13 Neuroendocrine Lesions of the Lung
The concept of a “diffuse neuroendocrine system” (DNS) is not a new one. Feyrter1 developed this paradigm in 1938, in a philosophical attempt to unify tumors in several anatomic locations that had potential secretory functions and similar morphologic characteristics. Pearse2 refined and renamed the cellular network in question 35 years later, coining the designation of “APUD” system (for amine precursor uptake and decarboxylation) to describe its shared biochemical attributes. Inherent in the latter scheme was the presumption that all “APUD” cells—and tumors deriving from them (“APUDomas”)—emanated from the remnants of the neural crest. In light of these and other observations, continuing nosologic revisionism over the last 10 years has pushed many pathologists away from such traditional diagnostic terms as “carcinoid” and “islet cell tumor” in describing certain potentially malignant but low-grade neoplasms of the neuroendocrine system,3,4 although “traditionalists” remain.5 The classification of poorly differentiated lesions has changed as well. This chapter outlines the current foundations of existing classification schemes for neuroendocrine tumors. The reader should come away with a simplified—and therefore practical—understanding of this confusing area of oncology.
Terminology Pertaining to Neuroendocrine Neoplasms
There is perhaps no other single aspect of neuroendocrine neoplasia that is as perplexing as the pathologic terminology that has been used to describe it. Such terms as “bronchial adenoma,” “carcinoid,” “atypical carcinoid,” “Kulchitsky cell carcinoma,” “argentaffinoma,” “APUDoma,” “atypical endocrine carcinoma,” “oat cell carcinoma,” “medullary thyroid carcinoma,” “islet cell tumor,” and “cutaneous Merkel cell carcinoma” (group 1) have all been employed historically in this context, in addition to “neuroblastoma,” “esthesioneuroblastoma,” “olfactory neuroblastoma,” “medulloblastoma,” “pineoblastoma,” “retinoblastoma,” “paraganglioma,” “pheochromocytoma,” “chemodectoma,” and “glomus jugulare tumor” (group 2).6–10
This diverse lexicon reflects a basic division of neuroendocrine tumors into two broad categories—epithelial (group 1) and neural (group 2).6 With that piece of information in hand, one can then go on to structure a much more user-friendly and straightforward classification scheme that has made significant inroads in the pathology literature.
Another crucial concept in understanding the categorization and clinical behavior of neuroendocrine neoplasms is that all of them are at least potentially malignant tumors, regardless of whether they belong to group 1 or group 2.9 Moreover, in selected subgroups (e.g., classic “carcinoid” tumor, extra-adrenal paraganglioma [PG], and pheochromocytoma [intra-adrenal PG]), one cannot reliably use the gross or microscopic characteristics of the tumors to predict whether they will behave innocuously or aggressively. Therefore, it follows logically that the modifier “benign” should not be applied in conjunction with any of the diagnostic terms noted earlier. For example, even with regard to appendiceal or classic bronchial “carcinoids”—generally regarded as defining the “low” end of the spectrum of biologic behavior in this context11–13—there are many well-documented examples of metastasizing lesions that can be found in the literature.
Current terminological recommendations are, therefore, different from those that might have pertained even 10 years ago, or from those with which some practitioners may feel “comfortable.” The designation of “neuroendocrine carcinoma” (NEC) has been proposed as a replacement for all of the various group 1 terms discussed earlier, with modifiers of “well-differentiated” (grade I/III); “moderately differentiated” (grade II/III); and “poorly differentiated” (grade III/III) being appended as appropriate.9,12,14 In contrast, most of the traditional rubric has been retained with reference to group 2 tumors, such that the recommended terms for the categorization of this constellation of lesions are “extra-adrenal PG,” “intra-adrenal PG” (pheochromocytoma), “sympatheticoadrenal neuroblastoma (NB)” (and congeners [e.g., ganglioneuroblastoma]), “olfactory NB,” “retinoblastoma,” and “primitive neuroectodermal tumor” (PNET).9 In reporting on a biopsy or resection specimen, the pathologist can then work within this framework—using further descriptive comments and summaries of the aggregate literature on each tumor—to provide the clinician with an outline of expected behavior for each neoplasm based on its nuances.
Distribution and Pathogenesis of Neuroendocrine Neoplasia
If one refers to basic textbooks on human embryogenesis, a common theme that is seen in all anatomic sites is that of a neuroendocrine or neuroectodermal stage of differentiation during early organ development.6,15,16 Because it is currently thought that oncogenesis partially (and aberrantly) recapitulates normal embryologic development, this information is central to our understanding of why neuroendocrine and neuroectodermal neoplasms have been reported in virtually every topographic location. Some of the latter are, by far, more commonly hosts to such tumors, for unknown reasons. For example, the lung is the most common site of neuroendocrine carcinogenesis, where the process is clearly related etiologically to cigarette smoking and is associated with partial deletion of the short arm of chromosome 3.17 However, identical sporadic primary neoplasms in other organs have not been linked with any definitive pathogenetic factors.18 Group 2 neoplasms in the “peripheral primitive neuroectodermal” category similarly demonstrate a uniform balanced translocation between chromosomes 11 and 22,19,20 with synthesis of a unique gene product (p30/32 glycoprotein) that is recognized by a particular set of monoclonal antibodies (e.g., 12E7 and O13).20
Other NECs and group 2 tumors (especially NB and retinoblastoma) occur in definite Mendelian-heritable—typically autosomal dominant—patterns, as seen in multiple endocrine neoplasia (MEN) type 1 (pancreatic and thymic group 1 tumors) and MEN2 (medullary thyroid carcinoma and also group 2 tumors [pheochromocytomas]).9,11,21,22 Ongoing work has elucidated the locations of at least some operative aberrant gene complexes in such disorders (e.g., the MEN2A locus on chromosome 10 and deletion of the Rb-1 “anti-oncogene” on chromosome 13 in heritable bilateral retinoblastoma).17,19,23 However, sporadic examples of the tumors discussed earlier do not necessarily exhibit the same karyotypic or gene sequence abnormalities.24 Clearly, more work is needed to provide a complete picture of the molecular disturbances at play,14,25 but this is an exciting area for current and future development because it may yield clinical tests that could be used in early diagnosis and treatment.
Other Pathologic Aspects of Neuroendocrine Neoplasia
Up to this point, this discussion has focused on “pure” neuroendocrine and neuroectodermal neoplasms. Increasingly in recent years, however, it has been recognized that human malignancies much more often show combined or “divergent” differentiation than was appreciated in the past. Accordingly, oncologists are now faced with such diagnoses as “adenocarcinoma/squamous carcinoma/transitional cell carcinoma with neuroendocrine features” or, more simply, “combined adenocarcinoma–small cell NEC.”26–33 In the former scenario, the pathologist is attempting to convey the concept that the tumor looks like conventional squamous carcinoma, adenocarcinoma, or transitional cell carcinoma with a routine hematoxylin and eosin stain, but that additional studies (e.g., ultrastructural or immunohistochemical) have demonstrated submicroscopic neuroendocrine differentiation in the neoplastic cells. In some organ systems, such as the lung, it is believed that such a constellation of findings portends a more aggressive course of certain tumor types (e.g., “large cell anaplastic pulmonary carcinoma with neuroendocrine features”).34 However, generic extrapolation of this model to other tissues would not be scientifically justified at this time and appears to be definitely invalid in some specific settings.31,33 When a truly combined carcinoma is seen at a light microscopic level, pathologists are describing the juxtaposition and admixture of two distinct histologic patterns, such as adenocarcinoma and small cell carcinoma (SCC).29,30 Again, using tumors of the lung as examples, it would be expected that the responses to therapy and the behavior of such combined lesions would also be a hybrid of those attending each component (i.e., adenocarcinoma or SCC) in pure form.35–38 Nonetheless, uniform validation of that premise and delineation of optimal therapeutic approaches for each of these “amalgam” tumors have yet to occur.
Pathologic Recognition of Neuroendocrine Differentiation
Standard Morphologic Examination
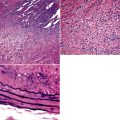
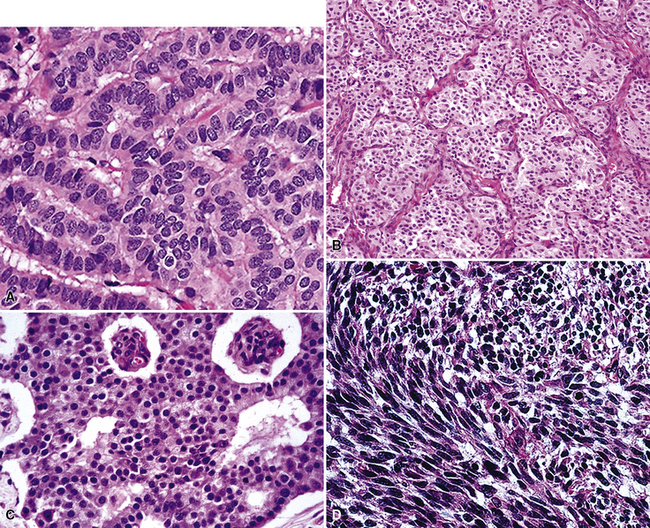
Because of the histologic appearance of some tumors, a neuroendocrine phenotype is obvious. Ready examples include classic pulmonary SCC and central bronchial “carcinoid” tumor. The standard cytomorphologic features of SCC are well known and include hyperchromatic nuclei with dispersed chromatin, inconspicuous nucleoli, and a tendency for the nuclei to mold to one another (Fig. 13-1). The cells are oval or carrot-shaped, with scant cytoplasm, and often there is marked crush artifact. Extensive necrosis is often observed, with either a geographic pattern or dropout of individual cells (i.e., apoptosis). Likewise, bronchial “carcinoid” features include stippled nuclear chromatin and a distinctly organoid growth pattern, with formation of rosettes, trabeculae, or ribbons (“festoons”)39 (Fig. 13-2).
Histochemical Methods
Histochemical techniques are still valuable in selected settings, although in current practice, they have been supplanted by newer technologies. With reference to neuroendocrine lesions, such techniques rely on the ability of endocrine cells to reduce silver solutions and form insoluble precipitates in tissue sections. Available methods are broadly subdivided into two groups—argentaffin and argyrophil stains.40–42
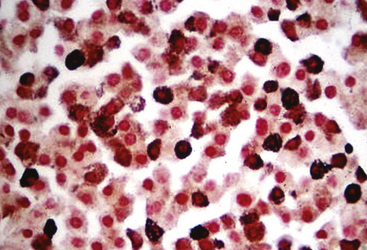
Argentaffin techniques depend on endogenous cellular reducing agents. The most widely used is the Fontana–Masson stain, which is only variably reactive with pulmonary neuroendocrine neoplasms. Argyrophilic methods are those in which an exogenous reducing agent is added, such as in the Grimelius or Churukian–Schenk procedure. They are typically positive in the majority of well-differentiated neuroendocrine tumors of the lung (Fig. 13-3), with much lesser reactivity in high-grade carcinomas, such as SCC.42
Electron Microscopy
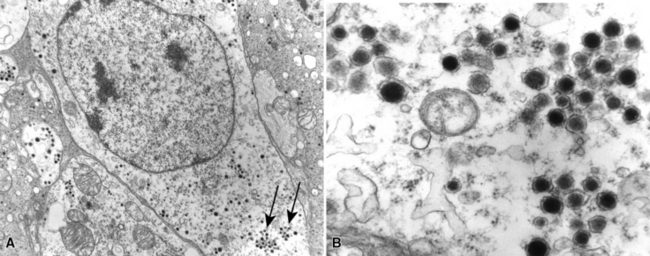
The ultrastructural hallmark of neuroendocrine neoplasms in the lung and elsewhere is the presence of cytoplasmic neurosecretory granules. These are rounded structures that consist of a central dense core, a peripheral lucent halo, and a single delimiting outer membrane. They vary from 30 to 300 nm in diameter, and may occur singly or in clusters42–55 (Fig. 13-4). Budding of neurosecretory granules from prominent Golgi apparatus is occasionally observed. One drawback of electron microscopy is that the number of granules and the number of cells containing granules tend to be greatest in well-differentiated tumors and lowest in poorly differentiated lesions (where they are most needed for diagnosis). Also, electron microscopy can examine only relatively few cells in any given tumor. However, it is still a highly useful technique if sufficient time and care are invested in a thorough search for dense core granules. In a study by Nagle and colleagues,40 ultrastructural analysis documented neuroendocrine differentiation in all of 41 putative neuroendocrine lesions, whereas a significant percentage of lesions were negative for endocrine markers using immunohistochemical techniques.
Immunohistology
Advances in immunohistochemistry over the last 15 years have yielded powerful tools for the pathologist in recognizing neuroendocrine differentiation.44,50,54–60 Advantages of this approach include a relatively low cost, rapid turnaround time, the ability to perform studies on routinely processed tissue, and the capacity to screen a large number of cells rapidly as opposed to the limited number that can be evaluated with electron microscopy. The following markers have the greatest utility in this context.
Intermediate Filament Proteins
Group I neuroendocrine neoplasms manifest uniform immunoreactivity for keratin, especially keratin classes 8 and 18.61 To a lesser extent, keratin 19 is also observed in NECs in general. Pulmonary NECs may label for keratin 20 as well, but only in fewer than 10% of cases.62 Keratin proteins are generally well recognized by several commercial monoclonal antibodies, especially CAM5.2 and MFN116. Monospecific reagents directly against only one keratin class polypeptide are also available, but are not necessary in this specific context. In our opinion, the optimal approach to the detection of keratin in any poorly differentiated neoplasm, neuroendocrine or otherwise, is to prepare a mixture of monoclonal antikeratin antibodies with partially overlapping and partially distinctive keratin class specificities. When reagents of this type are used on paraffin sections, together with properly performed proteolytic digestion or microwave (heat)-mediated epitope “retrieval” methods, the detectability of keratin in NECs should approximate 100%, even in essentially “undifferentiated” tumors (e.g., SCC).63 Another feature of keratin reactivity in many NECs is a distinctive punctate or globoid perinuclear zone of positivity.64 This result (Fig. 13-5) is simultaneously diagnostic of both epithelial and neuroendocrine differentiation in a small cell malignancy, and it obviates the need for other “neuroendocrine markers.” Keratin is not typically expected in group II neuroendocrine neoplasms, although some lesions in that group have shown its presence in an “aberrant” manner.65–68 The foremost example of that phenomenon is represented by PNET with “divergent” differentiation (also known as “polyphenotypic small cell tumor”), an example of which is the “desmoplastic small round cell tumor.”69–71 In most cases of keratin expression in more nondescript PNETs, reactivity for that protein is focal, unlike its pattern in NECs; in addition, vimentin expression tends to be mutually exclusive in PNETs and NECs. Nonetheless, problems in differential diagnosis may arise between those classes of neuroendocrine tumors, sometimes necessitating cytogenetic evaluation to distinguish between them.72
Neurofilament protein is variably coexpressed by NECs,73–76 but that determinant is seen as the sole intermediate filament in the majority of differentiated group II neuroendocrine tumors, such as PGs and pheochromocytomas, as well as some NBs.77–79 Unfortunately, neurofilament protein is not well visualized in paraffin sections, even with epitope retrieval technology, and it is most reliably evaluated in frozen material. Vimentin also may be evident in the cells of PGs in approximately 50% of cases,79 and it is the only intermediate filament that can be detected in primitive group II tumors, such as NB and PNET.68 That is also true in Ewing sarcoma, a tumor that is in the same family as PNETs. Vimentin is only exceptionally present in NECs, as stated earlier. Eusebi and coworkers80 recently described three NECs that coexpressed keratin and desmin as a reflection of divergent (sarcomatoid) rhabdomyoblastic differentiation. This same proclivity is regularly seen in desmoplastic small round cell tumors, in which keratin, desmin, and vimentin are commonly present concomitantly in the same neoplastic cells.69–71
Glial fibrillary acidic protein is not an expected reactant in either NEC or PNET. In the central nervous system, therefore, this marker is helpful in the differential diagnosis with small cell malignant gliomas,81 most of which are reactive for glial fibrillary acidic protein. That fact is all the more important because a proportion of glial tumors manifest aberrant keratin reactivity82 and also express vimentin.
Chromogranins
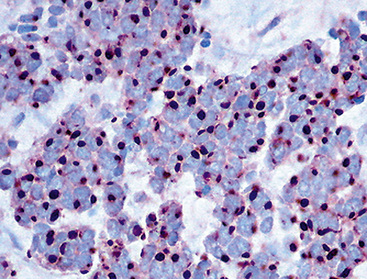
Chromogranins are matrical proteins that are associated with neurosecretory granules and are absolutely specific for neuroendocrine differentiation.83–85 These polypeptides have been subdivided biochemically into two groups, A and B,86,87 with the latter being synonymous with secretogranin-I. Conceptually, every cell that packages peptides into neurosecretory granules must synthesize either chromogranin-A (CGA) or chromogranin-B (CGB); hence, a screening reagent incorporating antibodies to both of those proteins would be extremely useful diagnostically. Unfortunately, reliable commercial anti-CGB products have not been introduced. The most widely used anti-CGA antibody is clone LK2H10.88 This reagent was raised against pheochromocytoma cells and shows excellent reactivity with paraffin sections (Fig. 13-6). The major shortcoming of anti-CGA is twofold. First, because neuroendocrine cells or tumors may preferentially synthesize CGB, CGA obviously cannot be regarded as a “universal” marker of such elements. Secondly, the detectability of both CGA and CGB is directly related to the number of neurosecretory granules in any given neuroendocrine cell population. If one is dealing with poorly differentiated malignancies that have only scant numbers of such organelles, immunostains for CGA and CGB will be predictably negative in the majority of cases. Therefore, one may legitimately conclude that a tumor has neuroendocrine properties if it can be labeled for a chromogranin, but negative results do not exclude that possibility, particularly in high-grade neoplasms.
Synaptophysin
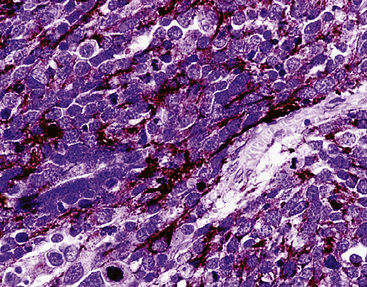
Synaptophysin is a 38 kDa molecule that is associated with the synaptic vesicles of neurons and cells with neuroendocrine or neuroectodermal characteristics.78,89–92 Monoclonal antibodies to this marker have been used widely in diagnostic surgical pathology and cytopathology, with good success, particularly in conjunction with epitope retrieval techniques (Fig. 13-7). In light of its subcellular associations, one might assume that synaptophysin would have a synonymous tissue distribution to that of the chromogranins; however, in practicality, that is not true. Thus, a sizable proportion of neuroendocrine neoplasms label for CGA or CGB, but not synaptophysin; the converse also applies.93 Thus, anti-synaptophysins and anti-chromogranins should be conceptualized as complementary reagents.
Loy and colleagues59 have challenged the specificity of chromogranins and synaptophysin as markers of neuroendocrine lineage, in a study using ultrastructure as the standard for definition of that lineage. However, in our opinion, the concept behind that analysis was flawed. Sampling bias (a significant problem in fine structural evaluations) probably accounted for those cases in which immunoreactivity was observed for the specified endocrine markers, but no neurosecretory granules were identified by electron microscopy.
CD57 (Leu-7)
CD57 was originally characterized as a marker for natural killer lymphocytes, and it is recognized by monoclonal antibody HNK-1,94 among others. Subsequently, shared epitopes of this molecule have been detected immunohistochemically in normal constituents and tumors of the brain, peripheral nervous system, soft tissues, prostate gland, thymus, and DNS.95–101 With specific reference to neuroendocrine neoplasms, CD57 appears to bind to a matrical component of neurosecretory granules that is distinct from both CGA and CGB.102 It both corroborates and extends the sensitivity of chromogranin immunostains in many clinical settings. Nonetheless, because of the imperfect specificity of Leu-7, it cannot be used as confidently as a “standalone” neuroendocrine marker. Moreover, it has the same failings in sensitivity as CGA and CGB that relate to the density of neurosecretory granules in poorly differentiated tumors.
Neural Cell Adhesion Molecule (NCAM; CD56)
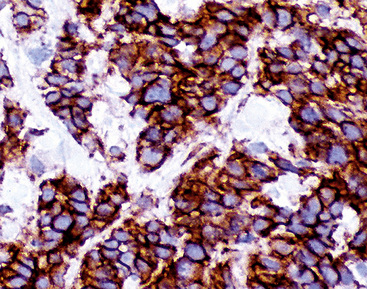
The monoclonal antibodies 123C3 and JLP5B9 recognize formalin-preserved epitopes of CD56, or neural cell adhesion molecule, a cell membrane protein that has a role in the cohesiveness of cells in the peripheral and central nervous systems.103–109 Like synaptophysin, NCAM is distributed among neuroendocrine and neuroectodermal cells and tumors (Fig. 13-8). Several studies have shown that NCAM is a sensitive marker of endocrine lineage in SCC of the lung as well as extrapulmonary sites.106–108 Nonetheless, it is not absolutely specific for neuroendocrine differentiation; a minority (up to 25%) of ovarian surface carcinomas, renal cell carcinomas, nonendocrine lung cancers, and endometrial carcinomas demonstrate CD56 immunoreactivity.108 Despite this caveat, antibodies to NCAM are useful additions to diagnostic panels for endocrine tumors. Reagents raised against the polysialylated form of CD56 are considered the most sensitive among this group.105
Neuron-Specific (Gamma-Dimer) Enolase
“Neuron-specific” enolase (NSE) is a gamma-dimeric form of 14-3-2 protein, a glycolytic enzyme that is present in neurons and cells of the DNS.110 It is thought to be associated with the formation of intercellular synapses in the nervous system. NSE was one of the first markers to be used in diagnostic immunohistochemistry as a general indicator of putative neuroendocrine differentiation.110–112 Heteroantisera to NSE are very sensitive in that regard, labeling virtually all neuronal and neuroendocrine proliferations, regardless of the level of cellular differentiation.112 Nevertheless, the specificity of those reagents is poor, owing to cross-reactivity between gamma-dimers and heterodimers (e.g., alpha-gamma, alpha-beta) of NSE that are expressed by non-neuroendocrine neoplasms of many types.113 In reaction to that problem, monoclonal antibodies to NSE have been developed and tested,114 but these have generally manifested a low degree of sensitivity and have not enjoyed widespread usage. An alternative approach to lessening the cross-reactivity of anti-NSE heteroantisera is to absorb them with acetone-fixed, pulverized human tissues known to contain high levels of the alpha or beta form of the molecule. However, that approach is time-consuming and somewhat demanding technically, and for this reason, has not been embraced in clinical practice. Currently, antibodies to NSE are primarily used as “screens” for endocrine differentiation,115–117 and reactivity with them is usually pursued further by applying other neuroendocrine immunostains, as described elsewhere in this chapter.
Protein Gene Product 9.5 (PGP9.5)
PGP9.5 is a protein that removes ubiquitin (an intermediate filament) from other proteins and protects them from degradation by proteases. It is expressed widely within cells and tumors of the DNS and therefore has been applied as an immunohistochemical marker of neuroendocrine differentiation.118–120 Nevertheless, the general distribution of PGP9.5 in human neoplasms has shown that it is not particularly specific for an endocrine lineage; in particular, non–small cell carcinomas of the lung that lack neuroendocrine morphologic patterns and contain no neurosecretory granules on ultrastructural analysis have been PGP9.5-positive in approximately 55% of cases in some series.121 Thus, the practical utility of this marker is similar to that of NSE.
Specific Neuropeptides
Specific neuropeptides, such as adrenocorticotropic hormone, gastrin, insulin, somatostatin, calcitonin, “whole” bombesin and its C-terminal flanking peptide, leu- and met- enkephalins, were the initial molecular moieties that were evaluated immunohistochemically in an effort to substantiate the neuroendocrine nature of selected epithelial human neoplasms.93 Today, these markers have been largely relegated to a secondary role in the pathologic assessment of tumors in the DNS. Antibodies to such peptides are of academic interest in correlating clinical endocrinopathies with histopathologic findings, but are not nearly sensitive enough to serve as screening reagents. Another potential but limited application is in the delineation of “occult” (non–endocrinopathy-related) peptide production by neuroendocrine tumors, which may serve as the basis for serologic monitoring of tumor growth and activity.
CD99 (MIC2; p30/32 Protein)
The membranocytoplasmic protein known as “CD99” in the hematopoietic antigen cluster designation is the same molecule that has been called “MIC2” or “p30/32 protein.”122 The function of this moiety is uncertain, but it is empirically known to be expressed in virtually all PNETs and Ewing sarcomas122,123 (Fig. 13-9). The specificity of CD99 antibodies for a neuroectodermal lineage is not absolute, however, because they may also label a minority (<15%) of alveolar rhabdomyosarcomas as well as the overwhelming majority (90%) of lymphoblastic lymphomas.124 CD99 is observed in up to 20% of NECs in some body sites as well.125,126 This is important because it may obscure the difference between NEC and PNET in selected instances; this is particularly true in light of the potential for keratin reactivity that both lesions have. On the other hand, differentiated group II neuroendocrine tumors, such as PGs, are nonreactive for this determinant.
Other Reagents
Several other antibodies have been developed that have immunohistochemical properties that overlap those of LK2H10 (anti-CGA). These include both polyclonal and hybridoma reagents. A monoclonal antibody designated “HISL-19” by Krisch and coworkers127 is believed to manifest neuroendocrine specificity. The determinant that it recognizes is proteinaceous, but seems to be nonidentical to chromogranin. This statement is based on the relative strength of reactivity of HISL-19 with several neuroendocrine tissues, which is dissimilar to that of LK2H10. Moreover, some nonendocrine tissues and tumors, such as the gallbladder epithelium and adenocarcinomas of the lung, stomach, and endometrium, are labeled by the former reagent but not by the latter. These differences notwithstanding, HISL-19 appears to bind to a neurosecretory granule-related moiety, and neoplasms with few of these granules (parathyroid adenoma, melanoma, NB, and oat cell lung cancer) are only weakly stained, but others with numerous granules (pituitary adenoma, pheochromocytoma, medullary thyroid carcinoma, carcinoid, and islet cell tumor) are labeled intensely. Western immunoblot analyses have shown that the target of HISL-19 is not neuron-specific enolase.127
Lloyd and colleagues128 employed monoclonal antibodies to adrenaline (epinephrine) and noradrenaline (norepinephrine) in the study of similar neuroendocrine neoplasms. In general, pheochromocytomas displayed adrenaline alone, but extra-adrenal PGs exhibited reactivity for adrenaline and noradrenaline, or noradrenaline only. NBs, carcinoids, pituitary adenomas, pancreatic endocrine tumors, and parathyroid adenomas also showed positivity for noradrenaline. These authors emphasized that catecholamine-positive neoplasms also expressed reactivity with LK2H10, implying that stains for adrenaline and noradrenaline did not add appreciably to the diagnostic information obtained with antichromogranin.
Haspel and colleagues129 found that mice infected with reovirus type I often had autoimmune syndromes featuring polyendocrinopathies. Spleen cells from such animals were used as the substrates for hybridoma production, and two of the resulting monoclonal antibodies (5B5 and 5B8) showed reactivity with human anterior pituitary cells in frozen and Bouin’s-fixed specimens.129 Further immunostaining results in normal and neoplastic human tissues were not provided, but it was believed that 5B5 and 5B8 recognized discrete hormonal products, such as growth hormone, based on competitive inhibition studies.
The absence of discussion of S-100 protein may seem surprising because of the still-common belief that this marker is associated with neuroendocrine tumors. In reality, that is not so. The only endocrine neoplasm that reproducibly contains cells positive for S-100 protein is PG, and the immunoreactive elements therein are actually “sustentacular” cells rather than tumor cells.79 It has been contended that sustentacular elements decrease in density when PGs undergo malignant change,130 but that is an uncertain tenet in reference to individual cases.
Several markers have been assessed with regard to their associations with neuroendocrine tumor grade; SOX2, PAX5, and CD117 are the principal analytes. Each demonstrates a tendency for greater reactivity in this context as the histologic grade increases, such that grade III lesions show the highest level of immunolabeling.131–133
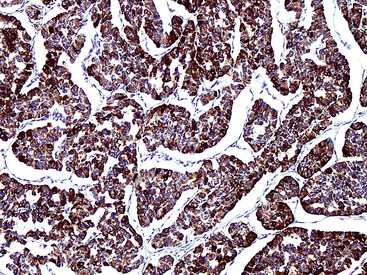
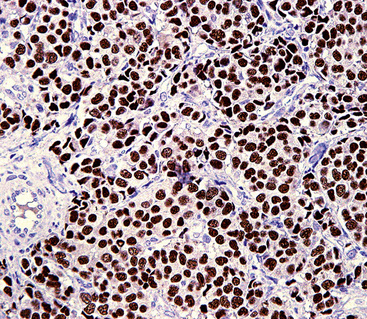
Yet another applicable reagent is antithyroid transcription factor-1 (TTF1).134–140 This intranuclear protein is expressed not only by thyroid epithelium but also by glandular cells of the lung, pulmonary adenocarcinomas, and NECs of the lung (Fig. 13-10). Among the last of these lesional groups, SCC and large cell NECs have the highest incidence of reactivity (in approximately 85% to 90% of cases). Interestingly, however, extrapulmonary high-grade NECs show a tendency to be TTF1-positive in many organ sites (bladder, uterine cervix, prostate, gastrointestinal tract), and therefore this marker can be considered to represent an “adjunct” neuroendocrine determinant, if additional studies for thyroid-related and lung-associated labels are negative. Obviously, then, TTF1 cannot be employed reliably to distinguish metastatic NEC of the lung from other neuroendocrine malignancies.
Specific Features of Pulmonary Neuroendocrine Proliferations
Before further discussion of specific neuroendocrine proliferations in the lung, a general consideration of the current nosology is in order. Travis et al.141,142 recently provided a classification scheme for pulmonary neoplasms in general through the auspices of the World Health Organization (WHO; Box 13-1).
Box 13-1 World Health Organization Classification of Pulmonary Neuroendocrine Lesions
From Travis WD, Colby TV, Corrin B, et al: Histological Typing of Lung & Pleural Tumours: International Histological Classification of Tumours. Geneva: World Health Organization; 1999:1–55.
As discussed earlier, we believe that all neuroendocrine proliferations of the lung are at least potentially malignant. The only exceptions are pulmonary tumorlets and neuroepithelial bodies, which are considered hyperplastic rather than neoplastic. As others have done,143 we espoused the scheme shown in Box 13-2 as more appropriate than the WHO paradigm in the general categorization of neuroendocrine lesions.
Box 13-2 Alternative Classification of Neuroendocrine Proliferations of the Lung
The following points constitute our rationale for this type of classification:
At this time, we make the diagnosis of “NEC,” followed by its grade, for all type I (epithelial) neuroendocrine tumors of the lung.144 Parenthetically, the traditional terminology is usually used as well to facilitate transitions in nomenclature.145 At some point, it should be possible to omit the older terminology altogether.
Grade I Neuroendocrine Carcinoma (“Classic Carcinoid”)
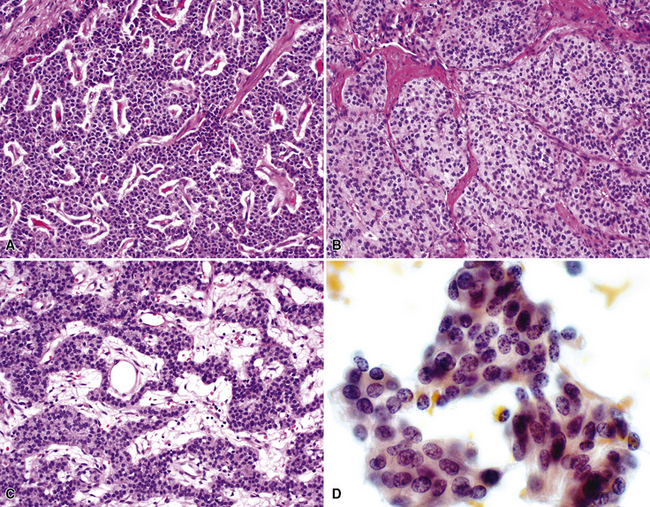
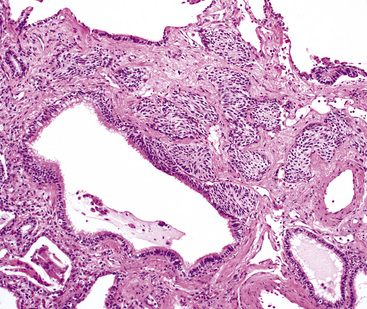
Bronchopulmonary “carcinoid” (grade I NEC) was initially considered an “adenoma” of the bronchus,146 a term that unfortunately persists in the lexicon of some individuals. The similarity of this lesion to gastrointestinal carcinoids, or “carcinoma-like tumors,” described 30 years earlier, was noted. Although the majority of classic carcinoids are centrally located, approximately 10% to 20% are found in the periphery (Fig. 13-11) of the lung.11,12,15,45,147–152 An anatomic landmark that is commonly used to distinguish central and peripheral tumors is the cartilaginous airways; neoplasms associated with such structures are considered central, and others without that relationship are considered peripheral.148
Clinical Features
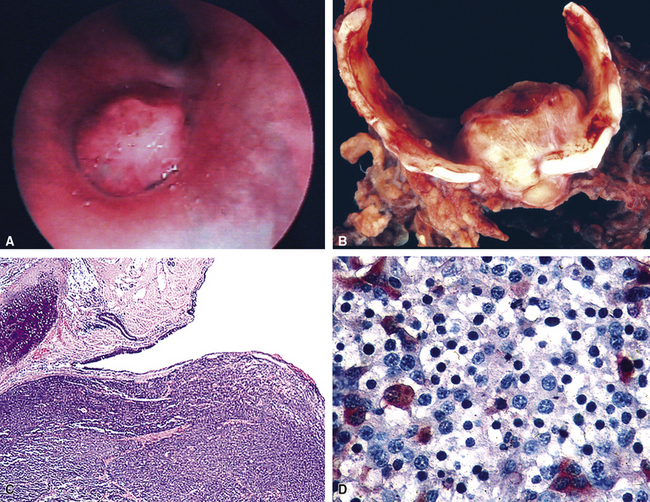
Grade I NECs with typical histologic features are rarely diagnostic problems. They usually grow as polypoid intraluminal masses with an intact overlying epithelium (Fig. 13-12) or one demonstrating squamous metaplasia.45,147,148 This pattern explains a common clinical presentation, which is localized airway obstruction. Patients manifest with wheezing, cough, or pneumonia. Carcinoid syndrome almost never develops, presumably because the tumors in question are incapable of synthesizing the biochemical substances responsible for its pathogenesis. Rare individuals with grade I NEC have associated Cushing syndrome or other endocrinopathies as a result of ectopic neuropeptide production by the neoplasm (see Fig. 13-12D).153–159 A proposed association between pulmonary carcinoids and sarcoidosis has also been made.160
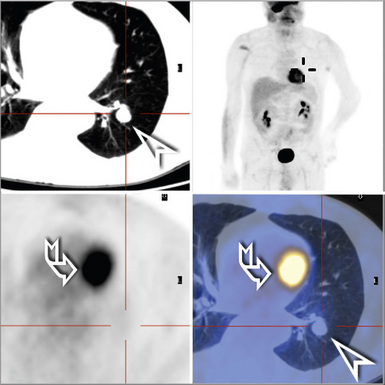
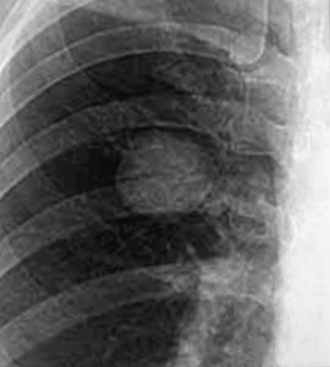
Localized obstruction also dominates the radiographic picture, with evidence of obstructive pneumonia or atelectasis (Fig. 13-13). Occasionally, a central mass with a dumbbell-like configuration is seen on plain films, and computed tomography usually demonstrates a lesion within and adjacent to a large airway.161
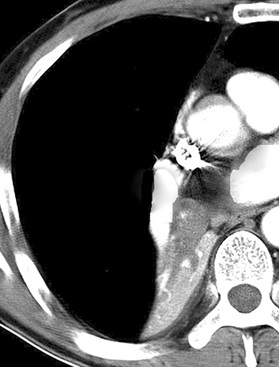
Peripheral carcinoids are subpleural, small, and well circumscribed.148,151,152 They often present as incidental findings, lacking the propensity to produce the obstructive changes of their central counterparts. Instead, the differential diagnosis of a “coin lesion” is encountered. Radiographically, such entities as granulomas, hamartomas, or peripheral adenocarcinomas enter consideration. Some studies have shown a predominance of peripheral grade I NECs in the right middle lobe, and a greater number of women have those tumors compared with central NECs of the lung.151,152
Rarely, grade I NECs may be synchronously multifocal.162,163 That eventuality is troublesome because of the possibility of metastasis to the lung from an extrapulmonary source. Extensive clinical evaluations and appropriate immunohistologic studies (discussed later) are usually required.
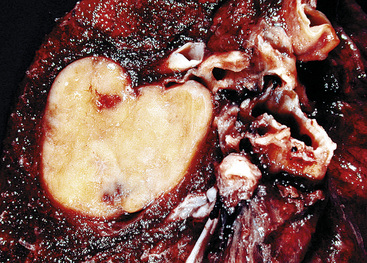
Gross and Microscopic Pathologic Findings
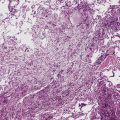
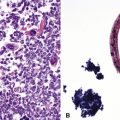
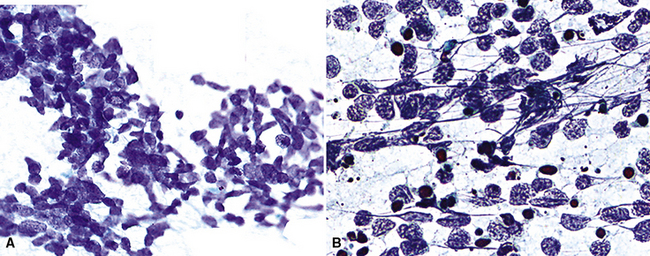
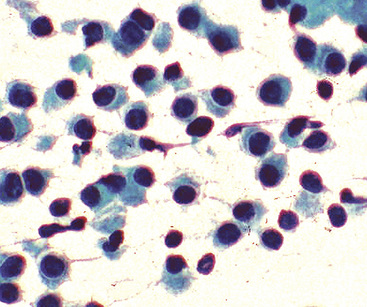
“Classic carcinoids” are typically 2 to 4 cm in maximum dimension. The lesions vary in color from tan-yellow to dark red, and they lack obvious internal necrosis and hemorrhage. Central tumors display diverse growth patterns, including trabecular, ribboning, insular, and solid sheet-like configurations11,147,148 (Fig. 13-14). A delicate fibrovascular stroma is present, sometimes with associated matrical amyloid deposition.164 Rare lesions may contain metaplastic bone or cartilage.11 The latter findings are postulated to reflect the production of factors related to tumor growth factor beta or various bone morphogenetic proteins.148 Tumor cell cytoplasm is relatively abundant, and it may be strikingly granular and oncocytic or even clear.165–169 Eccentricity of the nuclei can yield a plasmacytoid cellular image, particularly in fine-needle aspiration biopsy specimens (Fig. 13-15). A papillary configuration has also been reported.11,170 Other variants of grade I NEC produce melanin,171,172 and some are said to contain sustentacular cells immunoreactive to S-100 protein, as is more typically seen in PGs.173 An exceptional case presented by Skinner and Ewen174 featured diffuse replacement of the lung parenchyma by carcinoid tumor cells.
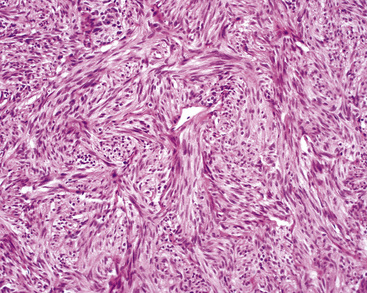
Peripheral carcinoids may be morphologically identical to central tumors, in which case the diagnosis is usually made without difficulty. Nonetheless, peripheral grade I NECs often demonstrate a prominent spindle cell growth pattern, which is associated with a somewhat different set of diagnostic alternatives (discussed later)151,152,175 (Fig. 13-16).
Differential Diagnostic Considerations
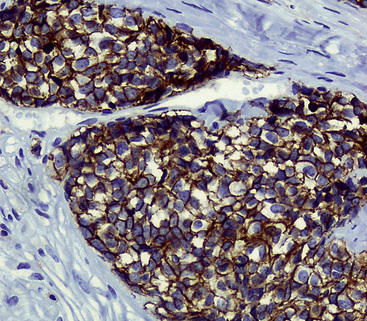
The intraluminal growth pattern of grade I pulmonary NEC can be simulated by salivary gland-like tumors, such as mucoepidermoid carcinoma; mesenchymal lesions, including peripheral nerve sheath tumors and rare inflammatory pseudotumors; and some metastatic neoplasms. Most of these possibilities present little difficulty histologically. Nonetheless, problems may arise occasionally in differentiating carcinoids from well-differentiated adenocarcinomas, higher-grade NECs, primary pulmonary PGs, and “solid” adenoid cystic carcinomas, particularly in small transbronchial biopsy specimens, which may demonstrate significant artifactual distortion. Distinction from “atypical” carcinoids is discussed in more detail later. However, uniform nuclear features, the lack of significant mitotic activity, abundant cytoplasm, and the absence of necrosis are all indicative of grade I NEC rather than a grade II tumor.3,5,149 Because of the well-differentiated nature of classic carcinoids, one can expect almost all of them to be diffusely reactive for keratin, CGA, synaptophysin, and CD57,4,173 whereas the other diagnostic alternatives do not show that immunophenotypic profile. The p53 gene, as studied by immunohistochemical or genetic analysis, is infrequently mutated in grade I NEC,176 as opposed to higher-grade neuroendocrine tumors.177
Another differential diagnostic consideration in this specific context is metastatic grade I neuroendocrine NEC from a source outside the lungs. Srivastava and Hornick178 immunohistologically compared pulmonary with nonpulmonary well-differentiated NECs, finding that the presence of thyroid transcription factor 1 was restricted to primary tumors of the lung. Classic pulmonary carcinoids also lacked reactivity for CDX2 and PDX1, both of which are alimentary tract–related markers.
Spindle cell growth in grade I NECs raises a dissimilar set of differential diagnostic considerations. These include fibrous and smooth muscle neoplasms, fibrous pseudotumors, primary or metastatic spindle cell melanomas, and metastatic medullary thyroid carcinomas. Careful attention to nuclear detail in spindle cell carcinoids shows retention of the typical stippled chromatin pattern, and nucleoli are small and inconspicuous. The lesions also express the majority of immunohistologic markers seen in central carcinoids, as discussed earlier. A combination of morphologic and adjunctive studies should be capable of excluding most of the differential diagnostic considerations mentioned earlier, but metastatic medullary thyroid carcinoma is virtually indistinguishable from grade I pulmonary NEC on pathologic grounds.164 Likewise, Eyden and coworkers179 have shown that metastatic melanomas sometimes exhibit partial neuroendocrine differentiation in ultrastructural or immunohistochemical studies.
A similar issue is the separation of tumorlets (Fig. 13-17) from peripheral carcinoids. This is an arbitrary distinction because immunohistochemical and ultrastructural studies indicate that the cells of these two lesions are essentially identical.180–182 Also, there is debate over whether tumorlets represent true neoplasms or are instead hyperplasias of respiratory neuroendocrine cells.15,181,183 They generally occur within and around small bronchioles, and they typically have a spindle cell composition. The small nests of tumor cells in a neuroendocrine tumorlet tend to be divided by a fibrous stroma into small packets, and this feature helps to differentiate neuroendocrine tumorlets from small peripheral carcinoid tumors, where tumor cell growth is more sheetlike, with variable fibrous stands interlaced. Although such tumorlets, which may number in the hundreds in some cases, are said to occur most often in diseased lungs with such associated lesions as bronchiectasis or pulmonary fibrosis,180–184 they also may arise in otherwise normal lungs.162 Rizvi and colleagues185 and Ferolla and colleagues186 have also shown that neuroendocrine hyperplasia is much more common in patients with pulmonary NECs than in others with non-neuroendocrine neoplasms. In any event, an arbitrary size of 4 mm or less has been suggested for tumorlets; larger lesions are considered peripheral carcinoids.181 A continuum for such proliferations was suggested by Miller and Muller,187 who found that peripheral carcinoid tumors were often associated with diffuse neuroendocrine cell hyperplasia and airway obstruction. A particular pitfall associated with tumorlets is that they may be misdiagnosed as much more aggressive lesions in small biopsy specimens, especially when clinical data are ignored or unavailable.188,189
Treatment and Outcome
The clinical outcome in cases of central grade I NECs of the lung is generally excellent.5,11,45 Complete excision is the treatment of choice, possibly necessitating lobectomy or sleeve resection.190 Conservative endobronchial excision is generally associated with an unacceptable rate of local recurrence.190–193
A peripheral location or a spindle cell growth pattern does not, in and of itself, equate with “atypical” morphology in grade I NECs of the lung. The prognosis for low-grade spindle cell peripheral lesions is equivalent to that of their central relatives, despite a slightly higher rate of lymph node metastasis in some series.45,151,152
Figures for the incidence of metastasis differ widely and depend on two major variables. These include the definition of peribronchial lymph node metastasis (i.e., direct invasion of nodes vs. embolic metastatic involvement) and the histologic purity of the primary lesion. Some studies with high rates of metastasis for classic “carcinoid” have improperly included higher-grade neuroendocrine lesions. Quoted metastasis rates vary from 1% to 20%; a figure of approximately 5% to 10% is probably closest to the actual incidence, and most secondary implants involve adjacent peribronchial or hilar lymph nodes.5,192–196 It is this behavioral attribute that leads us, as well as others, to consider central pulmonary carcinoids irrefutable carcinomas.143 Distant metastases also occur rarely, and there is a tendency for spread to the bones, liver, skin, or brain.45,193,194 Interestingly, osseous metastases are typically blastic.197 Flow cytometric measurement of DNA content in grade I NECs is not helpful in predicting their metastatic potential.149,198
The overall survival rate for patients with grade I NEC of the lung is greater than 95% at 5 years.5,45,148,192–196,199 Metastatic disease may be palliated with interleukin or somatostatin analogs,38,200 and surgical excision of secondary implants can be considered, given the slow growth of this tumor type.
Grade II Neuroendocrine Carcinoma (“Atypical Carcinoid”)
The term “atypical carcinoid” was first used by Arrigoni and coworkers in 1972.201 Those authors reviewed 216 bronchial carcinoids treated at the Mayo Clinic and found 23 with unusual features, including pleomorphism, mitotic activity, nuclear hyperchromatism with increased nucleocytoplasmic ratios, and evidence of spontaneous necrosis or hemorrhage. In the original series, 70% of the lesions metastasized and seven patients (30%) died of their tumors. Several terms have subsequently entered the literature on this lesion, including “Kulschitzky cell carcinoma”202 and “well-differentiated NEC,”12,15,194,202–209 and the criteria for their definition are still being debated.3,5,35
Clinical Findings
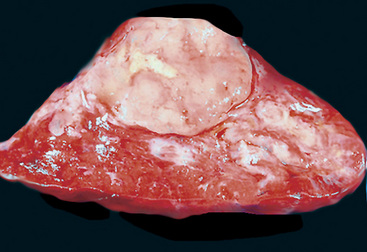
“Atypical carcinoids” of the lung are usually greater than 3 cm in maximum dimension. Other characteristics include an etiologic link to cigarette smoking, differing from the epidemiologic features of grade I NECs.5,201–216 Grade II NECs are more likely to be peripherally located in the lung (Fig. 13-18) compared with “classic” carcinoids; the former lesions have been so situated in the majority of cases in several studies.201–216 Potential associations with Cushing syndrome217 and Eaton-Lambert syndrome also apply to these lesions.
Pathologic Findings
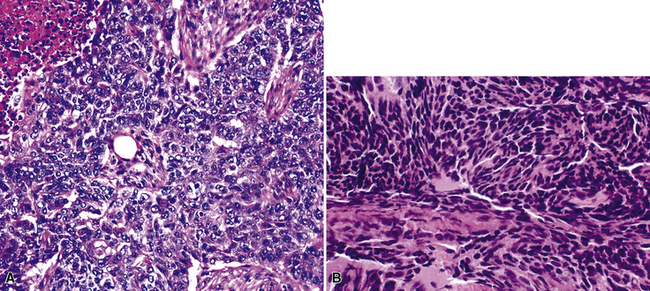
With the exception of the possible gross identification of hemorrhage or necrosis and a tendency to be slightly larger, the lesions were difficult, if not impossible, to distinguish from typical carcinoids by clinical, radiographic, or gross pathologic evaluation (Fig. 13-19). The pathologic requirements for a diagnosis of “atypical carcinoid” have varied from study to study, but most have included mitotic activity and necrosis. We currently use the criteria of El-Naggar and colleagues149 to define grade II NEC of the lung (Fig. 13-20), preferring that term to “atypical carcinoid,” but stipulating that the two are conceptually synonymous. Those requirements center on the following points:
Spindle cell change in grade II NECs of the lung is potentially seen but uncommon.215 As observed by Yousem and Taylor,3,218 a requirement for at least two of these criteria is more reasonable than basing the diagnosis of grade II NEC on only one feature in a lesion that is otherwise a classic carcinoid of the lung.
Immunohistochemical studies show some differences between grade I NEC and grade II NEC of the lung. As expected of more poorly differentiated tumors, “atypical carcinoids” tend to express fewer markers of neuroendocrine differentiation or to demonstrate more focal labeling for them; such markers as synaptophysin, CGA, and CD57 are usually less impressive in grade II lesions.92,117,120,219,220 Reactivity for specific neuropeptides—such as bombesin, calcitonin, and adrenocorticotropic hormone (ACTH)—is relatively uncommon.221 Mutant p53 protein is demonstrable by immunostaining in some atypical carcinoids, as opposed to its absence in the great majority of typical carcinoids but like its presence in most SCCs.176,222,223
Ultrastructural analysis of grade II NEC shows fewer neurosecretory granules compared with grade I neuroendocrine neoplasms.207 Cytogenetic analysis also has demonstrated multiple karyotypic abnormalities in grade II NEC but not in classic carcinoid,224–227 paralleling the flow cytometric incidences of aneuploidy in the two tumor types.149,218,228
Differential Diagnosis
In addition to its distinction from other neuroendocrine lesions, the differential diagnosis of grade II NEC includes poorly differentiated non-neuroendocrine carcinomas of the lung.44,50,53,210 In particular, selected examples of high-grade adenocarcinoma may simulate the microscopic configuration of “atypical carcinoids,” and “basaloid carcinoma” (basaloid squamous cell carcinoma)229 is regularly confused with grade II NEC. The nuclear characteristics of those lesions differ from one another in that non-neuroendocrine tumors do not manifest the dispersed chromatin seen in “atypical carcinoids,” instead showing more vesicular nuclei with often-prominent nucleoli (Fig. 13-21). Immunohistochemical studies may assist with the differential diagnosis in this context, but because some grade II NECs lack reactivity for endocrine markers and conversely selected non-neuroendocrine carcinomas of the lung demonstrate their presence, this technique is problematic. Electron microscopy is still probably the surest technique for differentiating grade II NEC from its non-neuroendocrine diagnostic simulators.
Treatment and Clinical Outcome
A vexing problem is which diagnostic label to assign a neuroendocrine pulmonary tumor with only one indicator of potentially aggressive behavior, such as angiolymphatic invasion, brisk mitotic activity, aneuploidy, or large size. None of these features independently appears to warrant adjuvant therapy, but it is probably prudent to suggest that they may be associated with an adverse outcome and to monitor the patient carefully for recurrence. Special techniques may provide additional information. In a flow cytometric analysis by El-Naggar and colleagues,149 approximately 80% of “atypical carcinoids” were aneuploid, whereas 80% of classic carcinoids were diploid. Also, grade II lesions were more likely to have an S-phase fraction of greater than 7%. Both of these factors were statistically linked to a worse outcome. However, in that analysis, the most significant predictor was accurate morphologic separation into grade I and grade II categories. Rush and colleagues,206 Jackson-York and coworkers,228 and Travis and colleagues230 concluded that accurate classification of these tumors is more important in the prognosis than DNA content. Other factors said to significantly decrease survival rates in cases of “atypical carcinoid” include lymph node metastases, vascular invasion, and overall tumor size of greater than 3 cm.5,148,201–209,210–216,230
Lymph node metastases are found at diagnosis in 30% to 50% of cases, and approximately 25% of patients with grade II NEC of the lung have remote metastatic disease at presentation.5,215,216,231 As in cases of SCC, the brain is a common site for metastasis and recurrence. Importantly, the mortality rate for this neoplasm is 30% to 50% at 2 years.5,148,201–209,210–215 Rush and colleagues206 have reported 5- and 10-year survival rates of 60% and 40%, respectively. Obviously, these figures reflect a different biologic potential than those of classic carcinoid and SCC. The use of adjunctive therapy in the management of grade II NEC of the lung is unresolved.232,233 Some researchers have suggested that chemotherapy, irradiation, or both can be beneficial in this setting, but there has been no consensus on that point, much less on which pharmacologic agents to use. The difficulty in securing diagnostically “pure” study populations for treatment evaluations has likely contributed to this uncertainty.
Grade III Neuroendocrine Carcinoma, Small Cell Type
Small cell carcinoma is probably the best recognized neuroendocrine neoplasm in this discussion, accounting for approximately 25% of all lung carcinomas.32 That diagnostic entity is generally traced to Barnard’s 1926 description of “oat cell carcinoma”234; although the tumor had been believed to represent a lymphoma or sarcoma before that time, he accurately recognized its epithelial nature. In 1959, Azzopardi refined the pathologic description of SCC.235
Clinical Features
Small cell carcinoma of the lung has many potential modes of clinical presentation. These include symptoms and signs similar to those of other common carcinomas of the lung, such as cough, hemoptysis, weight loss, anorexia, fatigue, and the syndrome of hypertrophic osteoarthropathy.236,237 In addition, however, unusual findings, such as hyponatremia, hypercalcemia, or Cushing syndrome, may emanate from ectopic production by the tumor of antidiuretic hormone, parathyroid hormone-like peptides, and ACTH, respectively.155,159,236,238,239 Rarely, individuals with SCC manifest Eaton-Lambert (pseudomyasthenia) syndrome owing to paraneoplastic interference with neuromuscular function.240,241 Retinopathy, central and peripheral neuropathies, encephalitis, glomerulonephritis, cutaneous reactions, sarcoid-like granulomatous lesions, and systemic vasculitis also have been reported in this context.241–248 Massive hepatomegaly with liver failure,249 myelophthisic anemia,250,251 or seizure activity and headaches252 are additional clinical constellations related to the effects of distant visceral metastases at initial diagnosis.
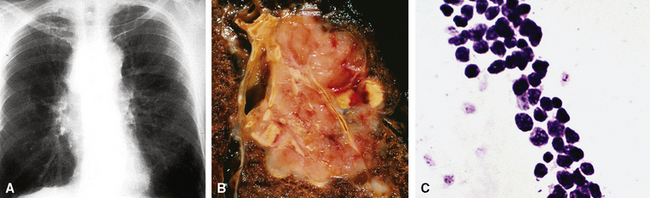
Chest radiographic findings may be relatively unremarkable or may demonstrate a central hilar (or, more rarely, a peripheral intrapulmonary) mass (Fig. 13-22). Obstructive pneumonia is a potential complication; bulky peribronchial or mediastinal lymphadenopathy is common and may be massive.161,236 Pleural effusions typically signify serosal metastases of SCC, and rarely, the tumor may so markedly involve the pleura that it produces a peripulmonary “rind” similar to that seen in association with mesotheliomas.253
Pathologic Findings
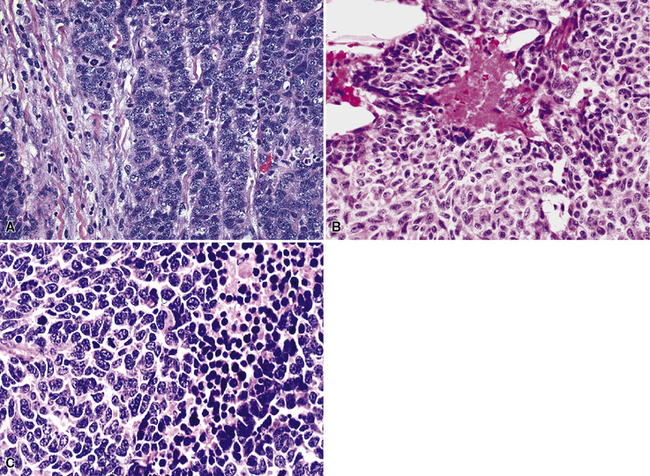
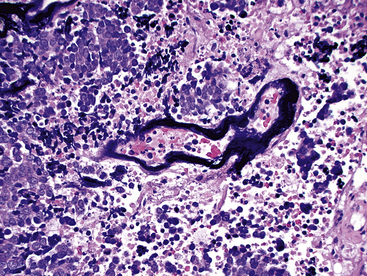
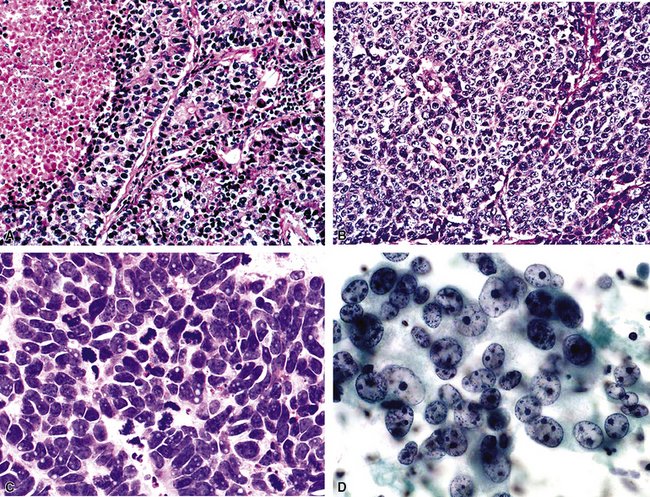
Microscopically, SCC exhibits a spectrum of morphologic appearances.235,254–263 Classic “oat cell” tumors are relatively rare, accounting for only 10% to 20% of cases.254,255 Oat cell morphology (Fig. 13-23), with bluntly fusiform, carrot-shaped cells, nuclear molding, and crush artifact, is more often seen in bronchial biopsy specimens or fine-needle aspiration biopsy specimens than in resected neoplasms or lymph node metastases. Tumor cell size is in the range of 1.5 to 3 times the diameter of non-neoplastic lymphocytes.263 The now defunct term “intermediate cell variant” of SCC (Fig. 13-24) accounts for approximately 75% of cases, and may be combined with oat cell areas.256–259 Cell size in the former subtype is more variable, but may be as much as twice that seen in oat cell SCC. Nuclear molding and crush artifact are less conspicuous; the tumor cells also may show small nucleoli, but they maintain the marked hyperchromatism and granular chromatin that are seen in other forms of SCC. There may be blunt spindle cell change, and the cells may show pseudorosette formation or may grow in distinct ribbons. Other recognized types are combined small cell and large cell NEC260,264,265 and mixtures of SCC with either adenocarcinoma or squamous carcinoma (Fig. 13-25).27 All variations share the tendency to show prominent apoptosis, brisk mitotic activity, scant amphophilic cytoplasm, and the “Azzopardi phenomenon,” which is the accretion of basophilic nucleic acid around intratumoral blood vessels254,255 (Fig. 13-26). Interestingly, D’Adda and coworkers266 found that both neoplastic components were genetically homologous in tumors showing a combination of SCC and large cell NEC.
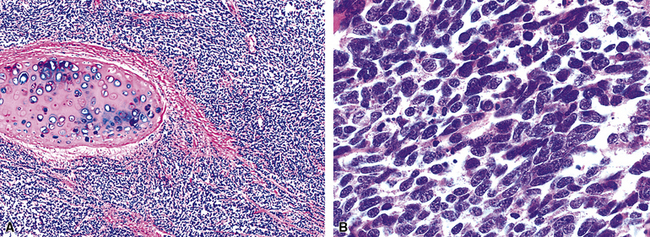
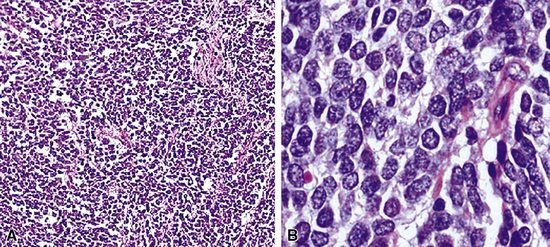
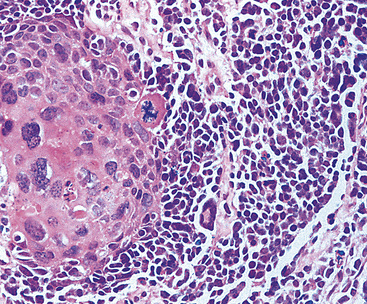
Figure 13-25 Divergent squamous differentiation is apparent in this small cell neuroendocrine carcinoma (left upper center).
In our opinion, differences in cytomorphology in SCC have little or no prognostic importance, based on a synthesis of the aggregated literature.236,256–259,267,268 Instead, it is necessary to be familiar with the microscopic subtypes to avoid misdiagnosis. Some variation in nuclear size, small nucleoli, and a modest amount of cytoplasm should not prevent one from making a diagnosis of SCC, recognizing that these features are seen regularly in the intermediate variant of SCC, especially in cytologic preparations.265 This is particularly true in fine-needle aspirate material.262,269–271
Another difficulty that one may face is the identification of SCC in biopsy specimens of peripheral lung nodules, as seen in approximately 10% of cases.272 In that setting, the fear among many pathologists is that the lesion may represent a lower-grade neuroendocrine lesion. This difficulty is best resolved by paying close attention to cytologic details. Marked nuclear hyperchromatism, brisk apoptosis, and scanty cytoplasm usually allow one to comfortably exclude a lower-grade lesion. Secondly, and more importantly, evidence has emerged that suggests that surgical therapy is appropriate for peripheral high-grade but low-stage NECs of the lung.272–278 As stated earlier, rigid adherence to the dogma of “surgery for non–small cell carcinoma; no surgery for SCC” is probably improperly restrictive. Smit and colleagues275 studied 20 patients with resected SCC of stages I, II, and III. The median survival in that series was 29 months for stage I and II tumors and 20 months for stage III lesions. Another review of this topic was published by Mentzer and colleagues.274 Other contextual topics of debate include whether surgery should precede or follow chemotherapy or be accompanied by irradiation.236,279,280 Nonetheless, we believe that sufficient data are available to suggest that all low-stage NECs of the lung should be resected, regardless of grade.
Another conundrum in fine-needle aspirates, limited biopsy specimens, or frozen sections is the distinction between SCC and high-grade large cell NEC of the lung.281,282 The two forms of grade III may coexist in the same tumor mass, yielding the neoplastic variant known as “combined SCC.”263–265,283 As discussed earlier, the separation of these two forms of high-grade NEC has dubious significance, in our opinion, with regard to the suitability of surgical management. Use of the designation “high-grade NEC” is an equitable solution; whether the lesion is then excised should be decided by the clinical stage rather than by cytologic details.
Differential Diagnosis
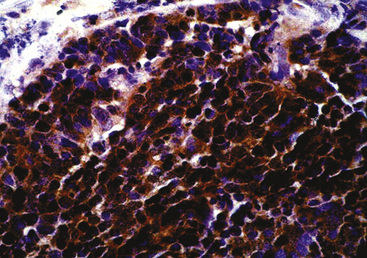
The diagnosis of SCC in biopsy material involves the exclusion of lymphoid infiltrates as well as other types of carcinoma that may be composed of small basaloid cells.254,255,284–286 As stated previously, broadly reactive keratin antibody mixtures label essentially all SCCs, in many cases with a characteristic dot-like pattern of cytoplasmic staining. Hence, a simple two-antibody panel for cytokeratin and CD45 is effective in showing that a morphologically indeterminate small cell tumor is epithelial.56 Extremely rare reports of apparent CD45 reactivity in SCC287 underscore the inadvisability of relying on one immunostain diagnostically. In our experience, a more difficult question is the exclusion of basaloid squamous cell carcinoma, particularly in a small biopsy specimen.229 The keratin labeling pattern described earlier may be helpful, and an extended panel of antibodies to CGA, neural cell adhesion molecule (NCAM; CD56), synaptophysin (Fig. 13-27), and CD57 can be applied as well. That set of reagents will identify approximately 80% of all SCCs.56,219 Incidentally, we do not subscribe to the premise that there are “small cell neuroendocrine” and “small cell undifferentiated” carcinomas, but rather believe that some poorly differentiated tumors simply do not express overt neuroendocrine differentiation to a degree detectable by current methods.56,288 Again, clinical stage is the best determinant of whether surgery is advisable, and in that framework, it becomes less crucial to separate SCC from basaloid squamous carcinoma in biopsy specimens.
Treatment and Clinical Outcome
Over the years, SCC has emerged as a clinically distinctive entity. As virtually all physicians are aware, long-term survival of patients with this neoplasm is rare; it is reported in fewer than 5% of cases in most centers,233,236,279,280,289,290 with rare exceptions.291 A staging system centered on the terms “limited” and “extensive” SCC has been supplanted by American Joint Committee on Cancer staging, in which stages I, II, and III correspond to the old “limited” stages and “extensive” disease is stage IV.236 Staging is a powerful parameter and is virtually the only reliable indicator of clinical outcome for pulmonary SCC. As we have stated repeatedly, low-stage tumors are potentially resectable272–278; chemotherapy with or without irradiation can be given postoperatively as “consolidation” treatment.236
Naturally, many factors have been investigated as possible prognostic indicators in SCC. As stated earlier, we do not believe that cytologic subdivision into “oat cell” and “intermediate cell” SCC is prognostically contributory, despite the claims of some other authors.268 Combined SCC (e.g., SCC admixed with squamous carcinoma, adenocarcinoma) and combined SCC (SCC admixed with large cell NEC) do, however, appear to be more refractory to therapy.236,292 Vollmer and colleagues263 have suggested that an ultrastructural loss of desmosomes was associated with more aggressive behavior in SCC, but that observation has not been the focus of practical application. DNA ploidy analysis similarly does not seem to contribute significant information. Numerous cytogenetic abnormalities are now reported in SCC of the lung; one of the most common is a deletion of 3p,293–296 suggesting that that area may harbor potential tumor suppressor loci. Other abnormalities that have commonly been seen include deletions of 5q, 9p, 11p, l3q, and 17p.296,297 Proto-oncogenes implicated include c-myc, n-myc, myb, c-kit, c-src, and c-jun.294,296 At least two tumor suppressor genes—p53 and the retinoblastoma gene—may play a role in the carcinogenesis of SCC.25,294,296 As mentioned in the discussion of grade I and grade II NECs of the lung, the p53 gene is more often mutated in SCC than it is in lower-grade lesions. Other molecules, including p-glycoprotein (the multidrug resistance protein), CD99, and bcl-2 protein have likewise been investigated as possible prognosticators.294,296 Duncavage and coworkers298 have shown that tumor cells in primary and metastatic SCC of the lung lack the Merkel cell polyoma virus, separating it biologically from primary SCC of the skin. Conversely, Ralston and colleagues299 found nuclear MASH1 immunoreactivity in more than 80% of cases of pulmonary SCC, but that marker was not apparent in primary cutaneous NECs.
Grade III Neuroendocrine Carcinoma, Large Cell Type (Large Cell Neuroendocrine Carcinoma)
The diagnosis of “large cell neuroendocrine carcinoma” (LCNC) is a relatively new addition to the nomenclature pertaining to pulmonary carcinomas. Travis and coworkers230 proposed that this term be used for tumors that show morphologically overt features of neuroendocrine differentiation by light microscopy, but do not fit into the categories of grade I NEC, grade II NEC, or SCC. Thus, LCNCs are, by definition, different lesions than non–small cell lung cancers that demonstrate evidence of neuroendocrine differentiation only by immunohistochemical or ultrastructural studies26,34; those are discussed later. LCNC is also synonymous with neoplasms that have been called “intermediately differentiated NEC” by Warren and coworkers.12,55 As we have already stated, we believe that use of the modifier “intermediate” for these cases is ill-chosen because it introduces confusion with the “intermediate” cellular variant of SCC. We prefer the term “grade III NEC, large cell type” to refer to LCNC, for reasons that are specified later. Other authors have endorsed that usage.
Clinical Features
The clinical attributes of LCNCs are hybrids of those attending adenocarcinoma of the lung and SCC. In series by Travis and colleagues5,230 and others,233,289,290,297,300–303 LCNCs almost always occur in heavy smokers, as does SCC. Symptoms and signs are generally most like those of non-neuroendocrine carcinoma; however, as in SCC, examples of Eaton-Lambert syndrome304 and paraneoplastic retinopathy305 also have been reported in connection with grade III large cell NEC. Although a minority of cases of LCNC present as central masses, they tend to be situated in the mid-to-peripheral lung fields (Fig. 13-28). Oddly, despite a high histologic grade, most LCNCs present as T1 or T2 tumors without lymph node metastases or evidence of systemic spread.5,300
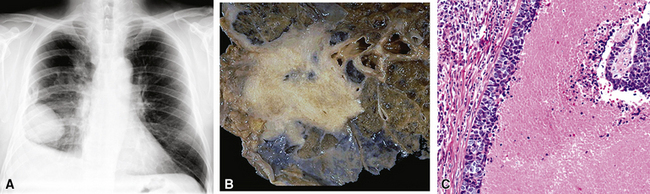
Figure 13-28 A, A large mass is seen in the lower right lung field, representing a large cell neuroendocrine carcinoma (LCNC). B, The gross appearance of LCNC is much like that of small cell neuroendocrine carcinoma (see Fig. 13-22B). C, Prominent infarct-like necrosis is a common finding in large cell neuroendocrine carcinoma of the lung.
Pathologic Findings
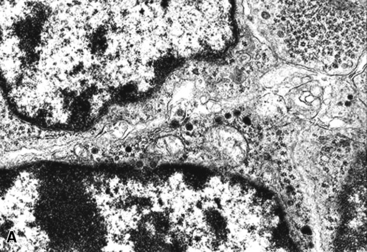
Grossly, pulmonary grade III NEC of the large cell type varies in maximum dimension from 1 to more than 10 cm and often demonstrates central necrosis, with or without dystrophic calcification.5,297,300 Microscopically, it typically shows extensive coagulative necrosis that is obvious on low-power examination (see Fig. 13-28). At slightly higher magnification, one often sees an insular or ribboning growth pattern in the tumor between the necrotic zones. The individual neoplastic cells are larger than those of grade II lesions, with moderate-to-abundant amphophilic cytoplasm. In some instances, tumor cells are granular and eosinophilic; Chetty and coworkers306 have reported rare examples with “rhabdoid” cytologic features, in which globular hyaline cytoplasmic inclusions were apparent in the tumor cells, and Khalifa and colleagues307 documented an example of LCNC with divergent sarcomatoid (pleomorphic and spindle cell) differentiation. Nuclei in LCNC are more heterogeneous than those in SCC or grade II NEC; nucleoli are variable in prominence and nuclear chromatin is more heterogeneous, varying from granular to vesicular (Fig. 13-29). The mitotic rate in LCNC is brisk, usually measuring in excess of 10 division figures per 10 high-power microscopic fields, and sometimes being in the range of 50 to 100.5 All cases show reactivity for cytokeratin, and immunoreactivity for at least one neuroendocrine determinant is observed in almost all cases.5,297 Nevertheless, we believe that a diagnosis of LCNC can be made confidently, even if all such markers are absent. In those instances, the cytomorphologic attributes of the tumor are so classically those of a neuroendocrine neoplasm that no interpretative doubt exists. Electron microscopy shows dense core granules in large cell grade III NEC.5,230,302
Differential Diagnosis
These lesions may be challenging to recognize. In the past, examples of LCNC were probably assigned to one of three other diagnostic categories: “atypical carcinoid”; “non–small cell carcinoma, not further specified”; or “large cell undifferentiated carcinoma.” A distinction from grade II NEC can usually be made by paying attention to several features. First, the cells of LCNC tend to be larger, and there is more nuclear pleomorphism. Chromatin in “atypical carcinoid” tends to be granular and nucleoli are small308; in contrast, the chromatin of LCNC is not uncommonly vesicular, and nucleoli are often prominent. Nucleocytoplasmic ratios in grade II tumors are actually higher than those of LCNCs. Necrosis in the latter tumor type is generally extensive and infarct-like, in contrast to grade II NEC, in which more punctate or limited confluent necrosis is seen. Mitotic activity is another key feature in this diagnostic comparison. As outlined by Travis and colleagues,5,230,308 LCNC has a high mitotic rate (>50/10 high-power fields). Accordingly, those authors suggested that cases with more than 10 mitoses per 10 high-power fields probably represent LCNC, whereas “atypical carcinoid” is more likely to show a figure of 10 or fewer. However, we prefer to assess the overall microscopic configuration of the lesion in making that distinction, rather than relying on one pathologic parameter. Despite that caveat, some examples of pulmonary neuroendocrine tumors are still encountered in which it is virtually impossible to assign a label of grade II NEC versus LCNC with certainty.
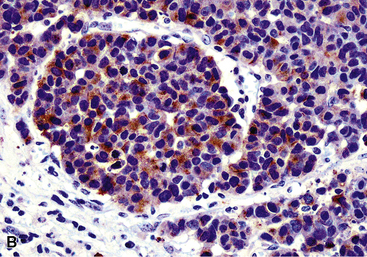
Differentiation of LCNC from non-neuroendocrine non–small cell carcinomas depends first and foremost on endocrine morphologic features, enumerated earlier. As referenced earlier, we believe that the low-power appearance of grade III large cell NEC is one of the most helpful indicators of the proper diagnosis. Extensive zonal necrosis, producing a “jigsaw puzzle” pattern, is often visible. Prompted by this feature, which, of course, can be mimicked by the appearance of necrotic squamous cell carcinoma or poorly differentiated adenocarcinoma, one then examines the lesion for organoid growth and neuroendocrine nuclear features. In contrast to our discussion of SCC, special techniques sometimes play an important part in the diagnosis of LCNC of the lung, and ultrastructural studies are useful in demonstrating neurosecretory granules.5,309 By immunohistochemical analysis, all cases of grade III large cell NEC should demonstrate keratin reactivity and many are carcinoembryonic antigen-positive.5,230,297 Virtually all LCNCs react with antibodies to NSE and CD56; among more “specific” markers, CGA is the most consistently detected antigen. Antibodies to CD57 and synaptophysin stain fewer cases. Rossi and coworkers310 showed that a three-marker panel comprising synaptophysin, CGA, and CD56 was effective in separating LCNC from nonendocrine tumors; if any two of those determinants were seen, the diagnosis was secure.
This discussion raises several points. First, given the vagaries of immunohistochemistry and neoplasia, one could predict that cases will be encountered with morphologic features that are consistent with—but not diagnostic of—LCNC, but do not demonstrate any “neuroendocrine” markers. Electron microscopic examination can be used in such cases (Fig. 13-30A and B). It is advisable to provide some corroborating evidence of neuroendocrine differentiation before making a diagnosis of LCNC in this specific context, because some basaloid squamous cell carcinomas229 and other non-neuroendocrine large cell carcinomas may closely mimic LCNC.26,34 Conversely, however, if no light microscopic features support the diagnosis of LCNC, immunostaining results and ultrastructural analysis should not be used to make the diagnosis. It has been well documented that generic non–small cell lung neoplasms may contain cells with endocrine characteristics,26,34 as considered subsequently.
Even though they are both forms of grade III NEC, LCNC and SCC can usually be distinguished from one another readily in surgical material. The fact that they are different tumor entities is supported by the observations of Ullmann and colleagues,311 showing that LCNC and SCC have dissimilar genotypes. The larger cell size, polygonal shape, lower nucleocytoplasmic ratios, nucleolation, and more irregular chromatin in LCNCs should make this distinction relatively straightforward in most cases. Nevertheless, the two cytologic forms of high-grade NEC occasionally coexist in “combined SCCs.”35,263,264 Moreover, in cytologic material, the cited distinction between SCC and LCNC is sometimes very challenging.312 Yang and coworkers269 have described three potential images of LCNC in fine-needle aspiration specimens, one of which closely simulates SCC. The presence of prominent nucleoli is a helpful clue to the recognition of LCNC in that setting.269,270
Treatment and Clinical Outcome
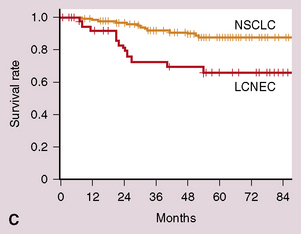
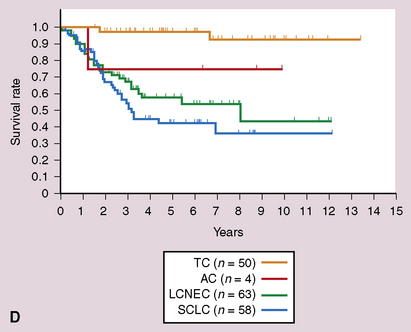
The aggressive behavior of grade III large cell NECs of the lung cannot be overstated313–317 (see Fig. 13-30C and D). Of 10 patients studied by Warren and colleagues,12 only 1 was alive at 2 years’ follow-up, despite the fact that all of the cases were stage I (T1 or 2/N0/M0). All of the patients in series by Travis and colleagues5,230 had either died of their tumors or were likely to do so at the same time point. Another report by Rush and coworkers206 quoted respective 5- and 10-year survival rates of 33% and 11% for LCNC. Dresler and colleagues300 studied a series of 40 non–small cell NECs, including 23 LCNC as defined here. The survival rate for stage I cases in that series was 18% at 40 months, significantly worse than that for those with similarly staged adenocarcinomas or squamous carcinomas. Surprisingly, the survival of patients with resected low-stage SCCs,272–278 which are usually peripheral, is better than that of patients with LCNC. Likewise, the latter tumor type is more aggressive than “atypical carcinoid.”5,206,230,300
Travis and colleagues5 and Rush and co-workers206 performed flow cytometric DNA analysis on cases of grade III large cell NEC of the lung. Both groups found no prognostic value in those studies.
The optimal therapy for these lesions is still in evolution, in large part because there has been a frustrating tendency for both pathologists and clinicians to push LCNC inappropriately into generic non–small cell carcinoma treatment protocols rather than evaluating them as a separate tumor entity. Because LCNCs tend to present as low-stage lesions,300–303,318 most can—and should—be surgically resected.315–317,319,320 However, there have been few randomized trials to address the value of specific chemotherapy regimens or irradiation. In most patients with grade III large cell NEC, therapy appears to fail, with either distant metastases or intrathoracic recurrence occurring.5,230,297,300 Therefore, the use of adjunctive treatment modalities would appear to have merit.315–317 Nonetheless, accrued experience suggests that LCNCs respond relatively unimpressively to standard chemotherapy regimens used for SCC.321 With selected exceptions, tumors representing “combined SCCs” (SCC/LCNC in the same tumor mass) have also had only a modest response to such treatment.35,236,263,264 In that regard, it is intriguing that expression of the multidrug resistance protein seems to be relatively common in grade III large cell NECs.322 Unfortunately, pulmonary NECs do not appear to manifest mutations in the epidermal growth factor receptor gene. Therefore, they are not likely to respond to epidermal growth factor receptor inhibitors.323 Faggiano and colleagues324 have found that a high mitotic count (>10 mitoses/high-power [×400] microscopic field), an absence of immunohistologic neuroendocrine markers, and an immunohistochemical bcl-2/bax ratio of greater than 1 were adverse prognosticators for LCNC at a pathologic level of evaluation.
Composite (Combined) Neuroendocrine/Non-Neuroendocrine Carcinomas
The concept of overtly “combined” or “divergent” differentiation has been recognized increasingly in the last few years, using conventional light microscopy.27,325 Although this pattern is perhaps most frequently encountered in the gut and the lung,258,325,326 composite epithelial tumors with a partial neuroendocrine phenotype have been observed in a wide variety of organ sites. The biologic significance of composite tumors with neuroendocrine differentiation is still being studied, but it appears to depend on the location of the particular neoplasm being considered as well as the level of cytologic anaplasia inherent in that tumor.282,325
A bewildering array of terms has been used to describe these neoplasms, and many are still in use. These include “stem cell carcinoma,” “amphicrine carcinoma,” “composite carcinoma (SCC/adenocarcinoma or squamous cell carcinoma),” and “carcinoid with glandular differentiation.”325,327–338 Needless to say, many people are confused by this diverse lexicon and are uncertain as to how to treat the neoplasms. In our view, because mixtures of NEC (with variable levels of differentiation, including high-grade tumors) and squamous cell carcinoma, adenocarcinoma, transitional carcinoma, or spindle cell carcinoma have been documented in malignant epithelial neoplasms of the lung, pancreas, stomach, duodenum, small bowel, colon, rectum, pancreas, liver, biliary tree, urinary bladder, prostate, uterine cervix and endometrium, ovaries, breast, skin, thyroid, salivary glands, and larynx,51,327–338 it would seem most straightforward to use descriptive terminology diagnostically, such as “combined NEC–adenocarcinoma” (Fig. 13-31) or “combined NEC–squamous cell carcinoma,” along with comments in surgical pathology reports that summarize the morphologic details and expected behavioral attributes of the lesion.
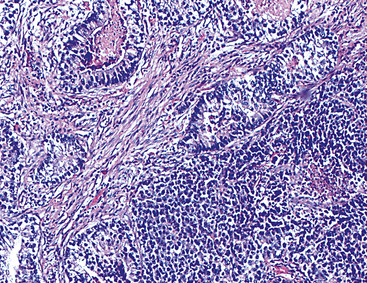
Figure 13-31 Combined adenocarcinoma (left) and grade II neuroendocrine carcinoma (right) of the lung.
Clinical Findings
Virtually any carcinoma in the lung can potentially have a neuroendocrine component, which can be grade I, II, or III NEC. Hence, there are no specific clinical features that can be ascribed to “combined” neuroendocrine/non-neuroendocrine tumors. Nonetheless, mixtures of squamous carcinoma or adenocarcinoma with NEC are most commonly seen in biopsy specimens that are taken after therapy,339 suggesting that effective treatment of the neuroendocrine cell population may allow for “overgrowth” of a minor nonendocrine component.
Pathologic Features
Unlike poorly differentiated carcinomas in which neuroendocrine differentiation is “occult,” and therefore discernible only by application of immunohistology or electron microscopy,340 the tumors considered in this section are recognizable as mixed by conventional microscopy. All of the neuroendocrine tumor types (i.e., grade I NEC, grade II NEC, small cell or large cell grade III NEC) that have been discussed up to this point may be components of combined carcinomas in the lung.
Special studies are likely to lead to a spectrum of findings. Some tumors demonstrate truly bifid differentiation—exhibiting, for example, true glandular and neuroendocrine features in the same neoplastic cells.325,327 Others display admixtures of cellular elements with mutually exclusive ultrastructural or immunophenotypic properties. These differences do not appear to have any meaning from mechanistic or clinical points of view. There are discussions in the literature about whether divergent neuroendocrine lesions are “collision” tumors.325 Because virtually all neoplasms derive from transformed stem cells, regardless of anatomic site, it would appear much more logical to conclude that divergent growth simply emanates from dissimilar (and largely unknown) post-transformational modulators of differentiation.341,342
Differential Diagnosis
Because of the unique histologic appearance of combined carcinomas with neuroendocrine elements, differential diagnostic considerations are limited. These generally concern making a distinction between poorly differentiated NEC components and high-grade portions of “pure” adenocarcinomas or squamous cell carcinomas with variable microscopic patterns that simulate neuroendocrine differentiation.284 Ultrastructural and immunohistochemical markers of neuroendocrine differentiation should be pursued if that question arises.
Treatment and Clinical Outcome
Combined neuroendocrine/non-neuroendocrine carcinomas generally have more adverse prognoses than histologically “pure” tumors,27,236,292,337,343 including lesions with grade I NEC components, which are very rare.338 Thus, therapy must be chosen to address all of the cellular elements in these composite neoplasms, but with the expectation that response to treatment will likely be blunted and survival will be worse than that of patients with pathologically homogeneous lesions.
Non–Small Cell Lung Carcinomas with Occult Neuroendocrine Differentiation
Beginning in the 1980s, several investigators have noted neuroendocrine features (e.g., neurosecretory granules, immunoreactivity for endocrine peptides) in lung tumors that otherwise had the microscopic appearance of poorly differentiated squamous cell carcinoma and adenocarcinoma or large cell “undifferentiated” carcinoma.26,340,344–355 With those observations, controversies began over diagnostic terms that should be appended to such neoplasms, as well as their behavioral attributes.
The notion that there may be a variety of cell types in any given neoplasm has steadily gained recognition over the last decade. Tumors of the skin, genitourinary tract, gastrointestinal tract, lung, and many other sites all share this potential, which is best termed “multidirectional differentiation.”325 “Occult” neuroendocrine lesions are no different than others with glandular or squamous differentiation in this context, in the sense that very poorly differentiated neoplasms may not show light microscopic patterns that indicate any of these cellular lineages.356 Their “hidden” characteristics can only be detected by adjunctive pathologic techniques; as stated previously, that practical point of difference nosologically separates LCNC (which has microscopically overt neuroendocrine features) and “large cell carcinoma with occult neuroendocrine differentiation” (which does not).
Pathologic Features
As discussed earlier, pulmonary squamous cell carcinomas and adenocarcinomas with OND are no different grossly or histologically than their counterparts that lack endocrine features.340,345–347,357 With specific reference to large cell “anaplastic” carcinomas of the lung with OND,26,34,341,350,351 the tumor cells are at least twice as large as those of SCC. By definition, those lesions lack overt glandular or squamous differentiation and have rather monomorphic nuclei with dispersed or vesicular chromatin and prominent nucleoli. Small foci of necrosis may be seen, but these lesions lack the infarct-like configuration seen in LCNC. Similarly, manifestations of organoid growth (e.g., insulae, ribbons, cords, rosettes) are absent in large cell carcinomas with OND. Still other tumors with occult neuroendocrine elements may show more unusual histologic patterns, such as sarcomatoid differentiation273 or a blastomatous configuration (e.g., as in “well-differentiated adenocarcinoma simulating fetal lung”).358,359
There is some debate over the “best” method to document OND in lung carcinomas.59,360 Some observers argue that ultrastructure should be the standard technique,59 but as discussed earlier, sampling problems interfere with the reliability of that procedure. Putative nonspecificity of neuroendocrine determinants, such as chromogranin and synaptophysin, is not a problem in our view, because we believe that publications that raised such concerns59 contained conceptual flaws. They were based on electron microscopy as the “validating” technology, ignoring the effects of sampling just cited. In our view, positivity for either of these two immunomarkers (Fig. 13-32) reliably predicts neuroendocrine differentiation. With reference to the incidence of neuroendocrine reactivity in nonendocrine carcinomas, Sørhaug and coworkers361 have posited that it is steadily increasing as immunohistochemical techniques become more sensitive.
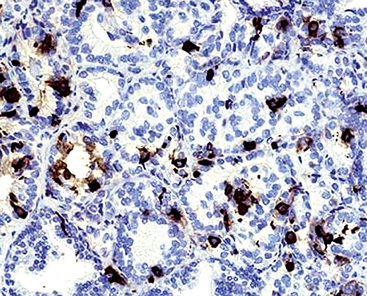
Differential Diagnosis
The differential diagnosis of squamous cell carcinomas or adenocarcinomas with OND principally includes similar tumors that lack endocrine features. On the other hand, large cell carcinoma with OND must be distinguished from both large cell “undifferentiated” lung carcinoma and LCNC, primarily by electron microscopy and immunohistology.34 In addition, large cell carcinomas may be sufficiently nebulous morphologically that metastatic melanoma, poorly differentiated sarcoma with epithelioid features, and large cell lymphoma enter into diagnostic consideration. Again, adjunctive pathologic studies are usually required.362
Treatment and Clinical Outcome
Some investigators have concluded that neuroendocrine differentiation justifies the use of modified therapeutic approaches, such as those employed in SCC, whereas other authors have demurred on that point.363–367 Similarly, there is no consensus as to whether any prognostic value may be derived from the identification of “occult” neuroendocrine differentiation in lung cancers of various pathologic types, with contradictions in published studies.365–369 Currently, all that can be said with confidence is that surgical excision is still the foundation of treatment in low-stage cases.34,367
Primary Intrapulmonary Paraganglioma
Paraganglioma is a distinctly uncommon lesion in the population at large,370 and it is vanishingly rare as a primary pulmonary tumor, with fewer than 25 reported cases in the world literature.371–381 The demographic profiles of patients with this neoplasm are variable, depending on the topographic location of the lesion and its possible occurrence in genetic syndrome complexes.
Clinical Features
In sporadic cases of PG in the lung and elsewhere, men predominate and usually come to diagnostic attention in middle life (40–50 years of age).370–380 In contrast, women outnumber men in the context of the MEN type 2 syndromes, and they are recognized as having a PG approximately 15 years earlier.370 The latter observation may simply reflect the fact that family members in kindreds with MEN are usually regularly screened for constituent tumors from childhood onward.382 PGs have been described in virtually all organ sites, including the orbits, nasal cavity, thyroid, heart, urinary bladder, gallbladder, liver, biliary system, kidneys, prostate, urethra, spermatic cord, uterus, ovaries, vagina, vulva, cauda equina, and lungs; primary pulmonary tumors are among the rarest.371
Functionality of PGs in these diverse locations is sporadic. Biosynthetic tumors may present with episodic or sustained hypertension; hypertensive crisis or “malignant hyperthermia” on induction of general anesthesia; episodic nausea, weakness, cardiac arrhythmias, pallor, flushing, headache, diaphoresis, anxiety, or localized pain; or cardiovascular decompensation with heart failure. Reflections of neuropeptide production may include Cushing syndrome with PGs that synthesize ACTH or watery diarrhea–hypokalemia complex (Verner-Morrison syndrome) with neoplasms that manufacture vasoactive intestinal polypeptide. Occasional examples also have apparently produced a parathyroid hormone–related peptide, with associated hypercalcemia, or an erythropoietin-like moiety in linkage with paraneoplastic polycythemia. Finally, rare patients with PG and associated systemic abnormalities may have von Recklinghausen disease (neurofibromatosis) or Beckwith-Wiedemann syndrome (hemihypertrophy and macroglossia).370
Nonfunctional tumors become manifest only through the appearance of a steadily enlarging mass, with other symptoms and signs dependent on the anatomic location of the lesion. In the lung, PGs that impinge on large airways produce symptoms related to obstruction (e.g., cough or stridor), but peripheral nonfunctional lesions are typically found incidentally on screening chest radiographs371–380 (Fig. 13-33). Rarely, multiple synchronous intrapulmonary PGs may be encountered,381 simulating metastases on chest radiographs.
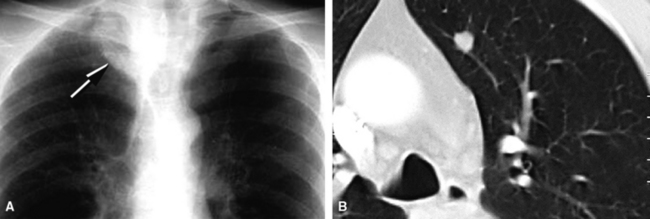
Pathologic Features
Grossly, PGs are typically spherical or slightly lobulated masses that range from a few millimeters to several centimeters in greatest dimension. They may be either centrally or peripherally located in the lung. Their cut surfaces are bloody in most cases because of dense intralesional vascularity, and the tumor tissue itself may be gray, pink, lavender, brown, or mottled (Fig. 13-34). Characteristically, immersion of fresh tissue in Bouin’s fixative or another picric acid–containing solution causes the specimen to assume a brownish appearance. In general, PG is a circumscribed lesion with a partial or complete fibrous capsule; hence, the surgeon will report that the lesion was relatively easy to dissect from contiguous structures. However, approximately 10% to 20% of tumors demonstrate local infiltration of adjacent tissues,371 and these may be submitted to the surgical pathology laboratory in a fragmented state or with adherent structures attached to their peripheral aspects. An important step in the initial pathologic evaluation of PG is not only to perform standard three-dimensional measurement of the lesion but also to weigh it after dissection of attached extraneous soft tissue. Secondly, one should pay special attention to whether a PG appears to be multinodular or multifocal. Multicentricity of such neoplasms correlates well with a syndromic association.
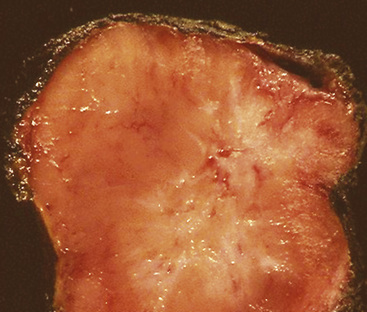
The histopathologic characteristics of pulmonary PGs are also variable. The peculiar nesting configuration of the tumor cells, separated by prominent fibrovascular stromal septa (“zellballen”; Fig. 13-35) is poorly developed in many of these lesions. In the lung in particular, this feature leads to considerable difficulty in distinguishing PG from carcinoid tumors (which, of course, are far more common). Aside from the organoid growth pattern of PGs, they are marked by a tendency toward nuclear pleomorphism, the common presence of intranuclear “pseudoinclusions” (invaginations of cytoplasm), intercellular hyaline globules, accumulations of intercellular proteinaceous material resembling thyroid colloid, potential spindle cell or oncocytic change, and elements resembling ganglion cells383,384 (Fig. 13-36). Mitotic figures are seen in approximately 45% of benign PGs and 65% of malignant lesions, regardless of location; hence, they are not useful in and of themselves in predicting tumor behavior. Similarly, although vascular invasion is apparent in one-fifth of malignant PGs, it is also evident in 5% to 6% of benign tumors.384 Thus, requests for a definitive diagnosis of PG in the frozen section laboratory are impossible to satisfy, particularly with reference to pulmonary tumors.
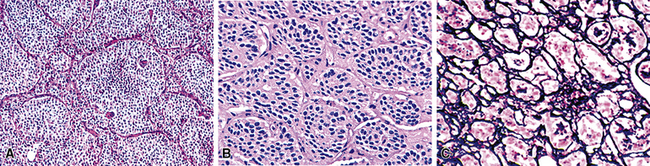
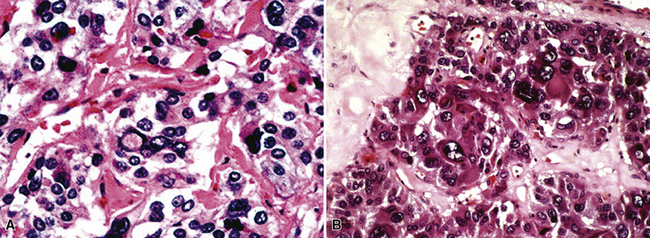
Relatively few PGs have been subjected to fine-needle aspiration and cytologic assessment.385–387 However, a report on this topic by Gonzalez-Campora and colleagues386 showed that such tumors commonly exhibited marked anisokaryosis, a tendency to form acini or follicles, and intranuclear invaginations of cytoplasm similar to those seen in papillary thyroid carcinoma. Nuclear pleomorphism has not been correlated with adverse behavior; in fact, malignant PGs have tended to display less nuclear variability than did their benign counterparts.384
Differential Diagnosis
Special studies are typically needed to bolster the histologic diagnosis of PG in visceral locations, especially the lung. Other considerations in pulmonary cases center on NEC as well as metastatic malignant PG originating at another site. Electron microscopy is still a useful modality in this context, and the neurosecretory granules of paraganglion cell tumors generally differ from those of other neuroendocrine cells and neoplasms.370 They feature an unusual “blister”-like configuration, where the internal submembranous “halos” of the granules are obviously eccentric and appear to emanate from the dense core in a bubble-like fashion. Otherwise, the cells of PGs are similar to those of the dispersed neuroendocrine system, showing prominent Golgi complexes and rough endoplasmic reticulum, macular intercellular junctions, and incomplete pericellular basal lamina.388
Immunohistologically, most examples of PG have a distinctive intermediate filament protein profile that is not shared by other neoplasms except for NBs.76,389 This features neurofilament protein, with or without vimentin, to the exclusion of other intermediate filament proteins. Neurofilament protein may be difficult to demonstrate in formalin-fixed, paraffin-embedded tissues, so in practical terms, the majority of routinely processed PGs do not manifest detectable intermediate filament protein.389 This observation is useful in making the distinction between PG (and other “type 2” [neural] neuroendocrine tumors) and “type 1” (epithelial) neoplasms that often are considered in the differential diagnosis and that uniformly exhibit keratin positivity.56 We have not found keratin proteins in PGs, in the lung or elsewhere. This is in stark contrast to the rate of 30% that has been cited by some authors for keratin positivity in PG66—a rate we believe to be a reflection of procedural shortcomings. Occasional examples of PG may also show focal immunoreactivity for glial fibrillary acidic protein, but they uniformly lack desmin.76 Virtually all PGs are diffusely and intensely positive for chromogranin-A and synaptophysin, whereas beta-tubulin and microtubule-associated protein are only focally seen in such lesions and are instead characteristic of neuroblastic and ganglioneuromatous neoplasms.56,83,89,90 Other immunodeterminants that can be detected in PGs include ACTH (approximately 30%), vasoactive intestinal polypeptide (40%), Leu- or Met-enkephalins (50% to 60%), calcitonin (<5%), CD56 (90%), CD57 (50%), and beta-endorphin (10%).75,79,93
A word is in order regarding “minute pulmonary chemodectoma” as a differential diagnostic consideration in this context. “Chemodectoma” is a term that was formerly used in reference to PG.370 In fact, minute pulmonary chemodectomas have no relationship to the paraganglion system. They are small peripheral pulmonary parenchymal aggregations of polygonal or bluntly fusiform cells, often with a concentric configuration. Immunohistochemical studies of minute pulmonary chemodectomas have demonstrated a similarity to meningothelial rather than neuroendocrine tissues, with reactivity for vimentin and epithelial membrane antigen.390 Moreover, analyses of cellular clonality in such lesions have demonstrated that they are polyclonal and probably reactive,391 rather than neoplastic, as is true of PGs.
One other recent development should be mentioned in reference to PGs. It has now been shown conclusively that both familial (MEN2-related) and selected sporadic examples of this tumor demonstrate mutations in the ret gene on chromosome 10.382 These take the form of (Cys634 → Arg) in MEN2A and (Met918 → Thr) in MEN2B. Whether this information enters the diagnostic sphere in the near future must await further technical developments and clinical correlation.
Treatment and Clinical Outcome
The most contentious aspect of PGs is the prediction of their often-capricious behavior. In the lung, the great majority of primary paraganglionic tumors have been benign biologically, but occasional examples have metastasized to regional lymph nodes or other viscera.371–380
At one extreme, some authors have stated that a diagnosis of malignant PG can only be made after metastasis has occurred; others have claimed that other findings can be correlated with aggressive behavior. These include pathologic mitotic figures or vascular invasion,384 decreased immunoreactivity for selected neuropeptides (particularly neuropeptide Y),392 and loss of intratumoral sustentacular cells positive for S-100 protein.130,393 These concepts are admittedly still in evolution. Some reports concerning the biology of PG have provided additional useful information. Based on the results of logistical regression analyses, mitotic activity, nuclear atypia, and vascular or capsular invasion are of little or no use as individual parameters in the prognostication of PGs.384 Similarly, Lack,370 Linnoila and colleagues,384 and Gonzalez-Campora and colleagues394 found no statistically significant association between static or flow cytometric DNA aneuploidy and behavior in paraganglion cell tumors, with only Pang and Tsao395 demurring on the latter point. Multiparametric assessment of 16 nonmicroscopic and histologic features by Linnoila and associates384 showed that 4 of them were the most predictively useful. These included extra-adrenal location, coarse gross nodularity of the tumor, confluent tumor necrosis on microscopic examination, and absence of intercellular hyaline globules. Among 120 PGs in that series, 71% of the biologically malignant lesions showed two or three of the four specified features, whereas 89% of the benign tumors manifested zero or one of them. Accordingly, there was a greater than 95% probability that more than 70% of PGs could be correctly classified using this paradigm.384 In our opinion, such an approach is recommended. It can be used by any pathologist and does not require special equipment.
Primary Primitive Neuroectodermal Tumors of the Lung
Primitive neuroectodermal tumors are small round cell neoplasms that are most commonly seen as primary soft tissue tumors. In the thorax, they typically affect the pleura and chest wall and are known as “Askin tumors” (Figs. 13-37 and 13-38). In the lung, only a few examples of PNET have been well documented as primary tumors by Imamura and coworkers,396 Mikami and coworkers,397 Verfaillie and colleagues,398 Suárez Antelo and coworkers,399 Lee and coworkers,400 and Takahashi and colleagues.401 Accordingly, specific clinicopathologic information on primary intrapulmonary lesions is anecdotal. These neoplasms have been seen in patients between 17 and 67 years of age who presented with nodular intrapulmonary masses. Characteristic t(11;22) chromosomal translocations and expression of CD99 or FLI-1402,403 (Fig. 13-39) were observed in each neoplasm, and they lacked immunoreactivity for keratin, myogenic markers, and S-100 protein. After surgical excision and appropriate chemotherapy, three of the patients were alive and free of disease; the others were lost to follow-up or were still being treated.
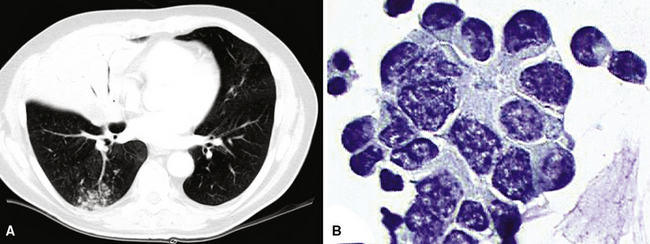
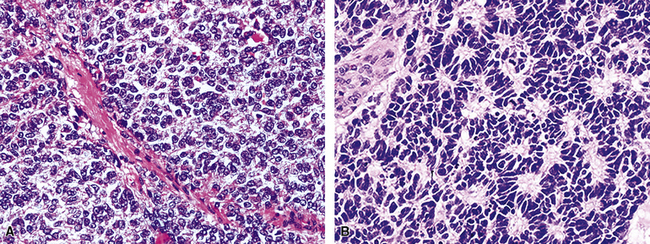
Figure 13-38 Primitive neuroectodermal tumor (A) comprises monomorphic small round cells (B), which may focally form rosettes.
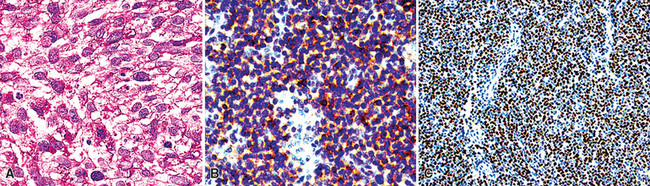
The differential diagnosis principally involves SCC—which should be uniformly keratin-positive56 and lack t(11;22)—and metastatic PNET arising in other locations and involving the lung secondarily. Another possibility is small cell primary or metastatic pulmonary synovial sarcoma, which is a particular interpretative trap because it shares potential immunoreactivity for CD99 and FLI-1402 with PNET.398 However, the cytogenetic profiles of those lesions are mutually exclusive; synovial sarcoma lacks t(11;22) and consistently exhibits a t(X;18) chromosomal translocation. Those genotypes can be identified using either in situ hybridization techniques or polymerase chain reaction–based assays.404
Primary Neuroblastoma of the Lung
Neuroblastoma continues to represent an important neoplasm in pediatric oncology. It is the fourth most frequently encountered malignancy in children, behind leukemia, lymphoma, and aggressive central nervous system tumors.370,405,406 The majority of NBs (and congeners thereof) are encountered in the first decade of life, with no particular predilection for either sex and only rare cases in adults.407,408 Paradoxically, however, the only examples of primary intrapulmonary NB that have thus far been reported have been in three patients older than 20 years of age.409,410
Clinical Features
Symptoms and signs of these neoplasms typically relate to the presence of an enlarging mass (Fig. 13-40). In the lung, they are therefore most closely allied to interference with the function of structures on which the lesions impinge, such as the major bronchi. In unusual instances, paraneoplastic phenomena such as those seen with more differentiated autonomic neural tumors—i.e., PGs—may be seen at presentation in association with NBs.411–413 In one case of pulmonary ganglioneuroblastoma reported by Hochholzer and associates,410 signs of a MEN syndrome were also observed.
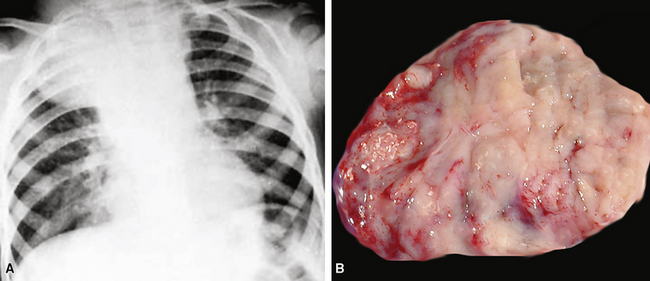
Pathologic Findings
The gross pathologic attributes of neuroblastic tumors are variable. Undifferentiated NB has a prototypical gray-white, encephaloid appearance. In lesions with partial ganglionic differentiation, nodules of more “fleshy” tissue are noted as discrete foci in the cut surfaces of the lesion. Other potential gross features of neuroblastic neoplasms include a hemangioma-like image because of extreme intratumoral hemorrhage, extensive cystic change, and diffuse or localized calcification that is often readily apparent on sectioning the mass.414
The microscopic characteristics of these tumors likewise form a continuum.414–418 At one pole, one encounters classic undifferentiated small round cell tumors with little or no discernible stroma and no attempts at neural rosette formation (Fig. 13-41). Further along the spectrum of differentiation, the next group of NBs begins to exhibit background “neuropil,” a fibrillary eosinophilic matrix that represents the elaboration of numerous cytoplasmic extensions by the tumor cells (Fig. 13-42). Often, Homer Wright rosettes—having a fibrillary nonluminal center—are also encountered. The separation between NB and ganglioneuroblastoma requires definable ganglion cell differentiation in the latter neoplasms, but determining where they begin and “differentiating NB” ends is more art than science. This statement is limited to “diffuse ganglioneuroblastomas” (“differentiating stromal-poor NBs” reported by Shimada and colleagues418) because, as noted earlier, stromal-rich tumors look most like ganglioneuromas (composed of mature ganglion cells and spindled Schwann cell–like elements) rather than predominantly small cell proliferations. Stromal-rich neoplasms that are not nodular (i.e., composite ganglioneuroblastoma) are further subdivided into “well-differentiated” lesions, with only a few randomly dispersed neuroblastic cells punctuating the image of a ganglioneuroma and “intermixed” tumors that contain small nests of neuroblasts having sharp interfaces with the surrounding ganglionic/Schwannian tissue.
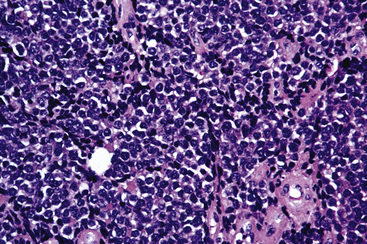
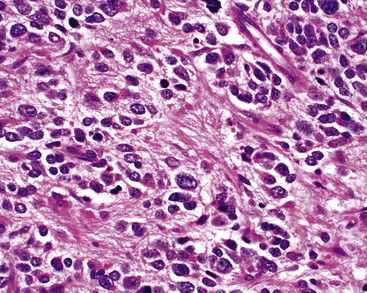
Figure 13-42 Differentiating neuroblastoma, exhibiting the formation of fibrillar eosinophilic cytoplasmic extensions (neuropil).
Rarely, primitive NBs exhibit a striking degree of nuclear pleomorphism; those lesions have been termed “anaplastic NBs.”419 Whether the pleomorphic appearance of such neoplasms correlates with a worse prognosis is uncertain.
Differential Diagnosis
The differential diagnosis with other small round cell tumors is principally a problem in “undifferentiated” NB.414 In that setting, electron microscopy demonstrates characteristically elongated cell processes containing microtubular complexes in neuroblastic tumors, with or without presynaptic vesicles or dense-core granules as well420 (Fig. 13-43).
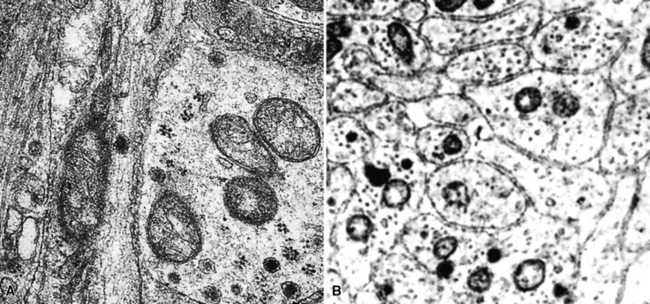
Immunohistologically, most neuroblastic neoplasms express only neurofilament protein among all of the intermediate filament types. Because that marker is usually not well preserved in formalin-fixed tissues, no intermediate filaments are demonstrable in the majority of cases.421 In contrast, SCC, an important consideration in adults, is uniformly keratin-positive.63,64 Synaptophysin is usually present in NB, ganglioneuroblastomas, and ganglioneuromas,89,90 but CGA is uncommonly observed in those neoplasms.83 Neuron-specific enolase and CD56 are sensitive markers for NB (and may have limited utility in identifying bone marrow micrometastases422), but they have a lack of specificity and may also be seen in PNETs as well as some cases of rhabdomyosarcoma.112 NB84 is another marker showing selective reactivity with NB and PNET, to the exclusion of other small cell tumors423 (Fig. 13-44). The once-difficult distinction between NB and PNET has been facilitated by the availability of antibodies to CD99 and beta-2-microglobulin, both of which are seen in PNET but not NB.362 Likewise, immunoreactivity for muscle-specific actin or desmin characterizes rhabdomyoblastic neoplasms and is not expected in NB.362 This observation principally comes into play in examples of “anaplastic” NB that may resemble embryonal rhabdomyosarcoma on conventional microscopy.424
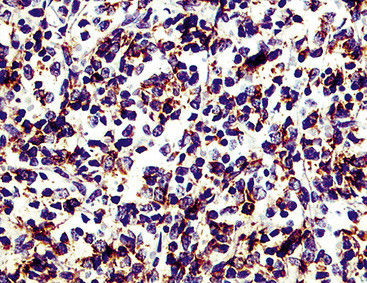
Treatment and Clinical Outcome
With reference to the three reported cases of primary intrapulmonary NB, two patients were alive and well 1 and 2.5 years, respectively, after surgical resection and chemotherapy.410,411 The other patient died shortly after admission to the hospital.
In much more general terms, two histologic parameters with prognostic significance should be additionally recorded in evaluating neuroblastic neoplasms. These include the level of ganglionic differentiation (<5% or ≥5%)414–418 and the “mitotic-to-karyorrhectic index” (MKI).418,425,426 The MKI is the number of mitotic or karyorrhectic nuclei counted in an evaluation of 5000 tumor cells in “neuropil-free” areas of any given tumor. The daunting prospect of assessing that many cells has dissuaded many pathologists from embracing the Shimada system with enthusiasm. However, common practice has shown that a reasonable estimation of the MKI can be obtained with simple pattern-matching approaches. With data on differentiation and MKI, prognostic substratification of stromal-poor neoplasms can be accomplished within appropriately age-, location-, and stage-matched tumor groups.427
Accurate classification and prognostication of neuroblastic tumors mandates the availability of detailed data on clinical, gross, and histologic levels. Moreover, biochemical observations—as derived from the clinical chemistry laboratory—may provide additional nuances in this context.428 The presence of vanillacetic acid in the urine (as opposed to homovanillic acid or vanillyolmandelic acid) worsens the clinical outlook, as do elevated serum levels of ferritin, lactate dehydrogenase, NSE, CGA, and creatine kinase isozyme BB.428 On the other hand, elevated somatostatin levels in serum or tumor tissue have been linked to a better prognosis.429
With regard to other adjunctive modalities of pathologic evaluation as applied to neuroblastic tumors, three have been used increasingly in the last decade. Cytogenetic assessment (requiring submission of fresh tissue, although current fluorescence–in situ hybridization methods may change that) has shown aberrations (deletions and rearrangements) in chromosome 1p in most cases of NB and ganglioneuroblastoma,430 possibly aiding in the differential diagnosis with other small cell tumors that lack such abnormalities. Prognostically, aneuploidy, hyperdiploidy, and near-tetraploidy in neuroblastic neoplasms correlate with improved prognosis, in contrast to the norm for most other malignant tumors.414,425 Conversely, DNA diploid lesions tend to behave aggressively. Likewise, increased copy numbers of the oncogene N-myc are also biologically detrimental in NBs.431–434 This marker can be assessed directly by the Southern blot method or indirectly by Northern/Western blot or in situ hybridization. Immunohistology for the protein product of N-myc can also be performed on fresh tissue, with generally acceptable results. The RET gene, which is important in the prognosis of other neuroendocrine tumors, also appears to correlate with neuronal differentiation in NBs and, therefore, roughly with prognosis.435 Lastly, the immunolabeling index for Ki-67 (an indicator of cell replication) is inversely correlated with the length of survival in NB436; further, loss of expression of the Trk-A gene or CD44 by the tumor cells is associated with high stage at diagnosis and a poor outcome.434
1 Feyrter F. Uber diffuse endokrine epitheliale organe. Leipzig: JA Barth, 1938.
2 Pearse A.G.E. The APUD cell concept and its implications in pathology. Pathol Annu. 1974;9:27-41.
3 Yousem S.A. Pulmonary carcinoid tumors and well-differentiated neuroendocrine carcinomas: is there room for an atypical carcinoid? Am J Clin Pathol. 1991;95:763-764.
4 Hasleton P.S., Bostanci G. Pulmonary carcinoid and related tumors. Rocz Akad Med Bialymst. 1997;42(suppl 1):28-42.
5 Travis W.D., Rush W., Flieder D.B., et al. Survival analysis of 200 pulmonary neuroendocrine tumors with clarification of criteria for atypical carcinoid and its separation from typical carcinoid. Am J Surg Pathol. 1998;22:934-944.
6 Lloyd R.V. Endocrine Pathology. New York: Springer-Verlag, 1990.
7 Lee J.E., Evans D.B. Advances in the diagnosis and treatment of gastrointestinal neuroendocrine tumors. Cancer Treat Res. 1997;90:227-238.
8 Warner R.R. Gut neuroendocrine tumors. Curr Ther Endocrinol Metab. 1997;6:606-614.
9 Mendelsohn G. Diagnosis and Pathology of Endocrine Diseases. Philadelphia: JB Lippincott, 1988.
10 Argani P., Erlandson R.A., Rosai J. Thymic neuroblastoma in adults: report of three cases with special emphasis on its association with the syndrome of inappropriate secretion of antidiuretic hormone. Am J Clin Pathol. 1997;108:537-543.
11 Hasleton P.S., Al-Saffar N. The histological spectrum of bronchial carcinoid tumors. Appl Pathol. 1989;7:205-218.
12 Warren W.H., Faber L.P., Gould V.E. Neuroendocrine neoplasms of the lung: a clinicopathologic update. J Thorac Cardiovasc Surg. 1989;98:321-332.
13 Glasser C.M., Bhagavan B.S. Carcinoid tumors of the appendix. Arch Pathol Lab Med. 1980;104:272-275.
14 Rusch V.W., Klimstra D.S., Venkatraman E.S. Molecular markers help characterize neuroendocrine lung tumors. Ann Thorac Surg. 1996;62:798-809.
15 Gould V.E., Linnoila R.I., Memoli V.A., Warren W.H. Neuroendocrine components of the bronchopulmonary tract. Lab Invest. 1983;49:519-537.
16 Shackney S.E., Shankey T.V. Common patterns of genetic evolution in human solid tumors. Cytometry. 1997;29:1-27.
17 Yunis J.J. Genes and chromosomes in the pathogenesis and prognosis of human cancers. Adv Pathol Lab Med. 1989;2:147-188.
18 Galanis E., Frytak S., Lloyd R.V. Extrapulmonary small cell carcinoma. Cancer. 1997;79:1729-1736.
19 Fletcher J.A. Cytogenetic aberrations in malignant soft tissue tumors. Adv Pathol Lab Med. 1991;4:235-246.
20 Dehner L.P. Primitive neuroectodermal tumor and Ewing’s sarcoma. Am J Surg Pathol. 1993;17:1-13.
21 Simpson N.E., Kidd K.K., Goodfellow P.J., et al. Assignment of multiple endocrine neoplasia type 2A to chromosome 10 by linkage. Nature. 1987;328:528-530.
22 Komminoth P. Multiple endocrine neoplasia type 1 and 2: 1997 diagnostic guidelines & molecular pathology. Pathologe. 1997;18:286-300.
23 Komminoth P. Multiple endocrine neoplasia type 1 & 2: from morphology to molecular pathology 1997. Verh Dtsch Ges Pathol. 1997;81:125-138.
24 Eng C., Mulligan L.M. Mutations of the RET protooncogene in the multiple endocrine neoplasia type 2 syndromes, related sporadic tumors, & Hirschsprung’s disease. Hum Mutat. 1997;9:97-109.
25 Cagle P.T., El-Naggar A.K., Xu H.J., et al. Differential retinoblastoma protein expression in neuroendocrine tumors of the lung: potential diagnostic implications. Am J Pathol. 1997;150:393-400.
26 McDowell E.M., Wilson T.S., Trump B.F. Atypical endocrine tumors of the lung. Arch Pathol Lab Med. 1981;105:20-28.
27 Adelstein D.J., Tomashefski J.F. Mixed small-cell and non-small-cell lung cancer. Chest. 1986;89:699-704.
28 Hishima T., Fukayama M., Hayashi Y., et al. Neuroendocrine differentiation in thymic epithelial tumors, with special reference to thymic carcinoma & atypical thymoma. Hum Pathol. 1998;29:330-338.
29 Usuda H., Emura I., Naito M., Hirono T. Peripheral lung carcinomas associated with central fibrosis and mixed small cell and other histologic components. Pathol Int. 1995;45:940-946.
30 Robertson N.J., Rahamim J., Smith M.E. Carcinosarcoma of the esophagus showing neuroendocrine, squamous, and glandular differentiation. Histopathology. 1997;31:263-266.
31 McWilliam L.J., Manson C., George N.J. Neuroendocrine differentiation and prognosis in prostatic adenocarcinoma. Br J Urol. 1997;80:287-290.
32 Leslie K.O., Colby T.V. Pathology of lung cancer. Curr Opin Pulm Med. 1997;3:252-256.
33 Bosman F.T. Neuroendocrine cells in non-endocrine tumors: what does it mean? Verh Dtsch Ges Pathol. 1997;81:62-72.
34 Wick M.R., Berg L.C., Hertz M.I. Large cell carcinoma of the lung with neuroendocrine differentiation. Am J Clin Pathol. 1992;97:796-805.
35 Radice P.A., Matthews M.J., Ihde D.C., et al. The clinical behavior of mixed small cell/large cell bronchogenic carcinoma compared to pure small cell subtypes. Cancer. 1982;50:2894-2902.
36 Fushimi H., Kikui M., Morino H., et al. Histologic changes in small cell lung carcinoma after treatment. Cancer. 1996;77:278-283.
37 Wiseman G.A., Kvols L.K. Therapy of neuroendocrine tumors with radiolabeled MIBG and somatostatin analogues. Semin Nucl Med. 1995;25:272-278.
38 DiBartolomeo M., Bajetta E., Buzzoni R., et al. Clinical efficacy of octreotide in the treatment of metastatic neuroendocrine tumors: a study by the Italian Trials in Medical Oncology group. Cancer. 1996;77:402-408.
39 Saldiva P.H.N., Capelozzi V.L., Battlehner C.N. Neuroendocrine tumors of the lung. In: Corrin B., editor. Pathology of Lung Tumors. New York: Churchill-Livingstone; 1997:55-70.
40 Nagle R.B., Payne C.M., Clark V.A. Comparison of the usefulness of histochemistry and ultrastructural cytochemistry in the identification of neuroendocrine neoplasms. Am J Clin Pathol. 1986;85:289-296.
41 Samsonov V.A. Carcinoid lung tumors: clinicomorphologic characteristics and diagnosis. Arkh Patol. 1995;57:20-24.
42 Kogan E.A., Sekamova S.M., Mazurenko N.N., Bogatyrev V.N. Peripheral small cell carcinoma, atypical and typical lung carcinoids. Arkh Patol. 1991;53:42-48.
43 Erlandson R.A. Diagnostic Transmission Electron Microscopy of Tumors. Raven Press, New York, 1994;123-125.
44 Carey F.A., Save V.E. Neuroendocrine differentiation in lung cancer. J Pathol. 1997;182:9-10.
45 Soga J., Yakuwa Y. Bronchopulmonary carcinoids: an analysis of 1875 reported cases with special reference to a comparison between typical carcinoids and typical varieties. Ann Thorac Cardiovasc Surg. 1999;5:211-219.
46 Dardick I., Christensen H., Stratis M. Immunoelectron microscopy for chromogranin A in small cell neuroendocrine carcinoma of lung. Ultrastruct Pathol. 1996;20:361-368.
47 Yang G.C. Mixed small cell/large cell carcinoma of the lung: report of a case with cytologic features and ultrastructural correlation. Acta Cytol. 1995;39:1175-1181.
48 Taccagni G.L., Rovere E., Terreni M.R., et al. Divergent differentiative histogenetic lines in lung tumors: identification of histotypes with pure and mixed ultrastructural phenotype and their prognostic significance. Ultrastruct Pathol. 1995;19:61-73.
49 Mount S.L., Taatjes D.J., von Turkovich M., et al. Diagnostic immunoelectron microscopy in surgical pathology: assessment of various tissue fixation and processing protocols. Ultrastruct Pathol. 1993;17:547-556.
50 Przybylowski P., Mlynarczyk W., Blotna-Filipiak M., Biczysko W. Application of electron microscopy and immunocytochemistry in lung cancer diagnosis. Folia Morphol. 1993;52:191-200.
51 Tsubota Y.T., Kawaguchi T., Hoso T., et al. A combined small cell and spindle cell carcinoma of the lung: report of a unique case with immunohistochemical and ultrastructural studies. Am J Surg Pathol. 1992;16:1108-1115.
52 Pilotti S., Patriarca C., Lombardi L., et al. Well-differentiated neuroendocrine carcinoma of the lung: a clinicopathologic and ultrastructural study of 10 cases. Tumori. 1992;78:121-129.
53 Dardick I., Yazdi H.M., Brosko C., et al. A quantitative comparison of light and electron microscopic diagnoses in specimens obtained by fine needle aspiration biopsy. Ultrastruct Pathol. 1991;15:105-129.
54 Muller K.M., Fisseler-Eckhoff A. Small cell bronchial cancer – pathologic anatomy. Langenbecks Arch Chir Suppl Kongressbd. 1991:534-543.
55 Warren W.H., Gould V.E. Neuroendocrine tumors of the bronchopulmonary tract: a reappraisal of their classification after 20 years. Surg Clin North Am. 2002;82:525-540.
56 Wick MR: Immunohistology of neuroendocrine and neuroectodermal tumors. Semin Diagn Pathol. 19(4):207–218.
57 Brambilla E., Veale D., Moro D., et al. Neuroendocrine phenotype in lung cancers: comparison of immunohistochemistry with biochemical determination of enolase isoenzymes. Am J Clin Pathol. 1992;98:88-97.
58 Broers J.L., Mijnheere E.P., Rot M.K., et al. Novel antigens characteristic of neuroendocrine malignancies. Cancer. 1991;67:619-633.
59 Loy T.S., Darkow G.V.D., Quesenbery J.T. Immunostaining in the diagnosis of pulmonary neuroendocrine carcinomas. An immunohistochemical study with ultrastructural correlations. Am J Surg Pathol. 1995;19:173-182.
60 Tome Y., Hirohashi S., Noguchi M., et al. Immunocytologic diagnosis of small-cell lung cancer in imprint smears. Acta Cytologica. 1991;35:485-490.
61 Miettinen M. Keratin immunohistochemistry: update of applications and pitfalls. Pathol Annu. 1993;28(2):113-143.
62 Chan J.K.C., Suser S., Wenig B.M., et al. Cytokeratin 20 immunoreactivity distinguishes Merkel cell (primary cutaneous neuroendocrine) carcinomas and salivary gland small cell carcinomas from small cell carcinomas of various sites. Am J Surg Pathol. 1997;21:226-234.
63 Battifora H. Diagnostic uses of antibodies to keratins: a review and immunohistochemical comparison of seven monoclonal and three polyclonal antibodies. Prog Surg Pathol. 1988;8:1-16.
64 Battifora H., Silva E.G. The use of antikeratin antibodies in the immunohistochemical distinction between neuroendocrine (Merkel cell) carcinoma of the skin, lymphoma, and oat cell carcinoma. Cancer. 1986;58:1040-1046.
65 Labrousse F., Leboutet M.J., Petit B., et al. Cytokeratin expression in paragangliomas of the cauda equina. Clin Neuropathol. 1999;18:208-213.
66 Chetty R., Pillay P., Jaichand V. Cytokeratin expression in adrenal pheochromocytomas and extraadrenal paragangliomas. J Clin Pathol. 1998;51:477-478.
67 Marley E.F., Liapis H., Humphrey P.A., et al. Primitive neuroectodermal tumor of the kidney – another enigma: a pathologic, immunohistochemical, and molecular diagnostic study. Am J Surg Pathol. 1997;21:354-359.
68 Moll R., Lee I., Gould V.E., et al. Immunocytochemical analysis of Ewing’s tumors: patterns of expression of intermediate filaments and desmosomal proteins indicate cell type heterogeneity and pluripotential differentiation. Am J Pathol. 1987;127:288-304.
69 Thorner P. Intraabdominal polyphenotypic tumor. Pediatr Pathol Lab Med. 1996;16:161-169.
70 Gerald W.L., Ladanyi M., de Alava E., et al. Clinical, pathologic, and molecular spectrum of tumors associated with t(11;22)(p13;q12): desmoplastic small round cell tumor and its variants. J Clin Oncol. 1998;16:3028-3036.
71 Ordonez N.G. Desmoplastic small round cell tumor. II. An ultrastructural and immunohistochemical study with emphasis on new immunohistochemical markers. Am J Surg Pathol. 1998;22:1314-1327.
72 Winters J.L., Geil J.D., O’Connor W.N. Immunohistology, cytogenetics, and molecular studies of small round cell tumors of childhood: a review. Ann Clin Lab Sci. 1995;25:66-78.
73 Broers J.L., Carney D.N., de Ley L., et al. Differential expression of intermediate filament proteins distinguishes classic from variant small-cell lung cancer cell lines. Proc Natl Acad Sci U S A. 1985;82:4409-4413.
74 Merot Y., Margolis R.J., Dahl D., et al. Coexpression of neurofilament and keratin proteins in cutaneous neuroendocrine carcinoma cells. J Invest Dermatol. 1986;86:74-77.
75 Moran C.A., Suster S., Fishback N., Koss M.N. Mediastinal paragangliomas: a clinicopathologic and immunohistochemical study of 16 cases. Cancer. 1993;72:2358-2364.
76 Kimura N., Nakazato Y., Nagura H., Sasano N. Expression of intermediate filaments in neuroendocrine tumors. Arch Pathol Lab Med. 1990;114:506-510.
77 Hirose T., Scheithauer B.W., Lopes M.B., et al. Olfactory neuroblastoma: an immunohistochemical, ultrastructural, and flow cytometric study. Cancer. 1995;76:4-19.
78 Miettinen M. Synaptophysin and neurofilament proteins as markers for neuroendocrine tumors. Arch Pathol Lab Med. 1987;111:813-818.
79 Johnson T.L., Zarbo R.J., Lloyd R.V., Crissman J.D. Paragangliomas of the head and neck: immunohistochemical neuroendocrine and intermediate filament typing. Mod Pathol. 1988;1:216-223.
80 Eusebi V., Damiani S., Pasquinelli G., et al. Small cell neuroendocrine carcinoma with skeletal muscle differentiation. Am J Surg Pathol. 2000;24:223-230.
81 Friede R.L., Janzer R.C., Roessmann U. Infantile small cell gliomas. Acta Neuropathol. 1982;57:103-110.
82 Oh D., Prayson R.A. Evaluation of epithelial and keratin markers in glioblastoma multiforme: an immunohistochemical study. Arch Pathol Lab Med. 1999;123:917-920.
83 Wilson R.S., Lloyd R.V. Detection of chromogranin in neuroendocrine cells with a monoclonal antibody. Am J Pathol. 1984;115:458-468.
84 Settleman J., Fonseca R., Nolan J., Angeletti R.H. Relationship of multiple forms of chromogranin. J Biol Chem. 1985;260:1645-1651.
85 O’Connor D.T. Chromogranin: widespread immunoreactivity in polypeptide hormone-producing tissues and in serum. Regul Pept. 1983;6:263-280.
86 Eriksson B., Arnberg H., Oberg K., et al. A polyclonal antiserum against chromogranin A and B – a new sensitive marker for neuroendocrine tumors. Acta Endocrinol. 1990;122:145-155.
87 Woussen-Colle M.C., Gourlet P., Vandermeers A., et al. Identification of a new chromogranin B fragment (314–365) in endocrine tumors. Peptides. 1995;16:231-236.
88 Lloyd R.V., Wilson R.S. Specific endocrine marker defined by a monoclonal antibody. Science. 1983;222:628-630.
89 Gould V.E., Lee I., Wiedenmann B., et al. Synaptophysin: a novel marker for neurons, certain neuroendocrine cells, and their neoplasms. Hum Pathol. 1986;17:979-983.
90 Buffa R., Rindi G., Sessa F., et al. Synaptophysin immunoreactivity and small clear vesicles in neuroendocrine cells and related tumors. Mol Cell Probes. 1987;1:367-381.
91 Stridsberg M. The use of chromogranin, synaptophysin, and islet amyloid polypeptide as markers for neuroendocrine tumors. Ups J Med Sci. 1995;100:169-199.
92 Poola I., Graziano S.L. Expression of neuron-specific enolase, chromogranin A, synaptophysin, and Leu-7 in lung cancer cell lines. J Exp Clin Cancer Res. 1998;17:165-173.
93 Erlandson R.A., Nesland J.M. Tumors of the endocrine/neuroendocrine system: an overview. Ultrastruct Pathol. 1994;18:149-170.
94 Lipinski M., Braham K., Caillaud J.M., et al. HNK-1 antibody detects an antigen expressed on neuroectodermal cells. J Exp Med. 1983;158:1775-1780.
95 Tsutsumi Y. Leu-7 immunoreactivity as a histochemical marker for paraffin-embedded neuroendocrine tumors. Acta Histochem Cytochem. 1984;17:15-21.
96 Bunn P.A.Jr, Linnoila I., Minna J.D., et al. Small cell lung cancer, endocrine cells of the fetal bronchus, and other neuroendocrine cells express the Leu-7 antigenic determinant present on natural killer cells. Blood. 1985;65:764-768.
97 Perentes E., Rubinstein L.J. Immunohistochemical recognition of human nerve sheath tumors by anti-Leu 7 (HNK-1) monoclonal antibody. Acta Neuropathol. 1985;68:319-324.
98 Abenoza P., Manivel J.C., Swanson P.E., Wick M.R. Synovial sarcoma: ultrastructural study and immunohistochemical analysis by a combined PAP/ABC procedure. Hum Pathol. 1986;17:1107-1115.
99 Rusthoven J.J., Robinson J.B., Kolin A., Pinkerton P.H. The natural killer cell associated HNK-1 (Leu-7) antibody reacts with hypertrophic and malignant prostatic epithelium. Cancer. 1985;56:289-293.
100 May E.E., Perentes E. Anti-Leu 7 immunoreactivity with human tumors: its value in the diagnosis of prostatic adenocarcinoma. Histopathology. 1987;11:295-304.
101 Kodama T., Watanable S., Sato Y., et al. An immunohistochemical study of thymic epithelial tumors. I. Epithelial component. Am J Surg Pathol. 1986;10:26-33.
102 Tischler A.S., Mobtaker H., Mann K., et al. Anti-lymphocyte antibody Leu-7 (HNK-1) recognizes a constituent of neuroendocrine granule matrix. J Histochem Cytochem. 1986;34:1213-1216.
103 Lantuejoul S., Moro D., Michalides R.J., et al. Neural cell adhesion molecules (NCAM) and NCAM-PSA expression in neuroendocrine lung tumors. Am J Surg Pathol. 1998;22:1267-1276.
104 Kaufmann O., Georgi T., Dietel M. Utility of 123C3 monoclonal antibody against CD56 (NCAM) for the diagnosis of small cell carcinomas on paraffin sections. Hum Pathol. 1997;28:1373-1378.
105 Del Rio M., Demoly P., Koros A.M., et al. JLP5B9: new monoclonal antibody against polysialylated neural cell adhesion molecule is of value in phenotyping lung cancer. J Immunol Meth. 2000;233:21-31.
106 Hage R., Elbers H.R., Brutel de la Riviere A., van den Bosch J.M. Neural cell adhesion molecule expression: prognosis in 889 patients with resected non-small cell lung cancer. Chest. 1998;114:1316-1320.
107 Jaques G., Auerbach B., Pritsch M., et al. Evaluation of serum neural cell adhesion molecule as a new tumor marker in small cell lung cancer. Cancer. 1993;72:418-425.
108 Seldeslagh K.A., Lauweryns J.M. NCAM expression in the pulmonary neural and diffuse neuroendocrine cell system. Microsc Res Tech. 1997;37:69-76.
109 Kwa H.B., Verheijen M.G., Litvinov S.V., et al. Prognostic factors in resected non-small cell lung cancer: an immunohistochemical study of 39 cases. Lung Cancer. 1996;16:35-45.
110 Tapia F.J., Barbosa A.J.A., Marangos P.J., et al. Neuron-specific enolase is produced by neuroendocrine tumors. Lancet. 1981;2:808-811.
111 Wick M.R., Scheithauer B.W., Kovacs K. Neuron-specific enolase in neuroendocrine tumors of the thymus, bronchus, and skin. Am J Clin Pathol. 1983;79:703-707.
112 Carlei F., Polak J.M. Antibodies to neuron-specific enolase for the delineation of the entire diffuse neuroendocrine system in health and disease. Semin Diagn Pathol. 1984;1:59-70.
113 Haimoto H., Takahashi Y., Koshikawa T., et al. Immunohistochemical localization of gamma-enolase in normal human tissues other than nervous and neuroendocrine tissues. Lab Invest. 1985;52:257-263.
114 Thomas P., Battifora H., Manderino G.L., Patrick J. A monoclonal antibody against neuron-specific enolase: immunohistochemical comparison with a polyclonal antiserum. Am J Clin Pathol. 1987;88:146-152.
115 Vinores S.A., Bonnin J.M., Rubinstein L.J., Marangos P.J. Immunohistochemical demonstration of neuron-specific enolase in neoplasms of the CNS and other tissues. Arch Pathol Lab Med. 1984;108:536-540.
116 DeLellis R.A. Endocrine tumors. In: Colvin R.B., Bhan A.K., McCluskey R.T., editors. Diagnostic Immunopathology. 1st ed. New York: Raven Press; 1988:301-338.
117 Said J.W., Vimadalal S., Nash G., et al. Immunoreactive neuron-specific enolase, bombesin, and chromogranin-A as markers for neuroendocrine lung tumors. Hum Pathol. 1985;16:236-240.
118 Rode J. PGP 9.5 – a new marker for vertebrate neurons and neuroendocrine cells. Brain Res. 1983;278:224-228.
119 Gosney J.R., Gosney M.A., Lye M., Butt S.A. Reliability of commercially available immunocytochemical markers for identification of neuroendocrine differentiation in bronchoscopic biopsies of bronchial carcinoma. Thorax. 1995;50:116-120.
120 Addis B.J., Hamid Q., Ibrahim N.B., et al. Immunohistochemical markers of small cell carcinoma and related neuroendocrine tumors of the lung. J Pathol. 1987;153:137-150.
121 Hibi K., Westra W.H., Borges M., et al. PGP9.5 as a candidate tumor marker for non-small cell lung cancer. Am J Pathol. 1999;155:711-715.
122 Amann G., Zoubek A., Salzer-Kuntschik M., et al. Relation of neurological marker expression and EWS gene fusion types in MIC2/CD99-positive tumors of the Ewing family. Hum Pathol. 1999;30:1058-1064.
123 Dehner L.P. Primitive neuroectodermal tumors and Ewing’s sarcoma. Am J Surg Pathol. 1993;17:1-13.
124 Soslow R.A., Bhargava V., Warnke R.A. MIC2, TdT, bcl-2, and CD34 expression in paraffin-embedded high-grade lymphoma/acute lymphoblastic leukemia distinguishes between distinct clinicopathologic entities. Hum Pathol. 1997;28:1158-1165.
125 Lumadue J.A., Askin F.B., Perlman E.J. MIC2 analysis of small cell carcinoma. Am J Clin Pathol. 1994;102:692-694.
126 Nicholson S.A., McDermott M.B., Swanson P.E., Wick M.R. CD99 and cytokeratin-20 in small-cell and basaloid tumors of the skin. Appl Immunohistochem Mol Morphol. 2000;8(1):37-41.
127 Krisch K., Buxbaum P., Horvat G., et al. Monoclonal antibody HISL-19 as an immunocytochemical probe for neuroendocrine differentiation. Its application in diagnostic pathology. Am J Pathol. 1986;123:100-108.
128 Lloyd R.V., Sisson J.C., Shapiro B., Verhofstad A.A. Immunohistochemical localization of epinephrine, norepinephrine, catecholamine-synthesizing enzymes, and chromogranin in neuroendocrine cells and tumors. Am J Pathol. 1986;125:45-64.
129 Haspel M.V., Onodera T., Prabhakar B.S., et al. Multiple organ-reactive monoclonal autoantibodies. Nature. 1983;304:73-76.
130 Unger P., Hoffman K., Pertsemlidis D., et al. S100 protein-positive sustentacular cells in malignant and locally aggressive adrenal pheochromocytomas. Arch Pathol Lab Med. 1991;115:484-487.
131 Sholl L.M., Long K.B., Hornick J.L. Sox2 expression in pulmonary non-small cell and neuroendocrine carcinomas. Appl Immunohistochem Mol Morphol. 2010;18:55-61.
132 Sica G., Vazquez M.F., Altorki N., et al. PAX-5 expression in pulmonary neuroendocrine neoplasms: its usefulness in surgical and fine-needle aspiration biopsy specimens. Am J Clin Pathol. 2008;129:556-562.
133 LaPoint R.J., Bourne P.A., Wang H.L., Xu H. Coexpression of c-kit and bcl-2 in small cell carcinoma and large cell neuroendocrine carcinoma of the lung. Appl Immunohistochem Mol Morphol. 2007;15:401-406.
134 Sturm N., Rossi G., Lantuejoul S., et al. Expression of thyroid transcription factor-1 in the spectrum of neuroendocrine cell lung proliferations with special interest in carcinoids. Hum Pathol. 2002;33:175-182.
135 Oliveira A.M., Tazelaar H.D., Myers J.L., et al. Thyroid transcription factor-1 distinguishes metastatic pulmonary from well-differentiated neuroendocrine tumors of other sites. Am J Surg Pathol. 2001;25:815-819.
136 Ordonez N.G. Value of thyroid transcription factor-1 immunostaining in distinguishing small cell lung carcinomas from other small cell carcinomas. Am J Surg Pathol. 2000;24:1217-1223.
137 Kaufmann O., Dietel M. Expression of thyroid transcription factor-1 in pulmonary and extrapulmonary small cell carcinomas and other neuroendocrine carcinomas of various primary sites. Histopathology. 2000;36:415-420.
138 Agoff S.N., Lamps L.W., Philip A.T., et al. Thyroid transcription factor-1 is expressed in extrapulmonary small cell carcinomas but not in other extrapulmonary neuroendocrine tumors. Mod Pathol. 2000;13:238-242.
139 Folpe A.L., Gown A.M., Lamps L.W., et al. Thyroid transcription factor-1: immunohistochemical evaluation in pulmonary neuroendocrine tumors. Mod Pathol. 1999;12:5-8.
140 Zamecnik J., Kodet R. Value of thyroid transcription factor-1 and surfactant apoprotein A in the differential diagnosis of pulmonary carcinomas: a study of 109 cases. Virchows Arch A. 2002;440:353-361.
141 Travis W.D., Colby T.V., Corrin B., et al. Histological Typing of Lung and Pleural Tumours (International Histological Classification of Tumours). Geneva, Switzerland: World Health Organization, 1999;1-55.
142 Travis W.D. Lung tumours with neuroendocrine differentiation. Eur J Cancer. 2009;45(Suppl 1):251-266.
143 Hofler H. Neuroendocrine tumors of the lung. Verh Dtsch Ges Pathol. 1997;81:118-124.
144 Moran C.A., Suster S., Coppola D., Wick M.R. Neuroendocrine carcinomas of the lung: a critical analysis. Am J Clin Pathol. 2009;131:206-221.
145 Franks T.J., Galvin J.R. Lung tumors with neuroendocrine morphology: essential radiologic and pathologic features. Arch Pathol Lab Med. 2008;132:1055-1061.
146 Kramer R. Adenoma of the bronchus. Ann Otol Rhinol Laryngol. 1930;39:689-695.
147 Carter D., Eggleston J.C. Tumors of the lower respiratory tract. In: Atlas of Tumor Pathology. Washington, DC: Armed Forces Institute of Pathology; 1995:162-188. series 2, fascicle 17
148 Colby T.V., Koss M.N., Travis W.D. Tumors of the lower respiratory tract. In: Atlas of Tumor Pathology. Washington, DC: Armed Forces Institute of Pathology; 1995:287-318. series 2, fascicle 13
149 El-Naggar A.K., Ballance W., Abdul Karim F.W., et al. Typical and atypical bronchopulmonary carcinoids. Am J Clin Pathol. 1991;95:828-834.
150 McCaughan B.C., Martini N., Bains M.S. Bronchial carcinoids: review of 124 cases. J Thorac Cardiovasc Surg. 1985;89:8-17.
151 Abdi E.A., Goel R., Bishop S., Bain G.O. Peripheral carcinoid tumours of the lung: a clinicopathologic study. J Surg Oncol. 1988;39:190-196.
152 Ranchod M., Levine G.D. Spindle cell carcinoid tumors of the lung: a clinicopathologic study of 35 cases. Am J Surg Pathol. 1980;4:315-331.
153 Zarate A., Kovacs K., Flores M., et al. ACTH and CRF-producing bronchial carcinoid associated with Cushing’s syndrome. Clin Endocrinol. 1986;24:523-529.
154 Findling J.W., Tyrrell J.B. Occult ectopic secretion of corticotropin. Arch Intern Med. 1986;146:929-933.
155 Ankotche A., Raffin-Sanson M.L., Mosnier-Pudard H., et al. Ectopic ACTH secretion: a heterogeneous entity. Presse Med. 1997;26:1330-1333.
156 Shrager J.B., Wright C.D., Wain J.C., et al. Bronchopulmonary carcinoid tumors associated with Cushing’s syndrome: a more aggressive variant of typical carcinoid. J Thorac Cardiovasc Surg. 1997;114:367-375.
157 Oliaro A., Filosso P.L., Casadio C., et al. Bronchial carcinoid associated with Cushing’s syndrome. J Cardiovasc Surg. 1995;36:511-514.
158 White A., Clark A.J. The cellular and molecular basis of the ectopic ACTH syndrome. Clin Endocrinol. 1993;39:131-141.
159 Liu T.H., Liu H.R., Lu Z.L., et al. Thoracic ectopic ACTH-producing tumors with Cushing’s syndrome. Zentrabl Pathol. 1993;139:131-139.
160 Levy N.T., Rubin J., DeRemee R.A., et al. Carcinoid tumors and sarcoidosis – does a link exist? Mayo Clin Proc. 1997;72:112-116.
161 Flieder D.B., Vazquez V.F. Lung tumors with neuroendocrine morphology: a perspective for the new millenium. Radiol Clin North Am. 2000;38:563-577.
162 Aubry M.C., Thomas C.F.Jr, Jett J.R., et al. Significance of multiple carcinoid tumors and tumorlets in surgical lung specimens: analysis of 28 patients. Chest. 2007;131:1635-1643.
163 Davies S.J., Gosney J.R., Hansell D.M., et al. Diffuse idiopathic pulmonary neuroendocrine cell hyperplasia: an under-recognised spectrum of disease. Thorax. 2007;62:248-252.
164 Al-Kaisa N., Abdul-Karim F.W., Mendelsohn G., Jacobs G. Bronchial carcinoid tumor with amyloid stroma. Arch Pathol Lab Med. 1988;112:211-214.
165 Scharifker D., Marchevsky A. Oncocytic carcinoid tumor of lung: an ultrastructural analysis. Cancer. 1981;47:530-532.
166 Sklar J.L., Churg A., Bensch K.G. Oncocytic carcinoid tumor of the lung. Am J Surg Pathol. 1980;4:287-292.
167 Ritter J.H., Nappi O. Oxyphilic proliferations of the respiratory tract and paranasal sinuses. Semin Diagn Pathol. 1999;16:105-116.
168 Gaffey M.J., Mills S.E., Frierson H.F.Jr, et al. Pulmonary clear cell carcinoid tumor: another entity in the differential diagnosis of pulmonary clear cell neoplasia. Am J Surg Pathol. 1998;22:1020-1025.
169 Arora R., Mathur S.R., Aron M., et al. Oncocytic carcinoid tumor of the lung: a case report of diagnostic pitfall in filter membrane preparation of bronchial washings. Acta Cytol. 2007;51:907-910.
170 Mark E.J., Quay S.C., Dickerson G.R. Papillary carcinoid tumor of the lung. Cancer. 1981;48:316-324.
171 Grazer R., Cohen S.M., Jacobs J.B., Lucas P. Melanin-containing peripheral carcinoid tumor of the lung. Am J Surg Pathol. 1982;6:73-78.
172 Carlson J.A., Dickersin G.R. Melanotic paraganglioid carcinoid tumor: a case report and review of the literature. Ultrastruct Pathol. 1993;17:353-372.
173 Al-Khafaji B., Noffsinger A.E., Miller M.A., et al. Immunohistologic analysis of gastrointestinal and pulmonary carcinoid tumors. Hum Pathol. 1998;29:992-999.
174 Skinner C., Ewen S.W.B. Carcinoid lung: diffuse pulmonary infiltration by a multifocal bronchial carcinoid. Thorax. 1976;31:212-219.
175 Fulciniti F., La Vecchia F., Staiano M., et al. Spindle cell neuroendocrine carcinoma of the lung: report of a case with fine needle aspiration cytology and differential diagnostic considerations. Acta Cytol. 2007;51:227-230.
176 Lohmann D.R., Fesseler B., Putz B., et al. Infrequent mutations of the p53 gene in pulmonary carcinoid tumors. Cancer Res. 1993;53:5797-5801.
177 Jiang S.X., Kameya T., Shinada J., Yoshimura H. The significance of frequent and independent p53 and bcl-2 expression in large cell neuroendocrine carcinoma of the lung. Mod Pathol. 1999;12:362-369.
178 Srivastava A., Hornick J.L. Immunohistochemical staining for CDX-2, PDX-1, NESP-55, and TTF-1 can help distinguish gastrointestinal carcinoid tumors from pancreatic endocrine and pulmonary carcinoid tumors. Am J Surg Pathol. 2009;33:626-632.
179 Eyden B., Pandit D., Banerjee S.S. Malignant melanoma with neuroendocrine differentiation: clinical, histological, immunohistochemical and ultrastructural features of three cases. Histopathology. 2005;47:402-409.
180 Pelosi G., Zancanaro C., Sbabo L., et al. Development of innumerable neuroendocrine tumorlets in pulmonary lobe scarred by intralobar sequestration: immunohistochemical and ultrastructural study of an unusual case. Arch Pathol Lab Med. 1992;116:1167-1174.
181 Canessa P.A., Santini D., Zanelli M., Capecchi V. Pulmonary tumorlets and microcarcinoids in bronchiectasis. Monaldi Arch Chest Dis. 1997;52:138-139.
182 Ramon-Capilla M., Arnau-Obrer A., Navarro-Ibanez R., et al. Pulmonary tumorlet: report of 5 cases. Arch Bronchopneumonol. 1996;32:489-491.
183 Zanetta G., Zanoni M., Colombo F. Argentaffin pulmonary tumorlets. Tumori. 1979;65:761-766.
184 Watanabe H., Kobayashi H., Honma K., et al. Diffuse panbronchiolitis with multiple tumorlets: a quantitative study of the Kultschitzky cells and the clusters. Acta Pathol Jpn. 1985;35:1221-1231.
185 Rizvi S.M., Goodwill J., Lim E., et al. The frequency of neuroendocrine cell hyperplasia in patients with pulmonary neuroendocrine tumours and non-neuroendocrine cell carcinomas. Histopathology. 2009;55:332-337.
186 Ferolla P., Daddi N., Urbani M., et al. Regional Multidisciplinary Group for the Diagnosis and Treatment of Neuroendocrine Tumors, CRO, Umbria Region Cancer Network, Italy. Tumorlets, multicentric carcinoids, lymph-nodal metastases, and long-term behavior in bronchial carcinoids. J Thorac Oncol. 2009;4:383-387.
187 Miller R.R., Muller N.L. Neuroendocrine cell hyperplasia and obliterative bronchiolitis in patients with peripheral carcinoid tumors. Am J Surg Pathol. 1995;19:653-658.
188 Satoh Y., Fujiyama J., Ueno M., Ishikawa Y. High cellular atypia in a pulmonary tumorlet: report of a case with cytologic findings. Acta Cytol. 2000;44:242-246.
189 Higashiyama M., Doi O., Kodama K., et al. A case of pulmonary tumorlet with tuberculoma misdiagnosed as small cell lung carcinoma by transbronchial lung biopsy. Kyobu Geka. 1995;48:165-168.
190 Shah R., Sabanathan S., Mearns J., et al. Carcinoid tumor of the lung. J Cardiovasc Surg. 1997;38:187-189.
191 DiGiorgio A., Tocchi A., Puntillo G., et al. Tracheobronchial carcinoids: current therapeutic trends. Ann Ital Chir. 1990;61:405-409.
192 Huwer H., Kalweit G., Kruger B., et al. Bronchopulmonary carcinoids: surgical therapy and prognosis. Pneumonologie. 1996;50:786-789.
193 Ferguson M.K., Landreneau R.J., Hazelrigg S.R., et al. Long-term outcome after resection for bronchial carcinoid tumors. Eur J Cardiothorac Surg. 2000;18:156-161.
194 Warren W.H., Gould V.E., Faber L.P., et al. Neuroendocrine neoplasms of the bronchopulmonary tract: a classification of the spectrum of carcinoid to small cell carcinoma and intervening variants. J Thorac Cardiovasc Surg. 1985;89:819-825.
195 Skov B.G., Krasnik M., Lantuejoul S., et al. Reclassification of neuroendocrine tumors improves the separation of carcinoids and the prediction of survival. J Thorac Oncol. 2008;3:1410-1415.
196 Gustafsson B.I., Kidd M., Chan A., et al. Bronchopulmonary neuroendocrine tumors. Cancer. 2008;113:5-21.
197 Ashraf M.H. Bronchial carcinoid with osteoblastic metastases. Thorax. 1977;32:509-511.
198 Padberg B.C., Woenckhaus J., Hilger G., et al. DNA cytophotometry and prognosis in typical and atypical bronchopulmonary carcinoids: a clinicomorphologic study of 100 neuroendocrine lung tumors. Am J Surg Pathol. 1996;20:815-822.
199 Daddi N., Urbani M., Semeraro A., et al. Surgical treatment of well differentiated neuroendocrine tumours of the lung. G Chir. 2008;29:246-249.
200 Lissoni P., Barni S., Tacini G., et al. Immunoendocrine therapy with low-dose subcutaneous interleukin-2 plus melatonin of locally advanced or metastatic endocrine tumors. Oncology. 1995;52:163-166.
201 Arrigoni M.G., Woolner L.B., Bernatz P.E. Atypical carcinoid tumors of the lung. J Thorac Cardiovasc Surg. 1972;64:413-421.
202 Paladugu R.R., Benefield J.R., Pak H.Y., et al. Bronchopulmonary Kulchitzky cell carcinomas: a new classification scheme for typical and atypical carcinoids. Cancer. 1985;55:1303-1311.
203 Lequaglie C., Patriarca C., Cataldo I., et al. Prognosis of resected well-differentiated neuroendocrine carcinoma of the lung. Chest. 1991;100:1053-1056.
204 Memoli V. Well-differentiated neuroendocrine carcinoma: a designation comes of age. Chest. 1991;100:892.
205 Garcia-Yuste M., Matilla J.M., Alvarez-Gago F., et al. Prognostic factors in neuroendocrine lung tumors. Ann Thorac Surg. 2000;70:258-263.
206 Rush W., Zeren H., Griffin J.L., et al. Histologic subtypes of neuroendocrine carcinoma: prognostic correlations [abstract]. Lab Invest. 1995;153A:72.
207 Warren W.H., Memoli V.A., Gould V.E. Immunohistochemical and ultrastructural analysis of bronchopulmonary neuroendocrine neoplasms. II. Well-differentiated neuroendocrine carcinomas. Ultrastruct Pathol. 1984;7:185-199.
208 Warren W.H., Memoli V.A., Gould V.E. Well differentiated and small cell neuroendocrine carcinomas of the lung: two related but distinct clinicopathologic entities. Virch Arch B Cell Pathol. 1988;55:299-310.
209 Warren W.H., Memoli V.A., Jordan A.G., et al. Re-evaluation of pulmonary neoplasms resected as small cell carcinomas: significance of distinguishing between well-differentiated and small cell neuroendocrine carcinomas. Cancer. 1990;65:1003-1010.
210 Carter D., Yesner R. Carcinomas of the lung with neuroendocrine differentiation. Semin Diagn Pathol. 1985;2:235-254.
211 Oliaro A., Donati G., Filosso P.L., Ruffini E. Neuroendocrine tumors of the lung. Minerva Chir. 2000;66:7-16.
212 Slodkowska J., Langfort R., Rudzinski P., Kupis W. Typical and atypical pulmonary carcinoids: pathologic and clinical analysis of 77 cases. Pneumonol Alergol Pol. 1998;66:297-303.
213 Pareja E., Arnau A., Aartigues E., et al. Bronchial carcinoid tumors: a prospective study. Arch Bronchopneumonol. 1998;34:71-75.
214 Balli M., Fabris G.A., Dewar A., et al. Atypical carcinoid tumor: a study of 33 cases with prognostic features. Histopathology. 1994;24:363-369.
215 Mills S.E., Walker A.N., Cooper P.H., et al. Atypical carcinoid tumor of the lung: a clinicopathologic study of 17 cases. Am J Surg Pathol. 1982;6:643-654.
216 Grote T.H., Macon W.R., Davis B., et al. Atypical carcinoid of the lung: a distinct clinicopathologic entity. Chest. 1988;93:370-375.
217 Burns T.M., Juel V.C., Sanders D.B., Phillips L.H. Neuroendocrine lung tumors and disorders of the neuromuscular junction. Neurology. 1999;52:1490-1491.
218 Yousem S.A., Taylor S.R. Typical and atypical carcinoid tumors of lung. A clinicopathologic and DNA analysis of 20 tumors. Modern Pathol. 1990;3:502-507.
219 Guinee D.G.Jr, Fishback N.F., Koss M.N., et al. The spectrum of immunohistochemical staining of small cell lung carcinoma in specimens from transbronchial and open-lung biopsies. Am J Clin Pathol. 1994;102:406-414.
220 Slodkowska J. The value of immunohistochemical identification of neuroendocrine differentiation in non-small cell lung carcinoma. Rocz Akad Med Bialymst. 1997;42(suppl 1):23-27.
221 Al-Saffar N., White A., Moore M., Hasleton P.S. Immunoreactivity of various peptides in typical and atypical bronchopulmonary carcinoid tumors. Br J Cancer. 1988;58:762-766.
222 Couce M.E., Bautista D., Costa J., Carter D. Analysis of K-ras, N-ras, H-ras, and p53 in lung neuroendocrine neoplasms. Diagn Mol Pathol. 1999;8:71-79.
223 Roncalli M., Doglioni C., Springall D.R., et al. Abnormal p53 expression in lung neuroendocrine tumors. Diagnostic and prognostic implications. Diag Molec Pathol. 1992;1:129-135.
224 Anbazhagan R., Tihan T., Bornman D.M., et al. Classification of small cell lung cancer and pulmonary carcinoid by gene expression profiles. Cancer Res. 1999;59:5119-5122.
225 Onuki N., Wistuba I.I., Travis W.D., et al. Genetic changes in the spectrum of neuroendocrine lung tumors. Cancer. 1999;85:600-607.
226 Ullmann R., Schwendel A., Klemen H., et al. Unbalanced chromosomal aberrations in neuroendocrine lung tumors as detected by comparative genomic hybridization. Hum Pathol. 1998;29:1145-1149.
227 Johansson M., Heim S., Mandahl N., et al. Cytogentic analysis of six bronchial carcinoids. Cancer Gen Cytogen. 1993;66:33-38.
228 Jackson-York G.L., David B.H., Warren W.H., et al. Flow cytometric DNA content analysis in neuroendocrine carcinoma of the lung. Cancer. 1991;68:374-379.
229 Brambilla E., Moro D., Veale D., et al. Basal cell (basaloid) carcinoma of the lung: a new morphologic and phenotypic entity with separate prognostic significance. Hum Pathol. 1992;23:993-1003.
230 Travis W.D., Linnoila R.I., Tsokos M.G., et al. Neuroendocrine tumors of the lung with proposed criteria for large-cell neuroendocrine carcinoma: an ultrastructural, immunohistochemical, and flow cytometric study of 35 cases. Am J Surg Pathol. 1991;15:529-553.
231 McBurney R.P., Kirklin J.W., Woolner L.B. Metastasizing bronchial adenomas. Surg Gynecol Obstet. 1953;96:482-492.
232 Marty-Ane C.H., Costes V., Pujol J.L., et al. Carcinoid tumors of the lung: do atypical features require aggressive management? Annu Thorac Surg. 1995;59:78-83.
233 Carretta A., Ceresoli G.L., Arrigoni G., et al. Diagnostic and therapeutic management of neuroendocrine lung tumors: a clinical study of 44 cases. Lung Cancer. 2000;29:217-225.
234 Barnard W.G. The nature of the “oat cell sarcoma” of the mediastinum. J Pathol Bacteriol. 1926;29:241-244.
235 Azzopardi J.G. Oat cell carcinoma of the bronchus. J Pathol Bacteriol. 1959;78:513-519.
236 Cook R.M., Miller Y.E., Bunn P.A.Jr. Small cell lung cancer: etiology, biology, clinical features, staging, and treatment. Curr Probl Cancer. 1993;17:69-141.
237 Perkins P.J. Delayed onset of secondary hypertrophic osteoarthropathy. Am J Roentgenol. 1978;130:561-562.
238 Johnson B.E., Chute J.P., Rushin J., et al. A prospective study of patients with lung cancer and hyponatremia of malignancy. Am J Respir Crit Care Med. 1997;156:1669-1678.
239 Takai E., Yano T., Iguchi H., et al. Tumor-induced hypercalcemia and parathyroid hormone-related protein in lung carcinoma. Cancer. 1996;78:1384-1387.
240 Ferroir J.P., Milleron B., Ropert A., et al. Atypical paraneoplastic myasthenic syndrome: Lambert–Eaton syndrome or myasthenia? Rev Pneumonol Clin. 1999;55:168-170.
241 Posner J.B. Paraneoplastic syndromes: a brief review. Ann N Y Acad Sci. 1997;835:83-90.
242 Thirkill C.E. Lung cancer-induced blindness. Lung Cancer. 1996;14:253-264.
243 Usalan C., Emri S. Membranoproliferative glomerulonephritis associated with small cell lung carcinoma. Int Urol Nephrol. 1998;30:209-213.
244 Brenner S., Golan H., Gat A., Bialy-Golan A. Paraneoplastic subacute cutaneous lupus erythematosus: report of a case associated with cancer of the lung. Dermatology. 1997;194:172-174.
245 Hsu C.W., Wang H.C., Lu J.Y. Small cell lung carcinoma associated with paraneoplastic limbic encephalitis. J Formos Med Assoc. 1999;18:368-371.
246 Kamiyoshihara M., Hirai T., Kawashima O., et al. Sarcoid reactions in primary pulmonary carcinoma: report of seven cases. Oncol Rep. 1998;5:177-180.
247 Conejo-Mir J.S., Casals M., Carciandia C., et al. Cutaneous sarcoid granulomas with oat cell carcinoma of the lung. Dermatology. 1995;191:59-61.
248 Ponge T., Boutoille D., Moreau A., et al. Systemic vasculitis in a patient with small-cell neuroendocrine bronchial cancer. Eur Respir J. 1998;12:1228-1229.
249 Krauss E.A., Ludwig P.W., Sumner H.W. Metastatic carcinoma presenting as fulminant hepatic failure. Am J Gastroenterol. 1979;72:651-654.
250 Horlyck A., Henriques U., Jakobsen A. The value of bone marrow examination in small cell carcinoma. Acta Oncol. 1994;33:909-911.
251 Tritz D.B., Doll D.C., Ringenberg Q.S., et al. Bone marrow involvement in small cell lung cancer: clinical significance and correlation with routine laboratory values. Cancer. 1989;63:763-766.
252 Nguyen L.N., Maor M.H., Oswald M.J. Brain metastases as the only manifestation of an undetected primary tumor. Cancer. 1998;83:2181-2184.
253 Falconieri G., Zanconati F., Bussani R., DiBonito L. Small cell carcinoma of lung simulating pleural mesothelioma: report of 4 cases with autopsy confirmation. Pathol Res Pract. 1995;191:1147-1152.
254 Yesner R. Small cell tumors of the lung. Am J Surg Pathol. 1983;7:775-785.
255 Carter D. Small cell carcinoma of the lung. Am J Surg Pathol. 1983;7:787-795.
256 Aisner S.C., Finkelstein D.M., Ettinger D.S., et al. The clinical significance of variant morphology small cell carcinoma of the lung. J Clin Oncol. 1990;8:402-408.
257 Bepler G., Neumann K., Holle R., et al. Clinical relevance of histologic subtyping in small cell lung cancer. Cancer. 1980;45:74-79.
258 Hirsch F.R., Matthews M.J., Aisner S.C., et al. Histopathologic classification of small cell lung cancer: changing concepts and terminology. Cancer. 1988;62:973-977.
259 Hirsch F.R., Matthews M.J., Yesner R. Histopathologic classification of small cell carcinoma of the lung: comments based on an interobserver examination. Cancer. 1982;50:1360-1366.
260 Magum M.D., Greco F.A., Hainsworth J.D., et al. Combined small cell and non-small cell lung cancer. J Clin Oncol. 1989;7:607-612.
261 Sehested M., Hirsch F.R., Osterlind K., et al. Morphologic variations of small cell lung cancer: a histopathologic study of pretreatment and posttreatment specimens in 104 patients. Cancer. 1986;57:805-807.
262 Thomas J.S., Lamb D., Ashcroft T., et al. How reliable is the diagnosis of lung cancer using small biopsy specimens? Report of a UKCCCR Lung Cancer Working Party. Thorax. 1993;48:1135-1139.
263 Vollmer R.T. The effect of cell size on the pathologic diagnosis of small and large cell carcinomas of the lung. Cancer. 1982;50:1380-1383.
264 Fushimi H., Kukui M., Morino H., et al. Detection of large cell component in small cell lung carcinoma by combined cytologic and histologic examinations and its clinical implication. Cancer. 1992;70:599-605.
265 Nicholson S.A., Beasley M.B., Brambilla E., et al. Small cell lung carcinoma (SCLC): a clinicopathologic study of 100 cases with surgical specimens. Am J Surg Pathol. 2002;26:1184-1197.
266 D’Adda T., Pelosi G., Lagrasta C., et al. Genetic alterations in combined neuroendocrine neoplasms of the lung. Mod Pathol. 2008;21:414-422.
267 Facilone F., Cimmino A., Assennato G., et al. What is the prognostic significance of histomorphology in small cell lung carcinoma? Pathologica. 1993;85:387-393.
268 Fraire A.E., Johnson E.H., Yesner R., et al. Prognostic significance of histopathologic subtype and stage in small cell lung cancer. Hum Pathol. 1992;23:520-528.
269 Yang Y.J., Steele C.T., Ou X.L., et al. Diagnosis of high-grade pulmonary neuroendocrine carcinoma by fine-needle aspiration biopsy: nonsmall-cell or small-cell type? Diagn Cytopathol. 2001;25:292-300.
270 Nicholson S.A., Ryan M.R. A review of cytologic findings in neuroendocrine carcinomas including carcinoid tumors with histologic correlation. Cancer. 2000;90:148-161.
271 Tome Y., Hirohashi S., Noguchi M., et al. Immunocytologic diagnosis of small cell lung cancer in imprint smears. Acta Cytol. 1991;35:485-490.
272 Gephardt G.N., Grady K.J., Ahmad M., et al. Peripheral small cell undifferentiated carcinoma of the lung: clinicopathologic features of 17 cases. Cancer. 1988;61:1002-1008.
273 Hamzik J., Vrastyak J., Janik M., et al. Surgical treatment of small cell pulmonary carcinoma. Rozhledy V Chir. 1994;73:106-109.
274 Mentzer S.J., Reilly J.J., Sugarbaker D.J. Surgical resection in the management of small cell carcinoma of the lung. Chest. 1993;103(suppl):349S-351S.
275 Smit E.F., Croen H.J., Timens W., et al. Surgical resection for small cell carcinoma of the lung: a retrospective study. Thorax. 1994;49:20-22.
276 Ishida T., Nishino T., Oka T., et al. Surgical treatment of patients with small cell carcinoma of the lung: a histochemical and immunohistochemical study. J Surg Oncol. 1989;40:188-193.
277 Shepherd F.A., Ginsberg R.J., Feld R., et al. Surgical treatment for limited small-cell lung cancer: the University of Toronto Lung Oncology Group experience. J Thorac Cardiovasc Surg. 1991;101:385-393.
278 Shepherd F.A., Ginsberg R.J., Patterson G.A., et al. Is there ever a role for salvage operations in limited small cell lung cancer? J Thorac Cardiovasc Surg. 1991;101:196-200.
279 Bunn P.A.Jr, Carney D.N. Overview of chemotherapy for small cell lung cancer. Semin Oncol. 1997;24(2 suppl 7):S69-S74.
280 Comis R.L. Developments in therapy for extensive disease small cell lung cancer. Semin Oncol. 1992;19(6 suppl 13):45-50.
281 Marchevsky A.M., Gal A.A., Shah S., Koss M.N. Morphometry confirms the presence of considerable nuclear size overlap between “small cells” and “large cells” in high-grade pulmonary neuroendocrine neoplasms. Am J Clin Pathol. 2001;116:466-472.
282 Wiatrowska B.A., Krol J., Zakowski M.F. Large-cell neuroendocrine carcinoma of the lung: proposed criteria for cytologic diagnosis. Diagn Cytopathol. 2001;24:58-64.
283 Brambilla E., Lanteujoul S., Sturm N. Divergent differentiation in neuroendocrine lung tumors. Semin Diagn Pathol. 2000;17:138-148.
284 Warren W.H., Gould V.E. Differential diagnosis of small cell neuroendocrine carcinoma of the lung. Chest Surg Clin N Am. 1997;7:49-63.
285 Brambilla E., Travis W.D., Colby T.V., et al. The new World Health Organization classification of lung tumors. Eur Respir J. 2001;18:1059-1068.
286 Hammar S.P. Approach to the diagnosis of neuroendocrine lung neoplasms: variabilities and pitfalls. Semin Thorac Cardiovasc Surg. 2006;18:183-190.
287 Nandedkar M.A., Palazzo J., Abbondanzo S.L., et al. CD45 (leukocyte common antigen) immunoreactivity in metastatic undifferentiated and neuroendocrine carcinoma: a potential diagnostic pitfall. Mod Pathol. 1998;11:1204-1210.
288 Copple B., Wright S.E., Moatamed F. Electron microscopy in small cell lung carcinomas: clinical correlations. J Clin Oncol. 1984;2:910-916.
289 Garcia-Yuste M., Matilla J.M., Alvarez-Gago T., et al. Prognostic factors in neuroendocrine lung tumors: a Spanish multicenter study. Spanish Multicenter Study of neuroendocrine tumors of the lung of the Spanish Society of Pneumonology and Thoracic Surgery (EMETNE-SEPAR). Ann Thorac Surg. 2000;70:258-263.
290 Cooper W.A., Thourani V.H., Gal A.A., et al. The surgical spectrum of pulmonary neuroendocrine neoplasms. Chest. 2001;119:14-18.
291 Huang Q., Muzitansky A., Mark E.J. Pulmonary neuroendocrine carcinomas: a review of 234 cases and a statistical analysis of 50 cases treated at one institution using a simple clinicopathologic classification. Arch Pathol Lab Med. 2002;126:545-553.
292 Cakir E., Demirag E., Aydin M., Unsal E. Clinicopathologic features and prognostic significance of lung tumours with mixed histologic patterns. Acta Chir Belg. 2009;109:489-493.
293 Hosoe S., Shigedo Y., Ueno K., et al. Detailed deletion mapping of the short arm of chromosome 3 in small cell and non-small cell carcinoma of the lung. Lung Cancer. 1994;20:297-305.
294 Vuitch F., Sekido Y., Fong K., et al. Neuroendocrine tumors of the lung: pathology and molecular biology. Chest Surg Clin N Am. 1997;7:21-47.
295 Kovatich A., Friedland D.M., Druck T., et al. Molecular alterations to human chromosome 3q loci in neuroendocrine lung tumors. Cancer. 1998;83:1109-1117.
296 Williams C.L. Basic science of small cell lung cancer. Chest Surg Clin N Am. 1997;7:1-19.
297 Jiang S.X., Kameya T., Shoji M., et al. Large cell neuroendocrine carcinoma of the lung: a histologic and immunohistochemical study of 22 cases. Am J Surg Pathol. 1998;22:526-537.
298 Duncavage E.J., Le B.M., Wang D., Pfeifer J.D. Merkel cell polyomavirus: a specific marker for Merkel cell carcinoma in histologically similar tumors. Am J Surg Pathol. 2009;33:1771-1777.
299 Ralston J., Chiriboga L., Nonaka D. MASH1: a useful marker in differentiating pulmonary small cell carcinoma from Merkel cell carcinoma. Mod Pathol. 2008;21:1357-1362.
300 Dresler C.M., Ritter J.H., Patterson G.A., et al. Clinical-pathologic analysis of 40 patients with large cell neuroendocrine carcinoma of the lung. Ann Thorac Surg. 1997;63:180-185.
301 Takei H., Asamura H., Maeshima, et al. Large cell neuroendocrine carcinoma of the lung: a clinicopathologic study of eighty-seven cases. J Thorac Cardiovasc Surg. 2002;124:285-292.
302 Jung K.J., Lee K.S., Han J., et al. Large cell neuroendocrine carcinoma of the lung: clinical, CT, and pathologic findings in 11 patients. J Thorac Imaging. 2001;16:156-162.
303 Iyoda A., Hiroshima K., Toyozaki T., et al. Clinical characterization of pulmonary large-cell neuroendocrine carcinoma and large-cell carcinoma with neuroendocrine morphology. Cancer. 2001;91:1992-2000.
304 Demirer T., Ravits J., Aboulafia D. Myasthenic (Eaton–Lambert) syndrome associated with pulmonary large cell neuroendocrine carcinoma. South Med J. 1994;87:1186-1189.
305 Stanford M.R., Edelstein C.E., Hughes J.D., et al. Paraneoplastic retinopathy in association with large cell neuroendocrine bronchial carcinoma. Br J Ophthalmol. 1995;79:617-618.
306 Chetty R., Bhana B., Batitang S., Govender D. Lung carcinoma composed of rhabdoid cells. Eur J Surg Oncol. 1997;23:432-434.
307 Khalifa M., Hruby G., Ehrlich L., et al. Combined large cell neuroendocrine carcinoma and spindle cell carcinoma of the lung. Ann Diagn Pathol. 2001;5:240-245.
308 Oliaro A., Filosso P.L., Donati G., Ruffini E. Atypical bronchial carcinoids: review of 46 patients. J Cardiovasc Surg. 2000;41:131-135.
309 Franklin W.A. Diagnosis of lung cancer: pathology of invasive and preinvasive neoplasia. Chest. 2000;117(4 suppl 1):80S-89S.
310 Rossi G., Marchioni A., Milani M., et al. TTF-1, cytokeratin 7, 34betaE12, and CD56/NCAM immunostaining in the subclassification of large cell carcinomas of the lung. Am J Clin Pathol. 2004;122:884-893.
311 Ullmann R., Petzmann S., Sharma A., et al. Chromosomal aberrations in a series of large-cell neuroendocrine carcinomas: unexpected divergence from small-cell carcinoma of the lung. Hum Pathol. 2001;32:1059-1063.
312 Hiroshima K., Abe S., Ebihara Y., et al. Cytological characteristics of pulmonary large cell neuroendocrine carcinoma. Lung Cancer. 2005;48:331-337.
313 Sun L., Sakurai S., Sano T., et al. High-grade neuroendocrine carcinoma of the lung: comparative clinicopathological study of large cell neuroendocrine carcinoma and small cell lung carcinoma. Pathol Int. 2009;59:522-529.
314 Asamura H., Kameya T., Matsuno Y., et al. Neuroendocrine neoplasms of the lung: a prognostic spectrum. J Clin Oncol. 2006;24:70-76.
315 Fernandez F.G., Battafarano R.J. Large-cell neuroendocrine carcinoma of the lung: an aggressive neuroendocrine lung cancer. Semin Thorac Cardiovasc Surg. 2006;18:206-210.
316 Battafarano R.J., Fernandez F.G., Ritter J., et al. Large cell neuroendocrine carcinoma: an aggressive form of non-small cell lung cancer. J Thorac Cardiovasc Surg. 2005;130:166-172.
317 Veronesi G., Morandi U., Alloisio M., et al. Large cell neuroendocrine carcinoma of the lung: a retrospective analysis of 144 surgical cases. Lung Cancer. 2006;53:111-115.
318 Shin A.R., Shin B.K., Choi J.A., et al. Large-cell neuroendocrine carcinoma of the lung: radiologic and pathologic findings. J Comput Assist Tomogr. 2000;24:567-573.
319 García-Yuste M., Matilla J.M., González-Aragoneses F. Neuroendocrine tumors of the lung. Curr Opin Oncol. 2008;20:148-154.
320 Iyoda A., Hiroshima K., Nakatani Y., Fujisawa T. Pulmonary large cell neuroendocrine carcinoma: its place in the spectrum of pulmonary carcinoma. Ann Thorac Surg. 2007;84:702-707.
321 Saji H., Tsuboi M., Matsubayashi J., et al. Clinical response of large cell neuroendocrine carcinoma of the lung to perioperative adjuvant chemotherapy. Anticancer Drugs. 2010;21:89-93.
322 Lai S.L., Goldstein L.J., Gottesman M.M., et al. MDR1 gene expression in lung cancer. J Natl Cancer Inst USA. 1989;81:1144-1150.
323 Sartori G., Cavazza A., Sgambato A., et al. EGFR and K-ras mutations along the spectrum of pulmonary epithelial tumors of the lung and elaboration of a combined clinicopathologic and molecular scoring system to predict clinical responsiveness to EGFR inhibitors. Am J Clin Pathol. 2009;131:478-489.
324 Faggiano A., Sabourin J.C., Ducreux M., et al. Pulmonary and extrapulmonary poorly differentiated large cell neuroendocrine carcinomas: diagnostic and prognostic features. Cancer. 2007;110:265-274.
325 DeLellis R.A., Tischler A.S., Wolfe H.J. Multidirectional differentiation in neuroendocrine neoplasms. J Histochem Cytochem. 1984;32:899-904.
326 Gaffey M.J., Mills S.E., Lack E.E. Neuroendocrine carcinoma of the colon and rectum. Am J Surg Pathol. 1990;14:1010-1023.
327 Chejfec G., Capella C., Solcia E., et al. Amphicrine cells, dysplasias, and neoplasias. Cancer. 1985;56:2683-2690.
328 Eusebi V., Capella C., Bondi A., et al. Endocrine-paracrine cells in pancreatic exocrine carcinomas. Histopathology. 1981;9:599-613.
329 Groben P., Reddick R., Askin F.B. The pathologic spectrum of small cell carcinoma of the cervix. Int J Gynecol Pathol. 1985;4:42-47.
330 Hales M., Rosenau W., Okerlund M.D., Galante M. Carcinoma of the thyroid with a mixed medullary and follicular pattern. Cancer. 1982;50:1352-1359.
331 Lewin K. Carcinoid tumors and the mixed (composite) glandular-endocrine cell carcinomas. Am J Surg Pathol. 1987;11(suppl 1):71-76.
332 Manivel J.C., Wick M.R., Sibley R.K. Neuroendocrine differentiation in Mullerian neoplasms. Am J Clin Pathol. 1986;86:438-443.
333 McCluggage W.G., Napier S.S., Primrose W.J., et al. Sinonasal neuroendocrine carcinoma exhibiting amphicrine differentiation. Histopathology. 1995;27:79-82.
334 Mills S.E., Wolfe J.T., Weiss M.A., et al. Small cell undifferentiated carcinoma of the urinary bladder. Am J Surg Pathol. 1987;11:606-617.
335 Reid J.D., Yuh S.L., Petrelli M., Jaffe R. Ductoinsular tumors of the pancreas. Cancer. 1982;49:908-915.
336 Silva E.G., Mackay B., Goepfert H., et al. Endocrine carcinoma of the skin (Merkel cell carcinoma). Pathol Annu. 1984;19(1):1-30.
337 Brambilla E., Lanteujoul S., Sturm N. Divergent differentiation in neuroendocrine lung tumors. Semin Diagn Pathol. 2000;17:138-148.
338 Sen F., Borczuk A.C. Combined carcinoid tumor of the lung: a combination of carcinoid and adenocarcinoma. Lung Cancer. 1998;21:53-58.
339 Frank G.A., Trakhtenberg A.K., Boguslavskii V.M. The prognosis of small cell carcinoma and malignant carcinoid of the lung. Vopr Onkol. 1989;35:192-198.
340 Mooi W.J., Dewar A., Springall D., et al. Non-small cell lung carcinomas with neuroendocrine features: a light microscopic, immunohisto-chemical, and ultrastructural study of 11 cases. Histopathology. 1988;13:329-337.
341 Otto W.R. Lung stem cells. Int J Exp Pathol. 1997;78:291-310.
342 Huang J., Behrens C., Wistuba I.I., et al. Clonality of combined tumors. Arch Pathol Lab Med. 2002;126:437-441.
343 Cakir E., Demirag E., Aydin M., Unsal E. Clinicopathologic features and prognostic significance of lung tumours with mixed histologic patterns. Acta Chir Belg. 2009;109:489-493.
344 Piehl M.R., Gould V.E., Warren W.H., et al. Immunohistochemical identification of exocrine and neuroendocrine subsets of large cell lung carcinomas. Pathol Res Pract. 1988;183:675-682.
345 Neal M.H., Kosinski R., Cohen P., et al. Atypical endocrine tumors of the lung: a histologic, ultrastructural, and clinical study of 19 cases. Hum Pathol. 1986;17:1264-1277.
346 Visscher D.W., Zarbo R.J., Trojanowski J.Q., et al. Neuroendocrine differentiation in poorly differentiated lung carcinomas: a light microscopic and immunohistologic study. Mod Pathol. 1990;3:508-512.
347 Skov B.G., Sorenson J.B., Hirsch F.R., et al. Prognostic impact of histologic demonstration chromogranin A and neuron specific enolase in pulmonary adenocarcinoma. Ann Oncol. 1991;2:355-360.
348 Linnoila R.I., Gazdar A.F. Non-small cell lung carcinoma with neuroendocrine features. Anat Pathol. 1990;18:1-5.
349 Linnoila R.I., Mulshine J.L., Steinberg S.M., et al. Neuroendocrine differentiation in endocrine and nonendocrine lung carcinomas. Am J Clin Pathol. 1988;90:641-652.
350 Hammond M.E., Sause W.T. Large cell neuroendocrine tumors of the lung: clinical significance and histopathologic definition. Cancer. 1985;56:1624-1629.
351 Berendsen H.H., Deleij L., Poppema S. Clinical characterization of non-small cell lung cancer tumors showing neuroendocrine differentiation features. J Clin Oncol. 1989;7:1614-1629.
352 Hiroshima K., Iyoda A., Shibuya K., et al. Prognostic significance of neuroendocrine differentiation in adenocarcinoma of the lung. Ann Thorac Surg. 2002;73:1732-1735.
353 Carnaghi C., Rimassa L., Garassino I., Santoro A. Clinical significance of neuroendocrine phenotype in non-small-cell lung cancer. Ann Oncol. 2001;12(suppl 2):S119-S123.
354 Baldi A., Groger A.M., Esposito V., et al. Neuroendocrine differentiation in non-small-cell lung carcinomas. In Vivo. 2000;14:109-114.
355 Fresvig A., Qvigstad G., Halvorsen T.B., et al. Neuroendocrine differentiation in bronchial carcinomas of classic squamous cell type: an immunohistochemical study of 29 cases applying the tyramide signal amplification technique. Appl Immunohistochem Mol Morphol. 2001;9:9-13.
356 Churg A. The fine structure of large cell undifferentiated carcinoma of the lung: evidence for its relation to squamous cell carcinomas and adenocarcinomas. Hum Pathol. 1978;9:143-156.
357 Baldi A., Groger A.M., Esposito Y., et al. Neuroendocrine differentiation in non-small cell lung carcinomas. In Vivo. 2000;14:109-114.
358 Nakatani Y., Kitamura H., Inayama Y., et al. Pulmonary adenocarcinoma of the fetal lung type: a clinicopathologic study indicating differences in histology, epidemiology, and natural history of low-grade and high-grade forms. Am J Surg Pathol. 1998;22:399-411.
359 Chetritt J., Fiche M., Cassagnau E., et al. Pulmonary endodermal tumor resembling fetal lung: low grade adenocarcinoma of the fetal lung type. Ann Pathol. 1999;19:116-118.
360 Abbona G., Papotti M., Viberti L., et al. Chromogranin A gene expression in non-small cell lung carcinomas. J Pathol. 1998;186:151-156.
361 Sørhaug S., Steinshamn S., Haaverstad R., et al. Expression of neuroendocrine markers in non-small cell lung cancer. APMIS. 2007;115:152-163.
362 Leong A.S.Y., Wick M.R., Swanson P.E. Immunohistology and Electron Microscopy of Anaplastic and Pleomorphic Tumors. Cambridge, UK: Cambridge University Press, 1997;161-208.
363 Graziano S.L., Mazid R., Newman N., et al. The use of neuroendocrine immunoperoxidase markers to predict chemotherapy response in patients with non-small-cell lung cancer. J Clin Oncol. 1989;7:1398-1406.
364 Hainsworth J.D., Johnson D.H., Greco F.A. Poorly-differentiated neuroendocrine carcinoma of unknown primary site: a newly recognized clinicopathologic entity. Ann Intern Med. 1988;109:364-371.
365 Hainsworth J.D., Wright E.P., Johnson D.H., et al. Poorly differentiated carcinoma of unknown primary site: clinical usefulness of immunoperoxidase staining. J Clin Oncol. 1991;9:1931-1938.
366 Schleusener J., Tazelaar H., Jung S. Neuroendocrine differentiation correlates with survival in chemotherapy treated non-small cell lung cancer [abstract]. Lab Invest. 1995;72:153A.
367 Wertzel H., Grahmann P.R., Bansbach S., et al. Results after surgery in undifferentiated large cell carcinoma of the lung: the role of neuroendocrine expression. Eur J Cardiothorac Surg. 1997;12:698-702.
368 Segawa Y., Takata S., Fujii M., et al. Immunohistochemical detection of neuroendocrine differentiation in non-small-cell lung cancer and its clinical implications. J Cancer Res Clin Oncol. 2009;135:1055-1059.
369 Sterlacci W., Fiegl M., Hilbe W., et al. Clinical relevance of neuroendocrine differentiation in non-small cell lung cancer assessed by immunohistochemistry: a retrospective study on 405 surgically resected cases. Virchows Arch. 2009;455:125-132.
370 Lack E.E. Pathology of Adrenal and Extraadrenal Paraganglia. Philadelphia: WB Saunders, 1994;1-350.
371 Goodman M.L., Laforet E.G. Solitary primary chemodectomas of the lung. Chest. 1972;61:48-60.
372 Siingh G., Lee R.E., Brooks D.H. Primary pulmonary paraganglioma: report of a case and review of the literature. Cancer. 1977;40:2286-2289.
373 DeLuise V.P., Holman C.W., Gray G.F. Primary pulmonary paraganglioma. N Y State J Med. 1977;77:2270-2271.
374 Hangartner J.R., Loosemore T.M., Burke M., Pepper J.R. Malignant primary pulmonary paraganglioma. Thorax. 1989;44:154-156.
375 Dusseldorf M., Straaten H.G. Primary pulmonary paraganglioma. Zentralbl Chir. 1990;115:1575-1578.
376 Lemonick D.M., Pai P.B., Hines G.L. Malignant primary pulmonary paraganglioma with hilar metastasis. J Thorac Cardiovasc Surg. 1990;99:563-564.
377 Vuorela A.L., Anttinen J. Primary chemodectoma of the lung. Am J Roentgenol. 1993;161:1111-1112.
378 Hagemeyer O., Gabius H.J., Kayser K. Paraganglioma of the lung—developed after exposure to nuclear radiation by the Tschernobyl atomic reactor accident? Respiration. 1994;61:236-239.
379 Skodt V., Jacobsen G.K., Helsted M. Primary paraganglioma of the lung: report of two cases and review of the literature. APMIS. 1995;103:597-603.
380 Saeki T., Akiba T., Joh K., et al. An extremely large solitary primary paraganglioma of the lung: report of a case. Surg Today. 1999;29:1195-1200.
381 Sattar H.A., Yang D.L., Husain A.N., et al. Multiple paragangliomata of the lungs and temporal bone. Ear Nose Throat J. 2008;87:E4-E6.
382 Rossel M., Pasini A., Chappuis S., et al. Distinct biological properties of two RET isoforms activated by MEN2A and MEN2B mutations. Oncogene. 1997;14:265-275.
383 Hironaka M., Fukayama M., Takayashiki N., et al. Pulmonary gangliocytic paraganglioma: case report and comparative immunohistochemical study of related neuroendocrine neoplasms. Am J Surg Pathol. 2001;25:688-693.
384 Linnoila R.I., Keiser H.R., Steinberg S.M., Lack E.E. Histopathology of benign versus malignant sympathoadrenal paragangliomas: clinicopathologic study of 120 cases including unusual histologic features. Hum Pathol. 1990;21:1168-1180.
385 Engzell V., Franzen S., Zajicek J. Aspiration biopsy of tumors of the neck. II. Cytologic findings in 13 cases of carotid body tumors. Acta Cytol. 1971;15:25-30.
386 Gonzalez-Campora R., Otal-Salaverri C., Panea-Flores P., et al. Fine needle aspiration cytology of paraganglionic tumors. Acta Cytol. 1988;32:386-390.
387 Hood I.C., Qizilbash A.H., Young J.E.H., Archibald S.D. Fine needle aspiration biopsy cytology of paragangliomas. Acta Cytol. 1983;27:651-657.
388 Fournel P., Boucheron S., Baril A., et al. Intrapulmonary chemodectoma: a new case with ultrastructural study. Rev Pneumonol Clin. 1986;42:250-253.
389 Trojanowski J.Q. Neurofilament and glial filament proteins. In: Wick M.R., Siegal G.P., editors. Monoclonal Antibodies in Diagnostic Immunohistochemistry. New York: Marcel Dekker; 1988:115-146.
390 Gaffey M.J., Mills S.E., Askin F.B. Minute pulmonary meningothelial-like nodules: a clinicopathologic study of so-called minute pulmonary chemodectoma. Am J Surg Pathol. 1988;12:167-175.
391 Niho S., Yokose T., Nishiwaki Y., Mukai K. Immunohistochemical and clonal analysis of minute pulmonary meningothelial-like nodules. Hum Pathol. 1999;30:425-429.
392 Helman L.J., Cohen P.S., Averbuch S.D., et al. Neuropeptide Y distinguishes benign from malignant pheochromocytoma. J Clin Oncol. 1989;7:720-725.
393 Schroder H.D., Johannsen L. Demonstration of S100 protein in sustentacular cells of pheochromocytomas and paragangliomas. Histopathology. 1986;10:1023-1033.
394 Gonzalez-Campora R., Diaz-Cano S., Lerma-Puertas E., et al. Paragangliomas: static cytometric studies of nuclear DNA patterns. Cancer. 1993;71:820-824.
395 Pang L.C., Tsao K.C. Flow cytometric DNA analysis for the determination of malignant potential in adrenal and extra-adrenal pheochromocytomas or paragangliomas. Arch Pathol Lab Med. 1993;117:1142-1147.
396 Imamura F., Funakoshi T., Nakamura S., et al. Primary primitive neuroectodermal tumor of the lung: report of two cases. Lung Cancer. 2000;27:55-60.
397 Mikami Y., Nakajima M., Hashimoto H., et al. Primary pulmonary primitive neuroectodermal tumor (PNET): a case report. Pathol Res Pract. 2001;197:113-119.
398 Verfaillie G., Hoorens A., Lamote J. Primary primitive neuroectodermal tumour of the lung. Acta Chir Belg. 2009;109:381-384.
399 Suárez Antelo J., Rodríguez García C., Montero Martínez C., Verea Hernando H. Pulmonary Ewing sarcoma/primitive neuroectodermal tumor: a case report and a review of the literature. Arch Bronconeumol. 2010;46:44-46.
400 Lee Y.Y., Kim do H., Lee J.H., et al. Primary pulmonary Ewing’s sarcoma/primitive neuroectodermal tumor in a 67-year-old man. J Korean Med Sci. 2007;22(suppl):S159-S163.
401 Takahashi D., Nagayama J., Nagatoshi Y., et al. Primary Ewing’s sarcoma family tumors of the lung a case report and review of the literature. Jpn J Clin Oncol. 2007;37:874-877.
402 Rossi S., Orvieto E., Furlanetto A., et al. Utility of the immunohistochemical detection of FLI-1 expression in round cell and vascular neoplasm using a monoclonal antibody. Mod Pathol. 2004;17:547-552.
403 Llombart-Bosch A., Machado I., Navarro S., et al. Histological heterogeneity of Ewing’s sarcoma/PNET: an immunohistochemical analysis of 415 genetically confirmed cases with clinical support. Virchows Arch. 2009;455:397-411.
404 Jambhekar N.A., Bagwan I.N., Ghule P., et al. Comparative analysis of routine histology, immunohistochemistry, reverse transcriptase polymerase chain reaction, and fluorescence in situ hybridization in diagnosis of Ewing family of tumors. Arch Pathol Lab Med. 2006;130:1813-1818.
405 Jaffe N. Neuroblastoma: review of the literature and an examination of factors contributing to its enigmatic character. Cancer Treat Rev. 1976;3:61-82.
406 Young J.L.Jr, Miller R.W. Incidence of malignant tumors in children. J Pediatr. 1975;86:254-258.
407 Kilton L.J., Aschenbrener C., Burns C.P. Ganglioneuroblastoma in adults. Cancer. 1976;37:974-983.
408 Nagashima Y., Miyagi Y., Tanaka Y., et al. Adult ganglioneuroblastoma of the anterior mediastinum. Pathol Res Pract. 1997;193:727-732.
409 Cooney T.P. Primary pulmonary ganglioneuroblastoma in an adult: maturation, involution, and the immune response. Histopathology. 1981;5:451-463.
410 Hochholzer L., Moran C.A., Koss M.N. Primary pulmonary ganglioneuroblastoma: a clinicopathologic and immunohistochemical study of two cases. Ann Diagn Pathol. 1998;2:154-158.
411 Kaplan S.J., Holbrook C.T., McDaniel H.G., et al. Vasoactive intestinal peptide secreting tumors of childhood. Am J Dis Childhood. 1980;134:21-24.
412 Kenny F.M., Stavrides A., Voorhess M.L., Klein R. Cushing’s syndrome associated with adrenal neuroblastoma. Am J Dis Childhood. 1967;113:611-615.
413 Ikuno N., Shimokawa I., Nakamura T., et al. RET oncogene expression correlates with neuronal differentiation of neuroblastic tumors. Pathol Res Pract. 1995;191:92-99.
414 Dehner L.P. Pathologic anatomy of classic neuroblastoma, including prognostic factors and differential diagnosis. In: Pochedly C., editor. Neuroblastoma: Tumor Biology and Therapy. Boca Raton: CRC Press; 1990:111-143.
415 Hughes M., Marsden H.B., Palmer M.K. Histologic patterns of neuroblastoma related to prognosis and clinical stage. Cancer. 1974;34:1706-1711.
416 Makinen J. Microscopic patterns as a guide to prognosis of neuroblastomas in childhood. Cancer. 1972;29:1637-1646.
417 McLaughlin J.E., Urich H. Maturing neuroblastoma and ganglioneuroblastoma: a study of four cases with long survival. J Pathol. 1977;121:19-26.
418 Shimada H., Chatten J., Newton W.A.Jr, et al. Histopathologic prognostic factors in neuroblastic tumors. J Natl Cancer Inst. 1984;73:405-416.
419 Abramowsky C.R., Katzenstein H.M., Alvarado C.S., Shehata B.M. Anaplastic large cell neuroblastoma. Pediatr Dev Pathol. 2009;12:1-5.
420 Hicks M.J., Mackay B. Comparison of ultrastructural features among neuroblastic tumors: maturation from neuroblastoma to ganglioneuroma. Ultrastruct Pathol. 1995;19:311-322.
421 Molenaar W.M., Baker D.L., Pleasure D., et al. The neuroendocrine and neural profiles of neuroblastomas, ganglioneuroblastomas, and ganglioneuromas. Am J Pathol. 1990;136:375-382.
422 Crary G.S., Singleton T.P., Neglia J.P., et al. Detection of metastatic neuroblastoma in bone marrow biopsy specimens with an antibody to neuron-specific enolase. Mod Pathol. 1992;5:308-314.
423 Miettinen M., Chatten J., Paetau A., Stevenson A. Monoclonal antibody NB84 in the differential diagnosis of neuroblastoma and other small round cell tumors. Am J Surg Pathol. 1998;22:327-332.
424 Cozzutto C., Carbone A. Pleomorphic (anaplastic) neuroblastoma. Arch Pathol Lab Med. 1988;112:621-625.
425 Berthold F., Trechow R., Utsch S., Zieschang J. Prognostic factors in metastatic neuroblastoma: a multivariate analysis of 182 cases. Am J Pediatr Hematol Oncol. 1992;14:207-215.
426 Joshi V.V., Chatten J., Sather H.N., Shimada H. Evaluation of the Shimada classification in advanced neuroblastoma with a special reference to the mitosis-karyorrhexis index. Mod Pathol. 1991;4:139-147.
427 Coldman A.J., Fryer C.J.H., Elwood J.M., Sonley M.J. Neuroblastoma: influence of age at diagnosis, stage, tumor site, and sex on prognosis. Cancer. 1980;46:1896-1901.
428 Gitlow S.E., Dziedzic L.B., Strauss L., et al. Biochemical and histologic determinants in the prognosis of neuroblastoma. Cancer. 1973;32:898-905.
429 Kogner P., Borgstrom P., Bjellerup P., et al. Somatostatin in neuroblastoma and ganglioneuroma. Eur J Cancer. 1997;33:2084-2089.
430 Hayashi Y., Kanda N., Inaba T., et al. Cytogenetic findings and prognosis in neuroblastoma with emphasis on marker chromosome 1. Cancer. 1989;63:126-132.
431 Berthold F., Sahin K., Hero B., et al. The current contribution of molecular factors to risk estimation in neuroblastoma patients. Eur J Cancer. 1997;33:2092-2097.
432 Brodeur G.M., Seeger R.C., Sather H., et al. Clinical implications of oncogene activation in human neuroblastomas. Cancer. 1986;58:541-545.
433 Hashimoto H., Daimaru Y., Enjoji M., Nakagawara A. N-myc gene product expression in neuroblastoma. J Clin Pathol. 1989;42:52-55.
434 Kramer K., Cheung N.K., Gerald W.L., et al. Correlation of myc-N amplification, Trk-A and CD44 expression with clinical stage in 250 patients with neuroblastoma. Eur J Cancer. 1997;33:2098-2100.
435 Ikuno N., Shimokawa I., Nakamura T., et al. RET oncogene expression correlates with neuronal differentiation of neuroblastic tumors. Pathol Res Pract. 1995;191:92-99.
436 Graham D., Magee H., Kierce B., et al. Evaluation of Ki-67 reactivity in neuroblastoma using paraffin-embedded tissue. Pathol Res Pract. 1995;191:87-91.