Chapter 592 Neurodegenerative Disorders of Childhood
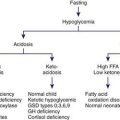
Neurodegenerative disorders of childhood encompass a large, heterogeneous group of diseases that result from specific genetic and biochemical defects, chronic viral infections, and varied unknown causes. Children with suspected neurodegenerative disorders were once subjected to brain and rectal (neural) biopsies, but with modern neuroimaging techniques and specific biochemical and molecular diagnostic tests, these invasive procedures are rarely necessary. The most important component of the diagnostic investigation continues to be a thorough history and physical examination. The hallmark of a neurodegenerative disease is regression and progressive deterioration of neurologic function with loss of speech, vision, hearing, or locomotion, often associated with seizures, feeding difficulties, and impairment of intellect. The age of onset, rate of progression, and principal neurologic findings determine whether the disease affects primarily the white or the gray matter. Upper motor neuron signs and progressive spasticity are the hallmarks of white matter disorders; convulsions, intellectual, and visual impairment that occur early in the disease course are the hallmarks of grey matter disorders. A precise history confirms regression of developmental milestones, and the neurologic examination localizes the process within the nervous system. Although the outcome of a neurodegenerative condition is usually fatal and available therapies are often limited in effect, it is important to make the correct diagnosis so that genetic counseling may be offered and prevention strategies can be implemented. Bone marrow transplantation and other novel therapies may prevent the progression of disease in certain presymptomatic individuals. For all conditions in which the specific enzyme defect is known, prevention by prenatal diagnosis (chorionic villus sampling or amniocentesis) is possible. Carrier detection is also often possible by enzyme assay. Table 592-1 summarizes selected inherited neurodegenerative and metabolic disorders by their age of onset.
Table 592-1 NEUROMETABOLIC CONDITIONS ASSOCIATED WITH DEVELOPMENTAL REGRESSION
| AGE AT ONSET (yr) | CONDITIONS | COMMENTS |
|---|---|---|
| <2, with hepatomegaly | Fructose intolerance | Vomiting, hypoglycemia, poor feeding, failure to thrive (when given fructose) |
| Galactosemia | Lethargy, hypotonia, icterus, cataract, hypoglycemia (when given lactose) | |
| Glycogenosis (glycogen storage disease) types I-IV | Hypoglycemia, cardiomegaly (type II) | |
| Mucopolysaccharidosis types I and II | Coarse facies, stiff joints | |
| Niemann-Pick disease, infantile type | Gray matter disease, failure to thrive | |
| Tay-Sachs disease | Seizures, cherry red macula, edema, coarse facies | |
| Zellweger syndrome | Hypotonia, high forehead, flat facies | |
| Gaucher disease (neuronopathic form) | Extensor posturing, irritability | |
| Carbohydrate-deficient glycoprotein syndromes | Dysmyelination, cerebellar hypoplasia | |
| <2, without hepatomegaly | Krabbe disease | Irritability, extensor posturing, optic atrophy and blindness |
| Rett syndrome | Girls with deceleration of head growth, loss of hand skills, hand wringing, impaired language skills, gait apraxia | |
| Maple syrup urine disease | Poor feeding, tremors, myoclonus, opisthotonos | |
| Phenylketonuria | Light pigmentation, eczema, seizures | |
| Menkes kinky hair disease | Hypertonia, irritability, seizures, abnormal hair | |
| Subacute necrotizing encephalopathy of Leigh | White matter disease | |
| Canavan disease | White matter disease, macrocephaly | |
| Neurodegeneration with brain iron accumulation disease | White matter disease, movement disorder | |
| 2-5 | Niemann-Pick disease types III and IV | Hepatosplenomegaly, gait difficulty |
| Wilson disease | Liver disease, Kayser-Fleischer ring; deterioration of cognition is late | |
| Gangliosidosis type II | Gray matter disease | |
| Neuronal ceroid lipofuscinosis | Gray matter disease | |
| Mitochondrial encephalopathies (e.g., myoclonic epilepsy with ragged red fibers [MERRF]) | Gray matter disease | |
| Ataxia-telangiectasia | Basal ganglia disease | |
| Huntington disease (chorea) | Basal ganglia disease | |
| Neurodegeneration with brain iron accumulation syndrome | Basal ganglia disease | |
| Metachromatic leukodystrophy | White matter disease | |
| Adrenoleukodystrophy | White matter disease, behavior problems, deteriorating school performance, quadriparesis | |
| 5-15 | Adrenoleukodystrophy | Same as for adrenoleukodystrophy in 2 to 5 yr olds |
| Multiple sclerosis | White matter disease | |
| Neuronal ceroid lipofuscinosis, juvenile and adult (Spielmeyer-Vogt and Kufs disease) | Gray matter disease | |
| Schilder disease | White matter disease, focal neurologic symptoms | |
| Refsum disease | Peripheral neuropathy, ataxia, retinitis pigmentosa | |
| Sialidosis II, juvenile form | Cherry red macula, myoclonus, ataxia, coarse facies | |
| Subacute sclerosing panencephalitis | Diffuse encephalopathy, myoclonus; may occur years after measles |
From Kliegman RM, Greenbaum LA, Lye PS: Practical strategies in pediatric diagnosis and therapy, ed 2, Philadelphia, 2004, Elsevier/Saunders, p 542.
592.1 Sphingolipidoses
The sphingolipidoses are characterized by intracellular storage of lipid substrates resulting from defective catabolism of the sphingolipids comprising cellular membranes (Fig. 592-1). The sphingolipidoses are subclassified into 6 categories: Niemann-Pick disease, Gaucher disease, GM1 gangliosidosis, GM2 gangliosidosis, Krabbe disease, and metachromatic leukodystrophy. Niemann-Pick disease and Gaucher disease are discussed in Chapter 80.4.
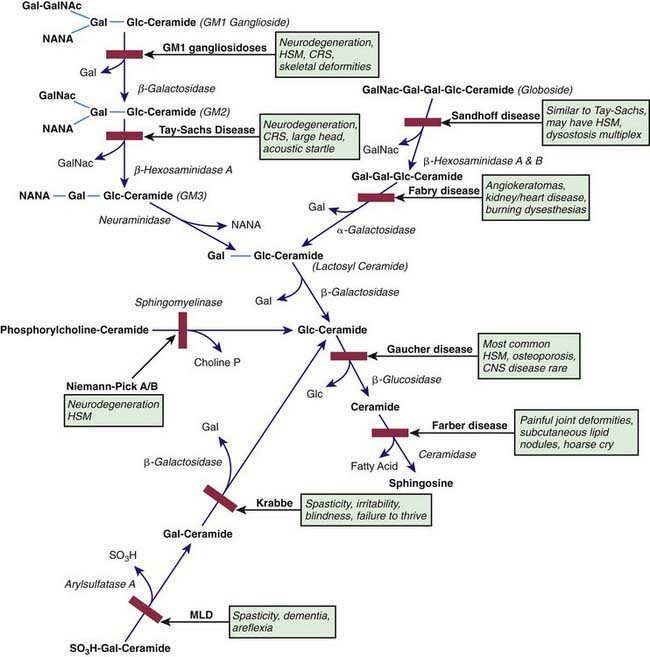
Gangliosidoses (Chapter 80.4)
GM1 Gangliosidoses
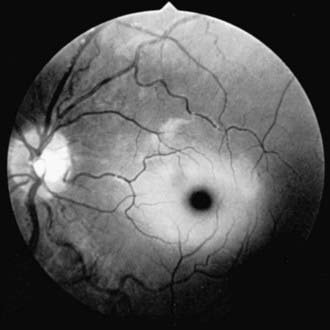
Infantile GM1 gangliosidosis presents at birth or during the neonatal period with anorexia, poor sucking, and inadequate weight gain. Development is globally retarded, and generalized seizures are prominent. The phenotype is striking and shares many characteristics with Hurler syndrome. The facial features are coarse, the forehead is prominent, the nasal bridge is depressed, the tongue is large (macroglossia), and the gums are hypertrophied. Hepatosplenomegaly is present early in the course as a result of accumulation of foamy histiocytes, and kyphoscoliosis is evident because of anterior beaking of the vertebral bodies. The neurologic examination is dominated by apathy, progressive blindness, deafness, spastic quadriplegia, and decerebrate rigidity. A cherry red spot in the macular region is visualized in approximately 50% of cases. The cherry red spot is characterized by an opaque ring (sphingolipid-laden retinal ganglion cells) encircling the normal red fovea (Fig. 592-2). Children rarely survive beyond age 2-3 yr, and death is due to aspiration pneumonia.
GM2 Gangliosidoses
The GM2 gangliosidoses are a heterogeneous group of autosomal recessive inherited disorders that consist of several subtypes, including Tay-Sachs disease (TSD), Sandhoff disease, juvenile GM2 gangliosidosis, and adult GM2 gangliosidosis. Tay-Sachs disease is most prevalent in the Ashkenazi Jewish population and has an approximate carrier rate of 1/30. TSD is due to mutations in the HEXA gene located on chromosome 15q23-q24. Affected infants appear normal until ≈6 mo of age, except for a marked startle reaction to noise that is evident soon after birth. Affected children then begin to lag in developmental milestones and, by 1 yr of age, they lose the ability to stand, sit, and vocalize. Early hypotonia develops into progressive spasticity, and relentless deterioration follows, with convulsions, blindness, deafness, and cherry red spots in almost all patients (see Fig. 592-2). Macrocephaly becomes apparent by 1 yr of age and results from the 200- to 300-fold normal content of GM2 ganglioside deposited in the brain. Few children live beyond 3-4 yr of age, and death is usually associated with aspiration or bronchopneumonia. A deficiency of the isoenzyme hexosaminidase A is found in tissues of patients with TSD. Mass screening for prenatal diagnosis of TSD is a reliable and cost-effective method of prevention because the condition occurs most frequently in a defined population (Ashkenazi Jews). Targeted screening is responsible for the fact that currently, the rare children with TSD born in the USA are most commonly born to non-Jewish parents who are not routinely screened. An accurate and inexpensive carrier detection test is available (serum or leukocyte hexosaminidase A), and the disease can be reliably diagnosed by chorionic villus sampling in the 1st trimester of pregnancy in couples at risk (heterozygote parents).
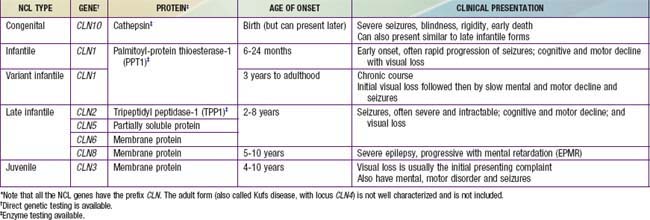
592.2 Neuronal Ceroid Lipofuscinoses
The neuronal ceroid lipofuscinoses (NCLs) are a group of inherited, neurodegenerative, lysosomal storage disorders characterized by visual loss, progressive dementia, seizures, motor deterioration, and early death. The NCLs are so named because of the intracellular accumulation of fluorescent lipopigments, ceroid and lipofuscin. They comprise a genetically and phenotypically heterogeneous group of disorders (the current number of NCL types is 10) that have traditionally been subclassified by age of onset, among other clinical features. They differ from one another in the associated ultrastructural patterns of the inclusions as seen by electron microscopy. Evaluation of neuronal biopsies (either brain, rectal, conjunctival, or skin) was once required for diagnosis (see Table 592-1). With the advent of enzymatic and molecular testing methods, clinicians can make specific NCL diagnoses using less invasive methods (Table 592-2).
592.5 Miscellaneous Disorders
Other Leukodystrophies
Metabolic and degenerative disorders can present with significant cerebral white matter changes, such as some mitochondrial disorders (Chapters 80.1 and 591.2) and glutaric aciduria type 1 (Chapter 79). In addition, the broader use of MRI has brought to light new leukodystrophies. One example is vanishing white matter disease or childhood ataxia with central nervous system hypomyelination (VWM/CACH) characterized by ataxia and spasticity. Some patients also have optic atrophy, seizures, and cognitive deterioration. The age of presentation and the rapidity of decline can be quite variable. In the early-onset forms, decline is usually rapid and followed quickly by death; in the later-onset forms, mental decline is usually slower and milder. Interestingly, acute demyelination in these disorders can be triggered by fever or fright. The diagnosis of VWM/CACH is based on clinical findings, characteristic abnormalities on cranial MRI, and autosomal recessive mutations in 1 of 5 causative genes (EIF2B1, EIF2B2, EIF2B3, EIF2B4, and EIF2B5) encoding the 5 subunits of the eucaryotic translation initiation factor, eIF2B.
Subacute Sclerosing Panencephalitis
This is a rare, progressive neurologic disorder caused by persistent measles virus infection of the CNS (Chapter 238). The number of reported cases has decreased dramatically to 0.06 cases/million population, paralleling the decline in reported measles cases. The initial clinical manifestations include personality changes, aggressive behavior, and impaired cognitive function in individuals who have been exposed to natural measles virus in early childhood. Myoclonic seizures soon dominate the clinical picture. Later, generalized tonic-clonic convulsions, hypertonia, and choreoathetosis become evident, followed by progressive bulbar palsy, hyperthermia, and decerebrate postures. Funduscopic examination early in the course of the disease reveals papilledema in approximately 20% of the cases. Optic atrophy, chorioretinitis, and macular pigmentation are observed in most patients. The diagnosis is established by the typical clinical course and 1 of the following: (1) measles antibody detected in the CSF, (2) a characteristic electroencephalogram consisting of bursts of high-voltage slow waves interspersed with a normal background that occur with a constant periodicity in the early stages of the disease, and (3) typical histologic findings in the brain biopsy or postmortem specimen. Treatment with a series of antiviral agents has been attempted without success. Death occurs usually within 1-2 yr from the onset of symptoms.
Boelens JJ. Trends in haematopoietic cell transplantation for inborn errors of metabolism. J Inherit Metab Dis. 2006;29:413-420.
Campbell H, Andrews N, Brown KE, et al. Review of the effect of measles vaccination on the epidemiology of SSPE. Int J Epidemiol. 2007;36:1334-1348.
Cartier N, Hacein-Bey-Abina S, Bartholomae CC, et al. Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science. 2009;326:818-823.
Chahrour M, Zoghbi HY. The story of Rett syndrome: from clinic to neurobiology. Neuron. 2007;56:422-437.
Christodoulou J, Ho G. MECP2-related disorders (website). http://www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=gene&part=rett. Accessed November 17, 2010
Fluharty AL. Arylsulfatase A deficiency (website). http://www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=gene&part=mld. Accessed November 17, 2010
Garbern JY. Pelizaeus-Merzbacher disease: pathogenic mechanisms and insights into the roles of proteolipid protein 1 in the nervous system. J Neurol Sci. 2005;228:201.
Garbern JY, Krajewski K, Hobson G. PLP1-related disorders (website). http://www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=gene&part=pmd. Accessed November 17, 2010
GeneReviews at GeneTests. Medical genetics information resource (database online). Copyright, University of Washington, Seattle. 1997–2009 http://www.genetests.org Accessed May 1, 2009
Gorospe JR. Alexander disease (website). http://www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=gene&part=alexander. Accessed November 17, 2010
Henneke M, Combes P, Diekmann S, et al. GJA12 mutations are a rare cause of Pelizaeus-Merzbacher-like disease. Neurology. 2008;70:748-754.
Heese BA. Current strategies in the management of lysosomal storage diseases. Semin Pediatr Neurol. 2008;15:119-126.
Jalanko A, Braulke T. Neuronal ceroid lipofuscinoses. Biochim Biophys Acta. 2009;1793:697-709.
Kaler SG. ATP7A-related copper transport disorders (website). http://www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=gene&part=menkes. Accessed November 17, 2010
Kaler SG, Holmes CS, Goldstein DS, et al. Neonatal diagnosis and treatment of Menkes disease. N Engl J Med. 2008;358:605-614.
Kohlschütter A, Schulz A. Towards understanding the neuronal ceroid lipofuscinoses. Brain Dev. 2009;31:499-502.
Mahmood A, Dubey P, Moser HW, et al. X-linked adrenoleukodystrophy: therapeutic approaches to distinct phenotypes. Pediatr Transplant. 2005;9(Suppl 7):55-62.
Moser HW. Therapy of X-linked adrenoleukodystrophy. NeuroRx. 2006;3:246-253.
Sawaishi Y. Review of Alexander disease: beyond the classical concept of leukodystrophy. Brain Dev. 2009;31:493-498.
Shahwan A, Farrell M, Delanty N. Progressive myoclonic epilepsies: a review of genetic and therapeutic aspects. Lancet Neurol. 2005;4:239-248.
Staretz-Chacham O, Lang TC, LaMarca ME, et al. Lysosomal storate disorders in the newborn. Pediatrics. 2009;123:1191-1207.
Tay G, Graham H, Graham HK, et al. Hip displacement and scoliosis in Rett syndrome—screening required. Dev Med Child Neurol. 2010;52:93-98.
Verity C, Winstone AM, Stellitano L, et al. The epidemiology of progressive intellectual and neurological deterioration in childhood. Arch Dis Child. 2001;95:361-364.
Vermeulen G, Seidl R, Mercimek-Mahmutoglu S, et al. Fright is a provoking factor in vanishing white matter disease. Ann Neurol. 2005;57:560-563.