[level-membership-for-neurosurgery-category]
CHAPTER 4 Neuroembryology
Classic neuroembryology dealt primarily with documentation of the timing and location of morphologic changes in embryonic development, both gross and microscopic, but an acceleration of genetic, molecular, and radiologic (i.e., neuroimaging) advances in recent years has fundamentally changed the way that we perceive and understand both normal formation and malformation of the brain. Completion of the first draft of the sequence of the human genome will have profound implications on our understanding of the genetics of brain development.1–4 An accelerating pace of genetic discovery is accompanied by the risk that reviews become obsolete before their publication. We now speak of genetics and genomics, but since the human genome project met its initial goals, we will necessarily move onto proteomics (i.e., study of all proteins expressed by the genome)5 and an ever-growing list of “omics” as we delve deeper into the complex molecular genetic interactions required for creating the brain.
Although we do not dispute the critical role of these advances in elucidating development, we strongly believe that this information is best used within the framework of a solid understanding of the timing of structural changes during brain development. In this chapter we offer a unified vision of neuroembryology, with molecular genetic details presented along with classic structural information. It is beyond the scope of this chapter to review the principles of genetics or the revolutionary techniques of gene cloning, polymerase chain reaction, and positional cloning or to attempt to catalog the genes responsible for neurological abnormalities.6 More information is available in review articles or book chapters on neurogenetics.7,8 We do provide examples of important genes relevant to neurodevelopment and extensive references that can aid in understanding basic concepts.
Because much of our knowledge about human brain development has resulted from searching for human homologues to developmental genes discovered in animals from Drosophila to mice, we include examples from these studies along with our discussions of the stages, processes, and abnormalities of human neuroembryology. Table 4-1 lists known gene mutations responsible for several important human central nervous system (CNS) malformations.9–44 When specifically referring to a human gene, we use the convention of denoting the gene symbol in italicized, capital letters.

We once thought of brain insults arising from either environmental or genetic factors, but we now recognize that these causes are interconnected and inseparable. Environmental factors act by influencing gene regulation and expression, and genetic differences determine responses to environmental agents, including toxins and transcription factors. The molecular details of how thousands of genes and the proteins that they encode work together to determine normal or abnormal structure and function are becoming clearer daily but are still overwhelmingly complex. Although the estimated number of genes in the human genome is only about 30,000 (far less than the previously estimated 100,000 and only 2.5 times larger than the fly genome), the human proteome is estimated to contain between 130,000 and 400,000 distinct proteins. Each has many potential ways of interacting with other proteins or genes and of being posttranslationally modified.4
Improvements in neuroradiologic techniques are helping to uncover relationships between genetic abnormalities and structural malformations and to provide another justification for learning more about the basics of embryologic and fetal development. As imaging has improved, so has our appreciation that many developmental disorders represent a spectrum of abnormalities much more complex than previously appreciated. The quality of the image and the speed of acquisition of fetal magnetic resonance imaging (MRI) are constantly improving, and it is already capable of very good anatomic definition of fetal brain malformations.45 The next major step, which will almost certainly occur in the near future, will be the development of MRI techniques for evaluating functional gene expression. In combination with the other molecular genetic advances described, improved imaging will provide an unprecedented opportunity to understand brain development and its disorders.
Classic Neuroembryology
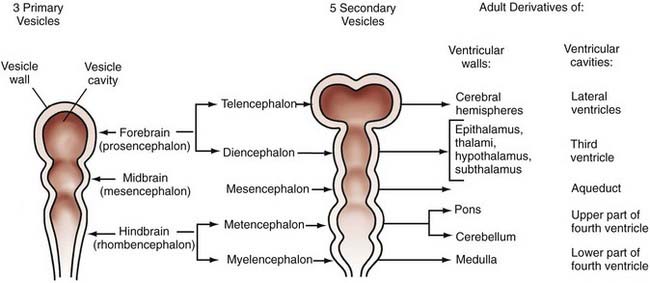
The technical meaning of the term embryology should restrict the study of development to the first 6 postconceptional weeks in humans, the embryonic period proper, but traditional use extends the term to include fetal life until birth, and this is the application that we use in this chapter. Although nonhuman embryologic studies have long been important to our understanding of brain development, meticulous descriptive morphogenic studies of serially sectioned human embryos over the past several decades have provided unique and invaluable information. Based on studies of internal and external morphology that originated from the Carnegie collection of embryos, the 8 weeks of embryonic development have been subdivided into 23 morphologic (Carnegie) stages.46 Stages 8 to 23 are relevant to neuroembryology, with the neural groove and folds first appearing in stage 8, which occurs at about 23 days’ gestation. At this time, the embryo is only about 1 mm long. No accepted morphologic staging system has been developed for the fetal period. These studies remain valuable in using known milestones in structural development to identify the termination period, or the gestational day beyond which the onset of a specific malformation could not have occurred. Knowledge of the exact times when defects such as anencephaly or meningomyelocele may occur is critical for molecular genetic and epidemiologic investigations and can provide important clues to pathophysiologic mechanisms. Further detail can be obtained from O’Rahilly and Muller’s updated classic atlas of developmental stages.46 From careful studies such as these, the adult derivatives of embryonic structures were determined long before we began to understand the signals and processing underlying their formation (Fig. 4-1).
Along with careful work over the past several decades that resulted in this embryonic staging system, similar analysis of the development of the cerebral vasculature led to a staging system usually referred to as Padgett stages.47–49 Knowledge of how the vascular system evolves leads to a clearer understanding of vascular malformations and common vascular anomalies (covered elsewhere in this book) and the patterns of secondary embryonic and fetal brain injuries and malformations. The arterial system essentially achieves an adult pattern by the end of the embryonic period, whereas the venous system develops much later in the fetal period. Although neither the seven stages of arterial evolution developed by Padgett (and still used) nor cerebral venous development will be considered further here, more information can be found in any of several excellent reviews of vascular development.50–53
Developmental Organization: Stages, Genes, and Regulatory Factors
Gastrulation
As the primitive streak extends forward, cells aggregate at one end, a collection designated the primitive node or Hensen’s node. Hensen’s node defines rostral. Cells of the epiblast on either side move toward the primitive streak, stream through it, and emerge beneath it to pass into the narrow cavity between the two sheets of cells, with the epiblast above and the hypoblast below; these migratory cells give rise to the mesoderm and endoderm internally, and some then replace the hypoblast.54
After extending about halfway across the blastoderm (epiblast), the primitive streak with Hensen’s node reverses the direction of its growth and retreats, moving posteriorly as the head fold and neural plate form anterior to Hensen’s node. As the node regresses, a notochordal process develops in the area rostral to it, and somites begin to form on either side of the notochord, with the more caudal somites differentiating first and successive ones differentiating anterior to the somites already formed. The notochord induces epiblast cells to form neuroectoderm (see “Induction”). Several genes essential in creating the fundamental architecture of the embryo and its nervous system are already expressed in the primitive node,55 and many reappear later to influence more advanced stages of ontogenesis.
Induction
Induction was discovered in 1924, when Hans Spemann and Hilde Mangold demonstrated that the dorsal lip of the newt gastrula was capable of inducing the formation of an ectopic second nervous system when transplanted to another site in a host embryo, into another individual of the same species, or to a ventral site of the same embryo.56 This dorsal lip of the amphibian gastrula, also called the Spemann organizer, is homologous with the Hensen node of embryonic birds and mammals.
The first gene isolated from the Spemann organizer was Gsc (goosecoid), which encodes a homeodomain protein (see the later section “Transcription Factors and Homeoboxes”) able to recapitulate transplantation of the dorsal lip tissue when injected into an ectopic site. It also normally induces the prechordal mesoderm and contributes to prosencephalic differentiation.55,57,58 In Hensen’s node in the chick, even before the primitive streak is fully formed, Wnt8c is expressed and is essential for the regulation of axis formation and later for hindbrain patterning in the region of the future rhombomere 4 (r4).59 The regulatory gene Cnot, with major domains in the primitive node, notochord, and prenodal and postnodal neural plate, is also involved in the induction of prechordal mesoderm and in formation of the notochord in particular.60
The specificity of induction lies not in the inductive molecule but rather in the receptor in the induced cell. This distinction is important because foreign molecules similar in structure to the natural inductor molecule may sometimes be erroneously recognized by the receptor as identical; such foreign molecules may act as teratogens if the embryo is exposed to such a toxin. Induction occurs during a very precise temporal window; the period of responsiveness of the induced cell is designated its competence, and the cell is incapable of responding before or after that precise time.61
Induction receptors are not necessarily in or on the plasma membrane of the cell but may be in the cytoplasm or in the nucleus. Retinoic acid is an example of a nuclear inducer. In some cases, the stimulus acts exclusively at the plasma membrane of target cells and does not require actual penetration of the cell.61,62 The receptors that represent the specificity of induction are also genetically programmed. Notch is a particularly important gene in regulating the competence of a cell to respond to inductive cues from within the neural tube and from surrounding embryonic tissues.63 Some mesodermal tissues, such as smooth muscle of the fetal gut, can act as mitogens on the neuroepithelium by increasing the rate of cellular proliferation,64,65 but this phenomenon is not true neural induction because the proliferating cells do not differentiate or mature. Some organizer and regulatory genes of the nervous system, such as Wnt1, also exhibit mitogenic effects,66 and insulin-like growth factor and basic fibroblast growth factor act as mitogens as well.67–69
Early formation of the neural plate is not accomplished exclusively by mitotic proliferation of neuroepithelial cells; surrounding cells are also converted to a neural fate. In amphibians, a gene known as Xash (achaete-scute) is expressed very early in the dorsal part of the embryo from the time of gastrulation and acts as a molecular switch to change the fate of undifferentiated cells to become neuroepithelium rather than surface ectodermal or mesodermal tissues.70 Some cells differentiate as specific types because they are actively inhibited from differentiating into others. All ectodermal cells are preprogrammed to form neuroepithelium, and neuroepithelial cells are preprogrammed to become neurons if not inhibited by genes that direct them along a different lineage, such as epidermal, glial, or ependymal.71–73
Neurulation
These forces arise in part from growth of the surrounding mesodermal tissues on either side of the neural tube, the future somites (Table 4-2).74 After surgical removal of mesoderm and endoderm from one side of the neuroepithelium in experimental animals, the neural tube still closes, but it is rotated and becomes asymmetric.75 The mesoderm appears to be important for orientation but not for closure of the neural tube. Expansion of the surface epithelium of the embryo is the principal extrinsic force for folding of the neuroepithelium to form the neural tube.76 Cells of the neural placode are mobile and migrate beneath the surface ectoderm, which causes the lateral margins of the placode to become raised toward the dorsal midline. Growth of the whole embryo itself does not appear to be an important factor because neurulation proceeds equally well in anamniotes (e.g., amphibians), which do not grow during this period, and in amniotes (e.g., mammals), which grow rapidly at this time.77
TABLE 4-2 Factors Involved in Closure of Neuroepithelium to Form the Neural Tube
From Menkes JH, Sarnat HB. Child Neurology, 6th ed. Philadelphia: Lippincott Williams & Wilkins; 2000:289.
Among the intrinsic forces of the neuroepithelium, the cells of the floor plate have a wedge shape—narrow at the apex and broad at the base—that facilitates bending.78 Although the width of the floor plate is small, its site in the ventral midline is crucial and sufficient to allow a significant influence. It represents yet another aspect of induction of the floor plate by the notochord, apart from its influence on the differentiation of neural cells.79 The ependymal cells that form the floor plate are the first neural cells to differentiate, and they induce growth of the parenchyma of the ventral zone more than the dorsal regions.80,81 This mechanical effect may also facilitate curving of the neural placode. The direction of proliferation of new cells in the mitotic cycle, determined in part by the orientation of the mitotic spindle, becomes another mechanical force shaping the neural tube.77,78 Adhesion molecules are also probably an important mechanical factor for neurulation. In later stages, the ependymal cell–lined central canal, which is much larger in the fetus than in the newborn, may have a role in exerting a centrifugal force to create the tubular shape, although in early spinal cord development the central canal is a tall, narrow, midline slit and only later in fetal life does it assume a rounded contour as seen in transverse sections.80
Neuroepithelial cells of the neural placode or plate downregulate the polarity of their plasma membrane so that the apical and basilar surfaces are not as distinct before neural tube closure. Cell differentiation in general involves such changes in cell polarity.82 The rostrocaudal orientation of most mitotic spindles of the neuroepithelium and the direction in which they push by the mass of daughter cells that they form also influence the shape of the neural tube (Fig. 4-2).83
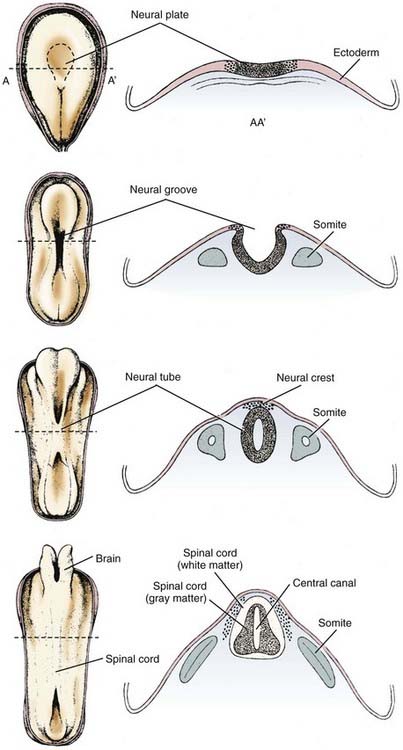
The neural tube closes in the dorsal midline first in the cervical region, with the closure then extending rostrally and caudally such that the anterior neuropore of the human embryo closes at 24 days and the posterior neuropore closes at 28 days, with the distances from the cervical region being unequal. This traditional view of a continuous zipper-like closure is an oversimplification. In the mouse embryo, the neural tube closes in the cranial region at four distinct sites, with the closure proceeding bidirectionally or unidirectionally and in general synchrony with somite formation.84,85 An intermittent pattern of anterior neural tube closure involving multiple sites has also been described in human embryos.86 In this closure pattern, the principal rostral neuropore closes bidirectionally87 to form the lamina terminalis, an essential primordium of the forebrain.80
Bending of the neural plate to form the neural tube is termed primary neurulation. Failure of the anterior neuropore to close by 24 days results in anencephaly. Because the lamina terminalis does not form, its derivatives (including the basal ganglia and other forebrain structures) do not develop. The lack of forebrain neuroectoderm results in failure of induction of the overlying mesoderm, and the cranium, meninges, and scalp fail to close in the midline.88 The term secondary neurulation refers only to the most caudal part of the spinal cord (i.e., conus medullaris), which develops from neuroepithelium caudal to the site of posterior neuropore closure. More details on abnormalities that occur because of problems with secondary neurulation are offered in other chapters in this textbook.
Neural crest cells arise from the dorsal midline of the neural tube at or shortly after the time of closure and migrate extensively along prescribed routes through the embryo to differentiate as the peripheral nervous system. This includes the dorsal root and sympathetic ganglia, adrenal medulla and carotid body chromaffin cells, melanocytes, and a few other cell types of ectodermal and mesodermal origin.89,90
Segmentation and Regionalization
Segmentation of the neural tube creates intrinsic compartments that restrict the movement of cells by physical and chemical boundaries between adjacent compartments. These embryonic compartments are known as neuromeres. The spinal cord has the appearance of a highly segmented structure; however, it is not intrinsically segmented in the embryo, fetus, or adult but rather corresponds in its entirety to the most caudal of the eight neuromeres that create the hindbrain. The apparent segmentation of the spinal cord results from clustering of nerve roots imposed by true segmentation of surrounding tissues derived from the mesoderm, tissues that form the neural arches of the vertebrae, somites, and associated structures. Neuromeres of the hindbrain are designated rhombomeres.91–94 The entire cerebellar cortex, vermis, flocculonodular lobe, and lateral hemispheres develop from rhombomere 1 (r1), with a small contribution to the anterior vermis from the mesencephalic neuromere, but the dentate and other deep cerebellar nuclei are formed in rhombomere 2 (r2).95,96 The rostral end of the neural tube forms a mesencephalic neuromere and probably six forebrain neuromeres (i.e., two diencephalic and four telencephalic prosomeres), although these may be subdivided further.97–99 The segmentation of the human embryonic brain into neuromeres is summarized in Table 4-3.
| NEUROMERE | DERIVED STRUCTURES IN MATURE CENTRAL NERVOUS SYSTEM |
|---|---|
| Rhombomere 8 (r8) | Entire spinal cord; caudal medulla oblongata; cranial nerves XI, XII |
| Rhombomere 7 (r7) | Medulla oblongata; cranial nerves IX, X; neural crest |
| Rhombomere 6 (r6) | Medulla oblongata; cranial nerves VIII, IX |
| Rhombomere 5 (r5) | Medulla oblongata; cranial nerves VI, VII; no neural crest |
| Rhombomere 4 (r4) | Medulla oblongata; cranial nervess VI, VII; neural crest |
| Rhombomere 3 (r3) | Caudal pons; cranial nerve V; no neural crest |
| Rhombomere 2 (r2) | Caudal pons; cranial nerves IV, V; cerebellar nuclei |
| Rhombomere 1 (r1) | Rostral pons; cerebellar cortex |
| Mesencephalic neuromere | Midbrain; cranial nerve III; neural crest |
| Diencephalic prosomere 2 | Dorsal diencephalons |
| Diencephalic prosomere 1 | Ventral diencephalon |
| Prosencephalic prosomere 2 | Telencephalic nuclei; olfactory bulb |
| Prosencephalic prosomere 1 | Cerebral cortex; hippocampus; corpus callosum |
From Menkes JH, Sarnat HB. Child Neurology, 6th ed. Philadelphia: Lippincott Williams & Wilkins; 2000:280.
The segments of the embryonic neural tube are distinguished by physical barriers formed by processes of early specializing cells that resemble the radial glial cells that appear later in development100,101 and by chemical barriers from secreted molecules that repel migratory cells. Cell adhesion is increased in the boundary zones between rhombomeres, which also contributes to the creation of barriers against cellular migration in the longitudinal axis. Limited mitotic proliferation of the neuroepithelium occurs in the boundary zones between rhombomeres. Although cells still divide in this zone, their nuclei remain near the ventricle during the mitotic cycle and do not move as far centrifugally within the elongated cell cytoplasm during the interkinetic gap phases as they generally do.101 The rhombomeres of the brainstem may also be visualized as a series of transverse ridges and grooves on the dorsal surface, the future floor of the fourth ventricle; these ridges are gross morphologic markers of the hindbrain compartments.94,102
The first evidence of segmentation is a boundary that separates the future mesencephalic neuromere from r1 of the hindbrain. More genes play a role in this initial segmentation of the neural tube than in any boundaries that subsequently form to separate other neuromeres. The mesencephalic-metencephalic region appears to develop early as a single, independent unit or “organizer” for other neuromeres rostral and caudal to that zone.103,104 The organizer genes recognized at the mesencephalic-metencephalic boundary for this earliest segmentation of the neural tube include Pax2, Wnt1, En1, En2, Pax5, Pax8, Otx1, Otx2, Gbx2, Nkx2-2, and Fgf8.
The earliest known gene with regional expression in the mouse is Pax2, and it is expressed even before the neural plate forms. It is the earliest gene recognized in the presumptive region of the midbrain-hindbrain boundary.105,106 In invertebrates, Pax2 is important for the activation of Wg (wingless) genes; this relationship is relevant because the first gene definitely associated with an identified midbrain-hindbrain boundary in vertebrates is Wnt1, a homologue of Wg. Regulation of Wnt1 may be divided into two phases. In the early phase (1 or 2 somites), the mesencephalon broadly expresses the gene throughout; in the later phase (15 to 20 somites), expression is restricted to the dorsal regions, the roof plate of the caudal diencephalon, the mesencephalon, the myelencephalon, and the spinal cord, but it is also expressed in a ring that extends ventrally just rostral to the midbrain-hindbrain boundary and in the ventral midline of the caudal diencephalon and mesencephalon.107–109 Wnt1 is essential in activating and preserving the function of the mouse engrailed genes En1 and En2. En1 is coexpressed with Wnt1 at the 1-somite stage in a domain only slightly caudal to Wnt1, which includes the midbrain and r1, the rostral half of the pons, and the cerebellar cortex but excludes the diencephalon.110 Activation of En2 begins at the 4-somite stage, and its function in mesencephalic and r1 development is similar, with differences in some details, particularly their roles in cerebellar development.111,112 The homeobox gene Otx2 appears early in the initial boundary zone of the midbrain-hindbrain, and as with Wnt1, it appears to be essential for the later expression of En1, En2, and Wnt1.113,114
Patterning of the Neural Tube
Development of the basic characteristics of the body plan is called patterning.91 These patterns are the anatomic expression of the genetic code within the nuclear DNA of every cell, but they may also result from signals from neighboring cells carried by molecules that are secretory translation products of various families of organizer genes, each in a highly precise and predictable temporal and spatial distribution.
Early development of the CNS in all vertebrates, even before closure of the neural placode or plate to form the neural tube, requires the establishment of a fundamental body plan of bilateral symmetry, with cephalization, or the identity of head and tail ends, and determination of the dorsal and ventral surfaces. These axes of the body itself and the CNS require the expression of genes that impose gradients of differentiation and growth. The genes that determine the polarity and gradients of the anatomic axes are called organizer genes. Many express themselves in the CNS and in other organs and tissues.54,109 The bilateral symmetry of many organs and programmed asymmetries, probably including neural structures such as the different targets of the left and right vagal nerves and left-right asymmetries in the cerebral cortex, is determined in large part by Pitx2, a gene expressed as early as in the primitive node.115 Some genes function to stimulate or inhibit the expression of others, or there is an antagonism or equilibrium between certain families of genes, as exemplified by those that exert dorsoventral or ventrodorsal gradients. The difference between an organizer gene and a regulator gene is its function, and the same gene often subserves both roles at different stages of development. The definitions and programs of these two groups are summarized in Table 4-4.
| Organizer Genes |
From Menkes JH, Sarnat HB. Child Neurology, 6th ed. Philadelphia: Lippincott Williams & Wilkins; 2000:281.
Transcription Factors and Homeoboxes
Transcription factors are proteins expressed by regulatory genes that bind to the regulatory regions of other genes and control transcription. These transcription factors are essential for functional expression of the gene, and several different protein structural motifs have been discovered to be highly evolutionarily conserved. One such motif is the basic helix-loop-helix structure, which is so fundamental to the evolution of life that it appears for the first time in certain bacteria even before evolution of a cell nucleus to concentrate the DNA.116
The zinc finger is another DNA-binding, gene-specific transcription factor motif. It consists of 28 amino acid repeats with pairs of cysteine and histidine residues and with each sequence folded around a zinc ion.117 Krox20 (this gene name is applied to the mouse; the human form is designated EGR2) is a zinc finger gene expressed in alternating rhombomeres, especially r3 and r5; neural crest tissue does not differentiate from these two rhombomeres, although it does in adjacent segments, including r4.118 Krox20 serves an additional function in the peripheral nervous system, where it regulates myelination by Schwann cells,119 and it also regulates the expression of some other genes, most notably those of the Hox family.118,120–123 Another developmentally important zinc finger gene is PLZF (human promyelocytic zinc finger). Studies of mouse and chick homologues of this gene have revealed that it is expressed in a restricted zone surrounding hindbrain rhombomere boundaries, thus suggesting an important functional role for it and other zinc finger genes in vertebrate hindbrain regionalization.124
Some transcription factor genes include homeoboxes. These restricted DNA sequences of 183 base pairs of nucleotides encode a class of proteins sharing a common or very similar 60–amino acid motif called the homeodomain.91 Homeoboxes or homeotic genes are classified into various families with common molecular structures and similar general expression during ontogenesis. They are especially associated with genes that program segmentation and the rostrocaudal gradients of the neural tube. Some of the families of homeobox genes important in development of the vertebrate nervous system are Gsc, Hox, En, Wnt, Shh, Nkx, Lim, and Otx.
Growth factors may also influence the pattern of the neural tube by behaving biologically as transcription factors: basic fibroblast growth factor behaves as an auxiliary inductor of the longitudinal axis with a rostrocaudal gradient during formation of the neural tube.125
Developmental Gene Families of the Central Nervous System
The genes that program the axes and gradients of the neural tube may be classified as families based on their similar nucleic acid sequences and their similar general functions, although important differences occur within a family in the site or neuromere where each gene is expressed and the anatomic structures that they form. A dorsalizing gene has a dorsal territory of expression and causes the ventral parts of the neural tube to differentiate as dorsal structures if influences from ventralizing genes do not antagonize them sufficiently and vice versa. In development of the somite, the sclerotome (which forms the cartilage and bone of the vertebral body) is normally situated ventral to the myotome (which forms muscle cells) and the dermatome. Ectopic cells of the floor plate or notochord implanted next to the somite of the chick embryo cause ventralization of the somite such that excess cartilage and bone are formed and there is a deficiency of muscle and dermis.126,127 The floor plate, or notochord in this instance, is the ventralizing inductor of the mesodermal somite, and this is caused by expression of Shh (Sonic Hedgehog), which also serves as a strong ventralizing gradient force in the neural tube.128–131 If a section of notochord is ectopically implanted dorsal or lateral to the neural tube, a second floor plate forms opposite the notochord, and motor neurons differentiate on either side of it despite the presence of a normal floor plate and motor neurons in the normal position.132 Shh, which is expressed as early as in the primitive node, induces ventralization of a dorsal region of the neural tube or duplicates the neural tube. Such an influence in the human fetus, the so-called split notochord, could be an explanation for the rare cases of diplomyelia or diastematomyelia.80 Excessive Shh expression, particularly its amino-terminal cleavage product, upregulates floor plate differentiation at the expense of motor neuron formation129 and may induce duplication of the neuraxis. Shh exerts a strong influence on differentiation of the ventral and medial structures of the prosencephalon,133 and defective expression of this gene has been found to be one molecular basis of human holoprosencephaly.134
To establish an equilibrium with genes with ventralizing influence, other genes exercise a dorsalizing influence. The Pax family is an example of genes that cause differentiation of the dorsal structures of the neural tube.135,136 The Wnt family is also dorsalizing in the hindbrain; in situ hybridization shows its transcription products to be expressed diffusely only in the early neuroepithelium and to be restricted to dorsal regions as the neural tube develops.137 The zinc finger gene Zic2 has a dorsalizing gradient in the forebrain. The rostrocaudal axis of the neural tube and segmentation, or the formation of neuromeres, are directed in large part by a family of 38 homeobox genes that are divided into four groups called Hox genes.92,93,138–141 Each of 13 Hox genes is expressed in certain rhombomeres and not in others (Table 4-5). Hox genes are not expressed in the forebrain. In addition to their functions in establishing the compartments or rhombomeres of the brainstem and effecting the differentiation of certain anatomic structures, Hox genes guide the growth cones forming the long descending and ascending pathways between the brain and spinal cord.158
Genes that direct the specific differentiation of structures are called regulator genes, and in many cases they have served as organizer genes in an earlier period. The most important families for development of the brainstem and midbrain in vertebrates are En, Wnt, Hox, Krox, and Pax. Table 4-5 summarizes the sites of expression and functions of selected important organizer and regulator genes. Many regulatory genes change their territories of expression in different stages of development; they can increase their territory to include more rhombomeres or have broader expression early in development and more restricted domains later. Sometimes, expression of a gene in the wrong neuromere, called ectopic expression, interferes with normal development (see Table 4-5).
Function of Retinoic Acid
Retinoic acid, the alcohol of which is vitamin A, is a hydrophobic molecule secreted by the notochord and by ependymal floor plate cells.226 Retinoid-binding proteins and receptors are already strongly expressed in mesenchymal cells of the primitive streak and in the preoptic region of the early developing hindbrain.227 Ependymal cells other than those of the floor plate and neuroepithelial cells have retinoic acid receptors but do not secrete this compound. Retinoic acid diffuses across the plasma membrane without requiring active transport and binds to intracellular receptors; it then enters the nucleus, where it binds to a specific nuclear receptor protein, changes the structural configuration of that protein, and thereby enables it to attach to a specific receptor on a target gene, where the complex formed by retinoic acid and its receptor then functions as a transcription factor for neural induction.228,229
Retinoic acid functions as a polarity molecule for determining the anterior and posterior surfaces of limb buds and, in the nervous system, is important in segmentation polarity. It produces a strong rostrocaudal polarity gradient.54,230,231 Excessive retinoic acid acts on the neural tube of amphibian tadpoles to transform anterior neural tissue to a posterior morphogenesis, which results in extreme microcephaly and suppression of optic cup formation.229 In vertebrates, optic cup formation may fail because of suppression of retinoic acid by genes of the Lim family, such as Islet3 and Lim1.160,165 Retinoic acid upregulates homeobox genes, those of the Hox family and Krox20 in particular, and can cause ectopic expression of these genes in rhombomeres where they are not normally expressed.122,232 An excess of retinoic acid, whether endogenous or exogenous (i.e., excess maternal intake of vitamin A or analogues during early gestation), results in severe malformations of the hindbrain and spinal cord. A single dose of retinoic acid administered intraperitoneally to maternal hamsters on embryonic day 9.5 results in the Chiari II malformation and frequently meningomyelocele in the fetuses.233–235 In cell cultures of cloned CNS stem cells from the mouse, retinoic acid enhances neuronal proliferation and astroglial differentiation.236
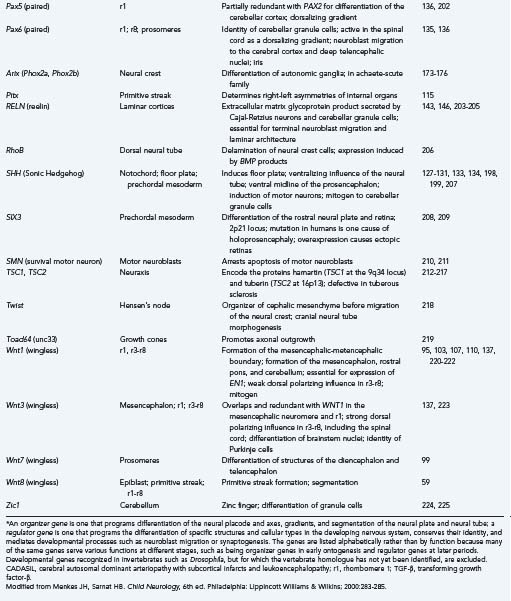
Flexures and Sulci of the Brain
The neural tube begins development as a straight structure, but further growth of surrounding structures exerts an external physical force that causes bending at precise points. The pontine and cervical flexures thus form. At 6 weeks’ gestation the telencephalic hemisphere is oval shaped, but ventral bending then occurs to create the operculum and eventually the sylvian fissure (Fig. 4-3). Because both the dorsal (frontal lobe) and ventral (temporal lobe) lips of the sylvian fissure correspond to the ventral surface of the primitive telencephalon, disorders of this region, such as schizencephaly and perisylvian syndromes of polymicrogyria at the lips of the sylvian fissure, follow a ventrodorsal genetic gradient in the vertical axis, and both lips are involved in these malformations.237
Developmental Patterns and Disorders
Holoprosencephaly
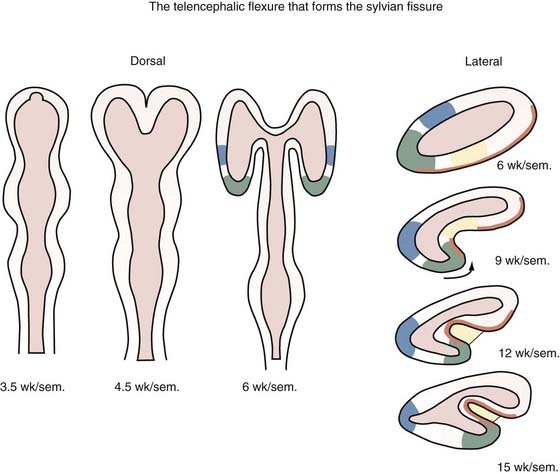
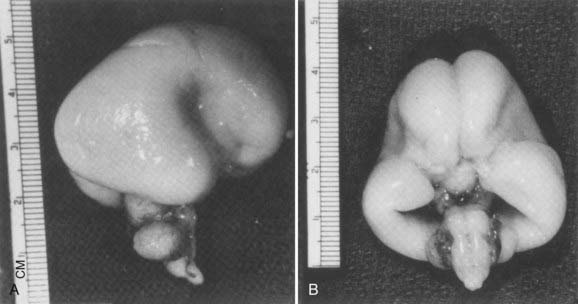
Holoprosencephaly (HPE) results from noncleavage of the midline ventral forebrain at approximately 33 days’ gestation and is classified by the degree of failure of hemispheric separation. This ventral failure of induction also leads to a wide spectrum of craniofacial defects ranging from cyclopia to milder defects such as iris colobomas, cleft palate, and single central incisors.238 In most cases, the severity of the brain abnormality parallels that of the face.239,240 Neuropathologic studies suggest that the correlation of severity between brain malformation and midfacial hypoplasia may be due to abnormalities in the expression of key developmental genes along their rostrocaudal gradients, which extend as far as the mesencephalic neuromere. These abnormally expressed genes may interfere with formation, migration, or apoptosis of the mesencephalic neural crest, which forms the membranous bones of the face, orbits, nose, and parts of the eyes.241 In alobar HPE, the most severe form, there is a single midline forebrain ventricle and a cerebral holosphere, with no separation at all into cerebral hemispheres. The interhemispheric fissure and corpus callosum are completely absent. In semilobar HPE, the defect is localized more anteriorly, and some portion of the posterior interhemispheric fissure and variable amounts of the splenium of the corpus callosum are present (Fig. 4-4). In lobar HPE, the mildest form, there is separation of most of the cerebral hemispheres, with only the most rostral aspects remaining noncleaved. The corpus callosum is absent in the region affected.
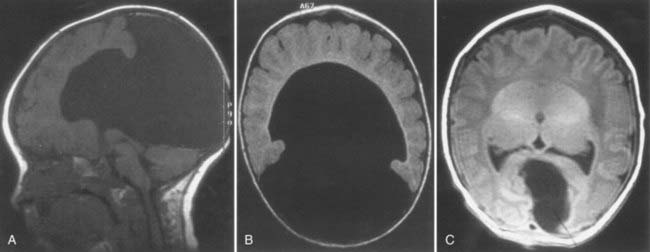
The middle interhemispheric variant, once thought to be rare,242 has in later large studies been identified quite frequently (Fig. 4-5).243 Radiologic studies have also revealed that HPE is much more variable than the simple classification system described earlier would suggest. There is greater variability in the pattern and degree of noncleavage of the deep gray nuclei than previously appreciated, with almost universal involvement of the hypothalamus.244 Thalamic noncleavage is correlated with the presence of a dorsal cyst, thus supporting the idea that the cyst arises because of blockage of rostral flow of cerebrospinal fluid, which then egresses dorsally.245,246
Human HPE is often seen as part of trisomy 13, but it has also been mapped to several other chromosomes.238 SHH was the first gene identified to cause HPE in humans.134 The active cleavage product of secreted Shh is covalently bound to cholesterol and then binds to its receptor Ptc (patched), which stops inhibiting the constitutive signaling activity of Smo (smoothened). Binding activates a cascade of molecular events that culminate in the transcription of dorsalizing genes such as Wnt and Bmp, which encode transcription factors. The Shh pathway normally keeps ventralizing and dorsalizing influences in appropriate balance. Interference with this complicated pathway explains why certain toxins cause HPE in animals.247 Mutations in two other genes, SIX3 and TGIF, can cause HPE through distinct signaling pathways.15,16 Mutations in the ZIC2 gene also cause HPE.14 ZIC2 promotes embryonic roof plate differentiation in the dorsal midline of the neural tube after closure. In mice, roof plate–specific properties such as an increased apoptotic rate and decreased mitotic rate are critical to formation of the interhemispheric fissure. Compromise of roof plate differentiation could therefore lead to HPE, and one patient with the middle interhemispheric variant has been shown to have this mutation.243
Because the telencephalic flexure fails to form in HPE, the hippocampus remains a dorsal structure and has the form of a U-shaped cord that extends across the midline and posteriorly to the caudal poles of the malformed forebrain.237
Neuronogenesis
Normal Proliferation of Neuroblasts
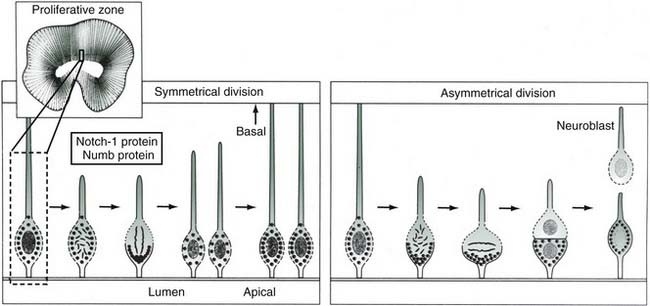
The neuroblast is histologically indistinguishable from other stem cells but is identified by its location in the primitive neural plate. After formation of the neural tube, neurons and glial cells are generated by proliferation of neuroepithelial cells in the ventricular zone with mitoses at the ventricular surface. The rate of division is greatest during the early first trimester in the spinal cord and brainstem and during the late first and early second trimester in the forebrain. Within the ventricular zone of the human fetal telencephalon, 33 mitotic cycles provide the total number of neurons required for the mature cerebral cortex. Most mitotic activity in the neuroepithelium occurs at the ventricular surface, and the orientation of the mitotic spindle determines the subsequent immediate fate of the daughter cells. If the cleavage plane is perpendicular to the ventricular surface, the two daughter cells become equal neuroepithelial cells preparing for further mitosis. If, however, the cleavage is parallel to the ventricular surface, the two daughter cells are unequal (i.e., asymmetric cleavage), and the one at the ventricular surface becomes another neuroepithelial cell, whereas the one away from the ventricular surface is separated from its ventricular attachment and becomes a postmitotic neuroblast ready to migrate to the cortical plate. These asymmetric cleavages are mediated by the products of two proteins that determine cell fate, Notch1 and Numb, which are localized to the basal and apical regions, respectively, of the neuroepithelial cell. With symmetrical cleavage, both daughter cells receive the same amount of each, but with asymmetric cleavage, the cells receive unequal ratios of each, thereby resulting in a migratory neuroblast (Fig. 4-6).195,197,248
Disorders of Neuronogenesis
Destructive processes may destroy so many neuroblasts that regeneration of the full complement of cells is impossible. This happens when the insult persists for a long time or is repetitive, with each subsequent generation of dividing cells being destroyed. Inadequate mitotic proliferation of neuroblasts results in hypoplasia of the brain. The entire brain may be affected, or portions may be selectively involved. Similarly, cerebellar hypoplasia often results from selective interference with proliferation of the external granular layer. In some cases, cerebral hypoplasia and microcephaly are the result of precocious development of the ependyma before all mitotic cycles of the neuroepithelium are complete because ependymal differentiation arrests mitotic activity at the ventricular surface.81 A mutation of a gene programming neuronogenesis may be another explanation for failure to generate sufficient neuroepithelial cells in some cases, although this pathogenesis remains hypothetical in humans.
Excessive neuroblasts are formed in every part of the nervous system by normal mitotic proliferation. This abundance is reduced by programmed cell death (i.e., apoptosis) until the definitive number of immature neurons is achieved. Apoptosis continues to play an important role in later stages of synaptogenesis and neuronal plasticity. Given the complexity of apoptotic regulation, we do not review it further here, but more information is available in several excellent reviews.249–254
Neuroblast Migration
Normal Development
No neurons of the mature human brain occupy a site in which they were generated from the neuroepithelium. They are born in the subependymal germinal matrix, the subventricular zone that develops from the lumen of the neural tube, and they migrate to their adult location along tracks of long, specialized fetal astrocytes (Figs. 4-7 and 4-8).
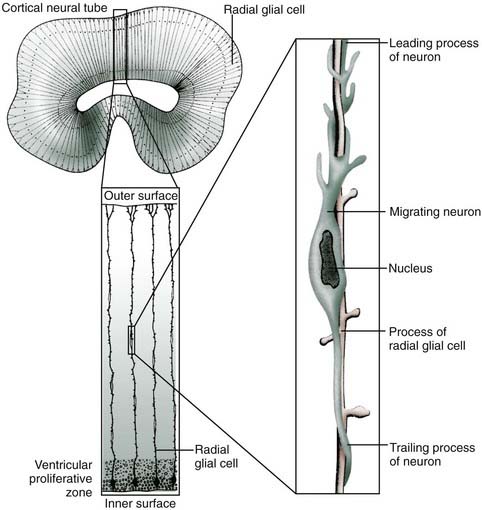
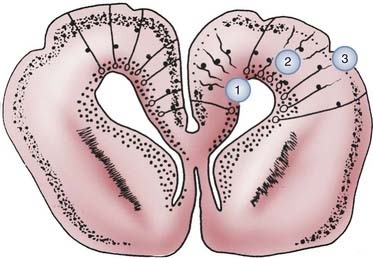
These radial glial fibers span the entire wall of the fetal cerebral hemisphere, with their cell bodies located in the periventricular region and their end-feet on the limiting pial membrane at the brain’s surface (Fig. 4-8). From about 6 to 34 weeks’ gestation, forebrain neuroblasts migrate in waves, with each successive wave moving past earlier born neurons to add a more superficial layer of cortex in an inside-out fashion. When complete, the cortex has six layers, and each contains specific types of neurons that must have successfully migrated to establish appropriate synaptic contacts with other neurons.
In addition to radial migration to the cerebral cortex, tangential migration also occurs, but the number of neuroblasts is far smaller.255,256 These migrations perpendicular to the radial fibers probably use axons rather than glial processes as guides for migratory neuroblasts, and this explains why all cells in a given region of cortex are not all from the same clone or vertical column. Neurons arriving at the neocortical plate by tangential migration are GABAergic (secreting γ-aminobutyric acid [GABA]) interneurons and represent about 10% of the total cortical neurons; they are distributed in all layers of the cortex but predominate in sensory granular cell layers 2 and 4 (Fig. 4-9). In contrast, in the cerebellar cortex, neuronal migration differs in that external granule cells first spread over the surface of the cerebellum and then migrate into the folia. They also have a much longer migratory period, which is not complete until after the first year of life.
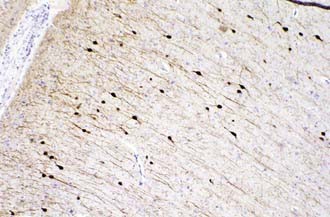
 -year-old girl. This protein is a marker of GABAergic interneurons that arrive in the cortical plate by tangential rather than radial migration. They represent about 10% of the total cortical neurons and are distributed in all layers, but predominantly in sensory layers 2 and 4 (calretinin stain, ×250).
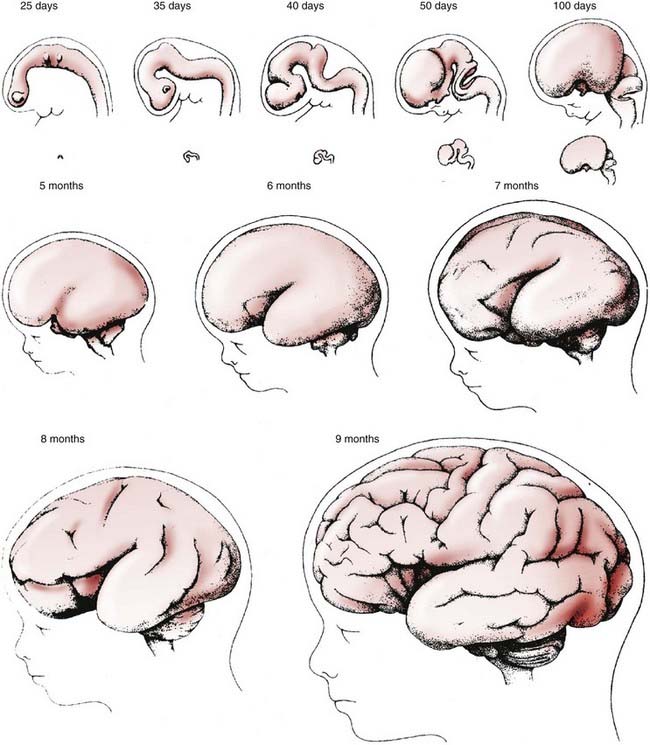
-year-old girl. This protein is a marker of GABAergic interneurons that arrive in the cortical plate by tangential rather than radial migration. They represent about 10% of the total cortical neurons and are distributed in all layers, but predominantly in sensory layers 2 and 4 (calretinin stain, ×250).The laminated structure of the mammalian cerebral cortex requires a large cortical surface area to accommodate increasing numbers of migrating neuroblasts and glioblasts. Convolutions provide this large surface area without incurring a concomitant increase in cerebral volume. The formation of gyri and sulci is a direct result of migration. Because most gyri form in the second half of gestation, a period of predominantly gliogenesis and glial cell migration, the proliferation of glia in the cortex and subcortical white matter may be more important than neuroblast migration in the formation of convolutions, but the growth of dendrites and synaptogenesis also probably influence gyration by contributing mass to the neuropil. The timing and sequence of gyral formation in the human brain are as predictable as other aspects of cerebral maturation (Figs. 4-10 and 4-11).
Disorders of Neuroblast Migration
Lissencephaly is the condition of a smooth cerebral cortex without convolutions (Fig. 4-12). At midgestation, the brain is normally smooth, with only the interhemispheric, sylvian, and calcarine fissures formed. Gyri and sulci develop between 20 and 36 weeks’ gestation, and the mature pattern of gyration is evident at term. In lissencephaly type 1, the cerebral cortex remains smooth, and the histopathologic pattern is that of a four-layer cortex in which layer 2 corresponds to layers 2 through 6 of normal neocortex, layer 3 represents a persistent fetal subplate zone and is cell sparse, and layer 4 consists of incompletely migrated neurons in the subcortical intermediate zone. In lissencephaly type 2, poorly laminated cortex with disorganized and disoriented neurons is seen histologically, and the gross appearance of the cerebrum is that of a smooth brain or a few, poorly formed sulci. The cerebral mantle may be thin and suggest a disturbance in cell proliferation and neuroblast migration. Malformations of the brainstem and cerebellum are often present. Lissencephaly frequently has a genetic origin, but it may result from nongenetic disturbances in neuroepithelial proliferation or neuroblast migration, including destructive, encephaloclastic processes such as congenital infections during fetal life.
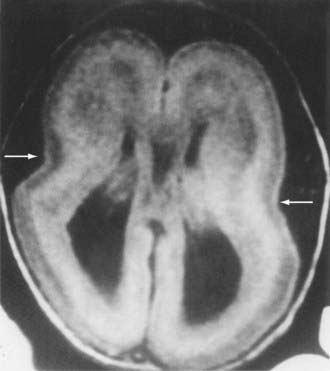
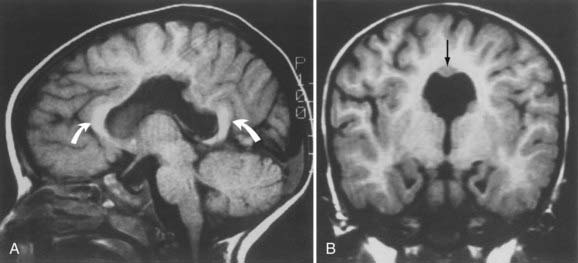
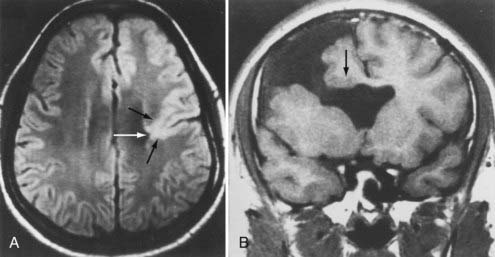
Mutations in two genes, LIS1 and DCX, have been associated with many cases of classic lissencephaly.19–21,33–35 They both appear to disrupt migration by interfering with microtubule function. Neuroimaging studies of gyral patterns in individuals with these mutations have shown distinct radiologic patterns, with more severe malformation posteriorly in individuals with LIS1 mutations and more severe malformation anteriorly in those with DCX mutations.257 DCX mutations result in classic X-linked lissencephaly in boys but cause a syndrome called double cortex or subcortical band heterotopia in girls.33 Through random X-chromosome inactivation early in embryogenesis, half of an affected girl’s cortical neurons inactivate the mutant allele, and half inactivate the normal allele. This population migrates abnormally and produces band heterotopia (Fig. 4-13).
Mutations in FCMD, which encodes a putative extracellular matrix molecule called fukutin, cause Fukuyama’s congenital muscular dystrophy, which includes lissencephaly.22 Mutation in RELN, the human gene encoding reelin, has been identified as a cause of lissencephaly with cerebellar hypoplasia.
Periventricular heterotopia (i.e., bilateral periventricular nodular heterotopia) differs from lissencephaly in that migration fails completely rather than being interrupted. The causative gene is FLNA (formerly FLN1), which encodes an actin-binding protein called filamin.26 Further molecular details of these complex disorders can be found in the outstanding review articles by Ross and Walsh258 and by Clark.259
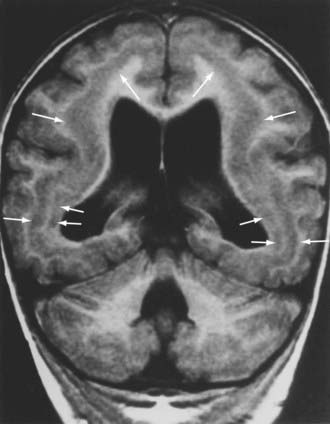
The term pachygyria signifies abnormally large, poorly formed gyri that may be isolated or associated with frankly lissencephalic regions elsewhere. Polymicrogyria refers to excessively numerous and abnormally small gyri that may coexist with pachygyria. Polymicrogyria does not necessarily denote a primary migratory disorder of genetic origin; small and poorly formed gyri may occur in zones of fetal ischemia and regularly surround porencephalic cysts caused by occlusion of the middle cerebral artery in fetal life. Schizencephaly is a unilateral or bilateral deep cleft encompassing the full thickness of the hemispheric wall between the meninges and the lateral ventricle (i.e., pial-ependymal seam). If the cerebral cortical walls on either side of the deep cleft are in contact, the condition is called closed lip, and if a wide subarachnoid space separates the two walls, it is called open lip (Fig. 4-14). Mutations in EMX2, a homeobox gene expressed in the developing forebrain, were initially identified in a small number of patients with severe schizencephaly,31 but genetic studies of a large cohort of patients with this malformation did not confirm this genetic defect.260 Because the telencephalic flexure results in both the dorsal and ventral lips of the sylvian fissure being derived from the ventral part of the primitive telencephalon, a genetic defect in schizencephaly would have to follow a ventrodorsal gradient in the vertical axis.237 Some cases may instead represent a form of porencephaly caused by fetal infarction, and in other cases, the cause of unilateral schizencephaly may be asymmetric mechanical stress in utero without implicating a genetic mechanism.237
Anatomic lesions such as periventricular leukomalacia, intracerebral hemorrhages and abscesses, hydrocephalus, and traumatic injuries may disrupt the delicate radial glial guide fibers and prevent normal migration, even though the migrating cell itself may escape the focally destructive lesion (Fig. 4-15).
The result may be the development of heterotopic nodules, with neurons of the various cortical types differentiating without laminar organization and with haphazard orientations of their processes. Postmigration CNS development includes the highly regulated and complex organizational processes of neurite outgrowth and synaptogenesis. Because of limited space, discussion of these nodules is not included in this chapter, but more information may be found in Volpe’s thorough introduction to these processes.261
Synaptogenesis
Formation of synapses within the CNS follows as precise a temporal and spatial distribution as other developmental processes do. Neocortical synaptogenesis is indirectly reflected in maturation of the electroencephalogram in preterm infants but represents large fields of thousands of synapses. In tissue sections of fetal and neonatal brain at autopsy, an immunocytochemical marker of the synaptic vesicle protein synaptophysin can be used to demonstrate this process in individual neurons.262 Synaptophysin is nonspecific for the type of synapse—excitatory or inhibitory, axodendritic or axosomatic—or the type of neurotransmitter that the vesicles contain but shows the overall pattern of synapse formation and can be combined with other immunoreactivities to define this specificity. Synaptogenesis requires the terminal axonal projection with mature synaptic vesicles, a postsynaptic membrane with receptors to respond to the chemical stimulus of depolarization, and maturity of the energy-producing Na+,K+-ATPase system to maintain a resting membrane potential and electrical excitability of the plasma membrane.
Myelination
Myelination is the process of acquiring a specialized myelin membrane around axons, which is elaborated from the plasma membranes of adjacent oligodendrocytes in the CNS and Schwann cells in the peripheral nervous system (Tables 4-6 and 4-7). As with the other processes of nervous system development, programming of differentiation of these myelin-producing cells and myelin formation is under precise genetic regulation.263,264 Myelination cycles are specific for each tract and occur during precise time windows. The overall period for myelination is very long; it begins as early as 16 weeks’ gestation and continues in some areas past 30 years of age.
TABLE 4-6 Myelination Cycles in the Human Central Nervous System Based on Myelin Tissue Stains
| PATHWAY* | BEGINS† | COMPLETED† |
|---|---|---|
| Fetal Onset | ||
| Spinal motor roots | 16 wk | 42 wk |
| Cranial motor nerves III, IV, V, VI | 20 wk | 28 wk |
| Spinal sensory roots | 20 wk | 5 mo |
| Medial longitudinal fasciculus | 24 wk | 28 wk |
| Acoustic nerve | 24 wk | 36 wk |
| Ventral commissure, spinal cord | 24 wk | 4 mo |
| Trapezoid body and lateral lemniscus | 25 wk | 36 wk |
| Inferior cerebellar peduncle (inner part) | 26 wk | 36 wk |
| Inferior cerebellar peduncle (outer part) | 32 wk | 4 mo |
| Superior cerebellar peduncle | 28 wk | 6 mo |
| Middle cerebellar peduncle (pontocerebellar) | 42 wk | 3 yr |
| Habenulopeduncular tract | 28 wk | 34 wk |
| Dorsal columns, spinal cord | 28 wk | 36 wk |
| Ansa reticularis | 28 wk | 8 mo |
| Medial lemniscus | 32 wk | 12 mo |
| Optic nerve | 38 wk | 6 mo |
| Corticospinal (pyramidal) tract | 38 wk | 2 yr |
| Optic radiations (geniculocalcarine) | 40 wk | 6 mo |
| Acoustic radiations (thalamocortical) | 40 wk | 3 yr |
| Postnatal Onset | ||
| Fornix | 2 mo | 2 yr |
| Thalamocortical radiations | 2 mo | 7 yr |
| Corpus callosum | 2 mo | 17 yr |
| Ipsilateral intracortical association fibers, frontotemporal and frontoparietal | 3 mo | 32 yr |
| Mamillothalamic tract | 8 mo | 6 yr |
* Myelination as determined by light microscopy using Luxol fast blue and other myelin stains.
† Gestational age is stated in weeks; postnatal age is stated in months and years.
From Sarnat HB. Cerebral Dysgenesis: Embryology and Clinical Expression. New York: Oxford University Press; 1992:61.
TABLE 4-7 Appearance of Myelination by Magnetic Resonance Imaging
| ANATOMIC REGION | T1WI (BRIGHT)* | T2WI (DARK) |
|---|---|---|
| Middle cerebellar peduncle | Birth | Birth-2 mo |
| Cerebellar white matter | Birth-4 mo | 3-5 mo |
| Posterior limb, internal capsule | ||
| Anterior portion | Birth-1 mo | 4-7 mo |
| Posterior portion | Birth | Birth-2 mo |
| Anterior limb, internal capsule | 2-3 mo | 7-11 mo |
| Genu, corpus callosum | 4-6 mo | 5-8 mo |
| Splenium, corpus callosum | 3-4 mo | 4-6 mo |
| Occipital white matter | ||
| Central | 3-5 mo | 9-14 mo |
| Peripheral | 4-7 mo | 11-15 mo |
| Frontal white matter | ||
| Central | 3-6 mo | 11-16 mo |
| Peripheral | 7-11 mo | 14-18 mo |
| Centrum semiovale | 2-6 mo | 7-11 mo |
* From birth to 3 months, the pattern on T1-weighted imaging (T1WI) is opposite that of the adult brain (gray matter appears brighter than white matter). The adult pattern is achieved by 2 years of age. It is not clear why myelination changes on T1WI and T2-weighted imaging (T2WI) occur at different times. T1WI is most useful for assessing myelination in the first year. Myelination generally progresses from caudad to cephalad, from dorsal to ventral, and from central to peripheral. The changes in signal reflect mainly increased bound water in myelin, which results in shortening of T1 (increased brightness on T1WI).
From Barkovich AJ. Pediatric Neuroimaging, 3rd ed. Philadelphia: Lippincott Williams & Wilkins; 2000:39.
Chronic hypoxia in premature infants is probably the most common cause of delayed myelination and contributes to delays in clinical neurological maturation.265 Unlike disorders of neuroblast migration, delay in myelination is not necessarily irreversible, and if the insult is removed, the process may catch up to the appropriate level of maturity over time.
Conclusion
Our growing understanding of the molecular genetic details of normal and abnormal brain development has made it clear that classification systems for disorders based on morphology or genetic mutations are inadequate. They should instead incorporate patterns of gene expression and detailed radiologic and clinical information.266,267 It is our hope that such approaches will foster collaborations among clinicians and scientists that will lead to a better understanding of brain malformations and creation of the tools to prevent, treat, and cure them.
Back SA, Volpe JJ. Cellular and molecular pathogenesis of periventricular white matter injury. Ment Retard Dev Disabil Res Rev. 1997;3:96-107.
Barkovich AJ, Kuzniecky RI, Jackson GD, et al. Classification system for malformations of cortical development: update 2001. Neurology. 2001;57:2168-2178.
Barkovich AJ, Quint DJ. Middle interhemispheric fusion: an unusual variant of holoprosencephaly. AJNR Am J Neuroradiol. 1993;14:431-440.
Chao MV, Bothwell M. Neurotrophins: to cleave or not to cleave. Neuron. 2002;33:9-12.
Chong SS, Pack SD, Roschke AV, et al. A revision of the lissencephaly and Miller-Dieker syndrome critical regions in chromosome 17p13.3. Hum Mol Genet. 1997;6:147-155.
Clark GD. Cerebral gyral dysplasias: molecular genetics and cell biology. Curr Opin Neurol. 2001;14:157-162.
de la Rosa EF, de Pablo F. Cell death in early neural development: beyond the neurotrophic theory. Trends Neurosci. 2000;23:454-458.
DeMyer W, Zeman W, Palmer CG. The face predicts the brain: diagnostic significance of median facial anomalies for holoprosencephaly (arhinencephaly). Pediatrics. 1964;34:256-263.
Dobyns WB, Truwit CL, Ross ME, et al. Differences in the gyral pattern distinguish chromosome 17–linked and X-linked lissencephaly. Neurology. 1999;53:270-277.
Eksioglu YZ, Scheffer IE, Cardena P, et al. Periventricular heterotopia: an X-linked dominant epilepsy locus causing aberrant cerebral cortical development. Neuron. 1996;16:77-87.
Flores-Sarnat L, Sarnat HB. Axes and gradients of the neural tube for a morphological/molecular genetic classification of central nervous system malformations. In: Sarnat HB, Curatolo P, editors. Handbook of Clinical Neurology. Vol. 87: Malformations of the Nervous System. London: Elsevier; 2008:4-11.
Hong SE, Shugart YY, Huang DT, et al. Autosomal recessive lissencephaly with cerebellar hypoplasia is associated with human RELN mutations. Nat Genet. 2000;26:93-96.
Incardona JP, Roelink H. The role of cholesterol in Shh signaling and teratogen-induced holoprosencephaly. Cell Mol Life Sci. 2000;57:1709-1719.
Kobayashi K, Nakahori Y, Miyake M, et al. An ancient retrotransposal insertion causes Fukuyama-type congenital muscular dystrophy. Nature. 1998;394:388-392.
Kuan CY, Roth KA, Flavell RA, et al. Mechanisms of programmed cell death in the developing brain. Trends Neurosci. 2000;23:291-297.
Lemke G. The molecular genetics of myelination: an update. Glia. 1993;7:263-271.
Lo Nigro C, Chong SS, Smith ACM, et al. Point mutations and an intragenic deletion in LIS1, the lissencephaly causative gene in isolated lissencephaly sequence and Miller-Dieker syndrome. Hum Mol Genet. 1997;6:157-164.
Miller FD, Kaplan DR. Neurotrophin signalling pathways regulating neuronal apoptosis. Cell Mol Life Sci. 2001;58:1045-1053.
Mione MC, Cavanaugh JF, Harris B, et al. Cell fate specification and symmetrical/asymmetrical divisions in the developing cerebral cortex. J Neurosci. 1997;17:2018-2029.
O’Rourke NA, Sullivan DP, Kaznowski CE, et al. Tangential migration of neurons in the developing cerebral cortex. Development. 1995;121:2165-2176.
Rakic P. Radial versus tangential migration of neuronal clones in the developing cerebral cortex. Proc Natl Acad Sci U S A. 1995;92:11323-11327.
Ross ME, Walsh CA. Human brain malformations and their lessons for neuronal migration. Annu Rev Neurosci. 2001;24:1041-1070.
Roth KA, D’Sa C. Apoptosis and brain development. Ment Retard Dev Disabil Res Rev. 2001;7:261-266.
Sarnat HB, Benjamin DR, Siebert JR, et al. Agenesis of the mesencephalon and metencephalon with cerebellar hypoplasia: putative mutation in the EN2 gene—report of 2 cases in early infancy. Pediatr Dev Pathol. 2002;5:54-68.
Sarnat HB, Curatolo P, editors. Handbook of Clinical Neurology, Vol. 87. Edinburgh: Elsevier, 2009. Series 3: Malformations of the Nervous System
Sarnat HB, Flores-Sarnat L, Trevenen CL. Synaptophysin immunoreactivity in the human hippocampus and neocortex from 6 to 41 weeks of gestation. J Neuropathol Exp Neurol. 2010;69:234-235.
Sarnat HB, Flores-Sarnat L. The telencephalic flexure and developmental disorders of the Sylvian fissure. 2010, submitted.
Sarnat HB, Flores-Sarnat L. Neuropathologic research strategies in holoprosencephaly. J Child Neurol. 2001;16:918-931.
Simon EM, Barkovich AJ. Holoprosencephaly: new concepts. Magn Reson Imaging Clin N Am. 2001;9:149-164. viii-ix
Simon EM, Hevner RF, Pinter JD, et al. The middle interhemispheric variant of holoprosencephaly. AJNR Am J Neuroradiol. 2002;23:151-156.
Simon EM, Hevner RF, Pinter JD, et al. The dorsal cyst in holoprosencephaly and the role of the thalamus in its formation. Neuroradiology. 2001;43:787-791.
Simon EM, Hevner RF, Pinter JD, et al. Assessment of the deep gray nuclei in holoprosencephaly. AJNR Am J Neuroradiol. 2000;21:1955-1961.
Tietjen I, Bodell A, Apse K, et al. Comprehensive EMX2 genotyping of a large schizencephaly case series. Am J Med Genet. 2007;143:1313-1316.
Volpe JJ. Neuronal proliferation, migration, organization and myelination. Neurology of the Newborn. 4th ed. Philadelphia: WB Saunders. 2001. 45-99
Yu WP, Collarini EJ, Pringle NP, et al. Embryonic expression of myelin genes: evidence for a focal source of oligodendrocyte precursors in the ventricular zone of the neural tube. Neuron. 1994;12:1353-1362.
Yuan J, Yankner BA. Apoptosis in the nervous system. Nature. 2000;407:802-809.
1 International Human Genome Sequencing Consortium. Initial sequencing and analysis of the human genome. Nature. 2001;409:860-921.
2 Venter JC, Adams MD, Myers EW, et al. The sequence of the human genome. Science. 2001;291:1304-1351.
3 Bird TD. Thoughts on the relationship of the human genome project to neurology. Arch Neurol. 2001;58:1764-1765.
4 Rosenberg RN. Genomic neurology: a new beginning. Arch Neurol. 2001;58:1739-1741.
5 Husi H, Grant SGN. Proteomics of the nervous system. Trends Neurosci. 2001;24:259-266.
6 Cravchik A, Subramanian G, Broder S, et al. Sequence analysis of the human genome: implications for the understanding of nervous system function and disease. Arch Neurol. 2001;58:1772-1778.
7 Dobyns WB. Introduction to Genetics. In: Swaiman KF, Ashwal S, editors. Pediatric Neurology: Principles and Practice. 3rd ed. St. Louis: CV Mosby; 1999:325-353.
8 Bird TD, Tapscott SJ. Clinical Neurogenetics. In: Bradley WG, Daroff RB, Fenichel GM, et al, editors. Neurology in Clinical Practice. Boston: Butterworth-Heinemann; 2000:777-803.
9 Qin J, Mizuguchi M, Itoh M, et al. Immunohistochemical expression of doublecortin in the human cerebrum: comparison of normal development and neuronal migration disorders. Brain Res. 2000;863:225-232.
10 Tsuru A, Mizuguchi M, Uyemura K, et al. Immunohistochemical expression of cell adhesion molecule L1 in hemimegalencephaly. Pediatr Neurol. 1997;16:45-49.
11 Gurrieri F, Trask BJ, van den Engh G, et al. Physical mapping of the holoprosencephaly critical region on chromosome 7q36. Nat Genet. 1993;3:247-251.
12 Roessler E, Belloni E, Gaudenz K, et al. Mutations in the human Sonic hedgehog gene cause holoprosencephaly. Nat Genet. 1996;14:357-360.
13 Vance GH, Nickerson C, Sarnat L, et al. Molecular cytogenetic analysis of patients with holoprosencephaly and structural rearrangements of 7q. Am J Med Genet. 1998;76:51-57.
14 Brown SA, Warburton D, Brown LY, et al. Holoprosencephaly due to mutations in ZIC2, a homologue of Drosophila oddpaired. Nat Genet. 1998;20:180-183.
15 Wallis DE, Roessler E, Hehr U, et al. Mutations in the homeodomain of the human SIX3 gene cause holoprosencephaly. Nat Genet. 1999;22:196-198.
16 Gripp KW, Wotton D, Edwards MC, et al. Mutations in TGIF cause holoprosencephaly and link NODAL signalling to human neural axis determination. Nat Genet. 2000;25:205-208.
17 Franco B, Guioli S, Pragliola A, et al. A gene deleted in Kallmann’s syndrome shares homology with neural cell adhesion and axonal path-finding molecules. Nature. 1991;353:529-536.
18 Legouis R, Hardelin JP, Levilliers J, et al. The candidate gene for the X-linked Kallmann syndrome encodes a protein related to adhesion molecules. Cell. 1991;67:423-435.
19 Dobyns WB, Reiner O, Carrozzo R, et al. Lissencephaly: a human brain malformation associated with deletion of the LIS1 gene located at chromosome 17p13. JAMA. 1993;270:2838-2842.
20 Chong SS, Pack SD, Roschke AV, et al. A revision of the lissencephaly and Miller-Dieker syndrome critical regions in chromosome 17p13.3. Hum Mol Genet. 1997;6:147-155.
21 Lo Nigro C, Chong SS, Smith ACM, et al. Point mutations and an intragenic deletion in LIS1, the lissencephaly causative gene in isolated lissencephaly sequence and Miller-Dieker syndrome. Hum Mol Genet. 1997;6:157-164.
22 Kobayashi K, Nakahori Y, Miyake M, et al. An ancient retrotransposal insertion causes Fukuyama-type congenital muscular dystrophy. Nature. 1998;394:388-392.
23 Hong SE, Shugart YY, Huang DT, et al. Autosomal recessive lissencephaly with cerebellar hypoplasia is associated with human RELN mutations. Nat Genet. 2000;26:93-96.
24 Sarnat HB, Benjamin DR, Siebert JR, et al. Agenesis of the mesencephalon and metencephalon with cerebellar hypoplasia: putative mutation in the EN2 gene—report of 2 cases in early infancy. Pediatr Dev Pathol. 2002;5:54-68.
25 Eksioglu YZ, Scheffer IE, Cardena P, et al. Periventricular heterotopia: an X-linked dominant epilepsy locus causing aberrant cerebral cortical development. Neuron. 1996;16:77-87.
26 Fox JW, Lamperti ED, Eksioglu YZ, et al. Mutations in filamin 1 prevent migration of cerebral cortical neurons in human periventricular heterotopia. Neuron. 1998;21:1315-1325.
27 Amir RE, Van den Veyver IB, Schultz R, et al. Influence of mutation type and X chromosome inactivation on Rett syndrome phenotypes. Ann Neurol. 2000;47:670-679.
28 Lynch SA, Bond PM, Copp AJ, et al. A gene for autosomal dominant sacral agenesis maps to the holoprosencephaly region at 7q36. Nat Genet. 1995;11:93-95.
29 Savage NM, Maclachlan NA, Joyce CA, et al. Isolated sacral agenesis in a fetus monosomic for 7q36.1(qter). J Med Genet. 1997;34:866-868.
30 Ross AJ, Ruiz-Perez V, Wang Y, et al. A homeobox gene, HLXB9, is the major locus for dominantly inherited sacral agenesis. Nat Genet. 1998;20:358-361.
31 Faina GT, Cardini FA, D’Incerti L, et al. Familial schizencephaly associated with EMX2 mutation. Neurology. 1997;48:1403-1406.
32 Dattani MT, Martinez-Barbera JP, Thomas PQ, et al. Mutations in the homeobox gene HESX1/Hesx1 associated with septooptic dysplasia in human and mouse. Nat Genet. 1998;19:125-133.
33 Gleeson JG, Allen KM, Fox JW, et al. Doublecortin, a brain-specific gene mutated in human X-linked lissencephaly and double cortex syndrome, encodes a putative signaling protein. Cell. 1998;92:63-72.
34 Gleeson JG, Minnerath SR, Fox JW, et al. Characterization of mutations in the gene doublecortin in patients with double cortex syndrome. Ann Neurol. 1999;45:146-153.
35 des Portes V, Pinard JM, Billuart P, et al. A novel CNS gene required for neuronal migration and involved in X-linked subcortical laminar heterotopia and lissencephaly syndrome. Cell. 1998;92:51-61.
36 Menchine M, Emeline JK, Mischel PS, et al. Tissue and cell-type specific expression of the tuberous sclerosis gene, TSC2, in human tissues. Mod Pathol. 1996;9:1071-1080.
37 Wienecke R, Maize JCJr, Reed JA, et al. Expression of the TSC2 product tuberin and its target Rap1 in normal human tissues. Am J Pathol. 1997;150:43-50.
38 van Slegtenhorst M, de Hoogt R, Hermans C, et al. Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science. 1997;277:805-808.
39 Ali JB, Sepp T, Ward S, et al. Mutations in the TSC1 gene account for a minority of patients with tuberous sclerosis. J Med Genet. 1998;35:969-972.
40 Au KS, Rodriguez JA, Finch JL, et al. Germ-line mutational analysis of the TSC2 gene in 90 tuberous sclerosis patients. Am J Hum Genet. 1998;62:286-294.
41 Kwiatkowska J, Jozwiak S, Hall F, et al. Comprehensive mutational analysis of the TSC1 gene: observations on frequency of mutation, associated features, and non-penetrance. Ann Hum Genet. 1998;18:9365-9375.
42 Jouet M, Kenwrick S. Gene analysis of L1 neural cell adhesion molecule in prenatal diagnosis of hydrocephalus. Lancet. 1995;345:161-162.
43 Graf WD, Born DE, Sarnat HB. The pachygyria-polymicrogyria spectrum of cortical dysplasia in X-linked hydrocephalus. Eur J Pediatr Surg. 1998;8(suppl 1):10-14.
44 Kenwrick S, Watkins A, De Angelis E. Neural cell recognition molecule L1: relating biological complexity to human disease mutations. Hum Mol Genet. 2000;9:879-886.
45 Simon EM, Goldstein RB, Coakley FV, et al. Fast MR imaging of fetal CNS anomalies in utero. AJNR Am J Neuroradiol. 2000;21:1688-1698.
46 O’Rahilly R, Muller F. The Embryonic Human Brain: An Atlas of Developmental Stages, 2nd ed. New York: Wiley-Liss; 1999.
47 Streeter G. The developmental alterations in the vascular system of the brain in the human embryo. Contrib Embryol Carneg Inst. 1918;8:5-8.
48 Padgett DH. The development of the cranial arteries in the human embryo. Contrib Embryol Carneg Inst. 1948;32:205-261.
49 Padgett DH. The development of the cranial venous system in man, from the viewpoint of comparative anatomy. Contrib Embryol Carneg Inst. 1957;36:79-140.
50 Truwit CL. Embryology of the cerebral vasculature. Neuroimaging Clin N Am. 1994;4:663-689.
51 Macdonald RL, Johns LM. Developmental vascular embryology of the central nervous system. In: Jafar JJ, Awad IA, Rosenwasser RH, editors. Vascular Malformations of the Central Nervous System. Philadelphia: Lippincott Williams & Wilkins; 1999:47-60.
52 Roach ES, Riela AR. Development of the cerebral vasculature. In: Roach ES, Riela AR, editors. Pediatric Cerebrovascular Disorders. Armonk, NY: Futura; 1995:13-33.
53 Truwit CL, Barkovich AJ. Disorders of brain development. In: Atlas SW, editor. MRI of the Brain and Spine. Philadelphia: Lippincott-Raven; 1996:179-264.
54 Wolpert L, Beddington R, Brocken J, et al. Principles of Development. Oxford: Oxford University Press; 1998.
55 De Robertis EM, Kim S, Leyns L, et al. Patterning by genes expressed in Spemann’s organizer. Cold Spring Harb Symp Quant Biol. 1997;62:169-175.
56 Spemann H, Mangold H. Über Induktion von Embryonalanlagen durch Implantation aftfremder Organisatoren. Wilhelm Roux Arch Entwick. 1924;100:599-638.
57 Cho KWY, Blumberg B, Steinbeisser H, et al. Molecular nature of Spemann’s organizer: the role of the Xenopus homeobox gene goosecoid. Cell. 1991;67:1111-1120.
58 Wakamiya M, Rivera-Pérez JA, Baldini A, et al. Goosecoid and Goosecoid-related genes in mouse embryogenesis. Cold Spring Harb Symp Quant Biol. 1997;62:145-149.
59 Hume CR, Dodd J. Cwnt-8C: a novel Wnt gene with a potential role in primitive streak formation and hindbrain organization. Development. 1993;119:1147-1160.
60 Stein S, Kessel M. A homeobox gene involved in node, notochord and neural plate formation of chick embryos. Mech Dev. 1995;49:37-48.
61 Duprat AM, Gualandris L, Kan P, et al. Review: neural induction. Arch Anat Microsc Morphol Exp. 1987;75:211-227.
62 Tiedemann H. The molecular mechanism of neural induction: neural differentiation of Triturus ectoderm exposed to HEPES buffer. Roux Arch Dev Biol. 1986;195:399-402.
63 Fortini ME, Artavanis-Tsakonas S. Notch: neurogenesis is only part of the picture. Cell. 1993;75:1245-1247.
64 Fontaine-Pérus JC, Chanconie M, Le Douarin NM, et al. Mitogenic effect of muscle on the neuroepithelium of the developing spinal cord. Development. 1989;107:413-422.
65 Fontaine-Pérus J. Migration of crest-derived cells from gut: gut influences on spinal cord development. Brain Res Bull. 1993;30:251-255.
66 Dickson ME, Krumlauf R, McMahon AP. Evidence for a mitogenic effect of Wnt-1 in the developing mammalian central nervous system. Development. 1994;120:1453-1471.
67 DiCicco-Bloom E, Black IB. Insulin growth factors regulate the mitotic cycle in cultured rat sympathetic neuroblasts. Proc Natl Acad Sci U S A. 1988;85:4066-4070.
68 Tao Y, Black IB, DiCicco-Bloom E. Neurogenesis in neonatal rat brain is regulated by peripheral injection of basic fibroblast growth factor (bFGF). J Comp Neurol. 1996;376:653-663.
69 Tao Y, Black IB, DiCicco-Bloom E. In vivo neurogenesis is inhibited by neutralizing antibody to basic fibroblast growth factor. J Neurobiol. 1997;33:289-296.
70 Turner DL, Weintraub H. Expression of achaete-scute homolog 3 in Xenopus embryos converts ectodermal cells to a neural fate. Genes Dev. 1994;8:1434-1447.
71 Anderson DJ. A molecular switch for the neuron-glia developmental decision. Neuron. 1995;15:1219-1222.
72 Hemmati-Brivantou A, Melton D. Vertebrate embryonic cells will become nerve cells unless told otherwise. Cell. 1997;88:13-17.
73 Tanabe Y, Jessell TM. Diversity and pattern in the developing spinal cord. Science. 1996;274:1115-1123.
74 Smith JL, Schoenwolf GC. Neurulation: coming to closure. Trends Neurosci. 1997;20:510-517.
75 Schoenwolf GC, Smith JL. Mechanisms of neurulation: traditional viewpoint and recent advances. Development. 1990;109:243-270.
76 Alvarez I, Schoenwolf GC. Expansion of surface epithelium provides the major extrinsic force for bending of the neural plate. J Exp Zool. 1992;261:3404-3408.
77 Jacobson AG. Experimental analysis of the shaping of the neural plate and tube. Am Zool. 1991;31:628-643.
78 Schoenwolf GC, Franks MV. Quantitative analyses of changes in cell shapes during bending of the avian neural plate. Dev Biol. 1984;105:257-272.
79 Smith J, Schoenwolf GC. Notochordal induction of cell wedging in the chick neural plate and its role in neural tube formation. J Exp Zool. 1989;25:49-62.
80 Sarnat HB. Cerebral Dysgenesis: Embryology and Clinical Expression. New York: Oxford University Press; 1992. 61
81 Sarnat HB. Role of the human fetal ependyma. Pediatr Neurol. 1992;8:163-178.
82 Aaku-Saraste E, Oback B, Hellwig A, et al. Neuroepithelial cells downregulate their plasma membrane polarity prior to neural tube closure and neurogenesis. Mech Dev. 1997;69:71-78.
83 Sausedo RA, Smith JL, Schoenwolf GC. Role of randomly oriented cell division in shaping and bending of the neural plate. J Comp Neurol. 1997;381:473-488.
84 Juriloff DM, Harris JM, Tom C, et al. Normal mouse strains differ in the site of initiation of closure of the cranial neural tube. Teratology. 1991;44:225-233.
85 Golden JA, Chernoff GF. Intermittent pattern of neural tube closure in two strains of mice. Teratology. 1993;47:73-80.
86 Busam KJ, Roberts DJ, Golden JA. Clinical teratology counseling and consultation. Case report: Two distinct anterior neural tube defects in a human fetus: evidence for an intermittent pattern of neural tube closure. Teratology. 1993;48:399-403.
87 O’Rahilly R, Müller F. Bidirectional closure of the rostral neuropore in the human embryo. Am J Anat. 1989;184:259-268.
88 Siebert JR. Anencephaly. MedLink Neurology. Available at <http://www.medlink.com>
89 Bronner-Fraser M. Neural crest formation and migration in the developing embryo. FASEB J. 1994;8:699-706.
90 Bronner-Fraser M. Origins and developmental potential of the neural crest. Exp Cell Res. 1995;218:405-417.
91 Keynes R, Lumsden A. Segmentation and the origin of regional diversity in the vertebrate central nervous system. Neuron. 1990;2:1-9.
92 McGinnis W, Krumlauf R. Homeobox genes and axial patterning. Cell. 1992;68:283-302.
93 Keynes R, Krumlauf R. Hox genes and regionalization of the nervous system. Annu Rev Neurosci. 1994;17:109-132.
94 Guthrie S. Patterning the hindbrain. Curr Opin Neurobiol. 1996;6:41-48.
95 Wassef M, Joyner AL. Early mesencephalon/metencephalon patterning and development of the cerebellum. Perspect Dev Neurobiol. 1997;5:3-16.
96 Goldowitz D, Hamre K. The cells and molecules that make a cerebellum. Trends Neurosci. 1998;21:375-382.
97 Figdor MC, Stem CD. Segmental organization of embryonic diencephalon. Nature. 1993;363:630-634.
98 Rubenstein JLR, Shimamura K, Martínez S. Regionalization of the prosencephalic neural plate. Annu Rev Neurosci. 1998;21:445-477.
99 Rubenstein JLR, Beachy PA. Patterning of the embryonic forebrain. Curr Opin Neurobiol. 1998;8:18-26.
100 Mai JK, Andressen C, Ashwell KWS. Demarcation of prosencephalic regions by CD15-positive radial glia. Eur J Neurosci. 1998;10:746-751.
101 Guthrie S, Butcher M, Lumsden A. Patterns of cell division and interkinetic nuclear migration in the chick embryo hindbrain. J Neurobiol. 1991;22:742-754.
102 McClure CFW. The segmentation of the primitive vertebrate brain. J Morphol. 1980;4:35-56.
103 Joyner AL. Engrailed, Wnt and Pax genes regulate midbrain-hindbrain development. Trends Genet. 1996;12:15-20.
104 Wassarman KM, Lewandoski M, Campbell J, et al. Specification of the anterior hindbrain organizer is dependent on Gbx2 gene function. Development. 1997;124:2923-2934.
105 Nomes HO, Dressler GR, Knapik EW, et al. Spatially and temporally restricted expression of Pax-2 during neurogenesis. Development. 1990;109:797-809.
106 Puschel AW, Westerfeld M, Dressler G. Comparative analysis of Pax-2 protein distributions during neurulation in mice and zebrafish. Mech Dev. 1992;38:197-208.
107 Rowitch DH, McMahon AP. Pax-2 expression in the murine neural plate precedes and encompasses the expression domains of Wnt-1 and En-1. Mech Dev. 1995;52:3-8.
108 Rowitch DH, Danielian PS, Lee SMK, et al. Cell interactions in patterning the mammalian midbrain. Cold Spring Harb Symp Quant Biol. 1997;62:535-544.
109 Wurst W, Auerback AB, Joyner AL. Multiple developmental defects in Engrailed-1 mutant mice: an early mid-hindbrain deletion and patterning defects in forelimbs and sternum. Development. 1994;120:2065-2075.
110 McMahon AP, Joyner AL, Bradley A, et al. The midbrain-hindbrain phenotype of Wnt-1−/Wnt-1 mice results from stepwise deletion of engrailed-expressing cells by 9.5 days postcoitum. Cell. 1992;69:581-595.
111 Millen KJ, Hui C-C, Joyner AL. A role for En-2 and other murine homologues of Drosophila segment polarity genes in regulating position information in the developing cerebellum. Development. 1995;121:3935-3945.
112 Kuemerle B, Zanjani H, Joyner A, et al. Pattern deformities and cell loss in Engrailed-2 mutant mice suggest two separate patterning events during cerebellar development. J Neurosci. 1997;17:7881-7889.
113 Rhinn M, Dierich A, Shawlot W, et al. Sequential roles for Otx2 in visceral endoderm and neuroectoderm for forebrain and midbrain induction and specification. Development. 1989;125:845-856.
114 Acampora D, Simeone A. Understanding the roles of Otx1 and Otx2 in the control of brain morphogenesis. Trends Neurosci. 1999;22:116-122.
115 Ryan AK, Blumberg B, Rodríguez-Estaban C, et al. Pitx2 determines left-right asymmetry of internal organs in vertebrates. Nature. 1998;394:545-551.
116 De Pomerai D. From Gene to Animal: An Introduction of the Molecular Biology of Animal Development, 2nd ed. Cambridge, United Kingdom: Cambridge University Press; 1990.
117 El-Baradi T, Pieler T. Zinc finger proteins: What we know and what we would like to know. Mech Dev. 1991;35:155-169.
118 Schneider-Maunoury S, Topilko P, Seitanidou T, et al. Disruption of Krox-20 results in alteration of rhombomeres 3 and 5 in the developing hindbrain. Cell. 1993;75:1199-1214.
119 Topilko P, Schneider-Maunoury S, Levi G, et al. Krox-20 controls myelination in the peripheral nervous system. Nature. 1994;371:796-799.
120 Nieto MA, Sechrist J, Wilkinson DG, et al. Relationship between spatially restricted Krox-20 gene expression in branchial neural crest and segmentation in the chick embryo hindbrain. EMBO J. 1995;14:1697-1710.
121 Wilkinson DG, Krumlauf R. Molecular approaches to the segmentation of the hindbrain. Trends Neurosci. 1990;13:335-339.
122 Morriss-Kay GM, Murphy P, Hill RE, et al. Effects of retinoic acid excess on expression of Hox-2.9 and Krox-20 and on morphological segmentation in the hindbrain of mouse embryos. EMBO J. 1991;10:2985-2995.
123 Nonchev S, Maconochie M, Vesque C, et al. The conserved role of Krox-20 in directing Hox gene expression during vertebrate hindbrain segmentation. Proc Natl Acad Sci U S A. 1996;93:9339-9345.
124 Cook M, Gould A, Brand N, et al. Expression of the zincfinger gene PLZF at rhombomere boundaries in the vertebrate hindbrain. Proc Natl Acad Sci U S A. 1995;92:2249-2253.
125 Doniach T. Basic FGF as an inducer of anteroposterior neural pattern. Cell. 1995;83:1067-1070.
126 Brand-Saberi B, Ebensperger C, Wilting J, et al. The ventralizing effect of the notochord on somite differentiation in chick embryos. Anat Embryol. 1993;188:239-245.
127 Pourquié O, Coltey M, Teillet M-A, et al. Control of dorsoventral patterning of somite derivatives by notochord and floor plate. Proc Natl Acad Sci U S A. 1993;90:5242-5246.
128 Martí E, Bumcrot DA, Takada R, et al. Requirement of 19K form of Sonic hedgehog for induction of distinct ventral cell types in CNS explants. Nature. 1995;375:322-325.
129 Roelink H, Porter JA, Chiang C, et al. Floor plate and motor neuron induction by different concentrations of the amino-terminal cleavage product of Sonic hedgehog autoproteolysis. Cell. 1995;81:445-455.
130 Tabin CJ, McMahon AP. Recent advances in hedgehog signalling. Trends Cell Biol. 1997;7:442-446.
131 Goulding M. Specifying motor neurons and their connections. Neuron. 1998;21:943-946.
132 van Straaten HWM, Hekking JWM, Wiertz-Hoessels EJLM, et al. Effect of the notochord on the differentiation of the floor plate area in the neural tube of the chick embryo. Anat Embryol. 1988;177:317-324.
133 Ericson J, Muhr J, Placzek M, et al. Sonic hedgehog induces the differentiation of ventral forebrain neurons: a common signal for ventral patterning within the neural tube. Cell. 1995;81:747-756.
134 Roessler E, Belloni E, Gaudenz K, et al. Mutations in the human Sonic hedgehog gene cause holoprosencephaly. Nat Genet. 1996;14:357-360.
135 Callaerts P, Halder G, Gehring WJ. Pax-6 in development and evolution. Annu Rev Neurosci. 1997;20:483-532.
136 Stoykova A, Gruss P. Roles for Pax genes in developing and adult brain as suggested by expression patterns. J Neurosci. 1994;14:1395-1412.
137 Saint-Jeannet J-P, He X, Varmus HE, et al. Regulation of dorsal fate in the neuraxis by Wnt-1 and Wnt-3a. Proc Natl Acad Sci U S A. 1997;94:13713-13718.
138 Murphy P, Hill RE. Expression of the mouse labial-like homeobox-containing genes, Hox-2.9 and Hox-1.6, during segmentation of the hindbrain. Development. 1991;111:61-74.
139 Boncinelli E, Somma R, Acampora D, et al. Organization of human homeobox genes. Hum Reprod. 1988;3:880-886.
140 Capecchi MR. Hox genes and mammalian development. Cold Spring Harb Symp Quant Biol. 1997;62:273-281.
141 Stern CD, Foley AC. Molecular dissection of Hox gene induction and maintenance in the hindbrain. Cell. 1998;94:143-145.
142 Zhao Q, Behringer RR, de Crombrugghe B. Prenatal folic acid treatment suppresses acrania and meroanencephaly in mice mutant for the Cart1 homeobox gene. Nat Genet. 1996;13:275-283.
143 Howell BW, Herick TM, Cooper JA. Reelin-induced tyrosine phosphorylation of Disabled-1 during neuronal positioning. Genes Dev. 1999;13:643-648.
144 Glinka A, Wu W, Delius H, et al. Dickkopf-1 is a member of a new family of secreted proteins and functions in head induction. Nature. 1998;391:357-362.
145 Anderson SA, Eisenstat DD, Shi L, et al. Interneuron migration from basal forebrain to neocortex: dependence on Dlx genes. Science. 1997;278:474-476.
146 Basler K, Edlund T, Jessell TM, et al. Control of cell pattern in the neural tube: regulation of cell differentiation by dorsalin-1, a novel TGF beta family member. Cell. 1993;73:687-702.
147 Gleeson JG, Allen KM, Fox JW, et al. Doublecortin, a brain-specific gene mutated in human X-linked lissencephaly and double cortex syndrome, encodes a putative signaling protein. Cell. 1998;92:63-72.
148 des Portes V, Pinard JM, Billuart P, et al. A novel CNS gene required for neuronal migration and involved in X-linked subcortical laminar heterotopia and lissencephaly syndrome. Cell. 1998;92:51-61.
149 Gleeson JG, Minnerath SR, Fox JW, et al. Characterization of mutations in the gene doublecortin in patients with double cortex syndrome. Ann Neurol. 1999;45:146-153.
150 Faina GT, Cardini FA, D’Incerti L, et al. Familial schizencephaly associated with EMX2 mutation. Neurology. 1997;48:1403-1406.
151 Millen KJ, Wurst W, Herrup K, et al. Abnormal embryonic cerebellar development and patterning of postnatal foliation in two mouse Engrailed-2 mutants. Development. 1994;120:695-706.
152 Zec N, Rowitch DH, Bitgood MJ, et al. Expression of the homeobox-containing genes EN1 and EN2 in human fetal midgestational medulla and cerebellum. J Neuropathol Exp Neurol. 1997;56:236-242.
153 Condron BG, Patel NH, Zinn K. Engrailed controls glial/neuronal cell fate decisions at the midline of the central nervous system. Neuron. 1994;13:541-554.
154 Eksioglu YZ, Scheffer IE, Cardena P, et al. Periventricular heterotopia: an X-linked dominant epilepsy locus causing aberrant cerebral cortical development. Neuron. 1996;16:77-87.
155 Fox JW, Lamperti ED, Eksioglu YZ, et al. Mutations in filamin 1 prevent migration of cerebral cortical neurons in human periventricular heterotopia. Neuron. 1998;21:1315-1325.
156 Alvarez-Bolado G, Cecconi F, Wehr R, et al. The fork head transcription factor Fkh5/Mf3 is a developmental marker gene for superior colliculus layers and derivatives of the hindbrain somatic afferent zone. Dev Brain Res. 1999;112:205-215.
157 Sasaki H, Hogan BLM. HNF-3β as a regulator of floor plate development. Cell. 1994;76:103-115.
158 Kessel M. Reversal of axonal pathways from rhombomere 3 correlates with extra Hox expression domains. Neuron. 1993;10:379-393.
159 Appel B, Korzh V, Glasgow E, et al. Motoneuron fate specification revealed by patterned LIM homeobox gene expression in embryonic zebrafish. Development. 1995;121:4117-4125.
160 Kikuchi Y, Segawa H, Tokumoto M, et al. Ocular and cerebellar defects in zebrafish induced by overexpression of the LIM domains of the Islet-3 LIM/homeodomain protein. Neuron. 1997;18:369-382.
161 Hirate Y, Mieda M, Harada T, et al. Identification of ephrin-A3 and novel genes specific to the midbrain-MHB in embryonic zebrafish by ordered differential display. Mech Dev. 2001;107:83-96.
162 Jouet M, Kenwrick S. Gene analysis of L1 neural cell adhesion molecule in prenatal diagnosis of hydrocephalus. Lancet. 1995;345:161-162.
163 Tsuru A, Mizuguchi M, Uyemura K, et al. Immunohistochemical expression of cell adhesion molecule L1 in hemimegalencephaly. Pediatr Neurol. 1997;16:45-49.
164 Cohen NR, Taylor JSH, Scott LB, et al. Errors in corticospinal axon guidance in mice lacking the neural cell adhesion molecule L1. Curr Biol. 1997;8:26-33.
165 Porter FD, Drago J, Xu Y, et al. Lhx2, a LIM homeobox gene, is required for eye, forebrain, and definitive erythrocyte development. Development. 1997;124:2935-2944.
166 Bertuzzi S, Porter FD, Pitts A, et al. Characterization of Lhx9, a novel LIM/homeobox gene expressed by the pioneer neurons in the mouse cerebral cortex. Mech Dev. 1999;80:193-198.
167 Karavanov AA, Saint-Jeannet JP, Karavanova I, et al. The LIM homeodomain protein Lim-1 is widely expressed in neural, neural crest and mesoderm derivatives in vertebrate development. Int J Dev Biol. 1996;40:453-461.
168 Clark DC, Mizuguchi M, Antalffy B, et al. Predominant localization of the LIS family of gene products to Cajal-Retzius cells and ventricular neuroepithelium in the developing human cortex. J Neuropathol Exp Neurol. 1997;56:1044-1052.
169 Dobyns WB, Reiner O, Carrozzo R, et al. Lissencephaly: a human brain malformation associated with deletion of the LIS1 gene located at chromosome 17p13. JAMA. 1993;270:2838-2842.
170 Lo Nigro C, Chong SS, Smith ACM, et al. Point mutations and an intragenic deletion in LIS1, the lissencephaly causative gene in isolated lissencephaly sequence and Miller-Dieker syndrome. Hum Mol Genet. 1997;6:157-164.
171 Chong SS, Pack SD, Roschke AV, et al. A revision of the lissencephaly and Miller-Dieker syndrome critical regions in chromosome 17p13.3. Hum Mol Genet. 1997;6:147-155.
172 Casarosa S, Fode C, Guillemot F. Mash1 regulates neurogenesis in the ventral telencephalon. Development. 1999;126:525-534.
173 Hirsch M-R, Tiveron M-C, Guillemot F, et al. Control of noradrenergic differentiation and Phox2a expression by MASH1 in the central and peripheral nervous system. Cell. 1993;75:463-476.
174 Tiveron M-C, Hirsch M-R, Brunet J-F. The expression pattern of the transcription factor Phox2a and Phox2b delineates synaptic pathways of the autonomic nervous system. J Neurosci. 1996;16:7649-7660.
175 Pattyn A, Morin X, Cremer H, et al. The homeobox gene Phox2b is essential for the development of autonomic neural crest derivatives. Nature. 1999;399:366-370.
176 Lo L, Tiveron M-C, Anderson DJ. MASH1 activates expression of the paired homeodomain transcription factor Phox2a, and couples pan-neuronal and subtype-specific components of autonomic neuronal identity. Development. 1998;125:609-620.
177 Helms AW, Gowan K, Abney A, et al. Overexpression of MATH1 disrupts the coordination of neural differentiation in cerebellum development. Mol Cell Neurosci. 2001;17:671-682.
178 Brown NL, Kanekar S, Vetter ML, et al. Math5 encodes a murine basic helix-loop-helix transcription factor expressed during early stages of retinal neurogenesis. Development. 1998;125:4621-4633.
179 Tanabe Y, William C, Jessell TM. Specification of motor neuron identity by the MNR2 homeodomain protein. Cell. 1998;95:67-80.
180 Lee JE, Hollenberg SM, Snider I, et al. Conversion of Xenopus ectoderm into neurons by NeuroD, a basic helix-loop-helix protein. Science. 1995;268:836-844.
181 Yokoyama M, Nishi Y, Miyamoto Y, et al. Molecular cloning of a human neuroD from a neuroblastoma cell line specifically expressed in the fetal brain and adult cerebellum. Brain Res Mol Brain Res. 1996;42:135-139.
182 Lee JE. NeuroD and neurogenesis. Dev Neurosci. 1997;19:27-32.
183 Katayama M, Mizuta I, Sakayama Y, et al. Differential expression of neuroD in primary cultures of cerebral cortical neurons. Exp Cell Res. 1997;236:412-417.
184 Morrow EM, Furukawa T, Lee JE, et al. NeuroD regulates multiple functions in the developing neural retina in rodent. Development. 1998;126:23-36.
185 Yan RT, Wang SZ. NeuroD induces photoreceptor cell overproduction in vivo and de novo generation in vitro. J Neurobiol. 1998;15:485-496.
186 Yamimi R, Steingrimsson E, Copeland NG, et al. The NEUROD gene maps to human chromosome 2q32 and mouse chromosome 2. Genomics. 1996;34:418-421.
187 Yamimi RM, Steingrimsson E, Montgomery-Dyer K, et al. NEUROD2 and NEUROD3 genes map to human chromosomes 17q12 and 5q23-q31 and mouse chromosomes 11 and 13, respectively. Genomics. 1997;40:355-357.
188 Ma Q, Kinter C, Anderson DJ. Identification of neurogenin, a vertebrate neuronal determination gene. Cell. 1996;87:43-52.
189 Sommer I, Ma Q, Anderson DJ. Neurogenins, a novel family of atonal-related bHLH transcription factors, are putative mammalian neuronal determination genes that reveal progenitor cell heterogeneity in the developing CNS and PNS. Mol Cell Neurosci. 1996;8:221-241.
190 Ma Q, Chen Z, del Barco Barrantes I, et al. Neurogenin1 is essential for the determination of neuronal precursors for proximal cranial sensory ganglia. Neuron. 1998;20:469-482.
191 Qiu M, Shimamuira K, Sussel L, et al. Control of anteroposterior and dorsoventral domains of Nkx-6.1 gene expression relative to other Nkx genes during vertebrate CNS development. Mech Dev. 1998;72:77-88.
192 Price M, Lazzaro D, Pohl T, et al. Regional expression of the homeobox gene Nkx-2.2 in the developing mammalian forebrain. Neuron. 1992;8:241-255.
193 Zhong W, Jiang M-M, Weinmaster G, et al. Differential expression of mammalian Numb, Numblike and Notch1 suggests distinct roles during mouse cortical neurogenesis. Development. 1997;124:1887-1897.
194 McMahon JA, Takada S, Zimmerman LB, et al. Noggin-mediated antagonism of BMP signaling is required for growth and patterning of the neural tube and somite. Genes Dev. 1998;12:1438-1452.
195 Chenn A, McConnell SK. Cleavage orientation and the asymmetric inheritance of Notch1 immunoreactivity in mammalian neurogenesis. Cell. 1995;82:6313-6314.
196 Joutel A, Corpechot C, Ducros A, et al. Notch3 mutations in CADASIL, a hereditary adult-onset condition causing stroke and dementia. Nature. 1996;383:707-710.
197 Zhong W, Feder JN, Jiang M-M, et al. Asymmetrical localization of a mammalian numb homolog during mouse cortical neuronogenesis. Neuron. 1996;17:43-53.
198 Goodrich LV, Mienkovic L, Higgins KM, et al. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science. 1997;277:1109-1113.
199 Traiffort E, Charaytoniuk DA, Faure H, et al. Regional distribution of Sonic hedgehog, Patched and Smoothened mRNA in the adult rat brain. J Neurochem. 1998;70:1327-1330.
200 Wechsler-Reya RJ, Scott MP. Control of neuronal precursor proliferation in the cerebellum by Sonic hedgehog. Neuron. 1999;22:103-114.
201 Goodrich LV, Scott MP. Hedgehog and Patched in neural development. Neuron. 1998;21:1243-1257.
202 Urbánek P, Fetka I, Meisler MH, et al. Cooperation of Pax2 and Pax5 in midbrain and cerebellum development. Proc Natl Acad Sci U S A. 1997;94:5703-5708.
203 Ogawa M, Miyata T, Nakajima K, et al. The reeler gene–associated antigen on Cajal-Retzius neurons is a crucial molecule for laminar organization of cortical neurons. Neuron. 1995;14:899-912.
204 Curran T, D’Arcangelo G. Role of reelin in the control of brain development. Brain Res Rev. 1998;26:285-294.
205 Hirotsune S, Takahara T, Sasaki N, et al. The reeler gene encodes a protein with an EGF-like motif expressed by pioneer neurons. Nat Genet. 1995;10:77-83.
206 Jeh-Ping L, Jessell TM. A role for rhoB in the delamination of neural crest cells from the dorsal neural tube. Development. 1998;125:5055-5067.
207 Ben-Arie N, Bellen HJ, Armstrong DL, et al. Math1 is essential for genesis of cerebellar granule neurons. Nature. 1997;390:169-172.
208 Wallis DE, Roessler E, Hehr U, et al. Mutations in the homeodomain of the human SIX3 gene cause holoprosencephaly. Nat Genet. 1999;22:196-198.
209 Loosli F, Winkler S, Wittbrodt J. Six3 overexpression initiates the formation of ectopic retina. Genes Dev. 1999;13:649-654.
210 Lefebvre S, Burglen L, Reboullet S, et al. Identification and characterization of a spinal muscular atrophy determining gene. Cell. 1995;80:155-165.
211 Roy N, Mahadevan MS, McLean M, et al. The gene for neuronal apoptosis inhibitory protein is partially deleted in individuals with spinal muscular atrophy. Cell. 1995;80:167-178.
212 Menchine M, Emeline JK, Mischel PS, et al. Tissue and cell-type specific expression of the tuberous sclerosis gene, TSC2, in human tissues. Mod Pathol. 1996;9:1071-1080.
213 Wienecke R, Maize JCJr, Reed JA, et al. Expression of the TSC2 product tuberin and its target Rap1 in normal human tissues. Am J Pathol. 1997;150:43-50.
214 van Slegtenhorst M, de Hoogt R, Hermans C, et al. Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science. 1997;277:805-808.
215 Ali JB, Sepp T, Ward S, et al. Mutations in the TSC1 gene account for a minority of patients with tuberous sclerosis. J Med Genet. 1998;35:969-972.
216 Au KS, Rodriguez JA, Finch JL, et al. Germ-line mutational analysis of the TSC2 gene in 90 tuberous sclerosis patients. Am J Hum Genet. 1998;62:286-294.
217 Kwiatkowska J, Jozwiak S, Hall F, et al. Comprehensive mutational analysis of the TSC1 gene: observations on frequency of mutation, associated features, and non-penetrance. Ann Hum Genet. 1998;18:9365-9375.
218 Chen Z-F, Behringer RR. Twist is required in head mesenchyme for cranial neural tube morphogenesis. Genes Dev. 1995;9:686-699.
219 Minturn JE, Fryer HJL, Geschwing DH, et al. TOAD-64, a gene expressed early in neuronal differentiation in the rat, is related to unc-33, a C. elegans gene involved in axon outgrowth. J Neurosci. 1995;15:6757-6766.
220 Mastick GS, Fan C-M, Tessier-Lavigne M, et al. Early deletion of neuromeres in Wnt-1−/− mutant mice: evaluation by morphological and molecular markers. J Comp Neurol. 1996;374:246-258.
221 Ericson J, Briscoe J, Rashbass P, et al. Graded Sonic hedgehog signaling and the specification of cell fate in the ventral neural tube. Cold Spring Harb Symp Quant Biol. 1997;62:451-466.
222 Fritzsch B, Nichols DH, Echelard Y, et al. Development of midbrain and anterior hindbrain ocular motoneurons in normal and Wnt-1 knockout mice. J Neurobiol. 1995;27:457-469.
223 Salinas PC, Fletcher C, Copeland NG, et al. Maintenance of Wnt-3 expression in Purkinje cells of the mouse cerebellum depends on interaction with granule cells. Development. 1994;120:1277-1286.
224 Aruga J, Minowa O, Yaginuma H, et al. Mouse Zic1 is involved in cerebellar development. J Neurosci. 1998;18:284-293.
225 Aruga J, Yokota N, Hashimoto M, et al. A novel zinc finger protein, Zic, is involved in neuronogenesis, especially in the cell lineage of cerebellar granule cells. J Neurochem. 1994;63:1880-1890.
226 Wagner M, Thaller C, Jessell T, et al. Polarizing activity and retinoid synthesis in the floor plate of the neural tube. Nature. 1990;345:819-822.
227 Ruberte E, Dolle P, Chambon P, et al. Retinoid acid receptors and cellular retinoid binding proteins. II. Their differential pattern of transcription during early morphogenesis in mouse. Development. 1991;111:45-60.
228 Summerbell D, Maden M. Retinoic acid, a developmental signalling molecule. Trends Neurosci. 1990;13:142-147.
229 Momoi M, Yamagata T, Ichihashi K, et al. Expression of cellular retinoic-acid binding protein in the developing nervous system of mouse embryos. Dev Brain Res. 1990;54:161-167.
230 Thaller C, Eichele G. Isolation of 3,4-didehydroretinoic acid, a novel morphogenetic signal in the chick wing bud. Nature. 1990;345:815-819.
231 Brockes J. Reading the retinoid signals. Nature. 1990;345:766-768.
232 Durston AJ, Timmermans JPM, Hage WJ, et al. Retinoic acid causes an anteroposterior transformation in the developing central nervous system. Nature. 1989;340:140-144.
233 Marín-Padilla M, Marín-Padilla MT. Morphogenesis of experimentally induced Arnold-Chiari malformation. J Neurol Sci. 1981;50:29-55.
234 Marín-Padilla M. Embryology and pathology of the axial skeleton and neural dysraphic disorders. Can J Neurol Sci. 1991;18:153-169.
235 Alles AJ, Sulik KK. Retinoic acid–induced spina bifida: evidence for a pathogenetic mechanism. Development. 1990;108:73-81.
236 Wohl CA, Weiss S. Retinoic acid enhances neuronal proliferation and astroglial differentiation in cultures of CNS stem cell–derived precursors. J Neurobiol. 1998;37:281-290.
237 Sarnat HB, Flores-Sarnat L. The telencephalic flexure and developmental disorders of the Sylvian fissure. 2010, submitted.
238 Ming JE, Muenke M. Holoprosencephaly: from Homer to Hedgehog. Clin Genet. 1998;53:155-163.
239 Sarnat HB, Flores-Sarnat L. Neuropathologic research strategies in holoprosencephaly. J Child Neurol. 2001;16:918-931.
240 DeMyer W, Zeman W, Palmer CG. The face predicts the brain: diagnostic significance of median facial anomalies for holoprosencephaly (arhinencephaly). Pediatrics. 1964;34:256-263.
241 Sarnat HB, Flores-Sarnat L. Neuropathologic research strategies in holoprosencephaly. J Child Neurol. 2001;16:918-931.
242 Barkovich AJ, Quint DJ. Middle interhemispheric fusion: an unusual variant of holoprosencephaly. AJNR Am J Neuroradiol. 1993;14:431-440.
243 Simon EM, Hevner RF, Pinter JD, et al. The middle interhemispheric variant of holoprosencephaly. AJNR Am J Neuroradiol. 2002;23:151-156.
244 Simon EM, Hevner RF, Pinter JD, et al. Assessment of the deep gray nuclei in holoprosencephaly. AJNR Am J Neuroradiol. 2000;21:1955-1961.
245 Simon EM, Hevner RF, Pinter JD, et al. The dorsal cyst in holoprosencephaly and the role of the thalamus in its formation. Neuroradiology. 2001;43:787-791.
246 Simon EM, Barkovich AJ. Holoprosencephaly: new concepts. Magn Reson Imaging Clin N Am. 2001;9:149-164. viii-ix
247 Incardona JP, Roelink H. The role of cholesterol in Shh signaling and teratogen-induced holoprosencephaly. Cell Mol Life Sci. 2000;57:1709-1719.
248 Mione MC, Cavanaugh JF, Harris B, et al. Cell fate specification and symmetrical/asymmetrical divisions in the developing cerebral cortex. J Neurosci. 1997;17:2018-2029.
249 Kuan CY, Roth KA, Flavell RA, et al. Mechanisms of programmed cell death in the developing brain. Trends Neurosci. 2000;23:291-297.
250 de la Rosa EF, de Pablo F. Cell death in early neural development: beyond the neurotrophic theory. Trends Neurosci. 2000;23:454-458.
251 Roth KA, D’Sa C. Apoptosis and brain development. Ment Retard Dev Disabil Res Rev. 2001;7:261-266.
252 Chao MV, Bothwell M. Neurotrophins: to cleave or not to cleave. Neuron. 2002;33:9-12.
253 Miller FD, Kaplan DR. Neurotrophin signalling pathways regulating neuronal apoptosis. Cell Mol Life Sci. 2001;58:1045-1053.
254 Yuan J, Yankner BA. Apoptosis in the nervous system. Nature. 2000;407:802-809.
255 O’Rourke NA, Sullivan DP, Kaznowski CE, et al. Tangential migration of neurons in the developing cerebral cortex. Development. 1995;121:2165-2176.
256 Rakic P. Radial versus tangential migration of neuronal clones in the developing cerebral cortex. Proc Natl Acad Sci U S A. 1995;92:11323-11327.
257 Dobyns WB, Truwit CL, Ross ME, et al. Differences in the gyral pattern distinguish chromosome 17–linked and X-linked lissencephaly. Neurology. 1999;53:270-277.
258 Ross ME, Walsh CA. Human brain malformations and their lessons for neuronal migration. Annu Rev Neurosci. 2001;24:1041-1070.
259 Clark GD. Cerebral gyral dysplasias: molecular genetics and cell biology. Curr Opin Neurol. 2001;14:157-162.
260 Tietjen I, Bodell A, Apse K, et al. Comprehensive EMX2 genotyping of a large schizencephaly case series. Am J Med Genet. 2007;143:1313-1316.
261 Volpe JJ. Neuronal proliferation, migration, organization and myelination. Neurology of the Newborn. 4th ed. Philadelphia: WB Saunders. 2001. 45-99
262 Sarnat HB, Flores-Sarnat L, Trevenen CL. Synaptophysin immunoreactivity in the human hippocampus and neocortex from 6 to 41 weeks of gestation. J Neuropathol Exp Neurol. 2010;69:234-235.
263 Lemke G. The molecular genetics of myelination: an update. Glia. 1993;7:263-271.
264 Yu WP, Collarini EJ, Pringle NP, et al. Embryonic expression of myelin genes: evidence for a focal source of oligodendrocyte precursors in the ventricular zone of the neural tube. Neuron. 1994;12:1353-1362.
265 Back SA, Volpe JJ. Cellular and molecular pathogenesis of periventricular white matter injury. Ment Retard Dev Disabil Res Rev. 1997;3:96-107.
266 Flores-Sarnat L, Sarnat HB. Axes and gradients of the neural tube for a morphological/molecular genetic classification of central nervous system malformations. In: Sarnat HB, Curatolo P, editors. Handbook of Clinical Neurology. Vol. 87: Malformations of the Nervous System. London: Elsevier; 2008:4-11.
267 Barkovich AJ, Kuzniecky RI, Jackson GD, et al. Classification system for malformations of cortical development: update 2001. Neurology. 2001;57:2168-2178.
[/level-membership-for-neurosurgery-category][not-level-membership-for-neurosurgery-category]
CHAPTER 4 Neuroembryology
Classic neuroembryology dealt primarily with documentation of the timing and location of morphologic changes in embryonic development, both gross and microscopic, but an acceleration of genetic, molecular, and radiologic (i.e., neuroimaging) advances in recent years has fundamentally changed the way that we perceive and understand both normal formation and malformation of the brain. Completion of the first draft of the sequence of the human genome will have profound implications on our understanding of the genetics of brain development.1–4 An accelerating pace of genetic discovery is accompanied by the risk that reviews become obsolete before their publication. We now speak of genetics and genomics, but since the human genome project met its initial goals, we will necessarily move onto proteomics (i.e., study of all proteins expressed by the genome)5 and an ever-growing list of “omics” as we delve deeper into the complex molecular genetic interactions required for creating the brain.
Although we do not dispute the critical role of these advances in elucidating development, we strongly believe that this information is best used within the framework of a solid understanding of the timing of structural changes during brain development. In this chapter we offer a unified vision of neuroembryology, with molecular genetic details presented along with classic structural information. It is beyond the scope of this chapter to review the principles of genetics or the revolutionary techniques of gene cloning, polymerase chain reaction, and positional cloning or to attempt to catalog the genes responsible for neurological abnormalities.6 More information is available in review articles or book chapters on neurogenetics.7,8 We do provide examples of important genes relevant to neurodevelopment and extensive references that can aid in understanding basic concepts.
Because much of our knowledge about human brain development has resulted from searching for human homologues to developmental genes discovered in animals from Drosophila to mice, we include examples from these studies along with our discussions of the stages, processes, and abnormalities of human neuroembryology. Table 4-1 lists known gene mutations responsible for several important human central nervous system (CNS) malformations.9–44 When specifically referring to a human gene, we use the convention of denoting the gene symbol in italicized, capital letters.
We once thought of brain insults arising from either environmental or genetic factors, but we now recognize that these causes are interconnected and inseparable. Environmental factors act by influencing gene regulation and expression, and genetic differences determine responses to environmental agents, including toxins and transcription factors. The molecular details of how thousands of genes and the proteins that they encode work together to determine normal or abnormal structure and function are becoming clearer daily but are still overwhelmingly complex. Although the estimated number of genes in the human genome is only about 30,000 (far less than the previously estimated 100,000 and only 2.5 times larger than the fly genome), the human proteome is estimated to contain between 130,000 and 400,000 distinct proteins. Each has many potential ways of interacting with other proteins or genes and of being posttranslationally modified.4
Improvements in neuroradiologic techniques are helping to uncover relationships between genetic abnormalities and structural malformations and to provide another justification for learning more about the basics of embryologic and fetal development. As imaging has improved, so has our appreciation that many developmental disorders represent a spectrum of abnormalities much more complex than previously appreciated. The quality of the image and the speed of acquisition of fetal magnetic resonance imaging (MRI) are constantly improving, and it is already capable of very good anatomic definition of fetal brain malformations.45 The next major step, which will almost certainly occur in the near future, will be the development of MRI techniques for evaluating functional gene expression. In combination with the other molecular genetic advances described, improved imaging will provide an unprecedented opportunity to understand brain development and its disorders.
Classic Neuroembryology
The technical meaning of the term embryology should restrict the study of development to the first 6 postconceptional weeks in humans, the embryonic period proper, but traditional use extends the term to include fetal life until birth, and this is the application that we use in this chapter. Although nonhuman embryologic studies have long been important to our understanding of brain development, meticulous descriptive morphogenic studies of serially sectioned human embryos over the past several decades have provided unique and invaluable information. Based on studies of internal and external morphology that originated from the Carnegie collection of embryos, the 8 weeks of embryonic development have been subdivided into 23 morphologic (Carnegie) stages.46 Stages 8 to 23 are relevant to neuroembryology, with the neural groove and folds first appearing in stage 8, which occurs at about 23 days’ gestation. At this time, the embryo is only about 1 mm long. No accepted morphologic staging system has been developed for the fetal period. These studies remain valuable in using known milestones in structural development to identify the termination period, or the gestational day beyond which the onset of a specific malformation could not have occurred. Knowledge of the exact times when defects such as anencephaly or meningomyelocele may occur is critical for molecular genetic and epidemiologic investigations and can provide important clues to pathophysiologic mechanisms. Further detail can be obtained from O’Rahilly and Muller’s updated classic atlas of developmental stages.46 From careful studies such as these, the adult derivatives of embryonic structures were determined long before we began to understand the signals and processing underlying their formation (Fig. 4-1).
Along with careful work over the past several decades that resulted in this embryonic staging system, similar analysis of the development of the cerebral vasculature led to a staging system usually referred to as Padgett stages.47–49 Knowledge of how the vascular system evolves leads to a clearer understanding of vascular malformations and common vascular anomalies (covered elsewhere in this book) and the patterns of secondary embryonic and fetal brain injuries and malformations. The arterial system essentially achieves an adult pattern by the end of the embryonic period, whereas the venous system develops much later in the fetal period. Although neither the seven stages of arterial evolution developed by Padgett (and still used) nor cerebral venous development will be considered further here, more information can be found in any of several excellent reviews of vascular development.50–53
Developmental Organization: Stages, Genes, and Regulatory Factors
Gastrulation
As the primitive streak extends forward, cells aggregate at one end, a collection designated the primitive node or Hensen’s node. Hensen’s node defines rostral. Cells of the epiblast on either side move toward the primitive streak, stream through it, and emerge beneath it to pass into the narrow cavity between the two sheets of cells, with the epiblast above and the hypoblast below; these migratory cells give rise to the mesoderm and endoderm internally, and some then replace the hypoblast.54
After extending about halfway across the blastoderm (epiblast), the primitive streak with Hensen’s node reverses the direction of its growth and retreats, moving posteriorly as the head fold and neural plate form anterior to Hensen’s node. As the node regresses, a notochordal process develops in the area rostral to it, and somites begin to form on either side of the notochord, with the more caudal somites differentiating first and successive ones differentiating anterior to the somites already formed. The notochord induces epiblast cells to form neuroectoderm (see “Induction”). Several genes essential in creating the fundamental architecture of the embryo and its nervous system are already expressed in the primitive node,55 and many reappear later to influence more advanced stages of ontogenesis.
Induction
Induction was discovered in 1924, when Hans Spemann and Hilde Mangold demonstrated that the dorsal lip of the newt gastrula was capable of inducing the formation of an ectopic second nervous system when transplanted to another site in a host embryo, into another individual of the same species, or to a ventral site of the same embryo.56 This dorsal lip of the amphibian gastrula, also called the Spemann organizer, is homologous with the Hensen node of embryonic birds and mammals.
The first gene isolated from the Spemann organizer was Gsc (goosecoid), which encodes a homeodomain protein (see the later section “Transcription Factors and Homeoboxes”) able to recapitulate transplantation of the dorsal lip tissue when injected into an ectopic site. It also normally induces the prechordal mesoderm and contributes to prosencephalic differentiation.55,57,58 In Hensen’s node in the chick, even before the primitive streak is fully formed, Wnt8c is expressed and is essential for the regulation of axis formation and later for hindbrain patterning in the region of the future rhombomere 4 (r4).59 The regulatory gene Cnot, with major domains in the primitive node, notochord, and prenodal and postnodal neural plate, is also involved in the induction of prechordal mesoderm and in formation of the notochord in particular.60
The specificity of induction lies not in the inductive molecule but rather in the receptor in the induced cell. This distinction is important because foreign molecules similar in structure to the natural inductor molecule may sometimes be erroneously recognized by the receptor as identical; such foreign molecules may act as teratogens if the embryo is exposed to such a toxin. Induction occurs during a very precise temporal window; the period of responsiveness of the induced cell is designated its competence, and the cell is incapable of responding before or after that precise time.61
Induction receptors are not necessarily in or on the plasma membrane of the cell but may be in the cytoplasm or in the nucleus. Retinoic acid is an example of a nuclear inducer. In some cases, the stimulus acts exclusively at the plasma membrane of target cells and does not require actual penetration of the cell.61,62 The receptors that represent the specificity of induction are also genetically programmed. Notch is a particularly important gene in regulating the competence of a cell to respond to inductive cues from within the neural tube and from surrounding embryonic tissues.63 Some mesodermal tissues, such as smooth muscle of the fetal gut, can act as mitogens on the neuroepithelium by increasing the rate of cellular proliferation,64,65 but this phenomenon is not true neural induction because the proliferating cells do not differentiate or mature. Some organizer and regulatory genes of the nervous system, such as Wnt1, also exhibit mitogenic effects,66 and insulin-like growth factor and basic fibroblast growth factor act as mitogens as well.67–69
Early formation of the neural plate is not accomplished exclusively by mitotic proliferation of neuroepithelial cells; surrounding cells are also converted to a neural fate. In amphibians, a gene known as Xash (achaete-scute) is expressed very early in the dorsal part of the embryo from the time of gastrulation and acts as a molecular switch to change the fate of undifferentiated cells to become neuroepithelium rather than surface ectodermal or mesodermal tissues.70 Some cells differentiate as specific types because they are actively inhibited from differentiating into others. All ectodermal cells are preprogrammed to form neuroepithelium, and neuroepithelial cells are preprogrammed to become neurons if not inhibited by genes that direct them along a different lineage, such as epidermal, glial, or ependymal.71–73
Neurulation
These forces arise in part from growth of the surrounding mesodermal tissues on either side of the neural tube, the future somites (Table 4-2).74 After surgical removal of mesoderm and endoderm from one side of the neuroepithelium in experimental animals, the neural tube still closes, but it is rotated and becomes asymmetric.75 The mesoderm appears to be important for orientation but not for closure of the neural tube. Expansion of the surface epithelium of the embryo is the principal extrinsic force for folding of the neuroepithelium to form the neural tube.76 Cells of the neural placode are mobile and migrate beneath the surface ectoderm, which causes the lateral margins of the placode to become raised toward the dorsal midline. Growth of the whole embryo itself does not appear to be an important factor because neurulation proceeds equally well in anamniotes (e.g., amphibians), which do not grow during this period, and in amniotes (e.g., mammals), which grow rapidly at this time.77
TABLE 4-2 Factors Involved in Closure of Neuroepithelium to Form the Neural Tube
From Menkes JH, Sarnat HB. Child Neurology, 6th ed. Philadelphia: Lippincott Williams & Wilkins; 2000:289.
Among the intrinsic forces of the neuroepithelium, the cells of the floor plate have a wedge shape—narrow at the apex and broad at the base—that facilitates bending.78 Although the width of the floor plate is small, its site in the ventral midline is crucial and sufficient to allow a significant influence. It represents yet another aspect of induction of the floor plate by the notochord, apart from its influence on the differentiation of neural cells.79 The ependymal cells that form the floor plate are the first neural cells to differentiate, and they induce growth of the parenchyma of the ventral zone more than the dorsal regions.80,81 This mechanical effect may also facilitate curving of the neural placode. The direction of proliferation of new cells in the mitotic cycle, determined in part by the orientation of the mitotic spindle, becomes another mechanical force shaping the neural tube.77,78 Adhesion molecules are also probably an important mechanical factor for neurulation. In later stages, the ependymal cell–lined central canal, which is much larger in the fetus than in the newborn, may have a role in exerting a centrifugal force to create the tubular shape, although in early spinal cord development the central canal is a tall, narrow, midline slit and only later in fetal life does it assume a rounded contour as seen in transverse sections.80
Neuroepithelial cells of the neural placode or plate downregulate the polarity of their plasma membrane so that the apical and basilar surfaces are not as distinct before neural tube closure. Cell differentiation in general involves such changes in cell polarity.82 The rostrocaudal orientation of most mitotic spindles of the neuroepithelium and the direction in which they push by the mass of daughter cells that they form also influence the shape of the neural tube (Fig. 4-2).83
The neural tube closes in the dorsal midline first in the cervical region, with the closure then extending rostrally and caudally such that the anterior neuropore of the human embryo closes at 24 days and the posterior neuropore closes at 28 days, with the distances from the cervical region being unequal. This traditional view of a continuous zipper-like closure is an oversimplification. In the mouse embryo, the neural tube closes in the cranial region at four distinct sites, with the closure proceeding bidirectionally or unidirectionally and in general synchrony with somite formation.84,85 An intermittent pattern of anterior neural tube closure involving multiple sites has also been described in human embryos.86 In this closure pattern, the principal rostral neuropore closes bidirectionally87 to form the lamina terminalis, an essential primordium of the forebrain.80
Bending of the neural plate to form the neural tube is termed primary neurulation. Failure of the anterior neuropore to close by 24 days results in anencephaly. Because the lamina terminalis does not form, its derivatives (including the basal ganglia and other forebrain structures) do not develop. The lack of forebrain neuroectoderm results in failure of induction of the overlying mesoderm, and the cranium, meninges, and scalp fail to close in the midline.88 The term secondary neurulation refers only to the most caudal part of the spinal cord (i.e., conus medullaris), which develops from neuroepithelium caudal to the site of posterior neuropore closure. More details on abnormalities that occur because of problems with secondary neurulation are offered in other chapters in this textbook.
Neural crest cells arise from the dorsal midline of the neural tube at or shortly after the time of closure and migrate extensively along prescribed routes through the embryo to differentiate as the peripheral nervous system. This includes the dorsal root and sympathetic ganglia, adrenal medulla and carotid body chromaffin cells, melanocytes, and a few other cell types of ectodermal and mesodermal origin.89,90
Segmentation and Regionalization
Segmentation of the neural tube creates intrinsic compartments that restrict the movement of cells by physical and chemical boundaries between adjacent compartments. These embryonic compartments are known as neuromeres. The spinal cord has the appearance of a highly segmented structure; however, it is not intrinsically segmented in the embryo, fetus, or adult but rather corresponds in its entirety to the most caudal of the eight neuromeres that create the hindbrain. The apparent segmentation of the spinal cord results from clustering of nerve roots imposed by true segmentation of surrounding tissues derived from the mesoderm, tissues that form the neural arches of the vertebrae, somites, and associated structures. Neuromeres of the hindbrain are designated rhombomeres.91–94 The entire cerebellar cortex, vermis, flocculonodular lobe, and lateral hemispheres develop from rhombomere 1 (r1), with a small contribution to the anterior vermis from the mesencephalic neuromere, but the dentate and other deep cerebellar nuclei are formed in rhombomere 2 (r2).95,96 The rostral end of the neural tube forms a mesencephalic neuromere and probably six forebrain neuromeres (i.e., two diencephalic and four telencephalic prosomeres), although these may be subdivided further.97–99 The segmentation of the human embryonic brain into neuromeres is summarized in Table 4-3.
| NEUROMERE | DERIVED STRUCTURES IN MATURE CENTRAL NERVOUS SYSTEM |
|---|---|
| Rhombomere 8 (r8) | Entire spinal cord; caudal medulla oblongata; cranial nerves XI, XII |
| Rhombomere 7 (r7) | Medulla oblongata; cranial nerves IX, X; neural crest |
| Rhombomere 6 (r6) | Medulla oblongata; cranial nerves VIII, IX |
| Rhombomere 5 (r5) | Medulla oblongata; cranial nerves VI, VII; no neural crest |
| Rhombomere 4 (r4) | Medulla oblongata; cranial nervess VI, VII; neural crest |
| Rhombomere 3 (r3) | Caudal pons; cranial nerve V; no neural crest |
| Rhombomere 2 (r2) | Caudal pons; cranial nerves IV, V; cerebellar nuclei |
| Rhombomere 1 (r1) | Rostral pons; cerebellar cortex |
| Mesencephalic neuromere | Midbrain; cranial nerve III; neural crest |
| Diencephalic prosomere 2 | Dorsal diencephalons |
| Diencephalic prosomere 1 | Ventral diencephalon |
| Prosencephalic prosomere 2 | Telencephalic nuclei; olfactory bulb |
| Prosencephalic prosomere 1 | Cerebral cortex; hippocampus; corpus callosum |
From Menkes JH, Sarnat HB. Child Neurology, 6th ed. Philadelphia: Lippincott Williams & Wilkins; 2000:280.
The segments of the embryonic neural tube are distinguished by physical barriers formed by processes of early specializing cells that resemble the radial glial cells that appear later in development100,101 and by chemical barriers from secreted molecules that repel migratory cells. Cell adhesion is increased in the boundary zones between rhombomeres, which also contributes to the creation of barriers against cellular migration in the longitudinal axis. Limited mitotic proliferation of the neuroepithelium occurs in the boundary zones between rhombomeres. Although cells still divide in this zone, their nuclei remain near the ventricle during the mitotic cycle and do not move as far centrifugally within the elongated cell cytoplasm during the interkinetic gap phases as they generally do.101 The rhombomeres of the brainstem may also be visualized as a series of transverse ridges and grooves on the dorsal surface, the future floor of the fourth ventricle; these ridges are gross morphologic markers of the hindbrain compartments.94,102
The first evidence of segmentation is a boundary that separates the future mesencephalic neuromere from r1 of the hindbrain. More genes play a role in this initial segmentation of the neural tube than in any boundaries that subsequently form to separate other neuromeres. The mesencephalic-metencephalic region appears to develop early as a single, independent unit or “organizer” for other neuromeres rostral and caudal to that zone.103,104 The organizer genes recognized at the mesencephalic-metencephalic boundary for this earliest segmentation of the neural tube include Pax2, Wnt1, En1, En2, Pax5, Pax8, Otx1, Otx2, Gbx2, Nkx2-2, and Fgf8.
The earliest known gene with regional expression in the mouse is Pax2, and it is expressed even before the neural plate forms. It is the earliest gene recognized in the presumptive region of the midbrain-hindbrain boundary.105,106 In invertebrates, Pax2 is important for the activation of Wg (wingless) genes; this relationship is relevant because the first gene definitely associated with an identified midbrain-hindbrain boundary in vertebrates is Wnt1, a homologue of Wg. Regulation of Wnt1 may be divided into two phases. In the early phase (1 or 2 somites), the mesencephalon broadly expresses the gene throughout; in the later phase (15 to 20 somites), expression is restricted to the dorsal regions, the roof plate of the caudal diencephalon, the mesencephalon, the myelencephalon, and the spinal cord, but it is also expressed in a ring that extends ventrally just rostral to the midbrain-hindbrain boundary and in the ventral midline of the caudal diencephalon and mesencephalon.107–109 Wnt1 is essential in activating and preserving the function of the mouse engrailed genes En1 and En2. En1 is coexpressed with Wnt1 at the 1-somite stage in a domain only slightly caudal to Wnt1, which includes the midbrain and r1, the rostral half of the pons, and the cerebellar cortex but excludes the diencephalon.110 Activation of En2 begins at the 4-somite stage, and its function in mesencephalic and r1 development is similar, with differences in some details, particularly their roles in cerebellar development.111,112 The homeobox gene Otx2 appears early in the initial boundary zone of the midbrain-hindbrain, and as with Wnt1, it appears to be essential for the later expression of En1, En2, and Wnt1.113,114
Patterning of the Neural Tube
Development of the basic characteristics of the body plan is called patterning.91 These patterns are the anatomic expression of the genetic code within the nuclear DNA of every cell, but they may also result from signals from neighboring cells carried by molecules that are secretory translation products of various families of organizer genes, each in a highly precise and predictable temporal and spatial distribution.
Early development of the CNS in all vertebrates, even before closure of the neural placode or plate to form the neural tube, requires the establishment of a fundamental body plan of bilateral symmetry, with cephalization, or the identity of head and tail ends, and determination of the dorsal and ventral surfaces. These axes of the body itself and the CNS require the expression of genes that impose gradients of differentiation and growth. The genes that determine the polarity and gradients of the anatomic axes are called organizer genes. Many express themselves in the CNS and in other organs and tissues.54,109 The bilateral symmetry of many organs and programmed asymmetries, probably including neural structures such as the different targets of the left and right vagal nerves and left-right asymmetries in the cerebral cortex, is determined in large part by Pitx2, a gene expressed as early as in the primitive node.115 Some genes function to stimulate or inhibit the expression of others, or there is an antagonism or equilibrium between certain families of genes, as exemplified by those that exert dorsoventral or ventrodorsal gradients. The difference between an organizer gene and a regulator gene is its function, and the same gene often subserves both roles at different stages of development. The definitions and programs of these two groups are summarized in Table 4-4.
| Organizer Genes |
From Menkes JH, Sarnat HB. Child Neurology, 6th ed. Philadelphia: Lippincott Williams & Wilkins; 2000:281.
Transcription Factors and Homeoboxes
Transcription factors are proteins expressed by regulatory genes that bind to the regulatory regions of other genes and control transcription. These transcription factors are essential for functional expression of the gene, and several different protein structural motifs have been discovered to be highly evolutionarily conserved. One such motif is the basic helix-loop-helix structure, which is so fundamental to the evolution of life that it appears for the first time in certain bacteria even before evolution of a cell nucleus to concentrate the DNA.116
The zinc finger is another DNA-binding, gene-specific transcription factor motif. It consists of 28 amino acid repeats with pairs of cysteine and histidine residues and with each sequence folded around a zinc ion.117 Krox20 (this gene name is applied to the mouse; the human form is designated EGR2) is a zinc finger gene expressed in alternating rhombomeres, especially r3 and r5; neural crest tissue does not differentiate from these two rhombomeres, although it does in adjacent segments, including r4.118 Krox20 serves an additional function in the peripheral nervous system, where it regulates myelination by Schwann cells,119 and it also regulates the expression of some other genes, most notably those of the Hox family.118,120–123 Another developmentally important zinc finger gene is PLZF (human promyelocytic zinc finger). Studies of mouse and chick homologues of this gene have revealed that it is expressed in a restricted zone surrounding hindbrain rhombomere boundaries, thus suggesting an important functional role for it and other zinc finger genes in vertebrate hindbrain regionalization.124
Some transcription factor genes include homeoboxes. These restricted DNA sequences of 183 base pairs of nucleotides encode a class of proteins sharing a common or very similar 60–amino acid motif called the homeodomain.91 Homeoboxes or homeotic genes are classified into various families with common molecular structures and similar general expression during ontogenesis. They are especially associated with genes that program segmentation and the rostrocaudal gradients of the neural tube. Some of the families of homeobox genes important in development of the vertebrate nervous system are Gsc, Hox, En, Wnt, Shh, Nkx, Lim, and Otx.
Growth factors may also influence the pattern of the neural tube by behaving biologically as transcription factors: basic fibroblast growth factor behaves as an auxiliary inductor of the longitudinal axis with a rostrocaudal gradient during formation of the neural tube.125
Developmental Gene Families of the Central Nervous System
The genes that program the axes and gradients of the neural tube may be classified as families based on their similar nucleic acid sequences and their similar general functions, although important differences occur within a family in the site or neuromere where each gene is expressed and the anatomic structures that they form. A dorsalizing gene has a dorsal territory of expression and causes the ventral parts of the neural tube to differentiate as dorsal structures if influences from ventralizing genes do not antagonize them sufficiently and vice versa. In development of the somite, the sclerotome (which forms the cartilage and bone of the vertebral body) is normally situated ventral to the myotome (which forms muscle cells) and the dermatome. Ectopic cells of the floor plate or notochord implanted next to the somite of the chick embryo cause ventralization of the somite such that excess cartilage and bone are formed and there is a deficiency of muscle and dermis.126,127 The floor plate, or notochord in this instance, is the ventralizing inductor of the mesodermal somite, and this is caused by expression of Shh (Sonic Hedgehog), which also serves as a strong ventralizing gradient force in the neural tube.128–131 If a section of notochord is ectopically implanted dorsal or lateral to the neural tube, a second floor plate forms opposite the notochord, and motor neurons differentiate on either side of it despite the presence of a normal floor plate and motor neurons in the normal position.132 Shh, which is expressed as early as in the primitive node, induces ventralization of a dorsal region of the neural tube or duplicates the neural tube. Such an influence in the human fetus, the so-called split notochord, could be an explanation for the rare cases of diplomyelia or diastematomyelia.80 Excessive Shh expression, particularly its amino-terminal cleavage product, upregulates floor plate differentiation at the expense of motor neuron formation129 and may induce duplication of the neuraxis. Shh exerts a strong influence on differentiation of the ventral and medial structures of the prosencephalon,133 and defective expression of this gene has been found to be one molecular basis of human holoprosencephaly.134
To establish an equilibrium with genes with ventralizing influence, other genes exercise a dorsalizing influence. The Pax family is an example of genes that cause differentiation of the dorsal structures of the neural tube.135,136 The Wnt family is also dorsalizing in the hindbrain; in situ hybridization shows its transcription products to be expressed diffusely only in the early neuroepithelium and to be restricted to dorsal regions as the neural tube develops.137 The zinc finger gene Zic2 has a dorsalizing gradient in the forebrain. The rostrocaudal axis of the neural tube and segmentation, or the formation of neuromeres, are directed in large part by a family of 38 homeobox genes that are divided into four groups called Hox genes.92,93,138–141 Each of 13 Hox genes is expressed in certain rhombomeres and not in others (Table 4-5). Hox genes are not expressed in the forebrain. In addition to their functions in establishing the compartments or rhombomeres of the brainstem and effecting the differentiation of certain anatomic structures, Hox genes guide the growth cones forming the long descending and ascending pathways between the brain and spinal cord.158
Genes that direct the specific differentiation of structures are called regulator genes, and in many cases they have served as organizer genes in an earlier period. The most important families for development of the brainstem and midbrain in vertebrates are En, Wnt, Hox, Krox, and Pax. Table 4-5 summarizes the sites of expression and functions of selected important organizer and regulator genes. Many regulatory genes change their territories of expression in different stages of development; they can increase their territory to include more rhombomeres or have broader expression early in development and more restricted domains later. Sometimes, expression of a gene in the wrong neuromere, called ectopic expression, interferes with normal development (see Table 4-5).
Function of Retinoic Acid
Retinoic acid, the alcohol of which is vitamin A, is a hydrophobic molecule secreted by the notochord and by ependymal floor plate cells.226 Retinoid-binding proteins and receptors are already strongly expressed in mesenchymal cells of the primitive streak and in the preoptic region of the early developing hindbrain.227 Ependymal cells other than those of the floor plate and neuroepithelial cells have retinoic acid receptors but do not secrete this compound. Retinoic acid diffuses across the plasma membrane without requiring active transport and binds to intracellular receptors; it then enters the nucleus, where it binds to a specific nuclear receptor protein, changes the structural configuration of that protein, and thereby enables it to attach to a specific receptor on a target gene, where the complex formed by retinoic acid and its receptor then functions as a transcription factor for neural induction.228,229
Retinoic acid functions as a polarity molecule for determining the anterior and posterior surfaces of limb buds and, in the nervous system, is important in segmentation polarity. It produces a strong rostrocaudal polarity gradient.54,230,231 Excessive retinoic acid acts on the neural tube of amphibian tadpoles to transform anterior neural tissue to a posterior morphogenesis, which results in extreme microcephaly and suppression of optic cup formation.229 In vertebrates, optic cup formation may fail because of suppression of retinoic acid by genes of the Lim family, such as Islet3 and Lim1.160,165 Retinoic acid upregulates homeobox genes, those of the Hox family and Krox20 in particular, and can cause ectopic expression of these genes in rhombomeres where they are not normally expressed.122,232 An excess of retinoic acid, whether endogenous or exogenous (i.e., excess maternal intake of vitamin A or analogues during early gestation), results in severe malformations of the hindbrain and spinal cord. A single dose of retinoic acid administered intraperitoneally to maternal hamsters on embryonic day 9.5 results in the Chiari II malformation and frequently meningomyelocele in the fetuses.233–235 In cell cultures of cloned CNS stem cells from the mouse, retinoic acid enhances neuronal proliferation and astroglial differentiation.236
Flexures and Sulci of the Brain
The neural tube begins development as a straight structure, but further growth of surrounding structures exerts an external physical force that causes bending at precise points. The pontine and cervical flexures thus form. At 6 weeks’ gestation the telencephalic hemisphere is oval shaped, but ventral bending then occurs to create the operculum and eventually the sylvian fissure (Fig. 4-3). Because both the dorsal (frontal lobe) and ventral (temporal lobe) lips of the sylvian fissure correspond to the ventral surface of the primitive telencephalon, disorders of this region, such as schizencephaly and perisylvian syndromes of polymicrogyria at the lips of the sylvian fissure, follow a ventrodorsal genetic gradient in the vertical axis, and both lips are involved in these malformations.237
[/not-level-membership-for-neurosurgery-category]
