support body functions include promoting pulmonary hygiene, maintaining skin integrity, initiating range-of-motion exercises, managing bowel and bladder functions, and ensuring adequate nutritional support.1
Eye Care
The blink reflex is often diminished or absent in the comatose patient. The eyelids may be flaccid and may depend on body positioning to remain in a closed position, and edema may prevent complete closure. Loss of these protective mechanisms results in drying and ulceration of the cornea, which can lead to permanent scarring and blindness.1
Two interventions that are commonly used to protect the eyes are instilling saline or methylcellulose lubricating drops and taping the eyelids in the shut position. Evidence suggests that an alternative technique may be more effective in preventing corneal epithelial breakdown. In addition to instillation of saline drops every 2 hours, a polyethylene film is taped over the eyes, extending beyond the orbits and eyebrows. The film creates a moisture chamber around the cornea and assists in keeping the eyes moist and in the closed position. This technique also prevents damage to the eyes that results from placement of tape or gauze directly on the delicate skin of the eyelids.10
Collaborative management of the patient in a coma is outlined in the Collaborative Management box on Coma.
Stroke
Stroke is a descriptive term for the sudden onset of acute neurological deficit persisting for more than 24 hours and caused by the interruption of blood flow to the brain. Stroke is the third leading cause of death in the United States, preceded by heart disease and cancer, and the leading cause of adult disability.11 Approximately 795,000 people have a stroke each year; 610,000 of these are first attacks, and 185,000 are recurrent attacks.11
Strokes are classified as ischemic or hemorrhagic (Concept Map on Stroke). Hemorrhagic strokes can be further categorized as subarachnoid hemorrhages (SAHs) and intracerebral hemorrhages. Approximately 87% of all strokes are ischemic, 10% are ICHs, and 3% are SAHs.11 Although less common, hemorrhagic strokes (ICHs and SAHs) have a higher mortality rate than ischemic strokes. Approximately 8% to 12% of ischemic strokes and 37% to 45% of hemorrhagic strokes result in death within 30 days.11 The annual cost for care and loss of productivity was estimated to be $73.7 billion in 2009.11
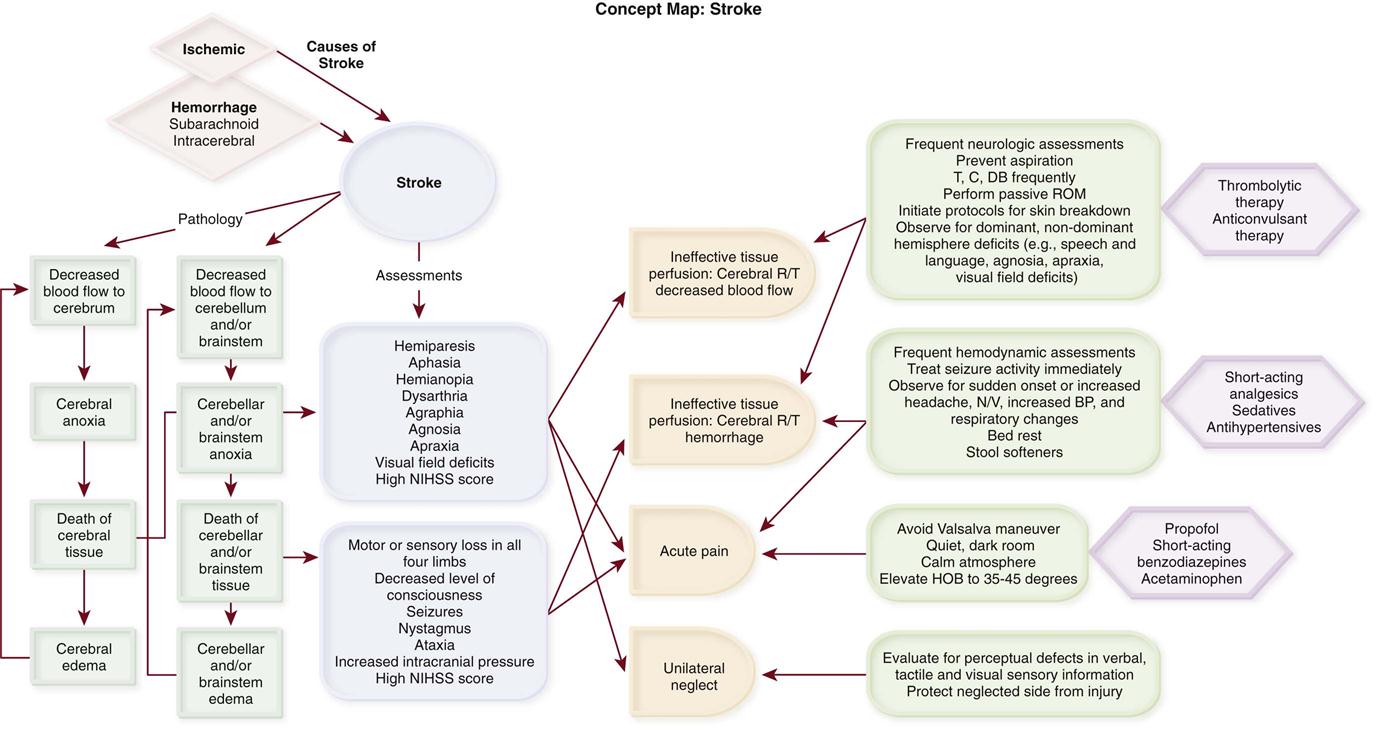
Ischemic Stroke
Ischemic stroke results from interruption of blood flow to the brain and accounts for 80% to 85% of all strokes. The interruption can be the result of a thrombotic or embolic event. Thrombosis can form in large vessels (large-vessel thrombotic strokes) or small vessels (small-vessel thrombotic strokes). Embolic sources include the heart (cardioembolic strokes) and atherosclerotic plaques in larger vessels (atheroembolic strokes). In 30% of the cases, the underlying cause of the stroke is unknown (cryptogenic strokes).12
Strokes are preventable. Most thrombotic strokes are the result of the accumulation of atherosclerotic plaque in the vessel lumen, especially at bifurcations or curves of the vessel. The pathogenesis of cerebrovascular disease is identical to that of coronary vasculature disease. The greatest risk factor for ischemic stroke is hypertension.12,13 Other risk factors are dyslipidemia, diabetes, smoking, and carotid atherosclerotic disease.11,14 Common sites of atherosclerotic plaque are the bifurcation of the common carotid artery, the origins of the middle and anterior cerebral arteries, and the origins of the vertebral arteries.13 Ischemic strokes resulting from vertebral artery dissection have been reported after chiropractic manipulation of the cervical spine.15
Etiology
An embolic stroke occurs when an embolus from the heart or lower circulation travels distally and lodges in a small vessel, obstructing the blood supply. At least 20% of ischemic strokes are attributed to a cardioembolic phenomenon.12 The most common cause of cardiac emboli is atrial fibrillation. It is responsible for about 50% of all cardiac emboli.16 Other sources of cardiac emboli are mitral stenosis, mechanical valves, atrial myxoma, endocarditis, and recent myocardial infarction.13 Researchers hypothesize that a patent foramen ovale or atrial septal aneurysms may be the cause of cryptogenic stroke.17
Pathophysiology
Ischemic stroke is a cerebral hemodynamic insult. When cerebral blood flow is reduced to a level insufficient to maintain neuronal viability, ischemic injury occurs. In focal stroke, an area of hypoperfused tissue, the ischemic penumbra, surrounds a core of ischemic cells. The ischemic penumbra can be salvaged with return of blood flow. However, sustained anoxic insult initiates a chain of biochemical events leading to apoptosis, or cellular death.18
The phenomenon of a focal ischemic stroke is identical to that associated with myocardial infarction, which is why brain attack is used in public education strategies. Often, a history of transient ischemic attacks (TIAs), brief episodes of neurological symptoms that last less than 24 hours, offers a warning that stroke is likely to occur. Sudden onset indicates embolism as the final insult to flow.12,13 The size of the stroke depends on the size and location of the occluded vessel and the availability of collateral blood flow.12 Global ischemia results when severe hypotension or cardiopulmonary arrest provokes a transient drop in blood flow to all areas of the brain.18
Cerebral edema sufficient to produce clinical deterioration develops in 10% to 20% of patients with ischemic stroke and can result in intracranial hypertension. The edema results from a loss of normal metabolic function of the cells and peaks at 4 days.12 This process is commonly the cause of death during the first week after a stroke.19 Secondary hemorrhage at the site of the stroke lesion, known as hemorrhagic conversion,19 and seizures20 are the two other major acute neurological complications of ischemic stroke.
Assessment and Diagnosis
The characteristic sign of an ischemic stroke is the sudden onset of focal neurological signs persisting for more than 24 hours.12 These signs usually occur in combination. Box 18-2 lists common patterns of neurological symptoms associated with an ischemic stroke. Hemiparesis, aphasia, and hemianopia are common. Changes in the level of consciousness usually occur only with brainstem or cerebellar involvement, seizure, hypoxia, hemorrhage, or elevated intracranial pressure (ICP). These changes may be exhibited as stupor, coma, confusion, and agitation.1 The reported frequency of seizures in patients with ischemic stroke ranges from 3% to 8%. If seizures occur, they are usually seen within 24 hours of an insult.20
The National Institutes of Health Stroke Scale (NIHSS) is often used as the basis of the focused neurological examination. The score ranges from 0 to 42 points; the higher the score, the more neurologically impaired the patient. A change of 4 points on the scale indicates significant neurological change. The components of the NIHSS include level of consciousness (LOC); LOC questions; LOC commands; gaze; visual fields; face, arm, and leg strength; sensation; limb ataxia; and language function.12 A copy of the NIHSS with complete instructions can be retrieved at http://www.ninds.nih.gov/disorders/stroke/strokescales.htm.
Confirmation of the diagnosis of ischemic stroke is the first step in the emergency evaluation of these patients. Differentiation from intracranial hemorrhage is vital. Noncontrast CT scanning is the method of choice for this purpose and is considered the most important initial diagnostic study. In addition to excluding intracranial hemorrhage, CT can assist in identifying early neurological complications and the cause of the insult.12 MRI can demonstrate infarction of cerebral tissue earlier than CT but is less useful in the emergency differential diagnosis.21 Because of the strong correlation between acute ischemic stroke and heart disease, 12-lead electrocardiography, chest radiography, and continuous cardiac monitoring are suggested to detect a cardiac cause or coexisting condition. Echocardiography is valuable in identifying a cardioembolic phenomenon when a sufficient index of suspicion warrants its use.1 Laboratory evaluation of hematological function, electrolyte and glucose levels, and renal and hepatic function is also recommended. Arterial blood gas analysis is performed if hypoxia is suspected, and an electroencephalogram is obtained if seizures are suspected. Lumbar puncture is performed only if SAH is suspected and the CT appearance is normal.1
Medical Management
Major changes have taken place in the medical management of ischemic stroke since 1996. On the basis of results of the National Institute of Neurologic Disorders and Stroke (NINDS) rtPA Stroke Study, thrombolytic therapy with intravenous recombinant tissue-type plasminogen activator (rtPA) was initially recommended within 3 hours of onset of ischemic stroke. This time frame has now been expanded from 3 hours to 4.5 hours.22 Patients who should be considered for thrombolysis are listed in Box 18-3. Confirmation of diagnosis with CT must be accomplished before rtPA administration. The recommended dose of rtPA is 0.9 mg/kg up to a maximum dose of 90 mg. Ten percent of the total dose is administered as an initial intravenous bolus, and the remaining 90% is administered by intravenous infusion over 60 minutes.17,21
The desired result of thrombolytic therapy is to dissolve the clot and reperfuse the ischemic brain. The goal is to reverse or minimize the effects of stroke. The major risk and complication of rtPA therapy is bleeding, especially intracranial hemorrhage. Unlike thrombolytic protocols for acute myocardial infarction, subsequent therapy with anticoagulant or antiplatelet agents is not recommended after rtPA administration in ischemic stroke. Patients receiving thrombolytic therapy for stroke should not receive aspirin, heparin, or warfarin for at least 24 hours after treatment.12,17
The major barriers to effective application of thrombolytic therapy for ischemic stroke are prehospital and in-hospital delays. To help decrease delays, the public needs to be educated about stroke symptoms and activation of the emergency medical system (EMS). EMS responders need adequate education and training on managing a patient with an acute ischemic stroke, focusing on stabilization and quick transport of the patient to the emergency department. The receiving hospital should ideally be primary stroke certified and have expert staff and the infrastructure to care for the complex stroke patient.21
Other emergency care of the patient with ischemic stroke must include airway protection and ventilatory assistance to maintain adequate tissue oxygenation.19 Hypertension is often present in the early period as a compensatory response, and in most cases, blood pressure (BP) must not be lowered (Table 18-2). For the patient who has not received thrombolytic therapy, antihypertensive therapy is considered only if the diastolic blood pressure is greater than 120 mm Hg or the systolic blood pressure is greater than 220 mm Hg.12 Criteria are different for patients who have received rtPA. Their blood pressure is kept below 180/105 mm Hg to prevent intracranial hemorrhage. Intravenous labetalol or nicardipine is used to achieve blood pressure control. If these agents are not effective, nitroprusside, hydralazine, or enalaprilat should be considered.12 Body temperature and glucose levels also must be normalized.12,19
TABLE 18-2
BLOOD PRESSURE MANAGEMENT FOR STROKE ACCORDING TO THE AMERICAN STROKE ASSOCIATION GUIDELINES
| BLOOD PRESSURE* | TREATMENT |
| Nonthrombolytic Candidates | |
| DBP >140 mm Hg | Sodium nitroprusside (0.5 mcg/kg/min); aim for 10%-20% reduction in DBP |
| SBP >220 mm Hg, DBP 121-140 mm Hg, or MAP† >130 mm Hg | 10-20 mg of labetalol‡ given by IVP over 1-2 min; may repeat or double labetalol every 20 min to a maximum dose of 300 mg |
| SBP <220 mm Hg, DBP = 120 mm Hg, or MAP† <130 mm Hg | Emergency antihypertensive therapy is deferred in the absence of aortic dissection, acute myocardial infarction, severe congestive heart failure, or hypertensive encephalopathy |
| Thrombolytic Candidates | |
| Pretreatment | |
| SBP >185 mm Hg or DBP >110 mm Hg | 1-2 inches of nitroglycerine paste (Nitropaste) or 1-2 doses of 10-20 mg of labetalol‡ given by IVP; if BP is not reduced and maintained to <185/110 mm Hg, the patient should not be treated with tPA |
| During and After Treatment | |
| Monitor BP | BP is monitored every 15 min for 2 hr, then every 30 min for 6 hr, and then hourly for 16 hr |
| DBP >140 mm Hg | Sodium nitroprusside (0.5 mcg/kg/min) |
| SBP >230 mm Hg or DBP 121-140 mm Hg | 10 mg of labetalol‡ given by IVP over 1-2 min; may repeat or double labetalol every 10 min to a maximum dose of 300 mg or give initial labetalol bolus and then start a labetalol drip at 2-8 mg/min |
| If BP not controlled by labetalol, consider sodium nitroprusside | |
| SBP 180-230 mm Hg or DBP 105-120 mm Hg | 10 mg of labetalol‡ given by IVP; may repeat or double labetalol every 10-20 min to a maximum dose of 300 mg or give initial labetalol bolus and then start a labetalol drip at 2-8 mg/min |
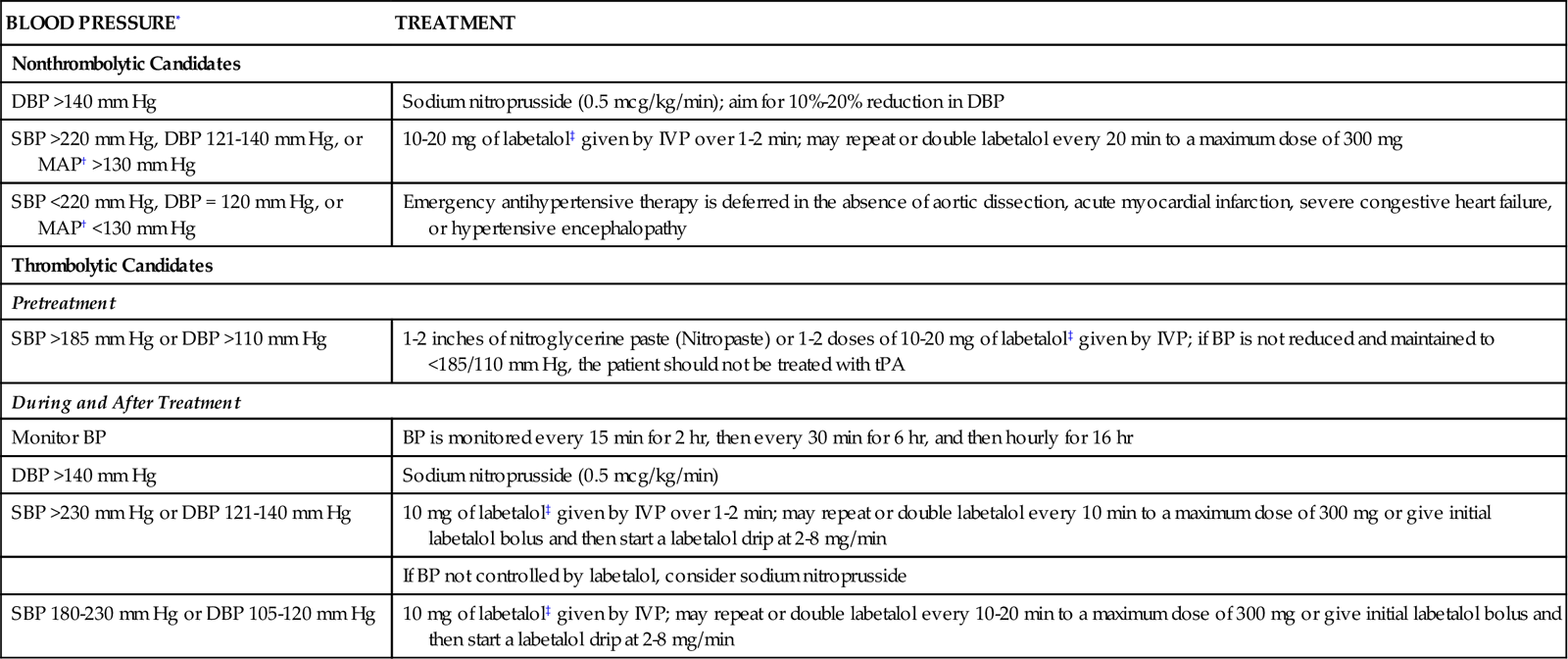
*All initial blood pressures should be verified before treatment by repeating reading in 5 minutes.
†As estimated by one third of the sum of systolic and double diastolic pressure.
‡Labetalol should be avoided in patients with asthma, cardiac failure, or severe abnormalities in cardiac conduction. For refractory hypertension, alternative therapy with sodium nitroprusside or enalapril may be considered.
From Bader MK, Littlejohns LR: AANN core curriculum for neuroscience nursing, ed 4, St Louis, 2004, Elsevier.
Medical management also includes the identification and treatment of acute complications such as cerebral edema and seizure activity. Prophylaxis for these complications is not recommended. Deep vein thrombosis (DVT) prophylaxis, however, should be initiated to decrease the risk of pulmonary embolism.12 One study demonstrated that improved outcomes for ischemic stroke patients can be achieved by managing swallowing issues, initiating DVT prophylaxis, and treating hypoxemia.23 Surgical decompression is recommended if a large cerebellar infarction compresses the brainstem.19
Subarachnoid Hemorrhage
Subarachnoid hemorrhage is bleeding into the subarachnoid space, which usually is caused by rupture of a cerebral aneurysm or arteriovenous malformation (AVM).19 At the time of autopsy, approximately 4% of the population has been found to have one or more aneurysms.24 Aneurysmal SAH is associated with a mortality rate of 25% to 50%, with most patients dying on the first day after the insult.24 Hemorrhage due to AVM rupture has a better chance of survival and is associated with an overall mortality rate of 10% to 15%.25
Etiology
Cerebral aneurysm rupture accounts for approximately 85% of all cases of spontaneous SAH.24 An aneurysm is an outpouching of the wall of a blood vessel that results from weakening of the wall of the vessel (Table 18-3).24 Ninety percent of aneurysms are congenital—the cause of which is unknown. The other 10% can be the result of traumatic injury (that stretches and tears the muscular middle layer of the arterial vessel) or infectious material (most often from infectious vegetation on valves of the left side of the heart after bacterial endocarditis) that lodges against a vessel wall and erodes the muscular layer, or they are of undetermined cause.26 Multiple aneurysms occur in approximately 30% of the cases and often are bilateral, occurring in the same location on both sides of the cerebral vascular system.27
TABLE 18-3
ANEURYSM CLASSIFICATION ACCORDING TO TYPE, SHAPE, LOCATION, AND COMMON CHARACTERISTICS
| TYPES OF ANEURYSMS | CHARACTERISTICS |
Berry or saccular
 |
Most common type, usually congenital; appears at a bifurcation in the anterior circulation, primarily at the base of the brain or the circle of Willis and its branches; grows from the base of the arterial wall with a neck or stem; contains blood; thinned dome is usually the site of rupture |
Giant or fusiform
 |
Can have an irregular shape and can be larger than 2.5 cm and atherosclerotic; involves mainly the internal carotid or vertebrobasilar artery; rarely ruptures; has no stem; can act like a space-occupying lesion in the brain; difficult to manage |
Mycotic
 |
Rare form; usually occurs from septic emboli, usually results from bacterial infection, which weaken the vessel wall, causing dilation involving the distal branches of the middle cerebral arteries |
Dissecting
 |
May occur during angiography; caused by trauma, syphilis, or arteriosclerosis, or when blood is forced between layers of the arterial wall; intima is pulled away from the medial layer, allowing blood to enter |
| Traumatic | Sometimes called a pseudoaneurysm, which may resolve after trauma |
| Charcot-Bouchard | Small aneurysm that can be seen in the area of the basal ganglia or the brainstem in individuals with a history of hypertension; chronic hypertension causes fibrinoid necrosis in the penetrating and subcortical arteries, weakening the arterial walls and causing formation of small aneurysmal outpouching |
AVM rupture is responsible for roughly 6% of all SAHs.27An arteriovenous malformation is a tangled mass of arterial and venous blood vessels that shunt blood directly from the arterial side into the venous side, bypassing the capillary system. They may be small, focal lesions or large, diffuse lesions that occupy almost an entire hemisphere.26 AVMs are always congenital, although the exact embryonic cause for these malformations is unknown. They also occur in the spinal cord and the renal, gastrointestinal, and integumentary systems.27 In contrast to SAH from aneurysm, which occurs in the middle-aged population, SAH from an AVM usually occurs in the second to fourth decades of life.27
Pathophysiology
The pathophysiology of the two most common causes of SAH is distinctly different.
Cerebral Aneurysm.
As the individual with a congenital cerebral aneurysm matures, blood pressure rises, and more stress is placed on the poorly developed and thin vessel wall. Ballooning of the vessel occurs, giving the aneurysm a berry-like appearance. Most cerebral aneurysms are saccular or berry-like with a stem or neck. Aneurysms are usually small, 2 to 7 mm in diameter, and often occur at the base of the brain on the circle of Willis.28 Figure 18-1 illustrates the usual distribution between the vessels. Most cerebral aneurysms occur at the bifurcations of blood vessels.24,25,28
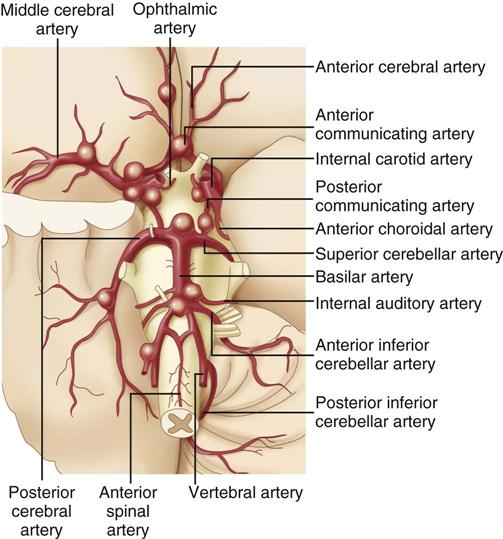
The aneurysm becomes clinically significant when the vessel wall becomes so thin that it ruptures, sending arterial blood at a high pressure into the subarachnoid space. For a brief moment after the aneurysm ruptures, ICP is thought to approach mean arterial pressure (MAP), and cerebral perfusion decreases.28 In other situations, the unruptured aneurysm expands and places pressure on surrounding structures. This is particularly true with posterior communicating artery aneurysms, because they put pressure on the oculomotor nerve (cranial nerve III), causing ipsilateral pupil dilation and ptosis.27
Arteriovenous Malformation.
The pathophysiologic features of an AVM are related to the size and location of the malformation. One or more cerebral arteries, also known as feeders, supply an AVM. These feeder arteries tend to enlarge over time, increasing both the volume of blood shunted through the malformation and the overall mass effect. Large, dilated, tortuous draining veins develop as a result of increasing arterial blood flow being delivered at a higher than normal pressure. Normal vascular flow has an MAP of 70 to 80 mm Hg, a mean arteriole pressure of 35 to 45 mm Hg, and a mean capillary pressure that drops from 35 to 10 mm Hg as it connects with the venous side. Lack of this capillary bridge allows blood with an MAP of 35 to 45 mm Hg to flow into the venous system. Unlike arteries, veins have no muscular layer, and the veins become extremely engorged and rupture easily. Some patients with AVMs also have cerebral atrophy. It is the result of chronic ischemia because of the shunting of blood through the AVM and away from normal cerebral circulation.29
Assessment and Diagnosis
The patient with an SAH characteristically has an abrupt onset of pain, described as the “worst headache of my life.” A brief loss of consciousness, nausea, vomiting, focal neurological deficits, and a stiff neck may accompany the headache.24,26–28 The SAH may result in coma or death.
The patient’s history may reveal one or more incidences of sudden onset of headache with vomiting in the weeks preceding a major SAH. These are small “warning leaks” of an aneurysm in which small amounts of blood ooze from the aneurysm into the subarachnoid space. The presence of blood is an irritant to the meninges, particularly the arachnoid membrane, and the irritation causes headache, stiff neck, and photophobia. These warning leaks seldom are detected because the condition is not severe enough for the patient to seek medical attention.28 If a neurological deficit, such as third cranial nerve palsy, develops before aneurysm rupture, medical intervention is sought, and the aneurysm may be surgically secured before the devastation of a rupture can occur. Symptoms of unruptured AVM—headaches with dizziness or syncope or fleeting neurological deficits—also may be found in the history.27
Diagnosis of SAH is based on clinical presentation, CT findings, and lumbar puncture results. Noncontrast CT is the cornerstone of definitive SAH diagnosis. In 95% of the cases, CT can demonstrate blood in the subarachnoid space if performed within 48 hours of the hemorrhage.24,28 On the basis of the appearance and the location of the SAH, diagnosis of the cause—aneurysm or AVM—may be made from the CT scan.26 MRI is not routinely used, but it may provide greater sensitivity for detecting areas of SAH clot and potential location of bleed.28
If the initial CT finding is negative, a lumbar puncture is performed to obtain cerebrospinal fluid (CSF) for analysis. CSF after SAH appears bloody and has a red blood cell count greater than 1000 cells/mm3. If the lumbar puncture is performed more than 5 days after the SAH, the CSF fluid is xanthochromic (dark amber) because the blood products have broken down.29 Cloudy CSF usually indicates some type of infectious process, such as bacterial meningitis, not SAH.28
After the SAH has been documented, cerebral angiography is necessary to identify the exact cause of the hemorrhage. If a cerebral aneurysm rupture is the cause, angiography is essential for identifying the exact location of the aneurysm in preparation for surgery.27,29,30 After the aneurysm has been located, it is graded using the Hunt and Hess classification scale. This scale categorizes the patient on the basis of the severity of the neurological deficits associated with the hemorrhage (Box 18-4).31 If AVM rupture is the cause, angiography is necessary to identify the feeding arteries and draining veins of the malformation.26
Medical Management
SAH is a medical emergency, and time is of the essence. Preservation of neurological function is the goal, and early diagnosis is crucial. Initial treatment must always support vital functions. Airway management and ventilatory assistance may be necessary.19 A ventriculostomy is performed to control ICP if the patient’s level of consciousness is depressed.30
Evidence suggests that only 19% of the deaths attributable to aneurysmal SAH are related to the direct effects of the initial hemorrhage.32 Rebleeding accounts for 22% of deaths from aneurysmal SAH, cerebral vasospasm for 23%, and nonneurological medical complications for 23%.32 Principal nonneurological causes of death are systemic inflammatory response syndrome (SIRS) and secondary organ dysfunction.33 After initial intervention has provided necessary support for vital physiologic functions, medical management of acute SAH is aimed primarily toward prevention and treatment of the complications of SAH that can produce further neurological damage and death.28
Rebleeding.
Rebleeding is the occurrence of a second SAH in an unsecured aneurysm or, less commonly, an AVM.6 The incidence of rebleeding during the first 24 hours after the first bleed is 4%, with a 1% to 2% chance per day for the following month. The mortality rate associated with aneurysmal rebleeding is approximately 70%.26,27
Historically, conservative measures to prevent rebleeding have included blood pressure control and SAH precautions (see “Nursing Management”). An elevation in blood pressure is a normal compensatory response to maintain adequate cerebral perfusion after a neurological insult. In the belief that hypertension contributes to rebleeding, intravenous antihypertensive agents are used to maintain a systolic blood pressure no greater than 140 mm Hg.28 Individualized guidelines must be determined on the basis of the clinical condition and preexisting values of the patient. Evidence suggests that rebleeding has more to do with variations in blood pressure than it does with absolute values and that blood pressure control does not lower the incidence of rebleeding.30
Surgical Clipping of Aneurysms.
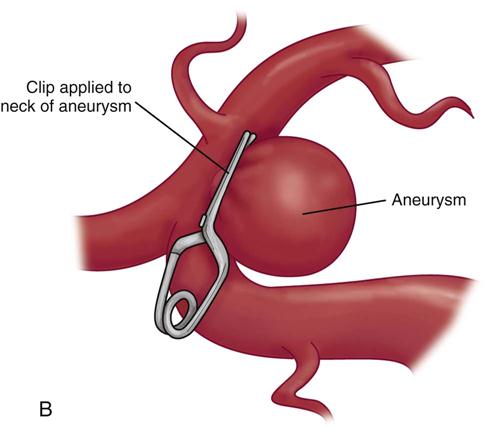
Definitive treatment for the prevention of rebleeding is surgical clipping or endovascular coiling with complete obliteration of the aneurysm.27,28 Timing of the operation is a key medical management issue. Since the introduction of microsurgery and improved surgical techniques, patients are commonly taken to the operating room within the first 48 hours after rupture.28 This early surgical intervention to secure the aneurysm eliminates the risk of rebleeding and allows more aggressive therapy to be used in the postoperative period for the treatment of vasospasm.26 Early surgery also allows the neurosurgeon to flush out the excess blood and clots from the basal cisterns (reservoir of CSF around the base of the brain and circle of Willis) to reduce the risk of vasospasm.33 Careful consideration of the patient’s clinical situation is necessary in determining the optimal time for surgery.
The surgical procedure involves a craniotomy to expose and isolate the area of aneurysm. A clip is placed over the neck of the aneurysm to eliminate the area of weakness (Figure 18-2). This is a technically difficult procedure that requires the skill of an experienced neurosurgeon. It is not uncommon, particularly in early surgery, for the clot to break away from the aneurysm as it is surgically exposed. Extensive hemorrhage into the craniotomy site results, and cessation of the hemorrhage often causes increased neurological deficits. Deficits also may occur as a result of surgical manipulation to gain access to the site of the aneurysm.28
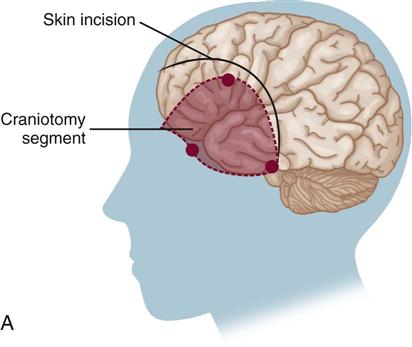
Surgical Excision of Arteriovenous Malformations.
Management of AVMs has traditionally involved surgical excision or conservative management of such symptoms as seizures and headache. The decision for surgical excision depends on the location and size of the AVM. Some malformations are located so deep in the cerebral structures (thalamus or midbrain) that attempts to remove the AVM would cause severe neurological deficits. History of a previous hemorrhage and the patient’s age and overall condition are also taken into account in the decision regarding surgical intervention.28
Surgical excision of large AVMs includes the risk of reperfusion bleeding. As feeding arteries of the AVM are clamped off, the arterial blood that usually flowed into the AVM is diverted into the surrounding circulation. In many cases, the surrounding tissue has been in a state of chronic ischemia, and the arterial vessels feeding these areas are maximally dilated. As arterial blood begins to flow at a higher volume and pressure into these dilated arteries, blood may seep from the vessels. Evidence of reperfusion bleeding in the operating room is an indication that no more arterial blood can be diverted from the AVM without risk of serious ICH. In the postoperative phase, a low blood pressure is maintained to prevent further reperfusion bleeding. For large AVMs, two to four stages of surgery may be required over 6 to 12 months.28
Embolization.
Embolization is used to secure a cerebral aneurysm or AVM that is surgically inaccessible because of size or location or because of the medical instability of the patient. Embolization involves several new interventional neuroradiology techniques. All of the techniques use a percutaneous transfemoral approach in a manner similar to an angiogram. Under fluoroscopic guidance, the catheter is threaded up to the internal carotid artery. Specially developed microcatheters are then manipulated into the area of the vascular anomaly, and embolic materials are placed endovascularly. Three embolization techniques are used, depending on the underlying pathologic derangement.26
The first type of embolization is used to embolize an AVM. Small polymeric silicone (Silastic) beads or glue is slowly introduced into the vessels feeding the AVM. Blood flow carries the material to the site, and embolization is achieved. This procedure may be used in combination with surgery. One to three sessions of embolization of the feeding vessels are performed to reduce the size of the lesion before a craniotomy is performed for total excision. The primary risk of this procedure is lodging of the embolic substance in a vessel that feeds normal tissue, which creates an embolic stroke with the immediate onset of neurological symptoms.26
The second type of embolization involves placement of one or more detachable coils into an aneurysm to produce an endovascular thrombus (Figure 18-3). The advantage of this technique is that an electrical current creates a positive charge on the coil, which induces electrothrombosis. Complications include embolic stroke, coil migration, overproduction of the clot, subtotal occlusion and intraprocedural rupture of the vasculature, and death.26
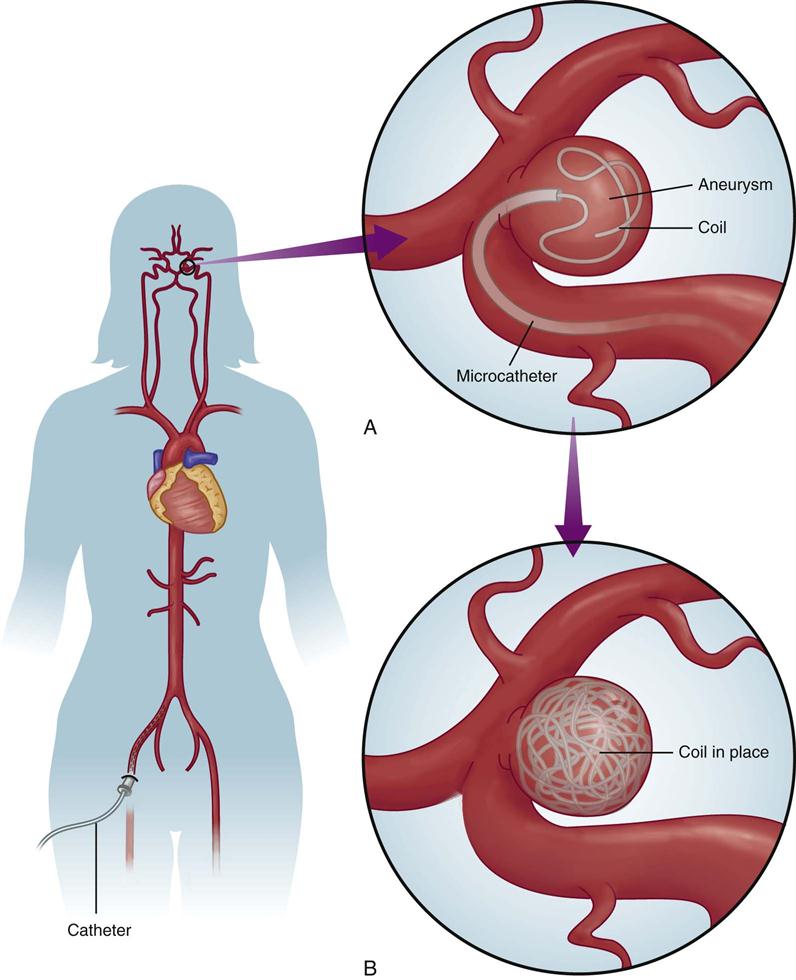
Cerebral Vasospasm.
The presence or absence of cerebral vasospasm significantly affects the outcome of aneurysmal SAH. This complication does not occur with SAH resulting from AVM rupture. Cerebral vasospasm is a narrowing of the lumen of the cerebral arteries, possibly in response to subarachnoid blood clots coating the outer surface of the blood vessels. Because aneurysms usually occur at the circle of Willis, the major vessels responsible for feeding the cerebral circulation are affected by vasospasm. Depending on the arterial vessels involved in the vasospasm reaction, decreased arterial flow occurs in large areas of the cerebral hemispheres.6
It is estimated that vasospasm, which is demonstrable by angiography, develops in 70% of all patients with SAH.34 Thirty-two percent of these patients have symptomatic vasospasm, resulting in ischemic stroke or death for up to 23% of them despite the use of maximal therapy.34 The onset of vasospasm is usually 3 to 12 days after the initial hemorrhage.19 Three treatments are commonly used: induced hypertensive, hypervolemic, hemodilution (HHH) therapy; oral nimodipine; and transluminal cerebral angioplasty.34
Hypertensive, Hypervolemic, Hemodilution Therapy.
HHH therapy involves increasing the patient’s blood pressure and cardiac output with vasoactive medications and diluting the patient’s blood with fluid and volume expanders. The goal of the therapy is to maintain systolic blood pressure between 150 and 160 mm Hg. The increase in volume and pressure forces blood through the vasospastic area at higher pressures. Hemodilution facilitates flow through the area by reducing blood viscosity.26 The Stroke Council of the American Heart Association (AHA) has recommended this therapy for prevention and treatment of vasospasm.30
The obvious deterrent to the use of induced hypertension is the risk of rebleeding in an unsecured aneurysm. Securing the aneurysm before HHH therapy is preferred. Cerebral edema, elevated ICP, cardiac failure, and electrolyte imbalance are also risks of HHH therapy. Careful monitoring of the patient’s neurological status, hemodynamic parameters, ICP, and serum electrolytes is necessary.28
Nimodipine.
Nimodipine is strongly recommended to reduce the poor outcomes associated with vasospasm. The exact nature of the effect of nimodipine is not clear, but use of the drug has demonstrated consistently positive effects on outcome without any demonstrable effect on the incidence or severity of vasospasm.30,34 A dose of 60 mg of nimodipine is given orally every 4 hours for 14 to 21 days. Nimodipine may produce hypotension, especially when administered concurrently with other antihypertensive agents.34
Cerebral Angioplasty.
Cerebral angioplasty is used when pharmacological management of cerebral vasospasm has failed. It is performed only when CT or MRI provides evidence that infarction has not occurred. An interventional neuroradiologist performs the procedure, and a local, general, or neuroleptic analgesia is used. The technique of cerebral angioplasty is very similar to that used in the coronary vasculature. Risks include intimal perforation or rupture, cerebral artery thrombosis or embolism, recurrence of stenosis, and severe, diffuse vasospasm unresponsive to therapy. Hemorrhage at the femoral site also may occur. This procedure is recommended when conventional therapy is unsuccessful.30,34
Hyponatremia.
Hyponatremia develops in 10% to 43% of patients with SAH as the result of a central salt-wasting syndrome. It usually occurs during the same period as vasospasm, several days after the initial hemorrhage.26 The use of fluid restriction to treat hyponatremia in the patient with SAH is associated with a poor outcome. The AHA Stroke Council strongly recommends that fluid restriction not be used in this instance and instead recommends sodium replenishment with isotonic fluids.30
Hydrocephalus.
Hydrocephalus is a late complication that occurs in approximately 25% of patients after SAH.30 Blood that has circulated in the subarachnoid space and has been absorbed by the arachnoid villi may obstruct the villi and reduce the rate of CSF absorption. Over time, increasing volumes of CSF in the intracranial space produce communicating hydrocephalus. Treatment consists of placing a drain to remove CSF. This can be accomplished temporarily by insertion of a ventriculostomy24 or permanently by placement of a ventriculoperitoneal shunt.27,30
Intracerebral Hemorrhage
Intracerebral hemorrhage is bleeding directly into cerebral tissue.35 ICH destroys cerebral tissue, causes cerebral edema, and increases ICP. The source of intracerebral bleeding is usually a small artery, but it can result also from rupture of an AVM or aneurysm. The most important cause of spontaneous ICH is hypertension, and this discussion concentrates on spontaneous hypertensive ICH.36
Spontaneous ICH accounts for at least 10% of all stroke admissions.35 The likelihood of death or disability is higher with ICH than with ischemic stroke or SAH. The mortality rate for hemorrhagic stroke is up to 50% within 1 month.37 Only 20% of patients with ICH return to a functional life at 6 months.37 The key risk factors for ICH are age-associated cerebral amyloid angiopathy and hypertension.35
Etiology
ICH is most often caused by hypertensive rupture of a cerebral vessel, resulting from a long-standing history of hypertension.35 Other possible causes of spontaneous ICH are anticoagulation or thrombolytic therapy, coagulation disorders, drug abuse, and hemorrhage into cerebral infarct or brain tumors.27,38 Often on questioning, the patient with a hypertensive hemorrhage admits to having discontinued antihypertensive medication 2 to 3 weeks before the hemorrhage.
Pathophysiology
The pathophysiology of ICH is caused by continued elevated blood pressure exerting force against smaller arterial vessels that have become damaged from arteriosclerotic changes. Eventually, these arteries break, and blood bursts from the vessels into the surrounding cerebral tissue, creating a hematoma. ICP rises precipitously in response to the increase in overall intracranial volume.38
Assessment and Diagnosis
Initial assessment usually reveals a critically ill patient who often is unconscious and requires ventilatory support. History from a relative or significant other describes a sudden onset of focal deficit often accompanied by severe headache, nausea, vomiting, and rapid neurological deterioration.39 Signs and symptoms vary depending on the location of the ICH.38 One third of the patients have maximal symptoms at onset. Assessment of vital signs usually reveals a severely elevated blood pressure (200/100 to 250/150 mm Hg). Signs of increased ICP are often present by the time the patient arrives in the emergency department. Diagnosis is established with CT. Angiography is recommended for patients considered surgical candidates without a clear cause of hemorrhage.35–39
Medical Management
ICH is a medical emergency. Initial management requires attention to airway, breathing, and circulation. Intubation is usually necessary. Blood pressure management must be based on individual factors. Reduction in blood pressure is usually necessary to decrease ongoing bleeding, but lowering blood pressure too much or too rapidly may compromise cerebral perfusion pressure (CPP), especially in the patient with elevated ICP. National guidelines recommend keeping the mean arterial blood pressure below 130 mm Hg in patients with a history of hypertension by means of moderate blood pressure reduction to a mean arterial pressure below 110 mm Hg.39 Vasopressor therapy after fluid replenishment is recommended if systolic blood pressure falls below 90 mm Hg.36
Increased ICP is common with ICH and is a major contributor to mortality. Recommended management includes mannitol when indicated, hyperventilation, and neuromuscular blockade with sedation. Steroids are avoided. The CPP must be kept higher than 70 mm Hg.36,37
The goal for fluid management is euvolemia, with a recommended pulmonary artery occlusion pressure (PAOP) of 10 to 14 mm Hg. Body temperature is maintained at less than 38.5° C through the use of acetaminophen or cooling blankets. Euglycemia, a blood glucose level less than 140 mg/dL, is maintained with insulin therapy, but hypoglycemia should be avoided. Use of short-acting benzodiazepines or propofol is recommended to treat agitation or hyperactivity. Pneumatic compression devices are used to decrease risk of pulmonary embolism. Anticonvulsant therapy is initiated if the patient experiences seizures. Coagulation disorders should be treated, and any anticoagulation medications should be withheld.36,39
The benefit of surgical treatment of spontaneous ICH is unclear. Recommendations for surgical removal of the clot depend on the size and location of the hematoma, the patient’s ICP, and other neurological symptoms.39 Surgical evacuation of the clot is recommended for patients with cerebellar hemorrhage greater than 3 cm with neurological deterioration or hydrocephalus with brainstem compression, as well as for young patients with moderate or large lobar hemorrhage with clinical deterioration. Numerous techniques are being investigated to lessen the risk of brain damage associated with craniotomy for ICH.39
Evidence-based guidelines for the management of the patient with ICH are listed in the Evidence-Based Practice box on Spontaneous Intracerebral Hemorrhage Management Guidelines.
Nursing Management
Nursing management of the patient with stroke incorporates a variety of nursing diagnoses (Nursing Diagnoses Priorities box on Stroke). Nursing priorities are directed toward (1) monitoring for changes in neurological and hemodynamic status, (2) maintaining surveillance for complications, (3) providing comfort and emotional support, and (4) educating the patient and family.
Monitoring for Changes in Neurological and Hemodynamic Status
The goal of frequent assessments is early recognition of neurological or hemodynamic deterioration. Close monitoring of the patient’s neurological signs and vital signs is essential and requires almost continuous observation. Automatic noninvasive devices such as a blood pressure cuff and a pulse oximeter are helpful. Seizure activity must be identified and treated immediately. It is essential that all personnel working with the patient be aware of the desired hemodynamic and neurological parameters set by the physician and that the physician be notified at the first sign of any changes.
Maintaining Surveillance for Complications
The patient with stroke should be monitored closely for signs of bleeding, vasospasm, and increased ICP. Other complications of stroke include aspiration, malnutrition, pneumonia, DVT, pulmonary embolism, pressure ulcers, contractures, and joint abnormalities.29 Nursing measures to prevent these complications are well known.
Additional complications that may be seen in the patient with stroke are related to the area of the brain that has been damaged. Damage to the temporoparietal area can create a variety of disturbances that affect the patient’s ability to interpret sensory information. Damage to the dominant hemisphere (usually left) produces problems with speech and language and abstract and analytic skills. Damage to the nondominant hemisphere (usually right) produces problems with spatial relationships. The resulting deficits include agnosia, apraxia, and visual field defects. Perceptual deficits are not as readily noticeable as motor deficits but may be more debilitating and can lead to the inability to perform skilled or purposeful tasks. The patient also may have impaired swallowing.26,29
Bleeding and Vasospasm.
Sudden onset of or an increase in headache and nausea and vomiting, increased blood pressure, and changes in respiration herald the onset of rebleeding and indicate a second SAH in an unsecured aneurysm. The first indication of vasospasm is usually the appearance of new focal or global neurological deficits.
SAH precautions must be implemented to prevent any stress or straining that could potentially precipitate rebleeding. Precautions include blood pressure control; bed rest; a dark, quiet environment; and stool softeners. Short-acting analgesics and sedatives are used to relieve pain and anxiety. The patient must be kept calm. Limb restraints cause straining and must be avoided. The head of the bed should be elevated to 35 to 45 degrees at all times. The patient is taught to avoid any activities that correspond to performance of the Valsalva maneuver, such as pushing with the legs to move up in bed, straining for a bowel movement, or holding his or her breath during procedures or discomfort. DVT precautions are routinely implemented. Collaboration with the patient and family is used to establish a visitation plan to meet patient and family needs. Often, family members at the bedside can help the patient remain calm.26,28,29
Increased Intracranial Pressure.
Numerous signs and symptoms of increased ICP can be observed. A change in the level of consciousness is the most sensitive indicator. Others include unequal pupil size, decreased pupillary response to light, headache, projectile vomiting, altered breathing patterns, Cushing’s triad (bradycardia, systolic hypertension, and bradypnea), diminished brainstem reflexes, papilledema, and abnormal extension (decerebrate posturing) or flexion (decorticate posturing).26,28,29
Impaired Swallowing.
Normal swallowing occurs in four phases that are controlled by the cranial nerves. Damage to the brain, brainstem, or cranial nerves can result in a variety of swallowing deficits that can place the patient at risk for aspiration. The stroke patient is observed for signs of dysphagia, including drooling; difficulty handling oral secretions; absence of gag, cough, or swallowing reflex; moist, gurgling voice quality; decreased mouth and tongue movements; and the presence of dysarthria. A speech therapy consultation is initiated if any of these signs is present, and the patient must not be orally fed. In the absence of these warning signs, the patient may be fed, as ordered by the physician, although he or she must be continually monitored for signs of aspiration.26,28,29
Educating the Patient and Family
Rehabilitation starts in the critical care area, with a multidisciplinary team designing and implementing an individualized plan to maximize the patient’s potential for neurological rehabilitation. Early in the patient’s hospital stay, the patient and family must be taught about stroke, its causes, and its treatment. As the patient moves toward discharge, teaching focuses on the interventions necessary for preventing the recurrence of the event and on maximizing the patient’s rehabilitation potential. The patient’s family must be encouraged to participate in the patient’s care; learn how to feed, dress, and bathe the patient; and learn some basic rehabilitation techniques. The importance of participating in a neurological rehabilitation program or a support group, or both, must be stressed (Patient Education box on Stroke).
Collaborative management of the patient with a stroke is outlined in the Collaborative Management box on Stroke.
Collaborative Management
Stroke
• Differentiate the cause of the stroke:
• Ischemic
• AVM
• Implement treatment according to cause of bleed:


 Surgical aneurysm clipping or AVM excision
Surgical aneurysm clipping or AVM excision


• Provide ventilatory assistance as required.
• Perform frequent neurological assessments.
• Maintain surveillance for complications.



• Provide comfort and emotional support.
Guillain-Barré Syndrome
Guillain-Barré syndrome (GBS), once thought to be a single entity characterized by inflammatory peripheral neuropathy, is a combination of clinical features with various forms of presentation and multiple pathological processes. A full discussion of this complex condition is beyond the scope of this chapter. Most cases of GBS do not require admission to a critical care unit. However, the prototype of GBS, known as acute inflammatory demyelinating polyradiculoneuropathy (AIDP), involves a rapidly progressive, ascending peripheral nerve dysfunction leading to paralysis that may produce respiratory failure. Because of the need for ventilatory support, AIDP is one of the few peripheral neurological diseases that necessitates care in a critical care environment.40 In this discussion, all references to GBS pertain to the AIDP prototype.
The annual incidence of GBS is 1.2 to 2.3 cases per 100,000 persons.41 It occurs more often in males and is the most commonly acquired demyelinating neuropathy.41 Occasionally, clusters of cases are reported, such as occurred following the 1977 swine flu vaccinations.42
Etiology
The precise cause of GBS remains unknown, but the syndrome involves an immune-mediated response involving cell-mediated immunity and development of immunoglobulin G (IgG) antibodies. Most patients report a viral infection 1 to 3 weeks before the onset of clinical manifestations, usually involving the upper respiratory tract.41
Numerous antecedent causes, or triggering events, have been associated with GBS. They include viral infections (e.g., influenza; cytomegalovirus; hepatitis A, B, or C; Epstein-Barr virus; human immunodeficiency virus), bacterial infections (e.g., gastrointestinal Campylobacter jejuni, Mycoplasma pneumoniae), vaccines (e.g., rabies, tetanus, influenza), lymphoma, surgery, and trauma.41
Pathophysiology
GBS affects the motor and sensory pathways of the peripheral nervous system as well as the autonomic nervous system functions of the cranial nerves. The major finding in AIDP-type GBS is a segmental demyelination process of the peripheral nerves. GBS is thought to be an autoimmune response to antibodies formed in response to a recent physiological event. T cells migrate to the peripheral nerves, resulting in edema and inflammation. Macrophages then invade the area and break down the myelin. Inflammation around this demyelinated area causes further dysfunction. Some axonal damage also occurs.41
The myelin sheath of the peripheral nerves is generated by Schwann cells and acts as an insulator for the peripheral nerve. Myelin promotes rapid conduction of nerve impulses by allowing the impulses to jump along the nerve by means of the nodes of Ranvier. Disruption of the myelin fiber slows and may eventually stop the conduction of impulses along the peripheral nerves. In GBS, the more thickly myelinated fibers of motor pathways and the cranial nerves are more severely affected than are the thinly myelinated sensory fibers of cutaneous pain, touch, and temperature.41
After the temporary inflammatory reaction stops, myelin-producing cells begin the process of reinsulating the demyelinated portions of the peripheral nervous system. When remyelination occurs, normal neurological function should return. In some instances, the axon may be damaged during the inflammatory process. The degree of axonal damage is responsible for the degree of neurological dysfunction that persists after recovery.41
Assessment and Diagnosis
Symptoms of GBS include motor weakness, paresthesias and other sensory changes, cranial nerve dysfunction (especially oculomotor, facial, glossopharyngeal, vagal, spinal accessory, and hypoglossal), and some autonomic dysfunction. The usual course of GBS begins with an abrupt onset of lower extremity weakness that progresses to flaccidity and ascends over a period of hours to days. Motor loss usually is symmetric, bilateral, and ascending. In the most severe cases, complete flaccidity of all peripheral nerves, including spinal and cranial nerves, occurs.40,41,43
The patient is admitted to the hospital when lower extremity weakness prevents mobility. Admission to the critical care unit is necessary when progression of the weakness threatens respiratory muscles. As the patient’s weakness progresses, close observation is essential. Frequent assessment of the respiratory system, including ventilatory parameters such as inspiratory force and tidal volume, is necessary. The most common cause of death of patients with GBS is respiratory arrest. As the disease progresses and respiratory effort weakens, intubation and mechanical ventilation are necessary. Frequent assessment of neurological deterioration is continued until the patient reaches the peak of the disease, and a plateau occurs.41,43
The diagnosis of GBS is based on clinical findings plus the results of CSF analysis and nerve conduction studies. The diagnostic finding is elevated CSF protein with normal cell count.44 The increased protein count usually occurs after the first week but does not occur in approximately 10% of all cases. Nerve conduction studies that test the velocity at which nerve impulses are conducted show significant reduction, as the demyelinating process of the disease suggests.43
Medical Management
With no curative treatment available, the medical management of GBS is limited. The disease must run its course, which is characterized by ascending paralysis that advances over 1 to 3 weeks and then remains at a plateau for 2 to 4 weeks.41 The plateau stage is followed by descending paralysis and return to normal or near-normal function. The main focus of medical management is the support of bodily functions and the prevention of complications.41
Plasmapheresis and intravenous immune globulin (IVIG) are used to treat GBS.41 They have been shown to be equally effective.45 Plasmapheresis involves the removal of venous blood through a catheter, separation of plasma from blood cells, and reinfusion of the blood cells plus autologous plasma or another replacement solution. Although the number of exchanges may vary, the usual regimen is five exchanges over a 5-day period.41 IVIG has emerged as the preferred therapy because of convenience and availability. The usual dose is 0.4 mg/kg for five days.43
Nursing Management
The nursing management of the patient with GBS incorporates a variety of nursing diagnoses and interventions (Nursing Diagnoses Priorities box on Guillain-Barré syndrome). The goal of nursing management is to support all normal body functions until the patient can do so on his or her own. Although the condition is reversible, the patient with GBS requires extensive long-term care, because recovery can be a long process. Nursing priorities are directed toward (1) maintaining surveillance for complications, (2) initiating rehabilitation, (3) facilitating nutritional support, (4) providing comfort and emotional support, and (5) educating the patient and family.
Maintaining Surveillance for Complications
Continuous assessment of the progressive paralysis associated with GBS is essential to timely intervention and the prevention of respiratory arrest and further neurological insult. After the patient is intubated and started on mechanical ventilation, close observation for pulmonary complications, such as atelectasis, pneumonia, and pneumothorax, is necessary. Autonomic dysfunction (dysautonomia) in the GBS patient can produce variations in heart rate and blood pressure that can reach extreme values.40,41,46 Hypertension and tachycardia may require beta-blocker therapy. All patients with GBS must be observed for this phenomenon.
Initiating Rehabilitation
In patients with GBS, immobility may last for months. The usual course of the disease involves an average of 10 days of symptom progression and 10 days of maximal level of dysfunction, followed by 2 to 48 weeks of recovery. Although GBS usually is completely reversible, the patient requires physical and occupational rehabilitation because of the problems of long-term immobility. Rehabilitation starts in the critical care area, with a multidisciplinary team designing and implementing an individualized plan to maximize the patient’s potential for rehabilitation.46
Facilitating Nutritional Support
Nutritional support is implemented early in the course of the disease. Because recovery from GBS is a long process, adequate nutritional support will be a problem for an extended period. Nutritional support is usually accomplished through the use of enteral feeding.
Providing Comfort and Emotional Support
Pain control is another important component in the care of the patient with GBS. Although patients may have minimal to no motor function, most sensory functions remain, causing patients considerable muscle aching and pain. Because of the duration of this illness, a safe, effective, long-term solution to pain management must be identified.46 These patients also require extensive psychological support. Although the illness is almost 100% reversible, lack of control over the situation, constant pain or discomfort, and the long-term nature of the disorder create coping difficulties for the patient. GBS does not affect the level of consciousness or cerebral function. Patient interaction and communication are essential elements of the nursing management plan.
Educating the Patient and Family
Early in the patient’s hospital stay, the patient and family must be taught about GBS and its different treatments. As the patient moves toward discharge, teaching focuses on the interventions to maximize the patient’s rehabilitation potential. The patient’s family must be encouraged to participate in the patient’s care and to learn some basic rehabilitation techniques. The importance of participating in a neurological rehabilitation program (if necessary) must be stressed (Patient Education box on Guillain-Barré Syndrome).46
Collaborative management of the patient with Guillain-Barré syndrome is outlined in the Collaborative Management box on Guillain-Barré syndrome.
Craniotomy
A craniotomy is performed to gain access to portions of the central nervous system (CNS) inside the cranium, usually to allow removal of a space-occupying lesion such as a brain tumor (Table 18-4). Common procedures include tumor resection or removal, cerebral decompression, evacuation of hematoma or abscess, and clipping or removal of an aneurysm or AVM. Most patients who undergo craniotomy for tumor resection or removal do not require care in a critical care unit. Patients who do usually need intensive monitoring or are at greater risk for complications because of underlying cardiopulmonary dysfunction or the surgical approach used. Box 18-5 provides definitions of common neurosurgical terms.47
TABLE 18-4
TUMOR TYPES AND CHARACTERISTICS
| TUMOR | CLINICAL FEATURES | TREATMENT/PROGNOSIS |
| Glioblastoma multiforme | Often manifests with nonspecific complaints and increased ICP As tumor grows, focal deficits develop |
Rapidly progressive course, with poor prognosis |
| Total surgical removal usually not possible; response to radiation poor | ||
| Astrocytoma | Presentation similar to that of glioblastoma multiforme, but course more protracted, often over several years; cerebellar astrocytoma, especially in children, may have more benign course | Variable prognosis |
| By diagnosis, total excision usually impossible; tumor often not radiosensitive | ||
| In cerebellar astrocytoma, total excision often possible | ||
| Medulloblastoma | Glioma most often seen in children | Treatment consists of surgery with radiation therapy and chemotherapy |
| Usually arises from roof of fourth ventricle and leads to increased ICP, with brainstem and cerebellar signs; may seed subarachnoid space | ||
| Ependymoma | Glioma arising from ependyma of ventricle, especially fourth; leads early to signs of increased ICP; arises also from central canal of spinal cord | Tumor not radiosensitive and best treated surgically, if possible |
| Oligodendroglioma | Slow-growing glioma; usually arises in cerebral hemisphere in adults Calcification may be visible on radiograph |
Treatment is surgical and usually successful |
| Brainstem glioma | Manifests in childhood with cranial nerve palsies, then long-tract signs in limbs; signs of increased ICP occur late in course | Tumor is inoperable |
| Treatment with irradiation and with shunt for increased ICP | ||
| Cerebellar hemangioblastoma | Manifests with dysequilibrium, ataxia of trunk or limbs, and signs of increased ICP; sometimes familial; may be associated with retinal and spinal lesions, polycythemia, and hypernephroma | Treatment is surgical |
| Pineal tumor | Manifests with increased ICP, sometimes associated with impaired upward gaze (Parinaud’s syndrome) and other deficits indicating midbrain lesion | Ventricular decompression by shunting, followed by surgical approach to tumor |
| Irradiation if tumor malignant | ||
| Prognosis depends on histopathological findings and tumor extent | ||
| Craniopharyngioma | Originates from remnants of Rathke’s pouch above sella turcica, depressing optic chiasm | Treatment is surgical, but total removal may not be possible |
| May manifest at any age but usually in childhood with endocrine dysfunction and bitemporal field deficits | ||
| Acoustic neuroma | Most common initial symptom is ipsilateral hearing loss; subsequent symptoms may include tinnitus, headache, vertigo, facial weakness or numbness, and long-tract signs | Treatment is by excision using a translabyrinthine approach, craniectomy, or combination Prognosis usually good |
| May be familial and bilateral when related to neurofibromatosis | ||
| Most sensitive screening tests are magnetic resonance imaging and brainstem auditory-evoked potentials | ||
| Meningioma | Originates from dura mater or arachnoid; compresses rather than invades adjacent neural structures Increasingly common with advancing age Tumor size varies greatly; symptoms vary with tumor site* Tumor usually benign; readily detected by computed tomography; may lead to calcification and bone erosion visible on plain skull radiographs |
Treatment is surgical |
| Tumor may recur if removal is incomplete; patient may receive irradiation with incomplete excision to decrease risk of recurrence | ||
| Primary central lymphoma | Associated with AIDS and other immunodeficiency states May manifest with focal deficits or disturbances of cognition and consciousness; may be indistinguishable from cerebral toxoplasmosis |
Treatment is by whole-brain irradiation |
| Chemotherapy may have adjunctive role | ||
| Prognosis depends on CD4 cell count at diagnosis |
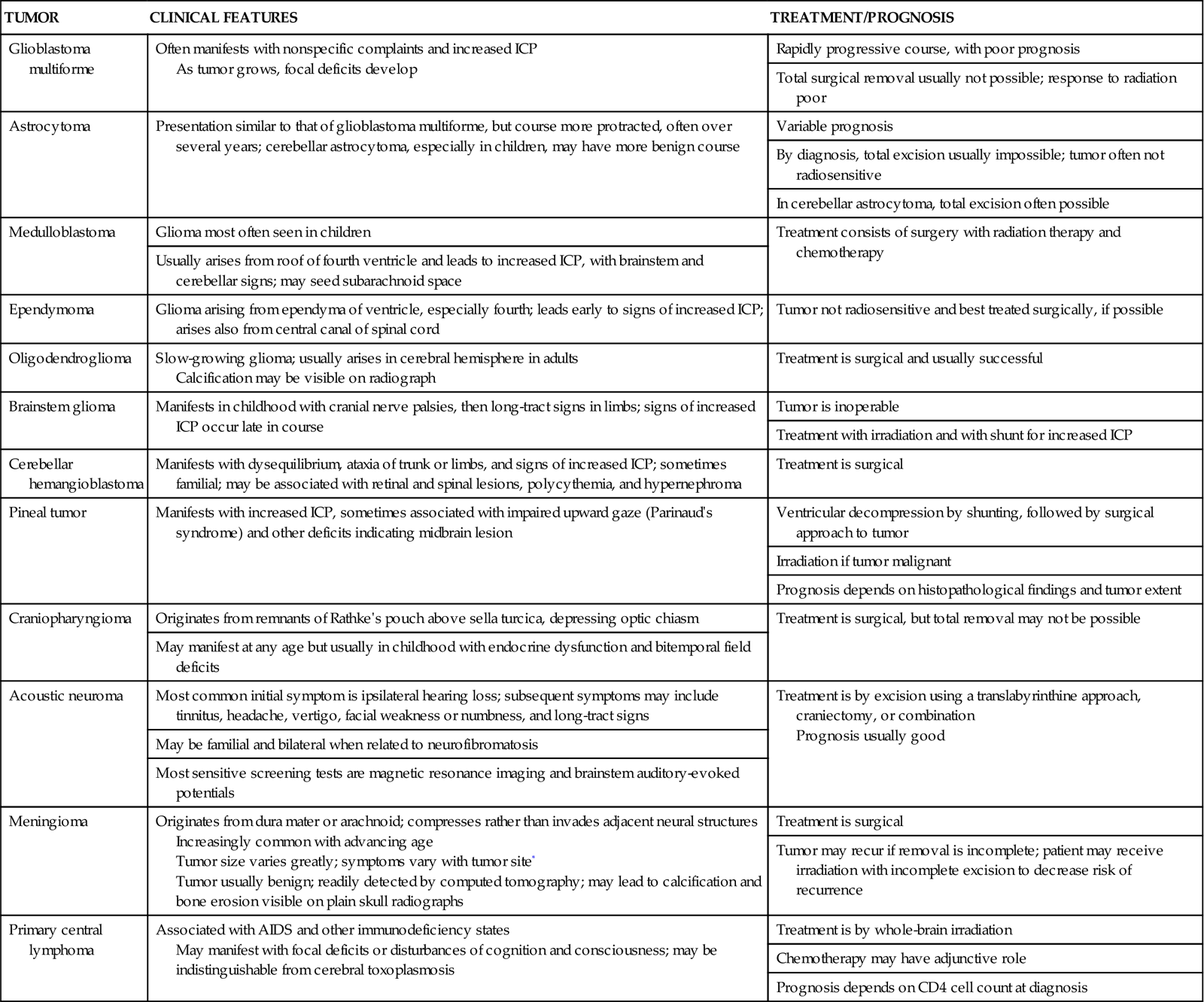
*For example, unilateral exophthalmos (sphenoidal ridge), anosmia, and optic nerve compression (olfactory groove).
From Gawlinski A, Hamwi D, editors: Acute care nurse practitioner: clinical curriculum and certification review, Philadelphia, 1999, Saunders.
Preoperative Care
Protection of the integrity of the CNS is a major priority of care for the patient awaiting a craniotomy. Optimal arterial oxygenation, hemodynamic stability, and cerebral perfusion are essential for maintaining adequate cerebral oxygenation. Management of seizure activity is essential for controlling metabolic needs.44
Detailed assessment and documentation of the patient’s preoperative neurological status are imperative for accurate postoperative evaluation. Attention is focused on identifying and describing the nature and extent of any preoperative neurological deficits. When pituitary surgery is planned, a thorough evaluation of endocrine function is necessary to prevent major intraoperative and postoperative complications.45
Trends in health care demand judicious use of routine preoperative studies. Depending on the type of surgery to be performed and the general health of the patient, preoperative screening may include a complete blood cell count (CBC); tests for blood urea nitrogen (BUN), creatinine, and fasting blood glucose (FBG); a chest radiograph; and an electrocardiogram. Blood type and crossmatch may also be ordered.
Preoperative teaching is necessary to prepare the patient and family for what to expect in the postoperative period. A description of the intravascular lines and intracranial catheters used during the postoperative period allows the family to focus on the patient, rather than be overwhelmed by masses of tubing. Some or all of the patient’s hair is shaved off in the operating room, and a large, bulky, turban-like craniotomy dressing is applied. Most patients experience some degree of postoperative eye or facial swelling and periorbital ecchymosis. An explanation of these temporary changes in appearance helps alleviate the shock and fear many patients and families experience in the immediate postoperative period.
All patients undergoing craniotomy require instruction to avoid activities known to provoke sudden changes in ICP. These activities include bending, lifting, straining, and the Valsalva maneuver. Patients commonly elicit the Valsalva maneuver during repositioning in bed by holding the breath and straining with a closed epiglottis. Teaching the patient to continue to breathe deeply through the mouth during all position changes is an effective deterrent.
The patient undergoing transsphenoidal surgery requires preparation for the sensations associated with nasal packing. The patient often awakens with alarm because of the inability to breathe through the nose. Preoperative instruction in mouth breathing and avoidance of coughing, sneezing, and blowing of the nose facilitates postoperative cooperation.
The psychosocial issues associated with the prospect of neurosurgery cannot be overemphasized. Few procedures are as threatening as those involving the brain or spinal cord. For some patients, the fear of permanent neurological impairment may be as ominous as or more so than the fear of death. Steps to meet the needs of the patient and the family include collaboration with clergy and social services personnel, patient-controlled visitation, and provision of as much privacy as the patient’s condition permits. The patient and family must be given the opportunity to express their fears and concerns jointly and apart from each other.47
Surgical Considerations
Whereas the emphasis in the surgical approach for most other types of surgery is to gain adequate exposure of the surgical site, the neurosurgeon must select a route that also produces the least amount of disruption to the intracranial contents. Neural tissue is unforgiving. A significant portion of neurological trauma and postoperative deficits is related to the surgical pathway through the brain tissue, rather than to the procedure performed at the site of pathology. Depending on the location of the lesion and the surgical route chosen, a transcranial or a transsphenoidal approach is used to open the skull.
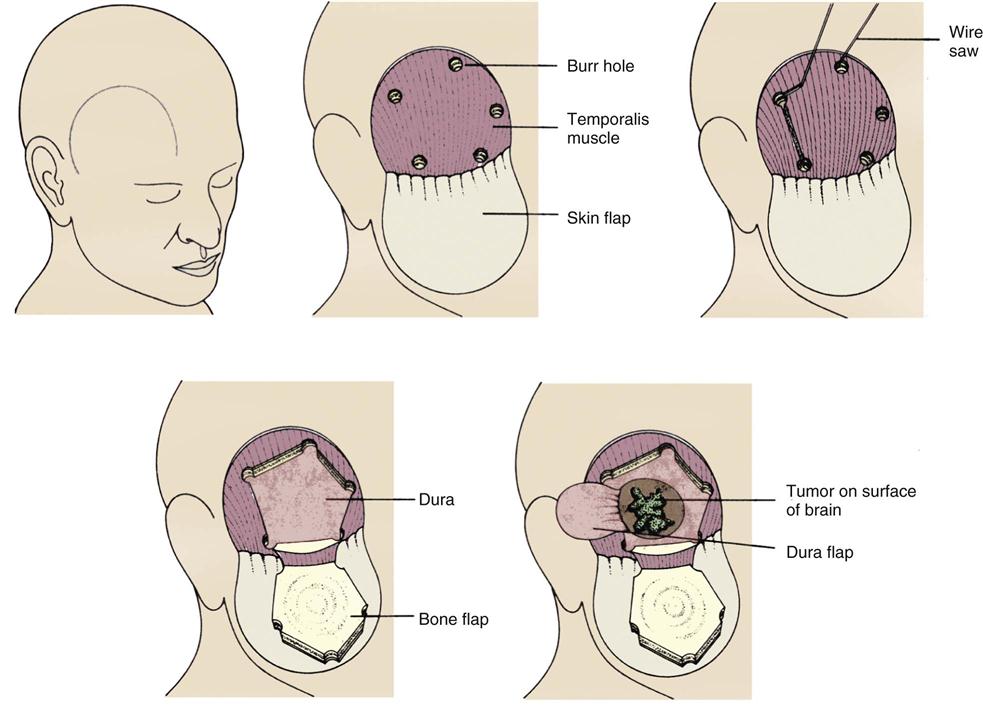
Transcranial Approach
In the transcranial approach, a scalp incision is made, and a series of burr holes is drilled into the skull to form an outline of the area to be opened (Figure 18-4). A special saw is then used to cut between the holes. In most cases, the bone flap is left attached to the muscle to create a hinge effect. In some cases, the bone flap is removed completely and stored for later retrieval and implantation or discarded and replaced with synthetic material. Next, the dura is opened and retracted. After the intracranial procedure, the dura and the bone flap are closed, the muscles and scalp are sutured, and a turban-like dressing is applied.44,47
Transsphenoidal Approach
The transsphenoidal approach is the technique of choice for removal of a pituitary tumor without extension into the intracranial vault (Figure 18-5).44,45 This approach involves making a microsurgical entrance into the cranial vault through the nasal cavity. The sphenoid sinus is entered to reach the anterior wall of the sella turcica. The sphenoid bone and the dura are then opened to gain intracranial access. After removal of the tumor, the surgical bed is packed with a small section of adipose tissue grafted from the patient’s abdomen or thigh. After closure of the intranasal structures, nasal splints and soft packing or nasal tampons impregnated with antibiotic ointment are placed in the nasal cavities. Occasionally, epistaxis balloons are used instead. A nasal drip pad or mustache-type dressing is placed at the base of the nose to catch surgical drainage.47
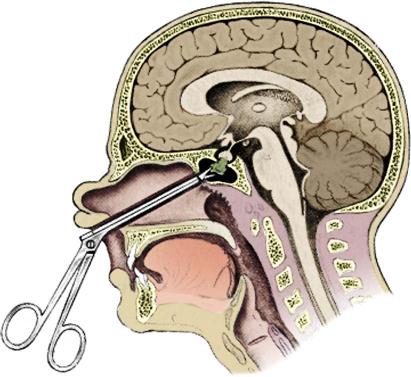
The patient may be placed in a supine, prone, or sitting position for a craniotomy procedure. A skull clamp connected to skull pins is used to position and secure the patient’s head throughout the operation. During a transsphenoidal approach or a transcranial approach into the infratentorial area, the patient’s head is elevated during surgery. This position puts the patient at risk for an air embolism. Air can enter the vascular system through the edges of the dura or a venous opening. Continuous monitoring of the patient’s heart sounds by Doppler ultrasonography signal allows immediate recognition of this complication. If it occurs, an attempt may be made to withdraw the embolus from the right atrium through a central line. Flooding the surgical field with irrigation fluid and placing a moistened sterile surgical sponge over the surgical site creates an immediate barrier to any further air entrance.44,47
Postoperative Medical Management
Definitive management of the postoperative neurosurgical patient varies, depending on the underlying reason for the craniotomy. During the initial postoperative period, management is usually directed toward the prevention of complications. Complications associated with a craniotomy include intracranial hypertension, surgical hemorrhage, fluid imbalance, CSF leak, and DVT.
Intracranial Hypertension
Postoperative cerebral edema is expected to peak 48 to 72 hours after surgery. If the bone flap is not replaced at the time of surgery, intracranial hypertension produces bulging at the surgical site. Close monitoring of the surgical site is important so that integrity of the incision can be maintained. Postcraniotomy management of intracranial hypertension is usually accomplished through CSF drainage, patient positioning, and steroid administration.44
Surgical Hemorrhage
Surgical hemorrhage after a transcranial procedure can occur in the intracranial vault and is manifested by signs and symptoms of increasing ICP. Hemorrhage after a transsphenoidal craniotomy may be evident from external drainage, the patient’s complaint of persistent postnasal drip, or excessive swallowing. Loss of vision after pituitary surgery indicates an evolving hemorrhage. Postoperative hemorrhage requires surgical reexploration.45
Fluid Imbalance
Fluid imbalance in the postcraniotomy patient usually results from a disturbance in production or secretion of antidiuretic hormone (ADH). ADH is secreted by the posterior pituitary (neurohypophysis) gland. It stimulates the renal tubules and collecting ducts to retain water in response to low circulating blood volume or increased serum osmolality. Inoperative trauma or postoperative edema of the pituitary gland or hypothalamus can result in insufficient ADH secretion. The outcome is unabated renal water loss even when blood volume is low and serum osmolality is high. This condition is known as diabetes insipidus (DI). The polyuria associated with DI is often more than 200 mL/hour. Urine specific gravity of 1.005 or less and elevated serum osmolality provide evidence of insufficient ADH. The loss of volume may provoke hypotension and inadequate cerebral perfusion. DI is usually self-limiting, and fluid replacement is the only required therapy. In some cases, however, it may be necessary to administer vasopressin intravenously to control the loss of fluid.44
The syndrome of inappropriate antidiuretic hormone [secretion] (SIADH) commonly occurs with neurological insult and results from excessive ADH secretion. SIADH is manifested by inappropriate water retention with hyponatremia in the presence of normal renal function. Urine specific gravity is elevated, and urine osmolality is greater than serum osmolality. The dangers associated with SIADH include circulating volume overload and electrolyte imbalance, both of which may impair neurological functioning. SIADH is usually self-limiting, with the mainstay of treatment being fluid restriction.44
Cerebrospinal Fluid Leak
Leakage of cerebrospinal fluid (CSF) results from an opening in the subarachnoid space, as evidenced by clear fluid draining from the surgical site. When this complication occurs after transsphenoidal surgery, it is signified by excessive, clear drainage from the nose or persistent postnasal drip. To differentiate CSF drainage from postoperative serous drainage, a specimen is tested for glucose content. A CSF leak is confirmed by glucose values of 30 mg/dL or greater. Management of the patient with a CSF leak includes bed rest and head elevation. Lumbar puncture or placement of a lumbar subarachnoid catheter may be used to reduce CSF pressure until the dura heals. The risk of meningitis associated with CSF leak often necessitates surgical repair to reseal the opening.44,45
Deep Vein Thrombosis
Research has demonstrated that neurosurgical patients have a higher risk for development of a DVT than non-neurosurgical patients. This difference is due to numerous additional risk factors significantly associated with neurosurgery, including preoperative leg weakness, longer preoperative critical care unit stay, longer recovery room time, longer postoperative critical care unit stay, more days of bed rest, and delay of postoperative mobility and activity.48 Clinical manifestations of DVT include leg or calf pain, edema, localized tenderness, and pain with dorsiflexion or plantar flexion (Homans’ sign). Unfortunately, the patient with a DVT is often asymptomatic and the diagnosis is not made until the patient experiences a pulmonary embolus.
The primary treatment for DVT is prophylaxis. In the neurosurgery patient, sequential (intermittent) pneumatic compression boots or stockings are effective in reducing the incidence of DVT. Effectiveness is enhanced when these devices are initiated in the preoperative period. Low-dose unfractionated heparin or low-molecular-weight heparin may also be used prophylactically in high-risk patients.44
Postoperative Nursing Management
The nursing management of the neurosurgical patient incorporates a variety of nursing diagnoses (Nursing Diagnosis Priorities box on Craniotomy). As in preoperative care, the primary goal of postcraniotomy nursing management is protection of the integrity of the CNS. Nursing priorities are directed toward (1) preserving adequate CPP, (2) promoting arterial oxygenation, (3) providing comfort and emotional support, (4) maintaining surveillance for complications, (5) initiating early rehabilitation, and (6) educating the patient and family. Frequent neurological assessment is necessary to evaluate accomplishment of these objectives and to identify problems and quickly intervene if complications do arise. Often, a ventriculostomy is placed to facilitate ICP monitoring and CSF drainage.
Preserving Adequate Cerebral Perfusion
Nursing interventions to preserve cerebral perfusion include patient positioning, fluid management, and avoidance of postoperative vomiting and fever.
Positioning.
Patient positioning is an important component of care for the craniotomy patient. The head of the bed should be elevated 30 to 45 degrees at all times to reduce the incidence of hemorrhage, facilitate venous drainage, and control ICP. Other positioning measures to control ICP include maintaining the patient’s head in a neutral position at all times and avoiding neck and hip flexion. These rules of positioning must be followed throughout all nursing activities, including linen changes and transporting of the patient for diagnostic evaluation. Most craniotomy patients can be turned from side to side within these restrictions, using pillows for support, except in some cases of extensive tumor removal, cranioplasty, and when the bone flap is not replaced. Specific orders from the surgeon must be obtained in these instances. The patient with an infratentorial incision may be restricted to only a very small pillow under the head to prevent strain on the incision. Avoidance of anterior and lateral neck flexion also protects the integrity of this type of incision.
Fluid Management.
Fluid management is another important component of postcraniotomy care. Hourly monitoring of fluid intake and output facilitates early identification of fluid imbalance. Urine specific gravity must be measured if DI is suspected. Fluid restriction may be ordered as a routine measure to lessen the severity of cerebral edema or as treatment for the fluid and electrolyte imbalances associated with SIADH.
Vomiting and Fever.
Postoperative vomiting must be avoided to prevent sharp spikes in ICP and possibly surgical hemorrhage. Antiemetics are administered as soon as nausea is apparent. Early resumption of nutrition in the neurosurgical patient is beneficial. If the patient is unable to eat, enteral hyperalimentation delivered through a feeding tube is the preferred method of nutritional support and can be initiated as early as 24 hours after surgery.50 Postoperative fever may also adversely affect ICP and increase the metabolic needs of the brain. Acetaminophen is administered orally, rectally, or through a feeding tube. External cooling measures, such as a hypothermia blanket, may be necessary.
Promoting Arterial Oxygenation
Routine pulmonary care is used to maintain airway clearance and prevent pulmonary complications. To prevent dangerous elevations in ICP, this care must be performed with the use of proper technique and at time intervals that are adequately spaced from other patient care activities. If pulmonary complications do arise, consideration must be given to maintaining adequate oxygenation during repositioning. It may be necessary to restrict turning the patient to only the side that places the good lung down.
Providing Comfort and Emotional Support
Pain management in the postcraniotomy patient primarily involves control of headache. Small doses of intravenous morphine are used in the critical care setting. As soon as oral analgesics can be tolerated, acetaminophen with codeine is used. Both analgesics cause constipation. Administration of stool softeners and initiation of a bowel program are important components of postcraniotomy care. Constipation is hazardous, because straining to have a bowel movement can create significant elevations in blood pressure and ICP.
Maintaining Surveillance for Complications
The postoperative neurosurgical patient is at risk for infection, corneal abrasions, and injury from falls or seizures.
Infection.
Care of the incision and surgical dressings is specific to the institution and physician. The rule of thumb for a craniotomy dressing is to reinforce it as needed and change it only with a physician’s order. Often, a drain is left in place to facilitate decompression of the surgical site. If a ventriculostomy is present, it is treated as a component of the surgical site. All drainage devices must be secured to the dressing to prevent unintentional displacement with patient movement. Sterile technique is required to prevent infection and resultant meningitis.
Corneal Abrasions.
Routine eye care may be necessary to prevent corneal drying and ulceration. Periorbital edema interferes with normal blinking and eyelid closure, which are essential to adequate corneal lubrication. Saline drops are instilled every 2 hours. If the patient remains in a coma state, covering the eyes with a polyethylene film extending over the orbits and eyebrows may be beneficial.10
Injury.
The postcraniotomy patient may experience periods of altered mentation. Protection from injury may require the use of restraint devices. The side rails of the bed must be padded to protect the patient from injury. Having a family member stay at the bedside or use of music therapy is often helpful to keep the patient calm during periods of restlessness. In rare circumstances, neuromuscular blockade and sedation may be necessary to control patient activity and metabolic needs on a short-term basis.
Initiating Early Rehabilitation
Increased activity, including ambulation, is begun as soon in the postoperative period as tolerated by the patient. Rehabilitation measures and discharge planning may begin in the critical care unit but are beyond the scope of this chapter. Transfer to a general care or rehabilitation unit is usually accomplished as soon as the patient is considered to be stable and free of complications.
Educating the Patient and Family
Preoperatively, the patient and family should be taught about the precipitating event necessitating the need for the craniotomy and its expected outcome. The severity of the disease and the need for critical care management postoperatively must be stressed. As the patient moves toward discharge, teaching focuses on medication instructions; incisional care, including the signs of infection; and the signs and symptoms of increased ICP. If the patient has neurological deficits, teaching focuses on the interventions to maximize the patient’s rehabilitation potential, and the patient’s family members must be encouraged to participate in the patient’s care and to learn some basic rehabilitation techniques. The importance of participating in a neurological rehabilitation program must be stressed (Patient Education box on Craniotomy).
Intracranial Hypertension
Pathophysiology
The intracranial space comprises three components: brain substance (80%), CSF (10%), and blood (10%). Under normal physiological conditions, the mean ICP is maintained below 15 mm Hg.29,49 Essential to understanding the pathophysiology of ICP, the Monro-Kellie hypothesis proposes that an increase in volume of one intracranial component must be compensated by a decrease in one or more of the other components so that total volume remains fixed. This compensation, although limited, includes displacing CSF from the intracranial vault to the lumbar cistern, increasing CSF absorption, and compressing the low-pressure venous system.50,51 Pathophysiological alterations that can elevate ICP are outlined in Table 18-5.
TABLE 18-5
MECHANISMS OF INTRACRANIAL PRESSURE ELEVATION
| PATHOPHYSIOLOGY | EXAMPLE | TREATMENT |
| Disorders of CSF Space | ||
| Overproduction of CSF | Choroid plexus papilloma | Diuretics, surgical removal |
| Communicating hydrocephalus from obstructed arachnoid | Old subarachnoid hemorrhage | Surgical drainage from lumbar drain |
| Noncommunicative hydrocephalus | Posterior fossa tumor obstructing aqueduct | Surgical drainage by ventricular drain |
| Interstitial edema | Any of above | Surgical drainage of CSF |
| Disorders of Intracranial Blood | ||
| Intracranial hemorrhage causing increased ICP | Epidural hematoma | Surgical drainage |
| Vasospasm | Subarachnoid hemorrhage | Hypervolemia and hypertensive therapy |
| Vasodilation | Elevated PaCO2 | Hyperventilation |
| Increasing cerebral blood volume and ICP | Hypoxia | Adequate oxygenation |
| Disorders of Brain Substance | ||
| Expanding mass lesion with local vasogenic edema causing increased ICP | Brain tumor | Steroids |
| Surgical removal | ||
| Ischemic brain injury with cytotoxic edema increasing ICP | Anoxic brain injury from cardiac or respiratory arrest | Resistant to therapy |
| Increased cerebral metabolic rate increasing cerebral blood flow and ICP | Seizures, hyperthermia | Anticonvulsant medications to control fever |
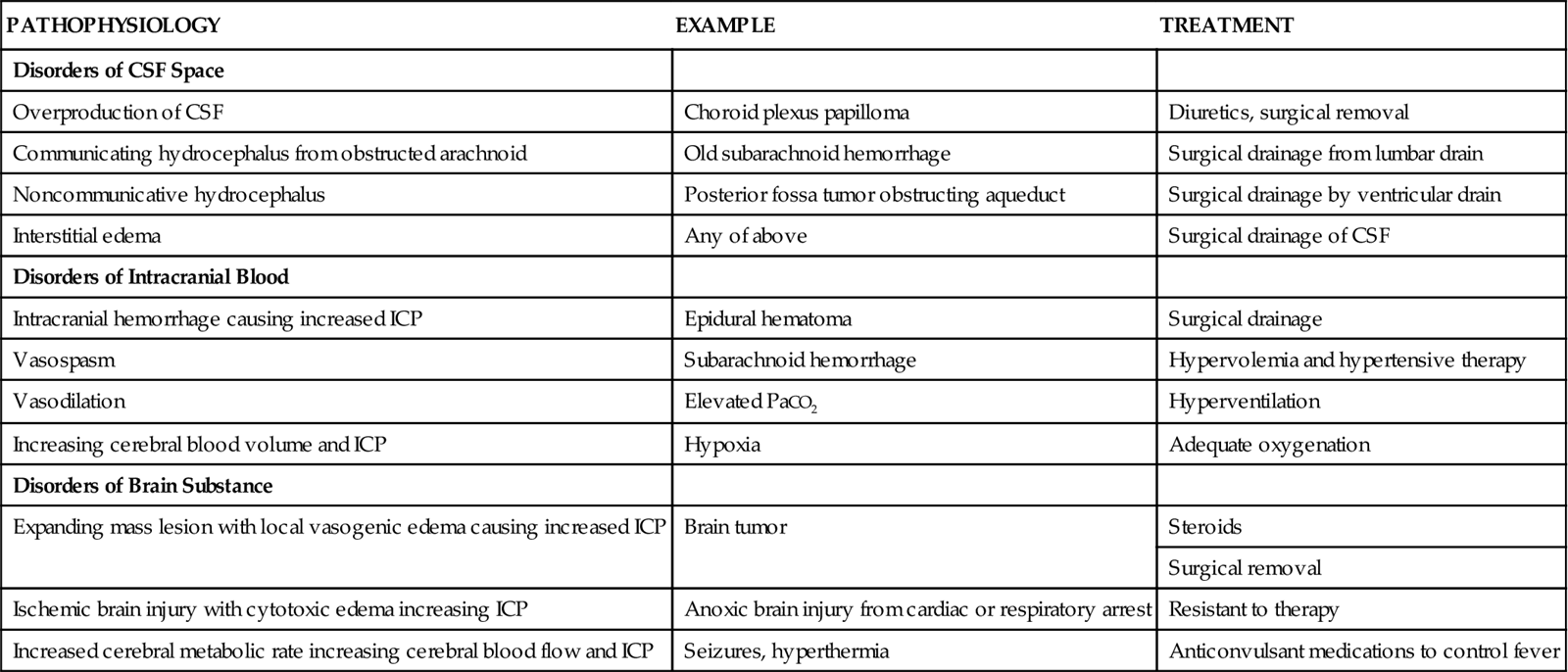
Modified from Helfaer MA, Kirsch JR: Intracranial vault pathophysiology, Crit Care Rep 1:12, 1989.
Volume-Pressure Curve
When capable of compliance, the brain can tolerate significant increases in intracranial volume without much increase in ICP. The amount of intracranial compliance, however, does have a limit. After this limit has been reached, a state of decompensation with increased ICP results. As the ICP rises, the relationship between volume and pressure changes, and small increases in volume may cause major elevations in ICP (Figure 18-6).29,50 The exact configuration of the volume-pressure curve and the point at which the steep rise in pressure occurs vary among patients. The configuration of this curve is also influenced by the cause and the rate of volume increases within the intracranial vault; for example, neurological deterioration occurs more rapidly in a patient with an acute epidural hematoma than in a patient with a meningioma of the same size.49 Regardless of how fast the pressure increases, intracranial hypertension occurs when ICP is greater than 20 mm Hg.50
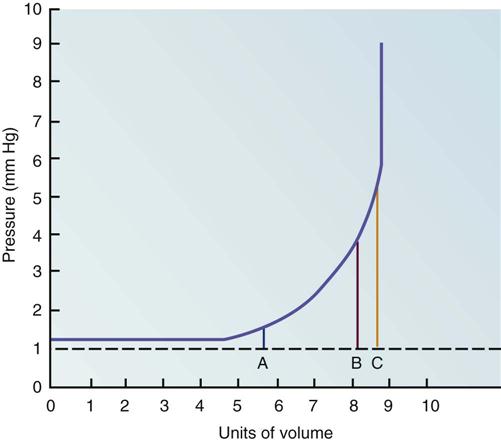
Cerebral Blood Flow and Autoregulation
Cerebral blood flow (CBF) corresponds to the metabolic demands of the brain and is normally 50 mL/100 g of brain tissue/min. Although the brain makes up only 2% of body weight, it requires 15% to 20% of the resting cardiac output and 15% of the body’s oxygen demands. The normal brain has a complex capacity to maintain constant CBF, despite wide ranges in systemic arterial pressure—an effect known as autoregulation. A mean arterial pressure of 50 to 150 mm Hg does not alter CBF when autoregulation is functioning. Outside the limits of this autoregulation, CBF becomes passively dependent on the perfusion pressure.50
Factors other than arterial blood pressure that affect CBF are conditions that result in acidosis, alkalosis, and changes in metabolic rate. Conditions that cause acidosis (e.g., hypoxia, hypercapnia, ischemia) result in cerebrovascular dilation. Conditions causing alkalosis (e.g., hypocapnia) result in cerebrovascular constriction. Normally, a reduction in metabolic rate (e.g., from hypothermia or barbiturates) decreases CBF, and rises in metabolic rate (e.g., from hyperthermia) increase CBF.49,52
Arterial blood gases exert a profound effect on CBF. Carbon dioxide, which affects the pH of the blood, is a potent vasoactive substance. Carbon dioxide retention (hypercapnia) leads to cerebral vasodilation, with increased cerebral blood volume, whereas hypocapnia leads to cerebral vasoconstriction and a reduction in cerebral blood volume. Prolonged hypocapnia, however, can lead to cerebral ischemia.53 Low arterial partial pressure of oxygen (PaO2) levels, especially below 40 mm Hg, lead to cerebral vasodilation, which increases the intracranial blood volume and can contribute to increased ICP. High PaO2 levels have not been shown to affect CBF in either direction.49,52
Assessment and Diagnosis
The numerous signs and symptoms of increased ICP include decreased level of consciousness, Cushing’s triad, diminished brainstem reflexes, papilledema, decerebrate posturing (abnormal extension), decorticate posturing (abnormal flexion), unequal pupil size, projectile vomiting, decreased pupillary reaction to light, altered breathing patterns, and headache.29 Patients may exhibit one or all of these symptoms, depending on the underlying cause of the elevation in ICP. One of the earliest and most important signs of increased ICP is a decrease in the level of consciousness. This change must be reported immediately to the physician.52
In the patient with suspected intracranial hypertension, a monitoring device may be placed within the cranium to quantify ICP. Under normal physiological conditions, the mean ICP is maintained below 15 mm Hg. The device is used to monitor ICP serially and assist with the management of intracranial hypertension. An increase in ICP can decrease blood flow to the brain, causing brain damage. The monitoring device can also provide a sterile access for draining excess CSF. The four sites for monitoring ICP are the intraventricular space, the subarachnoid space, the epidural space, and the parenchyma. Each site has advantages and disadvantages for monitoring ICP. The type of monitor chosen depends on the suspected pathological condition and the physician’s preferences.49,51,52,54 Chapter 17 provides a more detailed discussion of ICP monitoring.
Medical and Nursing Management
After intracranial hypertension is documented, therapy must be prompt to prevent secondary insults (Concept Map on Intracranial Hypertension). Although the exact pressure level denoting intracranial hypertension remains uncertain, most current evidence suggests that ICP generally must be treated when it exceeds 20 mm Hg.49,52 All therapies are directed toward reducing the volume of one or more of the components (e.g., blood, brain, CSF) that lie within the intracranial vault. A major goal of therapy is to determine the cause of the elevated pressure and, if possible, to remove the cause.49,55 In the absence of a surgically treatable mass lesion, intracranial hypertension is treated medically. Nursing priorities focus on (1) rapid assessment and (2) implementation of appropriate therapies for reducing ICP.
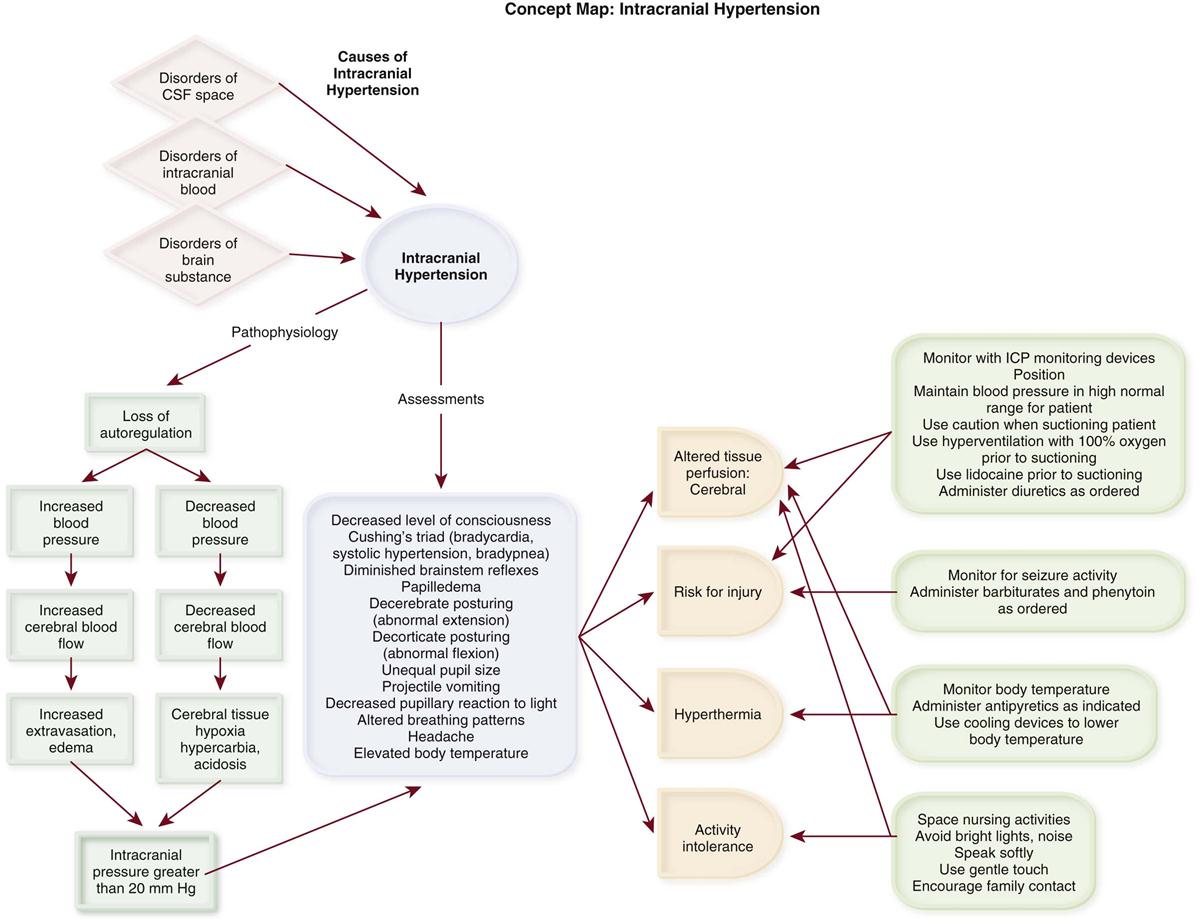
Positioning and Other Nursing Activities
Positioning of the patient is a significant factor in the prevention and treatment of intracranial hypertension. Head elevation has long been advocated as a conventional nursing intervention to control ICP, presumably by increasing venous return. However, this may decrease CPP. Close monitoring of ICP and CPP should be done with positioning, which should be customized to maximize CPP and minimize ICP.51
Positions that impede venous return from the brain cause elevations in ICP. Obstruction of jugular veins or an increase in intrathoracic or intraabdominal pressure is communicated as increased pressure throughout the open venous system, thereby impeding drainage from the brain and increasing ICP. Positions that decrease venous return from the head (e.g., Trendelenburg position, prone position, extreme flexion of the hips, angulation of the neck) must be avoided if possible. If changes to positions such as Trendelenburg are necessary to provide adequate pulmonary care, critical care nurses must closely monitor ICP and vital signs.49
Some routine nursing activities do affect ICP and can be harmful. Use of positive end-expiratory pressures (PEEP) greater than 20 cm H2O, coughing, suctioning, tight tracheostomy tube ties, and the Valsalva maneuver have been associated with increases in ICP. Cumulative increases in ICP have been reported when care activities are performed one after another. Conversely, family contact and gentle touch have been associated with decreases in ICP.49
Hyperventilation
Controlled hyperventilation has been an important adjunct of therapy for the patient with increased ICP. The rationale employed in hyperventilation is that if the PaCO2 can be reduced from its normal level of 35 to 40 mm Hg to a range of 25 to 30 mm Hg in the patient with intracranial hypertension, vasoconstriction of cerebral arteries, reduction of CBF, and increased venous return will result. This practice is being reexamined. Additional research has indicated that severe or prolonged hyperventilation can reduce cerebral perfusion and lead to cerebral ischemia and infarction. The current trend is to maintain PaCO2 levels on the lower side of normal (35 ± 2 mm Hg) by carefully monitoring arterial blood gas measurements and by adjusting ventilator settings.53,55
Although hypoxemia must be avoided, excessively high levels of oxygen offer no benefits, and increasing inspired oxygen concentrations above 60% may lead to toxic changes in lung tissue. The use of pulse oximetry has led to greater awareness of the circumstances, such as pain and anxiety, that can cause oxygen desaturation and therefore elevate ICP.29,49
Temperature Control
Directly proportional to body temperature, cerebral metabolic rate increases 10% per 1° C of increase in body temperature.51 This fact is significant because as the cerebral metabolic rate increases, blood flow to the brain must increase to meet the tissue demands. To avoid the increase in blood volume associated with a greater cerebral metabolic rate, nurses must prevent hyperthermia in the patient with a brain injury. Antipyretics and cooling devices must be used when appropriate while the source of the fever is being determined.51,52
Blood Pressure Control
Maintenance of arterial blood pressure in the high-normal range is essential in the brain-injured patient. Inadequate perfusion pressure decreases the supply of nutrients and oxygen requirements for cerebral metabolic needs. However, a blood pressure that is too high increases cerebral blood volume and may raise ICP.52 Figure 18-7 shows the relationship between blood pressure and ICP.
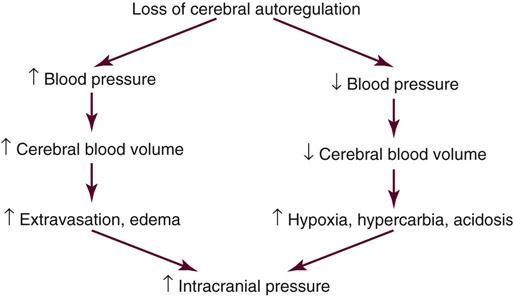
Control of systemic hypertension may require nothing more than the administration of a sedative agent. Small, frequent doses may be sufficient to blunt noxious stimuli and prevent them from triggering increases in blood pressure. When sedation proves inadequate in controlling systemic arterial hypertension, antihypertensive agents are used. Care must be taken in choosing these agents because many of the peripheral vasodilators (e.g., nitroprusside, nitroglycerin) also are cerebral vasodilators. All antihypertensives are believed to cause some degree of cerebral vasodilation. To reduce this vasodilating effect, concurrent treatment with beta-blockers (e.g., metoprolol, labetalol) may be beneficial.49,52
Systemic hypotension should be treated aggressively with fluids to maintain a systolic blood pressure greater than 90 mm Hg. Crystalloids, colloids, and blood products can be used, depending on the patient’s condition. Studies have demonstrated a positive effect on ICP and CPP with hypertonic saline. If fluids fail to adequately elevate the patient’s blood pressure, the use of inotropic agents may be necessary.49
Seizure Control
The incidence of posttraumatic seizures in the head-injured population has been estimated at 15% to 20%. Because of the risk of a secondary ischemic insult associated with seizures, many physicians prescribe anticonvulsant medications prophylactically. Seizures cause metabolic requirements to increase, resulting in elevation of CBF, cerebral blood volume, and ICP, even in paralyzed patients. If blood flow cannot match demand, ischemia develops, cerebral energy stores are depleted, and irreversible neuronal destruction occurs. The usual anticonvulsant regimen for seizure control includes phenytoin or phenobarbital, or both, in therapeutic doses.52 Fast-acting, short-duration agents such as lorazepam may be indicated for breakthrough seizures until therapeutic levels of the longer-acting drugs can be achieved.
Cerebrospinal Fluid Drainage
CSF drainage for intracranial hypertension may be used with other treatment modalities (Figures 18-8 and 18-9). CSF drainage is accomplished by the insertion of a pliable catheter into the anterior horn of the lateral ventricle (ventriculostomy), preferably on the nondominant side. This drainage can help support the patient through periods of cerebral edema by controlling spikes in ICP. One of the major advantages of the ventriculostomy is its dual role as a monitoring device and a treatment modality. Care should be taken to avoid infection. However, cleansing ointment such as bacitracin or povidone is not recommended.56
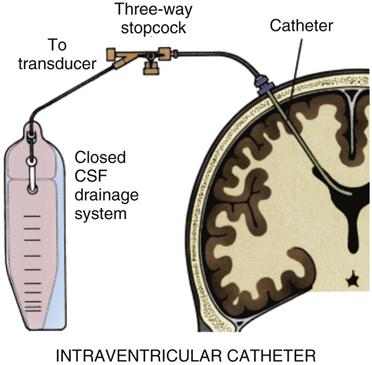
Hyperosmolar Therapy
Osmotic diuretics and hypertonic saline have also been used to reduce increased ICP. In the presence of an intact blood-brain barrier, hyperosmolar therapy is used to draw water from brain tissue into the intravascular compartment. The direction of flow is from the hypoconcentrated tissue to the hyperconcentrated cerebral vasculature.52
The most widely used osmotic diuretic is mannitol, a large-molecule agent that is retained almost entirely in the extracellular compartment and has little or none of the rebound effect observed with other osmotic diuretics. Administration of mannitol increases cerebral blood flow and thus induces cerebral vasoconstriction as part of the brain’s autoregulatory response to maintain blood flow constant.52
Two major complications associated with osmotic diuretics are electrolyte disturbances and dehydration. Careful attention must be paid to body weight and fluid and electrolyte stability. Serum osmolality should be kept between 300 and 320 mOsm/L. Hypernatremia and hypokalemia often are associated with repeated administration of osmotic agents. Central venous pressure readings should be monitored to assess for hypovolemia. Smaller doses of mannitol simplify fluid and electrolyte management, and their use is encouraged whenever possible.49,52
Hypertonic saline, given in concentrations ranging from 3% to 23.4%, can also be used to treat increased ICP. Hypertonic saline can be used in hypovolemic patients when mannitol is contraindicated.52 Adverse effects include electrolyte abnormalities,52 hypotension, pulmonary edema, acute renal failure, hemolysis, central pontine myelinolyisis, coagulopathy, and dysrhythmias.57
Control of Metabolic Demand
Any treatment modality that increases the incidence of noxious stimulation to the patient carries with it the potential for increasing ICP. Noxious stimuli include pain, the presence of an endotracheal tube, coughing, suctioning, repositioning, bathing, and many other routine nursing interventions. Agents used to reduce metabolic demands include the use of benzodiazepines such as midazolam and lorazepam, intravenous sedative-hypnotics such as propofol, opioid narcotics such as fentanyl and morphine, and neuromuscular blocking agents such as vecuronium and atracurium. These agents may be administered separately or in combination by continuous drip or as an intravenous bolus on an as-needed basis.49
The preferred treatment regimen begins with the administration of benzodiazepines for sedation and narcotics for analgesia. If these agents fail to blunt the patient’s response to noxious stimuli, propofol or a neuromuscular blocking agent is added. The use of these medications is recommended only in patients with an ICP monitor in place, because sedatives, opioids, and neuromuscular blocking agents affect the reliability of neurological assessment. The use of neuromuscular blocking agents without sedation is not recommended because these agents can cause skeletal muscle paralysis and because they have no analgesic effect and do not adequately protect the patient from pain and the physiological responses that can occur from pain-producing procedures.58 If these agents fail to control the patient’s ICP, barbiturate therapy is considered.52
Barbiturate Therapy.
Barbiturate therapy is a treatment protocol developed for the management of uncontrolled intracranial hypertension that has not responded to the conventional treatments previously described.52 The two most commonly used drugs in high-dose barbiturate therapy are pentobarbital and thiopental. The goal with either drug is a reduction of ICP to 15 to 20 mm Hg, while a mean arterial pressure of 70 to 80 mm Hg is maintained. Patients are maintained on high-dose barbiturate therapy until ICP has been controlled within the normal range for 24 hours. Barbiturates must never be stopped abruptly; they are tapered slowly over approximately 4 days. Despite the theoretical reasons for barbiturate use, clinical trials of its use have not shown improved outcome.49
Complications of high-dose barbiturate therapy can be disastrous unless a specific and organized approach is used. The most common complications are hypotension, hypothermia, and myocardial depression. If any complications occur and are allowed to persist unchecked, they may cause secondary insults to an already damaged brain. Hypotension, the most common complication, results from peripheral vasodilation and can be compounded in an already dehydrated patient who has received large doses of an osmotic diuretic in an attempt to control ICP. Careful monitoring of fluid status by central venous pressure or a pulmonary artery catheter can help prevent this complication. Myocardial depression results from cardiac muscle suppression and can be avoided by frequent monitoring of fluid status, cardiac output, and serum drug levels. If an adequate cardiac output cannot be maintained in the presence of normothermia, barbiturate doses must be reduced, regardless of serum levels.49,52
Collaborative management of the patient’s intracranial hypertension is outlined in the Collaborative Management box on Intracranial Hypertension.
Herniation Syndromes
The goal of neurological evaluation, ICP monitoring, and treatment of increased ICP is to prevent herniation. Herniation of intracerebral contents results in the shifting of tissue from one compartment of the brain to another and places pressure on cerebral vessels and vital function centers of the brain. If unchecked, herniation rapidly causes death as a result of the cessation of CBF and respirations.49
Supratentorial Herniation
The four types of supratentorial herniation syndrome are uncal; central, or transtentorial; cingulate; and transcalvarial (Figure 18-10).
Uncal Herniation.
Uncal herniation is the most common herniation syndrome. In uncal herniation, a unilateral, expanding mass lesion, usually of the temporal lobe, increases ICP, causing lateral displacement of the tip of the temporal lobe (uncus). Lateral displacement pushes the uncus over the edge of the tentorium, puts pressure on the oculomotor nerve (cranial nerve III) and the posterior cerebral artery ipsilateral to the lesion, and flattens the midbrain against the opposite side. Clinical manifestations of uncal herniation include ipsilateral pupil dilation, decreased level of consciousness, respiratory pattern changes leading to respiratory arrest, and contralateral hemiplegia leading to decorticate or decerebrate posturing. If no intervention occurs, uncal herniation results in fixed and dilated pupils, flaccidity, and respiratory arrest.49,59
Central Herniation.
In central, or transtentorial, herniation, an expanding mass lesion of the midline, frontal, parietal, or occipital lobe results in downward displacement of the hemispheres, basal ganglia, and diencephalon through the tentorial notch. Central herniation often is preceded by uncal and cingulate herniation. Clinical manifestations of central herniation include loss of consciousness; small, reactive pupils progressing to fixed, dilated pupils; respiratory changes leading to respiratory arrest; and decorticate posturing progressing to flaccidity. In the late stages, uncal and central herniation syndromes affect the brainstem similarly.49,59
Cingulate Herniation.
Cingulate herniation occurs when an expanding lesion of one hemisphere shifts laterally and forces the cingulate gyrus under the falx cerebri. Cingulate herniation occurs often. When a lateral shift is observed on the CT scan, cingulate herniation has occurred. Little is known about the effects of cingulate herniation, and there are no accompanying clinical manifestations that assist in its diagnosis. Cingulate herniation is not in itself a life-threatening condition, but if the expanding mass lesion that caused cingulate herniation is not controlled, uncal or central herniation will follow.49,59
Transcalvarial Herniation.
Transcalvarial herniation is the extrusion of cerebral tissue through the cranium. In the presence of severe cerebral edema, transcalvarial herniation occurs through an opening from a skull fracture or craniotomy site.49
Infratentorial Herniation
The two infratentorial herniation syndromes are upward transtentorial herniation and downward cerebellar herniation.
Upward Transtentorial Herniation.
Upward transtentorial herniation occurs when an expanding mass lesion of the cerebellum causes protrusion of the vermis (central area) of the cerebellum and the midbrain upward through the tentorial notch. Compression of the third cranial nerve and diencephalon occurs. Blockage of the central aqueduct and distortion of the third ventricle obstruct CSF flow. Deterioration progresses rapidly.49,59
Downward Cerebellar Herniation.
Downward cerebellar herniation occurs when an expanding lesion of the cerebellum exerts pressure downward, sending the cerebellar tonsils through the foramen magnum. Compression and displacement of the medulla oblongata occur, rapidly resulting in respiratory and cardiac arrest.49,59
Pharmacological Agents
Many pharmacological agents are used in the care of patients with neurological disorders. Table 18-6 reviews the various agents used and any special considerations necessary for administering them.60
TABLE 18-6
PHARMACOLOGICAL MANAGEMENT
Neurological Disorders
| DRUG | DOSAGE | ACTIONS | SPECIAL CONSIDERATIONS |
| Anticonvulsants | |||
| Phenytoin (Dilantin) | Loading dose: 10-20 mg/kg IV Maintenance dose: 100 mg q6-8h IV |
Prevents the influx of sodium at the cell membrane | Monitor serum levels closely; therapeutic level is 10-20 mg/L (if hypoalbuminuria, monitor free phenytoin serum levels: therapeutic level of 0.1–0.2 mg/L) |
| Infuse phenytoin no faster than 50 mg/min; administer with normal saline only because it precipitates with other solutions | |||
| Fosphenytoin (Cerebyx) | Loading dose: 15-20 mg/kg IV Maintenance dose: 4-6m g/kg/24 hr IV |
Prevents the influx of sodium at the cell membrane | Monitor serum levels closely; therapeutic level is 10-20 mg/L |
| Dosage, concentration, and infusion rate of fosphenytoin are expressed as phenytoin sodium equivalents (PE) | |||
| Barbiturates | |||
| Phenobarbital | Loading dose: 6-8 mg/kg IV Maintenance dose: 1-3 mg/kg/24 hr IV |
Produces central nervous system depression and reduces the spread of an epileptic focus | May depress cardiac and respiratory function |
| Administer phenobarbital at a rate of 60 mg/min; monitor serum level closely; therapeutic level is 15-40 mg/L | |||
| Pentobarbital | Loading dose: 3-10 mg/kg over 30 min | Induces barbiturate coma | Monitor serum level of pentobarbital closely; therapeutic level for coma is 15-40 mg/L |
| Maintenance dose: 0.5-3 mg/kg/hr IV | |||
| Osmotic Diuretics | |||
| Mannitol | 1-2 g/kg IV | Treats cerebral edema by pulling fluid from the extravascular space into the intravascular space; requires intact blood-brain barrier | Side effects include hypovolemia and increased serum osmolality |
| Monitor serum osmolality and notify the physician if >310 mOsm/L | |||
| Warm and shake before administering to ensure crystals are dissolved | |||
| Calcium Channel Blockers | |||
| Nimodipine (Nimotop) | 60 mg q4h NG or PO for 21 days | Decreases cerebral vasospasm | Side effects include hypotension, palpitations, headache, and dizziness |
| Monitor blood pressure frequently when implementing therapy | |||
| Local Anesthetics | |||
| Lidocaine | 50-10 0mg IV or 2 mL of 4% solution | Blunts the effects of tracheal stimulation on intracranial pressure | Must be administered not longer than 5 minutes before suctioning |
| Thrombolytics | |||
| Tissue-type plasminogen activator (tPA) | 0.9 mg/kg total, with 10% of the dose administered as IV bolus over 1 min and 90% of the dose administered as continuous IV infusion over 1 hr | Converts plasminogen to plasmin to dissolve clot | Treatment must start within 3 hr of the onset of the symptoms |
| Do not exceed 90 mg | |||
| Do not use anticoagulants during the first 24 hr | |||
| Monitor patient for bleeding | |||
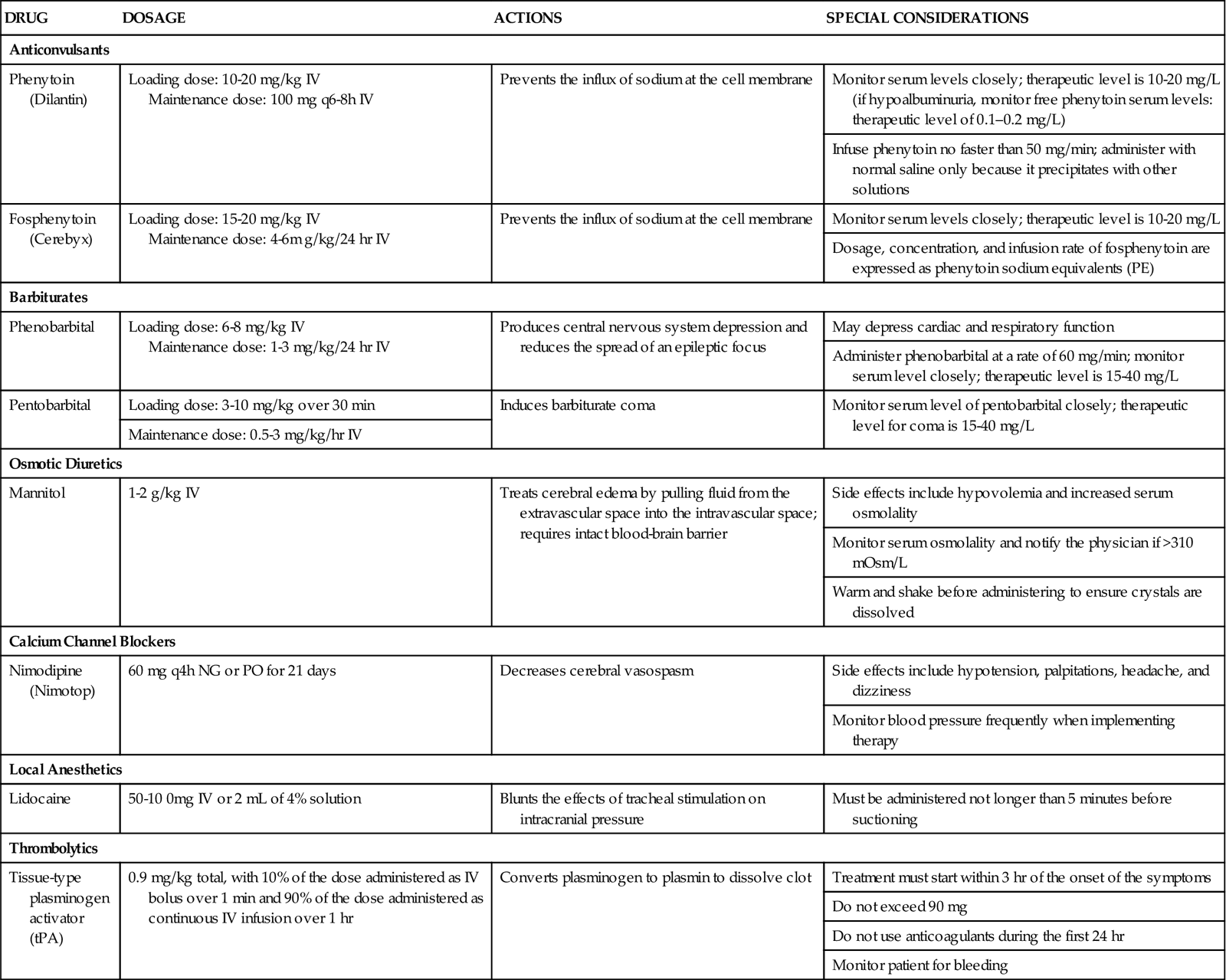
[/level-membership-for-critical-care-medicine-category][not-level-membership-for-critical-care-medicine-category]
Objectives
• Describe the etiology and pathophysiology of selected neurological disorders.
• Identify the clinical manifestations of selected neurological disorders.
• Explain the treatment of selected neurological disorders.
• Discuss the nursing priorities for managing a patient with selected neurological disorders.
• Discuss the concept of cerebral autoregulation.
• Describe the therapies commonly used to treat intracranial hypertension.
Be sure to check out the bonus material, including free self-assessment exercises, on the Evolve web site at
http://evolve.elsevier.com/Urden/priorities/.
To accurately anticipate and plan nursing interventions, the critical care nurse must have an understanding of the disease pathology, determine the areas of focused assessment, and be well acquainted with the medical management of the neurological patient. Fortunately, despite a wide array of neurological disorders, only a few routinely require the critical care environment.
Coma
Normal consciousness requires awareness and arousal. Awareness is the combination of cognition (mental and intellectual) and affect (mood) that can be construed based on the patient’s interaction with the environment.1 Alterations of consciousness may be the result of deficits in awareness, arousal, or both.2 There are four discrete disorders of consciousness: coma, vegetative state, minimally conscious state, and locked-in syndrome. Coma is characterized by the absence of both wakefulness and awareness, whereas a vegetative state is characterized by the presence of wakefulness with the absence of awareness. In the minimally conscious state, wakefulness is presence and awareness is severely diminished but not absent. Locked-in syndrome is characterized by the presence of wakefulness and awareness, but with quadriplegia and the inability to communicate verbally; thus the patient appears to be unconscious.3 Box 18-1 lists the disorders of consciousness in descending order of wakefulness.
Coma is the deepest state of unconsciousness; arousal and awareness are lacking.1–3 The patient cannot be aroused and does not demonstrate any purposeful response to the surrounding environment.4 Coma is a symptom rather than a disease, and it occurs as a result of some underlying process.1,2 The incidence of coma is difficult to ascertain because a wide variety of conditions can induce coma.1,2 This state of unconsciousness is unfortunately very common in critical care, and it is the focus of the following discussion.
Etiology
The causes of coma can be divided into two general categories: structural or surgical and metabolic or medical. Structural causes of coma include ischemic stroke, intracerebral hemorrhage (ICH), trauma, and brain tumors.5 Metabolic causes of coma include drug overdose, infectious diseases, endocrine disorders, and poisonings.5 Coma demands immediate attention, resulting in a high percentage of admissions to all hospital services.6 Table 18-1 provides a list of the possible causes of coma.
TABLE 18-1
| STRUCTURAL OR SURGICAL COMA | METABOLIC OR MEDICAL COMA |
| Trauma | Infection |
| Epidural hematoma | Meningitis |
| Subdural hematoma | Encephalitis |
| Metabolic encephalopathy | |
| Diffuse axonal injury | Metabolic conditions |
| Hypoglycemia | |
| Brain contusion | Hyperglycemia |
| Intracerebral hemorrhage | Hyperosmolar states |
| Uremia | |
| Subarachnoid hemorrhage | Hepatic encephalopathy |
| Hypertensive encephalopathy | |
| Posterior fossa hemorrhage | Hypoxic encephalopathy |
| Hyponatremia | |
| Supratentorial hemorrhage | Hypercalcemia |
| Myxedema | |
| Hydrocephalus | Intoxication |
| Ischemic stroke | Opioid overdose |
| Tumor | Alcohol |
| Other causes | Poisonings |
| Psychogenic causes |
Pathophysiology
Consciousness involves arousal, or wakefulness, and awareness. Neither of these functions is present in the patient in coma. Ascending fibers of the reticular activating system (ARAS) in the pons, hypothalamus, and thalamus maintain arousal as an autonomic function. Neurons in the cerebral cortex are responsible for awareness. Diffuse dysfunction of both cerebral hemispheres and diffuse or focal dysfunction of the reticular activating system can produce coma.1,6,7 Structural causes usually produce compression or dysfunction in the area of the ARAS, whereas most medical causes lead to general dysfunction of both cerebral hemispheres.8 Trauma, hemorrhage, and tumor can damage the ARAS, leading to coma. Destruction of large regions of bilateral cerebral hemispheres can be the result of seizures or viral agents. Toxic drugs, toxins, or metabolic abnormalities can suppress cerebral function.5–7
Assessment and Diagnosis
The clinical diagnosis of the coma state is readily established by assessment of the level of consciousness. However, determining the full nature and cause of coma requires a thorough history and physical examination. A medical history is essential, because events immediately preceding the change in level of consciousness can often provide valuable clues to the origin of the coma. When limited information is available and the coma is profound, the response of the patient to emergency treatment may provide clues to the underlying diagnosis; for example, the patient who becomes responsive with the administration of naloxone can be presumed to have ingested some type of opiate.6
Detailed serial neurological examinations are essential for all patients in coma. Assessment of pupillary size and reaction to light (normal, sluggish, or fixed), extraocular eye movements (normal, asymmetrical, or absent), motor response to pain (normal, decorticate, decerebrate, or flaccid), and breathing pattern yields important clues for determining whether the cause of the coma is structural or metabolic.1,6
The areas of the brainstem that control consciousness and pupillary responses are anatomically adjacent. The sympathetic and parasympathetic nervous systems control pupillary dilation and constriction, respectively. The anatomic directions of these pathways are known, and changes in pupillary responses can help identify where a lesion may be located. For example, if damage occurs in the midbrain region, pupils are slightly enlarged and unresponsive to light. Lesions that compress the third nerve result in a fixed and dilated pupil on the same side as the neurological insult. Pupillary responses are usually preserved when the cause of the coma is metabolic in origin. Pupillary light responses are often the key to differentiating between structural and metabolic causes of coma.1,6,7,9
Areas of the brainstem adjacent to those responsible for consciousness also control the oculomotor eye movement. The ability to maintain conjugate gaze requires preservation of the internuclear connections of cranial nerves III, VI, and VIII by means of the medial longitudinal fasciculus (MLF).9 As with pupillary responses, structural lesions that impinge on these pathways cause oculomotor dysfunction such as a disconjugate gaze. Deficits in extraocular eye movements usually accompany a structural cause.1,5,9
Focal or asymmetric motor deficits usually indicate structural lesions.1,5 Abnormal motor movements may also help pinpoint the location of a lesion. Decorticate posturing (abnormal flexion) can be seen with damage to the diencephalon. Decerebrate posturing (abnormal extension) can be seen with damage to the midbrain and pons. Flaccid posturing is an ominous sign and can be seen with damage to the medulla.9
Abnormal breathing patterns may also assist in differentiating structural from metabolic causes of coma. Cheyne-Stokes respirations are seen in patients with cerebral hemispheric dysfunction or metabolic suppression. Central neurogenic hyperventilation, or Kussmaul breathing, occurs with metabolic acidosis or damage to the midbrain and upper pons. Apneustic breathing may occur with damage to the pons, hypoglycemia, and anoxia. Ataxic breathing occurs with damage to the medulla. Agonal breathing occurs with failure of the respiratory centers in the medulla.6,9
In addition to physical assessment, laboratory studies and diagnostic procedures are done. Structural causes of coma are usually readily apparent with computed tomography (CT) or magnetic resonance imaging (MRI).4,8 Evoked potentials are also useful in facilitating a differential diagnosis between the disorders of consciousness and in evaluating a patient’s prognosis.3 Generally a patient in a coma with an absence of brainstem auditory evoked responses (BAERs) is considered to have a poor prognosis of recovery.3 Laboratory studies are also used to identify metabolic or endocrine abnormalities.7 Occasionally, the cause of coma is never clearly determined.
Medical Management
The goal of medical management of the patient in a coma is identification and treatment of the underlying cause of the condition. Initial medical management includes emergency measures to support vital functions and prevent further neurological deterioration. Protection of the airway and ventilatory assistance are often needed. Administration of thiamine (at least 100 mg), glucose, and an opioid antagonist is suggested when the cause of coma is not immediately known.1,6 Thiamine is administered before glucose, because the coma produced by thiamine deficiency, Wernicke’s encephalopathy, can be precipitated by a glucose load.1
The patient who remains in a coma after emergency treatment requires supportive measures to maintain physiological body functions and prevent complications. Intubation for continued airway protection and nutritional support are essential. Fluid and electrolyte management is often complex because of alterations in the neurohormonal system. Anticonvulsant therapy may be necessary to prevent further ischemic damage to the brain.1,5,6
The health care team and the patient’s family make decisions jointly regarding the level of medical management to be provided. Family members require informational support in terms of probable cause of the coma and prognosis for recovery of consciousness and function. Prognosis depends on the cause of the coma and the length of time unconsciousness persists. Only 15% of patients in nontraumatic coma make a satisfactory recovery.7 Metabolic coma usually has a better prognosis than coma caused by a structural lesion, and traumatic coma usually has a better outcome than nontraumatic coma.5,7
Nursing Management
Nursing management of the patient in a coma incorporates a variety of nursing diagnoses (Nursing Diagnosis Priorities box on Coma) and is directed by the specific cause of the coma, although some common interventions are used. The patient in a coma totally depends on the health care team. Nursing priorities are directed toward (1) monitoring for changes in neurological status and clues to the origin of the coma, (2) supporting all body functions, (3) maintaining surveillance for complications, (4) providing comfort and emotional support, and (5) initiating rehabilitation measures. Measures to support body functions include promoting pulmonary hygiene, maintaining skin integrity, initiating range-of-motion exercises, managing bowel and bladder functions, and ensuring adequate nutritional support.1
Eye Care
The blink reflex is often diminished or absent in the comatose patient. The eyelids may be flaccid and may depend on body positioning to remain in a closed position, and edema may prevent complete closure. Loss of these protective mechanisms results in drying and ulceration of the cornea, which can lead to permanent scarring and blindness.1
Two interventions that are commonly used to protect the eyes are instilling saline or methylcellulose lubricating drops and taping the eyelids in the shut position. Evidence suggests that an alternative technique may be more effective in preventing corneal epithelial breakdown. In addition to instillation of saline drops every 2 hours, a polyethylene film is taped over the eyes, extending beyond the orbits and eyebrows. The film creates a moisture chamber around the cornea and assists in keeping the eyes moist and in the closed position. This technique also prevents damage to the eyes that results from placement of tape or gauze directly on the delicate skin of the eyelids.10
Collaborative management of the patient in a coma is outlined in the Collaborative Management box on Coma.
Stroke
Stroke is a descriptive term for the sudden onset of acute neurological deficit persisting for more than 24 hours and caused by the interruption of blood flow to the brain. Stroke is the third leading cause of death in the United States, preceded by heart disease and cancer, and the leading cause of adult disability.11 Approximately 795,000 people have a stroke each year; 610,000 of these are first attacks, and 185,000 are recurrent attacks.11
Strokes are classified as ischemic or hemorrhagic (Concept Map on Stroke). Hemorrhagic strokes can be further categorized as subarachnoid hemorrhages (SAHs) and intracerebral hemorrhages. Approximately 87% of all strokes are ischemic, 10% are ICHs, and 3% are SAHs.11 Although less common, hemorrhagic strokes (ICHs and SAHs) have a higher mortality rate than ischemic strokes. Approximately 8% to 12% of ischemic strokes and 37% to 45% of hemorrhagic strokes result in death within 30 days.11 The annual cost for care and loss of productivity was estimated to be $73.7 billion in 2009.11
Ischemic Stroke
Ischemic stroke results from interruption of blood flow to the brain and accounts for 80% to 85% of all strokes. The interruption can be the result of a thrombotic or embolic event. Thrombosis can form in large vessels (large-vessel thrombotic strokes) or small vessels (small-vessel thrombotic strokes). Embolic sources include the heart (cardioembolic strokes) and atherosclerotic plaques in larger vessels (atheroembolic strokes). In 30% of the cases, the underlying cause of the stroke is unknown (cryptogenic strokes).12
Strokes are preventable. Most thrombotic strokes are the result of the accumulation of atherosclerotic plaque in the vessel lumen, especially at bifurcations or curves of the vessel. The pathogenesis of cerebrovascular disease is identical to that of coronary vasculature disease. The greatest risk factor for ischemic stroke is hypertension.12,13 Other risk factors are dyslipidemia, diabetes, smoking, and carotid atherosclerotic disease.11,14 Common sites of atherosclerotic plaque are the bifurcation of the common carotid artery, the origins of the middle and anterior cerebral arteries, and the origins of the vertebral arteries.13 Ischemic strokes resulting from vertebral artery dissection have been reported after chiropractic manipulation of the cervical spine.15
Etiology
An embolic stroke occurs when an embolus from the heart or lower circulation travels distally and lodges in a small vessel, obstructing the blood supply. At least 20% of ischemic strokes are attributed to a cardioembolic phenomenon.12 The most common cause of cardiac emboli is atrial fibrillation. It is responsible for about 50% of all cardiac emboli.16 Other sources of cardiac emboli are mitral stenosis, mechanical valves, atrial myxoma, endocarditis, and recent myocardial infarction.13 Researchers hypothesize that a patent foramen ovale or atrial septal aneurysms may be the cause of cryptogenic stroke.17
Pathophysiology
Ischemic stroke is a cerebral hemodynamic insult. When cerebral blood flow is reduced to a level insufficient to maintain neuronal viability, ischemic injury occurs. In focal stroke, an area of hypoperfused tissue, the ischemic penumbra, surrounds a core of ischemic cells. The ischemic penumbra can be salvaged with return of blood flow. However, sustained anoxic insult initiates a chain of biochemical events leading to apoptosis, or cellular death.18
The phenomenon of a focal ischemic stroke is identical to that associated with myocardial infarction, which is why brain attack is used in public education strategies. Often, a history of transient ischemic attacks (TIAs), brief episodes of neurological symptoms that last less than 24 hours, offers a warning that stroke is likely to occur. Sudden onset indicates embolism as the final insult to flow.12,13 The size of the stroke depends on the size and location of the occluded vessel and the availability of collateral blood flow.12 Global ischemia results when severe hypotension or cardiopulmonary arrest provokes a transient drop in blood flow to all areas of the brain.18
Cerebral edema sufficient to produce clinical deterioration develops in 10% to 20% of patients with ischemic stroke and can result in intracranial hypertension. The edema results from a loss of normal metabolic function of the cells and peaks at 4 days.12 This process is commonly the cause of death during the first week after a stroke.19 Secondary hemorrhage at the site of the stroke lesion, known as hemorrhagic conversion,19 and seizures20 are the two other major acute neurological complications of ischemic stroke.
Assessment and Diagnosis
The characteristic sign of an ischemic stroke is the sudden onset of focal neurological signs persisting for more than 24 hours.12 These signs usually occur in combination. Box 18-2 lists common patterns of neurological symptoms associated with an ischemic stroke. Hemiparesis, aphasia, and hemianopia are common. Changes in the level of consciousness usually occur only with brainstem or cerebellar involvement, seizure, hypoxia, hemorrhage, or elevated intracranial pressure (ICP). These changes may be exhibited as stupor, coma, confusion, and agitation.1 The reported frequency of seizures in patients with ischemic stroke ranges from 3% to 8%. If seizures occur, they are usually seen within 24 hours of an insult.20
The National Institutes of Health Stroke Scale (NIHSS) is often used as the basis of the focused neurological examination. The score ranges from 0 to 42 points; the higher the score, the more neurologically impaired the patient. A change of 4 points on the scale indicates significant neurological change. The components of the NIHSS include level of consciousness (LOC); LOC questions; LOC commands; gaze; visual fields; face, arm, and leg strength; sensation; limb ataxia; and language function.12 A copy of the NIHSS with complete instructions can be retrieved at http://www.ninds.nih.gov/disorders/stroke/strokescales.htm.
Confirmation of the diagnosis of ischemic stroke is the first step in the emergency evaluation of these patients. Differentiation from intracranial hemorrhage is vital. Noncontrast CT scanning is the method of choice for this purpose and is considered the most important initial diagnostic study. In addition to excluding intracranial hemorrhage, CT can assist in identifying early neurological complications and the cause of the insult.12 MRI can demonstrate infarction of cerebral tissue earlier than CT but is less useful in the emergency differential diagnosis.21 Because of the strong correlation between acute ischemic stroke and heart disease, 12-lead electrocardiography, chest radiography, and continuous cardiac monitoring are suggested to detect a cardiac cause or coexisting condition. Echocardiography is valuable in identifying a cardioembolic phenomenon when a sufficient index of suspicion warrants its use.1 Laboratory evaluation of hematological function, electrolyte and glucose levels, and renal and hepatic function is also recommended. Arterial blood gas analysis is performed if hypoxia is suspected, and an electroencephalogram is obtained if seizures are suspected. Lumbar puncture is performed only if SAH is suspected and the CT appearance is normal.1
Medical Management
Major changes have taken place in the medical management of ischemic stroke since 1996. On the basis of results of the National Institute of Neurologic Disorders and Stroke (NINDS) rtPA Stroke Study, thrombolytic therapy with intravenous recombinant tissue-type plasminogen activator (rtPA) was initially recommended within 3 hours of onset of ischemic stroke. This time frame has now been expanded from 3 hours to 4.5 hours.22 Patients who should be considered for thrombolysis are listed in Box 18-3. Confirmation of diagnosis with CT must be accomplished before rtPA administration. The recommended dose of rtPA is 0.9 mg/kg up to a maximum dose of 90 mg. Ten percent of the total dose is administered as an initial intravenous bolus, and the remaining 90% is administered by intravenous infusion over 60 minutes.17,21
The desired result of thrombolytic therapy is to dissolve the clot and reperfuse the ischemic brain. The goal is to reverse or minimize the effects of stroke. The major risk and complication of rtPA therapy is bleeding, especially intracranial hemorrhage. Unlike thrombolytic protocols for acute myocardial infarction, subsequent therapy with anticoagulant or antiplatelet agents is not recommended after rtPA administration in ischemic stroke. Patients receiving thrombolytic therapy for stroke should not receive aspirin, heparin, or warfarin for at least 24 hours after treatment.12,17
The major barriers to effective application of thrombolytic therapy for ischemic stroke are prehospital and in-hospital delays. To help decrease delays, the public needs to be educated about stroke symptoms and activation of the emergency medical system (EMS). EMS responders need adequate education and training on managing a patient with an acute ischemic stroke, focusing on stabilization and quick transport of the patient to the emergency department. The receiving hospital should ideally be primary stroke certified and have expert staff and the infrastructure to care for the complex stroke patient.21
Other emergency care of the patient with ischemic stroke must include airway protection and ventilatory assistance to maintain adequate tissue oxygenation.19 Hypertension is often present in the early period as a compensatory response, and in most cases, blood pressure (BP) must not be lowered (Table 18-2). For the patient who has not received thrombolytic therapy, antihypertensive therapy is considered only if the diastolic blood pressure is greater than 120 mm Hg or the systolic blood pressure is greater than 220 mm Hg.12 Criteria are different for patients who have received rtPA. Their blood pressure is kept below 180/105 mm Hg to prevent intracranial hemorrhage. Intravenous labetalol or nicardipine is used to achieve blood pressure control. If these agents are not effective, nitroprusside, hydralazine, or enalaprilat should be considered.12 Body temperature and glucose levels also must be normalized.12,19
TABLE 18-2
BLOOD PRESSURE MANAGEMENT FOR STROKE ACCORDING TO THE AMERICAN STROKE ASSOCIATION GUIDELINES
| BLOOD PRESSURE* | TREATMENT |
| Nonthrombolytic Candidates | |
| DBP >140 mm Hg | Sodium nitroprusside (0.5 mcg/kg/min); aim for 10%-20% reduction in DBP |
| SBP >220 mm Hg, DBP 121-140 mm Hg, or MAP† >130 mm Hg | 10-20 mg of labetalol‡ given by IVP over 1-2 min; may repeat or double labetalol every 20 min to a maximum dose of 300 mg |
| SBP <220 mm Hg, DBP = 120 mm Hg, or MAP† <130 mm Hg | Emergency antihypertensive therapy is deferred in the absence of aortic dissection, acute myocardial infarction, severe congestive heart failure, or hypertensive encephalopathy |
| Thrombolytic Candidates | |
| Pretreatment | |
| SBP >185 mm Hg or DBP >110 mm Hg | 1-2 inches of nitroglycerine paste (Nitropaste) or 1-2 doses of 10-20 mg of labetalol‡ given by IVP; if BP is not reduced and maintained to <185/110 mm Hg, the patient should not be treated with tPA |
| During and After Treatment | |
| Monitor BP | BP is monitored every 15 min for 2 hr, then every 30 min for 6 hr, and then hourly for 16 hr |
| DBP >140 mm Hg | Sodium nitroprusside (0.5 mcg/kg/min) |
| SBP >230 mm Hg or DBP 121-140 mm Hg | 10 mg of labetalol‡ given by IVP over 1-2 min; may repeat or double labetalol every 10 min to a maximum dose of 300 mg or give initial labetalol bolus and then start a labetalol drip at 2-8 mg/min |
| If BP not controlled by labetalol, consider sodium nitroprusside | |
| SBP 180-230 mm Hg or DBP 105-120 mm Hg | 10 mg of labetalol‡ given by IVP; may repeat or double labetalol every 10-20 min to a maximum dose of 300 mg or give initial labetalol bolus and then start a labetalol drip at 2-8 mg/min |
*All initial blood pressures should be verified before treatment by repeating reading in 5 minutes.
†As estimated by one third of the sum of systolic and double diastolic pressure.
‡Labetalol should be avoided in patients with asthma, cardiac failure, or severe abnormalities in cardiac conduction. For refractory hypertension, alternative therapy with sodium nitroprusside or enalapril may be considered.
From Bader MK, Littlejohns LR: AANN core curriculum for neuroscience nursing, ed 4, St Louis, 2004, Elsevier.
Medical management also includes the identification and treatment of acute complications such as cerebral edema and seizure activity. Prophylaxis for these complications is not recommended. Deep vein thrombosis (DVT) prophylaxis, however, should be initiated to decrease the risk of pulmonary embolism.12 One study demonstrated that improved outcomes for ischemic stroke patients can be achieved by managing swallowing issues, initiating DVT prophylaxis, and treating hypoxemia.23 Surgical decompression is recommended if a large cerebellar infarction compresses the brainstem.19
Subarachnoid Hemorrhage
Subarachnoid hemorrhage is bleeding into the subarachnoid space, which usually is caused by rupture of a cerebral aneurysm or arteriovenous malformation (AVM).19 At the time of autopsy, approximately 4% of the population has been found to have one or more aneurysms.24 Aneurysmal SAH is associated with a mortality rate of 25% to 50%, with most patients dying on the first day after the insult.24 Hemorrhage due to AVM rupture has a better chance of survival and is associated with an overall mortality rate of 10% to 15%.25
Etiology
Cerebral aneurysm rupture accounts for approximately 85% of all cases of spontaneous SAH.24 An aneurysm is an outpouching of the wall of a blood vessel that results from weakening of the wall of the vessel (Table 18-3).24 Ninety percent of aneurysms are congenital—the cause of which is unknown. The other 10% can be the result of traumatic injury (that stretches and tears the muscular middle layer of the arterial vessel) or infectious material (most often from infectious vegetation on valves of the left side of the heart after bacterial endocarditis) that lodges against a vessel wall and erodes the muscular layer, or they are of undetermined cause.26 Multiple aneurysms occur in approximately 30% of the cases and often are bilateral, occurring in the same location on both sides of the cerebral vascular system.27
TABLE 18-3
ANEURYSM CLASSIFICATION ACCORDING TO TYPE, SHAPE, LOCATION, AND COMMON CHARACTERISTICS
| TYPES OF ANEURYSMS | CHARACTERISTICS |
| Berry or saccular
|
Most common type, usually congenital; appears at a bifurcation in the anterior circulation, primarily at the base of the brain or the circle of Willis and its branches; grows from the base of the arterial wall with a neck or stem; contains blood; thinned dome is usually the site of rupture |
| Giant or fusiform
|
Can have an irregular shape and can be larger than 2.5 cm and atherosclerotic; involves mainly the internal carotid or vertebrobasilar artery; rarely ruptures; has no stem; can act like a space-occupying lesion in the brain; difficult to manage |
| Mycotic
|
Rare form; usually occurs from septic emboli, usually results from bacterial infection, which weaken the vessel wall, causing dilation involving the distal branches of the middle cerebral arteries |
| Dissecting
|
May occur during angiography; caused by trauma, syphilis, or arteriosclerosis, or when blood is forced between layers of the arterial wall; intima is pulled away from the medial layer, allowing blood to enter |
| Traumatic | Sometimes called a pseudoaneurysm, which may resolve after trauma |
| Charcot-Bouchard | Small aneurysm that can be seen in the area of the basal ganglia or the brainstem in individuals with a history of hypertension; chronic hypertension causes fibrinoid necrosis in the penetrating and subcortical arteries, weakening the arterial walls and causing formation of small aneurysmal outpouching |
AVM rupture is responsible for roughly 6% of all SAHs.27An arteriovenous malformation is a tangled mass of arterial and venous blood vessels that shunt blood directly from the arterial side into the venous side, bypassing the capillary system. They may be small, focal lesions or large, diffuse lesions that occupy almost an entire hemisphere.26 AVMs are always congenital, although the exact embryonic cause for these malformations is unknown. They also occur in the spinal cord and the renal, gastrointestinal, and integumentary systems.27 In contrast to SAH from aneurysm, which occurs in the middle-aged population, SAH from an AVM usually occurs in the second to fourth decades of life.27
Pathophysiology
The pathophysiology of the two most common causes of SAH is distinctly different.
Cerebral Aneurysm.
As the individual with a congenital cerebral aneurysm matures, blood pressure rises, and more stress is placed on the poorly developed and thin vessel wall. Ballooning of the vessel occurs, giving the aneurysm a berry-like appearance. Most cerebral aneurysms are saccular or berry-like with a stem or neck. Aneurysms are usually small, 2 to 7 mm in diameter, and often occur at the base of the brain on the circle of Willis.28 Figure 18-1 illustrates the usual distribution between the vessels. Most cerebral aneurysms occur at the bifurcations of blood vessels.24,25,28
The aneurysm becomes clinically significant when the vessel wall becomes so thin that it ruptures, sending arterial blood at a high pressure into the subarachnoid space. For a brief moment after the aneurysm ruptures, ICP is thought to approach mean arterial pressure (MAP), and cerebral perfusion decreases.28 In other situations, the unruptured aneurysm expands and places pressure on surrounding structures. This is particularly true with posterior communicating artery aneurysms, because they put pressure on the oculomotor nerve (cranial nerve III), causing ipsilateral pupil dilation and ptosis.27
Arteriovenous Malformation.
The pathophysiologic features of an AVM are related to the size and location of the malformation. One or more cerebral arteries, also known as feeders, supply an AVM. These feeder arteries tend to enlarge over time, increasing both the volume of blood shunted through the malformation and the overall mass effect. Large, dilated, tortuous draining veins develop as a result of increasing arterial blood flow being delivered at a higher than normal pressure. Normal vascular flow has an MAP of 70 to 80 mm Hg, a mean arteriole pressure of 35 to 45 mm Hg, and a mean capillary pressure that drops from 35 to 10 mm Hg as it connects with the venous side. Lack of this capillary bridge allows blood with an MAP of 35 to 45 mm Hg to flow into the venous system. Unlike arteries, veins have no muscular layer, and the veins become extremely engorged and rupture easily. Some patients with AVMs also have cerebral atrophy. It is the result of chronic ischemia because of the shunting of blood through the AVM and away from normal cerebral circulation.29
Assessment and Diagnosis
The patient with an SAH characteristically has an abrupt onset of pain, described as the “worst headache of my life.” A brief loss of consciousness, nausea, vomiting, focal neurological deficits, and a stiff neck may accompany the headache.24,26–28 The SAH may result in coma or death.
The patient’s history may reveal one or more incidences of sudden onset of headache with vomiting in the weeks preceding a major SAH. These are small “warning leaks” of an aneurysm in which small amounts of blood ooze from the aneurysm into the subarachnoid space. The presence of blood is an irritant to the meninges, particularly the arachnoid membrane, and the irritation causes headache, stiff neck, and photophobia. These warning leaks seldom are detected because the condition is not severe enough for the patient to seek medical attention.28 If a neurological deficit, such as third cranial nerve palsy, develops before aneurysm rupture, medical intervention is sought, and the aneurysm may be surgically secured before the devastation of a rupture can occur. Symptoms of unruptured AVM—headaches with dizziness or syncope or fleeting neurological deficits—also may be found in the history.27
Diagnosis of SAH is based on clinical presentation, CT findings, and lumbar puncture results. Noncontrast CT is the cornerstone of definitive SAH diagnosis. In 95% of the cases, CT can demonstrate blood in the subarachnoid space if performed within 48 hours of the hemorrhage.24,28 On the basis of the appearance and the location of the SAH, diagnosis of the cause—aneurysm or AVM—may be made from the CT scan.26 MRI is not routinely used, but it may provide greater sensitivity for detecting areas of SAH clot and potential location of bleed.28
If the initial CT finding is negative, a lumbar puncture is performed to obtain cerebrospinal fluid (CSF) for analysis. CSF after SAH appears bloody and has a red blood cell count greater than 1000 cells/mm3. If the lumbar puncture is performed more than 5 days after the SAH, the CSF fluid is xanthochromic (dark amber) because the blood products have broken down.29 Cloudy CSF usually indicates some type of infectious process, such as bacterial meningitis, not SAH.28
After the SAH has been documented, cerebral angiography is necessary to identify the exact cause of the hemorrhage. If a cerebral aneurysm rupture is the cause, angiography is essential for identifying the exact location of the aneurysm in preparation for surgery.27,29,30 After the aneurysm has been located, it is graded using the Hunt and Hess classification scale. This scale categorizes the patient on the basis of the severity of the neurological deficits associated with the hemorrhage (Box 18-4).31 If AVM rupture is the cause, angiography is necessary to identify the feeding arteries and draining veins of the malformation.26
Medical Management
SAH is a medical emergency, and time is of the essence. Preservation of neurological function is the goal, and early diagnosis is crucial. Initial treatment must always support vital functions. Airway management and ventilatory assistance may be necessary.19 A ventriculostomy is performed to control ICP if the patient’s level of consciousness is depressed.30
Evidence suggests that only 19% of the deaths attributable to aneurysmal SAH are related to the direct effects of the initial hemorrhage.32 Rebleeding accounts for 22% of deaths from aneurysmal SAH, cerebral vasospasm for 23%, and nonneurological medical complications for 23%.32 Principal nonneurological causes of death are systemic inflammatory response syndrome (SIRS) and secondary organ dysfunction.33 After initial intervention has provided necessary support for vital physiologic functions, medical management of acute SAH is aimed primarily toward prevention and treatment of the complications of SAH that can produce further neurological damage and death.28
Rebleeding.
Rebleeding is the occurrence of a second SAH in an unsecured aneurysm or, less commonly, an AVM.6 The incidence of rebleeding during the first 24 hours after the first bleed is 4%, with a 1% to 2% chance per day for the following month. The mortality rate associated with aneurysmal rebleeding is approximately 70%.26,27
Historically, conservative measures to prevent rebleeding have included blood pressure control and SAH precautions (see “Nursing Management”). An elevation in blood pressure is a normal compensatory response to maintain adequate cerebral perfusion after a neurological insult. In the belief that hypertension contributes to rebleeding, intravenous antihypertensive agents are used to maintain a systolic blood pressure no greater than 140 mm Hg.28 Individualized guidelines must be determined on the basis of the clinical condition and preexisting values of the patient. Evidence suggests that rebleeding has more to do with variations in blood pressure than it does with absolute values and that blood pressure control does not lower the incidence of rebleeding.30
Surgical Clipping of Aneurysms.
Definitive treatment for the prevention of rebleeding is surgical clipping or endovascular coiling with complete obliteration of the aneurysm.27,28 Timing of the operation is a key medical management issue. Since the introduction of microsurgery and improved surgical techniques, patients are commonly taken to the operating room within the first 48 hours after rupture.28 This early surgical intervention to secure the aneurysm eliminates the risk of rebleeding and allows more aggressive therapy to be used in the postoperative period for the treatment of vasospasm.26 Early surgery also allows the neurosurgeon to flush out the excess blood and clots from the basal cisterns (reservoir of CSF around the base of the brain and circle of Willis) to reduce the risk of vasospasm.33 Careful consideration of the patient’s clinical situation is necessary in determining the optimal time for surgery.
The surgical procedure involves a craniotomy to expose and isolate the area of aneurysm. A clip is placed over the neck of the aneurysm to eliminate the area of weakness (Figure 18-2). This is a technically difficult procedure that requires the skill of an experienced neurosurgeon. It is not uncommon, particularly in early surgery, for the clot to break away from the aneurysm as it is surgically exposed. Extensive hemorrhage into the craniotomy site results, and cessation of the hemorrhage often causes increased neurological deficits. Deficits also may occur as a result of surgical manipulation to gain access to the site of the aneurysm.28
Surgical Excision of Arteriovenous Malformations.
Management of AVMs has traditionally involved surgical excision or conservative management of such symptoms as seizures and headache. The decision for surgical excision depends on the location and size of the AVM. Some malformations are located so deep in the cerebral structures (thalamus or midbrain) that attempts to remove the AVM would cause severe neurological deficits. History of a previous hemorrhage and the patient’s age and overall condition are also taken into account in the decision regarding surgical intervention.28
Surgical excision of large AVMs includes the risk of reperfusion bleeding. As feeding arteries of the AVM are clamped off, the arterial blood that usually flowed into the AVM is diverted into the surrounding circulation. In many cases, the surrounding tissue has been in a state of chronic ischemia, and the arterial vessels feeding these areas are maximally dilated. As arterial blood begins to flow at a higher volume and pressure into these dilated arteries, blood may seep from the vessels. Evidence of reperfusion bleeding in the operating room is an indication that no more arterial blood can be diverted from the AVM without risk of serious ICH. In the postoperative phase, a low blood pressure is maintained to prevent further reperfusion bleeding. For large AVMs, two to four stages of surgery may be required over 6 to 12 months.28
Embolization.
Embolization is used to secure a cerebral aneurysm or AVM that is surgically inaccessible because of size or location or because of the medical instability of the patient. Embolization involves several new interventional neuroradiology techniques. All of the techniques use a percutaneous transfemoral approach in a manner similar to an angiogram. Under fluoroscopic guidance, the catheter is threaded up to the internal carotid artery. Specially developed microcatheters are then manipulated into the area of the vascular anomaly, and embolic materials are placed endovascularly. Three embolization techniques are used, depending on the underlying pathologic derangement.26
The first type of embolization is used to embolize an AVM. Small polymeric silicone (Silastic) beads or glue is slowly introduced into the vessels feeding the AVM. Blood flow carries the material to the site, and embolization is achieved. This procedure may be used in combination with surgery. One to three sessions of embolization of the feeding vessels are performed to reduce the size of the lesion before a craniotomy is performed for total excision. The primary risk of this procedure is lodging of the embolic substance in a vessel that feeds normal tissue, which creates an embolic stroke with the immediate onset of neurological symptoms.26
The second type of embolization involves placement of one or more detachable coils into an aneurysm to produce an endovascular thrombus (Figure 18-3). The advantage of this technique is that an electrical current creates a positive charge on the coil, which induces electrothrombosis. Complications include embolic stroke, coil migration, overproduction of the clot, subtotal occlusion and intraprocedural rupture of the vasculature, and death.26
[/not-level-membership-for-critical-care-medicine-category]
