Neuroendocrine Tumors of the Gastrointestinal and Pancreatobiliary Tracts
N. Volkan Adsay
David S. Klimstra
Neuroendocrine Cell System of the Gastrointestinal Tract
The neuroendocrine cells in the gastrointestinal (GI) tract constitute the largest endocrine system in the body. In addition to functioning as endocrine cells, they have autocrine, paracrine, and local neuromodulatory effects. They contain neurosecretory granules at the ultrastructural level, and some exhibit neuron-like cell processes. They have features of conventional endocrine cells and neural cells, which are reflected in the neoplasms derived from these cells. The term neuroendocrine is preferred to distinguish them from other endocrine cells, such as those of the thyroid, adrenal, or pituitary glands.
The neuroendocrine system of the GI and pancreatobiliary tract is heterogeneous. At least 14 different cell types are responsible for the production of more than 30 peptide hormones and bioamines. Enterochromaffin cells are the most abundant type and the presumed origin for most neuroendocrine tumors (NETs). They communicate with the crypt or gland lumen, and tumorigenesis may be induced by the luminal milieu. They are oriented to secrete their products basally toward the vessels, and NETs of these cells often produce secretory syndromes, such as carcinoid syndrome. In contrast, enterochromaffin-like (ECL) cells occurring in the stomach do not communicate with the lumen, but instead respond to circulating or local hormones. The neoplasms of neuroendocrine system in the GI tract were traditionally divided into foregut, midgut, and hindgut classifications, but this approach is no longer practical because of elucidation of the organ-specific characteristics of NETs.
Tumors of the neuroendocrine system are often identified by the hormone they secrete as functional NETs (e.g., gastrinoma, insulinoma). This approach is not based on immunohistochemical expression, but rather on serologic activity and consequent clinical manifestations. For example, strong immunohistochemical expression of gastrin in an NET is not considered sufficient to diagnose gastrinoma unless it is also clinically functional. One exception is ampullary and duodenal somatostatinomas, in which there are no measurable circulating levels of somatostatin. Tumors can secrete different hormones during the course of the disease, further complicating hormone-based classification. Similarly, although the specific type of a functional NET can have some loose association with specific histologic patterns, most of these associations are inconsistent and have not proved to have value in current practice.
Terminology and Classification
The terminology for neuroendocrine neoplasms has always been problematic. For well-differentiated tumors of the GI tract, the term carcinoid tumor has been widely used for more than a century, although some authorities advocate restricting the term to serotonin-producing NETs. Pancreatic tumors used to be designated as islet cell tumors. The preferred term,1 which is endorsed by the World Health Organization (WHO) 2010 classification,2 is well-differentiated neuroendocrine tumor (WDNET). WDNETs must be distinguished from poorly differentiated neuroendocrine carcinomas (PDNECs), such as small cell carcinoma and large cell neuroendocrine carcinoma. Designation as a WDNET is independent of the stage of the neoplasm, which represents a change from the WHO classifications of 2000 and 2004, which advocated the term well-differentiated neuroendocrine carcinoma for WDNETs with metastases.
Except for incipient neoplasms (i.e., tumorlets), all WDNETs should be considered malignant neoplasms. Low-grade and early-stage tumors often follow a protracted or benign clinical course after initial resection. Until recently, some authorities had classified these low-grade, early-stage lesions as benign (i.e., adenomas), citing their low incidence of recurrent or metastatic behavior, and this approach had been incorporated in an earlier WHO classification of pancreatic neuroendocrine tumors (PanNETs).3 Other investigators have maintained that even the rare occurrence of metastasis is an adequate sign of malignant potential, and they have proposed classifying the entire spectrum of NETs as carcinoma.4–6
As experience with the long-term behavior of these tumors has improved, it has become clear that WDNETs may be more aggressive than previously believed. In any case, they remain clinically, pathologically, and genetically distinct from PDNECs, and current recommendations restrict the use of the term carcinoma to the poorly differentiated group (not to metastasis or dissemination of WDNET) to avoid confusing these fundamentally distinct families of neuroendocrine neoplasms.
In reporting WDNETs of the GI tract (i.e., carcinoids) and pancreas (i.e., islet cell tumors), the preferred diagnostic expression is “WDNET (carcinoid tumor)” for the former and “WD pancreatic NET” for the latter. The types included in this group according to the WHO 2010 classification are NET grade 1 (Ki67 proliferation index less than 3% and mitotic index less than 2 mitoses per 10 high-power fields [HPFs]) and NET grade 2 (Ki67 index of 3% to 20% or 2 to 20 mitoses/10 HPFs).
Some morphologically well-differentiated NETs fall into the grade 3 category by virtue of their Ki67 and mitotic indices. Based on preliminary evidence,7 it appears that these rare examples are more closely related to WDNETs than to PDNECs. As such, the grade 3 category defined by the cutoff of 20% Ki67 or 20 mitoses per 10 HPFs includes two distinct but occasionally overlapping entities: highly proliferative WDNETs and true PDNECs of small cell and large cell neuroendocrine types similar to those in the lung. The latter group typically has a very high Ki67-labeling index (diffusely above 50%), very aggressive behavior, and favorable but short-lived responses to platinum-based chemotherapy. One study demonstrated that grade 3 neuroendocrine neoplasms with a Ki67 index in the 20% to 55% range had a better survival rate than those with a Ki67 index greater than 55%, but the less proliferative group did not respond to platinum-based chemotherapy.8
Further refinement of the grading cutoff points is needed. Data are being accumulated to allow better stratification based on the biologic, genetic, and therapeutic subgroups of WDNETs and PDNECs. The current approach has been endorsed by the WHO 2010 guidelines,2 College of American Pathologists (CAP),9 and American Joint Committee on Cancer (AJCC),10 and all NETs are graded and staged separately as for any other cancer type.
The most widely used staging systems for GI NETs are those from the CAP,9 AJCC,10 and European Neuroendocrine Tumor Society (ENETS).11,12 The prognostic value of these systems is still being verified. In reporting NETs, it is important to specify which staging system is used.
Neuroendocrine Tumor Differentiation
Neuroendocrine tumor differentiation manifests in various ways in the GI and pancreatobiliary tracts (Box 29.1). Many of the characteristics of these lesions are explored in this chapter, and more detailed discussions can be found in other chapters.
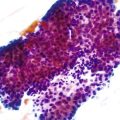
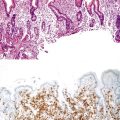
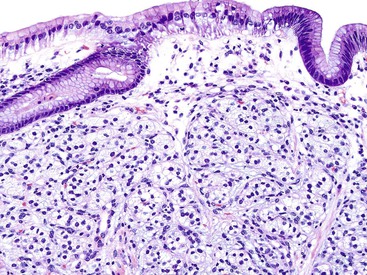
Early neuroendocrine cell proliferations can be seen throughout the GI tract. This phenomenon is best recognized in the stomach, where a spectrum of ECL cell proliferations occurs in the setting of hypergastrinemia and is usually compensatory for atrophic gastritis-related hypochlorhydria. In this condition, the trophic effects of gastrin lead to a spectrum of ECL cell proliferations, ranging from diffuse, linear, or nodular hyperplasia (Fig. 29.1) to dysplasia (i.e., in situ [Tis]) and to full-blown WDNETs. It is difficult to draw sharp lines between these processes, but criteria (albeit arbitrary) have been proposed. For microscopic proliferations, if there is nodular growth of ECL cells larger than 150 µm or a conglomeration of nodules, signs of microinfiltration, and new stroma, the lesion may be reported as “dysplasia or Tis.” These proliferations also can be designated “ECL cell proliferation with micronodule formation,” with further characteristics such as the size detailed in a comment. It is essential to look for signs of atrophy in the background and to investigate the clinical setting to determine the biologic significance of the lesion. The type 1 and 2 WDNETs that arise in this setting appear to be biologically different and more indolent than sporadic WDNETs (see Site-Specific Features, Stomach).
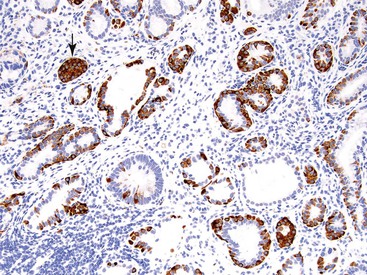
Another example of an incipient NET occurs in the pancreas. Multiple NETs may occur in patients with multiple endocrine neoplasia type I (MEN I) syndrome, but they can also occur without any syndromic background. Numerous microscopic nodules that appear to be precursor lesions often develop in these patients. Larger ones (<0.5 cm) are referred to as microadenomas (Fig. 29.2). They are often distinguished by morphology different from the background islets, a fibrous band around them, and clonal labeling for one of the pancreatic hormones; normal islets typically are shown to express multiple hormones in an established distribution by immunohistochemistry. As in the gastric ECL cell proliferations, it is often difficult to appreciate where the hyperplasia ends and true autonomous proliferation of the neoplasm begins.
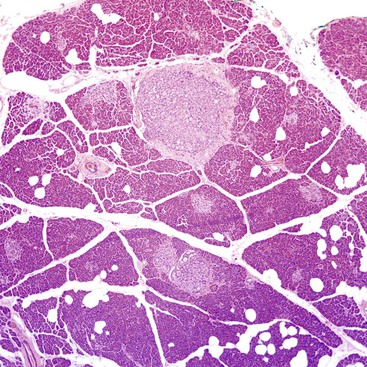
Incidental minute foci of neuroendocrine cell clusters encountered in the wall of the appendix are probably examples of the same proliferative phenomenon (Fig. 29.3). There are no established guidelines for the terminology or classification of these lesions, and there are no known underlying medical or genetic conditions that predispose to the proliferation. Patchy, small clusters that are less than a millimeter in the greatest dimension are typically reported as an “incidental neuroendocrine cell proliferation,” and a comment regarding their presumed benign nature should be provided.
True Neuroendocrine Tumors
Well-Differentiated Neuroendocrine Tumors
Clinical Features and Epidemiology
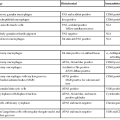
Most NETs are well differentiated (WDNETs). They are uncommon tumors, with an estimated annual incidence of 4 to 5 cases per 100,000 people. They represent 2% of all tumors of the GI tract. Gastroenteropancreatic NETs account for 75% of all NETs in the body. The increase in incidence of NETs is attributed to the improved diagnostic procedures and increased pathologic diagnoses of less differentiated tumors.13–16 The ileum and appendix are the most common sites, but NETs can occur in any part of the GI tract. However, WDNETs are very uncommon in the esophagus (Table 29.1).
Table 29.1
Staging of Gastrointestinal Neuroendocrine Tumors
| Site | Localized (%) | Regional (%) | Distant (%) |
| Stomach | 68* | 3 | 7 |
| Small intestine | 36 | 36 | 22 |
| Appendix | 55 | 29 | 10 |
| Colon | 39 | 27 | 25 |
| Rectum | 81 | 2 | 2 |

* Information is based on the U.S. Surveillance, Epidemiology, and End Results (SEER) Program analysis of 13,715 carcinoids. Totals are not 100% because some cases are not designated by site and stage.
Data from Modlin IM, Lye KD, Kidd M. A 5-decade analysis of 13,715 carcinoid tumors. Cancer. 2003;97:934-959.
Because they are slow growing and less infiltrative than ordinary carcinomas and often allow adaptive processes to take place, WDNETs are less likely than adenocarcinomas of the corresponding site to manifest with local symptoms. Instead, they often are identified by symptoms caused by the hormones they secrete, or they are detected incidentally during the workup for other conditions. Some come to attention at an advanced stage with metastasis.
The clinical manifestation of WDNETs depends on the site and the cell type. For example, appendiceal primaries are often detected incidentally during appendectomy as clinically silent, small tumors or as the cause of appendicitis. Ileal tumors are often not identified until they are metastatic, presumably because they do not produce local symptoms and remain undetected by routine endoscopy. They are associated with serotonin secretion, and when they metastasize, decreased hepatic metabolism may lead to the classic carcinoid syndrome: flushing, diarrhea, asthma, tricuspid regurgitation, and other symptoms.
Different functional NETs typically manifest at different clinical stages. For example, most pancreatic insulinomas manifest early with a set of symptoms and signs called the Whipple triad,17 and most pancreatic insulinomas are smaller than 2 cm at the time of diagnosis. In contrast, most pancreatic glucagonomas become symptomatic only when the tumor is large. The severity of the disease and clinical course (and prognosis) are significantly influenced by the type of hormone produced. For example, the diarrhea caused by VIPomas (i.e., WDNETs secreting vasoactive intestinal polypeptide, causing watery diarrhea, hypokalemia, and achlorhydria) can be debilitating and difficult to control and may even lead to death.
Some WDNETs come to clinical attention at another site based on the effects of their secreted hormone. For example, gastrinomas may lead to peptic ulcers of the duodenum or stomach (i.e., Zollinger-Ellison syndrome), or the gastrin they produce may exert a trophic effect on gastric ECL cells and lead to gastric WDNETs.
Gastrinomas may warrant a special note in the pathology report when they arise in the duodenum as an occult primary. They can manifest as very small primaries that lead to metastases and gastrinoma syndrome, in which the primary is often clinically and grossly undetectable because the lesion comprises a minute focus in the duodenal wall18 or within the gastrinoma triangle.19 In some cases, a gastrinoma may manifest as a large peripancreatic lymph node tumor (presumed to be a metastasis), and no primary can be documented by extensive sampling of the tissues from the triangle. This type of finding led to questioning whether there are true primary nodal gastrinomas.
Other characteristic clinical presentations should raise the possibility of WDNETs for the pathologist. For example, a large mass in the root of the mesentery without an obvious primary tumor elsewhere suggests a GI WDNET, especially one with an ileal origin. Similarly, a WDNET may be the culprit if a very large mass in the liver is encountered without finding an overt primary and the patient’s liver and general health are relatively good.
In addition to causing syndromes by secreting hormones, WDNETs may themselves be a manifestation of a genetic syndrome. PanNETs develop in 80% of MEN I patients, often in the background of multifocal proliferative changes in the islets. Neurofibromatosis may also lead to WDNETs, including the rare but distinctive ampullary somatostatinomas (i.e., glandular psammomatous WDNETs of the ampullary region). Twenty-five percent of patients with ampullary somatostatinomas have neurofibromatosis.20 For patients with synchronous NETs and gastrointestinal stromal tumors (GISTs) or neurofibromas, neurofibromatosis should be a strong consideration. In von Hippel-Lindau (VHL) syndrome, PanNETs often exhibit clear cytoplasm. VHL patients have a predilection for developing clear cell tumors with accompanying alterations in glycogen metabolism (Fig. 29.4). WDNETs have also been associated with inflammatory bowel disease.
The octreotide scan has changed the landscape greatly in making the clinical diagnosis of WDNETs. Targeting the type II somatostatin receptors that are widely expressed in most WDNETs, this scintigraphic scan has high specificity and sensitivity in detecting these tumors and their metastases.
Pathologic Features
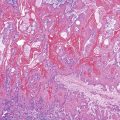
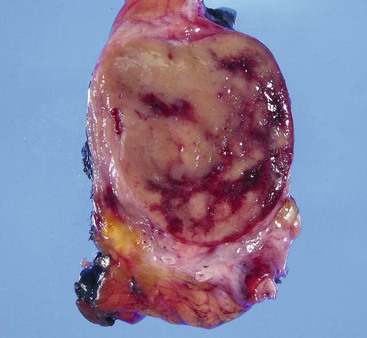
Grossly, WDNETs are usually well demarcated and grow with pushing borders. Rarely, PanNETs, especially early-stage and indolent tumors, may have capsule-like fibrous tissue surrounding the tumor. WDNETs are typically cellular, stroma-poor tumors, and cut sections reveal a fleshy, homogenous appearance (Fig. 29.5). The vascularity may lead to darker colors and hemorrhagic foci, especially if the tumor has been manipulated. In formalin-fixed specimens, the lesions become yellow to white. Necrosis and mucosal ulceration can be seen in more advanced cases and are typically signs of aggressive behavior. Some, particularly ileal NETs, can be associated with abundant sclerosis.
In the GI tract, WDNETs often form mucosa-covered, broad-based, polypoid lesions, with the bulk of the lesion in the submucosa and muscularis. PanNETs often protrude into the peripancreatic soft tissues, although rare examples may arise from the pancreatic duct wall and appear as sclerotic lesions constricting the duct, causing secondary dilation of upstream ducts, and mimicking intraductal papillary mucinous neoplasms (IPMNs).21,22
WDNETs typically are solid tumors. However, in the pancreas, as many as 10% manifest as a cystic mass caused by central degeneration, leaving a cuff of histologically conventional NET clinging to the cyst wall.23
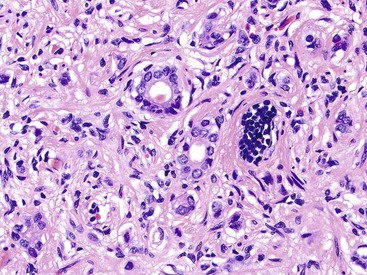
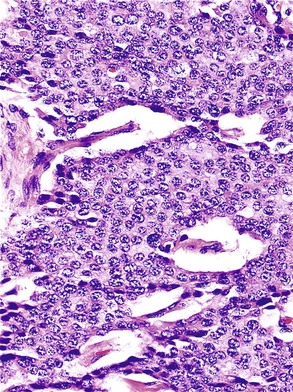
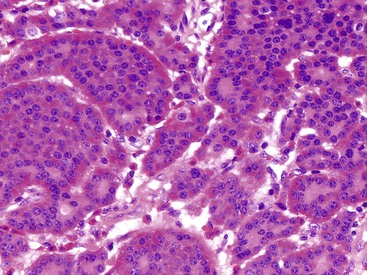
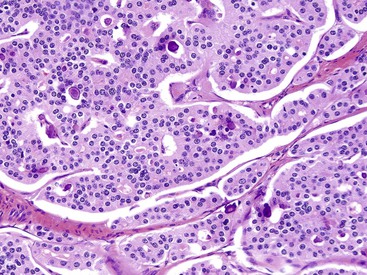
Microscopically, WDNETs are composed of uniform, round cells, with a moderate amount of cytoplasm and a coarsely granular, salt-and-pepper pattern of chromatin (Fig. 29.6). The latter finding is probably the most specific diagnostic feature of these tumors. Cytoplasmic granules may be evident and are especially prominent in midgut examples, along with melanin or lipofuscin pigments (Fig. 29.7). The cells grow in nests, acini, rosettes, ribbons, festoons, and trabeculae (Fig. 29.8). Gland formation by the tumor cells is common. Delicate vasculature is another hallmark, especially in cases with a prominent nested pattern (Fig. 29.9). Artifactual clefting around the nests is common, particularly in intestinal examples.
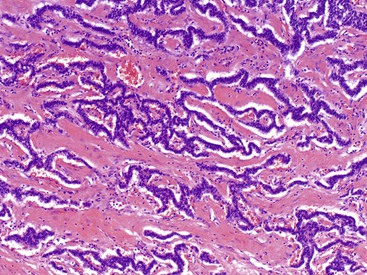
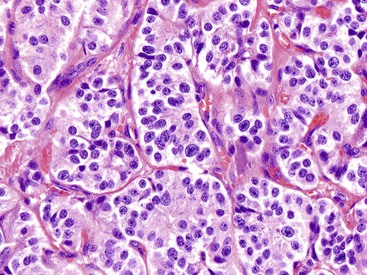
Morphologic Variants
In most of the numerous morphologic variants of WDNETs, at least one of the three classic morphologic characteristics (i.e., cellular monotony, cytoplasmic abundance, and salt-and-pepper chromatin pattern) is retained. Some WDNETs have prominent clear cytoplasm, a hallmark of VHL-associated PanNETs,24 although it is not entirely specific. In some clear cell NETs, the cytoplasm contains abundant lipid, producing a microvesicular cytoplasm and creating a picture reminiscent of adrenocortical cells. These lipid-rich variants (Fig. 29.10) often lack the nuclear features characteristic of NETs, potentially leading to misdiagnosis.25 In other NETs, the cytosolic contents, mostly intermediate filaments,26 may push the nucleus to the periphery and create a rhabdoid or plasmacytoid appearance (Fig. 29.11), mimicking signet ring cells, but intracellular mucin is lacking.
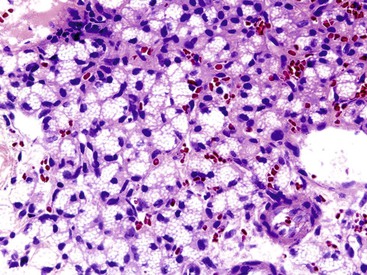
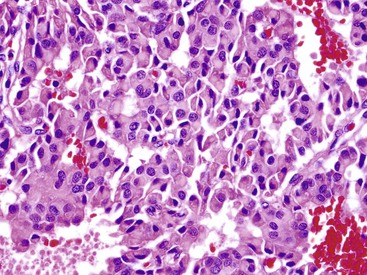
Some NETs, especially in the pancreas and occasionally in the stomach or rectum, have oncocytic features characterized by abundant acidophilic granular cytoplasm and single, prominent, eccentric nucleoli. When metastatic to liver, oncocytic NETs may be mistaken for hepatocellular carcinomas (Fig. 29.12). Some cases have a high nucleus-to-cytoplasm ratio that imparts a small, blue cell appearance. These cases often have a more diffuse growth pattern, which further accentuates the concern about a high-grade neoplasm, especially based on small biopsies. The Ki67 labeling index is helpful in establishing the well-differentiated nature of these samples.
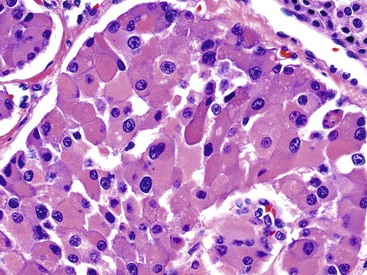
The endocrine atypia characteristic of normal endocrine organs may be seen in WDNETs as large, bizarre, pleomorphic nuclei with smudgy chromatin (Fig. 29.13).27 This pattern mimics the degenerative atypia seen in ancient schwannomas (i.e., rare, encapsulated, neural sheath tumor variants) and symplastic leiomyomas (i.e., atypical, bizarre, smooth muscle tumor variants). Pleomorphism in NETs can lead to their misdiagnosis as more aggressive neoplasms, but this finding has no prognostic importance unless accompanied by an elevated proliferative index rate.27
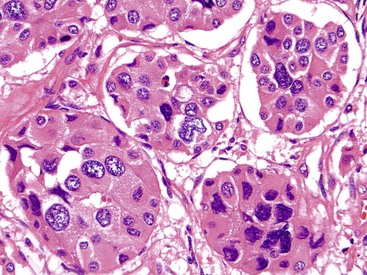
Stromal alterations may add variety to the morphologic spectrum of WDNETs. In some cases, these already hypervascular tumors become massively congested or hemorrhagic, creating a peliotic appearance. In others, particularly in the stomach, the stroma can exhibit myxoid features (Fig. 29.14). Although most WDNETs are fundamentally stroma-poor tumors, they can exhibit stromal sclerosis, especially the ileal NETs and PanNETs, which secrete serotonin (Fig. 29.15).28
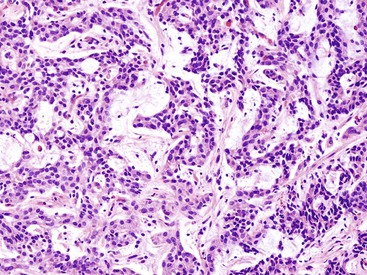
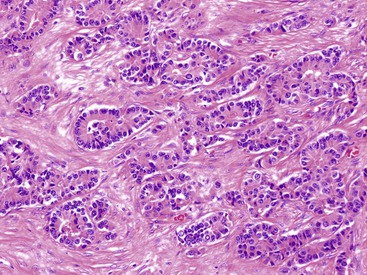
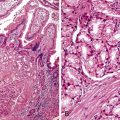
Rosette or glandular formations can be prominent in some WDNETs. In ampullary somatostatinomas, gland formation is so characteristic that the name glandular psammomatous NET of the duodenum has been applied (Fig. 29.16).18,29 They often entrap the ampullary ductules, which with the glands formed by the tumor can be mistaken for an adenocarcinoma. A WDNET variant in the appendix (i.e., tubular carcinoid) is also characterized by gland formation, and considering that neuroendocrine marker expression may be limited in this variant, it can be difficult to distinguish it from an adenocarcinoma. Ileal WDNETs also tend to have glands, which are often prominent in the superficial or mucosal component of the tumor and are often located at the periphery of the individual nests.
Unlike the pulmonary WDNETs, spindle cell morphology is uncommon in GI NETs and PanNETs. Occasionally, gastric WDNETs may have vague spindle cell morphology. Appendiceal tumors characterized by goblet cells form a distinct category (discussed later).
Immunohistochemistry and Electron Microscopy
WDNETs are defined by the presence of neuroendocrine granules, which usually can be highlighted by three widely available neuroendocrine markers: chromogranin, synaptophysin, and CD56. Chromogranin is the most specific, but its sensitivity is lower. Rectal and a subset of appendiceal NETs (i.e., tubular types) can be devoid of chromogranin A, which is the target of most available chromogranin antibodies. Synaptophysin is very sensitive but less specific, with a variety of mimics showing potential expression of this marker. CD56 is even less specific. These markers may not be needed for the diagnosis of WDNETs because the morphology is often distinctive enough. They may, however, become necessary for some of the morphologic variants and for the diagnosis of PDNECs (discussed later).
NETs show epithelial differentiation, and they express keratins in most cases. However, the type and degree of keratin positivity may vary. Although staining for a wide spectrum keratins is commonly positive, cytokeratin (CK) 7 and CK20 expression is relatively uncommon. CK19 occasionally is expressed in PanNETs and has been used as a marker for adverse outcomes.
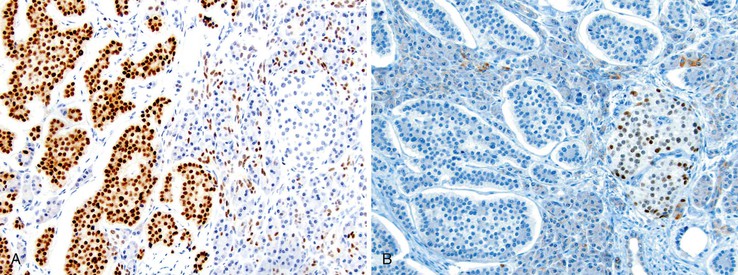
Nuclear transcription factors involved in the embryonic development of site-specific neuroendocrine cells have been used to determine the primary site of a WDNET,30 but they are not specific. For example, CDX2 is commonly expressed in the NETs of the GI tract (Fig. 29.17, A). The islet 1 transcription factor (ISL1) is expressed commonly in the PanNETs but is also common in rectal WDNETs (see Fig. 29-17, B).31,32 Pancreatic duodenal homeobox 1 (PDX1) is a marker of PanNETs at primary and metastatic sites,33 but the specificity of this marker is unknown. Thyroid transcription factor 1 (TTF-1) expression is uncommon in pancreatic and GI WDNETs, and its sensitivity for pulmonary carcinoid tumors is limited. TTF-1 is expressed in small cell carcinomas of any organ. The PAX8 transcription factor is commonly expressed in pancreatic and rectal NETs, but it is typically absent in ileal NETs. As in pancreatic islet cells, which express progesterone receptors and CD99 (presumably resulting from cross reactivity),34 PanNETs can also express these markers.
There are immunohistochemical caveats to consider. Ileal and rectal specimens can express prostate-specific acid phosphatase. Carcinoembryonic antigen (CEA) is expressed in 60% of WDNETs, and CA 19-9 positivity may also be seen. Staining for S100 can be positive in some WDNETs, especially in the appendix and some ampullary somatostatinomas.
The value of immunohistochemical analysis of hormone production is not clear. Correlation with functional activity is highly imperfect. Many tumors have multihormonal activity, and they may alter the predominant secretion over time.
Although somatostatin receptor scintigraphy is widely used in the clinical setting, immunohistochemical labeling for the somatostatin receptor is not typically performed in the United States. In Europe, it is more widely used in selected clinical conditions.35,36
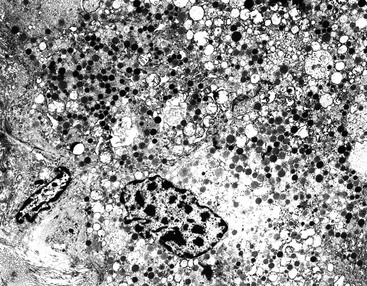
Ultrastructural detection of neurosecretory granules represented as membrane-bound, dense, core granules by electron microscopy is helpful in establishing the diagnosis of WDNETs. The pattern of the granules may be helpful in determining the specific cell type (Fig. 29.18). However, electron microscopy is seldom used in modern surgical pathology because of technical impracticalities and the utility of immunohistochemistry. Electron microscopy can help to demonstrate neuroendocrine differentiation in unusual settings, such as poorly differentiated or amphicrine tumors.
Molecular Biology
Neuroendocrine tumors that arise as a result of familial syndromes (i.e., MEN I, VHL, tuberous sclerosis, or neurofibromatosis type 1 [NF1]) have molecular alterations characteristic of the corresponding syndrome. Sporadic tumors may also show these alterations. For example, somatic mutations or loss of heterozygosity of the MEN1 gene (11q13) and its protein product menin can be seen in gastric and pancreatic WDNETs, and loss of NF1 (17q11) and its protein product neurofibromin may be found in somatostatinomas. WDNETs of different sites have different genetic backgrounds. For example, ileal NETs are not associated with MEN I and VHL syndromes.
Alterations of genes involved in the pathogenesis of adenocarcinomas of the GI and pancreatobiliary tracts, such as TP53, HER2, CDKN2A, and SMAD4 (DPC4) are much less common in PanNETs. However, almost one half of PanNETs have mutually exclusive inactivating somatic mutations of the DAXX (death-domain associated protein) and ATRX (α-thalassemia, mental retardation syndrome X-linked) genes. These genes are involved in a chromatin-remodeling complex critical in telomere maintenance. A subset of PanNETs show somatic mutations of the genes that encode for proteins in the mammalian target of rapamycin (mTOR) cell signaling pathway, including somatic mutations of PIK3CA, PTEN, and TSC2. Other molecules that seem to be abnormal in a smaller subset of cases include the X chromosome, heat shock protein 90 (HSP90), insulin-like growth factor 1 receptor (IGF1R), and epidermal growth factor receptor (EGFR).
Differential Diagnosis
Adequate morphologic fidelity among gastroenteropancreatic NETs helps to make their diagnosis relatively straightforward. However, a few situations can lead to diagnostic problems. Limited biopsies or preservation artifacts sometimes lead to misdiagnoses, especially considering that they can be located at the base of the mucosa and may be underrepresented in the biopsy. Rosettes and tubules can be mistaken as adenocarcinoma. In crushed specimens, WDNETs may mimic lymphoid tissue. More problematic are the rare and underrecognized variants. The pleomorphic variant can be mistaken as a high-grade carcinoma (see Fig. 29-13). Clear cell or lipid-rich variants (see Fig. 29-10) are often misdiagnosed as carcinomas from the kidney, adrenal, or liver. WDNETs with oncocytic features can be mistaken for a primary hepatocellular carcinoma in the liver (see Fig. 29-12) (see Site-Specific Features).
Pathologic Prognosticators and Clinical Outcomes
GI NETs and PanNETs are low-grade malignancies. WDNETs have a better prognosis than the conventional adenocarcinomas of the corresponding sites. However, as more data accumulate on the long-term prognosis of these tumors, it is clear that a significant proportion eventually recurs and metastasizes. The preferred sites of distant metastasis are liver, lung, peritoneum, and bone. Even cases with distant metastasis may have a protracted clinical course.
Outcomes are determined by the clinical setting, primary location, cell type, stage, grade, and other tumor features. The effect of the clinical setting is best exemplified in the stomach, where WDNETs arising because of hypergastrinemia against the background of autoimmune gastritis typically follow a benign clinical course. In contrast, sporadic WDNETs are often aggressive.
Because NETs in different locations often are studied separately from each other, site-specific differences in their behavior are coming to light. For example, appendiceal and rectal NETs are often small and localized when detected, whereas ileal tumors usually have metastasized (see Site-Specific Features).
Although part of explanation for the more benign conduct of NETs in some sites is their cell biology and anatomic milieu of these regions, part of the behavior is related to the stage of the tumors at detection. For example, most appendiceal and rectal WDNETs at diagnosis are small and benign, whereas those larger than 2 cm have malignant potential. Stage is proving to be the most powerful predictor of outcome.37
Different staging schemes have been proposed. In the United States, the tumor-node-metastasis (TNM) system put forth by the AJCC and Union for International Cancer Control (UICC) and endorsed by the CAP is in wide use, although a different TNM staging system proposed by the ENETS may prove to have more validity.38
In staging NETs, the same principles used for other cancers are applicable. For example, as a part of the recent paradigm shift,39,40 an intestinal WDNET should be classified as T4a if the surrogate serosal changes indicate clearcut tumor involvement, even if the tumor cells are not at the serosal surface. Staging of multiple, separate tumors (typically in ileal NETs) is challenging because it remains unknown whether they are synchronous primary tumors or metastases. In all cases, multicentricity should be documented, because these NETs are clearly more aggressive.41 The ENETS proposal, which is based on the size of the tumor rather than ambiguous parameters such as the peripancreatic soft tissue involvement used in the CAP, AJCC, and TNM systems, may prove to be more applicable and relevant for staging PanNETs.
Along with stage, cell biology influences outcome. Determination of the cell type is based on the clinical finding of a functional syndrome. Hormonal activity (i.e., cell type) correlates with behavior, although mostly indirectly and according to the stage at which tumors manifest. For example, clinically functioning insulinomas pursue an indolent clinical course in 90% of cases and have sometimes been classified as benign, but the favorable outcome of patients with insulinomas in part results from the relatively small size at which the tumors are detected because of the symptoms they cause. In contrast, other syndromic PanNETs result in recurrences or metastases in 50% to 70% of cases. Duodenal gastrinomas often have metastasized when the primary is less than 1 cm in diameter.
Authorities agree that NETs should be graded and staged separately. The grading system put forth by ENETS and endorsed by the Memorial Sloan-Kettering Cancer Center consensus group1 and the WHO guidelines2 classifies NETs in three categories (Table 29.2) based on mitotic activity (number of mitoses per 10 HPFs) and Ki67 proliferation index (percent of cells). This system is applied regardless of the primary location. As with other semiquantitative analyses used in surgical pathology, this grading system is subject to challenges created by heterogeneity, false positivity, and variations in methods, instruments, operators, and other extrinsic factors.42
Table 29.2
Grade (Subclassification) of Neuroendocrine Tumors
| Neuroendocrine Tumor Grade | Mitoses (per 10 HPFs)* | Ki67 Proliferation Index (%)† |
| 1 | <2 | <3 |
| 2 | 2-20 | 3-20 |
| 3 | >20 | >20 |
* Mitotic count in at least 50 high-power fields (HPFs); 1 HPF = 2 mm2.
† MIB1 antibody; percent of 500 to 2000 tumor cells in areas of highest nuclear labeling.
Data from Rindi G, Arnold R, Bosman FT, et al. Nomenclature and classification of neuroendocrine neoplasms of the digestive system. In: Bosman FT, Carneiro F, Hruban RH, Theise ND, eds. WHO Classification of Tumors of Digestive System. Lyon, France: International Agency for Research on Cancer (IARC) Press; 2010:13-14.
Image analysis systems for measuring the Ki67 index,43 which are not available in all centers, are user dependent. For Ki67 counting, visual inspection, which was once thought to be acceptable, is considered unreliable for differentiating grade 1 from grade 2.42,44 The preferable approach is manual or digital counting of a captured image of the proliferation hotspots.45 Guidelines in the original publications and CAP Web page are unclear about the fractional percentages that fall into the 2% to 3 % range, but most authorities assign cases with an index of less than 3% to grade 1.9,44 In the updated North American Neuroendocrine Tumor Society (NANETS) guidelines, this situation is clarified by making 3% or higher the cutoff for grade 2.46
Many studies have confirmed the value of grading metastatic tumors.47 If the Ki67 count is performed accurately, it often proves to be higher than the mitotic count, perhaps negating the need for the more tedious mitotic count. Studies have shown Ki67 to be valuable for limited cytologic specimens (i.e., cell blocks).48,49 In mitotic counting, it is imperative to correct for the microscope’s field area, which can vary greatly. Some studies are reassessing the mitotic cutoff points of the original proposal.50,51 Variations in optimal cutoff points may exist among anatomic sites, although more data are needed before modifications to the current grading scheme can be implemented. The 20% cutoff for Ki67 appears to lump two prognostically and biologically distinct groups into one category. Studies show that NETs with a Ki67 proliferation index of more than 55% are much more aggressive7 and closer to PDNECs8 than those with Ki67 values between 20% and 50%, which may prove to be merely highly proliferative versions of WDNETs. Because of this limited knowledge, we classify the former as PDNECs and the latter as WDNETs with an elevated proliferation index; however, more work is needed to clarify this issue.
In addition to grade and stage, other histologic findings may have prognostic significance for WDNETs (Boxes 29.2 and 29.3). Factors such as tumor invasion, necrosis, and ulceration usually should be included in pathology reports.
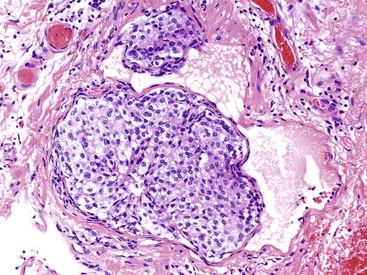
Although subjective, perineural invasion is thought to confer a worse prognosis. The guidelines are not clear about whether a nerve entrapped by the lesion should be regarded as invasion. It is important to recognize that non-neoplastic neuroendocrine cells can be found in a perineural location, particularly in the setting of chronic pancreatitis. In the appendix, neuroendocrine cell proliferations are intimately admixed with nerves. Similarly, vascular invasion is a sign of an aggressive tumor that can occupy a small blood or lymphatic vessel or directly extend into larger veins. Because WDNETs are highly vascularized tumors, vascular invasion can be difficult to recognize in the tumor. In the small intestine, the characteristic artifactual clefting may lead to the erroneous impression of vascular invasion (Fig. 29.19).
Necrosis is used as a criterion for identification of the intermediate-grade (atypical carcinoid) group of pulmonary NETs. The meaning of necrosis is less clear for GI and pancreatobiliary tumors. In some studies, it was found to be a strong prognosticator of outcome. It is an important feature of PDNECs, and it can identify more aggressive versions of WDNETs in the pancreas.52 Necrosis should be documented in reports about WDNETs. Similarly, mucosal ulceration is often a sign of aggressive behavior of gastrointestinal WDNETs, and it should be documented in the pathologist’s report.
In addition to the proliferation index, various immunohistochemical and molecular prognostic markers are being investigated. There is strong evidence from many studies that CK19 expression may have some adverse prognostic significance in PanNETs. C-KIT (CD117) has also been advocated as an independent adverse prognosticator.53 However, none of the markers under investigation is routinely used in clinical practice.
As in any malignant neoplasm, it is important to document the margin status for resections of WDNETs. If possible, the distance from the deepest tumor to the margin should be reported for polypectomies and mucosal resections.
The major morbidity for some functional WDNETs is not the direct effect of the tumor but rather the impact of the hormonal syndrome. For example, VIPomas may be lethal because of massive diarrhea, and serotonin-producing WDNETs may cause serious valvular heart disease. Given the multiple parameters affecting the outcomes of patients, decision support tools such as nomograms are being developed to stratify risk.54
Treatment
WDNETs are low-grade malignancies. Surgical removal can be curative for early-stage tumors in a significant number of cases.14 Resection, radiofrequency ablation, and chemoembolization of metastatic tumors are increasingly used, but the specific indications for these procedures are still being debated.46
If clinically feasible, an oncologic operation is preferred to enucleation or limited excision. Management varies by site and stage of the tumor. Although ileal tumors warrant resection regardless of tumor size, small appendiceal and rectal lesions can be managed by limited or local removal, appendectomy (if <2 cm), or polypectomy (if <0.5 cm).
Because they are relatively low-proliferative lesions, the response of WDNETs to cytotoxic agents is minimal or absent. Somatostatin analogues (e.g., octreotide) have shown some efficacy, but mostly leading to tumor stabilization rather than remission. Similar experience has been recorded for interferons. Among the targeted therapies being developed, everolimus and sunitinib have shown significant promise for PanNETs. Many new agents are being developed to target HSP90, IGF1R, EGFR, and mTOR pathways.46,55,56
Site-Specific Features
Although WDNETs show some common characteristics regardless of where they arise, there are also substantial differences based on their origin. These site-specific characteristics of WDNETs are also discussed in Chapters 24 through 28 and 40.
Esophagus
WDNETs are uncommon in the esophagus, which is the least common site for GI NETs. This may be related to the fact that the esophagus does not have a significant neuroendocrine cell population, although the mucosal glands in the distal esophagus have scattered neuroendocrine cells. Reported cases show a male predominance, and patients are most often in their 60s at diagnosis. Most cases are distal, particularly at the gastroesophageal junction. In one study, 50% of the cases were metastatic at the time of diagnosis. There does not seem to be any specific association or histopathologic or molecular findings that distinguish esophageal WDNETs.
Stomach
Gastric NETs represent 6% of all GI NETs. Their incidence appears to be on the rise, attributed partly to the increase in detection caused by widespread use of endoscopy.57–59 Although several GI neuroendocrine cell types are located in the stomach, 70% of tumors are derived from a single cell type (i.e., ECL cells), and less commonly, from G cells or enterochromaffin cells.
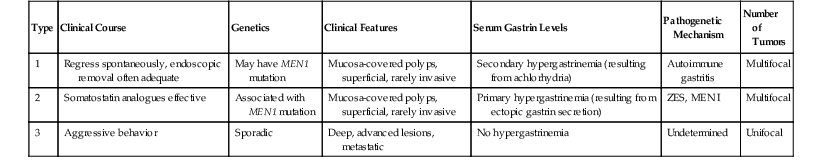
Gastric WDNETs arise in three distinct clinical settings, which impact the biology and management of the tumors (Table 29.3). Types 1 and 2 (or types A and B) arise in the fundic mucosa against a background of hypergastrinemia. Type 1 tumors are associated with secondary (compensatory) hypergastrinemia due to the hypochlorhydria of autoimmune gastritis and indirectly to pernicious anemia. Type 2 WDNETs are associated with primary hypergastrinemia (i.e., gastrin-secreting tumors in Zollinger-Ellison or MEN I syndromes). The fundamental process is the trophic effect of excessive gastrin on ECL cells, which leads to proliferations of this cell type. In Japan, similar cases have been associated with Helicobacter pylori infection, which can lead to the suppression of D cells and secondary hypergastrinemia, but this group of tumors has not been well characterized. Proton pump inhibitor use may cause neuroendocrine cell hyperplasia, mimicking the type 1 and 2 pathogenesis mechanism.60 Type 3 gastric WDNETs are sporadic tumors that can arise anywhere in the stomach.
Table 29.3
Clinical Features of Gastric Neuroendocrine Tumors
| Type | Clinical Course | Genetics | Clinical Features | Serum Gastrin Levels | Pathogenetic Mechanism | Number of Tumors |
| 1 | Regress spontaneously, endoscopic removal often adequate | May have MEN1 mutation | Mucosa-covered polyps, superficial, rarely invasive | Secondary hypergastrinemia (resulting from achlorhydria) | Autoimmune gastritis | Multifocal |
| 2 | Somatostatin analogues effective | Associated with MEN1 mutation | Mucosa-covered polyps, superficial, rarely invasive | Primary hypergastrinemia (resulting from ectopic gastrin secretion) | ZES, MEN I | Multifocal |
| 3 | Aggressive behavior | Sporadic | Deep, advanced lesions, metastatic | No hypergastrinemia | Undetermined | Unifocal |
MEN I, Multiple endocrine neoplasia syndrome, type I; ZES, Zollinger-Ellison syndrome.
Type 1 and 2 gastric WDNETs often occur in the background of or evolve from ECL cell proliferations (see Fig. 29-1). A system has been proposed to classify these lesions as hyperplasia (i.e., patchy, linear, and micronodular) if they do not form nodules larger than 150 µm in diameter, as dysplasia if nodules are larger than 150 µm (0.15 mm) or are fused units or infiltrative nodules, and as micro-NETs (i.e., microcarcinoids) if they are less than 500 µm (0.5 mm).61,62 The same group of investigators attempted to provide a more detailed definition of what qualifies as hyperplasia, but the work needs to be validated.63
For staging purposes, dysplasias and micro-NETs can be regarded as Tis, although many of them exhibit patchy involvement of the muscularis mucosa. Nodules larger than 0.5 mm are regarded as full-blown WDNET (carcinoid), but their behavior is distinctly better than that of the sporadic type (type 3) WDNETs, which have a metastasis rate of approximately 15%. It is important for the pathologist to recognize the background changes (i.e., autoimmune and precursor lesions) and clinical correlations. Although type 1 lesions appear to be benign and may regress spontaneously or after administration of somatostatin analogues,64 some patients require antrectomy to eliminate the source of excess gastrin.
Immunohistochemistry can help in the diagnosis of early proliferations, especially if the specific biopsy site is not verifiable by histologic examination alone. Neuroendocrine markers (i.e., chromogranin and synaptophysin) highlight the neuroendocrine cell proliferation and distinguish it from other cell populations. Gastrin immunostaining helps to establish that the suspect cells are not G cells. Evidence of autoimmune and atrophic gastritis in the background is also important. ECL cell proliferations can sometimes be difficult to distinguish from lymphoplasmacytic infiltrates, which are their common companions. Type 2 cases are characterized by loss of heterozygosity of 11q13 (location of the MEN1 gene), which can also be seen in some type 1 cases but not in type 3.
Most type 1 and 2 WDNETs manifest as mucosa-covered polyps, which are often multifocal and superficial at the time of diagnosis. Type 3 (sporadic) WDNETs are often advanced and involve the stomach transmurally. They also exhibit more aggressive behavior, with lymph node metastases in 60% and liver metastases in 50% of the cases. Some examples overlap with the PDNECs (discussed later).
Duodenum
WDNETs of the ampulla and duodenum have different presentation patterns. Ampullary region cases often manifest with obstructive jaundice. Delineation of duodenal WDNETs is based on the cell type and the corresponding clinical and pathologic characteristics. A significant proportion of gastrin-producing WDNETs occur in the gastrinoma triangle of the duodenum.65 One third are associated with Zollinger-Ellison syndrome, and these patients are typically younger, and the tumors have more indolent behavior than seen in other cases. Despite being small or occult, one third of the duodenal gastrinomas have lymph node metastases. Some syndromic gastrinomas appear as primaries within peripancreatic lymph nodes, although undetected minute duodenal primaries with large nodal metastases likely account for some of these cases.
Ampullary somatostatinoma (i.e., glandular psammomatous NET of the duodenum) is a distinctive type of WDNET (see Fig. 29-16) that appears to be restricted to the ampullary region.29,66–68 It is discussed in more detail in Chapter 41. A higher percentage of ampullary somatostatinomas is reported for African Americans. These NETs are called somatostatinomas not because the patients have somatostatin-related symptoms, but because they typically stain with somatostatin immunohistochemically, and they are fairly distinctive morphologically.
One fourth of these patients have neurofibromatosis, and some have concomitant GISTs or GI neurofibromas. Almost half have lymph node metastases at presentation, but they appear to be indolent. The serotonin-producing WDNETs that occur in the duodenum appear to be similar to their more common kindreds in the distal small intestine.
Jejunum and Ileum
The distal small bowel is the most common site of clinically relevant WDNETs. These tumors often manifest with nonspecific findings and may be misdiagnosed as irritable bowel syndrome because of the inaccessibility of the site to endoscopic examination. Some patients have intussusception. Small bowel WDNETs have been associated with a variety of conditions, including celiac disease, Crohn’s disease, duplications, and Meckel diverticulitis, but none has proved to be causal.
Most cases appear to be sporadic, and most produce serotonin (i.e., enterochromaffin cell type). Most of the attributes of the carcinoids and carcinoid syndrome refer to this group; however, a variety of hormones can be detected in the tumor and the serum of these patients. At clinical presentation, approximately 5% of patient have carcinoid syndrome, which is typically seen in cases with distant metastases that allow the serotonin secreted to bypass liver metabolism. Approximately one fourth of patients have multiple mucosal polyps. They may represent true synchronous neoplasms or intramucosal metastases from a single lesion; studies favor the latter theory.69
Small bowel WDNETs are notorious for manifesting with mesenteric metastatic disease leading to buckling or tethering of the bowel. For a patient with a large mesenteric mass, the possibility of an ileal or jejunal WDNET should be considered in addition to alternative diagnoses such as mesenchymal and lymphoid neoplasms. Fibrosis, presumably related to factors secreted by the tumor, can be striking and is thought to contribute to the obstructive symptoms. Some specimens have Crohn’s disease–like hypervascular polyps adjacent to the tumor, which are thought to be secondary and attributed to transforming growth factor. The reported 5-year survival rate for localized WDNETs is 60% to 70%, whereas the rate for those with distant metastasis is approximately 21%.
The typically mucosa-covered polyps are firm and homogenous on sections. The muscularis propria may be thickened, which is attributed to trophic factors. Microscopically, peripheral cytoplasmic granularity, subtle lipofuscin- or melanin-type pigment formation (see Fig. 29-7), rosettes (especially at the periphery of the nests), and monotony of the polygonal cells appear to be more common or more striking than at other sites. Retraction artifact accentuates the nested pattern. Microproliferations, which can be regarded as hyperplastic, may be seen in the adjacent mucosa. Perineural invasion is common. Lymphatics, especially those immediately adjacent to the mucosa, are often involved and may cause linear spread, which may be responsible for the multifocal mucosal growth in some cases. Medium-sized mesenteric arteries may exhibit a peculiar elastotic change similar to the cardiac changes seen in some patients with carcinoid syndrome. Alterations in chromosome 18 are a common finding in these tumors.
Appendix
If all the small neuroendocrine cell proliferations discovered in the appendix in autopsies or incidentally in appendectomies are also dignified as NETs, appendiceal NETs become one of the more common tumors in the body. These proliferations are reported in 0.7% to 2% of all appendices when sampled extensively. Approximately two thirds are located at the tip of the appendix and are not thought to cause the appendicitis that brings them to clinical attention. Those located in the tip can lead to the so-called “bell-clapper” configuration.
Patients with appendiceal NETs are approximately 2 decades younger than those with other NETs. Whether this is a reflection of the mean age of appendectomy or the peak density of subepithelial neuroendocrine cells known to occur in the same age group, or both, is an issue of debate. Appendiceal WDNETs are more commonly reported in women, but that may be related to incidental appendectomies performed during gynecologic operations.
More than 95% of appendiceal WDNETs are smaller than 2 cm in diameter. The incidence of metastasis is very low among these patients, and appendectomy is considered sufficient treatment. In contrast, in cases with tumors larger than 2 cm in diameter, nodal or distant metastases are identified in one third of patients. Most authorities agree on the need for right hemicolectomy for this group. However, whether this operation improves survival has not been proved. The prognostic role of mesoappendiceal involvement or perineural invasion has not been established. The natural occurrence of neuroendocrine cells and paraganglia in association with nerves in this region complicates defining the role of perineural invasion.
Appendiceal WDNETs that are presumably derived from L cells can have extensively or exclusively tubular architecture. This, combined with the paucity or total negativity of chromogranin A staining and variable CK7/CK20 profile,70 make tubular WDNETs difficult to distinguish from adenocarcinomas. Goblet cell carcinoid of the appendix is discussed later.
Colon
WDNETs are uncommon in the large intestines. Almost half are located in the cecum.71 They are more common in women and tend to be fairly large at the time of diagnosis, and appear to be aggressive. The 5-year survival rate is about 70% for localized tumors and 40% for those with regional spread. For tumors smaller than 2 cm in diameter, the reported metastasis rate is approximately 15%, whereas for those larger than 2 cm, the rate is 75%. Some WDNETs produce serotonin, and carcinoid syndrome is reported for 5%.
Rectum
Rectal WDNETs represent 17% of all GI NETs. They are typically diagnosed in the sixth decade of life. Approximately 50% are asymptomatic and found during routine endoscopy.72 More than two thirds are smaller than 0.5 cm in diameter at the time of diagnosis.73,74 In addition to early diagnosis, the small size is in part attributable to the biology of the tumors arising in this region from L cells of the hindgut.
The overall metastasis rate is 14%, and less than 5% of tumors smaller than 1 cm in diameter are metastatic. However, those larger than 2 cm are often metastatic. Some tumors are reported in cases of inflammatory bowel disease, and they can be multicentric and atypical.75,76 Carcinoid syndrome is rare.
Although most rectal WDNETs are straightforward diagnostically, a few pitfalls should be remembered: CEA positivity is common, and staining for chromogranin is negative in more than 50% of cases. Synaptophysin staining and characteristic morphology together are often conclusive. Specimens are commonly positive of prostate-specific acid phosphatase. The rectum is not the only site for positivity of this enzyme in NETs, but it is the most problematic because of the spread pattern of prostate cancer. However, staining for prostate-specific antigen is typically negative in WDNETs.
Most rectal NETs are initially diagnosed in polypectomy specimens, and if they are not properly processed, assessment of the margins can become a problem. Ulceration and invasion into the muscularis propria are regarded as signs of aggressiveness and are used as additional indications for a radical operation, especially if the tumor is between 1 and 2 cm in diameter. Those that are larger than 2 cm are typically treated with low anterior resection or abdominoperineal resection.
Pancreas
PanNETs constitute less than 5% of pancreatic tumors. Almost half are functional, showing serologic activity attributable to one of the six hormones that are produced by the islet cells (i.e., insulin, glucagon, gastrin, somatostatin, VIP, or pancreatic polypeptide). Most insulinomas follow a benign clinical course, likely because they typically are highly symptomatic even when they are small, which leads to their early detection.
Glucagonomas tend to be large at diagnosis and have a more aggressive course than other PanNETs. Tumors associated with MEN I or other syndromes tend to be multifocal and less aggressive; these cases also have numerous small nodules known as microadenomas (<0.5 cm in diameter). More than one half of PanNET patients have recurrences or metastases after resection, and many patients come to attention only after the development of metastatic disease that precludes resection. In staging PanNETs, the ENETS scheme based on the size of the lesion may prove to be more applicable and predictive than other systems.
PanNETs appear to have more morphologic versatility than GI WDNETs. Along with the lipid-rich, clear cell, pleomorphic, and other variants, which are more commonly seen in PanNETs, some PanNETs exhibit a pattern very similar to that of paragangliomas (see Fig. 29.13).20 These types also tend to have endocrine atypia. The location in the pancreas, keratin positivity, and lack of the basophilia and granularity of pheochromocytomas help to distinguish these tumors.
A subset of PanNETs has significant inclusion of entrapped, non-neoplastic ductules (Fig. 29.20). This variant has been reported under the misleading name ductulo-insular PanNET, which suggests that the ductules are an integral part of the neoplasm.60,77,78 The behavior and biology are not different from those of ordinary PanNETs. These tumors should not be confused with mixed neuroendocrine-adenocarcinomas, in which the glandular component is malignant.
Some PanNETs show prominent peliotic changes, including abundant blood and ectasia of vessels. Cystic degeneration is seen in approximately 10% of cases.
Gallbladder and Biliary Tract
WDNETs are uncommon in the gallbladder and biliary tract. Some are associated with MEN I or VHL syndromes. They are more common in the bile ducts than gallbladder. In the gallbladder, PDNECs are significantly more common than WDNETs. In the U.S. Surveillance Epidemiology, and End Results (SEER) database of the National Cancer Institute, the 5-year survival rate for WDNETs of the gallbladder is 40%. In the bile ducts, the common bile duct appears to be the preferred site, followed by cystic ducts.
Poorly Differentiated Neuroendocrine Carcinomas
Data on the PDNECs of the GI tract are limited.8,79–82 However, there is no question that they occur in essentially every component of the GI tract from esophagus to anus. Ampullary carcinomas seem to be more likely to have neuroendocrine components. In the esophagus, PDNECs are more common than WDNETs.
As in other organs, some PDNECs are almost identical to pulmonary small cell carcinomas (Fig. 29.21), but others are more like large cell neuroendocrine carcinomas. In any case, they are characterized and defined by high mitotic activity (>20 mitoses/10 HPFs and usually 40 to 50/10 HPFs) and necrosis in addition to their distinctive morphology and high-grade cytology. Immunoexpression of chromogranin and synaptophysin is typical and required for the diagnosis of large cell neuroendocrine carcinoma, but the intensity and extent of staining are usually less than in WDNETs. PDNECs are usually bulky or ulcerated tumors. Those with morphology as defined in the lung are uncommon in the pancreas. Pancreatic tumors suspected to be PDNECs often prove to be acinar cell carcinomas (see Chapter 40).83
In the 2010 WHO classification,2 PDNECs are unfortunately included in the grade 3 category along with WDNETs that have a Ki67 proliferative index greater than 20% or more than 20 mitoses per 10 HPFs. In one study, a Ki67 index value greater than 55% was found to more appropriately define PDNECs, identifying the group with rapidly progressive behavior and sensitivity to platinum-based chemotherapy.8 Although it is exceedingly uncommon to see a PDNEC developing in association with a WDNET, innocuous WDNETs can acquire more aggressive behavior in time, manifested as a higher proliferation index, but they typically maintain their WDNET morphology.
Almost half of the PDNECs of the GI tract are associated with conventional adenomas and ordinary carcinomas and may qualify as mixed adenoneuroendocrine carcinomas (MANECs). The amount of PDNEC component can vary from case to case. Based on limited evidence and extrapolating from other organs, it appears that even a small PDNEC component needs to be acknowledged because it may drive the overall tumor behavior and may warrant a different chemotherapy protocol.8 It is probably better to report these carcinomas as “PDNEC with a mixed adenocarcinoma component” and provide the relative proportions in the tumor in a comment.
PDNECs should be distinguished from other high-grade malignancies, and this can be a challenge at times. Poorly differentiated carcinomas (not otherwise specified or medullary types), melanomas, lymphomas, and metastatic tumors can display a nesting growth pattern and monotonous cytology similar to those of PDNECs. In those cases, nuclear chromatin pattern and immunohistochemical support by chromogranin, synaptophysin, and CD56 staining is necessary. Focal neuroendocrine cells are often seen in other malignancies. PDNECs, especially the small cell carcinomas, are commonly positive for TTF-1, regardless of their primary location, and this should not be used as evidence for a pulmonary origin. Similar to their pulmonary counterparts, PDNECs of the GI and pancreatobiliary tracts often show alterations in the retinoblastoma gene (RB1) and TP53.
PDNECs are highly aggressive tumors that disseminate rapidly. The survival range for patients with PDNECs cited is between 1 and 2 years, if not less. Current evidence favors treatment with small cell carcinoma protocols (i.e., platinum based).
Mixed Tumors
Mixed Adenoneuroendocrine Carcinomas
PDNECs are often encountered admixed with an adenocarcinoma or squamous cell carcinoma component. About one fourth of PDNECs in the GI tract have an adenocarcinoma component. Small cell carcinomas, which in their classic form preferentially arise in the esophagus or anus, can include squamous cell carcinoma components. Traditionally, terms such as composite, collision, and amphicrine have been used for these tumors based on the association between and distribution of the two components. Those lying side by side are designated collision, those in which the components are intimately intermingled are composite, and those with dual differentiation within the same cells are amphicrine. However, all of these forms most likely represent different facets of the same phenomenon in which the cells in these neoplasms show divergent differentiation. A more practical approach regards them as mixed carcinomas (Fig. 29.22).
Mixed Neuroendocrine Tumor with Non-neuroendocrine Carcinomas
In the GI tract, it is uncommon to see a WDNET mixed with an adenocarcinoma. In the pancreas, however, a distinct WDNET component is seen with an acinar cell carcinoma in addition to the common occurrence of scattered individual cells or small clusters (see Chapter 40).84,85 It is important to recognize the acinar component because it appears to be the main determinant of biologic behavior.
In the pancreas, pancreatoblastoma is another tumor type in which neuroendocrine differentiation can be prominent, along with acinar and ductal components. As in mixed acinar-neuroendocrine carcinomas, the amount and distribution of neuroendocrine cells can vary greatly in pancreatoblastoma (see Chapter 40).
Duodenal Gangliocytic Paraganglioma
Duodenal gangliocytic paraganglioma may also be considered a mixed tumor because there is a WDNET component admixed with two other seemingly independent cell types: a mesenchymal component that resembles Schwann cells and ganglion-like cells (Fig. 29.23). This tumor type appears to be specific for the duodenal region. It is discussed further in Chapter 41.
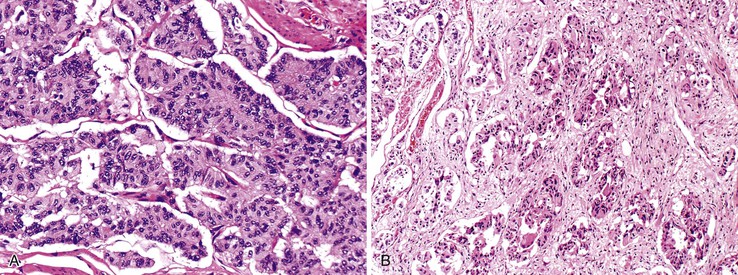
Incidental Neuroendocrine Cell Proliferations in Other Epithelial Tumors
Occasionally, small neuroendocrine cell clusters are identified incidentally in the vicinity of glandular epithelial tumors such as adenomas. The term adenoma-carcinoid has been applied in these cases (Fig. 29.24).86,87
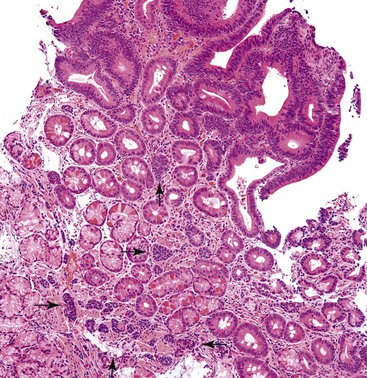
These cell clusters have been reported in the colon, but we have also seen examples in the ampulla. The WDNET (carcinoid) component of the lesions is typically small and composed of small cords and minute nests and does not form micronodules that would qualify them as full-blown tumors. They may represent incipient neoplasia, presumably arising by the induction of local factors and the tumorous milieu.
Chimeric Tumors with Incomplete Neuroendocrine Differentiation
Conventional-Type Goblet Cell Carcinoids
Although goblet cell carcinoids (GCCs) are classified in the generic category of carcinoids or neuroendocrine neoplasia, it is becoming clear that they should be regarded separately from ordinary carcinoids (i.e., WDNETs).88–91 GCCs appear to be amphicrine tumors arising from pluripotent stem cells with divergent neuroendocrine and goblet cell (mucinous) differentiation. The only justification for maintaining the GCC designation is that they behave more like carcinoids, especially if they are low stage.
With their specific morphology that includes a subtle infiltration pattern and glands that are highly similar to colonic crypts, these tumors are typically specific for the appendix (Fig. 29.25) (see Chapter 28). The dual (amphicrine) phenotype of classic GCCs is evidenced at ultrastructural and immunohistochemical levels by abundant intracellular mucin and neurosecretory granules, which can be highlighted by chromogranin and synaptophysin stains.
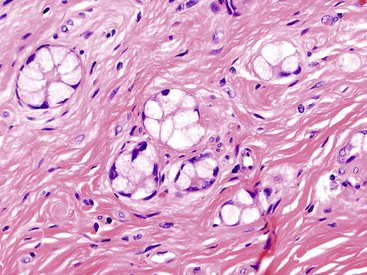
The distinctive pathologic features of GCCs translate into clinical behavior. In contrast with appendiceal WDNETs, which metastasize in less than 5% of cases, conventional GCCs metastasize in 15% to 30% of cases. The most common route of metastasis is through lymphatic vessels and transcoelomic and intraperitoneal invasion, whereas hematogenous metastasis to the liver or other distant organs is rare. GCC metastases most commonly target the ovary, followed by spread to the abdomen (i.e., carcinomatosis).
High-Grade Goblet Cell Carcinoids
GCCs come to clinical attention in two different settings88–91 In the first, tumors localize to the appendix and clinically manifest with appendicitis. These cases have fairly high cure rate and indolent behavior with a protracted clinical course. In the second, advanced tumors (occurring predominantly in women) manifest with peritoneal carcinomatosis, typically involving peritoneal surfaces, ovaries, and the uterus but rarely the liver. Gynecologic pathologists typically see widely disseminated examples of this entity, which is referred to as adenocarcinoma ex-GCC or dedifferentiated GCC.88–91 In addition to the conventional GCC pattern, specimens of these tumors show some areas with high-grade cytologic features and mixed patterns, including cordlike infiltration, goblet (signet ring) cells in cords or as individual cells, a nonmucinous microglandular pattern, an ordinary intestinal pattern, or extravasated mucin (Fig. 29.26).
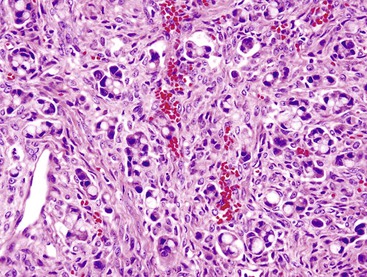
After dissemination, the tumors exhibit highly aggressive behavior. Small, round clusters of large goblet type cells or a rosette-like microglandular pattern is characteristic of these tumors and allows them to be recognized as appendiceal primaries even at metastatic sites.
Other Tumors with Neuroendocrine Differentiation
Nonepithelial Tumors with Neuroendocrine Differentiation
Small, blue cell tumors with neuroendocrine-type differentiation can occur in the gastrointestinal and pancreatobiliary tracts. Ewing sarcoma, primitive neuroectodermal tumors, and desmoplastic small cell tumors often show neuroendocrine differentiation to variable degrees. The latter often shows evidence of epithelial differentiation. The characteristic molecular and histomorphologic patterns of these tumors are beyond the scope of this text, but they should be kept in mind in the differential diagnosis that includes PDNECs.
Paragangliomas are tumors characterized by neuroendocrine differentiation and can manifest as a primary in the vicinity of the GI and pancreatobiliary tracts. These tumors are typically keratin negative. Evidence suggests a strong familial or genetic background in these cases, which may warrant immunohistochemical (for succinate dehydrogenase variants) and genetic testing. One PanNET variant shows a pattern highly reminiscent of paragangliomas.
Induced Neuroendocrine Differentiation
A peculiar phenomenon is the emergence of neuroendocrine features after therapy for conventional adenocarcinomas. For example, rectal adenocarcinomas that are treated with neoadjuvant chemotherapy and radiation therapy often show prominent neuroendocrine features in the residual tumor, including chromogranin positivity.92 Whether this represents clonal selection and evolution or induction by the therapy is not clear.
In the setting of graft-versus-host disease or similar patterns of injury such as those caused by the use of mycophenolate, which leads to the disappearance of glands, the remaining epithelium is often rich in neuroendocrine cells. In some cases, these residual neuroendocrine cells form cords and minute nests.
Focal Neuroendocrine Differentiation in Other Tumors
Scattered neuroendocrine cells (i.e., chromogranin-positive cells) are found in adenocarcinomas and poorly differentiated carcinomas of the GI and pancreatobiliary tracts. They are usually neglected unless immunohistochemical stains are performed. In contrast to the more diffuse (weak) immunoexpression of chromogranin or synaptophysin found in poorly differentiated neuroendocrine carcinomas, scattered neuroendocrine cells in conventional adenocarcinomas usually appear as individual cells with strong immunolabeling in a carcinoma that is otherwise negative for these markers. There are no established guidelines with proven clinical relevance for when these focal neuroendocrine cells may have significance, but most studies suggest they are not prognostically important.
Metastatic Tumors of Neuroendocrine Origin
NETs from other organs sometimes metastasize to the GI and pancreatobiliary tracts. The small intestine is notorious as the recipient of metastatic carcinomas, especially from the lung. Pulmonary PDNECs metastasize to virtually any site in the abdomen, including the pancreas.93 Because TTF-1 occurs in PDNECs of any organ, it is not helpful in the differential diagnosis. Evaluation of these cases requires careful clinical analysis. An adenoma or adenocarcinoma component or involvement of multiple local lymph nodes without significant lymph node involvement elsewhere favors a primary. A mucosacentric distribution also favors a primary, although it occasionally is seen in metastatic tumors.
Merkel cell carcinoma is another NET type that may manifest with GI or pancreatobiliary tract metastasis. Other PDNECs should be distinguished from Merkel cell carcinoma because its behavior is more indolent, even after metastasis has occurred. Distinctive overlapping of round nuclei with washed-off, even chromatin substantiated by the dotlike positivity pattern of CK20 staining helps to confirm the diagnosis.
Neuroendocrine Tumor Mimics
WDNETs usually have a distinctive morphology and are easy to recognize. However, certain tumor types can mimic NETs at any location. For example, glomus tumor, which can occur in the stomach, often exhibits a nesting pattern and cellular monotony that can closely mimic NETs. We have seen several examples of epithelioid GISTs that have been mistaken as WDNETs in the pancreas and GI tract.
Metastasis from the cribriform variant of prostate carcinoma and the nested or alveolar variant of mammary lobular carcinoma are likely to be mistaken for WDNETs. The mimicry of hepatocellular carcinoma and NETs is well documented, and many authorities advocate the liberal use of neuroendocrine markers to assess core-needle biopsies of the liver. Differentiation of PDNECs from other high-grade malignancies with a nested growth pattern and monotonous cytology requires careful morphologic and immunohistochemical evaluation.
Acknowledgment
We are indebted to Dr. Serdar Balci, Dr. Burcu Saka, Ms. Rhonda Everett, and Ms. Leslie Ducato for their extensive contributions in the preparation of this chapter.