[level-membership-for-neurology-category]
Chapter 41 Neuroimmunology
Immune System
The normal functions of the immune system and the disorders resulting from its dysfunction are listed in Box 41.1.
Adaptive and Innate Immunity
The innate immune system consists of the following components:
1. Skin—The exterior surface of the body, primarily the skin, is the body’s primary defense against foreign pathogens. Many inflammatory cells and antigen-presenting cells (APCs) line the epidermis and serve as the first line of defense.
2. Phagocytes are cells capable of phagocytosing foreign pathogens. They include polymorphonuclear cells, monocytes, and macrophages. These cells are present in the blood as well as in organs. Phagocytes recognize cell components or pathogen-associated molecular patterns (PAMPs) of a variety of microorganisms through families of pattern recognition receptors (PRRs) expressed on their cell surface. PRRs allow phagocytes to attach nonspecifically and phagocytose pathogens, which are then killed via intracellular lysosomes. Families of PRRs include the Toll-like receptors (TLRs) and the nucleotide-binding oligomerization domain (NOD) receptors.
3. Natural killer (NK) cells—NK cells recognize cell surface molecules on virally infected or tumor cells. They subsequently bind to the infected cells and kill them via cell-mediated cytotoxicity.
4. Acute-phase proteins—C-reactive protein is a model acute-phase protein whose concentration increases in response to infection. C-reactive protein binds to cell surface molecules on a variety of bacteria and fungi and acts as an opsonin, essentially increasing recognition of pathogens by phagocytic cells.
5. Complement system—The complement system is a cascade of serum proteins whose overall function is to enhance and mediate inflammation. The complement system has the intrinsic ability to lyse the cell membranes of many cells including bacteria. It functions in concert with components of both the innate and adaptive immune systems and can also act as an opsonin, facilitating phagocytosis. The complement cascade can be directly activated by certain microorganisms through the alternative pathway, or it can be activated by particular antibody subtypes through the classical pathway.
The adaptive immune response consists of the following components:
1. Antibodies—Otherwise known as immunoglobulins (Igs), antibodies are able to specifically recognize a variety of free antigens. Igs are produced by B cells and are present on their cell surface. In addition, Igs are secreted in large amounts in the serum. Antibodies recognize specific microbial and other antigens through their antigen-binding sites and bind phagocytes via their Fc receptors, thereby facilitating antigen removal. Some subclasses of Ig are capable of activating complement via their Fc portion, thereby lysing their targets.
2. B cells—The primary function of B cells is to produce antibody. Antigen binding to B cells stimulates proliferation and maturation of that particular B cell, with subsequent enhancement of antigen-specific antibody production, resulting in the development of antibody-secreting plasma cells. Most B cells express class II major histocompatibility complex (MHC) antigens and have the ability to function as APCs.
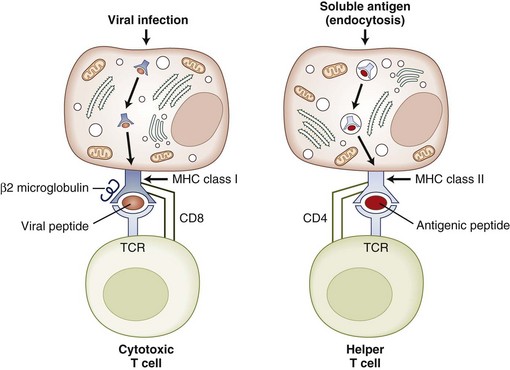
3. T cells, or thymus-derived cells, have the ability to recognize specific antigens via their T-cell receptors (TCRs). T cells may be classified into two main groups, T-helper (TH) cells expressing CD4 antigen on their cell surface and T-cytotoxic (TC) cells expressing CD8 on their surface. CD4 T cells recognize antigen presented in association with MHC class II on the surface of APCs. CD4 T cells help to promote B-cell maturation and antibody production and produce factors called cytokines to enhance the innate or nonspecific immune response. CD8 T cells recognize antigen in association with MHC class I antigen on the surface of most cells and play an important role in eliminating virus-infected cells. Cytotoxic T cells are capable of damaging target cells via the release of degrading enzymes and cytokines. Responses in which the T cell plays a major role are termed cell-mediated immunity (CMI). T cell–macrophage interactions often lead to delayed reactions, termed delayed-type hypersensitivity (DTH).
4. APCs are required to present antigen to T cells. They are found primarily in the skin, lymph nodes, spleen, and thymus. Unlike B cells that can recognize free antigen, T cells are only capable of recognizing antigen in the context of self-MHC molecules. APCs process antigen intracellularly and present antigen peptide in the groove of their MHC class II molecules. The primary APCs are macrophages, monocytes, dendritic cells, and Langerhans cells.
Principal Components of the Immune System
T-Cell Receptors
The TCR consists of two glycosylated polypeptide chains, alpha (α) and beta (β), of 45,000 and 40,000 dalton molecular weight, respectively. This heterodimer of an α and β chain is linked by disulfide bonds. Amino acid sequences show that each chain consists of variable (V), joining (J), and constant (C) regions closely resembling Igs (Fig. 41.1). There are about 102 TCR-variable genes grouped by homology into a small number of families, compared with 103 or greater for Igs (see later discussion). The principles governing generation of diversity in the TCR are very similar to those for Ig genes. T cells can only recognize short peptides that are associated with MHC molecules. In contrast, the Ig receptor can recognize peptides, whole proteins, nucleic acids, lipids, and small chemicals.
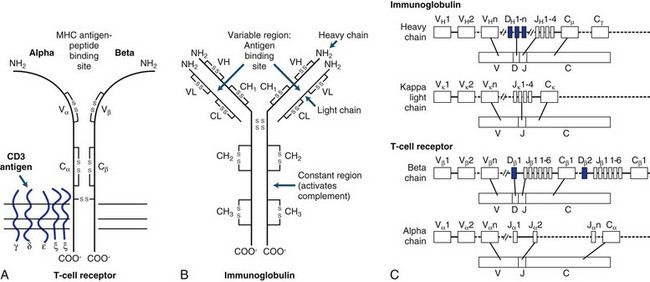
Immunoglobulins
Immunoglobulins are glycoproteins that are the secretory product of plasma cells. Their biochemical structure and genomic organization is shown in Fig. 41.1. All Ig molecules share a number of common features. Each molecule consists of two identical polypeptide light chains (kappa [κ] or lambda [λ]) linked to two identical heavy chains. The light and heavy chains are stabilized by intrachain and interchain disulfide bonds. According to the biochemical nature of the heavy chain, Igs are divided into five main classes: IgM, IgD, IgG, IgA, and IgE. These may be further divided into subclasses depending on differences in the heavy chain.
Genetics of the Immune System
Antigen Receptor Gene Rearrangements
Constant (C) gene segments are present in all receptors. The V, D, J, and C gene segments along with the intervening noncoding gene segments between the J and C regions are initially transcribed into mature RNA. Through a process of RNA splicing, the noncoding gene segments are excised, and the V(D)JC messenger RNA (mRNA) is translated into protein. After binding antigen, B cells undergo somatic mutations that further increase the diversity and the affinity of antigen binding (affinity maturation). This phenomenon does not occur in T cells. During isotype switching in B cells, further rearrangements lead to recombination of the same variable region gene with new constant region genes (see Fig. 41.1).
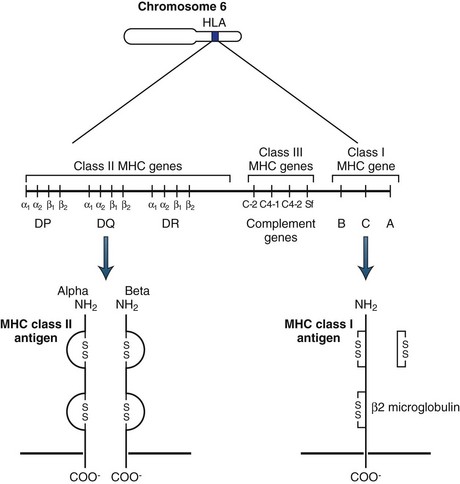
Major Histocompatibility and Human Leukocyte Antigens
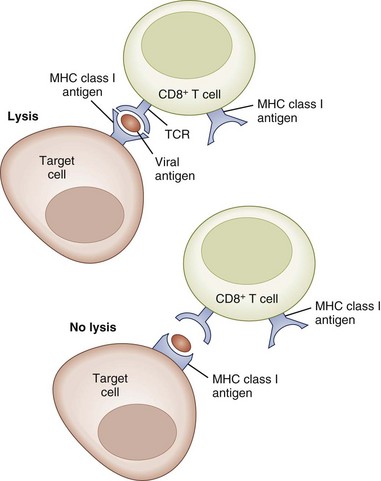
Class I antigens regulate the specificity of cytotoxic CD8+ T cells, which are responsible for killing cells bearing viral antigens or foreign transplantation antigens (Fig. 41.2). The target cells share class I MHC genes with the cytotoxic cell. Thus the cytotoxic cell that is specific for a particular virus is capable of recognizing the antigenic determinants of the virus only in association with a particular MHC class I gene product. The function of class II MHC gene products appears to be to regulate the specificity of T-helper cells, which in turn regulate DTH and antibody response to foreign antigens. Similarly, an immunized T-cell population will recognize a foreign antigen only if it is presented on the surface of an APC that shares the same class II MHC antigen specificity as the immunized T-cell population. Thus the functional specificity of the T-cell population is restricted by the MHC molecules they recognize. CD8+ T cells (cytotoxic) and CD4+ T cells (helper) are referred to as MHC class I and MHC class II restricted T cells, respectively (Fig. 41.3).
The analysis of the three-dimensional structure of the class I and class II molecules has confirmed the notion that these molecules are carriers of immunogenic peptides that are processed by APCs and presented on the cell surface (Fig. 41.4). Both MHC class I and class II molecules share similarities in crystal structure that allow them to accept and retain immunogenic peptides in grooves, or pockets, and present them to T cells.
Organization of the Immune Response
Initiation of the Immune Response
Accessory Molecules for T-Cell Activation
Costimulatory Molecules
Costimulatory molecules serve as a “second signal” to facilitate T-cell activation. Costimulatory pathways that are critical for T-cell activation include the B7-CD28 and CD40-CD154 pathways. Members of the integrin families including vascular cell adhesion molecule 1 (VCAM-1), intercellular adhesion molecule (ICAM-1), and leukocyte function antigen 3 (LFA-3) can provide costimulatory signals, but they also play critical roles in T-cell adhesion, facilitate interaction with the APCs, mediate adhesion to nonhematopoietic cells such as endothelial cells, and guide cell traffic (Fig. 41.5).
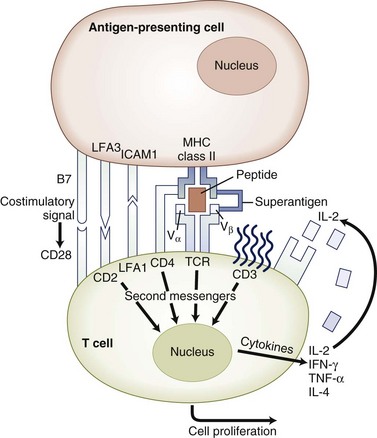
The B7-CD28 interaction is one of the most extensively studied costimulatory systems. The B7 molecules are expressed on antigen-presenting cells, and their expression is induced in activated cells. There are two forms of B7, B7-1 (CD80) and B7-2 (CD86), that share some homology but have different expression kinetics. The B7 molecules interact with their ligand, CD28, which is constitutively expressed on most T cells. Binding of the CD28 molecule mediates intracytoplasmic signals that increase expression of the growth factor, IL-2, and enhance expression of the anti-apoptotic molecule, Bcl-xL. An alternate ligand for B7 is CTLA-4, which is homologous to CD28 in structure, but in contrast to CD28, CTLA-4 functions to inhibit T-cell activation. Costimulatory molecules may deliver either a stimulatory (positive) or inhibitory (negative) signal for T-cell activation (Brunet et al., 1987). Examples of molecules delivering a positive costimulatory signal for T-cell activation include the B7-CD28, CD40-CD154 pathways. Examples of molecular pathways delivering a negative signal for T-cell activation include B7-CTLA4 and PD1-PD ligand (Khoury and Sayegh, 2004). The delicate balance between positive and negative regulatory signals can determine the outcome of a specific immune response.
Regulation of the Immune Response
Cytokines
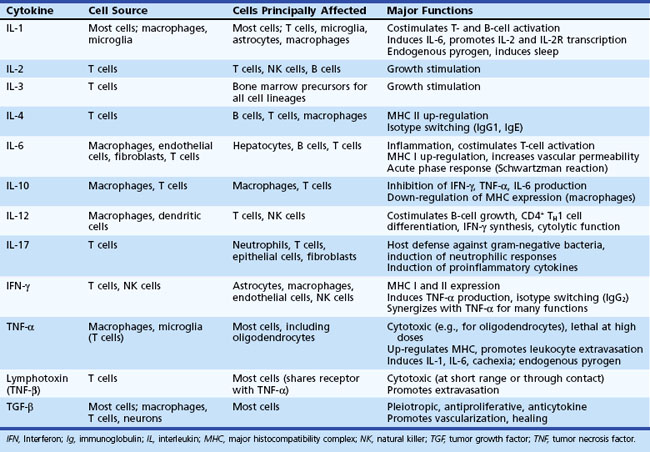
Cytokines are necessary for T-cell activation and for the amplification and modulation of the immune response. A limited representation of the cytokines that participate in the immune response is shown in Table 41.1. Secretion of IL-1 by macrophages results in stimulation of T cells. This leads to synthesis of IL-2 and IL-2 receptors and finally to the clonal expansion of T cells. Only activated T cells express the IL-2 receptor (CD25); therefore the cytokine-induced expansion favors antigen-activated cells only. T-cell activation causes secretion of interferon gamma (IFN-γ), which induces expression of MHC class I and class II molecules on many cell types including APCs. This in turn increases the T-cell response to the antigen. Secretion of IL-2 also results in activation of NK cells that mediate lysis of tumor cell targets. In addition, IL-3 is released, resulting in stimulation of hematopoietic stem cells. The signal for differentiation of B cells to form antibody-secreting cells involves clonal expansion and differentiation of virgin memory B cells. IL-4 and B-cell differentiation factors secreted by T cells induce differentiation and expansion of committed B cells to become plasma cells.
Table 41.1 An Abridged List of Cytokines Involved in Interactions Between the Immune and Nervous Systems
CD4+ T-helper cells differentiate into TH1 or TH2 phenotypes, as well as a recently described TH17 subset, which secrete characteristic cytokines and stimulate specific functions. TH1 cells secrete IFN-γ, IL-2, and TNF-α. These cytokines exert proinflammatory functions and, in TH1-mediated diseases such as MS, promote tissue injury. IL-2, TNF-α, and IFN-γ mediate activation of macrophages and induce DTH. TH1 cell differentiation is driven by IL-12, a cytokine produced by monocytes and macrophages. In contrast, the TH2 cytokines IL-4, IL-5, IL-6, IL-10, and IL-13 promote antibody production by B cells, enhance eosinophil functions, and generally suppress cell-mediated immunity (CMI). TH3 cells secrete TGF-β, which inhibits proliferation of T cells and inhibits activation of macrophages. Cytokines of the TH1 type may inhibit production of TH2 cytokines and vice versa. More recently, a subset of T cells that predominantly produce IL-17 have been described (Yao et al., 1995). These cells are believed to represent a distinct subset from IFN-γ-producing TH1 cells, evidenced by the dependence of THIL-17 cells on IL-6 and TGF-β for differentiation (Bettelli et al., 2006; Mangan et al., 2006; Veldhoen et al., 2006) and IL-23 for expansion (Aggarwal et al., 2003; Langrish et al., 2005), as opposed to TH1 cells, which are dependent on IL-12 and IL-2, respectively, for differentiation and expansion. Both TH1 and TH2 cytokines have been shown to suppress the development of TH17 cells (Harrington et al., 2005; Park et al., 2005). THIL-17 cells facilitate the recruitment of neutrophils and participate in the response to gram-negative organisms. These cells may also play a role in the initiation of autoimmune disease. Another effector T-cell subset, TH9 cells, has recently been described (Dardalhon et al., 2008; Veldhoen et al., 2008). Driven by the combined effects of TGF-β and IL-4, TH9 cells produce large amounts of IL-9 and IL-10. It has been shown that IL-9 combined with TGF-β can contribute to TH17 cell differentiation, and TH17 cells themselves can produce IL-9 (Elyaman et al., 2009).
Traditionally, TH cell subsets have been distinguished by their patterns of cytokine production, but identification of distinguishing surface molecule markers has been a major advance in the field. Tim (T cell, immunoglobulin and mucin-domain containing molecules) represent an important family of molecules that encode cell-surface receptors involved in the regulation of TH1 and TH2 cell–mediated immunity. Tim-3 is specifically expressed on TH1 cells and negatively regulates TH1 responses through interaction with the Tim-3 ligand, galactin-9, also expressed on CD4+ T cells (Monney et al., 2002; Sabatos et al., 2003; Zhu et al., 2005). Tim-2 is expressed on TH2 cells (Chakravarti et al., 2005), and appears to negatively regulate TH2 cell proliferation, although this has not been fully established. Tim-1 is expressed on TH2 cells > TH1 cells, and interacts with Tim-4 on APCs to induce T-cell proliferation (Meyers et al., 2005).
Self-Tolerance
Peripheral Tolerance
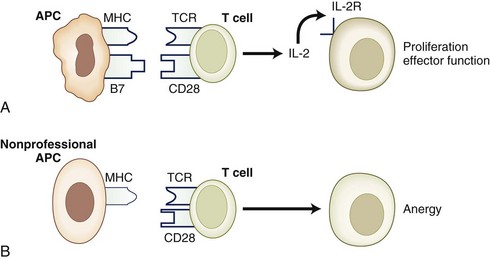
Anergy Due to Failure of T-cell Activation
In normal circumstances, an APC presents antigen as a peptide + MHC complex (signal one). In the absence of signal one, the T cell dies because of neglect. If signal one is presented in the absence of costimulatory signals (signal two), the T cell becomes anergic. An example of this situation occurs when an antigen is presented by nonprofessional APCs that lack the appropriate costimulatory molecules (Fig. 41.6). However, when a T cell is activated, it up-regulates the expression of an alternate costimulatory molecule, CTLA-4. CTLA4 engagement by CD80 and CD86 on the surface of APCs sends a negative signal to the T cell, inhibiting cell growth and proliferation. Animals deficient for CTLA-4 expression on their T lymphocytes have an uncontrolled lymphoproliferative phenotype with autoreactivity (Waterhouse et al., 1995).
Apoptosis
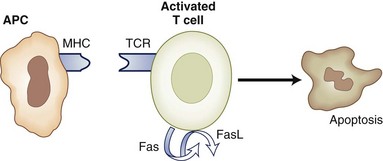
Repeated stimulation with an antigen may also induce apoptosis via the Fas/FasL pathway, a process termed activation-induced cell death (AICD). Therefore an autoreactive T lymphocyte may encounter large doses of self-antigen in the periphery and consequently may be deleted by AICD. Mice lacking Fas or FasL develop a lupus-like syndrome (Zhou et al., 1996), and mutations in the Fas gene were associated with an autoimmune disease with lymphoproliferation in humans (Drappa et al., 1996).
IL-2 is the prototypical growth factor, inducing clonal expansion of antigen-stimulated lymphocytes; paradoxically, disruption of the IL-2 gene leads to accumulation of activated lymphocytes and autoimmune syndromes (Sadlack et al., 1993). This is because IL-2 induces the transcription and surface expression of Fas ligand (FasL). Interactions of Fas with FasL lead to cell death (Fig. 41.7). Therefore IL-2 plays a dual role in T-cell regulation, reflecting a possible role for cytokine concentration and timing of exposure. Other cytokines that mediate apoptosis and cell death are TNF-α and IFN-γ. Complete absence of either of these cytokines results in deficient T-cell apoptosis, inability to terminate the immune response, and uncontrolled autoimmune disease.
Regulatory T Cells
Regulatory T cells (Treg) function to down-regulate CD4 and CD8 T-cell responses. Regulatory T cells can be of the CD4+ or CD8+ subtypes. Regulatory T cells have been found in several animal models of disease, and some are antigen specific. In vitro they can be generated under similar conditions used to generate anergic cells, and it has been postulated that they are the same entity (Lombardi et al., 1994). Several populations of regulatory or suppressor T cells have been described in humans. These include CD4+CD25+Foxp3 regulatory T cells (Baecher-Allan et al., 2001; Dieckmann et al., 2001; Levings et al., 2001; Stephens et al., 2001; Yagi et al., 2004), CD8+CD28− T cells (Koide and Engleman, 1990), IL-10-producing TH2 cells (Bacchetta et al., 1994), and TGF-β producing TH3 cells (Kitani et al., 2000; Roncarolo and Levings, 2000). In humans, there is little evidence for antigen-specific suppressor cell responses. Regulatory T cells suppress T-cell proliferation through a variety of mechanisms, including the production of immunosuppressive cytokines (TH2 or TGF-β) or through T-T cell interactions, including the expression of inhibitory molecules such as CTLA-4. Regulatory cells play an important role in the control of the immune response in autoimmune disorders, and the function of regulatory T cells may be enhanced by immunomodulatory therapies.
Immune System and Central Nervous System
Immune Privilege in the Central Nervous System
Important factors relevant to immunological responses in the CNS are: (1) absence of lymphatic drainage, limiting the immunological circulation; (2) the blood-brain barrier (BBB), which limits the passage of immune cells and factors; (3) the low level of expression of MHC factors, particularly MHC II in the resident cells of the CNS; (4) low levels of potent APCs, such as dendritic or Langerhans cells; and (5) the presence of immunosuppressive factors such as TGF-β (Wilbanks and Streilein, 1992) and CD200 (Webb and Barclay, 1984).
The CNS houses cells that are capable of antigen presentation under certain conditions in vitro, but to what extent this occurs in vivo remains under debate. In the CNS, endogenous expression of MHC class I and class II on APCs such as microglia is low, and in oligodendrocytes and astrocytes, it is almost undetectable. Neurons express MHC class I only when damaged and in the presence of IFN-γ (Neumann et al., 1995). Expression of MHC antigens on both microglia and astrocytes are enhanced by the presence of cytokines, TNF-α, and IFN-γ. Under certain conditions, microglial cells may play a role as APCs in the nervous system (Perry, 1994). More recently, populations of perivascular dendritic cells capable of antigen presentation have been identified in rodents (Greter et al., 2005), with analogous populations demonstrated in humans; however, their role in human disease is unclear.
Immune privilege in the CNS is also influenced by the constitutive expression of a number of immunoregulatory factors, some of which are common to immune privilege in the anterior chamber of the eye. Anterior chamber immune privilege is due in part to expression of TGF-β in the aqueous of the eye. In the CNS, TGF-β is produced by astrocytes and microglia and may play a role in down-regulating immune responses locally. Neurons are also capable of producing TGF-β, which in animal models has been shown to facilitate the differentiation of regulatory T cells (Liu et al., 2006). Increased expression of Fas ligand in the CNS compared with the peripheral nervous system (PNS) may increase apoptosis of T cells, thereby down-regulating the immune response (Moalem et al., 1999). Some CNS tumors express large amounts of TGF-β, which may play a role in protecting them from immune surveillance. CNS tumors may also express Fas or Fas ligand, facilitating protection from immune surveillance. Some populations of neurons express a cell surface marker named CD200. CD200 is a nonsignaling molecule but serves to inhibit activation of cells including microglia and macrophages that express the CD200 receptor (CD200R) (Hoek et al., 2000; Wright et al., 2000). CD200 has been shown to down-regulate inflammatory responses in models of MS (Liu et al., 2010) and uveitis (Banerjee and Dick, 2004; Broderick et al., 2002). Fractalkine (CXCL1) is a chemokine that is constitutively expressed on some populations of neurons. Interaction with its receptor, CX3CR1, present on microglia and NK cells, serves to down-regulate microglial-mediated neurotoxicity both in vitro and in animal models of Parkinson disease and ALS (Cardona et al., 2006). In the animal model of MS, absence of fractalkine or its receptor resulted in a reduction of NK cells in the CNS and exacerbation of disease, supporting the view that NK cells play an inhibitory role in CNS inflammation (Huang et al., 2006).
Neuroglial Cells and the Immune Response
Microglia are derived from bone marrow cells during ontogeny (Hickey and Kimura, 1988) and reside within the CNS as three principal types of cells: perivascular microglia, parenchymal microglia, and Kolmer cells, which reside in the choroid plexus. Microglia have mitotic potential and can differentiate from bone marrow–derived cells to perivascular microglia and parenchymal microglia. Compared to macrophages, microglia are relatively radioresistant. Microglia may exist either in a resting (ramified) form or activated or phagocytic forms within the CNS. Activated microglia express higher levels of MHC class II and produce higher levels of proinflammatory cytokines including TNF-α, IL-6, and IL-1, as well as nitric oxide and glutamate. Microglia express chemokine receptors and various pattern recognition receptors (PRRs) including Toll-like receptors. PRRs recognize pathogen-associated molecular patterns (PAMPs) expressed by a variety of microbes, and interaction results in microglial activation. The primary functions of microglia are immune surveillance for foreign antigens and phagocytic scavengers of cellular debris. Microglia, particularly perivascular microglia, may also participate in antigen presentation within the CNS under certain conditions. Microglia play a role in regulating the programmed elimination of neural cells during brain development and, in some cases, enhance neuronal survival by producing neurotrophic and antiinflammatory cytokines. Microglia may also play a role in neuroregeneration and repair. However, there is overwhelming evidence that microglia play a deleterious role in several neurodegenerative diseases: MS, ALS, Parkinson disease and HIV-associated dementia. Their role in Alzheimer disease (AD) is less clear. Overactivation of microglia, possibly by microbes or other environmental factors through PRRs, may result in chronic proinflammatory milieu in the CNS, leading to progressive neurodegeneration. Strategies to down-regulate such responses are under investigation (Block et al., 2007).
Putative Mechanisms of Human Autoimmune Disease
Genetic Factors
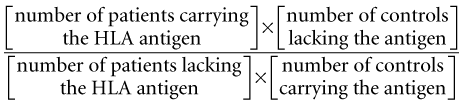
Association of a particular HLA haplotype with autoimmune disease may be due to the ability of a particular MHC molecule to bind and present autoantigen to the T cell, as in MS where the MHC class II allele, DRB1*1501, has been shown to be effective in presenting myelin basic protein (MBP peptide) to T-cell clones isolated from MS patients (Wucherpfennig et al., 1995; Wucherpfennig et al., 1997). Conversely, if an MHC molecule does not bind a particular self-antigen in the thymus, the developing T cell will not recognize that antigen as self and will escape negative selection. Therefore certain MHC haplotypes have an association with disease, whereas others protect against disease. Disease linkage tends to be with class II genes of the MHC rather than class I, suggesting a key role for T-cell autoimmunity.
Sex is one of the most important genetic determinants associated with autoimmune disease. Many autoimmune diseases are more frequent in females; systemic lupus erythematosus (SLE) is 10 times more common in women, and MS twice as common. Evidence from animal models has shown that females are more resistant to infections and reject foreign skin grafts sooner than their male counterparts. This is especially true during periods of high estrogen availability. Estrogen levels decrease after ovulation or during pregnancy, and this is associated with a progesterone surge. The lowering of estrogen ensures immunological tolerance toward the sperm and subsequently toward the fetus. Therefore estrogen’s effects on the immune system may predispose women toward autoimmune diseases. This is reflected in experimental disease models of autoimmunity. Only female (NZB × NZW)F1 mice develop the SLE-like disease, and this is abrogated by androgen treatment. Similarly, in experimental autoimmune encephalomyelitis (EAE), an experimental model for MS, female SJL mice are more susceptible to disease induction and are protected with testosterone (Dalal et al., 1997). Preliminary studies testing the effectiveness of a testosterone gel in males with MS have shown encouraging results but require additional validation. Initial studies investigating estriol effects in women with MS have shown a potent effect on reduction of new lesion formation, evident on gadolinium-enhanced magnetic resonance imaging (MRI) (Sicotte et al., 2002).
Environmental Factors
In many cases of molecular mimicry, the environmental antigen is a pathogen, and autoimmune disease follows the pathogen-caused disease. The classic example of this is streptococcal-induced endocarditis. Neurological diseases caused by this mechanism include streptococcal-induced chorea, Gd1b axonal neuropathy, Semple rabies vaccine-induced encephalomyelitis, and the anti-Hu paraneoplastic syndrome. Several studies have demonstrated that both adult (Ascherio and Munger, 2007) and pediatric (Alotaibi et al., 2004; Banwell et al., 2007; Lunemann et al., 2008; Pohl et al., 2006) MS patients more frequently demonstrate evidence of a remote infection with Epstein-Barr virus (EBV) than controls, implicating a role for this virus in disease pathogenesis. Interestingly, epitopes of EBV resemble myelin basic protein (MBP), supporting a role for molecular mimicry in disease pathogenesis (Lang et al., 2002). Despite these associations, however, it is clear that the majority of persons are infected with EBV without autoimmune sequelae. Recent studies have integrated risk factors in the pathogenesis of MS and found that the relative risk of MS among DR15-positive women with elevated (>1 : 320) anti-EBNA-1 titers was ninefold higher than that of DR15-negative women with low (<1 : 80) anti-EBNA-1 titers (De Jager et al., 2008).
Neuroimmunological Diseases
Multiple Sclerosis
MS is a complex polygenic disease. Monozygotic twins carry a concordance rate of 27%, whereas dizygotic twins of the same sex display a 2.3% concordance rate. The incidence for first-degree relatives of MS patients is 2% to 5%, whereas the incidence for the general population is under 0.1%. Genetic linkage studies have been performed, and several regions of interest have been found, but the most robust association remains with the HLA region. There is an increased incidence of MS in patients with the HLA-DR2 (DR1501) haplotype (Haines et al., 1998; Sawcer and Goodfellow, 1998; Stewart et al., 1981). More recently, a large genome-wide study identified single nucleotide polymorphism of IL-2R and IL-7R alleles as risk alleles for MS (Hafler et al., 2007). MS remains most prevalent among people of Northern European descent. There is a lower prevalence in other populations, such as Arabic and Mediterranean people, but among those with disease, there is a higher incidence of other disease-associated haplotypes such as DR4 and DR6.
Although genetic factors play an important role in pathogenesis, migration and other studies have demonstrated that environment also plays a critical role. Epidemiological studies have shown that residence in certain geographical areas and migration to these areas before the age of 15 increases the incidence of MS. In addition, there is a diminishing north-to-south gradient in MS prevalence in the Northern Hemisphere, with an opposite trend in the Southern Hemisphere. This led to the hypothesis and demonstration of an inverse association between sunlight exposure and MS (van der Mei et al., 2003).
An extension of this hypothesis has led to exploration of the role of vitamin D in MS, since vitamin D is metabolized in the skin by ultraviolet (UV) irradiation. A prospective study in army recruits found that 25-hydroxyvitamin D levels in the highest quintile (above 99.1 nmol/L) were associated with a lower risk of MS (odds ratio [OR], 0.38) (Munger et al., 2006). Treatment of animal models of MS with vitamin D ameliorates disease, and several studies have shown that the active form of vitamin D, calcitriol, can down-regulate proinflammatory dendritic cells (DC) and reduce TH1 lymphocyte responses while promoting antiinflammatory TH2 lymphocyte responses (Adorini et al., 2004; Griffin et al., 2001; Penna and Adorini, 2000; Penna et al., 2007). Studies exploring the therapeutic effects of vitamin D in MS are underway (Burton et al., 2010).
Pathologically, MS is characterized by inflammatory infiltrates in the CNS white matter, with resultant demyelination and axonal transections (Trapp et al., 1998) producing sclerotic plaques. Inflammation is generally perivenular, and lesions typically occur in the periventricular subcortical white matter, corpus callosum, optic nerve, brainstem, cerebellum, and spinal cord. Cortical lesions have also been described. The pathology of MS lesions has been classified into four distinct subtypes with the following predominant features: (1) cellular infiltration, (2) antibody deposition, (3) oligodendrocyte apoptosis, and (4) oligodendrocyte death without apoptosis (Lucchinetti et al., 2000). Observations to date show a single subtype of lesion in each patient, raising the possibility of distinct MS disease pathogenetic types. Activated microglia have been demonstrated in non-lesion, or otherwise normal-appearing white matter (NAWM) and may play a role in axonal damage and disease progression.
It is clear that the immune system plays a central role in mediating CNS damage in MS. Oligoclonal bands are commonly observed in the CNS of MS patients; however, the target of these antibodies has yet to be elucidated. Cell-mediated immunity, primarily involving T-helper cells, is believed to play an important role in initiating the disease, and immunological studies have substantiated the presence of activated T cells in MS. Most of the therapies for MS also target T cells. No clear autoantigen has been described in MS, and it therefore remains an immune-mediated disease rather than an autoimmune disease. Reactivity to various myelin antigens, including myelin basic protein (MBP), proteolipid protein (PLP), and myelin oligodendrocyte protein (MOG) has been investigated. MBP-reactive T cells are present in normal individuals; however, MS patients have a higher frequency of activated MBP-reactive T cells in the peripheral blood and the cerebrospinal fluid (CSF) (Zhang et al., 1994).
The immunopathogenesis of MS is thought to involve activation of myelin-specific T cells via molecular mimicry or by a superantigen presumably in the periphery. These cells are believed to predominantly be of the TH1 phenotype, as evidenced by increased production of IL-12 (the major inducer of TH1 cytokines) by APCs in the peripheral blood of MS patients with active disease, and the observation that a clinical trial of IFN-γ worsened MS. These cells then cross the BBB and get reactivated in the CNS when they are presented with their cognate antigen. Perivascular dendritic-like cells have been shown to play a role in T-cell reactivation in animal models of MS (Greter et al., 2005), but their role in the human disease is unclear. Adhesion molecules and their ligands are expressed on T cells and endothelial cells to facilitate passage through the BBB. VCAM-1 and its ligand, VLA-4, are up-regulated in T cells during chronic disease, thus perpetuating inflammation, and VLA-4 antibody therapy (natalizumab) has recently been shown to be effective in reducing MS relapses and lesion formation.
There is mounting evidence indicating that T cells play a key role in the relapsing-remitting phase of MS. However, recent work has demonstrated that disease progression in MS may be due to distinct mechanisms (Weiner, 2009). Epitope spreading may occur within the CNS, and in animal models can be facilitated by microglia, macrophages, and dendritic cells (McMahon et al., 2005). Activated microglia are found in progressive forms of the disease and have been associated with axonal damage and demyelination (Kutzelnigg et al., 2005). B-cell follicles have been demonstrated at autopsy in patients with chronic MS (Serafini et al., 2004) and may play a role in facilitating an autonomous inflammatory response within the CNS. Cortical demyelination is also associated with progressive forms of MS (Kutzelnigg et al., 2005).
Neuromyelitis optica (NMO), or Devic disease, is a rare subtype of MS characterized by clinical episodes of optic neuritis and transverse myelitis and demonstration of contiguous lesions in the spinal cord (Wingerchuk et al., 2006). The presence of serum antibodies targeting the aquaporin-4 water channel present on the surface of the glia limitans at the BBB has been shown to be a sensitive and specific marker of NMO (Lennon et al., 2004). Injection of aquaporin-4 antibodies into animal models of disease have demonstrated enhanced complement deposition around blood vessels, loss of aquaporin-4, and astrocyte and myelin damage (Kutzelnigg et al., 2005). This study, as well as others, indicate increasing recognition of the role of glial pathology in MS.
EAE is a T cell–mediated autoimmune demyelinating disease of the CNS. The disease can be induced in a number of experimental laboratory animals, including primates, by subcutaneous injection of whole-brain homogenate or of a purified preparation of MBP, PLP, or MOG emulsified in adjuvant. By altering the immunization protocols and animal strains, a relapsing-remitting or a chronic form of the disease may be induced. Various EAE models have demonstrated that mice with a TH1 cytokine profile are more susceptible to EAE than wild-type mice, whereas animals with a TH2 (IL-4, IL-10) cytokine profile are generally resistant to disease development (Chitnis et al., 2001). Transfer of TH17-producing cells induces a more severe form of EAE than transfer of TH1 cells (Jager et al., 2009); the role of these cells in MS is starting to be elucidated.
TMEV-IDD is induced by injecting TMEV picornavirus into the cerebral hemisphere. The virus infects neurons and glial cells. In some strains of mice, the host is unable to clear the virus, resulting in encephalitis and death; in other cases, the virus is cleared completely, and the host is resistant to demyelination. In TMEV-IDD-susceptible strains of mice, the virus is partially cleared, saving the host from death but inciting an immune response that results in demyelination. Thus the damage induced by the virus is due to a failure of the host to mount a fully protective response, which predisposes the pathogen to persistence, resulting in immunopathology. TMEV-IDD is a T cell–mediated disease; although the exact immunological mechanisms inducing demyelination remain unclear, damage may be a result of epitope spreading (Miller et al., 1997). Studies in both EAE and TMEV have contributed vastly to our understanding of MS, and these models offer a system for testing new therapeutics.
Details of available therapies in MS are discussed in Chapter 60. Here, we discuss the immunological mechanisms of currently used medications, as well as experimental therapeutic strategies.
β-Interferon (IFN-β) therapy for MS is one of the most important advances in the treatment of this disease. It is available in three different forms: subcutaneous IFN-β-1b (Betaseron), subcutaneous IFN-β-1a (Rebif), or intramuscular IFN-β-1a (Avonex). Interferons have many properties, including suppressing proliferation of viruses and T cells. The mechanisms of IFN-β action in MS has been attributed to several different mechanisms. Interferon-associated increased production of IL-10 by macrophages down-regulates the number of TH1 cells. IFN-β has also been shown to decrease production of IL-12 by dendritic cells, potential CNS APCs, further inhibiting TH1 cell formation. In addition, IFN-β modulates adhesion molecule expression, primarily by facilitating the conversion of cell-associated VCAM-1 into soluble VCAM-1. These drugs also down-regulate costimulatory molecule expression, thus decreasing T-cell activation and migration to the CNS (Yong, 2002).
Glatiramer acetate (GA), also known as copolymer-1 or Copaxone, is another class of drug used in the treatment of MS. In contrast to IFN-β, GA is a synthetic molecule that was originally designed to resemble MBP. It is composed of random repetitive sequences of the amino acids glutamic acid, lysine, alanine, and tyrosine (G-L-A-T). Its mechanism of action is unclear; however, it is thought to bind with high affinity to the MHC groove, leading to the generation of GA-specific T cells. Several studies have suggested that GA-specific T cells display a TH2 bias (Duda et al., 2000), and animal models have demonstrated that these cells can migrate to the CNS and ameliorate EAE disease through a local down-regulation of the immune responses (Aharoni et al., 1997). It has also been demonstrated that GA-specific T cells protect from optic nerve crush injuries, possibly mediated by the production of brain-derived neurotrophic factor (BDNF) (Kipnis et al., 2000).
Altered peptide ligands (APL) resembling MBP have been used in phase 1 trials for MS with little success. Two concurrent trials were initiated using the same compound, CGP77116. One showed an increased number of lesions on MRI in some patients after the initiation of treatment (Bielekova et al., 2000). In the other trial, 9% of patients developed allergic-type reactions associated with a TH2 deviation (Kappos et al., 2000). Both trials were stopped because of safety concerns.
Campath-1H targets the CD52 receptor present on lymphocytes and monocytes. Phase 2 studies have shown potent effects in MS, with depletion of peripheral lymphocytes. However, a quarter of treated patients develop autoimmune thyroid disease, and rarely immune thrombocytopenic purpura, suggesting that Campath enhances immune dysregulation in a subset of patients (Coles et al., 2008; Jones et al., 2009). Daclizumab blocks the IL-2R α chain (CD25) present in the high-affinity IL-2 receptor on T cells, thus inhibiting T-cell replication and making more IL-2 available to the low-affinity CD25 receptor present on NK cells, which induces a regulatory NK cell population (Bielekova et al., 2006).
Rituximab, an antibody that primarily targets activated B cells, has recently been shown to reduce disease activity in relapsing-remitting (Hauser et al., 2008) and a subset of primary progressive MS patients (Hawker et al., 2009). Interestingly, open-label studies have suggested that rituximab treatment is effective in patients with neuromyelitis optica, which is increasingly thought of as an antibody-mediated disease. Another therapy currently under investigation is CTLA4Ig, which blocks B7-CD28 costimulatory signals on T cells and may induce T-cell anergy in vivo.
Many therapeutic strategies that have been used in the past nonspecifically target components of the immune response. Nonspecific strategies include cyclophosphamide, mitoxantrone, and cladribine, which depress bone marrow production of cells, including T cells. Cyclophosphamide may also function by inducing a cytokine switch, with a decrease in IL-12 and an increase in IL-4, IL-5, and TGF-β (Comabella et al., 1998). A novel strategy to suppress immune responses in MS is a small molecule, fingolimod, which targets the sphingosine-1-phosphate receptor, which is necessary for lymphocyte egress from lymph nodes (Comi et al., 2010).
Acute Disseminated Encephalomyelitis
Acute disseminated encephalomyelitis (ADEM) is a monophasic demyelinating disease associated with vaccination or a systemic viral infection; it can affect both adults and children. ADEM was originally described in association with rabies and smallpox vaccines, both of which were prepared with neural tissues, suggesting a parallel with EAE, the animal model of MS. These vaccines have since been modified, using non-neural human diploid cell lines. ADEM has not been associated with any vaccines that are currently administered in the United States. The parainfectious variant of ADEM has been associated with measles infection, rubella, mumps, and several other viruses. Viral or bacterial epitopes resembling myelin antigens have the capacity to activate myelin-reactive T-cell clones through molecular mimicry (Wucherpfennig, Hafler, and Strominger, 1995) and can thereby elicit a CNS-specific autoimmune response. Thus, it has been suggested that microbial infections elicit a cross-reactive antimyelin response through molecular mimicry, resulting in ADEM. Myelin peptides have been shown to resemble several viral sequences, and in some cases, cross-reactive T-cell responses have been demonstrated. MBP is the prototypical inducer of EAE, but other myelin proteins like MOG and PLP have also been extensively studied. Examples of cross-reactive T cells with MBP antigens include HHV-6 (Tejada-Simon et al., 2003), coronavirus (Talbot et al., 1996), influenza virus hemagglutinin (Markovic-Plese et al., 2005), and EBV (Lang et al., 2002). PLP shares common sequences with Haemophilus influenzae (Olson et al., 2001). Semliki Forest virus (SFV) peptides mimic MOG (Mokhtarian et al., 1999).
The lesions in ADEM resemble those of MS. The CNS white matter contains perivascular inflammatory infiltrates as well as demyelination. The most likely mechanism by which this disease occurs is molecular mimicry. Experimental evidence has shown that T cells isolated from patients with ADEM are 10 times more likely to react with MBP than controls, likening this disease to EAE in animal models (Pohl-Koppe et al., 1998). We have recently found that 30% of patients with ADEM demonstrate serum antibodies to MOG, which were absent in MS patients (O’Connor K et al., 2007). Because of the monophasic nature of ADEM, it appears that the immunological response occurs acutely, but in contrast to MS, further amplification of inflammation within the CNS is suppressed.
Immune-Mediated Neuropathies
AIDP is the most common acute paralytic disease in the Western world, with a mean annual incidence of 1.8 per 100,000 persons. There is an increasing incidence with age. Mortality was generally due to respiratory failure and has now been significantly reduced with the introduction of positive-pressure ventilation. Epidemics have been found, most notably in northern China, where a high incidence has been associated with C. jejuni infections (McKhann et al., 1993).
AIDP or GBS is characterized pathologically by an endoneurial lymphocytic, monocytic, and macrophage infiltrate. Several autoantibodies to myelin glycolipids have been identified, including GM1, GD1a, and GD1b. Antibody-mediated demyelination due to complement fixation has been identified in pathology specimens. In some cases, axonal damage is present and is believed to be a result of bystander damage. Activation of calcium-dependent processes within the nerve, including calpain activation, has been shown in animal models to augment axonal degeneration (O’Hanlon et al., 2003). GBS is primarily an antibody-mediated disease, as evidenced by the fact that many patients improve after treatment with plasmapheresis, and that serum from GBS patients causes demyelination after transfer into experimental animals and peripheral nerve cultures. The Miller-Fisher variant of GBS is characterized by ophthalmoplegia, ataxia, and areflexia and is associated with the presence of GQ1b antibodies in the serum.
The occurrence of AIDP has been linked to many infectious diseases, including C. jejuni, herpesvirus, Mycoplasma pneumonia, and many other bacterial and viral infections, as well as vaccinations. The incidence of infection has been reported to be 90% in the 30 days before occurrence of GBS. C. jejuni is one of the most commonly identifiable agents, and molecular mimicry and host susceptibility play a role in disease pathogenesis. Autoantibodies not present in controls have been identified in the sera of GBS patients associated with C. jejuni, including autoantibodies to the gangliosides GM1, GD1a, GD1b, and GQ1b (Sheikh et al., 1998).
Treatment of AIDP involves supportive care and cardiac and respiratory monitoring. Plasmapheresis or intravenous immunoglobulin (IVIG) have been used for acute treatment of AIDP and have been shown to be equally effective in shortening recovery time. Plasmapheresis is a short-term immunotherapy that nonspecifically removes antibodies from the circulation. IVIG is an immunomodulating agent commonly used in the treatment of allergic and autoimmune diseases. It works in part through the presence of Fc fragments that interact with the inhibitory Fc receptor, FcγRIIB, which is also induced on macrophages following IVIG administration (Samuelsson et al., 2001). Additionally, IVIG may displace low-affinity autoantibodies from the nerve. High-dose steroids have not been found to be effective in AIDP.
Autoimmune Myasthenia Gravis
Although the B cell is the effector cell producing antibodies, experimental evidence has shown that autoreactive T cells are necessary for the disease to occur (Yi and Lefvert, 1994). Removal of the thymus results in improvement of disease in 80% to 90% of myasthenic patients. The role of thymic abnormalities remains unclear, and patients with thymomas have antibodies to additional skeletal muscle proteins such as the ryanodine receptor and titin, as well as the neuromuscular junctional protein, MuSK. Patients may also display symptoms of neuromyotonia. Antibodies directed towards α3-nAChR are associated with autoimmune autonomic neuropathy.
A large body of research is targeted at understanding the reasons for the failure of T-cell and subsequent failure of B-cell tolerance in MG. Both normal and myasthenic thymus glands contain myoid cells and epithelial cells that express the AChR. T cells expressing the Vβ5.1+ TCRs are overrepresented, both in the core of germinal centers and in perifollicular areas of hyperplastic thymuses, suggesting a role in the autoimmune response (Truffault et al., 1997). Failure of central or thymic tolerance may play an important role in disease pathogenesis.
Therapies in MG are targeted toward alleviating symptoms with acetylcholinesterase inhibitors and using strategies to reduce the damage being done by the immune system. Thymectomy is recommended for patients 15 to 65 years old, with 80% to 90% remission rate (Durelli et al., 1991). The thymus plays an important role in T-cell education in the developing human; therefore prepubertal thymectomy is discouraged. A variety of anticholinesterase inhibitors provide temporary symptomatic relief in most patients. Pyridostigmine bromide (Mestinon) and neostigmine bromide (Prostigmin) are the most commonly used agents and must be taken daily.
MG is an antibody-mediated disease and therefore responds to therapies that nonspecifically target antibodies. Both plasmapheresis and treatment with IVIG are used for acute MG exacerbations or in preparation for surgery (Gajdos et al., 1997). Because the autoantigen is known in MG, investigational therapies may target specific molecules such as the B-cell surface Ig or the TCR and deliver immunotoxins.
Inflammatory Muscle Diseases
PM is thought to result from a multitude of causes, including systemic autoimmune connective-tissue disorders and viral and bacterial infections. PM is characterized histopathologically by an endomysial inflammatory infiltrate containing predominantly CD8+ T cells. There is relative sparing of blood vessels. In one subtype of PM, T cells with γδ receptors have been identified surrounding non-necrotic muscle fibers (Hohlfeld et al., 1991).
In contrast, DM is characterized by perifascicular atrophy. There is hypoperfusion and subsequent degeneration of the muscle fibers in the periphery of the fascicle, secondary to microvascular damage. Damage to capillaries, resulting in muscle fiber ischemia, is mediated by complement. Immunofluorescence studies have revealed immune-complex deposition within the endothelium, indicating that this is an antibody-mediated disease; therefore the disease differs from PM (Kissel et al., 1986).
As with PM, IBM is mediated by CD8+ T cells. However, in contrast to PM, the muscle biopsy in IBM may also demonstrate the presence of characteristic autophagic “rimmed” vacuoles. Amyloid deposits may be demonstrated in the muscle, similar to those seen in AD, suggesting similarities in pathogenesis of these two diseases (Askanas et al., 1992).
Various autoantibodies directed against nuclear and cytoplasmic cell components are found in up to 30% of inflammatory myopathies. Most are nonspecific for connective-tissue disease. Viruses including coxsackie B are implicated in the pathogenesis of disease, and both PM and DM patients may have anti-Jo-1 antibodies to the viral enzyme, histidyl-tRNA synthetase (Mathews and Bernstein 1983). More recently, the presence of B cells, and in particular antibody-secreting plasma cells with V-D-J rearrangements, has been demonstrated in muscle biopsies from both IBM and PM and to a lesser extent in DM (Greenberg et al., 2005).
The mainstay of treatment of PM and DM is corticosteroids. Dosages may vary from 60 to 100 mg/day of prednisone, and duration is determined by clinical outcome. Alternative treatment options for the inflammatory myositis diseases include IVIG, methotrexate, azathioprine, cyclophosphamide, cyclosporine, and in extreme cases, total lymphoid or whole-body irradiation (Mastaglia et al., 1998). Mortality rates vary between 15% and 35% and are generally due to cardiac or respiratory failure. Because there is a higher incidence of malignancy with PM and DM, screening for breast, lung, hematological, ovary, stomach, and colon carcinoma should be performed on patients with these diagnoses. IBM may be more resistant to corticosteroid therapy and is often diagnosed after an assumed PM fails to respond to treatment.
Alzheimer Disease and Amyotrophic Lateral Sclerosis
β-Amyloid plaques and neurofibrillary tangles consisting of hyperphosphorylated tau protein are the hallmarks of AD. Clearance of amyloid plaques consisting of amyloid-β-fibrils is considered a primary goal of therapy. Amyloid plaques are often surrounded by activated microglia and reactive astrocytes, and are associated with complement activation, leading to the hypothesis that the immune response participates in the clearance of amyloid deposits. Further studies in animal models of AD demonstrated that immunization with amyloid-β peptide resulted in the induction of amyloid-β-specific antibodies, which enhanced the clearance of amyloid plaques (Janus et al., 2000; Morgan et al., 2000). Passive transfer of amyloid-β-specific antibodies yielded similar results. Amyloid plaque clearance is believed to occur through either microglial- and complement-mediated clearance or through direct antibody-amyloid interactions. These studies led to a clinical trial investigating an amyloid-β vaccine administered in conjunction with an adjuvant, which enhanced TH1 responses. Although cognitive testing results were favorable, 6% of patients developed meningoencephalitis, which is generally believed to be a result of T-cell responses to amyloid-β (Gilman et al., 2005; Orgogozo et al., 2003). Thus, induction of an antibody and microglial-mediated clearance of amyloid in the absence of a prominent TH1 response is the current goal of therapy. An intriguing study has recently demonstrated that nasal administration of glatiramer acetate (Copaxone), which induces a predominantly TH2 cellular response, to a murine model of AD resulted in the clearance of amyloid-β plaques in association with activated microglia, but in the absence of antibody formation (Frenkel et al., 2005). Future immunotherapeutic strategies for AD include modified vaccines and strategies to induce activation of microglial cells in the absence of deleterious side effects.
In ALS, a new avenue of investigation has emerged: exploring the role of microglia in disease pathogenesis and, in particular, on disease progression. Pathological analysis and neuroimaging using positron electron tomography (PET) studies have demonstrated activated microglia in areas of severe motor neuron loss. Studies in the animal model of ALS have demonstrated that the presence of the SOD1 mutation in microglia enhanced disease progression (Boillee et al., 2006). Use of minocycline, which acts in part by inhibiting microglial activation, has shown some initial promise in the treatment of ALS. Cyclooxygenase-2 (COX-2) inhibitors have reduced disease severity in animal models of ALS but have been ineffective in humans. Lymphocytes, including T cells, do not appear to play a significant role in this disease, and this is reinforced by the failure of studies using T cell–targeted therapies including total lymphoid irradiation (TLI) or cyclophosphamide. Thus, possible future therapies for ALS include strategies to down-regulate microglial activation.
Tumor Immunology
Tumors in the CNS have similar abilities to evade the immune system, and it has been shown that some gliomas produce high levels of TGF-β, an immunosuppressant. Down-regulation of class II MHC may also occur, but this is remains controversial. It has recently been established that gliomas may express high levels of FasL, allowing for local apoptosis of Fas-bearing cells including lymphocytes. Increased expression of the inhibitory costimulatory molecule, B7H1, on gliomas has been shown to play a role in down-regulating T-cell responses (Wintterle et al., 2003). Glioma patients have been shown to express increased frequencies of CD4+CD25+Foxp3+ regulatory T cells. Treatment with daclizumab (anti-CD25 antibody) in a murine model of glioma reduced regulatory T-cell function and enhanced host tumor immunity, so daclizumab therapy in CNS tumors merits additional investigation.
Paraneoplastic Syndromes
Neurological paraneoplastic disorders are defined as neurological syndromes arising in association with a distant cancer. These are mediated by antibodies produced by the immune system in reaction to a tumor antigen, which cross-react with neural tissue. It is likely that aberrant, primitive, or hamartomatous antigens are expressed by the tumor cells. Enhanced cellular infiltrates are found in tumors associated with paraneoplastic syndrome compared to those not associated with a paraneoplastic syndrome. Therefore one can postulate that the immune system is more active in these situations. Cancers associated with paraneoplastic syndromes are generally associated with a better outcome. Several autoantibodies associated with paraneoplastic syndromes have been identified. The anti-Hu antibody arises in association with small-cell cancer of the lung and cross-reacts with neurons; it is linked to a syndrome of encephalomyelitis and/or sensory neuropathy. Similarly, anti-Yo antibody produces cerebellar degeneration due to cross-reactivity with Purkinje cell cytoplasm and is associated with breast and ovarian cancer. Opsoclonus-myoclonus syndrome is associated with anti-Ri antibody and is found in cases of cancer of the ovary and breast. Cases of paraneoplastic and immune-mediated brainstem and limbic encephalitis have recently been reported to be caused by antibodies targeting LGI1, a neuronal-secreted protein that interacts with presynaptic ADAM23 and postsynaptic ADAM22 (Lai et al., 2010). NMDA receptor encephalitis typically presents with seizures and psychiatric symptoms, but a recent case report describes a patient presenting with optic neuritis and transverse myelitis mimicking neuromyelitis optica (Kruer et al., 2010). Antibodies directed against VGKC are also associated with acquired neuromyotonia, or Isaac syndrome, and Morvan syndrome, characterized by neuromyotonia and insomnia. Lambert-Eaton myasthenic syndrome is caused by antibodies directed against the P/Q type of voltage-gated calcium channels and is generally found in the setting of small-cell cancer of the lung. The same antibodies have also been associated with cases of lung cancer–associated cerebellar ataxia. Stiff person syndrome is associated with antibodies to glutamic acid decarboxylase (GAD), and both autoimmune and paraneoplastic forms, principally associated with breast cancer, have been described.
Antibody-Associated Neurological Syndromes
In the past 10 years, more sophisticated techniques have led to greater insights into the study of antibodies in CNS diseases. Antiphospholipid (APL) syndrome, or Hughes syndrome, results in CNS symptoms that include chorea, strokes, bleeding, migraine headaches, and epilepsy (Asherson, 2006). These result in part from the underlying systemic problems of coagulopathy and thrombocytopenia, but more direct effects of autoantibodies directed against neuronal antigens have been implicated both in APL syndrome and in CNS lupus. One study found that antibodies directed against the NR2A and NR2B subunits of the NMDA receptor were found in a subset of patients with CNS lupus and could facilitate apoptotic death of neurons (DeGiorgio et al., 2001).
Sydenham chorea is associated with Streptococcus pyogenes (β-hemolytic streptococcal) infections, and there is considerable evidence for a causative role of antibodies that cross-react with streptococcal antigens and neurons in the basal ganglia. This association has led to the postulation that molecular mimicry mechanisms related to streptococcal infections may result in other movement disorders, including Tourette syndrome and the clinical entity of pediatric autoimmune neuropsychiatric disorders associated with streptococcal infection (PANDAS), which encompasses tics and obsessive-compulsive disorder in children. Evidence for an immune-mediated mechanism is inconclusive (Harris and Singer, 2006).
Antibodies directed against glutamate receptor 3 (GluR3) are associated with Rasmussen encephalitis, a form of severe intractable epilepsy localized to one hemisphere and partially responsive to immunotherapy. The presence of antibodies directed against voltage-gated potassium channels in a small group of patients with intractable epilepsy has recently been demonstrated (Majoie et al., 2006) and raises the question of whether other immune-mediated forms of epilepsy may exist.
Immunology of Central Nervous System Transplant
Neural stem cells (NSCs) are increasingly being investigated in neurodegenerative diseases. In addition to their effects on repair, studies in the animal model of MS found that these cells suppress disease (Einstein et al., 2003; Einstein et al., 2007; Pluchino et al., 2003; Pluchino et al., 2005; Yang et al., 2009) through immunomodulatory mechanisms. Some studies suggested that NSCs can directly inhibit T-cell proliferation in response to concanavalin A (ConA) or to MOG peptide (Einstein et al., 2003; Pluchino et al., 2005) by inducing T-cell apoptosis (Pluchino et al., 2005; Yang et al., 2009) or through nitric oxide– and PGE2-mediated T-cell suppression (Wang et al., 2009). Neural stem cells can express costimulatory molecules, CD80 and CD86, particularly after exposure to the proinflammatory cytokines, IFN-γ and TNF-α (Imitola et al., 2004). Thus, NSCs are not conventional immune cells, but under certain conditions, they can interact with immune cells.
Adorini L., Penna G., Giarratana N., et al. Dendritic cells as key targets for immunomodulation by vitamin D receptor ligands. J Steroid Biochem Mol Biol. 2004;89-90:437-441.
Aggarwal S., Ghilardi N., Xie M.H., et al. Interleukin-23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin-17. J Biol Chem. 2003;278:1910-1914.
Aharoni R., Teitelbaum D., Sela M., et al. Copolymer 1 induces T cells of the T helper type 2 that crossreact with myelin basic protein and suppress experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. 1997;94:10821-10826.
Alotaibi S., Kennedy J., Tellier R., et al. Epstein-Barr virus in pediatric multiple sclerosis. JAMA. 2004;291:1875-1879.
Ascherio A., Munger K.L. Environmental risk factors for multiple sclerosis. Part I: the role of infection. Ann Neurol. 2007;61:288-299.
Asherson R.A. New subsets of the antiphospholipid syndrome in 2006: “PRE-APS” (probable APS) and microangiopathic antiphospholipid syndromes (“MAPS”). Autoimmun Rev. 2006;6:76-80.
Askanas V., Engel W.K., Alvarez R.B. Light and electron microscopic localization of beta-amyloid protein in muscle biopsies of patients with inclusion-body myositis. Am J Pathol. 1992;141:31-36.
Bacchetta R., Bigler M., Touraine J.L., et al. High levels of interleukin 10 production in vivo are associated with tolerance in SCID patients transplanted with HLA mismatched hematopoietic stem cells. J Exp Med. 1994;179:493-502.
Baecher-Allan C., Brown J.A., Freeman G.J., et al. CD4+CD25 high regulatory cells in human peripheral blood. J Immunol. 2001;167:1245-1253.
Banerjee D., Dick A.D. Blocking CD200-CD200 receptor axis augments NOS-2 expression and aggravates experimental autoimmune uveoretinitis in Lewis rats. Ocul Immunol Inflamm. 2004;12:115-125.
Banwell B., Krupp L., Kennedy J., et al. Clinical features and viral serologies in children with multiple sclerosis: a multinational observational study. Lancet Neurol. 2007;6:773-781.
Bettelli E., Carrier Y., Gao W., et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235-238.
Bielekova B., Catalfamo M., Reichert-Scrivner S., et al. Regulatory CD56(bright) natural killer cells mediate immunomodulatory effects of IL-2Ralpha-targeted therapy (daclizumab) in multiple sclerosis. Proc Natl Acad Sci U S A. 2006;103:5941-5946.
Bielekova B., Goodwin B., Richert N., et al. Encephalitogenic potential of the myelin basic protein peptide (amino acids 83-99) in multiple sclerosis: results of a phase II clinical trial with an altered peptide ligand. Nat Med. 2000;6:1167-1175.
Block M.L., Zecca L., Hong J.S. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8:57-69.
Boillee S., Yamanaka K., Lobsiger C.S., et al. Onset and progression in inherited ALS determined by motor neurons and microglia. Science. 2006;312:1389-1392.
Broderick C., Hoek R.M., Forrester J.V., et al. Constitutive retinal CD200 expression regulates resident microglia and activation state of inflammatory cells during experimental autoimmune uveoretinitis. Am J Pathol. 2002;161:1669-1677.
Brunet J.F., Denizot F., Luciani M.F., et al. A new member of the immunoglobulin superfamily–CTLA-4. Nature. 1987;328:267-270.
Burton J.M., Kimball S., Vieth R., et al. A phase I/II dose-escalation trial of vitamin D3 and calcium in multiple sclerosis. Neurology. 2010;74:1852-1859.
Cardona A.E., Pioro E.P., Sasse M.E., et al. Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci. 2006;9:917-924.
Chakravarti S., Sabatos C.A., Xiao S., et al. Tim-2 regulates T helper type 2 responses and autoimmunity. J Exp Med. 2005;202:437-444.
Chitnis T., Najafian N., Benou C., et al. Effect of targeted disruption of STAT4 and STAT6 on the induction of experimental autoimmune encephalomyelitis. J Clin Invest. 2001;108:739-747.
Coles A.J., Compston D.A., Selmaj K.W., et al. Alemtuzumab vs. interferon beta-1a in early multiple sclerosis. N Engl J Med. 2008;359:1786-1801.
Comabella M., Balashov K., Issazadeh S., et al. Elevated interleukin-12 in progressive multiple sclerosis correlates with disease activity and is normalized by pulse cyclophosphamide therapy. J Clin Invest. 1998;102:671-678.
Comi G., O’Connor P., Montalban X., et al. Phase II study of oral fingolimod (FTY720) in multiple sclerosis: 3-year results. Mult Scler. 2010;16:197-207.
Dalal M., Kim S., Voskuhl R.R. Testosterone therapy ameliorates experimental autoimmune encephalomyelitis and induces a T helper 2 bias in the autoantigen-specific T lymphocyte response. J Immunol. 1997;159:3-6.
Dardalhon V., Awasthi A., Kwon H., et al. IL-4 inhibits TGF-beta-induced Foxp3+ T cells and, together with TGF-beta, generates IL-9+ IL-10+ Foxp3(-) effector T cells. Nat Immunol. 2008;9:1347-1355.
De Jager P.L., Simon K.C., Munger K.L., et al. Integrating risk factors: HLA-DRB1*1501 and Epstein-Barr virus in multiple sclerosis. Neurology. 2008;70:1113-1118.
DeGiorgio L.A., Konstantinov K.N., Lee S.C., et al. A subset of lupus anti-DNA antibodies cross-reacts with the NR2 glutamate receptor in systemic lupus erythematosus. Nat Med. 2001;7:1189-1193.
Dieckmann D., Plottner H., Berchtold S., et al. Ex vivo isolation and characterization of CD4(+)CD25(+) T cells with regulatory properties from human blood. J Exp Med. 2001;193:1303-1310.
Drappa J., Vaishnaw A.K., Sullivan K.E., et al. Fas gene mutations in the Canale-Smith syndrome, an inherited lymphoproliferative disorder associated with autoimmunity. N Engl J Med. 1996;335:1643-1649.
Duda P.W., Schmied M.C., Cook S.L., et al. Glatiramer acetate (Copaxone) induces degenerate, Th2-polarized immune responses in patients with multiple sclerosis. J Clin Invest. 2000;105:967-976.
Durelli L., Maggi G., Casadio C., et al. Actuarial analysis of the occurrence of remissions following thymectomy for myasthenia gravis in 400 patients. J Neurol Neurosurg Psychiatry. 1991;54:406-411.
Einstein O., Fainstein N., Vaknin I., et al. Neural precursors attenuate autoimmune encephalomyelitis by peripheral immunosuppression. Ann Neurol. 2007;61:209-218.
Einstein O., Karussis D., Grigoriadis N., et al. Intraventricular transplantation of neural precursor cell spheres attenuates acute experimental allergic encephalomyelitis. Mol Cell Neurosci. 2003;24:1074-1082.
Elyaman W., Bradshaw E.M., Uyttenhove C., et al. IL-9 induces differentiation of TH17 cells and enhances function of FoxP3+ natural regulatory T cells. Proc Natl Acad Sci U S A. 2009;106:12885-12890.
Frenkel D., Maron R., Burt D.S., et al. Nasal vaccination with a proteosome-based adjuvant and glatiramer acetate clears beta-amyloid in a mouse model of Alzheimer disease. J Clin Invest. 2005;115:2423-2433.
Gajdos P., Chevret S., Clair B., et al. Clinical trial of plasma exchange and high-dose intravenous immunoglobulin in myasthenia gravis. Myasthenia Gravis Clinical Study Group. Ann Neurol. 1997;41:789-796.
Gilman S., Koller M., Black R.S., et al. Clinical effects of Abeta immunization (AN1792) in patients with AD in an interrupted trial. Neurology. 2005;64:1553-1562.
Greenberg S.A., Bradshaw E.M., Pinkus J.L., et al. Plasma cells in muscle in inclusion body myositis and polymyositis. Neurology. 2005;65:1782-1787.
Greter M., Heppner F.L., Lemos M.P., et al. Dendritic cells permit immune invasion of the CNS in an animal model of multiple sclerosis. Nat Med. 2005;11:328-334.
Griffin M.D., Lutz W., Phan V.A., et al. Dendritic cell modulation by 1alpha,25 dihydroxyvitamin D3 and its analogs: a vitamin D receptor-dependent pathway that promotes a persistent state of immaturity in vitro and in vivo. Proc Natl Acad Sci U S A. 2001;98:6800-6805.
Hafler D.A., Compston A., Sawcer S., et al. Risk alleles for multiple sclerosis identified by a genomewide study. N Engl J Med. 2007;357:851-862.
Haines J.L., Terwedow H.A., Burgess K., et al. Linkage of the MHC to familial multiple sclerosis suggests genetic heterogeneity. The Multiple Sclerosis Genetics Group. Hum Mol Genet. 1998;7:1229-1234.
Harrington L.E., Hatton R.D., Mangan P.R., et al. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6:1123-1132.
Harris K., Singer H.S. Tic disorders: neural circuits, neurochemistry, and neuroimmunology. J Child Neurol. 2006;21:678-689.
Hauser S.L., Waubant E., Arnold D.L., et al. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N Engl J Med. 2008;358:676-688.
Hawker K., O’Connor P., Freedman M.S., et al. Rituximab in patients with primary progressive multiple sclerosis: results of a randomized double-blind placebo-controlled multicenter trial. Ann Neurol. 2009;66:460-471.
Hickey W.F., Kimura H. Perivascular microglial cells of the CNS are bone marrow-derived and present antigen in vivo. Science. 1988;239:290-292.
Hoek R.M., Ruuls S.R., Murphy C.A., et al. Down-regulation of the macrophage lineage through interaction with OX2 (CD200). Science. 2000;290:1768-1771.
Hohlfeld R., Engel A.G., Ii K, et al. Polymyositis mediated by T lymphocytes that express the gamma/delta receptor. N Engl J Med. 1991;324:877-881.
Huang D., Shi F.D., Jung S., et al. The neuronal chemokine CX3CL1/fractalkine selectively recruits NK cells that modify experimental autoimmune encephalomyelitis within the central nervous system. FASEB J. 2006;20:896-905.
Imitola J., Comabella M., Chandraker A.K., et al. Neural stem/progenitor cells express costimulatory molecules that are differentially regulated by inflammatory and apoptotic stimuli. Am J Pathol. 2004;164:1615-1625.
Jager A., Dardalhon V., Sobel R.A., et al. Th1, Th17, and Th9 effector cells induce experimental autoimmune encephalomyelitis with different pathological phenotypes. J Immunol. 2009;183:7169-7177.
Janus C., Pearson J., McLaurin J., et al. A beta peptide immunization reduces behavioural impairment and plaques in a model of Alzheimer’s disease. Nature. 2000;408:979-982.
Jones J.L., Phuah C.L., Cox A.L., et al. IL-21 drives secondary autoimmunity in patients with multiple sclerosis, following therapeutic lymphocyte depletion with alemtuzumab (Campath-1H). J Clin Invest. 2009;119:2052-2061.
Kappos L., Comi G., Panitch H., et al. Induction of a non-encephalitogenic type 2 T helper-cell autoimmune response in multiple sclerosis after administration of an altered peptide ligand in a placebo-controlled, randomized phase II trial. The Altered Peptide Ligand in Relapsing MS Study Group. Nat Med. 2000;6:1176-1182.
Khoury S.J., Sayegh M.H. The roles of the new negative T cell costimulatory pathways in regulating autoimmunity. Immunity. 2004;20:529-538.
Kipnis J., Yoles E., Porat Z., et al. T cell immunity to copolymer 1 confers neuroprotection on the damaged optic nerve: possible therapy for optic neuropathies. Proc Natl Acad Sci U S A. 2000;97:7446-7451.
Kissel J.T., Mendell J.R., Rammohan K.W. Microvascular deposition of complement membrane attack complex in dermatomyositis. N Engl J Med. 1986;314:329-334.
Kitani A., Chua K., Nakamura K., et al. Activated self-MHC-reactive T cells have the cytokine phenotype of Th3/T regulatory cell 1 T cells. J Immunol. 2000;165:691-702.
Koide J., Engleman E.G. Differences in surface phenotype and mechanism of action between alloantigen-specific CD8+ cytotoxic and suppressor T cell clones. J Immunol. 1990;144:32-40.
Kruer M.C., Koch T.K., Bourdette D.N., et al. NMDA receptor encephalitis mimicking seronegative neuromyelitis optica. Neurology. 2010;74:1473-1475.
Kutzelnigg A., Lucchinetti C.F., Stadelmann C., et al. Cortical demyelination and diffuse white matter injury in multiple sclerosis. Brain. 2005;128:2705-2712.
Lai M., Huijbers M.G., Lancaster E., et al. Investigation of LGI1 as the antigen in limbic encephalitis previously attributed to potassium channels: a case series. Lancet Neurol. 2010;9:776-785.
Lang H.L., Jacobsen H., Ikemizu S., et al. A functional and structural basis for TCR cross-reactivity in multiple sclerosis. Nature Immunology. 2002;10:940-943.
Langrish C.L., Chen Y., Blumenschein W.M., et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233-240.
Lennon V.A., Wingerchuk D.M., Kryzer T.J., et al. A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet. 2004;364:2106-2112.
Levings M.K., Sangregorio R., Roncarolo M.G. Human CD25(+)CD4(+) T regulatory cells suppress naive and memory T cell proliferation and can be expanded in vitro without loss of function. J Exp Med. 2001;193:1295-1302.
Liu Y., Bando Y., Vargas-Lowy D., et al. CD200R1 agonist attenuates mechanisms of chronic disease in a murine model of multiple sclerosis. J Neurosci. 2010;30:2025-2038.
Liu Y., Teige I., Birnir B., et al. Neuron-mediated generation of regulatory T cells from encephalitogenic T cells suppresses EAE. Nat Med. 2006;12:518-525.
Lombardi G., Sidhu S., Batchelor R., et al. Anergic T cells as suppressor cells in vitro. Science. 1994;264:1587-1589.
Lucchinetti C., Bruck W., Parisi J., et al. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann Neurol. 2000;47:707-717.
Lunemann J.D., Huppke P., Roberts S., et al. Broadened and elevated humoral immune response to EBNA1 in pediatric multiple sclerosis. Neurology. 2008;71:1033-1035.
Majoie H.J., de Baets M., Renier W., et al. Antibodies to voltage-gated potassium and calcium channels in epilepsy. Epilepsy Res. 2006;71:135-141.
Mangan P.R., Harrington L.E., O’Quinn D.B., et al. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441:231-234.
Markovic-Plese S., Hemmer B., Zhao Y., et al. High level of cross-reactivity in influenza virus hemagglutinin-specific CD4+ T-cell response: implications for the initiation of autoimmune response in multiple sclerosis. J Neuroimmunol. 2005;169:31-38.
Mastaglia F.L., Phillips B.A., Zilko P.J. Immunoglobulin therapy in inflammatory myopathies. J Neurol Neurosurg Psychiatry. 1998;65:107-110.
Mathews M.B., Bernstein R.M. Myositis autoantibody inhibits histidyl-tRNA synthetase: a model for autoimmunity. Nature. 1983;304:177-179.
McKhann G.M., Cornblath D.R., Griffin J.W., et al. Acute motor axonal neuropathy: a frequent cause of acute flaccid paralysis in China. Ann Neurol. 1993;33:333-342.
McMahon E.J., Bailey S.L., Castenada C.V., et al. Epitope spreading initiates in the CNS in two mouse models of multiple sclerosis. Nat Med. 2005;11:335-339.
Meyers J.H., Chakravarti S., Schlesinger D., et al. TIM-4 is the ligand for TIM-1, and the TIM-1-TIM-4 interaction regulates T cell proliferation. Nat Immunol. 2005;6:455-464.
Miller S.D., Vanderlugt C.L., Begolka W.S., et al. Persistent infection with Theiler’s virus leads to CNS autoimmunity via epitope spreading. Nat Med. 1997;3:1133-1136.
Moalem G., Monsonego A., Shani Y., et al. Differential T cell response in central and peripheral nerve injury: connection with immune privilege. FASEB J. 1999;13:1207-1217.
Mokhtarian F., Zhang Z., Shi Y., et al. Molecular mimicry between a viral peptide and a myelin oligodendrocyte glycoprotein peptide induces autoimmune demyelinating disease in mice. J Neuroimmun. 1999;95:43-54.
Monney L., Sabatos C.A., Gaglia J.L., et al. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature. 2002;415:536-541.
Morgan D., Diamond D.M., Gottschall P.E., et al. A beta peptide vaccination prevents memory loss in an animal model of Alzheimer’s disease. Nature. 2000;408:982-985.
Munger K.L., Levin L.I., Hollis B.W., et al. Serum 25-hydroxyvitamin D levels and risk of multiple sclerosis. JAMA. 2006;296:2832-2838.
Neumann H., Cavalie A., Jenne D.E., et al. Induction of MHC class I genes in neurons. Science. 1995;269:549-552.
O’Connor K.C., McLaughlin K.A., De Jager P.L., et al. Self-antigen tetramers discriminate between myelin autoantibodies to native or denatured protein. Nat Med. 2007.
O’Hanlon G.M., Humphreys P.D., Goldman R.S., et al. Calpain inhibitors protect against axonal degeneration in a model of anti-ganglioside antibody-mediated motor nerve terminal injury. Brain. 2003;126:2497-2509.
Olson J.K., Croxford J.L., Calenoff M.A., et al. A virus-induced molecular mimicry model of multiple sclerosis. J Clin Invest. 2001;108:311-318.
Orgogozo J.M., Gilman S., Dartigues J.F., et al. Subacute meningoencephalitis in a subset of patients with AD after Abeta42 immunization. Neurology. 2003;61:46-54.
Park H., Li Z., Yang X.O., et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6:1133-1141.
Penna G., Adorini L. 1 Alpha,25-dihydroxyvitamin D3 inhibits differentiation, maturation, activation, and survival of dendritic cells leading to impaired alloreactive T cell activation. J Immunol. 2000;164:2405-2411.
Penna G., Amuchastegui S., Giarratana N., et al. 1,25-Dihydroxyvitamin D3 selectively modulates tolerogenic properties in myeloid but not plasmacytoid dendritic cells. J Immunol. 2007;178:145-153.
Perry V.H. Modulation of microglia phenotype. Neuropathol Appl Neurobiol. 1994;20:177.
Pluchino S., Quattrini A., Brambilla E., et al. Injection of adult neurospheres induces recovery in a chronic model of multiple sclerosis. Nature. 2003;422:688-694.
Pluchino S., Zanotti L., Rossi B., et al. Neurosphere-derived multipotent precursors promote neuroprotection by an immunomodulatory mechanism. Nature. 2005;436:266-271.
Pohl D., Krone B., Rostasy K., et al. High seroprevalence of Epstein-Barr virus in children with multiple sclerosis. Neurology. 2006;67:2063-2065.
Pohl-Koppe A., Burchett S.K., Thiele E.A., et al. Myelin basic protein reactive Th2 T cells are found in acute disseminated encephalomyelitis. J Neuroimmunol. 1998;91:19-27.
Roncarolo M.G., Levings M.K. The role of different subsets of T regulatory cells in controlling autoimmunity. Curr Opin Immunol. 2000;12:676-683.
Sabatos C.A., Chakravarti S., Cha E., et al. Interaction of Tim-3 and Tim-3 ligand regulates T helper type 1 responses and induction of peripheral tolerance. Nat Immunol. 2003;4:1102-1110.
Sadlack B., Merz H., Schorle H., et al. Ulcerative colitis-like disease in mice with a disrupted interleukin-2 gene. Cell. 1993;75:253-261.
Samuelsson A., Towers T.L., Ravetch J.V. Anti-inflammatory activity of IVIG mediated through the inhibitory Fc receptor. Science. 2001;291:484-486.
Sawcer S., Goodfellow P.N. Inheritance of susceptibility to multiple sclerosis. Curr Opin Immunol. 1998;10:697-703.
Serafini B., Rosicarelli B., Magliozzi R., et al. Detection of ectopic B-cell follicles with germinal centers in the meninges of patients with secondary progressive multiple sclerosis. Brain Pathol. 2004;14:164-174.
Sheikh K.A., Nachamkin I., Ho T.W., et al. Campylobacter jejuni lipopolysaccharides in Guillain-Barre syndrome: molecular mimicry and host susceptibility. Neurology. 1998;51:371-378.
Sicotte N.L., Liva S.M., Klutch R., et al. Treatment of multiple sclerosis with the pregnancy hormone estriol. Ann Neurol. 2002;52:421-428.
Stephens L.A., Mottet C., Mason D., et al. Human CD4(+)CD25(+) thymocytes and peripheral T cells have immune suppressive activity in vitro. Eur J Immunol. 2001;31:1247-1254.
Stewart G.J., McLeod J.G., Basten A., et al. HLA family studies and multiple sclerosis: A common gene, dominantly expressed. Hum Immunol. 1981;3:13-29.
Talbot P.J., Paquette J.S., Ciurli C., et al. Myelin basic protein and human coronavirus 229E cross-reactive T cells in multiple sclerosis. Annals of Neurology. 1996;39:233-240.
Tejada-Simon M.V., Zang Y.C., Hong J., et al. Cross-reactivity with myelin basic protein and human herpesvirus-6 in multiple sclerosis. Annals of Neurology. 2003;53:189-197.
Trapp B.D., Peterson J., Ransohoff R.M., et al. Axonal transection in the lesions of multiple sclerosis. N Engl J Med. 1998;338:278-285.
Truffault F., Cohen-Kaminsky S., Khalil I., et al. Altered intrathymic T-cell repertoire in human myasthenia gravis. Ann Neurol. 1997;41:731-741.
van der Mei I.A., Ponsonby A.L., Dwyer T., et al. Past exposure to sun, skin phenotype, and risk of multiple sclerosis: case-control study. BMJ. 2003;327:316.
Veldhoen M., Hocking R.J., Atkins C.J., et al. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179-189.
Veldhoen M., Uyttenhove C., van Snick J., et al. Transforming growth factor-beta ‘reprograms’ the differentiation of T helper 2 cells and promotes an interleukin 9-producing subset. Nat Immunol. 2008;9:1341-1346.
Wang L., Shi J., van Ginkel F.W., et al. Neural stem/progenitor cells modulate immune responses by suppressing T lymphocytes with nitric oxide and prostaglandin E2. Exp Neurol. 2009;216:177-183.
Waterhouse P., Penninger J.M., Timms E., et al. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science. 1995;270:985-988.
Webb M., Barclay A.N. Localisation of the MRC OX-2 glycoprotein on the surfaces of neurones. J Neurochem. 1984;43:1061-1067.
Weiner H.L. The challenge of multiple sclerosis: how do we cure a chronic heterogeneous disease? Ann Neurol. 2009;65:239-248.
Wilbanks G.A., Streilein J.W. Fluids from immune privileged sites endow macrophages with the capacity to induce antigen-specific immune deviation via a mechanism involving transforming growth factor-beta. Eur J Immunol. 1992;22:1031-1036.
Wingerchuk D.M., Lennon V.A., Pittock S.J., et al. Revised diagnostic criteria for neuromyelitis optica. Neurology. 2006;66:1485-1489.
Wintterle S., Schreiner B., Mitsdoerffer M., et al. Expression of the B7-related molecule B7-H1 by glioma cells: a potential mechanism of immune paralysis. Cancer Res. 2003;63:7462-7467.
Wright G.J., Puklavec M.J., Willis A.C., et al. Lymphoid/neuronal cell surface OX2 glycoprotein recognizes a novel receptor on macrophages implicated in the control of their function. Immunity. 2000;13:233-242.
Wucherpfennig K.W., Catz I., Hausmann S., et al. Recognition of the immunodominant myelin basic protein peptide by autoantibodies and HLA-DR2-restricted T cell clones from multiple sclerosis patients. Identity of key contact residues in the B-cell and T-cell epitopes. J Clin Invest. 1997;100:1114-1122.
Wucherpfennig K.W., Hafler D.A., Strominger J.L. Structure of human T-cell receptors specific for an immunodominant myelin basic protein peptide: positioning of T-cell receptors on HLA-DR2/peptide complexes. Proc Natl Acad Sci U S A. 1995;92:8896-8900.
Yagi H., Nomura T., Nakamura K., et al. Crucial role of FOXP3 in the development and function of human CD25+CD4+ regulatory T cells. Int Immunol. 2004;16:1643-1656.
Yang J., Jiang Z., Fitzgerald D.C., et al. Adult neural stem cells expressing IL-10 confer potent immunomodulation and remyelination in experimental autoimmune encephalitis. J Clin Invest. 2009;119:3678-3691.
Yao Z., Painter S.L., Fanslow W.C., et al. Human IL-17: a novel cytokine derived from T cells. J Immunol. 1995;155:5483-5486.
Yi Q, Lefvert A.K. Idiotype- and anti-idiotype-reactive T lymphocytes in myasthenia gravis. Evidence for the involvement of different subpopulations of T helper lymphocytes. J Immunol. 1994;153:3353-3359.
Yong V.W. Differential mechanisms of action of interferon-beta and glatiramer acetate in MS. Neurology. 2002;59:802-808.
Zhang J., Markovic-Plese S., Lacet B., et al. Increased frequency of interleukin 2-responsive T cells specific for myelin basic protein and proteolipid protein in peripheral blood and cerebrospinal fluid of patients with multiple sclerosis. J Exp Med. 1994;179:973-984.
Zhou T., Edwards C.K.3rd, Yang P., et al. Greatly accelerated lymphadenopathy and autoimmune disease in lpr mice lacking tumor necrosis factor receptor I. J Immunol. 1996;156:2661-2665.
Zhu C., Anderson A.C., Schubart A., et al. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat Immunol. 2005;6:1245-1252.
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]
Chapter 41 Neuroimmunology
Immune System
The normal functions of the immune system and the disorders resulting from its dysfunction are listed in Box 41.1.
Adaptive and Innate Immunity
The innate immune system consists of the following components:
1. Skin—The exterior surface of the body, primarily the skin, is the body’s primary defense against foreign pathogens. Many inflammatory cells and antigen-presenting cells (APCs) line the epidermis and serve as the first line of defense.
2. Phagocytes are cells capable of phagocytosing foreign pathogens. They include polymorphonuclear cells, monocytes, and macrophages. These cells are present in the blood as well as in organs. Phagocytes recognize cell components or pathogen-associated molecular patterns (PAMPs) of a variety of microorganisms through families of pattern recognition receptors (PRRs) expressed on their cell surface. PRRs allow phagocytes to attach nonspecifically and phagocytose pathogens, which are then killed via intracellular lysosomes. Families of PRRs include the Toll-like receptors (TLRs) and the nucleotide-binding oligomerization domain (NOD) receptors.
3. Natural killer (NK) cells—NK cells recognize cell surface molecules on virally infected or tumor cells. They subsequently bind to the infected cells and kill them via cell-mediated cytotoxicity.
4. Acute-phase proteins—C-reactive protein is a model acute-phase protein whose concentration increases in response to infection. C-reactive protein binds to cell surface molecules on a variety of bacteria and fungi and acts as an opsonin, essentially increasing recognition of pathogens by phagocytic cells.
5. Complement system—The complement system is a cascade of serum proteins whose overall function is to enhance and mediate inflammation. The complement system has the intrinsic ability to lyse the cell membranes of many cells including bacteria. It functions in concert with components of both the innate and adaptive immune systems and can also act as an opsonin, facilitating phagocytosis. The complement cascade can be directly activated by certain microorganisms through the alternative pathway, or it can be activated by particular antibody subtypes through the classical pathway.
The adaptive immune response consists of the following components:
1. Antibodies—Otherwise known as immunoglobulins (Igs), antibodies are able to specifically recognize a variety of free antigens. Igs are produced by B cells and are present on their cell surface. In addition, Igs are secreted in large amounts in the serum. Antibodies recognize specific microbial and other antigens through their antigen-binding sites and bind phagocytes via their Fc receptors, thereby facilitating antigen removal. Some subclasses of Ig are capable of activating complement via their Fc portion, thereby lysing their targets.
2. B cells—The primary function of B cells is to produce antibody. Antigen binding to B cells stimulates proliferation and maturation of that particular B cell, with subsequent enhancement of antigen-specific antibody production, resulting in the development of antibody-secreting plasma cells. Most B cells express class II major histocompatibility complex (MHC) antigens and have the ability to function as APCs.
3. T cells, or thymus-derived cells, have the ability to recognize specific antigens via their T-cell receptors (TCRs). T cells may be classified into two main groups, T-helper (TH) cells expressing CD4 antigen on their cell surface and T-cytotoxic (TC) cells expressing CD8 on their surface. CD4 T cells recognize antigen presented in association with MHC class II on the surface of APCs. CD4 T cells help to promote B-cell maturation and antibody production and produce factors called cytokines to enhance the innate or nonspecific immune response. CD8 T cells recognize antigen in association with MHC class I antigen on the surface of most cells and play an important role in eliminating virus-infected cells. Cytotoxic T cells are capable of damaging target cells via the release of degrading enzymes and cytokines. Responses in which the T cell plays a major role are termed cell-mediated immunity (CMI). T cell–macrophage interactions often lead to delayed reactions, termed delayed-type hypersensitivity (DTH).
4. APCs are required to present antigen to T cells. They are found primarily in the skin, lymph nodes, spleen, and thymus. Unlike B cells that can recognize free antigen, T cells are only capable of recognizing antigen in the context of self-MHC molecules. APCs process antigen intracellularly and present antigen peptide in the groove of their MHC class II molecules. The primary APCs are macrophages, monocytes, dendritic cells, and Langerhans cells.
Principal Components of the Immune System
T-Cell Receptors
The TCR consists of two glycosylated polypeptide chains, alpha (α) and beta (β), of 45,000 and 40,000 dalton molecular weight, respectively. This heterodimer of an α and β chain is linked by disulfide bonds. Amino acid sequences show that each chain consists of variable (V), joining (J), and constant (C) regions closely resembling Igs (Fig. 41.1). There are about 102 TCR-variable genes grouped by homology into a small number of families, compared with 103 or greater for Igs (see later discussion). The principles governing generation of diversity in the TCR are very similar to those for Ig genes. T cells can only recognize short peptides that are associated with MHC molecules. In contrast, the Ig receptor can recognize peptides, whole proteins, nucleic acids, lipids, and small chemicals.
Immunoglobulins
Immunoglobulins are glycoproteins that are the secretory product of plasma cells. Their biochemical structure and genomic organization is shown in Fig. 41.1. All Ig molecules share a number of common features. Each molecule consists of two identical polypeptide light chains (kappa [κ] or lambda [λ]) linked to two identical heavy chains. The light and heavy chains are stabilized by intrachain and interchain disulfide bonds. According to the biochemical nature of the heavy chain, Igs are divided into five main classes: IgM, IgD, IgG, IgA, and IgE. These may be further divided into subclasses depending on differences in the heavy chain.
Genetics of the Immune System
Antigen Receptor Gene Rearrangements
Constant (C) gene segments are present in all receptors. The V, D, J, and C gene segments along with the intervening noncoding gene segments between the J and C regions are initially transcribed into mature RNA. Through a process of RNA splicing, the noncoding gene segments are excised, and the V(D)JC messenger RNA (mRNA) is translated into protein. After binding antigen, B cells undergo somatic mutations that further increase the diversity and the affinity of antigen binding (affinity maturation). This phenomenon does not occur in T cells. During isotype switching in B cells, further rearrangements lead to recombination of the same variable region gene with new constant region genes (see Fig. 41.1).
Major Histocompatibility and Human Leukocyte Antigens
Class I antigens regulate the specificity of cytotoxic CD8+ T cells, which are responsible for killing cells bearing viral antigens or foreign transplantation antigens (Fig. 41.2). The target cells share class I MHC genes with the cytotoxic cell. Thus the cytotoxic cell that is specific for a particular virus is capable of recognizing the antigenic determinants of the virus only in association with a particular MHC class I gene product. The function of class II MHC gene products appears to be to regulate the specificity of T-helper cells, which in turn regulate DTH and antibody response to foreign antigens. Similarly, an immunized T-cell population will recognize a foreign antigen only if it is presented on the surface of an APC that shares the same class II MHC antigen specificity as the immunized T-cell population. Thus the functional specificity of the T-cell population is restricted by the MHC molecules they recognize. CD8+ T cells (cytotoxic) and CD4+ T cells (helper) are referred to as MHC class I and MHC class II restricted T cells, respectively (Fig. 41.3).
The analysis of the three-dimensional structure of the class I and class II molecules has confirmed the notion that these molecules are carriers of immunogenic peptides that are processed by APCs and presented on the cell surface (Fig. 41.4). Both MHC class I and class II molecules share similarities in crystal structure that allow them to accept and retain immunogenic peptides in grooves, or pockets, and present them to T cells.
Organization of the Immune Response
Initiation of the Immune Response
Accessory Molecules for T-Cell Activation
Costimulatory Molecules
Costimulatory molecules serve as a “second signal” to facilitate T-cell activation. Costimulatory pathways that are critical for T-cell activation include the B7-CD28 and CD40-CD154 pathways. Members of the integrin families including vascular cell adhesion molecule 1 (VCAM-1), intercellular adhesion molecule (ICAM-1), and leukocyte function antigen 3 (LFA-3) can provide costimulatory signals, but they also play critical roles in T-cell adhesion, facilitate interaction with the APCs, mediate adhesion to nonhematopoietic cells such as endothelial cells, and guide cell traffic (Fig. 41.5).
The B7-CD28 interaction is one of the most extensively studied costimulatory systems. The B7 molecules are expressed on antigen-presenting cells, and their expression is induced in activated cells. There are two forms of B7, B7-1 (CD80) and B7-2 (CD86), that share some homology but have different expression kinetics. The B7 molecules interact with their ligand, CD28, which is constitutively expressed on most T cells. Binding of the CD28 molecule mediates intracytoplasmic signals that increase expression of the growth factor, IL-2, and enhance expression of the anti-apoptotic molecule, Bcl-xL. An alternate ligand for B7 is CTLA-4, which is homologous to CD28 in structure, but in contrast to CD28, CTLA-4 functions to inhibit T-cell activation. Costimulatory molecules may deliver either a stimulatory (positive) or inhibitory (negative) signal for T-cell activation (Brunet et al., 1987). Examples of molecules delivering a positive costimulatory signal for T-cell activation include the B7-CD28, CD40-CD154 pathways. Examples of molecular pathways delivering a negative signal for T-cell activation include B7-CTLA4 and PD1-PD ligand (Khoury and Sayegh, 2004). The delicate balance between positive and negative regulatory signals can determine the outcome of a specific immune response.
Regulation of the Immune Response
Cytokines
Cytokines are necessary for T-cell activation and for the amplification and modulation of the immune response. A limited representation of the cytokines that participate in the immune response is shown in Table 41.1. Secretion of IL-1 by macrophages results in stimulation of T cells. This leads to synthesis of IL-2 and IL-2 receptors and finally to the clonal expansion of T cells. Only activated T cells express the IL-2 receptor (CD25); therefore the cytokine-induced expansion favors antigen-activated cells only. T-cell activation causes secretion of interferon gamma (IFN-γ), which induces expression of MHC class I and class II molecules on many cell types including APCs. This in turn increases the T-cell response to the antigen. Secretion of IL-2 also results in activation of NK cells that mediate lysis of tumor cell targets. In addition, IL-3 is released, resulting in stimulation of hematopoietic stem cells. The signal for differentiation of B cells to form antibody-secreting cells involves clonal expansion and differentiation of virgin memory B cells. IL-4 and B-cell differentiation factors secreted by T cells induce differentiation and expansion of committed B cells to become plasma cells.
Table 41.1 An Abridged List of Cytokines Involved in Interactions Between the Immune and Nervous Systems
CD4+ T-helper cells differentiate into TH1 or TH2 phenotypes, as well as a recently described TH17 subset, which secrete characteristic cytokines and stimulate specific functions. TH1 cells secrete IFN-γ, IL-2, and TNF-α. These cytokines exert proinflammatory functions and, in TH1-mediated diseases such as MS, promote tissue injury. IL-2, TNF-α, and IFN-γ mediate activation of macrophages and induce DTH. TH1 cell differentiation is driven by IL-12, a cytokine produced by monocytes and macrophages. In contrast, the TH2 cytokines IL-4, IL-5, IL-6, IL-10, and IL-13 promote antibody production by B cells, enhance eosinophil functions, and generally suppress cell-mediated immunity (CMI). TH3 cells secrete TGF-β, which inhibits proliferation of T cells and inhibits activation of macrophages. Cytokines of the TH1 type may inhibit production of TH2 cytokines and vice versa. More recently, a subset of T cells that predominantly produce IL-17 have been described (Yao et al., 1995). These cells are believed to represent a distinct subset from IFN-γ-producing TH1 cells, evidenced by the dependence of THIL-17 cells on IL-6 and TGF-β for differentiation (Bettelli et al., 2006; Mangan et al., 2006; Veldhoen et al., 2006) and IL-23 for expansion (Aggarwal et al., 2003; Langrish et al., 2005), as opposed to TH1 cells, which are dependent on IL-12 and IL-2, respectively, for differentiation and expansion. Both TH1 and TH2 cytokines have been shown to suppress the development of TH17 cells (Harrington et al., 2005; Park et al., 2005). THIL-17 cells facilitate the recruitment of neutrophils and participate in the response to gram-negative organisms. These cells may also play a role in the initiation of autoimmune disease. Another effector T-cell subset, TH9 cells, has recently been described (Dardalhon et al., 2008; Veldhoen et al., 2008). Driven by the combined effects of TGF-β and IL-4, TH9 cells produce large amounts of IL-9 and IL-10. It has been shown that IL-9 combined with TGF-β can contribute to TH17 cell differentiation, and TH17 cells themselves can produce IL-9 (Elyaman et al., 2009).
Traditionally, TH cell subsets have been distinguished by their patterns of cytokine production, but identification of distinguishing surface molecule markers has been a major advance in the field. Tim (T cell, immunoglobulin and mucin-domain containing molecules) represent an important family of molecules that encode cell-surface receptors involved in the regulation of TH1 and TH2 cell–mediated immunity. Tim-3 is specifically expressed on TH1 cells and negatively regulates TH1 responses through interaction with the Tim-3 ligand, galactin-9, also expressed on CD4+ T cells (Monney et al., 2002; Sabatos et al., 2003; Zhu et al., 2005). Tim-2 is expressed on TH2 cells (Chakravarti et al., 2005), and appears to negatively regulate TH2 cell proliferation, although this has not been fully established. Tim-1 is expressed on TH2 cells > TH1 cells, and interacts with Tim-4 on APCs to induce T-cell proliferation (Meyers et al., 2005).
Self-Tolerance
Peripheral Tolerance
Anergy Due to Failure of T-cell Activation
In normal circumstances, an APC presents antigen as a peptide + MHC complex (signal one). In the absence of signal one, the T cell dies because of neglect. If signal one is presented in the absence of costimulatory signals (signal two), the T cell becomes anergic. An example of this situation occurs when an antigen is presented by nonprofessional APCs that lack the appropriate costimulatory molecules (Fig. 41.6). However, when a T cell is activated, it up-regulates the expression of an alternate costimulatory molecule, CTLA-4. CTLA4 engagement by CD80 and CD86 on the surface of APCs sends a negative signal to the T cell, inhibiting cell growth and proliferation. Animals deficient for CTLA-4 expression on their T lymphocytes have an uncontrolled lymphoproliferative phenotype with autoreactivity (Waterhouse et al., 1995).
