Chapter 49B Neurological Complications of Systemic Disease
Children
Cardiac Disorders and the Nervous System
Congenital Heart Disease
Cerebral Dysgenesis and Malformations
Cerebral dysgenesis is a consideration to explain neurological symptoms in children with CHD. Autopsy studies reveal a 10% to 29% prevalence of cerebral malformations. Patients with hypoplastic left heart syndrome are at a higher risk for associated brain dysgenesis. In one series of 41 patients with hypoplastic left heart syndrome, 29% had associated brain malformations of variable severity; 27% had microcephaly, 21% had immature cortical mantle, and the remainder had other malformations, one with holoprosencephaly. Other reports include agenesis of the corpus callosum, Dandy-Walker syndrome, and aqueductal stenosis. Lutterman and colleagues (1998) described the association of moyamoya disease and structural CHD, including ventricular septal defect (VSD), aortic and mitral valve stenosis, and tetralogy of Fallot.
Chromosomal and Genetic Disorders
The combination of CHD and neurological disorders, mainly developmental delay, are sometimes manifestations of genetic conditions combining both cardiac and central nervous system (CNS) involvement. Such conditions include trisomy 21, trisomy 13, trisomy 18, Williams syndrome, DiGeorge syndrome, and velocardiofacial syndrome. Chromosomal microarray analysis, also known as array-based comparative genomic hybridization (CGH), has recently become an extremely valuable diagnostic tool, allowing the detection of subtle genomic imbalances undetected by conventional chromosome analysis. Lu et al. (2008) studied 101 patients with CHD with or without other malformations such as cleft palate, club foot, and polydactyly, and array-based CGH detected significant abnormalities in 21.8% of patients. Richards et al. (2008) also found that children with CHD and other anomalies, specifically neurological problems, had a higher incidence of cryptic chromosomal abnormalities detected by CGH, which were not detected by conventional karyotyping. The authors advocated screening patients with CHD and neurological abnormalities such as developmental delay with chromosomal microarray analysis.
Neurological Complications Unrelated to Intervention and Cardiac Surgery
Infective Endocarditis
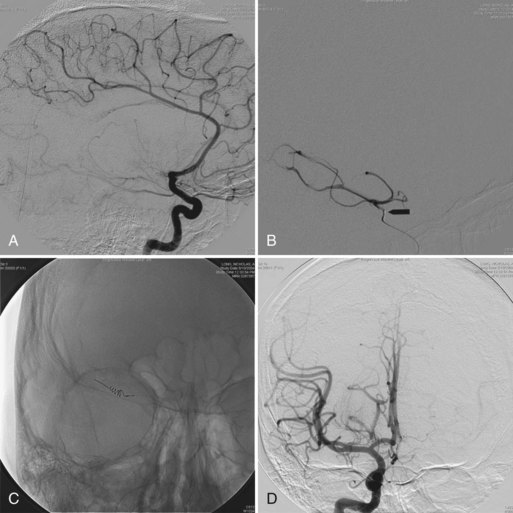
The implementation of subacute endocarditis prophylaxis before dental and surgical procedures in patients with CHD has greatly reduced the incidence of bacterial endocarditis. Approximately one-third of cases of infective endocarditis are associated with neurological complications. These include cerebral embolization, usually in the middle cerebral artery territory, and meningitis, brain abscess, and seizures (Fig. 49B.1). Cerebral mycotic aneurysms complicate 1.2% to 5% of cases of infective endocarditis and carry a high mortality rate of 60%. The risk of hemorrhagic transformation of septic infarctions is high and associated with a mortality rate of 80% to 90%.
Neurological Complications of Intervention and Cardiac Surgery
Fallon and associates (1995) reviewed data for 523 cardiac surgery patients and found neurological events or deficits in 31 patients in the immediate postoperative period. Seizures occurred in 16, pyramidal signs (hemiparesis-quadriparesis) in 11, extrapyramidal signs in 8, and neuro-ophthalmic deficits (gaze palsies, visual field defects) in 6. Six patients were unconscious, and four demonstrated miscellaneous neurological changes such as development of Horner syndrome secondary to brachial plexus injury, vocal cord palsy, isolated bulbar palsy, and transient ischemic episodes. A period of low perfusion pressure, either intraoperatively or postoperatively, was present in more patients who had an adverse neurological event than in those who were normal. The highest frequency of adverse neurological events was in the cardiac diagnostic group of arch anomaly. The likely pathogenesis of CNS injury is microembolization and ischemia during bypass or development of intracranial hemorrhage (Du Plessis et al., 1999). Corrective surgery for coarctation of the aorta is especially associated with CVAs.
A postoperative encephalopathy characterized by choreoathetosis and developmental delay is a well-defined complication after cardiac surgery in children, but not after cardiac surgery in adults. The incidence has dropped from 18% to 0.6% in recent reports (du Plessis et al., 2002). A mild transitory form can follow cardiac surgery in infants. The severe form occurs in children who undergo such surgery after infancy. In the severe postpump choreoathetosis, the early mortality rate approaches 40%. Most of the patients have residual involuntary movements and severe long-term neurological disturbances years later. The mild form is associated with cognitive and behavioral disturbances despite complete resolution of choreoathetosis. The mechanisms underlying pathogenesis remain unclear, with the usual proposed explanations being deep hypothermia and intraoperative hypoxic injury (Wessel et al., 1995). Brain imaging in these cases usually reveals nonspecific changes such as cerebral atrophy. Neuropathological data are limited; however, the external globus pallidus is the most consistent locus of injury, with evidence of gliosis, neuronal loss, nerve fiber degeneration, and capillary proliferation (Kupsky et al., 1995).
Open heart surgery is associated with several risk factors for stroke (Fig. 49B.2). The risks include altered intravascular endothelial surfaces, thrombus formation facilitated by the use of prosthetic devices, gaseous emboli originating from the cardiopulmonary bypass, global hypoperfusion, inflammatory cascades and microvascular inflammatory changes, and occurrence of prothrombotic state during surgery, owing to consumptive coagulopathy and decreased protein C and antithrombin levels.
Spinal cord injury occurs especially after aortic coarctation repair. Peripheral neuromuscular complications include plexopathies (mostly brachial), pressure palsies (peroneal and ulnar nerves), myopathy, “critical care neuropathy,” and polyneuropathy developing after withdrawal of neuromuscular blocking agents. Dittrich et al. (2003) reviewed data for 90 patients younger than 1 year of age who underwent cardiac surgery. These patients had no brain anomalies or syndromes associated with delayed mental development, but 32% had evidence of psychomotor impairment. Neurological sequelae were more frequent after palliative surgery than after corrective surgery.
Cardiac Transplantation
Perez-Miralles and associates (2005) reported neurological complications after cardiac transplantation in 13.7% of patients. Other studies, however, have reported an incidence of 50% to 70%, mostly in the perioperative period.
In the series of Cemillan and colleagues (2004), 48% of transplant recipients suffered neurological complications such as encephalopathy (16.6%), seizures (13.6%), neuromuscular disorders (10.6%), headaches (10.6%), CVA (10.1%), psychiatric problems (2.2%), and CNS infections (2.2%). Signs and symptoms of cyclosporine toxicity include tremor, seizures, and encephalopathy. Risk factors for encephalopathy were renal and hepatic failure and hemodynamic instability. Risk factors for stroke were the presence of systemic hypoperfusion, arrhythmias, coagulopathies, and hypertension.
Connective Tissue Diseases and Vasculitides
Polyarteritis Nodosa
Neurological manifestations can develop in 50% to 70% of children. Mononeuritis multiplex, a characteristic feature of the disease in adults, is much less frequent in children, whereas CNS manifestations are more frequent in children than adults. Focal neurological deficits secondary to ischemia, infarction, and hemorrhage are common. The signs and symptoms include unilateral blindness, visual field defect, seizures, headache, encephalopathy, cognitive decline, cranial neuropathies, and aseptic meningitis. In the brain, changes are mainly seen in the small meningeal arteries (Nadeau et al., 2002).
Corticosteroid therapy improves life expectancy and decreases the incidence of hypertension and renal complications. In severe cases, lack of response to steroids is an indication for use of oral or intravenous-pulse cyclophosphamide. Plasmapheresis has not improved survival. Methotrexate, azathioprine, mycophenolate mofetil, intravenous immunoglobulin (IVIG) and more recently tumor necrosis factor (TNF) inhibitors (infliximab) and anti-CD20 monoclonal antibodies (rituximab) have been used successfully in children (Gedalia et al., 2009).
Kawasaki Disease
The most common neurological manifestations consist of extreme irritability, probably caused by aseptic meningitis, headaches, and encephalopathy. Cerebral infarction, seizures, polyneuropathy, myositis, cranial neuropathies, and retinal vasculitis are rare complications. Muneuchi and associates (2006) described a single patient with a silent right cerebellar infarct and suggested the need to consider the possibility of brain lesions in all children with Kawasaki disease with or without neurological symptoms.
Juvenile Rheumatoid Arthritis
The neurological complications of the systemic form include acute encephalopathy, which can be lethal as a result of the macrophage activation syndrome (Ueno et al., 2002). The cause of this syndrome is disruption of the macrophage-lymphocyte interaction, causing uncontrolled proliferation of highly activated macrophages and T lymphocytes, with consequent sepsis-like symptoms often resulting in multiple organ failure. High-grade fever, hepatosplenomegaly, pancytopenia, consumption coagulopathy, and low erythrocyte sedimentation rate are other features. Treatment is with high-dose steroids and cyclosporine (Stabile et al., 2006).
Systemic Lupus Erythematosus
CNS involvement occurs in 30% to 60% of children with SLE during the course of their illness. Neuropsychiatric abnormalities occur in as many as 95% of patients. Patients with CNS involvement usually have a more severe clinical course. The prevalence of recurrent headaches is 71%, migraine 36%, cognitive disorders 55%, isolated seizures 47%, epilepsy 15%, acute confusional state 35%, dysesthesia or paresthesia 14%, transient ischemic attacks (TIAs) 12%, and CVA 8%. Chorea and myositis are rare (Ghosh et al., 2005). Parkinsonism has been reported in three patients (Kwong et al., 2000). Corticosteroid-related myopathy can complicate the course of the disease. Ophthalmoplegia, diplopia, sudden blindness, or ptosis can occur, and findings of papilledema, optic neuritis, retinal hemorrhage, and vasculitis (“cotton wool spots”) have been described. Possible neurological complications of SLE include ataxia, vertigo, sensorineural hearing loss, aseptic meningitis, transverse myelopathy, and peripheral neuropathy with predominantly sensory deficits. The most common clinical manifestations of neuropsychiatric lupus are depression, memory problems, emotional lability, trouble with concentration, and psychosis. Psychiatric assessment is important in the evaluation of children with SLE. Muscal et al. (2010) and others have described pediatric lupus patients presenting with seizures, altered mental status, and MRI findings suggestive of posterior reversible encephalopathy syndrome (PRES).
Vasculitis in SLE is rare and affects small arterioles and venules. Perivasculitis is more common. CVAs occur mainly in patients with hypertension or severe renal and cardiac disease (Fig. 49B.3) and have been associated with positive results on serological tests for syphilis and the presence of lupus anticoagulant (LA). Gattorno and colleagues (1995) found that 79% of pediatric patients with SLE had anticardiolipin antibodies (aCLs), and 42% had LA. These patients were at high risk for the development of deep vein thrombosis and other antiphospholipid antibody (APA)-related pathology. A statistically significant correlation also has been found between APA and neurological manifestations such as vascular events, seizures, and psychosis. Other antibody systems such as antiribosomal P antibodies, antineuronal antibodies, or lymphocytotoxic antibodies also may be associated with an increased risk for neurological involvement. The pathogenesis of neuropsychiatric lupus is likely to be multifactorial, including autoantibody production, microangiopathy, intrathecal production of proinflammatory cytokines, and premature atherosclerosis. It is possible that the autoantibodies associated with neuropsychiatric lupus may require a disrupted blood-brain barrier to exert their effect (Nishimura et al., 2008).
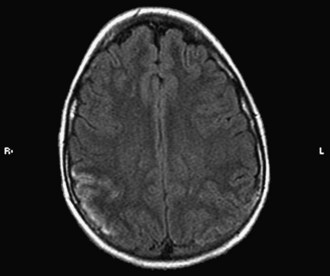
Laboratory abnormalities include high erythrocyte sedimentation rate, anemia, leukopenia, thrombocytopenia, low CH50, and low C3 and C4. ANAs are present in almost all patients. Antibodies to double-stranded DNA are pathognomonic for SLE and are present in almost all patients with active disease. Antibodies to extractable nuclear antigen (Sm, Ro/SS-A, La/SS-B, RNP) and antihistone antibodies are strongly associated with SLE. Anti-Sm is highly specific for the disease. Rheumatoid factor is present in 10% to 30% of the patients. The MRI abnormalities in neuropsychiatric lupus include small periventricular and subcortical white matter lesions, cortical atrophy, ventricular dilatation, and infarction. Data from newer imaging techniques such diffusion tensor imaging (DTI), magnetization transfer imaging (MTI), and quantitative volumetric studies are very promising in detecting CNS damage and could be used as biomarkers in clinical trials (Hughes et al., 2007).
Behçet Disease
The reported incidence of CNS involvement (“neuro-Behçet”) in children ranges from 5% to 15%. According to Saip and colleagues (2005), headache is the most common neurological symptom. Other neurological complications include encephalomyelitis, seizures, brainstem and basal ganglia involvement, aseptic meningitis, pseudotumor cerebri, and dural sinus thrombosis. Psychiatric manifestations include depression, psychosis, and dementia. A few children have suffered acute focal and generalized myositis. Vasculitis of both the arterial and venous systems, associated with thrombosis and arterial aneurysms, can occur.
Respiratory Disorders
Bronchopulmonary Dysplasia
Bronchopulmonary dysplasia (BPD) is a chronic lung disease of infancy that follows mechanical ventilation and oxygen therapy for acute respiratory distress or hyaline membrane disease in premature newborns. Multiple studies have shown that the neurological outcome of infants with BPD is closely associated with the presence of complications of prematurity such as intraventricular hemorrhage, periventricular leukomalacia, seizures, and prolonged ventilatory support, leading to the gross and fine motor delays and cerebral palsy seen in this population. The presence of severe BPD alone, however, poses an additional risk for neuromotor sequelae that include mild spasticity, microcephaly, and behavioral problems (Majnemer et al., 2000). When compared with a matched cohort of preterm peers, children with prematurity and BPD exhibited a higher frequency of subtle neurological signs such as involuntary movements, poor coordination, clumsiness, poor postural control, synkinesias, and dyspraxia and an increased need for special education.
Cystic Fibrosis
Chiari type I malformation seems to be more common in patients with CF than in the general population. Needleman and colleagues (2000) described five children and adolescents with CF and Chiari type I malformations with swallowing dysfunction, syncope, numbness, headaches, and recurrent emesis.
Sarcoidosis
The clinical presentation varies between younger (before 4 years of age) and older children (8 to 15 years) and is different from that in the adult population (Yanardag et al., 2006). Children younger than 4 years of age present mainly with a maculopapular rash, uveitis, and arthritis. In the 8- to 15-year-old age group, signs and symptoms include fever, cough, lymphadenopathy, malaise, ocular lesions, and abnormalities on the chest radiograph. In 1985, Edward Blau described families with autosomal dominant granulomatous disease similar to early-onset sarcoidosis. Recent data have suggested that Blau syndrome and early-onset sarcoidosis may represent the same disease. They both share the same genetic mutations in the NOD2 (nucleotide binding oligomerization domain 2), also known as CARD 15 (capsule recruitment domain family member 15). CNS abnormalities, known as neurosarcoidosis, occur in 30% of children, compared with 5% to 10% of affected adults. CNS complications include encephalopathy, seizures, cranial neuropathy (with nerve VII most frequently affected), optic neuritis, mass lesions, obstructive hydrocephalus, basilar granulomatous meningitis, aseptic meningitis, peripheral neuropathy, headaches, myelopathy, and pituitary-hypothalamic lesions. Children are more likely to develop seizures and space-occupying lesions and less likely to develop cranial nerve palsies (Baumann et al., 2003).
Hypertension
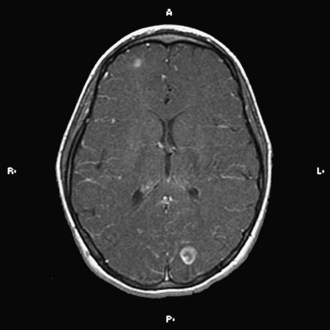
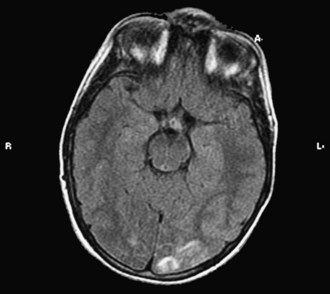
Whereas retinal arteriolar spasm is a more characteristic sign, papilledema is seen in only a third of cases. Posterior reversible leukoencephalopathy syndrome (PRES) is a syndrome in which abrupt rise in blood pressure leads to visual loss, hallucinations, headache, lethargy, transitory motor deficits, and seizures. The first description of this syndrome was in patients receiving cyclosporine. Brain imaging reveals increased T2 and fluid-attenuated inversion recovery signals, predominantly involving the posterior parietal-occipital regions, affecting both white and gray matter (Fig. 49B.4). Similar findings can be seen in other parts of the brain. It is postulated that dysfunction in cerebral autoregulation causes perivascular edema that compresses surrounding microvessels, leading to proliferative endarteritis. This process increases the vulnerability to infarction and petechial hemorrhage. Cases of PRES are reported in pediatric lupus, influenza A infections, and in association with severe infections, sepsis, and shock (Bartynski et al., 2009). Prompt management and safe reduction of blood pressure are very important in the management of hypertensive encephalopathy. Antiepileptic medications are used to treat persistent seizures, but chronic therapy usually is not required. Some reports of PRES in children with cancer have described irreversible MRI changes. In 3 of 11 patients in one series, epilepsy developed despite clinical and radiographic evidence of recovery, requiring chronic antiepileptic drug therapy (Morris et al., 2005).
Hematological Disorders
Sickle Cell Disease
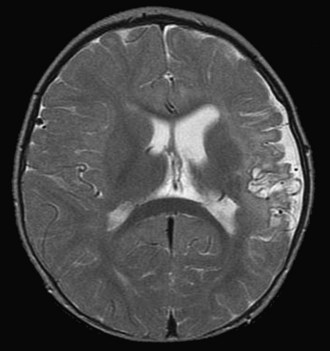
Sickle cell disease is the most common hematological disorder associated with CVAs. The incidence of stroke in sickle cell disease ranges from 7% to 33% (Fig. 49B.5). The prevalence of silent infarcts was 21.8% among 266 patients in the Cooperative Study of Sickle Cell Disease (Kinney et al., 1999). Silent infarcts occur in the distribution of small vessels. The most common underlying lesion is an intracranial arterial stenosis or obstruction, often seen in the proximal middle cerebral and anterior cerebral arteries. Sickled erythrocytes cause chronic injury to the vessel endothelium, resulting in a narrow lumen. Subarachnoid hemorrhage and intraparenchymal hemorrhage also can occur. Spontaneous acute epidural and subgaleal hematomas are rare complications of sickle cell crises (Dahdaleh et al., 2009).
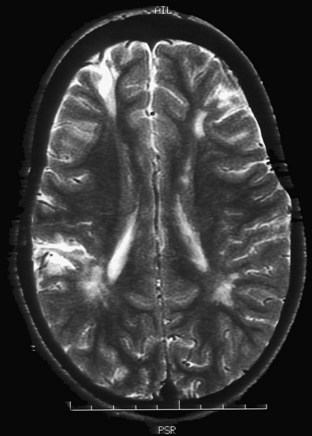
Prengler and coworkers (2005) evaluated 76 patients with sickle cell disease with transcranial Doppler studies and perfusion MRI. All patients with seizures had decreased cerebral perfusion ipsilateral to an electrocardiographic abnormality, suggesting that a complex mechanism of large- and small-vessel disease and hypoperfusion plays a role in the pathogenesis of seizures.
The Stroke Prevention Trial in Sickle Cell Anemia (STOP) reported in 1998 that long-term transfusion therapy to decrease the levels of hemoglobin S to less than 20% to 30% reduced the risk of stroke in high-risk patients (Adams et al., 1998). Silent cerebral infarction is the most frequent neurological complication in patients with sickle cell disease. It is reported in as many as 28% of children with sickle cell disease, who have suffered a clinically evident stroke and is associated with lower cognitive scores and poor academic achievement (Kwiatkowski et al., 2009). An ongoing silent cerebral infarct transfusion trial (SIT) is comparing blood transfusion versus observation, with the plan to determine new standard care practices to reduce neurological morbidity (Casella et al., 2010). Hydroxyurea has shown efficacy in preventing stroke in earlier studies. However, the pediatric hydroxyurea phase III clinical trial (BABY HUG) is facing many challenges (Thompson et al., 2010).
Children with sickle cell disease are susceptible to infections with Streptococcus pneumoniae, Haemophilus influenzae, and Mycoplasma pneumoniae, which can involve the CNS. Sinus venous thrombosis, posterior reversible leukoencephalopathy, and acute demyelination are other complications. Moyamoya disease is a relatively uncommon neurovascular complication of sickle cell disease and is a risk factor for stroke and transient ischemic episodes despite prophylactic blood transfusion. Encephaloduroarteriosynangiosis (EDAS) procedures constitute an effective treatment option for moyamoya disease, with promising results (Fryer et al., 2003).
Partial exchange transfusion is standard therapy in the setting of occlusive crisis and ischemic injury. Indications for anticoagulation are dissection and sinus venous thrombosis, especially with coexistent prothrombotic abnormalities and recurrent stroke. Bone marrow transplantation sometimes is offered. Emerging prophylactic regimens include citrulline, arginine, aspirin, and overnight oxygen supplementation (Kirkham et al., 2006).
Hemophilia
Studies show that 42% of patients with hemophilia B and 34% of patients with hemophilia A suffer neurological complications. These include intracranial hemorrhage, peripheral nerve lesions due to intramuscular hemorrhage after minor injuries, and spontaneous hematoma formation in the groin or other closed anatomical spaces. Femoral nerve involvement is the most commonly reported. Other affected nerves are the lumbosacral plexus, median nerve, radial nerve, and lateral cutaneous nerve. Hemophilic “pseudotumor” in the spinal canal and cranium have been described, as well as spinal epidural and subdural hematomas presenting as back pain and torticollis (Cuvelier et al., 2006).
Klinge and associates (1999) reported the incidence of intracranial hemorrhage in 42.5% of patients with hemophilia A or B. Bleeding occurred within 1 week of birth in 41% of cases. Trauma, the most important factor, occurred in 57% of cases either during birth (30%) or later (27%). Sixty-three percent had seizures, and 1 of the 33 patients died. Psychomotor retardation occurred in 59% and cerebral palsy in 45% of cases. Nelson and colleagues (2000) evaluated the incidence and sequelae of 2- to 5-mm focal white-matter hyperintensities on T2-weighted images in hemophiliac patients and reported no correlation with neurological factors; the investigators concluded that the findings were incidental. Prompt performance of CT is critical for early recognition of the presence of hemorrhage and institution of therapy.
Gastrointestinal Disorders
Hepatic Encephalopathy
Stage 1: Mild confusion, irritability, excessive crying, sleep disturbances, mental slowing, and short attention span
Stage 2: Excessive sleepiness, moderate confusion, personality changes, inappropriate behavior, intermittent periods of disorientation, and inability to perform mental tasks
Stage 3: Profound confusion, stupor, delirium, hyperreflexia, and an extensor plantar response sign
Stage 4: Coma with or without decerebrate or decorticate posturing (4a: response to pain present, 4b: no response to pain)
Liver Transplantation
Neurological problems occur in 48% of pediatric orthotopic liver transplant recipients. A majority involve seizures, mental status changes, or coma. Three-fourths of comatose patients had significant intracerebral hemorrhage on brain imaging. Neurological complications constitute a significant source of mortality and morbidity in these patients. Ghaus and associates (2001) reviewed data for 41 adults and pediatric patients who underwent liver transplantation. Encephalopathy occurred in 62%, with either immediate or delayed onset. Seizures (multifocal myoclonus, focal, or status epilepticus) occurred in 11% and were associated with the presence of encephalopathy. Three patients had neuropathy. Other complications were headache, tremor, fatigue, restlessness, enuresis, dizziness, critical illness myopathy, and detached retina. Erol et al. (2007) found seizures to be the most common neurological complications of liver transplantation. Brain imaging revealed atrophy, subarachnoid hemorrhage, intracerebral hemorrhage, focal cerebritis, meningitis and posterior reversible encephalopathy syndrome. Drug toxicity may occur with use of cyclosporine, tacrolimus, or corticosteroids, in addition to the development of infections (from Candida albicans, Pseudomonas aeruginosa, Staphylococcus aureus, vancomycin-resistant enterococci, Mycobacterium tuberculosis, and other opportunistic pathogens such as Aspergillus fumigatus and Listeria monocytogenes). Late posttransplantation complications include infections with CMV and Epstein-Barr virus (EBV), lymphoproliferative disease, chronic rejection, biliary strictures, and hepatic artery and vein thrombosis.
Endocrine Disorders
Thyroid Disorders
Hypothyroidism
Clinical evidence of hypothyroidism is difficult to appreciate in the newborn period. Many of the classic features such as large tongue, umbilical hernia, hoarse cry, facial puffiness, cold mottled hands and feet, hypotonia, constipation, feeding problems, somnolence, and apnea develop only with time. Prolongation of physiological jaundice may be one of the earliest signs. The anterior and posterior fontanels are large. A delay in diagnosis results in delayed subsequent linear growth, anemia, sensorineural hearing loss, cardiomegaly, and pericardial effusion. Retardation of physical and mental development becomes apparent by 3 to 6 months of age. Myxedema may involve the skin of the eyelids, backs of the hands, and genitalia. All affected children have hypotonia except for those with Kocher-Debré-Semelaigne syndrome, in which generalized muscular hypertrophy, predominating in the calf muscles, gives the patient an athletic appearance. Newborn screening programs detect these disorders in the United States and much of Western Europe, providing early detection and treatment. Despite the eradication of severe mental retardation, the intelligence quotient (IQ) of treated infants is 6 to 19 points lower than of controls. The need for special education increases fourfold in affected children. Sensorineural hearing loss, attention problems, and various neuropsychiatric problems can persist and may not be reversible with postnatal therapy (Morin et al., 2002).
Hashimoto encephalopathy manifests with acute to subacute evidence of cognitive impairment, variable psychiatric symptoms, alteration in consciousness, hallucinations, involuntary movements, seizures, myoclonus, opsoclonus, chorea, ataxia, strokelike episodes, and myelopathy. The literature contains many pediatric cases with this condition (Alink et al., 2005). Most affected patients are adolescent females. Most of the published cases presented with seizures, generalized tonic-clonic in 80% of the cases. The diagnosis requires the clinical triad of neuropsychiatric symptoms, detection of antimicrosomal or antithyroglobulin antibodies, and exclusion of other causes. The most frequently detected antibodies are antithyroid peroxidase, antithyroglobulin, and to a lesser extent, thyroid-stimulating hormone receptor–blocking antibodies. Recently, α-enolase, a novel autoantigen, was described and appears to be highly relevant for Hashimoto encephalopathy (Ochi et al., 2002) and might help in making a more precise diagnosis in the future. The proposed mechanism of pathogenesis is an autoimmune cerebral vasculitis perhaps related to immune complex deposition (Marshall and Doyle, 2006). Some patients have antithyroid antibodies in the cerebrospinal fluid and 60% show mild to moderate elevation of CSF protein levels. MRI findings are normal in most pediatric patients; however, some children show nonspecific prolongation of T2-weighted signals in the subcortical white matter. Spectroscopy studies in a few pediatric patients revealed evidence of hypoperfusion, mainly in the frontotemporal regions (Watemberg et al., 2006). The symptoms in Hashimoto encephalopathy often occur even if the patient is euthyroid. The condition does not respond to thyroxine replacement. Most children show a dramatic response to a short course of high-dose corticosteroids, with a slow taper over weeks to months. Full recovery was described in 55% of reported cases. Incomplete recovery is associated with neuropsychological difficulties, seizures, and behavioral problems. Cases with multiple recurrences or cases failing to respond to a short course of corticosteroids are placed on long-term treatment with prednisone, azathioprine, cyclophosphamide, Plaquenil, methotrexate, Cellcept, periodic IVIG, or plasma exchange, usually with good success.
Diabetes Mellitus
Nery Ferreira and colleagues (2005) evaluated 48 children with DM1 for periods ranging from 5 to 10 years. Neurological complaints such as lower extremity pain and numbness were reported by 6.3% of patients. Almost 65% exhibited abnormalities on examination consistent with neuropathy: absence of deep tendon reflexes and vibration sensation. Sixty percent showed changes in motor and sensory nerve conduction. El Bahri-Ben Mrad and colleagues (2000) reported a 10% incidence of clinical neuropathy, compared with 29% with neurophysiological evidence, predominantly in the legs.
A longitudinal epidemiological study of the evolution of diabetic microvascular disease and autonomic function revealed a substantial prevalence of microvascular and neurological abnormalities (Karavanaki and Baum, 1999). Reduced pupillary adaptation in darkness was noted in 7.9% of patients and impaired vibration sensation in the lower extremities in 6.2%. Low sensory nerve conduction and autonomic dysfunction were findings in 25% of newly diagnosed diabetic children, whose disease was not yet under good control (Solders et al., 1997). Turgut and associates (2004) suggested that monitoring of the dorsal sural sensory nerve action potential (SNAP) is a sensitive method for detection of peripheral neuropathy in early stages of diabetes in children.
Nordwall and coworkers (2006) found evidence of subclinical neuropathy in 59% of children and adolescents with duration of diabetes longer than 3 years, despite intensive insulin treatment and good control.
Diabetic ketoacidosis is the most serious complication of diabetes and the most common cause of death. Cerebral edema develops in 1% of the cases and occurs rarely in diabetics older than 20 years. The attributed etiology of cerebral edema is retention of intracellular osmolytes in the brain during hydration, causing a shift of water into the intracellular space. Signs of cerebral edema include agitation, confusion, lethargy, headache, emesis, and incontinence (Muir et al., 2004).
Rare cases of extrapontine and central pontine myelinosis have been described after treatment of diabetic ketoacidoses and were attributed to rapid correction of hyperosmolality (Sivaswamy et al., 2007). Hypoglycemia, a known complication of diabetic treatment, usually occurs at night or during early morning hours. Shehadeh and coworkers (1998) reported that 32% of pediatric diabetic patients experienced at least one severe hypoglycemic episode. Variable neurological signs and symptoms such as confusion, tremor, seizures, behavioral changes, blurred vision, transitory hemiparesis, and coma occurred in all of the cases. One patient had permanent hemiparesis.
Renal Disorders
Complications of Dialysis
Headaches
Headaches occur in up to 70% of patients on dialysis (Antoniazzi et al., 2003b). Approximately half of such patients have evidence of a migrainous disorder. The International Headache Society has defined criteria for the headaches related to hemodialysis. The headache must begin during hemodialysis and terminate within 24 hours (Antoniazzi et al., 2003a).
Renal Transplantation
Recent advances in surgical techniques and immunosuppression have greatly enhanced the outcome in renal transplantation and graft survival. Mir et al. (2004) analyzed data for 72 pediatric renal transplant recipients and reported hypertension in 31.9%, acute rejection in 27.8%, chronic rejection in 13.9%, and CMV infections in 20.8% of patients.
Qvist and colleagues (2002) studied neurodevelopmental outcome in 33 school-aged children who underwent transplantation before the age of 5 years; their mean IQ was 87. Neuropsychiatric testing was impaired in 6% to 24%. The children with learning problems had a greater number of hypertensive crises and seizures and a higher incidence of prematurity. Six of seven children attending a special school had evidence of brain infarcts on MRI. No significant difference existed in mean serum aluminum levels between these patients and children who had received transplants and who demonstrated normal school performance.
Adams R.J., McKie V.C., Hsu L., et al. Prevention of a first stroke by transfusions in children with sickle cell anemia and abnormal results on transcranial Doppler ultrasonography. N Engl J Med. 1998;339:5-11.
Alink J., de Vries T.W. Unexplained seizures, confusion or hallucinations: think Hashimoto encephalopathy. Acta Paediatrica. 2008;97:451-453.
Antoniazzi A.L., Bigal M.E., Bordini C.A., et al. Headache associated with dialysis: the International Headache Society criteria revisited. Cephalgia. 2003;23:146-149.
Antoniazzi A.L., Bigal M.E., Bordini C.A., et al. Headache and hemodialysis: a prospective study. Headache. 2003;43:99-102.
Bartynski W.S., Upadhyaya A.R., Boardman J.F. Posterior reversible encephalopathy syndrome and cerebral vasculopathy associated with influenza A infection: report of a case and review of the literature. J Comput Assist Tomogr. 2009;33(6):917-922.
Baumann R.J., Robertson W.C.Jr. Neurosarcoid presents differently in children than in adults. Pediatrics. 2003;112:480-486.
Casella J.F., King A.A., Barton B., et al. Design of the silent cerebral infarct transfusion (SIT) trial. Pediatr Hematol Oncol. 2010;27:69-89.
Cemillan C.A., Alonso-Pulpon L., Burgos-Lazaro R., et al. Neurological complications in a series of 205 orthotopic heart transplant patients. Rev Neurol. 2004;38:906-912.
Cuvelier G.D., Davis J.H., Purves E.C., et al. Torticollis as a sign of cervico-thoracic epidural haematoma in an infant with severe haemophilia A. Haemophilia. 2006;12:683-686.
Dahdaleh N.S., Lindley T.E., Kirby P.A., et al. A “neurosurgical crisis” of sickle cell disease. J Neurosurg Pediatr. 2009;4:532-535.
Dittrich H., Buhrer C., Grimmer I., et al. Neurodevelopment at 1 year of age in infants with congenital heart disease. Heart. 2003;89:365-366.
Du Plessis A.J. Mechanisms of brain injury during infant cardiac surgery. Semin Pediatr Neurol. 1999;6:32-47.
Du Plessis A.J., Bellinger D.C., Gauvreau K., et al. Neurologic outcome of choreoathetoid encephalopathy after cardiac surgery. Pediatr Neurol. 2002;27:9-17.
El Bahri-Ben Mrad F., Gouider R., Fredj M., et al. Childhood diabetic neuropathy: a clinical and electrophysiological study. Funct Neurol. 2000;15:35-40.
Erol I., Alehan F., Ozcay F., et al. Neurological complications of liver transplantation in pediatric patients: A single center experience. Pediatr Transplantation. 2007;11:152-159.
Fallon P., Aparicio J.M., Elliott M.J., et al. Incidence of neurological complications of surgery for congenital heart disease. Arch Dis Child. 1995;72:418-422.
Fryer R.H., Anderson R.C., Chiriboga C.A., et al. Sickle cell anemia with moyamoya disease: outcomes after EDAS procedure. Pediatr Neurol. 2003;29:124-130.
Gattorno M., Buoncompagni A., Molinari A.C., et al. Antiphospholipid antibodies in paediatric systemic lupus erythematosus, juvenile chronic arthritis and overlap syndrome: SLE patients with both lupus anticoagulant and high-titre anticardiolipin antibodies are at risk for clinical manifestations related to the antiphospholipid syndrome. Br J Rheumatol. 1995;34:873-881.
Gedalia A., Cuchacovich R. Systemic vasculitis in childhood. Curr Rheumatol Rep. 2009;11:402-409.
Ghaus N., Bohlega S., Rezeig M. Neurological complications in liver transplantation. J Neurol. 2001;248:1042-1048.
Ghosh J.B., Gupta D. Chorea and microphonia as presenting feature of SLE. Indian J Pediatr. 2005;72:85.
Hughes M., Sundgren P.C., Fan X., et al. Diffusion tensor imaging in patients with acute onset of neuropsychiatric lupus erythematosus: a prospective study of apparent diffusion coefficient, fractional anisotropy values, and eigen values in different regions of the brain. Acta Radiol. 2007;48:213-222.
Karavanaki K., Baum J.D. Prevalence of microvascular and neurologic abnormalities in a population of diabetic children. J Pediatr Endocrinol Metab. 1999;12:411-422.
Kinney T.R., Sleeper L.A., Wang W.C., et al. Silent cerebral infarcts in sickle cell anemia: a risk factor analysis. The cooperative study of sickle cell disease. Pediatrics. 1999;103:640-645.
Kirkham F.J., Lerner N.B., Noetzel M., et al. Trials in sickle cell disease. Pediatr Neurol. 2006;34:450-458.
Klinge J., Auberger K., Auerswald G., et al. Prevalence and outcome of intracranial haemorrhage in haemophiliacs—a survey of the Paediatric Group of the German Society of Thrombosis and Haemostasis (GTH). Eur J Pediatr. 1999;158(Suppl. 3):162-165.
Kupsky W.J., Drozd M.A., Barlow C.F. Selective injury of the globus pallidus in children with post-cardiac surgery choreic, syndrome. Dev Med Child Neurol. 1995;37:135-144.
Kwiatkowski J.L., Zimmerman R.A., Pollock A.N., et al. Silent infarcts in young children with sickle cell disease. Br J Haematol. 2009;146:300-305.
Kwong K.L., Chu R., Wong S.N. Parkinsonism as unusual neurological complication in childhood systemic lupus erythematous. Lupus. 2000;9:474-477.
Lu X.Y., Phung M.T., Shaw C.A., et al. Genomic imbalances in neonates with birth defects: high detection rates by using chromosomal microarray analysis. Pediatrics. 2008;122:1310-1318.
Lutterman J., Scott M., Nass R., et al. Moyamoya syndrome associated with congenital heart disease. Pediatrics. 1998;101:57-60.
Majnemer A., Riley P., Shevell M., et al. Severe bronchopulmonary dysplasia increases risk for later neurological and motor sequelae in preterm survivors. Dev Med Child Neurol. 2000;42:53-60.
Marshall G.A., Doyle J.J. Long-term treatment of Hashimoto’s encephalopathy. J Neuropsychiatry Clin Neurosci. 2006;18:14-20.
Mir S., Erdogan H., Serdaroglu E., et al. Pediatric renal transplantation: single center experience. Pediatr Transplant. 2005;9:56-61.
Morin A., Guimarey L., Apezteguia M., et al. Linear growth in children with congenital hypothyroidism detected by neonatal screening and treated early: a longitudinal study. J Pediatr Endocrinol Metab. 2002;15:973-977.
Morris E.B., Laningham F.H., Sandlund J.T., et al. Posterior reversible encephalopathy syndrome in children with cancer. Pediatr Blood Cancer. 2005;48:152-159.
Muir A.B., Quisling R.G., Yang M.C., et al. Cerebral edema in childhood diabetic ketoacidosis: natural history, radiographic findings, and early identification. Diabetes Care. 2004;27:1541-1546.
Muneuchi J., Kusuhara K., Kanaya Y., et al. Magnetic resonance studies of brain lesions in patients with Kawasaki disease. Brain Dev. 2006;28:30-33.
Muscal E., Traipe E., de Guzman M.M., et al. MR imaging findings suggestive of posterior reversible encephalopathy syndrome in adolescents with systemic lupus erythematosus. Pediatr Radiol. Jan 30, 2010.
Nadeau S.E. Neurologic manifestations of systemic vasculitis. Neurol Clin. 2002;20:123-150.
Needleman J.P., Panitch H.B., Bierbrauer K.S., et al. Chiari type I malformation in children and adolescents with cystic fibrosis. Pediatr Pulmonol. 2000;30:490-492.
Nelson M.D.Jr, Wilson D.A., Kisker C.T., et al. Incidence of focal white matter lesions in a population of hemophilic children and their normal siblings. Hemophilia growth and development study. Pediatr Radiol. 2000;30:705-709.
Nery Ferreira B.E., Silva I.N., de Oliveira J.T. High prevalence of diabetic polyneuropathy in a group of Brazilian children with type 1 diabetes mellitus. J Pediatr Endocrinol Metab. 2005;18:1087-1094.
Nishimura K., Harigai M., Omori M., et al. Blood-brain barrier damage as a risk factor for corticosteroid-induced psychiatric disorders in systemic lupus erythematosus. Psychoneuroendocrinology. 2008;33:395-403.
Nordwall M., Hyllienmark L., Ludvigsson J. Early diabetic complications in a population of young patients with type 1 diabetes mellitus despite intensive treatment. J Pediatr Endocrinol Metab. 2006;19:45-54.
Ochi H., Horiuchi I., Arali N. Proteomic analysis of human brain identifies α-enolase as a novel autoantigen in Hashimoto’s encephalopathy. FEBS Lett. 2002;528:197-202.
Perez-Miralles F., Sanchez-Manso J.C., Almenar-Bonet L., et al. Incidence of and risk factors for neurologic complications after heart transplantation. Transplant Proc. 2005;37:4067-4070.
Prengler M., Pavlakis S.G., Boyd S., et al. Sickle cell disease: ischemia and seizures. Ann Neurol. 2005;58:290-302.
Qvist E., Pihko H., Fagerudd P., et al. Neurodevelopmental outcome in high-risk patients after renal transplantation in early childhood. Pediatr Transplant. 2002;6:53-62.
Richards A.A., Santos L.J., Nichols H.A., et al. Cryptic chromosomal abnormalities identified in children with congenital heart disease. Pediatric Res. 2008;64:358-363.
Saip S., Siva A., Altintas A., et al. Headache in Behçet’s syndrome. Headache. 2005;45:911-919.
Shehadeh N., Kassem J., Tchaban I., et al. High incidence of hypoglycemic episodes with neurologic manifestations in children with insulin dependent diabetes mellitus. J Pediatr Endocrinol Metab. 1998;11(Suppl. 1)):183-187.
Sivaswamy L., Karia S. Extrapontine myelinolysis in a 4 year old with diabetic ketoacidosis. Eur J Paediatr Neurol. 2007;11:389-393.
Solders G., Thalme B., Aguirre-Aquino M., et al. Nerve conduction and autonomic nerve function in diabetic children. A 10-year follow-up study. Acta Paediatr. 1997;86:361-366.
Stabile A., Bertoni B., Ansuini V., et al. The clinical spectrum and treatment options of macrophage activation syndrome in the pediatric age. Eur Rev Med Pharmacol Sci. 2006;10:53-59.
Thompson B.W., Miller S.T., Rogers Z.R., et al. The pediatric hydroxyurea phase III clinical trial (BABY HUG): challenges of study design. Pediatr Blood Cancer. 2010;54:250-255.
Turgut N., Karasalihoglu S., Kucukugurluoglu Y., et al. Clinical utility of dorsal sural nerve conduction studies in healthy and diabetic children. Clin Neurophysiol. 2004;115:1452-1456.
Ueno H., Katamura K., Hattori H., et al. Acute lethal encephalopathy in systemic juvenile rheumatoid arthritis. Pediatr Neurol. 2002;26:315-317.
Watemberg N., Greenstein D., Levine A. Encephalopathy associated with Hashimoto thyroiditis: pediatric perspective. J Child Neurol. 2006;21:1-5.
Wessel D., du Plessis A.J. Choreoathetosis. In: Volpe J, editor. Brain Injury and Pediatric Cardiac Surgery. Boston: Butterworth-Heinemann; 1995:353.
Yanardag H., Pamuk O.N., Uygun S., et al. Sarcoidosis: child vs adult. Indian J Pediatr. 2006;73:143-145.